
a zvýšiť dávka Talzenny (po 3 – 5 polčasoch eliminácie inhibítora P-gp) na dávku používanú pred začiatkom podávania silného inhibítora P-gp (pozri časť 4.5).
Špeciálne populácie
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene (celkový bilirubín ≤ 1 × horný limit normálu [ULN]
a hladina aspartátaminotransferázy (AST) > ULN alebo celkový bilirubín > 1,0 až 1,5 × ULN
a akákoľvek hladina AST) nie je potrebná žiadna úprava dávky. Talzenna nebola skúmaná u pacientov
so stredne závažnou (celkový bilirubín > 1,5 až 3,0 × ULN a akákoľvek hladina AST) alebo závažnou poruchou funkcie pečene (celkový bilirubín > 3,0 × ULN a akákoľvek hladina AST) (pozri časť 5.2). Talzenna sa môže u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene používať len vtedy, keď prínos prevažuje nad možným rizikom. Funkcie pečene a nežiaduce udalosti majú byť
u pacientov starostlivo monitorované.
Porucha funkcie obličiek
U pacientov s miernou poruchou funkcie obličiek (60 ml/min ≤ klírens kreatinínu [CrCl] < 90 ml/min)
sa nevyžaduje žiadna úprava dávky. U pacientov so stredne závažnou poruchou funkcie obličiek (30 ml/min ≤ CrCl < 60 ml/min) sa odporúča začiatočná dávka Talzenny 0,75 mg jedenkrát denne. Talzenna nebola skúmaná u pacientov so závažnou poruchou funkcie obličiek (CrCl < 30 ml/min) ani u pacientov vyžadujúcich hemodialýzu. Použitie Talzenny sa neodporúča u pacientov so závažnou poruchou funkcie obličiek alebo vyžadujúcich hemodialýzu (pozri časť 5.2). Talzenna sa môže
u pacientov so závažnou poruchou funkcie obličiek používať len vtedy, keď prínos prevažuje nad možným rizikom. Funkcie obličiek a nežiaduce udalosti majú byť u pacientov starostlivo monitorované (pozri časť 5.2).
Starší pacienti
U starších pacientov (≥ 65 rokov) nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť Talzenny u detí a dospievajúcich vo veku < 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Talzenna je určená na perorálne použitie. Aby sa predišlo kontaktu s obsahom kapsuly, kapsuly sa
majú prehĺtať celé a nesmú sa otvárať ani rozpúšťať. Môžu sa užívať spolu s jedlom alebo bez neho
(pozri časť 5.2).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Myelosupresia
U pacientov liečených talazoparibom bola hlásená myelosupresia pozostávajúca z anémie,
leukopénie/neutropénie a/alebo trombocytopénie (pozri časť 4.8). Talazoparib sa nemá začať podávať, kým sa pacienti nezotavia z hematologickej toxicity spôsobenej predchádzajúcou liečbou (≤ stupeň 1).
U pacientov užívajúcich talazoparib je potrebné vykonať opatrenia spojené s rutinným monitorovaním hematologických parametrov a príznakov a prejavov spojených s anémiou, leukopéniou/neutropéniou a/alebo trombocytopéniou. Ak k takým udalostiam dôjde, odporúčajú sa úpravy dávok (zníženie alebo prerušenie) (pozri časť 4.2). Podľa potreby sa môže použiť podporná starostlivosť s transúziou krvi a/alebo trombocytov a/alebo podávaním kolónie stimulujúcich faktorov alebo bez nich.
Myelodysplastický syndróm/akútnamyeloblastováleukémia
U pacientov užívajúcich inhibítory poly (adenozíndifosftátribózo) polymerázy (PARP) vrátane
talazoparibu bol hlásený myelodysplastický syndróm/akútna myeloblastová leukémia (MDS/AML). Celkovo bol MDS/AML hlásený u 2 z 584 (0,3 %) pacientov so solídnym nádorom liečených talazoparibom v klinických štúdiách. Faktory potenciálne prispievajúce k rozvoju MDS/AML zahŕňajú predchádzajúce podanie chemoterapie obsahujúcej platinu, iné látky poškodzujúce DNA
alebo rádioterapiu. Na začiatku liečby sa má urobiť kompletné vyšetrenie krvného obrazu a počas liečby sa má mesačne monitorovať kvôli príznakom hematologickej toxicity. Ak sa potvrdí MDS/AML, talazoparib sa má vysadiť.
