
cie komôr. Neboli hlásené žiadne prípady náhlej smrti u novodiagnostikovaných pacientov
s CML v chronickej fáze v klinickej štúdii fázy III.
Retencia tekutín a edém
U novodiagnostikovaných pacientov s CML sa v klinickej štúdii fázy III menej často (0,1 až 1%)
pozorovali ťažké formy retencie tekutín ako pleurálny výpotok, pľúcny edém a perikardiálny výpotok. Podobné udalosti sa zaznamenali aj z hlásení po uvedení lieku na trh. Nečakaný a rýchly nárast
telesnej hmotnosti musí byť pozorne vyšetrený. Ak sa počas liečby nilotinibom objavia prejavy ťažkej
retencie tekutín, musí byť zhodnotená etiológia a pacienti majú byť náležite liečení (pozri časť 4.2,
pokyny k zvládaniu nehematologických toxicít).
Srdcovocievne príhody
U novodiagnostikovaných pacientov s CML v klinickej štúdii fázy III a z hlásení po uvedení lieku na
trh boli zaznamenané srdcovocievne príhody. V tomto klinickom skúšaní s mediánom trvania liečby
60,5 mesiacov sa vyskytli srdcovocievne príhody 3. až 4. stupňa vrátane periférnej arteriálnej
okluzívnej choroby (1,4% pri 300 mg a 1,1% pri 400 mg nilotinibu dvakrát denne), ischemická choroba srdca (2,2% pri 300 mg a 6,1% pri 400 mg nilotinibu dvakrát denne) a ischemické mozgovocievne príhody (1,1% pri 300 mg a 2,2% pri 400 mg nilotinibu dvakrát denne). Pacienti majú byť poučení, aby v prípade výskytu akútnych prejavov a príznakov srdcovocievnych príhod okamžite vyhľadali lekársku pomoc. Srdcovocievny stav pacienta musí byť zhodnotený a počas liečby nilotinibom majú byť podľa platných štandardných odporúčaní sledované a aktívne manažované srdcovocievne rizikové faktory. Na kontrolu srdcovocievnych rizík má byť predpísaná vhodná liečba (pozri časť 4.2, pokyny k zvládaniu nehematologických toxicít).
Reaktivácia hepatitídy B
Reaktivácia hepatitídy B u pacientov, ktorí sú chronickými prenášačmi tohto vírusu, sa vyskytla v
prípade, že títo pacienti užívali inhibítory BCR-ABL-tyrozínkinázy. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie.
Pacienti majú byť vyšetrení na HBV infekciu pred začatím liečby nilotinibom. Pred začatím liečby u pacientov s pozitívnym sérologickým testom na hepatitídu B (vrátane pacientov s aktívnym ochorením) a u pacientov s pozitívnym testom na HBV infekciu počas liečby je potrebné konzultovať s odborníkmi na ochorenia pečene a liečbu hepatitídy B. Prenášači vírusu HBV, ktorí potrebujú liečbu nilotinibom, majú byť pozorne sledovaní na prejavy a symptómy aktívnej HBV infekcie počas celej liečby a niekoľko mesiacov po ukončení liečby (pozri časť 4.8).
Osobitné monitorovanie pacientov s Ph+ CML v chronickej fáze, ktorí dosiahli trvalú hlbokú
molekulovú odpoveď
Vhodnosť na ukončenie li ečby
Pacienti, u ktorých sa potvrdila expresia typických BCR-ABL transkriptov, e13a2/b2a2 alebo
e14a2/b3a2, sa môžu považovať za vhodných na ukončenie liečby. Pacienti musia mať typické
BCR-ABL transkripty, ktoré umožnia kvantifikáciu BCR-ABL, vyhodnotenie hĺbky molekulovej
odpovede a stanovenie prípadnej straty molekulovej remisie po prerušení liečby nilotinibom.
Monitorovanie pacientov, kt orí ukončil i l ieč bu
U pacientov vhodných na ukončenie liečby sa musí vykonávať časté monitorovanie hladín BCR-ABL transkriptov spolu s kvantitatívnym diagnostickým testom validovaným na meranie hladín molekulovej odpovede s citlivosťou najmenej MR4,5 (BCR-ABL/ABL ≤0,0032% IS). Hladiny
BCR-ABL transkriptov sa musia stanoviť pred a počas ukončenia liečby (pozri časti 4.2 a 5.1).
Strata veľkej molekulovej odpovede (MMR=BCR-ABL/ABL ≤0,1% IS) alebo potvrdená strata MR4 (dve po sebe idúce merania s odstupom aspoň 4 týždne ukazujúce stratu MR4 (MR4=BCR-ABL/ABL
≤0,01% IS)) bude impulzom pre opätovné začatie liečby v priebehu 4 týždňov od doby, odkedy je známe, že došlo k strate remisie. Molekulový relaps sa môže vyskytnúť počas obdobia bez liečby
a údaje o dlhodobých výsledkoch ešte nie sú k dispozícii. Veľmi dôležité je preto časté monitorovanie
hladín BCR-ABL transkriptov a úplného krvného obrazu s diferenciálom, aby sa zistila prípadná strata
remisie (pozri časť 4.2). U pacientov, ktorí nedosiahnu MMR po troch mesiacoch od opätovného
začatia liečby, sa má vykonať test na mutáciu kinázy BCR-ABL.
Laboratórne testy a monitoring
Lipidy v krvi
V klinickom skúšaní fázy III u novo diagnostikovaných pacientov s CML sa u 1,1% pacientov
liečených 400 mg nilotinibu dvakrát denne vyskytlo zvýšenie celkového cholesterolu stupňa 3-4;
v skupine s dávkovaním 300 mg dvakrát denne však nebolo pozorované žiadne zvýšenie stupňa 3-4 (pozri časť 4.8). Pred začatím liečby nilotinibom, po 3 a 6 mesiacoch od začatia liečby a najmenej raz ročne pri dlhodobej liečbe sa odporúča stanoviť profily lipidov (pozri časť 4.2). Ak je potrebné podanie inhibítorov HMG-CoA reduktázy (liečiv znižujúcich hladinu lipidov), treba sa pred začatím liečby riadiť podľa časti 4.5, keďže určité inhibítory HMG-CoA reduktázy sú tiež metabolizované prostredníctvom CYP3A4.
Glukóza v krvi
V klinickom skúšaní fázy III u novo diagnostikovaných pacientov s CML sa u 6,9% pacientov
liečených 400 mg nilotinibu dvakrát denne a u 7,2% pacientov liečených 300 mg nilotinibu dvakrát
denne vyskytlo zvýšenie glukózy v krvi stupňa 3-4. Pred začatím liečby Tasignou sa odporúča stanoviť hladinu glukózy a podľa klinickej potreby ju monitorovať počas liečby (pozri časť 4.2). Ak výsledky testov oprávňujú liečbu, lekári sa majú riadiť miestnymi normami pre prax a smernicami pre liečbu.
I
nterakcie s inými liekmi
Je potrebné vyhnúť sa podávaniu Tasigny s látkami, ktoré sú silnými inhibítormi CYP3A4 (vrátane,
ale nielen s ketokonazolom, itrakonazolom, vorikonazolom, klaritromycínom, telitromycínom, ritonavirom). Ak sa vyžaduje liečba niektorou z uvedených látok, odporúča sa podľa možnosti prerušiť liečbu nilotinibom (pozri časť 4.5). Ak krátkodobé prerušenie liečby nie je možné, je
indikované dôsledné monitorovanie pacienta vzhľadom na predĺženie intervalu QT (pozri časti 4.2, 4.5
a 5.2).
Súčasné užívanie nilotinibu a liekov, ktoré sú silné induktory CYP3A4 (napr. fenytoín, rifampicín,
karbamazepín, fenobarbital a ľubovník bodkovaný), pravdepodobne zníži expozíciu nilotinibu
v klinicky významnej miere. Preto u pacientov, ktorí užívajú nilotinib, je potrebné zvoliť na súbežné podávanie alternatívne liečivá s nižším potenciálom indukovať CYP3A4 (pozri časť 4.5).
Vplyv jedla
Jedlo zvyšuje biologickú dostupnosť nilotinibu. Tasigna sa nesmie užívať spolu s jedlom (pozri časti
4.2 a 4.5) a má sa užívať 2 hodiny po jedle. Najmenej jednu hodinu po užití dávky sa nemá požiť žiadne jedlo. Je potrebné vyhýbať sa grapefruitovej šťave a iným jedlám, o ktorých je známe, že
inhibujú CYP3A4. Pre pacientov, ktorí nie sú schopní prehĺtať tvrdé kapsuly, obsah každej tvrdej
kapsuly možno zmiešať s jednou čajovou lyžičkou jablčného pyré a ihneď ho užiť. Nesmie sa použiť viac než jedna lyžička jablčného pyré a nesmie sa požiť iné jedlo než jablčné pyré (pozri časť 5.2).
Porucha funkciepečene
Porucha funkcie pečene má malý vplyv na farmakokinetiku nilotinibu. Podanie jednorazovej dávky
200 mg nilotinibu spôsobilo u osôb s ľahkou, stredne ťažkou a ťažkou poruchou funkcie pečene zväčšenie AUC o 35%, 35% a 19% v porovnaní s kontrolnou skupinou osôb s normálnou funkciou
pečene. Predpokladaná Cmax nilotinibu v rovnovážnom stave sa zvýšila o 29%, 18% a 22%. Z klinických skúšaní boli vylúčení pacienti so zvýšením alanínaminotransferázy (ALT) a/alebo aspartátaminotransferázy (AST) na >2,5-násobok (alebo na >5-násobok, ak súviselo s ochorením)
hornej hranice normálneho rozmedzia a/alebo celkového bilirubínu na >1,5-násobok hornej hranice normálneho rozmedzia. Metabolizmus nilotinibu prebieha prevažne v pečeni. U pacientov s poruchou funkcie pečene sa preto môže zvýšiť expozícia nilotinibu a pri ich liečbe je potrebná opatrnosť (pozri časť 4.2).
Sérová lipáza
Pozorovalo sa zvýšenie lipázy v sére. U pacientov s pankreatitídou v anamnéze sa odporúča opatrnosť.
Keď zvýšenie lipázy sprevádzajú abdominálne príznaky, liečba nilotinibom sa má vysadiť a majú sa
zvážiť vhodné diagnostické postupy na vylúčenie pankreatitídy.
Totálna gastrektómia
Biologická dostupnosť nilotinibu môže byť znížená u pacientov s totálnou gastrektómiou (pozri časť
5.2). U týchto pacientov sa majú zvážiť častejšie následné vyšetrenia.
Syndróm z rozpadu nádoru
Vzhľadom na možný výskyt syndrómu z rozpadu nádoru (TLS) sa pred začatím liečby nilotinibom
odporúča úprava klinicky významnej dehydratácie a liečba vysokých hladín kyseliny močovej (pozri časť 4.8).
Laktóza
Tvrdé kapsuly Tasigny obsahujú laktózu. Pacienti so zriedkavými dedičnými problémami
galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie
nesmú užívať tento liek.
Pediatrická populácia
Laboratórne abnormality vo forme ľahkých až stredne závažných prechodných zvýšení
aminotransferáz a celkového bilirubínu sa u detí pozorovali s vyššou frekvenciou ako u dospelých, čo naznačuje zvýšené riziko hepatotoxicity v pediatrickej populácii (pozri časť 4.8). Funkcia pečene
(hladiny bilirubínu a pečeňových aminotransferáz) sa má monitorovať raz za mesiac alebo podľa
klinickej indikácie. Zvýšenie bilirubínu a pečeňových aminotransferáz sa má manažovať dočasným prerušením podávania nilotinibu, znížením dávky a/alebo ukončením podávania nilotinibu (pozri
časť 4.2). Dlhodobé následky dlhotrvajúcej liečby nilotinibom u detí a dospievajúcich nie sú známe.
4.5 Liekové a iné interakcie
Tasigna sa môže podávať v kombinácii s hematopoetickými rastovými faktormi, ako sú erytropoetín alebo faktor stimulujúci kolónie granulocytov (G-CSF), ak je to klinicky indikované. Môže sa podávať s hydroxyureou alebo anagrelidom, ak je to klinicky indikované.
Nilotinib sa metabolizuje prevažne v pečeni a je tiež substrátom glykoproteínu P (Pg-p), efluxnej pumpy mnohých liečiv. Preto absorpciu a následnú elimináciu systémovo absorbovaného nilotinibu môžu ovplyvňovať látky, ktoré pôsobia na CYP3A4 a/alebo Pg-p.
Látky, ktoré môžuzvýšiťsérové koncentrácie nilotinibu
Súčasné podávanie nilotinibu s imatinibom (substrát a moderátor P-gp a CYP3A4) malo slabý
inhibičný účinok na CYP3A4 a/alebo P-gp. AUC imatinibu sa zvýšila o 18% až 39% a AUC
nilotinibu sa zvýšila o 18% až 40%. Nie je pravdepodobné, že tieto zmeny sú klinicky významné.
Expozícia nilotinibu u zdravých osôb vzrástla 3-násobne, keď sa podával spolu so silným inhibítorom CYP3A4 ketokonazolom. Preto je potrebné vyhýbať sa súbežnej liečbe silnými inhibítormi CYP3A4 vrátane ketokonazolu, itrakonazolu, vorikonazolu, ritonaviru, klaritromycínu a telitromycínu (pozri časť 4.4). Zvýšenú expozíciu nilotinibu možno očakávať aj pri stredne silných inhibítoroch CYP3A4. Majú sa zvážiť alternatívne súčasne podávané lieky, ktoré neinhibujú alebo len minimálne inhibujú CYP3A4.
Látky, ktoré môžuznížiťsérové koncentrácie nilotinibu
Rifampicín, silný induktor CYP3A4, znižuje Cmax nilotinibu o 64% a zmenšuje AUC nilotinibu o 80%.
Rifampicín a nilotinib sa nemajú používať súčasne.
Súčasné podávanie iných liekov, ktoré indukujú CYP3A4 (napr. fenytoín, karbamazepín, fenobarbital a ľubovník bodkovaný), pravdepodobne tiež zníži expozíciu nilotinibu v klinicky významnej miere.
U pacientov, ktorí majú indikovanú liečbu induktormi CYP3A4, sa majú zvoliť alternatívne látky
s menším potenciálom pre enzýmovú indukciu.
Rozpustnosť nilotinibu závisí od pH, pričom rozpustnosť je nižšia pri vyššom pH. U zdravých osôb, ktoré dostávali 40 mg ezomeprazolu raz denne počas 5 dní, sa pH žalúdka výrazne zvýšilo, ale absorpcia nilotinibu sa len mierne znížila (pokles Cmax o 27% a pokles AUC0-∞ o 34%). Nilotinib možno podľa potreby používať súčasne s ezomeprazolom alebo inými inhibítormi protónovej pumpy.
V štúdii u zdravých subjektov nebola po podaní jednorazovej dávky 400 mg nilotinibu 10 hodín po a 2 hodiny pred famotidínom pozorovaná žiadna významná zmena vo farmakokinetike nilotinibu.
V prípade, že je potrebná súčasná liečba H2 blokátormi, možno ich podať približne 10 hodín pred a 2 hodiny po podaní Tasigny.
Vo vyššie uvedenej štúdii nemalo podanie antacida (hydroxid hlinitý/hydroxid horečnatý/simetikón)
2 hodiny pred alebo 2 hodiny po podaní jednorazovej dávky 400 mg nilotinibu taktiež vplyv na
farmakokinetiku nilotinibu. V prípade potreby možno podať antacidá približne 2 hodiny po alebo
2 hodiny pred podaním Tasigny.
Látky, ktorých systémovékoncentráciemôžezmeniťnilotinib
In vitro je nilotinib pomerne silný inhibítor CYP3A4, CYP2C8, CYP2C9, CYP2D6 a UGT1A1,
s hodnotou Ki najnižšou pre CYP2C9 (Ki=0,13 mikroM).
Štúdia liekových interakcií u zdravých dobrovoľníkov pri jednorazovom podaní 25 mg warfarínu, citlivého substrátu CYP2C9, a 800 mg nilotinibu nemala za následok žiadne zmeny farmakokinetických parametrov warfarínu alebo farmakodynamiky warfarínu, stanovených ako protrombínový čas (PT) a medzinárodný normalizovaný pomer (INR). Nie sú údaje o rovnovážnom stave. Táto štúdia naznačuje, že klinicky významná lieková interakcia medzi nilotinibom a warfarínom je menej pravdepodobná do dávky 25 mg warfarínu. Vzhľadom na chýbajúce údaje pri rovnovážnom stave sa odporúča kontrola farmakodynamických markerov warfarínu (INR alebo PT) po začatí liečby nilotinibom (najmenej počas prvých 2 týždňov).
U pacientov s CML nilotinib podávaný v dávke 400 mg dvakrát denne po dobu 12 dní zvýšil 2,6 resp.
2-násobne systémovú expozíciu (AUC resp. Cmax) perorálne podaného midazolamu (substrát CYP3A4). Nilotinib je stredne silný inhibítor CYP3A4. Pri súčasnom podávaní spolu s nilotinibom preto môže byť systémová expozícia iných liekov primárne metabolizovaných cez CYP3A4 (napr. určité inhibítory HMG-CoA reduktázy) zvýšená. U liekov s úzkym terapeutickým indexom, ktoré sú substrátmi CYP3A4 (vrátane alfentanilu, cyklosporínu, dihydroergotamínu, ergotamínu, fentanylu, sirolimu a takrolimu ale nevynímajúc aj iné lieky), môže byť potrebný monitoring a úprava dávky, pokiaľ sa podávajú spoločne s nilotinibom.
Antiarytmiká a iné látky,ktorémôžupredlžovaťQTinterval
Nilotinib sa má používať opatrne u pacientov, ktorí majú alebo u ktorých sa môže vyvinúť predĺženie
QT intervalu, vrátane pacientov užívajúcich antiarytmiká ako amiodaron, disopyramid, prokaínamid,
chinidín a sotalol, alebo iné liečivá, ktoré spôsobujú predĺženie QT ako chlorochín, halofantrín,
klaritromycín, haloperidol, metadón a moxifloxacín (pozri časť 4.4).
Interakcie s jedlom
Absorpcia a biologická dostupnosť nilotinibu sa zvýšia, ak sa užíva s jedlom, čo má za následok jej
vyššie sérové koncentrácie (pozri časti 4.2, 4.4 a 5.2). Je potrebné vyhýbať sa grapefruitovej šťave
a iným jedlám, o ktorých je známe, že inhibujú CYP3A4.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ž
eny vofertilnomveku/Antikoncepcia
Ženy vo fertilnom veku musia používať vysoko účinnú antikoncepciu počas liečby nilotinibom a po
dobu do dvoch týždňov od ukončenia liečby.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití nilotinibu u gravidných žien.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Tasigna sa nemá užívať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu nilotinibom. Ak sa užíva počas gravidity, pacientka musí byť informovaná o prípadnom riziku pre plod.
Ak žena, ktorá sa lieči nilotinibom, uvažuje o gravidite, možno zvážiť prerušenie liečby na základe kritérií vhodnosti pre prerušenie liečby, ako sú opísané v častiach 4.2 a 4.4. Existuje obmedzené množstvo údajov o graviditách u pacientok počas pokusu o remisiu bez liečby (TFR, treatment-free remission). Ak sa plánuje gravidita počas fázy TFR, pacientka musí byť informovaná o možnej nutnosti opätovného začatia liečby nilotinibom počas gravidity (pozri časti 4.2 a 4.4).
Dojčenie
Nie je známe, či sa nilotinib vylučuje do ľudského mlieka. Dostupné toxikologické údaje u zvierat
preukázali vylučovanie nilotinibu do mlieka (pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť vylúčené. Tasigna nemá byť užívaná počas dojčenia.
Fertilita
Štúdie na zvieratách neukázali účinok na fertilitu samcov a samíc potkana (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tasigna nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Napriek tomu sa odporúča, aby pacienti, u ktorých sa vyskytnú závraty, únava, zhoršenie zraku alebo iné nežiaduce účinky, ktoré môžu ovplyvňovať schopnosť bezpečne viesť vozidlá alebo obsluhovať stroje, nevykonávali tieto činnosti, kým nežiaduce účinky pretrvávajú (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Údaje uvedené nižšie sú získané pri použití nilotinibu u celkovo u 717 dospelých pacientov
v randomizovanej klinickej štúdii fázy III s pacientmi s novodiagnostikovanou Ph+ CML v chronickej fáze, ktorí dostávali odporúčanú dávku 300 mg dvakrát denne (n=279), a v otvorenej multicentrickej štúdii fázy II s dospelými pacientmi s CML s rezistenciou alebo intoleranciou voči imatinibu
v chronickej fáze (n=321) a v akcelerovanej fáze (n=137), ktorí dostávali odporúčanú dávku 400 mg dvakrát denne. K dispozícií sú aj údaje o bezpečnosti z dvoch štúdií s Tasignou o ukončení liečby.
U dospelých pacientov s novodiagnostikovanou CML v chronickej fáze
Medián trvania expozície bol 60,5 mesiacov (rozmedzie 0,1-70,8 mesiacov).
Najčastejšími (≥10%) nehematologickými nežiaducimi reakciami boli exantém, pruritus, bolesť hlavy, nauzea, únava, alopécia, myalgia a bolesť v hornej časti brucha. Väčšina z týchto nežiaducich reakcií bola mierna až stredne závažná. Zápcha, suchosť kože, asténia, svalové kŕče, hnačka, artralgia, bolesť brucha, vracanie a periférny edém sa pozorovali menej často (<10% a ≥5%), boli mierne až stredne závažné, zvládnuteľné a spravidla si nevyžiadali zníženie dávky.
K príznakom hematologickej toxicity, ktoré sa objavili pri liečbe, patrí myelosupresia: trombocytopénia (18%), neutropénia (15%) a anémia (8%). Biochemické reakcie na liek zahŕňajú zvýšenú alaníntransaminázu (24%), hyperbilirubinémiu (16%), zvýšenú aspartáttransaminázu (12%), zvýšenú lipázu (11%), zvýšený bilirubín v krvi (10%), hyperglyklémiu (4%), hypercholesterolémiu (3%) a hypertriacylglycerolémiu (<1%). Pleurálny a perikardiálny výpotok sa bez ohľadu na príčinnú súvislosť vyskytli u 2% a <1% pacientov v uvedenom poradí, ktorí dostávali nilotinib 300 mg dvakrát denne. Gastrointestinálne krvácanie bez ohľadu na príčinnú súvislosť bolo hlásené u 3% z týchto pacientov.
Zmena stredného časovo spriemerovaného intervalu QTcF v rovnovážnom stave oproti východiskovej hodnote bola 6 ms. Ani u jedného pacienta sa počas liečby skúšaným liekom nevyskytla absolútna hodnota QTcF >500 ms. Predĺženie QTcF oproti východiskovej hodnote väčšie ako 60 ms sa počas liečby skúšaným liekom pozorovalo u <1% pacientov. Nezaznamenala sa náhla smrť alebo epizódy torsade de pointes (prechodné alebo pretrvávajúce). Nepozoroval sa pokles priemernej ejekčnej frakcie ľavej komory (LVEF) oproti východiskovej hodnote kedykoľvek počas liečby. Žiadny pacient nemal počas liečby LVEF <45%, ani absolútny pokles LVEF o viac než 15%.
Ukončenie liečby pre nežiaduce reakcie na liek sa zaznamenalo u 10% pacientov.
U dospelých pacientov s CML s rezi stenci ou al ebo i ntol er anciou voči i mati ni bu v chronickej fázea akcelerovanej fáze
Údaje uvedené nižšie sú získané pri použití nilotinibu u 458 dospelých pacientov v otvorenom
multicentrickom klinickom skúšaní fázy II u pacientov s rezistenciou alebo intoleranciou voči imatinibu s CML v chronickej fáze (n=321) a v akcelerovanej fáze (n=137) liečených odporúčanou dávkou 400 mg dvakrát denne.
Najčastejšími (≥10%) nehematologickými nežiaducimi udalosťami súvisiacimi s liekom boli exantém, svrbenie, nauzea, únava, bolesť hlavy, vracanie, myalgia, zápcha a hnačka. Väčšina z týchto nežiaducich udalostí bola mierna až stredne závažná. Alopécia, svalové kŕče, znížená chuť do jedenia, artralgia, bolesť brucha, bolesť kostí, periférny edém, asténia, bolesť v hornej časti brucha, suchosť kože, erytém a bolesť v končatine sa pozorovali menej často (<10% a ≥5%) a boli mierne až stredne závažné (stupeň 1 alebo 2). Ukončenie liečby pre nežiaduce reakcie na liek sa zaznamenalo u 16% pacientov v chronickej fáze a u 10% pacientov v akcelerovanej fáze.
K príznakom hematologickej toxicity, ktoré sa objavili pri liečbe, patrí myelosupresia: trombocytopénia (31%), neutropénia (17%) a anémia (14%). Pleurálny a perikardiálny výpotok, ako aj komplikácie spôsobené zadržiavaním tekutín sa vyskytli u <1% pacientov užívajúcich Tasignu. Zlyhávanie srdca sa pozorovalo u <1% pacientov. Gastrointestinálne krvácanie bolo hlásené u 1%
a krvácanie do CNS u <1% pacientov.
QTcF dlhší ako 500 ms sa pozoroval u <1% pacientov. Nepozorovali sa žiadne epizódy torsade de
pointes (prechodné alebo pretrvávajúce).
Tabuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie sú zoradené podľa frekvencie na základe nasledujúcej konvencie: veľmi časté
(≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až
<1/1 000), veľmi zriedkavé (<1/10 000) a neznáme (z dostupných údajov). V rámci jednotlivých
skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Než i aduce re akci e najčast ej šie hláse né v kli nick ých s kúšaniach s Tasignou
Nehematologické nežiaduce reakcie (okrem laboratórnych abnormalít), ktoré sú hlásené najmenej u
5% dospelých pacientov v klinických skúšaniach Tasigny, ktoré slúžia ako základ pre schválené indikácie, ukazuje Tabuľka 3.
Tabuľka 3 Nehematologické nežiaduce reakcie (≥5% všetkých pacientov)*
N
ovodiagnostikovaná
C
ML−CP
300 mg dvakrát denne n=279
C
ML–CP a CML−AP s rezistenciou alebo
i
ntoleranciou voči imatinibu
400 mg dvakrát denne n=458
A
nalýza po 60 mesiacoch Analýza po 24 mesiacoch
C
ML-
C
ML-
O
rgánový systém/ Nežiaduca reakcia
Frekvencia Všetky
stupne
Stupeň
3-4
Frekvencia Všetky
stupne
Stupeň
3-4
CP
n=321
Stupeň
3-4
AP
n=137
Stupeň
3-4
P
oruchy metabolizmu a výživy
% % % % % %
Znížená chuť
do jedenia **
Časté 4 0 Časté 8 <1 <1 0
P
oruchy nervového systému
Bolesť hlavy Veľmi časté 16 2 Veľmi časté 15 1 2 <1
Poruchy gastrointestinálneho traktuNauzea Veľmi časté 14 <1 Veľmi časté 20 <1 <1 <1
Zápcha Časté 10 0 Veľmi časté 12 <1 <1 0
Hnačka Časté 9 <1 Veľmi časté 11 2 2 <1