Antikoncepcia užienvofertilnomveku
Talazoparib bol v in vitro teste chromozómových aberácií v ľudských lymfocytoch z periférnej krvi
a v mikrojadrovom in vivo teste v kostnej dreni u potkanov klastogénny, ale v Amesovom teste nebol mutagénny (pozri časť 5.3), a pri podávaní gravidným ženám môže spôsobiť poškodenie plodu.
Gravidné ženy majú byť oboznámené s možným rizikom pre plod (pozri časť 4.6). Ženy vo fertilnom
veku nesmú počas užívania Talzenny otehotnieť a nesmú byť na začiatku liečby gravidné. Pred liečbou sa má všetkým ženám vo fertilnom veku urobiť tehotenský test.
U pacientok ženského pohlavia sa počas liečby Talzennou a aspoň 7 mesiacov po dokončení liečby vyžaduje vysokoúčinná metóda antikoncepcie. Keďže sa u pacientiek s rakovinou prsníka používanie hormonálnej antikoncepcie neodporúča, majú sa použiť dve nehormonálne a doplnkové antikoncepčné metódy (pozri časť 4.6).
Mužskí pacienti so ženskými partnerkami vo fertilnom veku alebo gravidnými partnerkami majú byť poučení, aby používali účinnú antikoncepciu počas liečby Talzennou a aspoň 4 mesiace po poslednej dávke (aj ak sú po vazektómii).
4.5 Liekové a iné interakcie
Talazoparib je substrát pre liekové prenášače P-gp a proteín spôsobujúci rezistenciu karcinómu prsníka (BCRP - Breast Cancer Resistance Protein) a je prevažne eliminovaný obličkami ako nezmenená zlúčenina.
Látky, ktorémôžuovplyvniťkoncentrácietalazoparibuvplazme
Inhibítory P-gp
Údaje zo štúdie liekových interakcií u pacientov s pokročilými solídnymi nádormi ukazujú, že súbežné podávanie viacerých denných dávok inhibítora P-gp, 100 mg itrakonazolu dvakrát denne
s jednorazovou 0,5 mg dávkou talazoparibu zvýšilo celkovú expozíciu (AUCinf) o približne 56%
a maximálnu koncentráciu (Cmax) talazoparibu o 40 %, v porovnaní s jednorazovou 0,5 mg dávkou
talazoparibu podanou samostatne. Populačná farmakokinetická (PK) analýza tiež ukázala, že súbežné
používanie silných inhibítorov P-gp zvýšilo expozíciu talazoparibu o 45 % v porovnaní s talazoparibom podávaným samostatne.
Je potrebné sa vyhýbať súbežnému používaniu silných inhibítorov P-gp (okrem iného vrátane amiodarónu, karvedilolu, klaritromycínu, kobicistátu, darunaviru, dronedarónu, erytromycínu, indinaviru, itrakonazolu, ketokonazolu, lapatinibu, lopinaviru, propafenónu, chinidínu, ranolazínu, ritonaviru, sakvinaviru, telapreviru, tipranaviru a verapamilu). Ak sa súbežnému podávaniu silného P- gp inhibítora nedá vyhnúť, dávka Talzenny sa má znížiť (pozri časť 4.2).
Induktory P-gp
Údaje zo štúdie liekových interakcií u pacientov s pokročilými solídnymi nádormi ukazujú, že súbežné podávanie jednorazovej 1 mg dávky talazoparibu s viacerými dennými dávkami induktora P-gp,
600 mg rifampínu, pričom sa rifampín podával 30 minút pred talazoparibom v deň podania dávky talazoparibu, zvýšilo Cmax talazoparibu o približne 37 % oproti jednorazovej 1 mg dávke talazoparibu podávanej samostatne, pričom AUCinf nebolo ovplyvnené. Toto je pravdepodobne čistý účinok oboch, indukcie P-gp aj inhibície rifampínom, za testovacích podmienok v štúdii liekových interakcií.
Pri súbežnom podávaní s rifampínom sa nevyžadujú žiadne úpravy dávok talazoparibu. Účinok iných induktorov P-gp na expozíciu talazoparibu však nebol skúmaný. Iné induktory P-gp (okrem iného vrátane karbamazepínu, fenytoínu a ľubovníka bodkovaného) môžu znížiť expozíciu talazoparibu.
Inhibítory BCRP
Účinok inhibítorov BCRP na PK talazoparibu nebol skúmaný in vivo. Súbežné podávanie talazoparibu s inhibítormi BCRP môže zvýšiť expozíciu talazoparibu. Je potrebné sa vyhýbať súbežnému používaniu silných inhibítorov BCRP (okrem iného vrátane kurkumínu a cyklosporínu). Ak sa nedá súbežnému podávaniu silných inhibítorov BCRP vyhnúť, pacienta je potrebné monitorovať ohľadom možného zvýšeného rizika nežiaducich reakcií.
Účinok látok redukujúcich kyseliny
Populačná PK analýza ukazuje, že súbežné podávanie látok redukujúcich kyseliny vrátane inhibítorov protónovej pumpy a antagonistov histamínového receptora 2 (H2RA) alebo iných látok redukujúcich kyseliny nemá žiaden významný vplyv na absorpciu talazoparibu.
Systémová hormonálna antikoncepcia
Štúdie liekových interakcií medzi talazoparibom a perorálnymi kontraceptívami sa neuskutočnili.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku/antikoncepciaumužovažien
Ženy vo fertilnom veku nesmú počas užívania Talzenny otehotnieť a nesmú byť na začiatku liečby
gravidné. Pred liečbou sa má všetkým ženám vo fertilnom veku urobiť tehotenský test (pozri časť 4.4).
Ženy vo fertilnom veku musia používať vysokoúčinné formy antikoncepcie (pozri časť 4.4) pred začiatkom liečby talazoparibom, počas liečby a 7 mesiacov po ukončení liečby talazoparibom. Keďže sa u pacientiek s rakovinou prsníka používanie hormonálnej antikoncepcie neodporúča, majú sa použiť dve nehormonálne a doplnkové antikoncepčné metódy. Poučte mužských pacientov so ženskými partnerkami vo fertilnom veku alebo gravidnými partnerkami, aby používali účinnú antikoncepciu počas liečby Talzennou a aspoň 4 mesiace po poslednej dávke (aj ak sú po vazektómii) (pozri časť 4.4).
Gravidita
Nie sú k dispozícii žiadne údaje o použití Talzenny u gravidných žien. Štúdie na zvieratách preukázali
embryofetálnu toxicitu (pozri časť 5.3). Talzenna môže spôsobiť poškodenie plodu, keď sa podáva gravidnej žene. Talzenna sa neodporúča užívať počas gravidity alebo u žien vo fertilnom veku nepoužívajúcich antikoncepciu (pozri časť 4.4).
Dojčenie
Nie je známe, či sa talazoparib vylučuje do ľudského materského mlieka. Riziko u dojčiat nemožno
vylúčiť, a preto sa počas liečby Talzennou a aspoň 1 mesiac po poslednej dávke dojčenie neodporúča.
Fertilita
Nie sú k dispozícii žiadne informácie o fertilite u pacientov. Na základe neklinických nálezov na
semenníkoch (čiastočne reverzibilných) a vaječníkoch (reverzibilných) môže Talzenna ovplyvniť fertilitu u mužov vo fertilnom veku (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Talzenna môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní talazoparibu sa môže vyskytnúť únava/asténia alebo závrat.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Celkový bezpečnostný profil Talzenny je založený na združených údajoch od 494 pacientov, ktorí
dostávali talazoparib v dávke 1 mg denne v klinických štúdiách so solídnymi nádormi, vrátane
286 pacientov z randomizovanej štúdie fázy 3 s HER-2 negatívnym lokálne pokročilým alebo metastatickým karcinómom prsníka so zárodočnou mutáciou BRCA (gBRCAm – germline BRCA
mutated) a 83 pacientov z nerandomizovanej štúdie fázy 2 u pacientov s lokálne pokročilým alebo metastatickým karcinómom prsníka so zárodočnou mutáciou BRCA.
Najčastejšie (≥ 25 %) nežiaduce reakcie u pacientov užívajúcich talazoparib v týchto klinických štúdiách boli únava (57,1 %), anémia (49,6 %), nevoľnosť (44,3 %), neutropénia (30,2 %), trombocytopénia (29,6 %) a bolesť hlavy (26,5 %). Najčastejšie (≥ 10 %) nežiaduce reakcie stupňa ≥ 3 na talazoparib boli anémia (35,2 %), neutropénia (17,4 %) a trombocytopénia (16,8 %).