Vracanie Časté 6 0 Veľmi časté 10 <1 <1 0
Bolesť hornej časti brucha
Veľmi časté 10 1 Časté 5 <1 <1 0
Bolesť brucha Časté 6 0 Časté 6 <1 <1 <1
Dyspepsia Časté 5 0 Časté 3 0 0 0
Poruchy kože a podkožného tkaniva
Exantém Veľmi časté 33 <1 Veľmi časté 28 1 2 0
Pruritus Veľmi časté 18 <1 Veľmi časté 24 <1 <1 0
Alopécia Veľmi časté 10 0 Časté 9 0 0 0
Suchosť kože Časté 10 0 Časté 5 0 0 0
Erytém Časté 3 0 Časté 5 <1 <1 0
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Myalgia Veľmi časté 10 <1 Veľmi časté 10 <1 <1 <1
Svalové kŕče Časté 9 0 Časté 8 <1 <1 0
Artralgia Časté 8 <1 Časté 7 <1 1 0
Bolesť kostí Časté 4 0 Časté 6 <1 <1 0
Bolesť končatín Časté 5 <1 Časté 5 <1 <1 <1
Celkové poruchy a reakcie v mieste podania
Únava Veľmi časté 12 0 Veľmi časté 17 1 1 <1
Asténia Časté 9 <1 Časté 6 <1 0 <1
Periférny edém Časté 5 0 Časté 6 0 0 0
* Za účelom uvedenia v tejto tabuľke sú percentá zaokrúhlené na celé čísla. Pre určenie termínov
s frekvenciou aspoň 5% a klasifikáciu termínov podľa kategórie frekvencií sa však používajú percentá
s presnosťou jedného desatinného miesta.
** Zahŕňa aj preferované označenie anorexia
Nasledujúce nežiaduce reakcie boli hlásené u dospelých pacientov v klinických štúdiách Tasigny, ktoré slúžia ako základ pre schválené indikácie, s frekvenciou nižšou ako 5%. Pri laboratórnych abnormalitách sú hlásené aj veľmi časté nežiaduce reakcie, ktoré nie sú zahrnuté do Tabuľky 3. Tieto nežiaduce reakcie sú uvedené podľa klinickej významnosti.
Infekcie a nákazy
Časté: folikulitída, infekcia horných dýchacích ciest (vrátane faryngitídy, nazofaryngitídy, nádchy). Menej časté: pneumónia, infekcia močových ciest, gastroenteritída, bronchitída, infekcia herpes
simplex, kandidóza (vrátane orálnej kandidózy).
Neznáma frekvencia: sepsa, subkutánny absces, análny absces, furunkul, tinea pedis, reaktivácia hepatitídy B.
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Časté: kožný papilóm.
Neznáma frekvencia: papilóm ústnej dutiny, paraproteinémia.
Poruchy krvi a lymfatického systému
Časté: leukopénia, eozinofília, febrilná neutropénia, pancytopénia, lymfopénia. Menej časté: trombocytémia, leukocytóza.
Poruchy imunitného systému
Neznáma frekvencia: hypersenzitivita.
Poruchy endokrinného systému
Menej časté: hypertyreóza, hypotyreóza.
Neznáma frekvencia: sekundárny hyperparatyreoidizmus, tyreoiditída.
Poruchy metabolizmu a výživy
Veľmi časté: hypofosfatémia (vrátane zníženia fosforu v krvi).
Časté: nerovnováha elektrolytov (vrátane hypomagneziémie, hyperkaliémie, hypokaliémie, hyponatriémie, hypokalciémie, hyperkalciémie, hyperfosfatémie), diabetes mellitus, hyperglykémia,
hypercholesterolémia, hyperlipidémia, hypertriacylglycerolémia.
Menej časté: dehydratácia, zvýšená chuť do jedenia, dna, dyslipidémia.
Neznáma frekvencia: hyperurikémia, hypoglykémia.
Psychické poruchy
Časté: depresia, nespavosť, úzkosť.
Neznáma frekvencia: dezorientácia, zmätenosť, amnézia, dysfória.
Poruchy nervového systému
Časté: závraty, periférna neuropatia, hypestézia, parestézia.
Menej časté: intrakraniálne krvácanie, ischemická mozgová príhoda, prechodný ischemický atak, mozgový infarkt, migréna, strata vedomia (vrátane synkopy), tremor, poruchy pozornosti,
hyperestézia.
Neznáma frekvencia: mozgovocievne príhody,, edém mozgu, zápal zrakového nervu, letargia, dyzestézia, syndróm nepokojných nôh.
Poruchy oka
Časté: krvácanie do oka, periorbitálny edém, očný pruritus, konjunktivitída, suchosť očí (vrátane
xeroftalmie).
Menej časté: zhoršenie zraku, neostré videnie, krvácanie do spojoviek, znížená zraková ostrosť, edém
mihalnice, fotopsia, hyperémia (skléry, spojoviek, očí), podráždenie očí.
Neznáma frekvencia: edém papily zrakového nervu, chorioretinopatia, diplopia, fotofóbia, opuch očí, blefaritída, bolesť očí, alergická konjunktivitída, ochorenie povrchu oka.
Poruchy ucha a labyrintu
Časté: vertigo.
Neznáma frekvencia: zhoršenie sluchu, bolesť ucha, tinnitus.
Poruchy srdca a srdcovej činnosti
Časté: angina pectoris, arytmia (zahŕňa átrioventrikulárnu blokádu, srdcový flutter, extrasystoly, tachykardiu, fibriláciu predsiení, bradykardiu), palpitácie, predĺženie QT na elektrokardiograme.
Menej časté: zlyhávanie srdca, infarkt myokardu, ischemická choroba srdca, srdcový šelest,
perikardiálny výpotok, cyanóza.
Neznáma frekvencia: dysfunkcia komôr, perikarditída, pokles ejekčnej frakcie.
Poruchy ciev
Časté: hypertenzia, návaly tepla, periférna arteriálna stenóza.
Menej časté: hypertenzná kríza, periférna arteriálna okluzívna choroba, intermitentná klaudikácia, stenóza končatinových tepien, hematóm, arterioskleróza.
Neznáma frekvencia: hemoragický šok, hypotenzia, trombóza.
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté: dyspnoe, námahové dyspnoe, epistaxa, kašeľ, dysfónia.
Menej časté: pľúcny edém, pleurálny výpotok, intersticiálna choroba pľúc, bolesť pohrudnice, pleuritída, faryngolaryngálna bolesť, podráždenie hrdla.
Neznáma frekvencia: pľúcna hypertenzia, sipot, orofaryngeálna bolesť.
Poruchy gastrointestinálneho traktu
Časté: pankreatitída, nepríjemné pocity v bruchu, distenzia brucha, dysgeúzia, flatulencia.
Menej časté: gastrointestinálne krvácanie, meléna, ulcerácia ústnej dutiny, gastroezofagálny reflux,
stomatitída, bolesť v ezofágu, suchosť v ústach, gastritída, citlivosť zubov.
Neznáma frekvencia: perforácia gastrointestinálneho vredu, retroperitoneálne krvácanie, hemateméza,
žalúdkový vred, ulcerózna ezofagitída, subileus, enterokolitída, hemoroidy, hiátová hernia, rektálne krvácanie, gingivitída.
Poruchy pečene a žlčových ciest
Veľmi časté: hyperbilirubinémia (vrátane zvýšenia bilirubínu v krvi). Časté: abnormálna funkcia pečene.
Menej časté: hepatotoxicita, toxická hepatitída, žltačka.
Neznáma frekvencia: cholestáza, hepatomegália.
Poruchy kože a podkožného tkaniva
Časté: nočné potenie, ekzém, urtikária, hyperhidróza, kontúzia, akné, dermatitída (vrátane alergickej,
exfoliatívnej a akneiformnej).
Menej časté: exfoliatívny exantém, lieková erupcia, bolestivosť kože, ekchymóza, opuch tváre.
Neznáma frekvencia: erythema multiforme, erythema nodosum, kožný vred, syndróm palmárno- plantárnej erytrodyzestézie, petechie, fotosenzitivita, pľuzgier, kožné cysty, sebaceózna hyperplázia, atrofia kože, zmena sfarbenia pokožky, exfoliácia kože, hyperpigmentácia kože, hypertrofia kože, hyperkeratóza, psoriáza.
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté: muskuloskeletálna bolesť hrudníka, muskuloskeletálna bolesť, bolesti chrbta, bolesť v boku,
bolesť šije, svalová slabosť.
Menej časté: muskuloskeletálna stuhnutosť, opuch kĺbov.
Neznáma frekvencia: artritída.
Poruchy obličiek a močových ciest
Časté: polakizúria.
Menej časté: dyzúria, nutkanie na močenie, noktúria.
Neznáma frekvencia: zlyhanie obličiek, hematúria, inkontinencia moču, chromatúria.
Poruchy reprodukčného systému a prsníkov
Menej časté: bolesť prsníkov, gynekomastia, erektilná dysfunkcia.
Neznáma frekvencia: stvrdnutie prsníkov, menorágia, opuch prsných bradaviek.
Celkové poruchy a reakcie v mieste podania
Časté: bolesť v hrudníku (vrátane bolesti v hrudníku nekardiálneho pôvodu), bolesť, pyrexia,
nepríjemné pocity v hrudníku, celková nevoľnosť.
Menej časté: edém tváre, gravitačný edém, ochorenie podobné chrípke, zimnica, pocit zmeny telesnej teploty (vrátane pocitu horúčavy, pocitu chladu).
Neznáma frekvencia: lokalizovaný edém.
Laboratórne a funkčné vyšetrenia
Veľmi časté: zvýšenie alanínaminotransferázy, zvýšenie aspartátaminotransferázy, zvýšenie lipázy, zvýšenie lipoproteínového cholesterolu (vrátane lipoproteínu s nízkou a vysokou hustotou), zvýšenie celkového cholesterolu, zvýšené triacylglyceroly.
Časté: zníženie hemoglobínu, zvýšenie amylázy v krvi, zvýšenie alkalickej fosfatázy v krvi, zvýšenie gamaglutamyltransferázy, zvýšenie kreatinínfosfokinázy v krvi, pokles telesnej hmotnosti, zvýšenie
telesnej hmotnosti, zvýšenie inzulínu v krvi, zníženie globulínov.
Menej časté: zvýšenie laktátdehydrogenázy v krvi, zníženie glukózy v krvi, zvýšenie močoviny v krvi.
Neznáma frekvencia: zvýšenie troponínu, zvýšenie nekonjugovaného bilirubínu v krvi, zníženie
inzulínu v krvi, zníženie C-peptidu pre inzulín, zvýšenie parathormónu v krvi.
Klinicky významné alebo závažné abnormality rutinných hematologických alebo biochemických laboratórnych hodnôt u dospelých pacientov sú uvedené v Tabuľke 4.
Tabuľka 4 Laboratórne abnormality stupňa 3-4*
N
ovodiagnostikovaná
C
ML-CP
300 mg dvakrát denne
C
ML-CP a CML-AP
s rezistenciou alebo intoleranciou voči
im
a
ti
n
i
bu
400 mg dvakrát denne
H
e
m
atologické parametre
Myelosupresia
n=279 (%)
CML-CP n=321 (%)
CML-AP n=137 (%)
- Neutropénia 12 31 42
- Trombocytopénia 10 30 42
- Anémia 4 11 27
Biochemické parametre- Zvýšený kreatinín 0 1 <1
- Zvýšená lipáza 9 18 18
- Zvýšená SGOT (AST) 1 3 2
- Zvýšená SGPT (ALT) 4 4 4
- Hypofosfatémia 7 17 15
- Zvýšený bilirubín (celkový) 4 7 9
- Zvýšená glukóza 7 12 6
- Zvýšený cholesterol (celkový) 0 ** **
- Zvýšené triacylglyceroly 0 ** **
*Za účelom uvedenia v tejto tabuľke sa používajú percentá s presnosťou jedného desatinného miesta, zaokrúhlené na celé číslo
**Údaje sa nezbierali
U
k
ončenie
li
e
čby
u pacientov s Ph+ CML v chronickej fáze, ktorí dosiahli trvalú hlbokú molekulovúodpoveďPo ukončení liečby nilotinibom v rámci pokusu dosiahnuť TFR môžu pacienti pocítiť
muskuloskeletálne symptómy častejšie ako pred ukončením liečby, napr. myalgiu, bolesť končatín, artralgiu, bolesť kostí, bolesť chrbtice alebo muskuloskeletálnu bolesť.
V klinickej štúdii fázy II u novodiagnostikovaných pacientov s Ph+ CML v chronickej fáze (N=190) boli muskuloskeletálne symptómy hlásené počas jedného roka od ukončenia liečby Tasignou u 24,7% oproti 16,3% počas predchádzajúceho roka pri liečbe nilotinibom.
V klinickej štúdii fázy II u pacientov s Ph+ CML v chronickej fáze liečených nilotinibom a predtým liečených imatinibom (N=126) boli muskuloskeletálne symptómy hlásené počas jedného roka od ukončenia liečby Tasignou u 42,1% oproti 14,3% počas predchádzajúceho roka pri liečbe nilotinibom.
Opis vybranýchnežiaducichreakcií Náhl a smrťMenej časté prípady (0,1 až 1%) náhlej smrti boli hlásené v klinických skúšaniach Tasigne a/alebo v programoch podávania z humanitárnych dôvodov u pacientov s CML s rezistenciou alebo intoleranciou voči imatinibu v chronickej fáze alebo akcelerovanej fáze, ktorí mali v anamnéze ochorenie srdca alebo významné srdcové rizikové faktory (pozri časť 4.4).
Reaktivácia hepatitídy BV súvislosti s inhibítormi BCR-ABL-tyrozínkinázy bola hlásená reaktivácia hepatitídy B. Niektoré
prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie (pozri časť 4.4).
Skúsenosti po uvedení na trhNasledujúce nežiaduce reakcie pochádzajú zo skúsenosti s Tasignou po uvedení na trh formou
spontánnych hlásení, prípadov publikovaných v literatúre, programov rozšíreného prístupu a z
klinických štúdií iných ako globálne klinické skúšania pre registráciu. Keďže tieto reakcie sú hlásené dobrovoľne u populácie neurčenej veľkosti, nie je vždy možné spoľahlivo odhadnúť ich frekvenciu alebo stanoviť príčinnú súvislosť s expozíciou nilotinibu.
Frekvencia zriedkavé: U pacientov liečených nilotinibom sa zaznamenali prípady syndrómu z rozpadu nádoru.
Pediatrická populáciaBezpečnosť nilotinibu u pediatrických pacientov (od 2 do <18 rokov) s CML s pozitívnym
chromozómom Philadelphia v chronickej fáze (n=69) sa skúmala v dvoch štúdiách (pozri časť 5.1). Frekvencia, typ a závažnosť nežiaducich reakcií pozorovaných u pediatrických pacientov sa vo všeobecnosti zhodovali s tými, ktoré sa pozorovali u dospelých, s výnimkou laboratórnych abnormalít hyperbilirubinémie (stupeň 3/4: 13,0%) a zvýšenia aminotransferáz (AST stupeň 3/4: 1,4%, ALT stupeň 3/4: 8,7%), ktoré boli hlásené častejšie ako u dospelých pacientov. Hladiny bilirubínu
a aminotrasferáz sa majú počas liečby monitorovať (pozri časti 4.2 a 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Zaznamenali sa ojedinelé správy o úmyselnom predávkovaní nilotinibom, keď sa nešpecifikovaný počet tvrdých kapsúl Tasigny požil v kombinácii s alkoholom a inými liekmi. Udalosti zahŕňali neutropéniu, vracanie a ospalosť. Zmeny EKG alebo hepatotoxicita neboli uvádzané. Podľa hlásení došlo následne k zotaveniu.
V prípade predávkovania má byť pacient pod dohľadom a má sa mu podať primeraná podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE08
Mechanizmus účinkuNilotinib je účinný inhibítor aktivity ABL-tyrozínkinázy onkoproteínu BCR-ABL v bunkových líniách
aj v primárnych leukemických bunkách s pozitívnym chromozómom Philadelphia. Látka sa viaže
s vysokou afinitou na väzbové miesto ATP takým spôsobom, že je účinným inhibítorom BCR-ABL divokého typu a zachováva si aktivitu voči 32/33 mutantných foriem BCR-ABL rezistentných voči imatinibu. V dôsledku tejto biochemickej aktivity nilotinib selektívne inhibuje proliferáciu a indukuje apoptózu bunkových línií a primárnych leukemických buniek s pozitívnym chromozómom Philadelphia od pacientov s CML. Na myšacích modeloch CML nilotinib ako jediná použitá látka po perorálnom podávaní znižuje nádorovú záťaž a predlžuje prežívanie.
Farmakodynamické účinkyNilotinib má malý alebo žiadny účinok na väčšinu iných skúmaných proteínkináz vrátane Src, okrem
kináz receptorov PDGF, KIT a efrínu, ktoré inhibuje v koncentráciách v rozmedzí dosahovanom po
perorálnom podaní terapeutických dávok odporúčaných na liečbu CML (pozri Tabuľku 5).
Tabuľka 5 Profil nilotinibu voči kinázam (fosforylácia IC50 nM)BCR-ABL PDGFR KIT

20 69 210
Klinickáúčinnosť Kl i ni cké š túdie pri novodi agnosti kovanej CM L v c hroni ck ej f áze Na stanovenie účinnosti nilotinibu oproti imatinibu sa uskutočnilo otvorené, multicentrické,
randomizované klinické skúšanie fázy III s 846 dospelými pacientmi s novodiagnostikovanou CML
s pozitívnym chromozómom Philadelphia v cytogeneticky potvrdenej chronickej fáze. Pacientom bolo ochorenie diagnostikované v priebehu posledných šiestich mesiacov a v minulosti neboli liečení,
s výnimkou hydroxyurey a/alebo anagrelidu. Pacienti boli randomizovaní v pomere 1:1:1, aby
dostávali buď nilotinib 300 mg dvakrát denne (n=282), nilotinib 400 mg dvakrát denne (n=281), alebo imatinib 400 mg raz denne (n=283). Randomizácia bola stratifikovaná podľa Sokalovho rizikového skóre v čase stanovenia diagnózy.
Východiskové charakteristiky boli dobre vyvážené medzi troma skupinami liečby. Medián veku bol
47 rokov v oboch skupinách nilotinibu a 46 rokov v skupine imatinibu, pričom 12,8% pacientov bolo vo veku ≥65 rokov v skupine liečby nilotinibu 300 mg dvakrát denne, 10,0% v skupine nilotinibu
400 mg dvakrát denne a 12,4% v skupine imatinibu 400 mg raz denne. Počet liečených mužov bol o
niečo vyšší než žien (56,0% v skupine nilotinibu 300 mg dvakrát denne, 62,3% v skupine nilotinibu
400 mg dvakrát denne a 55,8% v skupine imatinibu 400 mg raz denne). Viac než 60% všetkých pacientov boli belosi a 25% všetkých pacientov boli Ázijčania.
Primárna analýza údajov sa vykonala v čase, keď všetkých 846 pacientov ukončilo 12 mesiacov liečby
(alebo ju vysadilo skôr). Následné analýzy sa týkajú času, keď pacienti ukončili 24, 36, 48, 60 a
72 mesiacov liečby (alebo ju vysadili skôr). Medián trvania liečby bol približne 70 mesiacov
v skupinách liečby nilotinibom a 64 mesiacov v skupine imatinibu. Medián skutočnej veľkosti dávky
bol 593 mg/deň pri nilotinibe 300 mg dvakrát denne, 772 mg/deň pri nilotinibe 400 mg dvakrát denne a 400 mg/deň pri imatinibe 400 mg raz denne. Táto štúdia naďalej pokračuje.
Primárnym parametrom účinnosti bola veľká molekulová odpoveď (major molecular response, MMR) po 12 mesiacoch. MMR bola definovaná ako ≤0,1% BCR-ABL/ABL % podľa medzinárodnej škály (international scale, IS) pri stanovení RQ-PCR, čo zodpovedá poklesu BCR-ABL transkriptov ≥3 log oproti štandardizovanej východiskovej hodnote. Podiel MMR po 12 mesiacoch bol štatisticky významne vyšší pri nilotinibe 300 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne (44,3% oproti 22,3%, p<0,0001). Podiel MMR po 12 mesiacoch bol tiež štatisticky významne vyšší
pri nilotinibe 400 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne (42,7% oproti 22,3%, p<0,0001).
Podiel MMR po 3, 6, 9 a 12 mesiacoch bol 8,9%, 33,0%, 43,3% a 44,3% pri nilotinibe 300 mg dvakrát denne, 5,0%, 29,5%, 38,1% a 42,7% pri nilotinibe 400 mg dvakrát denne a 0,7%, 12,0%, 18,0%
a 22,3% pri imatinibe 400 mg raz denne.
Podiely MMR v 12., 24., 36., 48., 60. a 72. mesiaci sú uvedené v Tabuľke 6.
Tabuľka 6 Podiel MMR
MMR v 12. mesiaci
Nilotinib
300 mg dvakrát denne
n=282
(%)
Nilotinib
400 mg dvakrát denne
n=281
(%)
Imatinib
400 mg raz denne n=283
(%)
Odpoveď (95% CI) 44,31 (38,4; 50,3) 42,71 (36,8; 48,7) 22,3 (17,6; 27,6)
MMR v 24. mesiaciOdpoveď (95% CI) 61,71 (55,8; 67,4) 59,11 (53,1; 64,9) 37,5 (31,8; 43,4)
MMR v 36. mesiaci2Odpoveď (95% CI) 58,51 (52,5; 64,3) 57,31 (51,3; 63,2) 38,5 (32,8; 44,5)
MMR v 48. mesiaci3Odpoveď (95% CI) 59,91 (54,0; 65,7) 55,2 (49,1; 61,1) 43,8 (38,0; 49,8)
MMR v 60. mesiaci4Odpoveď (95% CI) 62,8 (56,8; 68,4) 61,2 (55,2; 66,9) 49,1 (43,2; 55,1)
MMR v 72. mesiaci5Odpoveď (95% CI) 52,5 (46,5; 58,4) 57,7 (51,6; 63,5) 41,7 (35,9; 47,7)
1 Hodnota p pre podiel odpovedí (oproti imatinibu 400 mg) <0,0001 testu podľa Cochrana-Mantela- Haenszela (CMH)
2 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 199 (35,2%) pacientov vyhodnotiť MMR v 36. mesiaci
(87 v skupine nilotinibu 300 mg dvakrát denne a 112 v skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=17), atypické východiskové transkripty (n=7) alebo pre ukončenie liečby pred 36. mesiacom (n=175).
3 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 305 (36,1%) pacientov vyhodnotiť MMR v 48. mesiaci
(98 v skupine nilotinibu 300 mg dvakrát denne, 88 v skupine nilotinibu 400 mg dvakrát denne a 119 v
skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=18), atypické východiskové transkripty (n=8) alebo pre ukončenie liečby pred 48. mesiacom (n=279).
4 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 322 (38,1%) pacientov vyhodnotiť MMR v 60. mesiaci
(99 v skupine nilotinibu 300 mg dvakrát denne, 93 v skupine nilotinibu 400 mg dvakrát denne a 130 v
skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=9), atypické východiskové transkripty (n=8) alebo pre ukončenie liečby pred 60. mesiacom (n=305).
5 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 395 (46,7%) pacientov vyhodnotiť MMR v 72. mesiaci
(130 v skupine nilotinibu 300 mg dvakrát denne, 110 v skupine nilotinibu 400 mg dvakrát denne a 155
v skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=25), atypické východiskové
transkripty (n=8) alebo pre ukončenie liečby pred 72. mesiacom (n=362).
Podiely MMR v rôznych časoch (vrátane pacientov, ktorí dosiahli MMR ako responderi v danom čase
alebo skôr) sú uvedené v kumulatívnej incidencii MMR (pozri Obrázok 1).
Obrázok 1 Kumulatívna incidencia MMR