K úpravám dávky (zníženiu alebo prerušeniu dávky) kvôli akejkoľvek nežiaducej reakcii došlo
u 62,3 % pacientov užívajúcich Talzennu. Najčastejšie nežiaduce reakcie vedúce k úpravám dávky boli anémia (33,0 %), neutropénia (15,8 %) a trombocytopénia (13,4 %).
K trvalému vysadeniu kvôli nežiaducej reakcii došlo u 3,6 % pacientov užívajúcich Talzennu. Medián trvania expozície bol 5,4 mesiaca (rozsah 0,03 – 61,1).
Tabuľkový zoznamnežiaducichreakcií
Tabuľka 3 zhŕňa nežiaduce reakcie na základe združeného súboru údajov uvedené v kategóriách podľa
triedy orgánových systémov a frekvencie. Kategórie frekvencií sú definované nasledovne: veľmi časté
(≥ 1/10) a časté (≥ 1/100 až < 1/10). V každej skupine frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
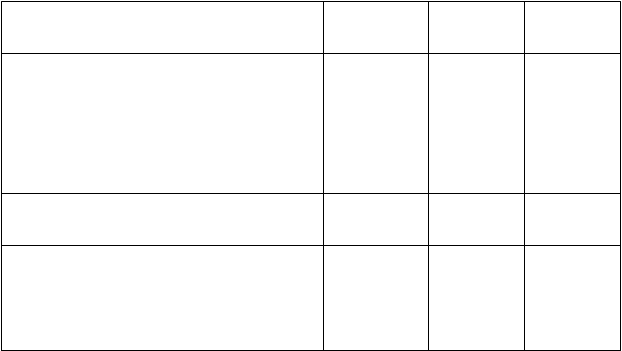
Tabuľka 3. Nežiaduce reakcie založené na združených súboroch údajov z 5 štúdií (N = 494)
Trieda orgánových systémov
Frekvencia
Preferovaný termín
Poruchy krvi a lymfatického systému
Veľmi časté Trombocytopéniaa Anémiab Neutropéniac Leukopéniad
Časté
Lymfopéniae
Poruchy metabolizmu a výživy
Veľmi časté
Všetky stupne* n (%)
146 (29,6)
245 (49,6)
149 (30,2)
77 (15,6)
30 (6,1)
Stupeň 3
n (%)
63 (12,8)
172 (34,8)
77 (15,6)
24 (4,9)
13 (2,6)
Stupeň 4
n (%)
20 (4,0)
2 (0,4)
9 (1,8)
1 (0,2)
0 (0,0)
Znížená chuť do jedla 100 (20,2) 2 (0,4) 0 (0,0)
Poruchy nervového systému
Veľmi časté
Závrat
Bolesť hlavy
ČastéDysgeúzia
69 (14,0)
131 (26,5)
42 (8,5)
1 (0,2)
5 (1,0)
0 (0,0)
N/A N/A
0 (0,0)
Trieda orgánových systémov
Frekvencia
Preferovaný termín
Poruchy gastrointestinálneho traktu
Veľmi časté Vracanie Hnačka Nevoľnosť Bolesť bruchaf
Časté Stomatitída Dyspepsia
Poruchy kože a podkožného tkaniva
Veľmi časté
Všetky stupne* n (%)
110 (22,3)
112 (22,7)
219 (44,3)
105 (21,3)
32 (6,5)
41 (8,3)
Stupeň 3
n (%)
7 (1,4)
3 (0,6)
4 (0,8)
8 (1,6)
0 (0,0)
0 (0,0)
Stupeň 4
n (%)
0 (0,0)
0 (0,0) N/A N/A
0 (0,0) N/A
Alopéciag 110 (22,3) N/A N/A
Celkové poruchy a reakcie v mieste podania
Veľmi časté
Únavah
Skratky: n = počet pacientov, N/A = neaplikovateľné.
* Nevyskytli sa žiadne nežiaduce reakcie na liek stupňa 5.