100
90
Nilotinib 300 mg dvakrát denne (n = 282)
Nilotinib 400 mg dvakrát denne (n = 281)
Imatinib 400 mg raz denne (n = 283)
Po 6 rokoch
P
o
3 rokoch
P
o
4 rokoch
P
o
5 rokoch

80
70
Po 1 roku60
Po 2 rokoch71%;
P < 0,0001
73%;
P < 0,0001

70%;
P < 0,0001
76%;
P < 0,0001 77%;
P < 0,0001
77%;
P < 0,0001
73%;
P < 0,0001
79%;
P < 0,0001
77%;
P < 0,0001
55%; P < 0,0001
50 51%;
P < 0,0001
40
30
67%;
P < 0,0001
44%
53%
56%
60%
61%
20
27%
10
0
0 6 12 18 24 30 36 42 48 54 60
Mesiace od randomizácie
66 72
Vo všetkých skupinách rizika podľa Sokala boli podiely MMR v každom čase trvale vyššie v oboch
skupinách nilotinibu ako v skupine imatinibu.
V retrospektívnej analýze 91% (234/258) pacientov liečených nilotinibom 300 mg dvakrát denne dosiahlo úroveň BCR-ABL ≤10% po 3 mesiacoch liečby v porovnaní so 67% (176/264) pacientov liečených nilotinibom 400 mg dvakrát denne. Pacienti s BCR-ABL na úrovni ≤10% po 3 mesiacoch liečby preukázali väčšiu mieru celkového prežívania v 72. mesiaci v porovnaní s tými, ktorí nedosiahli takúto úroveň molekulovej odpovede (94,5% oproti 77,1% v uvedenom poradí [p=0,0005]).
Na základe analýzy času do prvej MMR podľa Kaplana-Meiera bola pravdepodobnosť dosiahnutia MMR v rozličných časoch vyššia pri nilotinibe 300 mg aj 400 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne (HR=2,17 a stratifikovaný log-rank p<0,0001 medzi nilotinibom
300 mg dvakrát denne a imatinibom 400 mg raz denne, HR=1,88 a stratifikovaný log-rank p<0,0001
medzi nilotinibom 400 mg dvakrát denne a imatinibom 400 mg raz denne).
Podiel pacientov, ktorí mali molekulovú odpoveď ≤0,01% a ≤0,0032% podľa IS v rozličných časoch sú uvedené v Tabuľke 7 a podiel pacientov, ktorí mali molekulovú odpoveď ≤0,01% a ≤0,0032% podľa IS v rôznych časoch sú uvedené na Obrázkoch 2 a 3. Molekulové odpovede ≤0,01% a
≤0,0032% podľa IS zodpovedajú zníženiu transkriptov BCR-ABL ≥4 log a ≥4,5 log v uvedenom
poradí oproti štandardizovanej východiskovej hodnote.
Tabuľka 7 Podiely pacientov, ktorí mali molekulovú odpoveď ≤0,01% (zníženie 4 log) a
≤0,0032% (zníženie 4,5 log)
Nilotinib
300 mg dvakrát denne n=282
(%)
Nilotinib
400 mg dvakrát denne n=281
(%)
Imatinib
400 mg raz denne n=283
(%)
≤
0,01% ≤0,0032% ≤0,01% ≤0,0032% ≤0,01% ≤0,0032%
V 12. Mesiaci 11,7 4,3 8,5 4,6 3,9 0,4
V 24. Mesiaci 24,5 12,4 22,1 7,8 10,2 2,8
V 36. Mesiaci 29,4 13,8 23,8 12,1 14,1 8,1
V 48. Mesiaci 33,0 16,3 29,9 17,1 19,8 10,2
V 60. Mesiaci 47,9 32,3 43,4 29,5 31,1 19,8
V 72. Mesiaci 44,3 31,2 45,2 28,8 27,2 18,0
Obrázok 2 Kumulatívna incidencia molekulovej odpovede ≤0,01% (zníženie 4 log)

100
Nilotinib 300 mg dvakrát denne (n = 282)

Nilotinib 400 mg dvakrát denne (n = 281)

90 Imatinib 400 mg raz denne (n = 283)
80
70
60
50
Po 1 roku
40
20%;
P < 0,0001
30
15%;
P = 0,0004
20
Po 2 rokoch39%;
P < 0,0001
33%;
P < 0,0001
Po 3 rokoch50%;
P < 0,0001
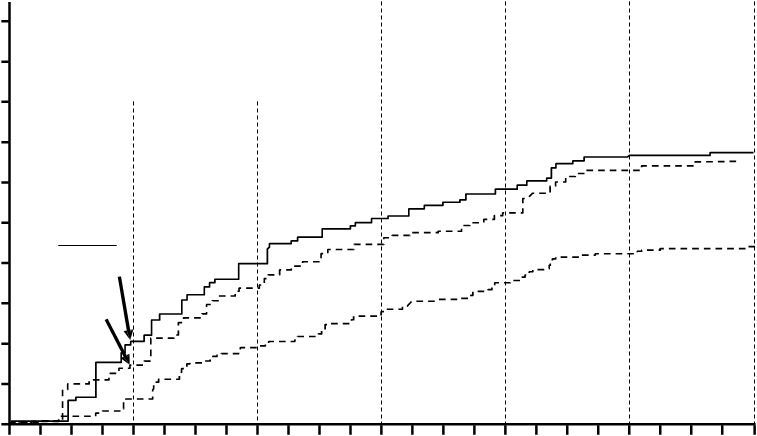
44%;
P < 0,0001
26%
Po 4 rokoch56%;
P < 0,0001
50%;
P < 0,0001
32%
Po5 rokoch66%;
P < 0,0001
63%;
P < 0,0001
42%
Po 6 rokoch67%;
P < 0,0001
65%;
P < 0,0001
43%
18%
10
0 6%
0 6 12 18 24 30 36 42 48 54 60
66 72
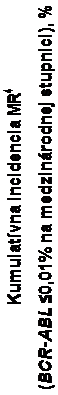 Mes
i
ac
e od randomizácie
Mes
i
ac
e od randomizácie
 O
brázok 3 Kumulatívna incidencia molekulovej odpovede ≤0,0032% (zníženie 4,5 log)
O
brázok 3 Kumulatívna incidencia molekulovej odpovede ≤0,0032% (zníženie 4,5 log)
100
90
Nilotinib 300 mg dvakrát denne (n = 282)

Nilotinib 400 mg dvakrát denne (n = 281)
Imatinib 400 mg raz denne (n = 283)
80
70
60
50
40
Po 1 roku30
11%;
P < 0,0001
20 7%;
P < 0,0001
Po 2 rokoch25%;
P < 0,0001
Po 3 rokoch32%;
P < 0,0001
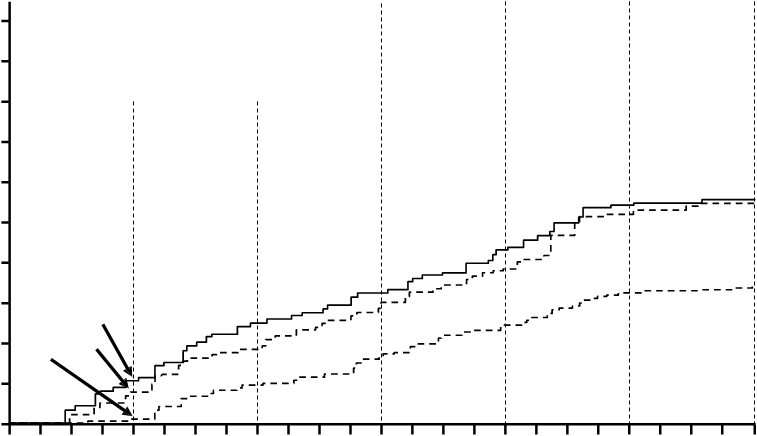
28%;
P = 0,0003
Po 4 rokoch40%;
P < 0,0001
37%;
P = 0,0002
23%
Po 5 rokoch54%;
P < 0,0001
52%;
P < 0,0001
31%
Po 6 rokoch56%;
P < 0,0001
55%;
P < 0,0001
33%
1% 19%;
10 P = 0,0006
9%
0
15%
0 6 12 18 24 30 36 42 48 54 60
66 72
 Mes
i
ac
e od randomizácie
Mes
i
ac
e od randomizácie
Na základe odhadov podľa Kaplana-Meiera pre trvanie prvej MMR boli podiely pacientov, ktorí si
udržali odpoveď počas 72 mesiacov spomedzi pacientov, ktorí dosiahli MMR 92,5% (95% CI:
88,6-96,4%) v skupine nilotinibu 300 mg dvakrát denne, 92,2% (95% CI: 88,5-95,9%) v skupine nilotinibu 400 mg dvakrát denne a 88,0% (95% CI: 83,0-93,1%) v skupine imatinibu 400 mg raz
denne.
Kompletná cytogenetická odpoveď (CCyR) bola definovaná ako 0% Ph+ metafáz v kostnej dreni na základe hodnotenia minimálne 20 metafáz. Najlepší podiel CCyR po 12 mesiacoch (vrátane pacientov, ktorí dosiahli CCyR ako responderi v 12. mesiaci alebo skôr) bol štatisticky vyšší pri nilotinibe
300 mg aj 400 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne, pozri Tabuľku 8.
Podiel CCyR v priebehu 24 mesiacov (vrátane pacientov, ktorí dosiahli CCyR ako responderi
v 24. mesiaci alebo skôr), bol štatisticky vyšší v°skupinách nilotinibu 300 mg dvakrát denne aj 400 mg dvakrát denne v porovnaní so skupinou imatinibu 400 mg raz denne.
Tabuľka 8 Najlepší podiel CCyR
V priebehu 12 mesiacov
Nilotinib
300 mg dvakrát denne n=282
(%)
Nilotinib
400 mg dvakrát denne n=281
(%)
Imatinib
400 mg raz denne n=283
(%)
Odpoveď (95% CI) 80,1 (75,0; 84,6) 77,9 (72,6; 82,6) 65,0 (59,2; 70,6)
Žiadna odpoveď 19,9 22,1 35,0
Hodnota p testu CMH pre podiel odpovede (oproti imatinibu 400 mg raz denne)
V priebehu 24 mesiacov
<0,0001 0,0005
Odpoveď (95% CI) 86,9 (82,4; 90,6) 84,7 (79,9; 88,7) 77,0 (71,7; 81,8)
Žiadna odpoveď 13,1 15,3 23,0
Hodnota p testu CMH pre podiel odpovede (oproti imatinibu 400 mg raz denne)
0,0018 0,0160

Na základe odhadov podľa Kaplana-Meiera boli podiely pacientov, ktorí si udržali odpoveď počas
72 mesiacov spomedzi pacientov, ktorí dosiahli CCyR 99,1% (95% CI: 97,9-100%) v skupine nilotinibu 300 mg dvakrát denne, 98,7% (95% CI: 97,1-100%) v skupine nilotinibu 400 mg dvakrát
denne a 97,0% (95% CI: 94,7-99,4%) v skupine imatinibu 400 mg raz denne.
Progresia do akcelerovanej fázy (accelerated phase, AP) alebo blastickej krízy (blast crisis, BC) počas liečby je definovaná ako čas od dátumu randomizácie do prvej dokumentovanej progresie ochorenia do akcelerovanej fázy alebo blastickej krízy alebo úmrtia súvisiaceho s CML. Progresia do akcelerovanej fázy alebo blastickej krízy počas liečby sa pozorovala celkovo u 17 pacientov: u
2 pacientov pri nilotinibe 300 mg dvakrát denne, u 3 pacientov pri nilotinibe 400 mg dvakrát denne a u
12 pacientov pri imatinibe 400 mg raz denne. Odhadované podiely pacientov bez progresie do akcelerovanej fázy alebo blastickej krízy v 72 mesiacoch boli 99,3%, 98,7% a 95,2% v uvedenom
poradí (HR=0,1599 a stratifikovaný log-rank p=0,0059 medzi nilotinibom 300 mg dvakrát denne
a imatinibom raz denne, HR=0,2457 a stratifikovaný log-rank p=0,0185 medzi nilotinibom 400 mg dvakrát denne a imatinibom raz denne). Žiadne nové prípady progresie do AP/BC neboli hlásené po
analýze 2-ročného obdobia.
Pri zahrnutí vývinu klonov ako kritéria progresie celkovo 25 pacientov progredovalo počas liečby do dátumu ukončenia zberu údajov do akcelerovanej fázy alebo blastickej krízy (3 v skupine nilotinibu
300 mg dvakrát denne, 5 v skupine nilotinibu 400 mg dvakrát denne a 17 v skupine imatinibu 400 mg raz denne). Odhadované podiely pacientov bez progresie do akcelerovanej fázy alebo blastickej krízy
vrátane vývinu klonov boli v 72 mesiacoch 98,7%, 97,9% a 93,2% v uvedenom poradí (HR=0,1626 a stratifikovaný log-rank p=0,0009 medzi nilotinibom 300 mg dvakrát denne a imatinibom raz denne, HR=0,2848 a stratifikovaný log-rank p=0,0085 medzi nilotinibom 400 mg dvakrát denne
a imatinibom raz denne).
Celkovo 55 pacientov zomrelo počas liečby alebo počas následného sledovania po ukončení liečby (21
v skupine nilotinibu 300 mg dvakrát denne, 11 v skupine nilotinibu 400 mg dvakrát denne a 23
v skupine imatinibu 400 mg raz denne). Dvadsaťšesť (26) z týchto 55 úmrtí súviselo s CML (6
v skupine nilotinibu 300 mg dvakrát denne, 4 v skupine nilotinibu 400 mg dvakrát denne a 16
v skupine imatinibu 400 mg raz denne). Odhadované podiely pacientov, ktorí žili v 72 mesiacoch, boli
91,6%, 95,8% a 91,4% v uvedenom poradí (HR=0,8934 a stratifikovaný log-rank p=0,7085 medzi nilotinibom 300 mg dvakrát denne a imatinibom, HR=0,4632 a stratifikovaný log-rank p=0,0314
medzi nilotinibom 400 mg dvakrát denne a imatinibom). Ak sa za udalosti považujú len úmrtia súvisiace s CML, odhadované podiely celkového prežívania v 72 mesiacoch boli 97,7%, 98,5%
a 93,9% v uvedenom poradí (HR=0,3694 a stratifikovaný log-rank p=0,0302 medzi nilotinibom
300 mg dvakrát denne a imatinibom, HR=0,2433 a stratifikovaný log-rank p=0,0061 medzi nilotinibom 400 mg dvakrát denne a imatinibom).
Kl i ni cké s kúšani a pri CML v chronickej a akcelerovanej fáze s rezist enciou alebo i nt ol eranci ou voči
imatinibu
Na stanovenie účinnosti nilotinibu u dospelých pacientov s CML s rezistenciou alebo intoleranciou
voči imatinibu sa uskutočnilo otvorené, nekontrolované, multicentrické klinické skúšanie fázy II
s oddelenými ramenami liečby chronickej a akcelerovanej fázy ochorenia. Účinnosť sa stanovila
u 321 CP pacientov a 137 AP pacientov zaradených do skúšania. Medián trvania liečby bol 561 dní
u CP pacientov a 264 dní u AP pacientov (pozri Tabuľku 9). Tasigna sa podávala kontinuálne (dvakrát denne 2 hodiny po jedle a bez požitia jedla najmenej jednu hodinu po podaní), kým sa nepreukázala neprimeraná odpoveď alebo progresia ochorenia. Dávka bola 400 mg dvakrát denne a bolo dovolené zvýšenie dávky na 600 mg dvakrát denne.
Tabuľka 9 Trvanie expozície nilotinibu
Medián trvania liečby v dňoch
(25.-75. percentil)
Chronická fáza
n=321
561 (196-852)
Akcelerovaná fáza
n=137
264 (115-595)

Rezistencia voči imatinibu zahŕňala nedosiahnutie kompletnej hematologickej odpovede (do
3 mesiacov), cytogenetickej odpovede (do 6 mesiacov) alebo veľkej cytogenetickej odpovede (do
12 mesiacov), alebo progresiu ochorenia po dosiahnutí predchádzajúcej cytogenetickej alebo
hematologickej odpovede. Intoleranciu voči imatinibu mali pacienti, ktorí prerušili liečbu imatinibom pre toxicitu a nedosiahli veľkú cytogenetickú odpoveď v čase zaradenia do klinického skúšania.
Celkovo 73% pacientov malo rezistenciu voči imatinibu a 27% intoleranciu voči imatinibu. Väčšina pacientov mala dlhodobú anamnézu CML, ktorá zahŕňala extenzívnu predchádzajúcu liečbu inými antineoplastikami vrátane imatinibu, hydroxyurey a interferónu a u niektorých dokonca neúspešnú orgánovú transplantáciu (Tabuľka 10). Medián najvyššej predchádzajúcej dávky imatinibu bol
600 mg/deň. Najvyššia predchádzajúca dávka imatinibu bola ³600 mg/deň u 74% všetkých pacientov, pričom 40% pacientov dostávalo dávky imatinibu ³800 mg/deň.
Tabuľka 10 Charakteristika anamnézy CML
Medián času od stanovenia diagnózy v
mesiacoch
(rozmedzie)
Imatinib
Rezistencia
Intolerancia bez MCyR Medián trvania liečby imatinibom v dňoch
(25.-75. percentil)
Chronická fáza
(n=321)
58
(5-275)
226 (70%)
95 (30%)
975 (519-1 488)
Akcelerovaná fáza
(n=137)*
71
(2-298)
109 (80%)
27 (20%)
857 (424-1 497)
Predchádzajúca liečba hydroxyureou 83% 91%
Predchádzajúca liečba interferónom 58% 50%
Predchádzajúca transplantácia kostnej drene
7% 8%
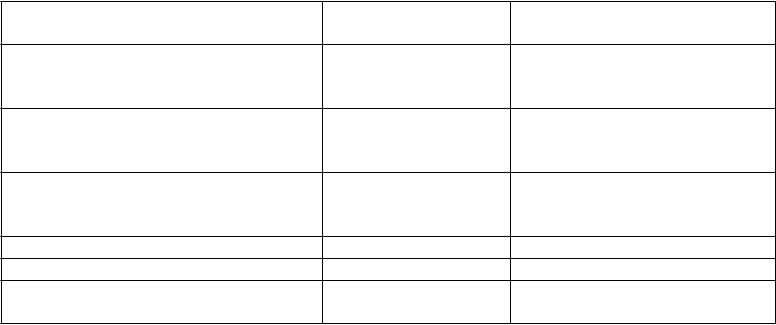
*U jedného pacienta chýbajú informácie o rezistencii/intolerancii voči imatinibu.
Primárnym ukazovateľom u CP pacientov bolo dosiahnutie veľkej cytogenetickej odpovede (MCyR),
definovanej ako eliminovanie (CCyR, kompletná cytogenetická odpoveď) alebo významné zníženie na
<35% Ph+ metafáz (parciálna cytogenetická odpoveď) hematopoetických buniek s Ph+. Kompletná hematologická odpoveď (CHR) u CP pacientov sa hodnotila ako sekundárny ukazovateľ. Primárny
ukazovateľ u AP pacientov bola celkovo potvrdená hematologická odpoveď (HR), definovaná buď
ako kompletná hematologická odpoveď, žiadny dôkaz leukémie, alebo návrat do chronickej fázy.
Chronická fázaPodiel MCyR u 321 CP pacientov bol 51%. Väčšina pacientov s odpoveďou na liečbu dosiahla MCyR rýchlo v priebehu 3 mesiacov (medián 2,8 mesiaca) od začiatku liečby nilotinibom a odpoveď u nich pretrvávala. Medián času do dosiahnutia CCyR bol niečo nad 3 mesiace (medián 3,4 mesiaca).
Z pacientov, ktorí dosiahli MCyR, 77% (95% CI: 70% - 84%) si udržalo odpoveď 24 mesiacov. Medián trvania MCyR sa nedosiahol. Z pacientov, ktorí dosiahli CCyR, 85% (95% CI: 78% - 93%) si
udržalo odpoveď do 24 mesiacov. Medián trvania CCyR sa nedosiahol. Pacienti s CHR pri zaradení
do klinického skúšania dosiahli MCyR rýchlejšie (1,9 oproti 2,8 mesiacom). Z CP pacientov bez CHR
pri zaradení do klinického skúšania 70% dosiahlo CHR, medián času do dosiahnutia CHR bol
1 mesiac a medián trvania CHR bol 32,8 mesiaca. Odhadované 24-mesačné celkové prežívanie
u CML-CP pacientov bol 87%.
Akcelerovaná fázaCelkový potvrdený podiel HR u 137 AP pacientov bol 50%. Väčšina pacientov s odpoveďou na liečbu dosiahla HR pri liečbe nilotinibom rýchlo (medián 1,0 mesiac) a odpoveď pretrvávala (medián trvania potvrdenej HR bol 24,2 mesiaca). Z pacientov, ktorí dosiahli HR, 53% (95% CI: 39% - 67%) si
udržalo odpoveď 24 mesiacov. Podiel MCyR bol 30% s mediánom času do odpovede 2,8 mesiaca. Z pacientov, ktorí dosiahli MCyR, 63% (95% CI: 45% - 80%) si udržalo odpoveď 24 mesiacov.
Medián trvania MCyR bol 32,7 mesiaca. Odhadované 24-mesačné celkové prežívanie u CML-AP
pacientov bolo 70%.
Podiely odpovedí v dvoch ramenách liečby sú uvedené v Tabuľke 11.
Tabuľka 11 Odpoveď pri CML
(Najlepší podiel
odpovedí)
Chronická fáza Akcelerovaná fáza
Intoleran- cia
(
n=95)
Hematologická odpoveď (%)
Rezisten- cia (n=226)
Celkovo
(n=321)
Intoleran- cia
(n=27)
Rezisten- cia (n=109)
Celkovo* (n=137)
Celkovo (95%CI) Kompletná
NEL
Návrat do CP
-
87 (74-94)
-
-
-
65 (56-72)
-
-
-
701 (63-76)
-
48 (29-68)
37
7
4
51 (42-61)
28
10
13
50 (42-59)
30
9
11
Cytogenetická odpoveď (%)
Veľká (95%CI) Kompletná Parciálna
57 (46-67)
41
16
49 (42-56)
35
14
51 (46-57)
37
15
33 (17-54)
22
11
29 (21-39)
19
10
30 (22-38)
20
10
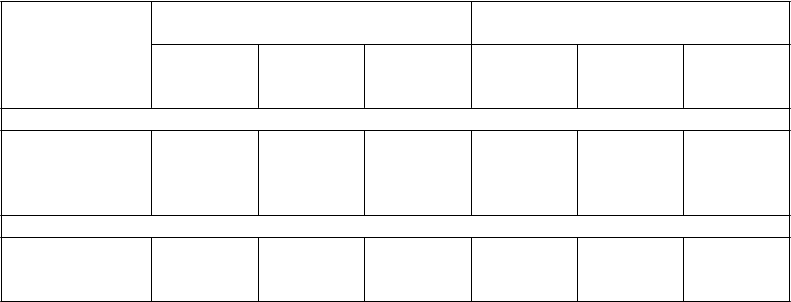
NEL = žiadny dôkaz leukémie/odpovede kostnej drene
1 114 CP pacientov malo CHR pri zaradení do klinického skúšania, preto u nich nebolo možné hodnotiť kompletnú hematologickú odpoveď
* U jedného pacienta chýbajú informácie o rezistencii/intolerancii voči imatinibu.
Údaje o účinnosti u pacientov v blastickej kríze CML zatiaľ nie sú dostupné. Klinické skúšanie fázy II, ktoré skúmalo Tasignu v skupine CP a AP pacientov extenzívne predliečených rôznymi druhmi liečby, vrátane ďalšieho inhibítora tyrozínkinázy popri imatinibe, malo tiež oddelené ramená liečby. Z týchto pacientov malo 30/36 (83%) voči liečbe rezistenciu a žiadnu intoleranciu. U
22 CP pacientov, u ktorých sa hodnotila účinnosť, bol podiel MCyR vyvolaných nilotinibom 32% a
CHR 50%. U 11 AP pacientov, u ktorých sa hodnotila účinnosť, bol celkový podiel HR vyvolaných liečbou 36%.
Po zlyhaní imatinibu sa zistilo 24 rôznych mutácií BCR-ABL u 42% pacientov s CML v chronickej fáze a u 54% v akcelerovanej fáze, u ktorých sa mutácie hodnotili. Potvrdila sa účinnosť Tasigny u pacientov s rozličnými mutáciami BCR-ABL spojenými s rezistenciou voči imatinibu, okrem T315I.
Ukonč eni e l ieč by u pacientov s Ph+ CML v chronickej fáze, ktorí dostávali nilotinib ako li eč bu prvej línie a ktorí dosiahli trvalú hlbokú molekulovú odpoveď V otvorenej štúdii s jedným ramenom bolo zaradených 215 dospelých pacientov s Ph+ CML
v chronickej fáze liečených nilotinibom v prvej línii počas ≥2 rokov, ktorí dosiahli MR4,5 meranú pomocou testu MolecularMD MRDx BCR-ABL, do pokračovania liečby nilotinibom počas ďalších
52 týždňov (konsolidačná fáza s nilotinibom). 190 z 215 pacientov (88,4%) vstúpilo do fázy TFR po
dosiahnutí trvalej hlbokej molekulovej odpovede počas konsolidačnej fázy definovanej podľa
nasledujúcich kritérií:
- 4 posledné štvrťročné vyhodnotenia (vykonané každých 12 týždňov) boli aspoň MR4
(BCR-ABL/ABL ≤0,01% IS) a udržali sa počas jedného roka
- posledné vyhodnotenie je MR4,5 (BCR-ABL/ABL ≤0,0032% IS)
- nie viac ako dve vyhodnotenia spadajú medzi MR4 a MR4,5 (0,0032% IS < BCR-ABL/ABL
≤0,01% IS).
Primárnym ukazovateľom bolo percento pacientov s MMR v 48. týždni od začatia fázy TFR (vzhľadom na každého pacienta, ktorý vyžadoval opätovné začatie liečby ako non-responder). Zo
190 pacientov, ktorí vstúpili do fázy TFR, bolo 98 pacientov (51,6% [95% IS: 44,2; 58,9]) s MMR
v 48. týždni.
Osemdesiatosem pacientov (46,3%) prerušilo fázu TFR kvôli strate MMR, 1 (0,5%) v dôsledku úmrtia z neznámej príčiny, 1 (0,5%) kvôli rozhodnutiu lekára a 3 pacienti (1,6%) z vlastného rozhodnutia.
Z týchto 88 pacientov znovu začalo liečbu nilotinibom 86 pacientov a 2 pacienti natrvalo prerušili
štúdiu. Osemdesiatpäť z týchto 86 pacientov (98,8%) znovu dosiahlo MMR (jeden pacient natrvalo prerušil štúdiu z vlastného rozhodnutia) a 76 pacientov (88,4%) znovu dosiahlo MR4,5 v čase ukončenia štúdie.
Odhadovaný medián trvania liečby nilotinibom podľa Kaplana-Meiera (KM) pre opätovné dosiahnutie MMR a MR4,5 bol 7,9 týždňov (95% IS: 5,1; 8,0) a 13,1 týždňov (95% IS: 12,3; 15,7), v uvedenom poradí. Odhadovaný výskyt MMR a MR4,5 podľa KM v 24. týždni od opätovného začatia bol 98,8% (95% IS: 94,2; 99,9) a 90,9% (95% IS: 83,2; 96,0), v uvedenom poradí.
Odhadovaný medián prežívania bez liečby (treatment-free survival, TFS) podľa KM nebol doteraz dosiahnutý (Obrázok 4); 99 zo 190 pacientov (52,1%) nemalo udalosť TFS.
Obrázok 4 Odhad prežívania bez liečby podľa Kaplana-Meiera od začiatku TFR (analýzacelého súboru)100
90
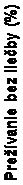
80
70
60
50
40
30
20
Pat
Evt Cen190 91 99
10
Cenzorované pozorovania
0
0 12
24 36
48 60 72
84 96
S rizikom: udalosti
Čas od TFR (týždne)
190:0
165:25
120:70
108:81
90:89
38:91
12:91
1:91
0:91
Ukonč eni e l ieč by u pacientov s CML v chronickej fáze, ktorí dosiahli trvalú hlbokú molekulovú
odpove ď pri li ečbe ni l ot inibom po predchádzajúcej li ečbe i mat ini bom
V otvorenej štúdii s jedným ramenom bolo zaradených 163 dospelých pacientov s Ph+ CML
v chronickej fáze užívajúcich inhibítory tyrozínkinázy (TKI, tyrosine kinase inhibitors) počas
≥3 rokov (imatinib ako úvodná liečba TKI počas viac ako 4 týždňov bez zdokumentovanej MR4,5 pri imatinibe v čase prechodu na nilotinib, potom prešli na nilotinib minimálne na dva roky), a ktorí dosiahli MR4,5 pri liečbe nilotinibom meranú pomocou testu MolecularMD MRDx BCR-ABL, do pokračovania liečby nilotinibom počas ďalších 52 týždňov (konsolidačná fáza s nilotinibom). 126 zo
163 pacientov (77,3%) vstúpilo do fázy TFR po dosiahnutí trvalej hlbokej molekulovej odpovede
počas konsolidačnej fázy definovanej podľa nasledujúcich kritérií:
- 4 posledné štvrťročné vyhodnotenia (vykonané každých 12 týždňov) nepreukázali žiadnu potvrdenú stratu MR4,5 (BCR-ABL/ABL ≤0,0032% IS) počas jedného roka
Primárnym ukazovateľom bolo percento pacientov bez potvrdenej straty MR4,0 alebo straty MMR počas 48 týždňov po ukončení liečby. Zo 126 pacientov, ktorí vstúpili do fázy TFR, bolo 73 pacientov (57,9% [95% IS: 48,8; 66,7]) bez straty MMR, bez potvrdenej straty MR4,0 a bez opätovného začatia liečby nilotinibom počas 48 týždňov.
Z 53 pacientov, ktorí prerušili fázu TFR kvôli potvrdenej strate MR4,0 alebo strate MMR,
51 pacientov znovu začalo liečbu nilotinibom a 2 pacienti prerušili štúdiu. Štyridsaťosem z týchto
51 pacientov (94,1%) znovu dosiahlo MR4,0 a 47 pacientov (92,2%) znovu dosiahlo MR4,5 v čase ukončenia štúdie.
Odhadovaný medián trvania liečby nilotinibom podľa Kaplana-Meiera (KM) pre opätovné dosiahnutie
MR4,0 a MR4,5 bol 12,0 týždňov (95% IS: 8,3; 12,7) a 13,1 týždňov (95% IS: 12,4; 16,1),
v uvedenom poradí. Odhadovaný výskyt MR4,0 a MR4,5 podľa KM v 48. týždni od opätovného začatia bol 100,0% (95% IS: neodhadnuté) a 94,8% (95% IS: 85,1; 99,0), v uvedenom poradí.
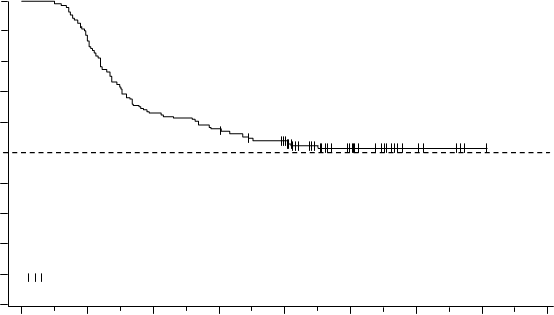
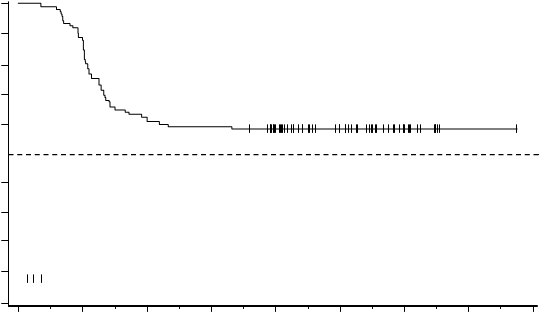
Medián TFS nebol doteraz dosiahnutý (Obrázok 5); 74 zo 126 pacientov (58,7%) nemalo udalosť
TFS.
Obrázok 5 Odhad prežívania bez liečby podľa Kaplana-Meiera od začiatku TFR (analýzacelého súboru)100
90

80
70
60
50
40
30
20
Pat126
Evt
52
Cen74
10 Cenzorované pozorovania
0
0 12 24 36 48 60 72 84 96
Čas od TFR (týždne)
S rizikom: udalosti
126:0 107:19 76:49 74:51 61:52 36:52 14:52 1:52 0:52
Pediatrická populácia
Bezpečnosť a účinnosť nilotinibu u pediatrických pacientov s Ph+ CML v chronickej fáze sa skúmali
v dvoch štúdiách. Celkovo 69 pediatrických pacientov (od 2 do <18 rokov) s buď
novodiagnostikovanou Ph+ CML v chronickej fáze (n=25), alebo Ph+ CML v chronickej fáze
s rezistenciou voči imatinibu/dasatinibu alebo intoleranciou voči imatinibu (n=44) dostalo liečbu nilotinibom v dávke 230 mg/m2 dvakrát denne, zaokrúhlenej na najbližších 50 mg dávky (až do maximálnej jednotlivej dávky 400 mg).
V zlúčenej populácii pacientov s CML bol medián skutočnej veľkosti dávky 435,5 mg/m2/deň (rozsah:
149 až 517 mg/m2/deň) a medián relatívnej veľkosti dávky 94,7% (rozsah: 32 až 112%). Štyridsať
pacientov (58,0%) malo relatívnu veľkosť dávky vyššiu ako 90%. Medián trvania liečby nilotinibom bol 13,80 mesiacov (rozsah: 0,7-30,9 mesiacov).
U pacientov s CML s rezistenciou alebo intoleranciou bola miera veľkej molekulovej odpovede
(MMR; BCR-ABL/ABL ≤0,1% IS) 40,9% (95% CI: 26,3; 56,8) po 12 cykloch, s 18 pacientmi
s MMR. U pacientov s novodiagnostikovanou CML bola miera MMR 60,0% (95% CI: 38,7; 78,9) po
12 cykloch, s 15 pacientmi, ktorí dosiahli MMR. U pacientov s CML s rezistenciou alebo intoleranciou bola kumulatívna miera MMR 47,7% po 12 cykloch. U pacientov
s novodiagnostikovanou CML bola kumulatívna miera MMR 64,0% po 12 cykloch.
U 21 pacientov s CML s rezistenciou alebo intoleranciou, ktorí dosiahli MMR kedykoľvek počas liečby, bol medián času do prvej MMR 2,76 mesiaca (95% CI: 0,03; 5,55).
U 17 novodiagnostikovaných pacientov s CML, ktorí dosiahli MMR, bol medián času do prvej MMR
5,55 mesiacov (95% CI: 5,52; 5,75).
Medzi pacientmi s CML s rezistenciou alebo intoleranciou bol percentuálny podiel pacientov, ktorí dosiahli BCR-ABL/ABL ≤0,01% IS (MR4.0) do dátumu ukončenia štúdie 11,4%, zatiaľ čo 4,5% pacientov dosiahlo BCR-ABL/ABL ≤0,0032% IS (MR4.5). Medzi novodiagnostikovanými pacientmi, ktorí dosiahli MR4.0, bol percentuálny podiel 32%, zatiaľ čo 28,0% dosiahlo MR4.5.
U žiadneho z 21 pacientov s CML s rezistenciou alebo intoleranciou, ktorí počas liečby dosiahli MMR, sa nepotvrdila strata MMR. Spomedzi 17 novodiagnostikovaných pacientov s CML, ktorí dosiahli MMR, sa u jedného pacienta potvrdila strata MMR (pacient stratil CHR v dôsledku zvýšeného počtu bazofilov, avšak neprogredoval do AP/BC).
Jeden pacient s CML s rezistenciou alebo intoleranciou progredoval do AP/BC po približne
10 mesiacoch na liečbe.
Počas liečby ani po ukončení liečby neboli v oboch štúdiách hlásené žiadne úmrtia.
5.2 Farmakokinetické vlastnosti
Absorpcia
Maximálne koncentrácie nilotinibu sa dosiahnu 3 hodiny po perorálnom podaní. Absorpcia nilotinibu
po perorálnom podaní bola približne 30%. Absolútna biologická dostupnosť nilotinibu nebola
stanovená. V porovnaní s perorálnym roztokom (pH 1,2 až 1,3) bola relatívna biologická dostupnosť nilotinibu v kapsliach približne 50%. Keď sa Tasigna užíva spolu s jedlom, u zdravých dobrovoľníkov sa Cmax nilotinibu zvýši o 112% a plocha pod krivkou sérovej koncentrácie v závislosti od času (AUC) o 82% v porovnaní so stavom nalačno. Podanie Tasigny 30 minút po jedle zvýšilo biologickú dostupnosť nilotinibu o 29%, 2 hodiny po jedle o 15% (pozri časti 4.2, 4.4 a 4.5).
Absorpcia nilotinibu (relatívna biologická dostupnosť) sa môže znížiť približne o 48% u pacientov s totálnou gastrektómiou a o 22% u pacientov s parciálnou gastrektómiou.
Distribúcia
Pomer nilotinibu v krvi a plazme je 0,71. Väzba na bielkoviny plazmy podľa pokusov in vitro je
približne 98%.
Biotransformácia
Hlavné metabolické dráhy zistené u zdravých osôb sú oxidácia a hydroxylácia. Nilotinib je hlavná
cirkulujúca zložka v sére. Žiadny z metabolitov neprispieva vo významnej miere k farmakologickej
aktivite nilotinibu. Nilotinib sa primárne metabolizuje CYP3A4, s možným menším podielom
CYP2C8.
Eliminácia
Po jednorazovej dávke rádioaktívne značeného nilotinibu podanej zdravým osobám sa viac ako 90%
dávky eliminovalo do 7 dní, prevažne stolicou (94% dávky). Nezmenený nilotinib tvoril 69% dávky.
Zdanlivý eliminačný polčas odhadnutý z farmakokinetiky pri opakovanom podávaní raz denne bol približne 17 hodín. Variabilita farmakokinetiky nilotinibu medzi pacientmi bola stredne vysoká až vysoká.
Linearita/nelinearita
Expozícia nilotinibu v rovnovážnom stave závisela od dávky, pričom zvyšovanie systémovej
expozície bolo nižšie, ako by bolo úmerné dávke pri dávkach vyšších ako 400 mg podávaných raz denne. Denná systémová expozícia nilotinibu pri podávaní 400 mg dvakrát denne bola v rovnovážnom
stave o 35% vyššia ako pri podávaní 800 mg raz denne. Systémová expozícia (AUC) nilotinibu
v rovnovážnom stave pri dávkach 400 mg dvakrát denne bola približne o 13,4% vyššia než pri
dávkach 300 mg dvakrát denne. Priemerné minimálne a maximálne koncentrácie nilotinibu počas
12 mesiacov boli približne o 15,7% a 14,8% vyššie pri podávaní 400 mg dvakrát denne v porovnaní s podávaním 300 mg dvakrát denne. Pri zvýšení dávky zo 400 mg dvakrát denne na 600 mg dvakrát
denne nedošlo k významnému zvýšeniu expozície nilotinibu.
Podmienky rovnovážneho stavu sa v podstate dosiahli do 8 dní. Zvýšenie sérovej expozície nilotinibu medzi prvou dávkou a rovnovážnym stavom bolo približne 2-násobné pri podávaní raz denne a
3,8-násobné pri podávaní dvakrát denne.
Biologická dostupnosť/bioekvivalenčnéštúdie
Preukázalo sa, že jednorazové podanie 400 mg nilotinibu pri použití 2 tvrdých kapsúl po 200 mg, keď
sa obsah každej tvrdej kapsuly zmiešal s jednou čajovou lyžičkou jablčného pyré, je bioekvivalentné
s jednorazovým podaním 2 neporušených tvrdých kapsúl po 200 mg.
Pediatrická populácia
Po podávaní nilotinibu pediatrickým pacientom v dávke 230 mg/m2 dvakrát denne, zaokrúhlenej na najbližšiu 50 mg dávku (až po maximálnu jednotlivú dávku 400 mg), boli expozícia v rovnovážnom stave a klírens nilotinibu podobné (do 2-násobku) ako u dospelých pacientov liečených dávkou
400 mg dvakrát denne. Farmakokinetická expozícia nilotinibu po jednej alebo viacerých dávkach sa
zdá byť porovnateľná medzi pediatrickými pacientmi od 2 rokov do <10 rokov a od ≥10 rokov do
<18 rokov.
5.3 Predklinické údaje o bezpečnosti
Nilotinib sa hodnotil v štúdiách farmakologickej bezpečnosti, toxicity po opakovanom podávaní,
genotoxicity, reprodukčnej toxicity, fototoxicity a karcinogenity (u potkanov a myší).
Nilotinib neovplyvnil funkcie CNS alebo dýchacej sústavy. V štúdiách kardiálnej bezpečnosti in vitro sa zistil predklinický signál predĺženia QT, ktoré bolo dôsledkom nilotinibom spôsobenej blokády prúdov hERG a predĺženia trvania akčného potenciálu v izolovaných srdciach králikov. Žiadne účinky v EKG meraniach sa nepozorovali u psov alebo opíc pri podávaní do 39 týždňov, ani u psov
v špeciálnej telemetrickej štúdii.
V štúdiách toxicity pri opakovanom podávaní trvajúcich do 4 týždňov u psov a do 9 mesiacov
u makakov krabožravých sa zistilo, že primárnym cieľovým orgánom toxicity nilotinibu je pečeň. Zmeny pozostávali zo zvýšenej aktivity alanínaminotransferázy a alkalickej fosfatázy
a histopatologických nálezov (najmä hyperplázie/hypertrofie sínusových buniek alebo Kupfferových
buniek, hyperplázie žlčovodov a periportálnej fibrózy). Vo všeobecnosti boli klinické biochemické zmeny plne reverzibilné po štvortýždňovom období zotavenia a histologické zmeny vykazovali čiastočnú reverzibilitu. Expozície pri najnižších dávkach, pri ktorých sa pozorovali účinky na pečeň, boli nižšie ako expozícia u ľudí pri dávke 800 mg/deň. Iba menšie zmeny v pečeni sa pozorovali u myší alebo potkanov pri podávaní do 26 týždňov. Prevažne reverzibilné zvýšenia hladiny cholesterolu sa pozorovali u potkanov, psov a opíc.
V skúšaniach genotoxicity v bakteriálnych systémoch in vitro a v cicavčích systémoch in vitro a in vivo s metabolickou aktiváciou alebo bez nej sa nenašiel dôkaz mutagénneho potenciálu nilotinibu.
V štúdii karcinogenity na potkanoch trvajúcej 2 roky bola maternica hlavným cieľovým orgánom
s inými ako neoplastickými léziami (dilatácia, rozšírenie ciev, hyperplázia endotelových buniek, zápal a/alebo hyperplázia epitelu). Karcinogenita sa nepreukázala pri podávaní nilotinibu 5, 15 a
40 mg/kg/deň. Expozícia (vyjadrená ako AUC) pri najvyššej dávke predstavovala približne 2- až
3-násobok dennej expozície nilotinibu v rovnovážnom stave (na základe AUC) u ľudí pri dávke
800 mg/deň.
V štúdii karcinogenity na myšiach Tg.rasH2 trvajúcej 26 týždňov, v ktorej sa nilotinib podával
v dávkach 30, 100 a 300 mg/kg/deň, boli zachytené papilómy/karcinómy kože pri 300 mg/kg, čo
predstavuje približne 30- až 40-násobok expozície u ľudí (na základe AUC) pri maximálnej schválenej dávke 800 mg/deň (podávanej dvakrát denne 400 mg). Hladina bez pozorovaného účinku pre kožné neoplastické lézie bola 100 mg/kg/deň, čo predstavuje približne 10- až 20-násobok expozície u ľudí
pri maximálnej schválenej dávke 800 mg/deň (podávanej dvakrát denne 400 mg). Hlavné cieľové orgány pre iné ako neoplastické lézie boli koža (epidermálna hyperplázia), rastúce zuby
(degenerácia/atrofia skloviny horných rezákov a zápal gingívy/odontogénneho epitelu rezákov)
a týmus (zvýšená incidencia a/alebo závažnosť poklesu lymfocytov).
Nilotinib nespôsoboval teratogenitu, ale vykazoval embryo- a fetotoxicitu pri dávkach, ktoré boli toxické aj pre samice. Zvýšené poimplantačné straty sa pozorovali v štúdii fertility pri podávaní samcom aj samiciam, aj v štúdii embryotoxicity pri podávaní samiciam. V skúšaniach embryotoxicity sa zistila letalita embryí a účinky na plod (predovšetkým znížená hmotnosť plodov, predčasné zrastanie kostí tváre (zrast maxily a lícnej kosti), viscerálne odchýlky a odchýlky skeletu) u potkanov a zvýšená resorpcia plodov a odchýlky skeletu u králikov. V štúdii pre- a postnatálneho vývinu
u potkanov spôsobila expozícia nilotinibu u samíc zníženú telesnú hmotnosť mláďat spojenú so zmenami parametrov telesného vývinu, ako aj znížené ukazovatele párenia a fertility u potomstva.
Expozícia nilotinibu u samíc pri hladinách bez pozorovaných nežiaducich účinkov bola spravidla
nižšia alebo rovnaká ako u ľudí pri 800 mg/deň.
V štúdii vývoja mláďat sa nilotinib podával pomocou perorálnej sondy mláďatám potkana od prvého týždňa po narodení do mladého dospelého veku (70. deň po narodení) v dávkach 2, 6 a 20 mg/kg/deň. Okrem štandardných parametrov štúdie sa vykonali hodnotenia vývojových medzníkov, účinkov na CNS, párenia a fertility. Na základe zníženia telesnej hmotnosti u oboch pohlaví a oneskoreného oddelenia predkožky u samcov (čo môže súvisieť s poklesom telesnej hmotnosti) sa za dávku bez pozorovaného účinku považovalo u dospievajúcich potkanov 6 mg/kg/deň. Dospievajúce zvieratá v porovnaní s dospelými nevykazovali zvýšenú citlivosť na nilotinib. Okrem toho bol profil toxicity u dospievajúcich potkanov porovnateľný s profilom toxicity, ktorý sa pozoroval u dospelých potkanov.
Nepozorovali sa žiadne účinky na počet/pohyblivosť spermií a fertilitu u potkaních samcov a samíc až do najvyššej testovanej dávky, približne 5-násobku odporúčaného dávkovania u ľudí.
Zistilo sa, že nilotinib absorbuje svetlo v rozmedzí UV-B a UV-A, distribuuje sa do kože a vykazuje fototoxický potenciál in vitro, nepozorovali sa však žiadne účinky in vivo. Riziko vyvolania fotosenzitivity nilotinibom u pacientov sa preto považuje za veľmi nízke.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
T
a
signa 50 mg tvrdé kapsuly
O
bsah kapsuly
monohydrát laktózy krospovidón typ A poloxamér 188
koloidný oxid kremičitý bezvodý
magnéziumstearát
Stena kapsuly
želatína
oxid titaničitý (E171)
červený oxid železitý (E172)
žltý oxid železitý (E172)
Farbi vo na potl ač
šelak (E904)
čierny oxid železitý (E172)
propylénglykol hydroxid amónny
Tasigna 200 mg tvrdé kapsuly
Obsah kapsuly
monohydrát laktózy krospovidón typ A
poloxamér 188
koloidný oxid kremičitý bezvodý
magnéziumstearát
Stena kapsuly
želatína
oxid titaničitý (E171)
žltý oxid železitý (E172)
Farbi vo na potl ač
šelak (E904)
červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Tasigna je dostupná v nasledujúcich veľkostiach balenia:
Tasigna 50 mg tvrdé kapsuly
Blistre PVC/PVDC/Alu
· Balenie obsahujúce 120 (3 balenia po 40) tvrdých kapsúl.
Tasigna 200 mg tvrdé kapsuly
Blistre PVC/PVDC/Alu
· Jednotlivé balenia obsahujúce 28 tvrdých kapsúl v puzdre.
· Jednotlivé balenia obsahujúce 28 tvrdých kapsúl (7 denných blistrov, z ktorých každý obsahuje
4 tvrdé kapsuly) alebo 40 tvrdých kapsúl (5 blistrov, z ktorých každý obsahuje 8 tvrdých kapsúl).
· Multibalenia obsahujúce 112 (4 puzdrá po 28) tvrdých kapsúl.
· Multibalenia obsahujúce 112 (4 balenia po 28) tvrdých kapsúl, 120 (3 balenia po 40) tvrdých kapsúl alebo 392 (14 balení po 28) tvrdých kapsúl.
Blistre PA/Alu/PVC/Alu
· Jednotlivé balenia obsahujúce 28 tvrdých kapsúl v puzdre.
· Multibalenia obsahujúce 112 (4 balenia po 28) tvrdých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLA
T
a
signa 50 mg tvrdé kapsuly
EU/1/07/422/015
Tasigna 200 mg tvrdé kapsulyEU/1/07/422/001-004
EU/1/07/422/007-008
EU/1/07/422/011-012
EU/1/07/422/014
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. novembra 2007
Dátum posledného predĺženia registrácie: 19. novembra 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
1. NÁZOV LIEKU
Tasigna 150 mg tvrdé kapsuly
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna tvrdá kapsula obsahuje 150 mg nilotinibu (ako monohydrát hydrochloridu). Pomocná látka soznámymúčinkom
Jedna tvrdá kapsula obsahuje 117,08 mg monohydrátu laktózy
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Tvrdá kapsula.
Biely až žltkastý prášok v červenej nepriehľadnej tvrdej želatínovej kapsule veľkosti 1, s čiernou
axiálnou potlačou „NVR/BCR“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Tasigna je indikovaná na liečbu:
- dospelých a pediatrických pacientov s novodiagnostikovanou chronickou myelocytovou leukémiou (CML) s pozitívnym chromozómom Philadelphia v chronickej fáze,
- pediatrických pacientov s CML s pozitívnym chromozómom Philadelphia v chronickej fáze s rezistenciou alebo intoleranciou voči predchádzajúcemu druhu liečby vrátane imatinibu.
4.2 Dávkovanie a spôsob podávania
Liečbu má začať lekár, ktorý má skúsenosti s diagnostikovaním a liečbou pacientov s CML.
Dávkovanie
Liečbamápokračovaťdovtedy,kýmsapozorujeklinickýprínos,alebokýmsanevyskytne
neakceptovateľná toxicita.
Privynechanídávkypacientnemáužiťdávkunavyše,alemáužiťobvyklúpredpísanúnajbližšiu
dávku.
Dávkovanie u dospelých pacientov s CML s pozitívnym chromozómom Philadelphia
Odporúčaná dávka je 300 mg dvakrát denne.
Pre dávku 400 mg raz denne (pozri úpravy dávky nižšie) sú dostupné 200 mg tvrdé kapsuly.
D
ávkovanie u pediatrických pacientov s CML s pozitívnym chromozómom Philadelphia
Dávkovanie sa u pediatrických pacientov stanovuje individuálne na základe plochy povrchu tela (mg/m2). Odporúčaná dávka nilotinibu je 230 mg/m2 dvakrát denne, zaokrúhlená na najbližšiu 50 mg dávku (až po maximálnu jednotlivú dávku 400 mg) (pozri Tabuľku 1). Na dosiahnutie želanej dávky sa môžu kombinovať tvrdé kapsuly Tasigny rôznych síl.
Nie sú skúsenosti s liečbou pediatrických pacientov mladších ako 2 roky. K dispozícii nie sú žiadne údaje od novodiagnostikovaných pediatrických pacientov mladších ako 10 rokov a iba obmedzené údaje od pediatrických pacientov mladších ako 6 rokov s rezistenciou alebo intoleranciou voči imatinibu.
Tabuľka 1 Schéma dávkovania nilotinibu 230 mg/m2 dvakrát denne
Plocha povrchu tela
(BSA)
Dávka v mg
(dvakrát denne)
Do 0,32 m2 50 mg
0,33 – 0,54 m2 100 mg