282 (57,1) 17 (3,4) 1 (0,2)
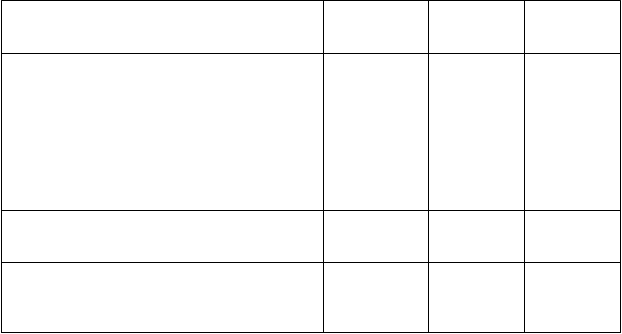
a. Zahŕňa preferované termíny trombocytopénia a znížený počet krvných doštičiek.
b. Zahŕňa preferované termíny anémia, znížený hematokrit a znížená hladina hemoglobínu.
c. Zahŕňa preferované termíny neutropénia a znížený počet neutrofilov.
d. Zahŕňa preferované termíny leukopénia a znížený počet bielych krviniek.
e. Zahŕňa preferované termíny znížený počet lymfocytov a lymfopénia.
f. Zahŕňa preferované termíny bolesť brucha, bolesť hornej časti brucha, nepríjemný pocit v bruchu a bolesť dolnej časti brucha.
g. Pre talazoparib je stupeň 1 21 % a stupeň 2 2 %.
h. Zahŕňa preferované termíny únava a asténia.
PopisvybranýchnežiaducichreakciíMyelosupresiaNežiaduce reakcie spojené s myelosupresiou, ako napríklad anémia, neutropénia a trombocytopénia, boli u pacientov liečených 1 mg talazoparibu denne hlásené veľmi často. Udalosti spojené
s myelosupresiou stupňa 3 a 4 boli hlásené nasledovne: anémia 34,8 % a 0,4 %, neutropénia 15,6 %
a 1,8 % a trombocytopénia 12,8 a 4,0 %. Neboli hlásené žiadne úmrtia kvôli nežiaducim reakciám spojeným s myelosupresiou. Nežiaduce udalosti spojené s myelosupresiou súvisiace s úpravami dávky boli hlásené až u približne 30 % pacientov v populácii liečenej talazoparibom 1 mg/deň a tie, ktoré súviseli s trvalým vysadením skúšaného lieku, boli hlásené u menej ako 1 % pacientov.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieS predávkovaním talazoparibom sú obmedzené skúsenosti. U jedného pacienta, ktorý si omylom sám podal tridsať kapsúl talazoparibu 1 mg v 1. deň a bol okamžite ošetrený dekontamináciou žalúdka, neboli hlásené žiadne nežiaduce reakcie. Prejavy predávkovania nie sú stanovené. V prípade predávkovania sa má liečba talazoparibom zastaviť a lekár má zvážiť dekontamináciu žalúdka, postupovať podľa všeobecných podporných opatrení a liečiť symptomaticky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: iné antineoplastické látky, ATC kód: L01XX60
Mechanizmus účinku
Talazoparib je inhibítorom enzýmov PARP, PARP1 a PARP2. Enzýmy PARP sú súčasťou signálnych
dráh odpovede na poškodenie bunkovej DNA, ako napríklad oprava DNA, génová transkripcia a bunková smrť. Inhibítory PARP (PARPi) uplatňujú cytotoxické účinky na nádorové bunky
2 mechanizmami – inhibíciou katalytickej aktivity PARP a zachytávaním PARP, pričom sa proteín
PARP naviazaný na PARPi nedokáže ľahko uvoľniť z lézie na DNA, čím zabraňuje oprave DNA, replikácii a transkripcii, čím vedie k apoptóze a/alebo bunkovej smrti. Liečba línií rakovinových
buniek nesúcich defekty v génoch opravy DNA talazoparibom v monoterapii vedie k zvýšeným
hladinám γH2AX (marker dvojreťazcových zlomov v DNA), čoho výsledkom je znížená proliferácia buniek a zvýšená apoptóza. Protinádorová aktivita talazoparibu bola tiež pozorovaná na modeli so xenograftom karcinómu prsníka s mutovaným génom BRCA odvodeným od pacienta (PDX – patient derived xenograft), kedy bol pacient predtým liečený režimom založeným na platine. V tomto PDX modeli znížil talazoparib rast nádoru a zvýšil hladinu γH2AX aj apoptózu v nádoroch.
Elektrofyziológia srdca
Účinok talazoparibu na srdcovú repolarizáciu bol hodnotený pomocou časovo zladených
elektrokardiogramov (EKG) pri hodnotení vzťahu medzi zmenou intervalu QT korigovaného podľa srdcovej frekvencie (QTc) oproti východiskovej hodnote a zodpovedajúcimi koncentráciami
talazoparibu v plazme u 37 pacientov s pokročilými solídnymi nádormi. Talazoparib nemal klinicky
významný účinok na predĺženie intervalu QTc pri maximálnej klinicky odporúčanej dávke 1 mg jedenkrát denne.
Klinická účinnosť a bezpečnosť
Randomizovaná štúdia fázy 3, EMBRACA
EMBRACA bola otvorená, randomizovaná, multicentrická štúdia s 2 paralelnými ramenami porovnávajúca Talzennu oproti chemoterapii (kapecitabín, eribulín, gemcitabín, vinorelbín)
u pacientov s HER2-negatívnym lokálne pokročilým alebo metastatickým karcinómom prsníka so zárodočnou mutáciou BRCA, ktorí dostávali nie viac ako 3 predchádzajúce režimy cytotoxickej
chemoterapie na ich metastatické alebo lokálne pokročilé ochorenie. U pacientov sa vyžadovalo, aby dostávali liečbu antracyklínom a/alebo taxánom (ak nebola kontraindikovaná) v neoadjuvantnom, adjuvantnom a/alebo metastatickom nastavení. Od pacientov s predchádzajúcou liečbou platinou pri
pokročilom ochorení sa vyžadovalo, aby nemali žiaden záznam o progresii ochorenia počas liečby platinou. Nebola povolená žiadna predchádzajúca liečba inhibítorom PARP.
Zo 431 pacientov randomizovaných v štúdii EMBRACA malo 408 (95 %) centrálne potvrdenú patogénnu alebo potenciálne patogénnu mutáciu gBRCAm pomocou testu v klinickom skúšaní,
z ktorých 354 (82 %) bolo potvrdených testom BRACAnalysis CDx®. Stav mutácie BRCA (pozitívny na gén náchylnosti na karcinóm prsníka 1 [BRCA1] alebo pozitívny na gén náchylnosti na karcinóm
prsníka 2 [BRCA2] ) bol podobný v oboch ramenách liečby.
Celkom bolo 431 pacientov randomizovaných v pomere 2 : 1 k podávaniu 1 mg kapsúl Talzenny jedenkrát denne alebo chemoterapie v štandardných dávkach, kým nedošlo k progresii alebo neprijateľnej toxicite. Zo 431 pacientov randomizovaných v štúdii EMBRACA bolo 287 randomizovaných do ramena s Talzennou a 144 do ramena s chemoterapiou. Randomizácia bola stratifikovaná podľa predchádzajúceho použitia chemoterapie na metastatické ochorenie (0 oproti 1, 2 alebo 3), podľa triple negatívneho stavu ochorenia (triple negative breast cancer [TNBC] oproti non- TNBC) a anamnézy metastáz v centrálnej nervovej sústave (áno oproti nie).
Demografické údaje pacientov, východiskové hodnoty a charakteristiky ochorenia boli medzi ramenami liečby v štúdii vo všeobecnosti podobné (pozri tabuľku 4).