0,55 – 0,76 m2 150 mg
0,77 – 0,97 m2 200 mg
0,98 – 1,19 m2 250 mg
1,20 – 1,41 m2 300 mg
1,42 – 1,63 m2 350 mg
≥1,64 m2 400 mg
Pacienti s CML s pozitívnym chromozómom Philadelphia v chronickej fáze, ktorí dostávali nilotinib ako li ečbu prve j lí nie a kt orí dosi ahl i t rval ú hl bokú molek ul ovú odpoveď ( MR4,5)Ukončenie liečby možno zvážiť u vybraných CML pacientov s pozitívnym chromozómom Philadelphia (Ph+) v chronickej fáze, ktorí sa liečili nilotinibom 300 mg dvakrát denne počas minimálne 3 rokov, ak sa hlboká molekulová odpoveď udržala počas minimálne jedného roka bezprostredne pred ukončením liečby. Ukončenie liečby nilotinibom má začať lekár, ktorý má skúsenosti s liečbou pacientov s CML (pozri časť 4.4 a 5.1).
U pacientov, ktorí sú vhodní na ukončenie liečby nilotinibom, sa musia monitorovať hladiny
BCR-ABL transkriptu a úplný krvný obraz s diferenciálom raz mesačne počas prvého roka, potom každých 6 týždňov počas druhého roka a potom každých 12 týždňov. Monitorovanie hladiny
BCR-ABL transkriptu sa musí vykonať kvantitatívnym diagnostickým testom, validovaným na
stanovenie hladiny molekulovej odpovede na Medzinárodnej škále (International Scale, IS),
s citlivosťou najmenej MR4,5 (BCR-ABL/ABL ≤0,0032% IS).
U pacientov, ktorí stratia MR4 (MR4=BCR-ABL/ABL ≤0,01% IS), ale nie MMR
(MMR=BCR-ABL/ABL ≤0,1% IS) počas fázy bez liečby, sa majú hladiny BCR-ABL monitorovať každé 2 týždne, kým sa hladiny BCR-ABLvrátia do rozsahu medzi MR4 a MR4,5. U pacientov, ktorí
si udržia hladiny BCR-ABL medzi MMR a MMR4 počas najmenej 4 po sebe nasledujúcich meraní, sa
možno vrátiť k pôvodnému monitorovaciemu plánu.
Pacienti, ktorí stratia MMR, musia znovu začať liečbu do 4 týždňov od doby, odkedy je známe, že došlo k strate remisie. Liečba s nilotinibom sa má opakovane začať dávkou 300 mg dvakrát denne alebo na úrovni zníženej dávky 400 mg jedenkrát denne v tom prípade, ak pacient mal zníženú dávku pred ukončením liečby. U pacientov, ktorí znovu začali liečbu nilotinibom, sa majú hladiny BCR-ABL transkriptov monitorovať raz mesačne, až kým sa obnoví MMR, a potom každých 12 týždňov (pozri časť 4.4).
Prispôsobenie alebo úprava dávky
Tasignu môže byť potrebné dočasne vysadiť a/alebo znížiť jej dávku pre príznaky hematologickej
toxicity (neutropénia, trombocytopénia), ktoré nesúvisia so základným leukemickým ochorením (pozri
Tabuľku 2).
Tabuľka 2 Úprava dávky pre neutropéniu a trombocytopéniu
Dospelí pacienti s novodiagnostikovanou CML v chronickej
fáze pri dávke 300 mg dvakrát denne
Pediatrickí pacienti s novodiagnostikovanou CML v chronickej
fáze pri dávke
230 mg/m2 dvakrát denne a CML
s rezistenciou alebo
intoleranciou voči imatinibu v chronickej fáze pri dávke
230 mg/m2 dvakrát denne
ANC* <1,0 x 109/l a/alebo počet
trombocytov <50 x 109/l
ANC* <1,0 x 109/l a/alebo počet
trombocytov <50 x 109/l
1. Liečba nilotinibom sa musí prerušiť a musí
sa monitorovať krvný obraz.
2. Liečba sa musí znovu začať do 2 týždňov
s predchádzajúcou dávkou, ak ANC
>1,0 x 109/l a/alebo trombocyty
>50 x 109/l.
3. Ak počty krviniek ostávajú nízke, môže byť potrebné znížiť dávku na 400 mg raz
denne.
1. Liečba nilotinibom sa musí prerušiť a musí
sa monitorovať krvný obraz.
2. Liečba sa musí znovu začať do 2 týždňov
s predchádzajúcou dávkou, ak ANC
>1,5 x 109/l a/alebo trombocyty
>75 x 109/l.
3. Ak počty krviniek ostávajú nízke, môže byť potrebné znížiť dávku na 230 mg/m2 raz denne.
4. Ak udalosť nastane po znížení dávky, zvážte ukončenie liečby.