Tabuľka 4. Demografické údaje, východiskové hodnoty a charakteristiky ochorenia –
štúdia EMBRACA
Talazoparib
(
N = 287)
Chemoterapia
(
N = 144)
Medián veku (roky, [rozsah]) 45,0 (27,0; 84,0) 50,0 (24,0; 88,0) Veková kategória (roky), n (%)
< 50 182 (63,4 %) 67 (46,5 %)
50 až < 65 78 (27,2 %) 67 (46,5 %)
≥ 65 27 (9,4 %) 10 (6,9 %) Pohlavie, n (%)
Ženy 283 (98,6 %) 141 (97,9 %) Muži 4 (1,4 %) 3 (2,1 %)
Rasa, n (%)
Aziati 31 (10,8 %) 16 (11,1 %) Černosi alebo Afroameričania 12 (4,2 %) 1 (0,7 %) Belosi 192 (66,9 %) 108 (75,0 %) Iné 5 (1,7 %) 1 (0,7 %) Neuvedené 47 (16,4 %) 18 (12,5 %)
Výkonnostný stav podľa ECOG, n (%)
0 153 (53,3 %) 84 (58,3 %)
1 127 (44,3 %) 57 (39,6 %)
2 6 (2,1 %) 2 (1,4 %) Chýba 1 (0,3 %) 1 (0,7 %)
Stav hormónálnych receptorov, n (%)
HER2-pozitívny 0 (0,0 %) 0 (0,0 %) Triple negatívny 130 (45,3 %) 60 (41,7 %)
Pozitívny na hormálne receptory (ER-pozitívny alebo
PgR-pozitívny)
Stav BRCA podľa hodnotenia centrálnym alebo miestnym laboratóriom, n (%)
157 (54,7 %) 84 (58,3 %)
287 (100,0 %) 144 (100,0 %)
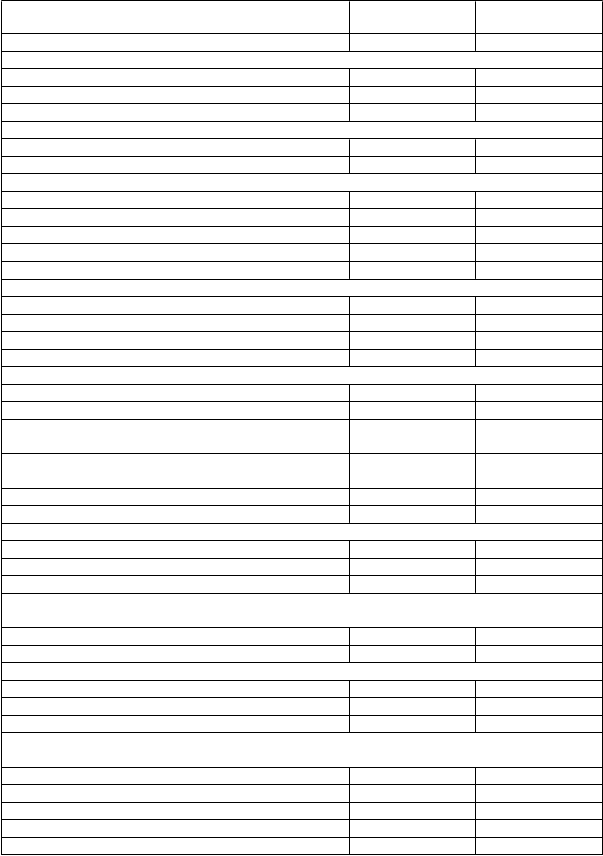
Pozitívny na mutáciu BRCA1 133 (46,3 %) 63 (43,8 %) Pozitívny na mutáciu BRCA2 154 (53,7 %) 81 (56,3 %)
Čas od počiatočnej diagnózy karcinómu prsníka po diagnózu pokročilého karcinómu prsníka (roky)
n 286 144
Medián 1,9 2,7
Minimum, maximum 0, 22 0, 24
Kategórie pre čas od počiatočnej diagnózy karcinómu prsníka po diagnózu pokročilého karcinómu prsníka
< 12 mesiacov 108 (37,6 %) 42 (29,2 %)
≥ 12 mesiacov 178 (62,0 %) 102 (70,8 %) Počet predchádzajúcich cytotoxických režimov pre lokálne pokročilé alebo metastatické ochorenie
Priemer (štandardná odchýlka) 0,9 (1,01) 0,9 (0,89) Medián 1 1
Minimum, maximum 0, 4 0, 3
Počet pacientov, ktorí dostali predchádzajúce cytotoxické režimy na lokálne pokročilé alebo metastatické ochorenie, n (%)
0 111 (38,7 %) 54 (37,5 %)
1 107 (37,3 %) 54 (37,5 %)
2 57 (19,9 %) 28 (19,4 %)
3 11 (3,8 %) 8 (5,6 %)
≥ 4 1 (0,3 %) 0 (0,0 %)
Tabuľka 4. Demografické údaje, východiskové hodnoty a charakteristiky ochorenia –
štúdi
a EMBRACA
Talazoparib
 (
N = 287)
(
N = 287)
Počet pacientov, ktorí dostali nasledujúce predchádzajúce liečby, n (%)
Chemoterapia(N = 144)
Taxán 262 (91,3 %) 130 (90,3 %) Antracyklín 243 (84,7 %) 115 (79,9 %) Platina 46 (16,0 %) 30 (20,8 %)
Skratky: BRCA = gén náchylnosti na karcinóm prsníka, ER = estrogénový receptor, HER2 = receptor pre ľudský
epidermálny rastový faktor 2, N = počet pacientov, n = počet pacientov v kategórii, PgR = progesterónový receptor.