*ANC = absolútny počet neutrofilov
Ak sa vyvinú klinicky významné príznaky stredne ťažkej alebo ťažkej nehematologickej toxicity, podávanie sa má prerušiť a pacienti sa majú monitorovať a vhodne liečiť. Ak bola predchádzajúca dávka 300 mg dvakrát denne u dospelých pacientov alebo 230 mg/m2 dvakrát denne u pediatrických pacientov, podávanie sa má sa znova začať s dávkou 400 mg raz denne u dospelých pacientov alebo
230 mg/m2 dvakrát denne u pediatrických pacientov, keď toxické príznaky zmiznú. Ak bola predchádzajúca dávka 400 mg raz denne u dospelých pacientov alebo 230 mg/m2 raz denne u pediatrických pacientov, liečba sa má ukončiť. Ak je to klinicky vhodné, má sa zvážiť opätovné zvýšenie dávky na 300 mg dvakrát denne u dospelých pacientov alebo 230 mg/m2 dvakrát denne u pediatrických pacientov.
Zvýšenie sérovej lipázy: Pri zvýšení sérovej lipázy stupňa 3-4 sa má dávka u dospelých pacientov znížiť na 400 mg raz denne alebo prerušiť podávanie. U pediatrických pacientov sa liečba musí prerušiť až do zlepšenia príznakov na stupeň £1. Následne, ak bola predchádzajúca dávka 230 mg/m2 dvakrát denne, môže sa liečba znova začať s dávkou 230 mg/m2 raz denne. Ak bola predchádzajúca dávka 230 mg/m2 raz denne, liečba sa má ukončiť.Hladina sérovej lipázy sa má stanovovať každý mesiac alebo ak je to klinicky indikované (pozri časť 4.4).
Zvýšenie bilirubínu a pečeňových aminotransferáz: Pri zvýšení bilirubínu a pečeňových aminotransferáz stupňa 3-4 sa má dávka u dospelých pacientov znížiť na 400 mg raz denne alebo prerušiť podávanie. U pediatrických pacientov sa musí pri zvýšení bilirubínu stupňa ³2 alebo zvýšení pečeňových aminotransferáz stupňa ³3 liečba prerušiť, kým sa hladiny vrátia na stupeň £1. Následne, ak bola predchádzajúca dávka 230 mg/m2 dvakrát denne, môže sa liečba znova začať s dávkou
230 mg/m2 raz denne. Ak bola predchádzajúca dávka 230 mg/m2 raz denne a zlepšenie na stupeň £1
trvá dlhšie ako 28 dní, liečba sa má ukončiť.Hladina bilirubínu a pečeňových aminotransferáz sa má stanovovať každý mesiac alebo ak je to klinicky indikované.
O
sobitné skupiny
Starší ľudia
Približne 12% osôb v klinickej štúdii bolo vo veku 65 rokov alebo starších. Nepozorovali sa významné
rozdiely v bezpečnosti a účinnosti u pacientov vo veku ≥65 rokov v porovnaní s dospelými vo veku 18
až 65 rokov.
Porucha funkcie obličiek
Klinické skúšania sa nevykonali u pacientov so zhoršenou funkciou obličiek.
Vzhľadom na to, že nilotinib a jeho metabolity sa nevylučujú obličkami, u pacientov s poruchou funkcie obličiek sa nepredpokladá pokles celkového telesného klírensu.
Porucha funkcie pečene
Porucha funkcie pečene má mierny vplyv na farmakokinetiku nilotinibu. Úprava dávky sa u pacientov s poruchou funkcie pečene nepovažuje za potrebnú. Pri liečbe pacientov s poruchou funkcie pečene je
potrebná opatrnosť (pozri časť 4.4).
Poruchy srdca
Z klinických skúšaní boli vylúčení pacienti s nekompenzovaným alebo významným ochorením srdca
(napr. nedávnym infarktom myokardu, kongestívnym zlyhávaním srdca, nestabilnou angina pectoris alebo klinicky významnou bradykardiou). U pacientov so závažnými poruchami srdca je potrebná opatrnosť (pozri časť 4.4).
Počas liečby nilotinibom bolo zaznamenané zvýšenie hladiny celkového cholesterolu v sére (pozri časť 4.4). Pred začatím liečby nilotinibom, po 3 a 6 mesiacoch od začatia liečby a najmenej raz ročne pri dlhodobej liečbe je potrebné stanoviť profily lipidov.
Počas liečby nilotinibom bolo zaznamenané zvýšenie hladiny glukózy v krvi (pozri časť 4.4). Pred začatím liečby nilotinibom je potrebné stanoviť hladinu glukózy v krvi a monitorovať ju počas liečby.
Pediatrická populácia
Bezpečnosť a účinnosť Tasigny boli stanovené u pediatrických pacientov s CML s pozitívnym chromozómom Philadelphia v chronickej fáze vo veku od 2 do menej ako 18 rokov (pozri časti 4.8,
5.1 a 5.2). Nie sú skúsenosti u pediatrických pacientov mladších ako 2 roky alebo u pediatrických pacientov s CML s pozitívnym chromozómom Philadelphia v akcelerovanej fáze alebo blastickej kríze. K dispozícii nie sú žiadne údaje od novodiagnostikovaných pediatrických pacientov mladších
ako 10 rokov a iba obmedzené údaje od pediatrických pacientov mladších ako 6 rokov s rezistenciou
alebo intoleranciou voči imatinibu.
Spôsob podávania
Tasigna sa má užívať dvakrát denne s intervalom približne 12 hodín a nesmie sa užívať s jedlom.
Tvrdé kapsuly sa majú prehltnúť celé a zapiť vodou. Jedlo sa nemá požiť 2 hodiny pred užitím dávky
a jedlo sa nemá požiť najmenej jednu hodinu po užití dávky.
Pre pacientov, ktorí nie sú schopní prehĺtať tvrdé kapsuly, obsah každej tvrdej kapsuly možno zmiešať s jednou čajovou lyžičkou jablčného pyré a ihneď ho užiť. Nesmie sa použiť viac než jedna lyžička jablčného pyré a nesmie sa požiť iné jedlo než jablčné pyré (pozri časti 4.4 a 5.2).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Myelosupresia
Liečba nilotinibom sa spája s trombocytopéniou, neutropéniou a anémiou (všeobecné kritériá toxicity
stupňa 3-4 podľa National Cancer Institute). Kompletné vyšetrenie krvného obrazu je potrebné vykonať každé dva týždne počas prvých 2 mesiacov a potom každý mesiac, alebo ak je to klinicky indikované. Myelosupresia bola spravidla reverzibilná a zvyčajne ju bolo možné zvládnuť dočasným vysadením alebo znížením dávky Tasigny (pozri časť 4.2).
Predĺženie intervalu QT
Ukázalo sa, že nilotinib v závislosti od koncentrácie predlžuje u dospelých a pediatrických pacientov
repolarizáciu srdcových komôr, čo sa stanovilo prostredníctvom intervalu QT na povrchovom EKG.
V klinickej štúdii fázy III u pacientov s novodiagnostikovanou CML v chronickej fáze, ktorí dostávali
300 mg nilotinibu dvakrát denne, bola zmena stredného časovo spriemerovaného intervalu QTcF
v rovnovážnom stave oproti východiskovej hodnote 6 ms. Ani u jedného pacienta sa nevyskytol interval QTcF >480 ms. Nepozorovali sa epizódy torsade de pointes.
V štúdii so zdravými dobrovoľníkmi pri expozíciách porovnateľných s expozíciami pozorovanými
u pacientov bola časovo spriemerovaná stredná zmena QTcF po odrátaní placeba oproti východiskovej
hodnote 7 ms (CI ± 4 ms). Žiadny účastník nemal QTcF >450 ms. Okrem toho sa v priebehu skúšania nepozorovali klinicky významné arytmie. Predovšetkým sa nepozorovali epizódy torsade de pointes
(prechodné alebo pretrvávajúce).
Významné predĺženie intervalu QT sa môže vyskytnúť, keď sa nilotinib nesprávne užíva so silnými inhibítormi CYP3A4 a/alebo s liekmi so známym potenciálom predlžovať QT a/alebo s jedlom (pozri časť 4.5). Prítomnosť hypokaliémie a hypomagneziémie môže tento účinok ďalej zosilniť. Predĺženie intervalu QT môže pacientov vystaviť riziku úmrtia.
Tasigna sa má používať opatrne u pacientov, ktorí majú alebo u ktorých je významné riziko, že sa
u nich môže vyvinúť predĺženie QTc, ako sú pacienti:
- s vrodeným výrazným predĺžením QT.
- s nekompenzovaným alebo významným ochorením srdca vrátane nedávneho infarktu myokardu, kongestívneho zlyhávania srdca, nestabilnej angina pectoris alebo klinicky významnej
bradykardie.
- užívajúci antiarytmiká alebo iné látky, ktoré spôsobujú predĺženie QT.
Odporúča sa dôsledne sledovať účinok na interval QTc a vykonať východiskové EKG vyšetrenie pred začiatkom liečby nilotinibom a ak je to klinicky indikované. Hypokaliémia alebo hypomagneziémia sa
musia korigovať pred podaním Tasigny a majú sa periodicky monitorovať počas liečby.
Náhla smrť
Menej časté prípady (0,1 až 1%) náhlej smrti boli hlásené u pacientov s CML v chronickej alebo
akcelerovanej fáze s rezistenciou alebo intoleranciou voči imatinibu, ktorí v anamnéze mali ochorenie srdca alebo významné srdcové rizikové faktory. Popri základnej malignite boli často prítomné aj sprievodné ochorenia, ako aj súbežne podávané lieky. Prispievajúcimi faktormi mohli byť poruchy repolarizácie komôr. Neboli hlásené žiadne prípady náhlej smrti u novodiagnostikovaných pacientov
s CML v chronickej fáze v klinickej štúdii fázy III.
R
etencia tekutín a edém
U novodiagnostikovaných pacientov s CML sa v klinickej štúdii fázy III menej často (0,1 až 1%)
pozorovali ťažké formy retencie tekutín ako pleurálny výpotok, pľúcny edém a perikardiálny výpotok. Podobné udalosti sa zaznamenali aj z hlásení po uvedení lieku na trh. Nečakaný a rýchly nárast
telesnej hmotnosti musí byť pozorne vyšetrený. Ak sa počas liečby nilotinibom objavia prejavy ťažkej
retencie tekutín, musí byť zhodnotená etiológia a pacienti majú byť náležite liečení (pozri časť 4.2, pokyny k zvládaniu nehematologických toxicít).
Srdcovocievne príhody
U novodiagnostikovaných pacientov s CML v klinickej štúdii fázy III a z hlásení po uvedení lieku na
trh boli zaznamenané srdcovocievne príhody. V tomto klinickom skúšaní s mediánom trvania liečby
60,5 mesiacov sa vyskytli srdcovocievne príhody 3. až 4. stupňa vrátane periférnej arteriálnej okluzívnej choroby (1,4% pri 300 mg a 1,1% pri 400 mg nilotinibu dvakrát denne), ischemická
choroba srdca (2,2% pri 300 mg a 6,1% pri 400 mg nilotinibu dvakrát denne) a ischemické
mozgovocievne príhody (1,1% pri 300 mg a 2,2% pri 400 mg nilotinibu dvakrát denne). Pacienti majú
byť poučení, aby v prípade výskytu akútnych prejavov a príznakov srdcovocievnych príhod okamžite vyhľadali lekársku pomoc. Srdcovocievny stav pacienta musí byť zhodnotený a počas liečby nilotinibom majú byť podľa platných štandardných odporúčaní sledované a aktívne manažované srdcovocievne rizikové faktory. Na kontrolu srdcovocievnych rizík má byť predpísaná vhodná liečba (pozri časť 4.2, pokyny k zvládaniu nehematologických toxicít).
Reaktivácia hepatitídy B
Reaktivácia hepatitídy B u pacientov, ktorí sú chronickými prenášačmi tohto vírusu, sa vyskytla v
prípade, že títo pacienti užívali inhibítory BCR-ABL-tyrozínkinázy. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie.
Pacienti majú byť vyšetrení na HBV infekciu pred začatím liečby nilotinibom. Pred začatím liečby u pacientov s pozitívnym sérologickým testom na hepatitídu B (vrátane pacientov s aktívnym ochorením) a u pacientov s pozitívnym testom na HBV infekciu počas liečby je potrebné konzultovať s odborníkmi na ochorenia pečene a liečbu hepatitídy B. Prenášači vírusu HBV, ktorí potrebujú liečbu liekom nilotinibom, majú byť pozorne sledovaní na prejavy a symptómy aktívnej HBV infekcie počas celej liečby a niekoľko mesiacov po ukončení liečby (pozri časť 4.8).
Osobitné monitorovanie pacientov s Ph+ CML v chronickej fáze, ktorí dosiahli trvalú hlbokú
molekulovú odpoveď
Vhodnosť na ukončenie li ečby
Pacienti, u ktorých sa potvrdila expresia typických BCR-ABL transkriptov, e13a2/b2a2 alebo e14a2/b3a2, sa môžu považovať za vhodných na ukončenie liečby. Pacienti musia mať typické BCR-ABL transkripty, ktoré umožnia kvantifikáciu BCR-ABL, vyhodnotenie hĺbky molekulovej
odpovede a stanovenie prípadnej straty molekulovej remisie po prerušení liečby nilotinibom.
Monitorovani e pacientov, kt orí ukončil i l ieč bu
U pacientov vhodných na ukončenie liečby sa musí vykonávať časté monitorovanie hladín BCR-ABL transkriptov spolu s kvantitatívnym diagnostickým testom validovaným na meranie hladín molekulovej odpovede s citlivosťou najmenej MR4,5 (MR4,5=BCR-ABL/ABL ≤0,0032% IS). Hladiny BCR-ABL transkriptov sa musia stanoviť pred a počas ukončenia liečby (pozri časti 4.2
a 5.1).
Strata veľkej molekulovej odpovede (MMR=BCR-ABL/ABL ≤0,1% IS) alebo potvrdená strata MR4 (dve po sebe idúce merania s odstupom aspoň 4 týždne ukazujúce stratu MR4 (MR4=BCR-ABL/ABL
≤0,01% IS)) bude impulzom pre opätovné začatie liečby v priebehu 4 týždňov od doby, odkedy je známe, že došlo k strate remisie. Molekulový relaps sa môže vyskytnúť počas obdobia bez liečby
a údaje o dlhodobých výsledkoch ešte nie sú k dispozícii. Veľmi dôležité je preto časté monitorovanie hladín BCR-ABL transkriptov a úplného krvného obrazu s diferenciálom, aby sa zistila prípadná strata remisie (pozri časť 4.2). U pacientov, ktorí nedosiahnu MMR po troch mesiacoch od opätovného
začatia liečby, sa má vykonať test na mutáciu kinázy BCR-ABL.
Laboratórne testy a monitoring
Lipidy v krvi
V klinickom skúšaní fázy III u novo diagnostikovaných pacientov s CML sa u 1,1% pacientov liečených 400 mg nilotinibu dvakrát denne vyskytlo zvýšenie celkového cholesterolu stupňa 3-4;
v skupine s dávkovaním 300 mg dvakrát denne však nebolo pozorované žiadne zvýšenie stupňa 3-4 (pozri časť 4.8). Pred začatím liečby nilotinibom, po 3 a 6 mesiacoch od začatia liečby a najmenej raz ročne pri dlhodobej liečbe sa odporúča stanoviť profily lipidov (pozri časť 4.2). Ak je potrebné
podanie inhibítorov HMG-CoA reduktázy (liečiv znižujúcich hladinu lipidov), treba sa pred začatím liečby riadiť podľa časti 4.5, keďže určité inhibítory HMG-CoA reduktázy sú tiež metabolizované
prostredníctvom CYP3A4.
Glukóza v krvi
V klinickom skúšaní fázy III u novo diagnostikovaných pacientov s CML sa u 6,9% pacientov liečených 400 mg nilotinibu dvakrát denne a u 7,2% pacientov liečených 300 mg nilotinibu dvakrát denne vyskytlo zvýšenie glukózy v krvi stupňa 3-4. Pred začatím liečby Tasignou sa odporúča stanoviť hladinu glukózy a podľa klinickej potreby ju monitorovať počas liečby (pozri časť 4.2). Ak výsledky testov oprávňujú liečbu, lekári sa majú riadiť miestnymi normami pre prax a smernicami pre liečbu.
Interakcie s inými liekmi
Je potrebné vyhnúť sa podávaniu Tasigny s látkami, ktoré sú silnými inhibítormi CYP3A4 (vrátane,
ale nielen s ketokonazolom, itrakonazolom, vorikonazolom, klaritromycínom, telitromycínom,
ritonavirom). Ak sa vyžaduje liečba niektorou z uvedených látok, odporúča sa podľa možnosti prerušiť liečbu nilotinibom (pozri časť 4.5). Ak krátkodobé prerušenie liečby nie je možné, je
indikované dôsledné monitorovanie pacienta vzhľadom na predĺženie intervalu QT (pozri časti 4.2, 4.5
a 5.2).
Súčasné užívanie nilotinibu a liekov, ktoré sú silné induktory CYP3A4 (napr. fenytoín, rifampicín,
karbamazepín, fenobarbital a ľubovník bodkovaný), pravdepodobne zníži expozíciu nilotinibu
v klinicky významnej miere. Preto u pacientov, ktorí užívajú nilotinib, je potrebné zvoliť na súbežné podávanie alternatívne liečivá s nižším potenciálom indukovať CYP3A4 (pozri časť 4.5).
Vplyv jedla
Jedlo zvyšuje biologickú dostupnosť nilotinibu. Tasigna sa nesmie užívať spolu s jedlom (pozri časti
4.2 a 4.5) a má sa užívať 2 hodiny po jedle. Najmenej jednu hodinu po užití dávky sa nemá požiť žiadne jedlo. Je potrebné vyhýbať sa grapefruitovej šťave a iným jedlám, o ktorých je známe, že inhibujú CYP3A4. Pre pacientov, ktorí nie sú schopní prehĺtať tvrdé kapsuly, obsah každej tvrdej kapsuly možno zmiešať s jednou čajovou lyžičkou jablčného pyré a ihneď ho užiť. Nesmie sa použiť viac než jedna lyžička jablčného pyré a nesmie sa požiť iné jedlo než jablčné pyré (pozri časť 5.2).
Porucha funkcie pečene
Porucha funkcie pečene má malý vplyv na farmakokinetiku nilotinibu. Podanie jednorazovej dávky
200 mg nilotinibu spôsobilo u osôb s ľahkou, stredne ťažkou a ťažkou poruchou funkcie pečene zväčšenie AUC o 35%, 35% a 19% v porovnaní s kontrolnou skupinou osôb s normálnou funkciou
pečene. Predpokladaná Cmax nilotinibu v rovnovážnom stave sa zvýšila o 29%, 18% a 22%. Z klinických skúšaní boli vylúčení pacienti so zvýšením alanínaminotransferázy (ALT) a/alebo aspartátaminotransferázy (AST) na >2,5-násobok (alebo na >5-násobok, ak súviselo s ochorením)
hornej hranice normálneho rozmedzia a/alebo celkového bilirubínu na >1,5-násobok hornej hranice normálneho rozmedzia. Metabolizmus nilotinibu prebieha prevažne v pečeni. U pacientov s poruchou funkcie pečene sa preto môže zvýšiť expozícia nilotinibu a pri ich liečbe je potrebná opatrnosť (pozri časť 4.2).
Sérová lipáza
Pozorovalo sa zvýšenie lipázy v sére. U pacientov s pankreatitídou v anamnéze sa odporúča opatrnosť.
Keď zvýšenie lipázy sprevádzajú abdominálne príznaky,liečba nilotinibom sa má vysadiť a majú sa
zvážiť vhodné diagnostické postupy na vylúčenie pankreatitídy.
Totálna gastrektómia
Biologická dostupnosť nilotinibu môže byť znížená u pacientov s totálnou gastrektómiou (pozri časť
5.2). U týchto pacientov sa majú zvážiť častejšie následné vyšetrenia.
Syndróm z rozpadu nádoru
Vzhľadom na možný výskyt syndrómu z rozpadu nádoru (TLS) sa pred začatím liečby nilotinibom
odporúča úprava klinicky významnej dehydratácie a liečba vysokých hladín kyseliny močovej (pozri časť 4.8).
Laktóza
Tvrdé kapsuly Tasigny obsahujú laktózu. Pacienti so zriedkavými dedičnými problémami
galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie
nesmú užívať tento liek.
Pediatrická populácia
Laboratórne abnormality vo forme ľahkých až stredne závažných prechodných zvýšení
aminotransferáz a celkového bilirubínu sa u detí pozorovali s vyššou frekvenciou ako u dospelých, čo naznačuje zvýšené riziko hepatotoxicity v pediatrickej populácii (pozri časť 4.8). Funkcia pečene (hladiny bilirubínu a pečeňových aminotransferáz) sa má monitorovať raz za mesiac alebo podľa klinickej indikácie. Zvýšenie bilirubínu a pečeňových aminotransferáz sa má manažovať dočasným prerušením podávania nilotinibu, znížením dávky a/alebo ukončením podávania nilotinibu (pozri
časť 4.2). Dlhodobé následky dlhotrvajúcej liečby nilotinibom u detí a dospievajúcich nie sú známe.
4.5 Liekové a iné interakcie
Tasigna sa môže podávať v kombinácii s hematopoetickými rastovými faktormi, ako sú erytropoetín alebo faktor stimulujúci kolónie granulocytov (G-CSF), ak je to klinicky indikované. Môže sa podávať s hydroxyureou alebo anagrelidom, ak je to klinicky indikované.
Nilotinib sa metabolizuje prevažne v pečeni a je tiež substrátom glykoproteínu P (Pg-p), efluxnej pumpy mnohých liečiv. Preto absorpciu a následnú elimináciu systémovo absorbovaného nilotinibu môžu ovplyvňovať látky, ktoré pôsobia na CYP3A4 a/alebo Pg-p.
Látky, ktoré môžuzvýšiťsérové koncentrácie nilotinibu
Súčasné podávanie nilotinibu s imatinibom (substrát a moderátor P-gp a CYP3A4) malo slabý
inhibičný účinok na CYP3A4 a/alebo P-gp. AUC imatinibu sa zvýšila o 18% až 39% a AUC
nilotinibu sa zvýšila o 18% až 40%. Nie je pravdepodobné, že tieto zmeny sú klinicky významné.
Expozícia nilotinibu u zdravých osôb vzrástla 3-násobne, keď sa podával spolu so silným inhibítorom CYP3A4 ketokonazolom. Preto je potrebné vyhýbať sa súbežnej liečbe silnými inhibítormi CYP3A4 vrátane ketokonazolu, itrakonazolu, vorikonazolu, ritonaviru, klaritromycínu a telitromycínu (pozri časť 4.4). Zvýšenú expozíciu nilotinibu možno očakávať aj pri stredne silných inhibítoroch CYP3A4. Majú sa zvážiť alternatívne súčasne podávané lieky, ktoré neinhibujú alebo len minimálne inhibujú CYP3A4.
Látky, ktoré môžuznížiťsérové koncentrácie nilotinibu
Rifampicín, silný induktor CYP3A4, znižuje Cmax nilotinibu o 64% a zmenšuje AUC nilotinibu o 80%.
Rifampicín a nilotinib sa nemajú používať súčasne.
Súčasné podávanie iných liekov, ktoré indukujú CYP3A4 (napr. fenytoín, karbamazepín, fenobarbital a ľubovník bodkovaný), pravdepodobne tiež zníži expozíciu nilotinibu v klinicky významnej miere.
U pacientov, ktorí majú indikovanú liečbu induktormi CYP3A4, sa majú zvoliť alternatívne látky
s menším potenciálom pre enzýmovú indukciu.
Rozpustnosť nilotinibu závisí od pH, pričom rozpustnosť je nižšia pri vyššom pH. U zdravých osôb, ktoré dostávali 40 mg ezomeprazolu raz denne počas 5 dní, sa pH žalúdka výrazne zvýšilo, ale absorpcia nilotinibu sa len mierne znížila (pokles Cmax o 27% a pokles AUC0-∞ o 34%). Nilotinib možno podľa potreby používať súčasne s ezomeprazolom alebo inými inhibítormi protónovej pumpy.
V štúdii u zdravých subjektov nebola po podaní jednorazovej dávky 400 mg nilotinibu 10 hodín po a 2 hodiny pred famotidínom pozorovaná žiadna významná zmena vo farmakokinetike nilotinibu.
V prípade, že je potrebná súčasná liečba H2 blokátormi, možno ich podať približne 10 hodín pred a 2 hodiny po podaní Tasigny.
Vo vyššie uvedenej štúdii nemalo podanie antacida (hydroxid hlinitý/hydroxid horečnatý/simetikón)
2 hodiny pred alebo 2 hodiny po podaní jednorazovej dávky 400 mg nilotinibu taktiež vplyv na'
farmakokinetiku nilotinibu. V prípade potreby možno podať antacidá približne 2 hodiny po alebo
2 hodiny pred podaním Tasigny.
Látky, ktorých systémovékoncentráciemôžezmeniťnilotinib
In vitro je nilotinib pomerne silný inhibítor CYP3A4, CYP2C8, CYP2C9, CYP2D6 a UGT1A1,
s hodnotou Ki najnižšou pre CYP2C9 (Ki=0,13 mikroM).
Štúdia liekových interakcií u zdravých dobrovoľníkov pri jednorazovom podaní 25 mg warfarínu, citlivého substrátu CYP2C9, a 800 mg nilotinibu nemala za následok žiadne zmeny farmakokinetických parametrov warfarínu alebo farmakodynamiky warfarínu, stanovených ako protrombínový čas (PT) a medzinárodný normalizovaný pomer (INR). Nie sú údaje o rovnovážnom stave. Táto štúdia naznačuje, že klinicky významná lieková interakcia medzi nilotinibom a warfarínom je menej pravdepodobná do dávky 25 mg warfarínu. Vzhľadom na chýbajúce údaje pri rovnovážnom stave sa odporúča kontrola farmakodynamických markerov warfarínu (INR alebo PT) po začatí liečby nilotinibom (najmenej počas prvých 2 týždňov).
U pacientov s CML nilotinib podávaný v dávke 400 mg dvakrát denne po dobu 12 dní zvýšil 2,6 resp.
2-násobne systémovú expozíciu (AUC resp. Cmax) perorálne podaného midazolamu (substrát CYP3A4). Nilotinib je stredne silný inhibítor CYP3A4. Pri súčasnom podávaní spolu s nilotinibom preto môže byť systémová expozícia iných liekov primárne metabolizovaných cez CYP3A4 (napr. určité inhibítory HMG-CoA reduktázy) zvýšená. U liekov s úzkym terapeutickým indexom, ktoré sú substrátmi CYP3A4 (vrátane alfentanilu, cyklosporínu, dihydroergotamínu, ergotamínu, fentanylu, sirolimu a takrolimu ale nevynímajúc aj iné lieky), môže byť potrebný monitoring a úprava dávky, pokiaľ sa podávajú spoločne s nilotinibom.
Antiarytmiká a iné látky,ktorémôžupredlžovaťQT interval
Nilotinib sa má používať opatrne u pacientov, ktorí majú alebo u ktorých sa môže vyvinúť predĺženie
QT intervalu, vrátane pacientov užívajúcich antiarytmiká ako amiodaron, disopyramid, prokaínamid,
chinidín a sotalol, alebo iné liečivá, ktoré spôsobujú predĺženie QT ako chlorochín, halofantrín,
klaritromycín, haloperidol, metadón a moxifloxacín (pozri časť 4.4).
Interakcie s jedlom
Absorpcia a biologická dostupnosť nilotinibu sa zvýšia, ak sa užíva s jedlom, čo má za následok jej
vyššie sérové koncentrácie (pozri časti 4.2, 4.4 a 5.2). Je potrebné vyhýbať sa grapefruitovej šťave
a iným jedlám, o ktorých je známe, že inhibujú CYP3A4.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia
Ženy vo fertilnom veku musia používať vysoko účinnú antikoncepciu počas liečby nilotinibom a po
dobu do dvoch týždňov od ukončenia liečby.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití nilotinibu u gravidných žien.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Tasigna sa nemá užívať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu nilotinibom. Ak sa užíva počas gravidity, pacientka musí byť informovaná o prípadnom riziku pre plod.
Ak žena, ktorá sa lieči nilotinibom, uvažuje o gravidite, možno zvážiť prerušenie liečby na základe kritérií vhodnosti pre prerušenie liečby, ako sú opísané v častiach 4.2 a 4.4. Existuje obmedzené množstvo údajov o graviditách u pacientok počas pokusu o remisiu bez liečby (TFR, treatment-free remission). Ak sa plánuje gravidita počas fázy TFR, pacientka musí byť informovaná o možnej nutnosti opätovného začatia liečby nilotinibom počas gravidity (pozri časti 4.2 a 4.4).
Dojčenie
Nie je známe, či sa nilotinib vylučuje do ľudského mlieka. Dostupné toxikologické údaje u zvierat
preukázali vylučovanie nilotinibu do mlieka (pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť vylúčené. Tasigna nemá byť užívaná počas dojčenia.
Fertilita
Štúdie na zvieratách neukázali účinok na fertilitu samcov a samíc potkana (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tasigna nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Napriek tomu sa odporúča, aby pacienti, u ktorých sa vyskytnú závraty, únava, zhoršenie zraku alebo iné nežiaduce účinky, ktoré môžu ovplyvňovať schopnosť bezpečne viesť vozidlá alebo obsluhovať stroje, nevykonávali tieto činnosti, kým nežiaduce účinky pretrvávajú (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Údaje uvedené nižšie sú získané pri použití nilotinibu u 279 dospelých pacientov v randomizovanej
klinickej štúdii fázy III s pacientmi s novodiagnostikovanou Ph+ CML v chronickej fáze, ktorí dostávali dávku 300 mg nilotinibu dvakrát denne. K dispozícií sú aj údaje o bezpečnosti z jednej
štúdie s Tasignou o ukončení liečby u pacientov s CML, ktorí dostávali nilotinib ako liečbu prvej línie.
Medián trvania expozície bol 60,5 mesiacov (rozmedzie 0,1-70,8 mesiacov).
Najčastejšími (≥10%) nehematologickými nežiaducimi reakciami boli exantém, pruritus, bolesť hlavy, nauzea, únava, alopécia, myalgia a bolesť v hornej časti brucha. Väčšina z týchto nežiaducich reakcií bola mierna až stredne závažná. Zápcha, suchosť kože, asténia, svalové kŕče, hnačka, artralgia, bolesť brucha, vracanie a periférny edém sa pozorovali menej často (<10% a ≥5%), boli mierne až stredne závažné, zvládnuteľné a spravidla si nevyžiadali zníženie dávky.
K príznakom hematologickej toxicity, ktoré sa objavili pri liečbe, patrí myelosupresia: trombocytopénia (18%), neutropénia (15%) a anémia (8%). Biochemické reakcie na liek zahŕňajú zvýšenú alaníntransaminázu (24%), hyperbilirubinémiu (16%), zvýšenú aspartáttransaminázu (12%), zvýšenú lipázu (11%), zvýšený bilirubín v krvi (10%), hyperglyklémiu (4%), hypercholesterolémiu (3%) a hypertriacylglycerolémiu (<1%). Pleurálny a perikardiálny výpotok sa bez ohľadu na príčinnú súvislosť vyskytli u 2% a <1% pacientov v uvedenom poradí, ktorí dostávali nilotinib 300 mg dvakrát denne. Gastrointestinálne krvácanie bez ohľadu na príčinnú súvislosť bolo hlásené u 3% z týchto pacientov.
Zmena stredného časovo spriemerovaného intervalu QTcF v rovnovážnom stave oproti východiskovej hodnote bola 6 ms. Ani u jedného pacienta sa počas liečby skúšaným liekom nevyskytla absolútna hodnota QTcF >500 ms. Predĺženie QTcF oproti východiskovej hodnote väčšie ako 60 ms sa počas liečby skúšaným liekom pozorovalo u <1% pacientov. Nezaznamenala sa náhla smrť alebo epizódy torsade de pointes (prechodné alebo pretrvávajúce). Nepozoroval sa pokles priemernej ejekčnej frakcie ľavej komory (LVEF) oproti východiskovej hodnote kedykoľvek počas liečby. Žiadny pacient nemal počas liečby LVEF <45%, ani absolútny pokles LVEF o viac než 15%.
Ukončenie liečby pre nežiaduce reakcie na liek sa zaznamenalo u 10% pacientov.
Tabuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie sú zoradené podľa frekvencie na základe nasledujúcej konvencie: veľmi časté
(≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až
<1/1 000), veľmi zriedkavé (<1/10 000) a neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Než i aduce reakcie najčast ej š ie hlásené v klinických skúšaniach s Tasignou
Nehematologické nežiaduce reakcie (okrem laboratórnych abnormalít), ktoré sú hlásené najmenej u
5% dospelých pacientov liečených 300 mg nilotinibu dvakrát denne v randomizovanej štúdii fázy III
ukazuje Tabuľka 3.
Tabuľka 3 Nehematologické nežiaduce reakcie (≥5% všetkých pacientov)*
O
rgánový systém Frekvencia Nežiaduca reakcia Všetky
stupne
Stupeň
3-4
% % Poruchy nervového systému Veľmi časté Bolesť hlavy 16 2
P
oruchy gastrointestinálneho traktu
Veľmi časté Nauzea 14 <1
Veľmi časté Bolesť v hornej časti
brucha
10 1
P
oruchy kože a podkožného
t
kaniva
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
C
elkové poruchy a reakcie v mieste podania
Časté Zápcha 10 0
Časté Hnačka 9 <1
Časté Bolesť brucha 6 0
Časté Vracanie 6 0
Časté Dyspepsia 5 0
Veľmi časté Exantém 33 <1
Veľmi časté Pruritus 18 <1
Veľm časté Alopécia 10 0
Časté Suchosť kože 10 0
Veľmi časté Myalgia 10 <1
Časté Svalové kŕče 9 0
Časté Artralgia 8 <1
Časté Bolesť v končatine 5 <1
Veľmi časté Únava 12 0
Časté Asténia 9 <1
Časté Periférny edém 5 <1
* Za účelom uvedenia v tejto tabuľke sú percentá zaokrúhlené na celé čísla. Pre určenie termínov
s frekvenciou aspoň 5% a klasifikáciu termínov podľa kategórie frekvencií sa však používajú percentá
s presnosťou jedného desatinného miesta.
Nasledujúce nežiaduce reakcie boli hlásené u dospelých pacientov v štúdii fázy III Tasigny s frekvenciou nižšou ako 5%. Pri laboratórnych abnormalitách sú hlásené aj veľmi časté udalosti (³1/10), ktoré nie sú zahrnuté do Tabuľky 3. Tieto nežiaduce reakcie sú uvedené podľa klinickej významnosti a sú zoradené podľa klesajúcej závažnosti v rámci každej kategórie na základe nasledujúcej dohody: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100), neznáme (z dostupných údajov).
Infekcie a nákazy
Časté: folikulitída, infekcia horných dýchacích ciest (vrátane faryngitídy, nazofaryngitídy, nádchy). Neznáma frekvencia: infekcia vírusom herpes simplex, orálna kandidóza, subkutánny absces, análny
absces, tinea pedis, reaktivácia hepatitídy B.
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Časté: kožný papilóm.
Neznáma frekvencia: papilóm ústnej dutiny, paraproteinémia.
Poruchy krvi a lymfatického systému
Časté: leukopénia, eozinofília, lymfopénia.
Menej časté: pancytopénia.
Neznáma frekvencia: febrilná neutropénia.
Poruchy imunitného systému
Neznáma frekvencia: hypersenzitivita.
Poruchy endokrinného systému
Neznáma frekvencia: sekundárny hyperparatyreoidizmus.
Poruchy metabolizmu a výživy
Veľmi časté: hypofosfatémia (vrátane zníženia fosforu v krvi).
Časté: diabetes mellitus, hypercholesterolémia, hyperlipidémia, hypertriacylglycerolémia,
hyperglykémia, znížená chuť do jedenia, hypokalciémia, hypokaliémia.
Menej časté: hyperkaliémia, dyslipidémia, dna.
Neznáma frekvencia: hyperurikémia, hypoglykémia, poruchy chuti do jedenia.
Psychické poruchy
Časté: nespavosť, depresia, úzkosť.
Neznáma frekvencia: amnézia, dysfória.
Poruchy nervového systému
Časté: závraty, hypestézia, periférna neuropatia.
Menej časté: ischemická mozgová príhoda, mozgový infarkt, migréna, parestézia.
Neznáma frekvencia: mozgovocievne príhody, bazilárna arteriálna stenóza, synkopa, tremor, letargia, dyzestézia, syndróm nepokojných nôh, hyperestézia.
Poruchy oka
Časté: očný pruritus, konjunktivitída, suchosť očí (vrátane xeroftalmie).
Menej časté: edém mihalnice, fotopsia, krvácanie do spojoviek, hyperémia (skléry, spojoviek, očí). Neznáma frekvencia: periorbitálny edém, blefaritída, bolesť očí, chorioretinopatia, alergická konjunktivitída, ochorenie povrchu oka, neostré videnie.
Poruchy ucha a labyrintu
Časté: vertigo.
Poruchy srdca a srdcovej činnosti*
Časté: angina pectoris, arytmia (zahŕňa atrioventrikulárny blok, tachykardiu, fibriláciu predsiení, komorové extrasystoly, bradykardiu), predĺženie QT na elektrokardiograme, palpitácie, infarkt
myokardu.
Menej časté: zlyhávanie srdca, cyanóza.
Neznáma frekvencia: pokles ejekčnej frakcie, perikardiálny výpotok, perikarditída, diastolická
dysfunkcia, blokáda ľavého Tawarovho ramienka.
*hlásené v skupine liečby 300 mg dvakrát denne a/alebo 400 mg dvakrát denne štúdie fázy III
Poruchy ciev
Časté: hypertenzia, návaly tepla.
Menej časté: intermitentná klaudikácia, periférna arteriálna okluzívna choroba, arterioskleróza. Neznáma frekvencia: hematóm, periférna arteriálna stenóza.
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté: dyspnoe, kašeľ.
Menej časté: pleurálny výpotok.
Neznáma frekvencia: námahové dyspnoe, pleuritída, epistaxa, orofaryngeálna bolesť.
Poruchy gastrointestinálneho traktu
Časté: distenzia brucha, nepríjemné pocity v bruchu, dysgeúzia, flatulencia.
Menej časté: pankreatitída, gastritída, citlivosť zubov.
Neznáma frekvencia: vred pažeráka, žalúdkový vred, bolesť v ezofágu, stomatitída, suchosť v ústach, enterokolitída, hemoroidy, hiátová hernia, rektálne krvácanie, gingivitída.
Poruchy pečene a žlčových ciest
Veľmi časté: hyperbilirubinémia (vrátane zvýšenia bilirubínu v krvi). Časté: abnormálna funkcia pečene.
Menej časté: žltačka.
Neznáma frekvencia: toxická hepatitída.
Poruchy kože a podkožného tkaniva
Časté: erytém, hyperhidróza, kontúzia, akné, dermatitída (vrátane alergickej, exfoliatívnej a akneiformnej), nočné potenie, ekzém.
Menej časté: lieková erupcia, bolestivosť kože.
Neznáma frekvencia: erythema multiforme, urtikária, pľuzgier, kožné cysty, sebaceózna hyperplázia,
opuch tváre, atrofia kože, hypertrofia kože, exfoliácia kože, hyperpigmentácia kože, zmena sfarbenia pokožky, hyperkeratóza, psoriáza.
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté: bolesť kostí, bolesti chrbta, svalová slabosť.
Menej časté: bolesť svalov a kostí, bolesť v boku.
Poruchy obličiek a močových ciest
Neznáma frekvencia: dyzúria, polakizúria, chromatúria.
Poruchy reprodukčného systému a prsníkov
Menej časté: erektilná dysfunkcia.
Neznáma frekvencia: gynekomastia, stvrdnutie prsníkov, menorágia, opuch prsných bradaviek.
Celkové poruchy a reakcie v mieste podania
Časté: pyrexia, bolesť v hrudníku (vrátane bolesti v hrudníku nekardiálneho pôvodu), nepríjemné pocity v hrudníku.
Menej časté: bolesť, zimnica, pocit zmeny telesnej teploty (vrátane pocitu horúčavy, pocitu chladu),
celková nevoľnosť.
Neznáma frekvencia: edém tváre, lokalizovaný edém.
Laboratórne a funkčné vyšetrenia
Veľmi časté: zvýšenie alanínaminotransferázy, zvýšenie aspartátaminotransferázy, zvýšenie lipázy,
zvýšenie lipoproteínového cholesterolu (vrátane lipoproteínu s nízkou a vysokou hustotou), zvýšenie celkového cholesterolu, zvýšené triacylglyceroly.
Časté: zníženie hemoglobínu, zvýšenie amylázy v krvi, zvýšenie alkalickej fosfatázy v krvi, zvýšenie gamaglutamyltransferázy, zvýšenie telesnej hmotnosti, zvýšenie inzulínu v krvi, zníženie globulínov. Neznáma frekvencia: zvýšenie parathormónu v krvi, zníženie inzulínu v krvi, zníženie C-peptidu pre
inzulín, zníženie telesnej hmotnosti.
Klinicky významné alebo závažné abnormality rutinných hematologických alebo biochemických laboratórnych hodnôt u dospelých pacientov sú uvedené v Tabuľke 4.
Tabuľka 4 Laboratórne abnormality stupňa 3-4*
H
e
m
atologické parametre
Myelosupresia
n=279
(%)
- Neutropénia 12
- Trombocytopénia 10
- Anémia 4
Biochemické parametre
- Zvýšený kreatinín 0
- Zvýšená lipáza 9
- Zvýšená SGOT (AST) 1
- Zvýšená SGPT (ALT) 4
- Hypofosfatémia 7
- Zvýšený bilirubín (celkový) 4
- Zvýšená glukóza 7
- Zvýšený cholesterol (celkový) 0
- Zvýšené triacylglyceroly 0
*Za účelom uvedenia v tejto tabuľke sa používajú percentá s presnosťou jedného desatinného miesta, zaokrúhlené na celé číslo
Ukončenieliečbyu pacientov s Ph+ CML v chronickej fáze, ktorí dosiahli trvalú hlbokú molekulovúodpoveďPo ukončení liečby nilotinibom v rámci pokusu dosiahnuť TFR môžu pacienti pocítiť
muskuloskeletálne symptómy častejšie ako pred ukončením liečby, napr. myalgiu, bolesť končatín, artralgiu, bolesť kostí, bolesť chrbtice alebo muskuloskeletálnu bolesť.
V klinickej štúdii fázy II u novodiagnostikovaných pacientov s Ph+ CML v chronickej fáze (N=190) boli muskuloskeletálne symptómy hlásené počas jedného roka od ukončenia liečby Tasignou u 24,7% oproti 16,3% počas predchádzajúceho roka pri liečbe nilotinibom.
O
pis vybranýchnežiaducichreakciíReaktivácia hepatitídy BV súvislosti s inhibítormi BCR-ABL-tyrozínkinázy bola hlásená reaktivácia hepatitídy B. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie (pozri časť 4.4).
Skúsenosti po uvedení na trhNasledujúce nežiaduce reakcie pochádzajú zo skúsenosti s Tasignou po uvedení na trh formou
spontánnych hlásení, prípadov publikovaných v literatúre, programov rozšíreného prístupu a z klinických štúdií iných ako globálne klinické skúšania pre registráciu. Keďže tieto reakcie sú hlásené dobrovoľne u populácie neurčenej veľkosti, nie je vždy možné spoľahlivo odhadnúť ich frekvenciu alebo stanoviť príčinnú súvislosť s expozíciou nilotinibu.
Frekvencia zriedkavé: U pacientov liečených nilotinibom sa zaznamenali prípady syndrómu z rozpadu nádoru.
Pediatrická populáciaBezpečnosť nilotinibu u pediatrických pacientov (od 2 do <18 rokov) s CML s pozitívnym
chromozómom Philadelphia v chronickej fáze (n=69) sa skúmala v dvoch štúdiách (pozri časť 5.1).
Frekvencia, typ a závažnosť nežiaducich reakcií pozorovaných u pediatrických pacientov sa vo všeobecnosti zhodovali s tými, ktoré sa pozorovali u dospelých, s výnimkou laboratórnych abnormalít
hyperbilirubinémie (stupeň 3/4: 13,0%) a zvýšenia aminotransferáz (AST stupeň 3/4: 1,4%, ALT
stupeň 3/4: 8,7%), ktoré boli hlásené častejšie ako u dospelých pacientov. Hladiny bilirubínu a aminotrasferáz sa majú počas liečby monitorovať (pozri časti 4.2 a 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieZaznamenali sa ojedinelé správy o úmyselnom predávkovaní nilotinibom, keď sa nešpecifikovaný počet tvrdých kapsúl Tasigny požil v kombinácii s alkoholom a inými liekmi. Udalosti zahŕňali neutropéniu, vracanie a ospalosť. Zmeny EKG alebo hepatotoxicita neboli uvádzané. Podľa hlásení došlo následne k zotaveniu.
V prípade predávkovania má byť pacient pod dohľadom a má sa mu podať primeraná podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE08
Mechanizmus účinku
Nilotinib je účinný inhibítor aktivity ABL-tyrozínkinázy onkoproteínu BCR-ABL v bunkových líniách
aj v primárnych leukemických bunkách s pozitívnym chromozómom Philadelphia. Látka sa viaže
s vysokou afinitou na väzbové miesto ATP takým spôsobom, že je účinným inhibítorom BCR-ABL divokého typu a zachováva si aktivitu voči 32/33 mutantných foriem BCR-ABL rezistentných voči imatinibu. V dôsledku tejto biochemickej aktivity nilotinib selektívne inhibuje proliferáciu a indukuje apoptózu bunkových línií a primárnych leukemických buniek s pozitívnym chromozómom Philadelphia od pacientov s CML. Na myšacích modeloch CML nilotinib ako jediná použitá látka po perorálnom podávaní znižuje nádorovú záťaž a predlžuje prežívanie.
Farmakodynamické účinkyNilotinib má malý alebo žiadny účinok na väčšinu iných skúmaných proteínkináz vrátane Src, okrem
kináz receptorov PDGF, KIT a efrínu, ktoré inhibuje v koncentráciách v rozmedzí dosahovanom po
perorálnom podaní terapeutických dávok odporúčaných na liečbu CML (pozri Tabuľku 5).
Tabuľka 5 Profil nilotinibu voči kinázam (fosforylácia IC50 nM)BCR-ABL PDGFR KIT
20 69 210
Klinickáúčinnosť Kl i ni cké š túdie pri novodi agnosti kovanej CM L v c hroni ck ej f áze Na stanovenie účinnosti nilotinibu oproti imatinibu sa uskutočnilo otvorené, multicentrické, randomizované klinické skúšanie fázy III s 846 dospelými pacientmi s novodiagnostikovanou CML
s pozitívnym chromozómom Philadelphia v cytogeneticky potvrdenej chronickej fáze. Pacientom bolo
ochorenie diagnostikované v priebehu posledných šiestich mesiacov a v minulosti neboli liečení, s výnimkou hydroxyurey a/alebo anagrelidu. Pacienti boli randomizovaní v pomere 1:1:1, aby dostávali buď nilotinib 300 mg dvakrát denne (n=282), nilotinib 400 mg dvakrát denne (n=281), alebo imatinib 400 mg raz denne (n=283). Randomizácia bola stratifikovaná podľa Sokalovho rizikového skóre v čase stanovenia diagnózy.
Východiskové charakteristiky boli dobre vyvážené medzi troma skupinami liečby. Medián veku bol
47 rokov v oboch skupinách nilotinibu a 46 rokov v skupine imatinibu, pričom 12,8% pacientov bolo vo veku ≥65 rokov v skupine liečby nilotinibu 300 mg dvakrát denne, 10,0% v skupine nilotinibu
400 mg dvakrát denne a 12,4% v skupine imatinibu 400 mg raz denne. Počet liečených mužov bol o niečo vyšší než žien (56,0% v skupine nilotinibu 300 mg dvakrát denne, 62,3% v skupine nilotinibu
400 mg dvakrát denne a 55,8% v skupine imatinibu 400 mg raz denne). Viac než 60% všetkých
pacientov boli belosi a 25% všetkých pacientov boli Ázijčania.
Primárna analýza údajov sa vykonala v čase, keď všetkých 846 pacientov ukončilo 12 mesiacov liečby
(alebo ju vysadilo skôr). Následné analýzy sa týkajú času, keď pacienti ukončili 24, 36, 48, 60 a
72 mesiacov liečby (alebo ju vysadili skôr). Medián trvania liečby bol približne 70 mesiacov
v skupinách liečby nilotinibom a 64 mesiacov v skupine imatinibu. Medián skutočnej veľkosti dávky bol 593 mg/deň pri nilotinibe 300 mg dvakrát denne, 772 mg/deň pri nilotinibe 400 mg dvakrát denne a 400 mg/deň pri imatinibe 400 mg raz denne. Táto štúdia naďalej pokračuje.
Primárnym parametrom účinnosti bola veľká molekulová odpoveď (major molecular response, MMR)
po 12 mesiacoch. MMR bola definovaná ako ≤0,1% BCR-ABL/ABL % podľa medzinárodnej škály
(international scale, IS) pri stanovení RQ-PCR, čo zodpovedá poklesu BCR-ABL transkriptov ≥3 log oproti štandardizovanej východiskovej hodnote. Podiel MMR po 12 mesiacoch bol štatisticky významne vyšší pri nilotinibe 300 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne (44,3% oproti 22,3%, p<0,0001). Podiel MMR po 12 mesiacoch bol tiež štatisticky významne vyšší
pri nilotinibe 400 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne (42,7% oproti 22,3%, p<0,0001).
Podiel MMR po 3, 6, 9 a 12 mesiacoch bol 8,9%, 33,0%, 43,3% a 44,3% pri nilotinibe 300 mg dvakrát denne, 5,0%, 29,5%, 38,1% a 42,7% pri nilotinibe 400 mg dvakrát denne a 0,7%, 12,0%, 18,0%
a 22,3% pri imatinibe 400 mg raz denne.
Podiely MMR v 12., 24., 36., 48.,60. a 72. mesiaci sú uvedené v Tabuľke 6.
Tabuľka 6 Podiel MMR
MMR v 12. mesiaci
Nilotinib
300 mg dvakrát denne
n=282
(%)
Nilotinib
400 mg dvakrát denne
n=281
(%)
Imatinib
400 mg raz denne n=283
(%)
Odpoveď (95% CI) 44,31 (38,4; 50,3) 42,71 (36,8; 48,7) 22,3 (17,6; 27,6)
MMR v 24. mesiaciOdpoveď (95% CI) 61,71 (55,8; 67,4) 59,11 (53,1; 64,9) 37,5 (31,8; 43,4)
MMR v 36. mesiaci2Odpoveď (95% CI) 58,51 (52,5; 64,3) 57,31 (51,3; 63,2) 38,5 (32,8; 44,5)
MMR v 48. mesiaci3Odpoveď (95% CI) 59,91 (54,0; 65,7) 55,2 (49,1; 61,1) 43,8 (38,0; 49,8)
MMR v 60. mesiaci4Odpoveď (95% CI) 62,8 (56,8; 68,4) 61,2 (55,2; 66,9) 49,1 (43,2; 55,1)
MMR v 72. mesiaci5Odpoveď (95% CI) 52,5 (46,5; 58,4) 57,7 (51,6; 63,5) 41,7 (35,9; 47,7)
1 Hodnota p pre podiel odpovedí (oproti imatinibu 400 mg) <0,0001 testu podľa Cochrana-Mantela- Haenszela (CMH)
2 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 199 (35,2%) pacientov vyhodnotiť MMR v 36. mesiaci
(87 v skupine nilotinibu 300 mg dvakrát denne a 112 v skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=17), atypické východiskové transkripty (n=7) alebo pre ukončenie liečby pred 36. mesiacom (n=175).
3 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 305 (36,1%) pacientov vyhodnotiť MMR v 48. mesiaci
(98 v skupine nilotinibu 300 mg dvakrát denne, 88 v skupine nilotinibu 400 mg dvakrát denne a 119 v skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=18), atypické východiskové transkripty (n=8) alebo pre ukončenie liečby pred 48. mesiacom (n=279).
4 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 322 (38,1%) pacientov vyhodnotiť MMR v 60. mesiaci
(99 v skupine nilotinibu 300 mg dvakrát denne, 93 v skupine nilotinibu 400 mg dvakrát denne a 130 v skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=9), atypické východiskové transkripty (n=8) alebo pre ukončenie liečby pred 60. mesiacom (n=305).
5 Len pacienti, ktorí dosiahli MMR v uvedenom čase, sú zahrnutí ako responderi v danom čase. Zo všetkých pacientov nebolo možné u celkovo 395 (46,7%) pacientov vyhodnotiť MMR v 72. mesiaci
(130 v skupine nilotinibu 300 mg dvakrát denne, 110 v skupine nilotinibu 400 mg dvakrát denne a 155
v skupine imatinibu) pre chýbajúce/nevyhodnotiteľné vyšetrenie PCR (n=25), atypické východiskové
transkripty (n=8) alebo pre ukončenie liečby pred 72. mesiacom (n=362).
Podiely MMR v rôznych časoch (vrátane pacientov, ktorí dosiahli MMR ako responderi v danom čase
alebo skôr) sú uvedené v kumulatívnej incidencii MMR (pozri Obrázok 1).
Obrázok 1 Kumulatívna incidencia MMR
100
90
Nilotinib 300 mg dvakrát denne (n = 282)
Nilotinib 400 mg dvakrát denne (n = 281)
Imatinib 400 mg raz denne (n = 283)
Po 6 rokoch
P
o
3 rokoch
P
o
4 rokoch
P
o
5 rokoch
80
70
Po 1 roku60
Po 2 rokoch71%;
P < 0,0001
73%;
P < 0,0001
70%;
P < 0,0001
76%;
P < 0,0001 77%;
P < 0,0001
77%;
P < 0,0001
73%;
P < 0,0001
79%;
P < 0,0001
77%;
P < 0,0001
55%;
P < 0,0001
50 51%;
P < 0,0001
40
30
67%;
P < 0,0001
44%
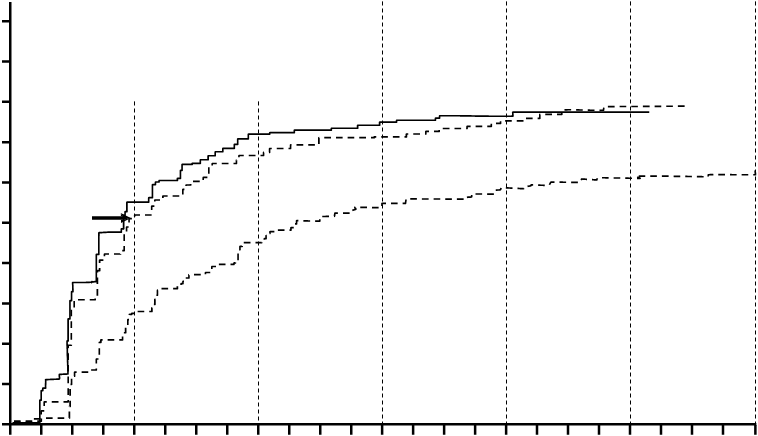
53%
56%
60%
61%
20
27%
10
0
0 6 12 18 24 30 36 42 48 54 60
Mesiace od randomizácie
66 72
Vo všetkých skupinách rizika podľa Sokala boli podiely MMR v každom čase trvale vyššie v oboch
skupinách nilotinibu ako v skupine imatinibu.
V retrospektívnej analýze 91% (234/258) pacientov liečených nilotinibom 300 mg dvakrát denne dosiahlo úroveň BCR-ABL ≤10% po 3 mesiacoch liečby v porovnaní so 67% (176/264) pacientov liečených nilotinibom 400 mg dvakrát denne. Pacienti s BCR-ABL na úrovni ≤10% po 3 mesiacoch liečby preukázali väčšiu mieru celkového prežívania v 72. mesiaci v porovnaní s tými, ktorí nedosiahli takúto úroveň molekulovej odpovede (94,5% oproti 77,1% v uvedenom poradí [p=0,0005]).
Na základe analýzy času do prvej MMR podľa Kaplana-Meiera bola pravdepodobnosť dosiahnutia MMR v rozličných časoch vyššia pri nilotinibe 300 mg aj 400 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne (HR=2,17 a stratifikovaný log-rank p<0,0001 medzi nilotinibom
300 mg dvakrát denne a imatinibom 400 mg raz denne, HR=1,88 a stratifikovaný log-rank p<0,0001
medzi nilotinibom 400 mg dvakrát denne a imatinibom 400 mg raz denne).
Podiel pacientov, ktorí mali molekulovú odpoveď ≤0,01% a ≤0,0032% podľa IS v rozličných časoch sú uvedené v Tabuľke 7 a podiel pacientov, ktorí mali molekulovú odpoveď ≤0,01% a ≤0,0032% podľa IS v rôznych časoch sú uvedené na Obrázkoch 2 a 3. Molekulové odpovede ≤0,01% a
≤0,0032% podľa IS zodpovedajú zníženiu transkriptov BCR-ABL ≥4 log a ≥4,5 log v uvedenom poradí oproti štandardizovanej východiskovej hodnote.
T
abuľka 7 Podiely pacientov, ktorí mali molekulovú odpoveď ≤0,01% (zníženie 4 log) a
≤
0,0032% (zníženie 4,5 log)
Nilotinib
300 mg dvakrát denne n=282
(%)
Nilotinib
400 mg dvakrát denne n=281