Primárnym cieľom účinnosti bolo prežívanie bez progresie (PFS) hodnotené v súlade s kritériami RECIST (Response Evaluation Criteria in Solid Tumors) verziou 1.1, podľa zaslepeného nezávislého centrálneho hodnotenia (BICR). Sekundárnymi cieľmi boli miera objektívna odpovede (ORR), celkové prežívanie (OS), bezpečnosť a PK.
Štúdia preukázala štatisticky významné zlepšenie PFS pre Talzennu v porovnaní s chemoterapiou (pozri tabuľku 5). Predbežná analýza OS sa vykonala pri 51 % z celkového počtu plánovaných udalostí. Údaje o účinnosti zo štúdie EMBRACA založené na primárnej analýze podľa nezávislého hodnotenia
a skúšajúceho lekára sú zhrnuté v tabuľke 5. Kaplan-Meierove krivky pre PFS sú zobrazené na obrázku 1.
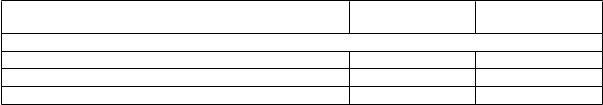
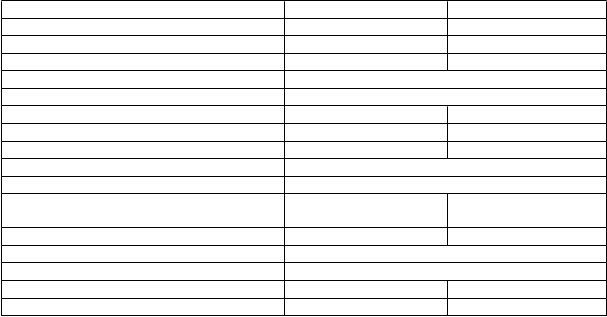 Tabuľka 5. Súhrn výsledkov účinnosti – štúdia EMBRACATalazoparib Chemoterapia
Tabuľka 5. Súhrn výsledkov účinnosti – štúdia EMBRACATalazoparib ChemoterapiaPrežívanie bez progresie podľa BICR N = 287 N = 144
Udalosti, počet (%) 186 (65 %) 83 (58 %) Medián (95 % IS), mesiace 8,6 (7,2; 9,3) 5,6 (4,2; 6,7) Miera rizikaa (95 % IS) 0,54 (0,41; 0,71)
2-stranná p-hodnotab p < 0,0001
Celkové prežívanie (predbežná analýza)c N = 287 N = 144
Udalosti, počet (%) 108 (38 %) 55 (38 %) Medián (95 % IS), mesiace 22,3 (18,1; 26,2) 19,5 (16,3; 22,4) Miera rizika (95 % IS) 0,76 (0,55; 1,06)
2-stranná p-hodnotab p = 0,1053
Objektívna odpoveď podľa skúšajúceho lekárad, e
N = 219 N = 114
ORR (%, 95 % IS) 62,6 (55,8; 69,0) 27,2 (19,3; 36,3) Pomer šancí (95 % IS) 4,99 (2,93; 8,83)
2-stranná p-hodnotaf p < 0,0001
Trvanie odpovede podľa skúšajúceho lekárad N = 137 N = 31
Medián (IQR), mesiace 5,4 (2,8; 11,2) 3,1 (2,4; 6,7) Skratky: BICR = zaslepené nezávislé centrálne hodnotenie, IS = interval spoľahlivosti, CMH = Cochran- Mantel-Haenszel, CR = kompletná odpoveď, IQR = interkvartilový rozsah, ITT = populácia „s úmyslom liečiť“, ORR = miera objektívnej odpovede, OS = celkové prežívanie, PR = parciálna odpoveď, RECIST 1.1
= kritériá hodnotenia odpovede u solídnych nádorov verzia 1.1.
a. Miera rizika bola založená na stratifikovanom Coxovom regresnom modeli s liečbou ako jediným kovariátom (stratifikačné faktory: počet predchádzajúcich režimov cytotoxickej chemoterapie, triple
negatívny stav, anamnéza metastáz v centrálnej nervovej sústave) a relatívne k chemoterapii všeobecne s
< 1 favorizoval talazoparib.
b. Stratifikovaný log-rank test.
c. nastalo 51 % projektovaného koncového počtu udalostí OS (