(%)
Imatinib
400 mg raz denne n=283
(%)
≤
0,01% ≤0,0032% ≤0,01% ≤0,0032% ≤0,01% ≤0,0032%
V 12. mesiaci 11,7 4,3 8,5 4,6 3,9 0,4
V 24. mesiaci 24,5 12,4 22,1 7,8 10,2 2,8
V 36. mesiaci 29,4 13,8 23,8 12,1 14,1 8,1
V 48. mesiaci 33,0 16,3 29,9 17,1 19,8 10,2
V 60. mesiaci 47,9 32,3 43,4 29,5 31,1 19,8
V 72. mesiaci 44,3 31,2 45,2 28,8 27,2 18,0
Obrázok 2 Kumulatívna incidencia molekulovej odpovede ≤0,01% (zníženie 4 log)
100
Nilotinib 300 mg dvakrát denne (n = 282)
Nilotinib 400 mg dvakrát denne (n = 281)
90 Imatinib 400 mg raz denne (n = 283)
80
70
60
50
Po 1 roku
40
20%;
P < 0,0001
30
15%;
P = 0,0004
20
Po 2 rokoch39%;
P < 0,0001
33%;
P < 0,0001
Po 3 rokoch50%;
P < 0,0001

44%;
P < 0,0001
26%
Po 4 rokoch56%;
P < 0,0001
50%;
P < 0,0001
32%
Po 5 rokoch66%;
P < 0,0001
63%;
P < 0,0001
42%
Po 6 rokoch67%;
P < 0,0001
65%;
P < 0,0001
43%
18%
10
0 6%
0 6 12 18 24 30 36 42 48 54 60
66 72
Mes
i
ac
e od randomizácie
O
brázok 3 Kumulatívna incidencia molekulovej odpovede ≤0,0032% (zníženie 4,5 log)
100
90
Nilotinib 300 mg dvakrát denne (n = 282)
Nilotinib 400 mg dvakrát denne (n = 281)
Imatinib 400 mg raz denne (n = 283)
80
70
60
50
40
Po 1 roku30
11%;
P < 0,0001
20 7%;
P < 0,0001
Po 2 rokoch25%;
P < 0,0001
Po 3 rokoch32%;
P < 0,0001
28%;
P = 0,0003
Po 4 rokoch40%;
P < 0,0001
37%;
P = 0,0002
23%
Po 5 rokoch54%;
P < 0,0001
52%;
P < 0,0001
31%
Po 6 rokoch56%;
P < 0,0001
55%;
P < 0,0001
33%
1% 19%;
10 P = 0,0006
9%
0
15%
0 6 12 18 24 30 36 42 48 54 60
66 72
Mes
i
ac
e od randomizácie
Na základe odhadov podľa Kaplana-Meiera pre trvanie prvej MMR boli podiely pacientov, ktorí si
udržali odpoveď počas 72 mesiacov spomedzi pacientov, ktorí dosiahli MMR 92,5% (95% CI:
88,6-96,4%) v skupine nilotinibu 300 mg dvakrát denne, 92,2% (95% CI: 88,5-95,9%) v skupine nilotinibu 400 mg dvakrát denne a 88,0% (95% CI: 83,0-93,1%) v skupine imatinibu 400 mg raz
denne.
Kompletná cytogenetická odpoveď (CCyR) bola definovaná ako 0% Ph+ metafáz v kostnej dreni na základe hodnotenia minimálne 20 metafáz. Najlepší podiel CCyR po 12 mesiacoch (vrátane pacientov, ktorí dosiahli CCyR ako responderi v 12. mesiaci alebo skôr) bol štatisticky vyšší pri nilotinibe
300 mg aj 400 mg dvakrát denne v porovnaní s imatinibom 400 mg raz denne, pozri Tabuľku 8.
Podiel CCyR v priebehu 24 mesiacov (vrátane pacientov, ktorí dosiahli CCyR ako responderi
v 24. mesiaci alebo skôr), bol štatisticky vyšší v skupinách nilotinibu 300 mg dvakrát denne aj 400 mg dvakrát denne v porovnaní so skupinou imatinibu 400 mg raz denne.
Tabuľka 8 Najlepší podiel CCyR
V priebehu 12 mesiacov
Nilotinib
300 mg dvakrát denne n=282
(%)
Nilotinib
400 mg dvakrát denne n=281
(%)
Imatinib
400 mg raz denne n=283
(%)
Odpoveď (95% CI) 80,1 (75,0; 84,6) 77,9 (72,6; 82,6) 65,0 (59,2; 70,6)
Žiadna odpoveď 19,9 22,1 35,0
Hodnota p testu CMH pre podiel odpovede (oproti imatinibu 400 mg raz denne)
V priebehu 24 mesiacov
<0,0001 0,0005
Odpoveď (95% CI) 86,9 (82,4; 90,6) 84,7 (79,9; 88,7) 77,0 (71,7; 81,8)
Žiadna odpoveď 13,1 15,3 23,0
Hodnota p testu CMH pre podiel odpovede (oproti imatinibu 400 mg raz denne)
0,0018 0,0160
Na základe odhadov podľa Kaplana-Meiera boli podiely pacientov, ktorí si udržali odpoveď počas
72 mesiacov spomedzi pacientov, ktorí dosiahli CCyR 99,1% (95% CI: 97,9-100%) v skupine nilotinibu 300 mg dvakrát denne, 98,7% (95% CI: 97,1-100%) v skupine nilotinibu 400 mg dvakrát
denne a 97,0% (95% CI: 94,7-99,4%) v skupine imatinibu 400 mg raz denne.
Progresia do akcelerovanej fázy (accelerated phase, AP) alebo blastickej krízy (blast crisis, BC) počas liečby je definovaná ako čas od dátumu randomizácie do prvej dokumentovanej progresie ochorenia do akcelerovanej fázy alebo blastickej krízy alebo úmrtia súvisiaceho s CML. Progresia do akcelerovanej fázy alebo blastickej krízy počas liečby sa pozorovala celkovo u 17 pacientov: u
2 pacientov pri nilotinibe 300 mg dvakrát denne, u 3 pacientov pri nilotinibe 400 mg dvakrát denne a u
12 pacientov pri imatinibe 400 mg raz denne. Odhadované podiely pacientov bez progresie do akcelerovanej fázy alebo blastickej krízy v 72 mesiacoch boli 99,3%, 98,7% a 95,2% v uvedenom
poradí (HR=0,1599 a stratifikovaný log-rank p=0,0059 medzi nilotinibom 300 mg dvakrát denne
a imatinibom raz denne, HR=0,2457 a stratifikovaný log-rank p=0,0185 medzi nilotinibom 400 mg dvakrát denne a imatinibom raz denne). Žiadne nové prípady progresie do AP/BC neboli hlásené po
analýze 2-ročného obdobia.
Pri zahrnutí vývinu klonov ako kritéria progresie celkovo 25 pacientov progredovalo počas liečby do dátumu ukončenia zberu údajov do akcelerovanej fázy alebo blastickej krízy (3 v skupine nilotinibu
300 mg dvakrát denne, 5 v skupine nilotinibu 400 mg dvakrát denne a 17 v skupine imatinibu 400 mg raz denne). Odhadované podiely pacientov bez progresie do akcelerovanej fázy alebo blastickej krízy
vrátane vývinu klonov boli v 72 mesiacoch 98,7%, 97,9% a 93,2% v uvedenom poradí (HR=0,1626 a stratifikovaný log-rank p=0,0009 medzi nilotinibom 300 mg dvakrát denne a imatinibom raz denne, HR=0,2848 a stratifikovaný log-rank p=0,0085 medzi nilotinibom 400 mg dvakrát denne
a imatinibom raz denne).
Celkovo 55 pacientov zomrelo počas liečby alebo počas následného sledovania po ukončení liečby (21
v skupine nilotinibu 300 mg dvakrát denne, 11 v skupine nilotinibu 400 mg dvakrát denne a 23
v skupine imatinibu 400 mg raz denne). Dvadsaťšesť (26) z týchto 55 úmrtí súviselo s CML (6
v skupine nilotinibu 300 mg dvakrát denne, 4 v skupine nilotinibu 400 mg dvakrát denne a 16
v skupine imatinibu 400 mg raz denne). Odhadované podiely pacientov, ktorí žili v 72 mesiacoch, boli
91,6%, 95,8% a 91,4% v uvedenom poradí (HR=0,8934 a stratifikovaný log-rank p=0,7085 medzi nilotinibom 300 mg dvakrát denne a imatinibom, HR=0,4632 a stratifikovaný log-rank p=0,0314
medzi nilotinibom 400 mg dvakrát denne a imatinibom). Ak sa za udalosti považujú len úmrtia
súvisiace s CML, odhadované podiely celkového prežívania v 72 mesiacoch boli 97,7%, 98,5%
a 93,9% v uvedenom poradí (HR=0,3694 a stratifikovaný log-rank p=0,0302 medzi nilotinibom
300 mg dvakrát denne a imatinibom, HR=0,2433 a stratifikovaný log-rank p=0,0061 medzi nilotinibom 400 mg dvakrát denne a imatinibom).
Ukončenie liečby u pacientov s Ph+ CML v chronickej fáze, ktorí dostávali nilotinib ako liečbu prvej
línie a dosiahli trvalú hlbokú molekulovú odpoveď
V otvorenej štúdii s jedným ramenom bolo zaradených 215 dospelých pacientov s Ph+ CML
v chronickej fáze liečených nilotinibom v prvej línii počas ≥2 rokov, ktorí dosiahli MR4,5 meranú pomocou testu MolecularMD MRDxTM BCR-ABL, do pokračovania liečby nilotinibom počas ďalších
52 týždňov (konsolidačná fáza s nilotinibom). 190 z 215 pacientov (88,4%) vstúpilo do TFR po dosiahnutí trvalej hlbokej molekulovej odpovede počas konsolidačnej fázy definovanej podľa nasledujúcich kritérií:
- 4 posledné štvrťročné vyhodnotenia (vykonané každých 12 týždňov) boli aspoň MR4
(BCR-ABL/ABL ≤0,01% IS) a udržali sa počas jedného roka
- posledné vyhodnotenie je MR4,5 (BCR-ABL/ABL ≤0,0032% IS)
- nie viac ako dve vyhodnotenia spadajú medzi MR4 a MR4,5 (0,0032% IS < BCR-ABL/ABL
≤0,01% IS).
Primárnym ukazovateľom bolo percento pacientov s MMR v 48. týždni od začatia fázy TFR (vzhľadom na každého pacienta, ktorý vyžadoval opätovné začatie liečby ako non-responder). Zo
190 pacientov, ktorí vstúpili do fázy TFR, bolo 98 pacientov (51,6% [95% IS: 44,2; 58,9]) s MMR
v 48. týždni.
Osemdesiatosem pacientov (46,3%) prerušilo fázu TFR kvôli strate MMR, 1 (0,5%) v dôsledku úmrtia z neznámej príčiny, 1 (0,5%) kvôli rozhodnutiu lekára a 3 pacienti (1,6%) z vlastného rozhodnutia.
Z týchto 88 pacientov znovu začalo liečbu nilotinibom 86 pacientov a 2 pacienti natrvalo prerušili
štúdiu. Osemdesiatpäť z týchto 86 pacientov (98,8%) znovu dosiahlo MMR (jeden pacient natrvalo prerušil štúdiu z vlastného rozhodnutia) a 76 pacientov (88,4%) znovu dosiahlo MR4,5 v čase ukončenia štúdie.
Odhadovaný medián trvania liečby nilotinibom podľa Kaplana-Meiera (KM) pre opätovné dosiahnutie MMR a MR4,5 bol 7,9 týždňov (95% IS: 5,1; 8,0) a 13,1 týždňov (95% IS: 12,3; 15,7), v uvedenom poradí. Odhadovaný výskyt MMR a MR4,5 podľa KM v 24. týždni od opätovného začatia bol 98,8% (95% IS: 94,2; 99,9) a 90,9% (95% IS: 83,2; 96,0), v uvedenom poradí.
Odhadovaný medián prežívania bez liečby (treatment-free survival, TFS) podľa KM nebol doteraz
dosiahnutý (Obrázok 4); 99 zo 190 pacientov (52,1%) nemalo udalosť TFS.
Obrázok 4 Odhad prežívania bez liečby podľa Kaplana-Meiera od začiatku TFR (analýzacelého súboru)100
90
80
70
60
50
40
30
20
Pat
Evt Cen190 91 99
10
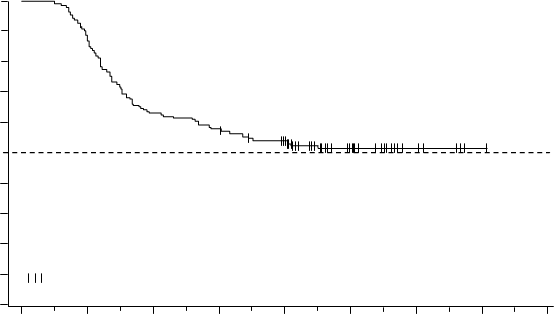
Cenzorované pozorovania
0
0 12
24 36
48 60 72
84 96
S rizikom: udalosti
Čas od TFR (týždne)
190:0
165:25
120:70
108:81
90:89
38:91
12:91
1:91
0:91
Pediatrická populácia
Bezpečnosť a účinnosť nilotinibu u pediatrických pacientov s Ph+ CML v chronickej fáze sa skúmali
v dvoch štúdiách. Celkovo 69 pediatrických pacientov (od 2 do <18 rokov) s buď
novodiagnostikovanou Ph+ CML v chronickej fáze (n=25), alebo Ph+ CML v chronickej fáze
s rezistenciou voči imatinibu/dasatinibu alebo intoleranciou voči imatinibu (n=44) dostalo liečbu nilotinibom v dávke 230 mg/m2 dvakrát denne, zaokrúhlenej na najbližších 50 mg dávky (až do maximálnej jednotlivej dávky 400 mg).
V zlúčenej populácii pacientov s CML bol medián skutočnej veľkosti dávky 435,5 mg/m2/deň (rozsah:
149 až 517 mg/m2/deň) a medián relatívnej veľkosti dávky 94,7% (rozsah: 32 až 112%). Štyridsať pacientov (58,0%) malo relatívnu veľkosť dávky vyššiu ako 90%. Medián trvania liečby nilotinibom bol 13,80 mesiacov (rozsah: 0,7-30,9 mesiacov).
U pacientov s CML s rezistenciou alebo intoleranciou bola miera veľkej molekulovej odpovede
(MMR; BCR-ABL/ABL ≤0,1% IS) 40,9% (95% CI: 26,3; 56,8) po 12 cykloch, s 18 pacientmi
s MMR. U pacientov s novodiagnostikovanou CML bola miera MMR 60,0% (95% CI: 38,7; 78,9) po
12 cykloch, s 15 pacientmi, ktorí dosiahli MMR. U pacientov s CML s rezistenciou alebo intoleranciou bola kumulatívna miera MMR 47,7% po 12 cykloch. U pacientov
s novodiagnostikovanou CML bola kumulatívna miera MMR 64,0% po 12 cykloch.
U 21 pacientov s CML s rezistenciou alebo intoleranciou, ktorí dosiahli MMR kedykoľvek počas liečby, bol medián času do prvej MMR 2,76 mesiaca (95% CI: 0,03; 5,55).
U 17 novodiagnostikovaných pacientov s CML, ktorí dosiahli MMR, bol medián času do prvej MMR
5,55 mesiacov (95% CI: 5,52; 5,75).
Medzi pacientmi s CML s rezistenciou alebo intoleranciou bol percentuálny podiel pacientov, ktorí dosiahli BCR-ABL/ABL ≤0,01% IS (MR4.0) do dátumu ukončenia štúdie 11,4%, zatiaľ čo 4,5% pacientov dosiahlo BCR-ABL/ABL ≤0,0032% IS (MR4.5). Medzi novodiagnostikovanými pacientmi, ktorí dosiahli MR4.0, bol percentuálny podiel 32%, zatiaľ čo 28,0% dosiahlo MR4.5.
U žiadneho z 21 pacientov s CML s rezistenciou alebo intoleranciou, ktorí počas liečby dosiahli MMR, sa nepotvrdila strata MMR. Spomedzi 17 novodiagnostikovaných pacientov s CML, ktorí dosiahli MMR, sa u jedného pacienta potvrdila strata MMR (pacient stratil CHR v dôsledku zvýšeného počtu bazofilov, avšak neprogredoval do AP/BC).
Jeden pacient s CML s rezistenciou alebo intoleranciou progredoval do AP/BC po približne
10 mesiacoch na liečbe.
Počas liečby ani po ukončení liečby neboli v oboch štúdiách hlásené žiadne úmrtia.
5.2 Farmakokinetické vlastnosti
Absorpcia
Maximálne koncentrácie nilotinibu sa dosiahnu 3 hodiny po perorálnom podaní. Absorpcia nilotinibu
po perorálnom podaní bola približne 30%. Absolútna biologická dostupnosť nilotinibu nebola
stanovená. V porovnaní s perorálnym roztokom (pH 1,2 až 1,3) bola relatívna biologická dostupnosť
nilotinibu v kapsliach približne 50%. Keď sa Tasigna užíva spolu s jedlom, u zdravých dobrovoľníkov sa Cmax nilotinibu zvýši o 112% a plocha pod krivkou sérovej koncentrácie v závislosti od času (AUC) o 82% v porovnaní so stavom nalačno. Podanie Tasigny 30 minút po jedle zvýšilo biologickú dostupnosť nilotinibu o 29%, 2 hodiny po jedle o 15% (pozri časti 4.2, 4.4 a 4.5).
Absorpcia nilotinibu (relatívna biologická dostupnosť) sa môže znížiť približne o 48% u pacientov s totálnou gastrektómiou a o 22% u pacientov s parciálnou gastrektómiou.
Distribúcia
Pomer nilotinibu v krvi a plazme je 0,71. Väzba na bielkoviny plazmy podľa pokusov in vitro je
približne 98%.
Biotransformácia
Hlavné metabolické dráhy zistené u zdravých osôb sú oxidácia a hydroxylácia. Nilotinib je hlavná
cirkulujúca zložka v sére. Žiadny z metabolitov neprispieva vo významnej miere k farmakologickej aktivite nilotinibu. Nilotinib sa primárne metabolizuje CYP3A4, s možným menším podielom
CYP2C8.
Eliminácia
Po jednorazovej dávke rádioaktívne značeného nilotinibu podanej zdravým osobám sa viac ako 90%
dávky eliminovalo do 7 dní, prevažne stolicou (94% dávky). Nezmenený nilotinib tvoril 69% dávky.
Zdanlivý eliminačný polčas odhadnutý z farmakokinetiky pri opakovanom podávaní raz denne bol približne 17 hodín. Variabilita farmakokinetiky nilotinibu medzi pacientmi bola stredne vysoká až vysoká.
Linearita/nelinearita
Expozícia nilotinibu v rovnovážnom stave závisela od dávky, pričom zvyšovanie systémovej
expozície bolo nižšie, ako by bolo úmerné dávke pri dávkach vyšších ako 400 mg podávaných raz denne. Denná systémová expozícia nilotinibu pri podávaní 400 mg dvakrát denne bola v rovnovážnom stave o 35% vyššia ako pri podávaní 800 mg raz denne. Systémová expozícia (AUC) nilotinibu
v rovnovážnom stave pri dávkach 400 mg dvakrát denne bola približne o 13,4% vyššia než pri
dávkach 300 mg dvakrát denne. Priemerné minimálne a maximálne koncentrácie nilotinibu počas
12 mesiacov boli približne o 15,7% a 14,8% vyššie pri podávaní 400 mg dvakrát denne v porovnaní s podávaním 300 mg dvakrát denne. Pri zvýšení dávky zo 400 mg dvakrát denne na 600 mg dvakrát denne nedošlo k významnému zvýšeniu expozície nilotinibu.
Podmienky rovnovážneho stavu sa v podstate dosiahli do 8 dní. Zvýšenie sérovej expozície nilotinibu medzi prvou dávkou a rovnovážnym stavom bolo približne 2-násobné pri podávaní raz denne a
3,8-násobné pri podávaní dvakrát denne.
Biologická dostupnosť/bioekvivalenčnéštúdie
Preukázalo sa, že jednorazové podanie 400 mg nilotinibu pri použití 2 tvrdých kapsúl po 200 mg, keď
sa obsah každej tvrdej kapsuly zmiešal s jednou čajovou lyžičkou jablčného pyré, je bioekvivalentné
s jednorazovým podaním 2 neporušených tvrdých kapsúl po 200 mg.
Pediatrická populácia
Po podávaní nilotinibu pediatrickým pacientom v dávke 230 mg/m2 dvakrát denne, zaokrúhlenej na
najbližšiu 50 mg dávku (až po maximálnu jednotlivú dávku 400 mg), boli expozícia v rovnovážnom
stave a klírens nilotinibu podobné (do 2 násobku) ako u dospelých pacientov liečených dávkou
400 mg dvakrát denne. Farmakokinetická expozícia nilotinibu po jednej alebo viacerých dávkach sa
zdá byť porovnateľná medzi pediatrickými pacientmi od 2 rokov do <10 rokov a od ≥10 rokov do
<18 rokov.
5.3 Predklinické údaje o bezpečnosti
Nilotinib sa hodnotil v štúdiách farmakologickej bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, reprodukčnej toxicity, fototoxicity a karcinogenity (u potkanov a myší).
Nilotinib neovplyvnil funkcie CNS alebo dýchacej sústavy. V štúdiách kardiálnej bezpečnosti in vitro sa zistil predklinický signál predĺženia QT, ktoré bolo dôsledkom nilotinibom spôsobenej blokády prúdov hERG a predĺženia trvania akčného potenciálu v izolovaných srdciach králikov. Žiadne účinky v EKG meraniach sa nepozorovali u psov alebo opíc pri podávaní do 39 týždňov, ani u psov
v špeciálnej telemetrickej štúdii.
V štúdiách toxicity pri opakovanom podávaní trvajúcich do 4 týždňov u psov a do 9 mesiacov
u makakov krabožravých sa zistilo, že primárnym cieľovým orgánom toxicity nilotinibu je pečeň. Zmeny pozostávali zo zvýšenej aktivity alanínaminotransferázy a alkalickej fosfatázy
a histopatologických nálezov (najmä hyperplázie/hypertrofie sínusových buniek alebo Kupfferových
buniek, hyperplázie žlčovodov a periportálnej fibrózy). Vo všeobecnosti boli klinické biochemické zmeny plne reverzibilné po štvortýždňovom období zotavenia a histologické zmeny vykazovali
čiastočnú reverzibilitu. Expozície pri najnižších dávkach, pri ktorých sa pozorovali účinky na pečeň,
boli nižšie ako expozícia u ľudí pri dávke 800 mg/deň. Iba menšie zmeny v pečeni sa pozorovali u myší alebo potkanov pri podávaní do 26 týždňov. Prevažne reverzibilné zvýšenia hladiny cholesterolu
sa pozorovali u potkanov, psov a opíc.
V skúšaniach genotoxicity v bakteriálnych systémoch in vitro a v cicavčích systémoch in vitro a in vivo s metabolickou aktiváciou alebo bez nej sa nenašiel dôkaz mutagénneho potenciálu nilotinibu.
V štúdii karcinogenity na potkanoch trvajúcej 2 roky bola maternica hlavným cieľovým orgánom
s inými ako neoplastickými léziami (dilatácia, rozšírenie ciev, hyperplázia endotelových buniek, zápal
a/alebo hyperplázia epitelu). Karcinogenita sa nepreukázala pri podávaní nilotinibu 5, 15 a
40 mg/kg/deň. Expozícia (vyjadrená ako AUC) pri najvyššej dávke predstavovala približne 2- až 3- násobok dennej expozície nilotinibu v rovnovážnom stave (na základe AUC) u ľudí pri dávke
800 mg/deň.
V štúdii karcinogenity na myšiach Tg.rasH2 trvajúcej 26 týždňov, v ktorej sa nilotinib podával
v dávkach 30, 100 a 300 mg/kg/deň, boli zachytené papilómy/karcinómy kože pri 300 mg/kg, čo predstavuje približne 30- až 40-násobok expozície u ľudí (na základe AUC) pri maximálnej schválenej
dávke 800 mg/deň (podávanej dvakrát denne 400 mg). Hladina bez pozorovaného účinku pre kožné
neoplastické lézie bola 100 mg/kg/deň, čo predstavuje približne 10- až 20-násobok expozície u ľudí
pri maximálnej schválenej dávke 800 mg/deň (podávanej dvakrát denne 400 mg). Hlavné cieľové orgány pre iné ako neoplastické lézie boli koža (epidermálna hyperplázia), rastúce zuby (degenerácia/atrofia skloviny horných rezákov a zápal gingívy/odontogénneho epitelu rezákov)
a týmus (zvýšená incidencia a/alebo závažnosť poklesu lymfocytov).
Nilotinib nespôsoboval teratogenitu, ale vykazoval embryo- a fetotoxicitu pri dávkach, ktoré boli toxické aj pre samice. Zvýšené poimplantačné straty sa pozorovali v štúdii fertility pri podávaní samcom aj samiciam, aj v štúdii embryotoxicity pri podávaní samiciam. V skúšaniach embryotoxicity sa zistila letalita embryí a účinky na plod (predovšetkým znížená hmotnosť plodov, predčasné zrastanie kostí tváre (zrast maxily a lícnej kosti), viscerálne odchýlky a odchýlky skeletu) u potkanov a zvýšená resorpcia plodov a odchýlky skeletu u králikov. V štúdii pre- a postnatálneho vývinu
u potkanov spôsobila expozícia nilotinibu u samíc zníženú telesnú hmotnosť mláďat spojenú so zmenami parametrov telesného vývinu, ako aj znížené ukazovatele párenia a fertility u potomstva.
Expozícia nilotinibu u samíc pri hladinách bez pozorovaných nežiaducich účinkov bola spravidla
nižšia alebo rovnaká ako u ľudí pri 800 mg/deň.
V štúdii vývoja mláďat sa nilotinib podával pomocou perorálnej sondy mláďatám potkana od prvého týždňa po narodení do mladého dospelého veku (70. deň po narodení) v dávkach 2, 6 a 20 mg/kg/deň. Okrem štandardných parametrov štúdie sa vykonali hodnotenia vývojových medzníkov, účinkov na CNS, párenia a fertility. Na základe zníženia telesnej hmotnosti u oboch pohlaví a oneskoreného oddelenia predkožky u samcov (čo môže súvisieť s poklesom telesnej hmotnosti) sa za dávku bez pozorovaného účinku považovalo u dospievajúcich potkanov 6 mg/kg/deň. Dospievajúce zvieratá v porovnaní s dospelými nevykazovali zvýšenú citlivosť na nilotinib. Okrem toho bol profil toxicity u dospievajúcich potkanov porovnateľný s profilom toxicity, ktorý sa pozoroval u dospelých potkanov.
Nepozorovali sa žiadne účinky na počet/pohyblivosť spermií a fertilitu u potkaních samcov a samíc až do najvyššej testovanej dávky, približne 5-násobku odporúčaného dávkovania u ľudí.
Zistilo sa, že nilotinib absorbuje svetlo v rozmedzí UV-B a UV-A, distribuuje sa do kože a vykazuje fototoxický potenciál in vitro, nepozorovali sa však žiadne účinky in vivo. Riziko vyvolania fotosenzitivity nilotinibom u pacientov sa preto považuje za veľmi nízke.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
O
bsah kapsuly
monohydrát laktózy
krospovidón typ A
poloxamér 188
koloidný oxid kremičitý bezvodý
magnéziumstearát
Stena kapsuly
želatína
oxid titaničitý (E171)
červený oxid železitý (E172)
žltý oxid železitý (E172)
Farbivo napotlač
šelak
čierny oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Blistre PVC/PVDC/Alu
Tasigna je dostupná v nasledujúcich veľkostiach balenia:
· Jednotlivé balenia obsahujúce 28 tvrdých kapsúl (7 denných blistrov, z ktorých každý obsahuje
4 tvrdé kapsuly) alebo 40 tvrdých kapsúl (5 blistrov, z ktorých každý obsahuje 8 tvrdých kapsúl).
· Spoločné balenia obsahujúce 112 (4 balenia po 28) tvrdých kapsúl, 120 (3 balenia po 40)
tvrdých kapsúl alebo 392 (14 balení po 28) tvrdých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/07/422/005-006
EU/1/07/422/009-010
EU/1/07/422/013
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. novembra 2007
Dátum posledného predĺženia registrácie: 19. novembra 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu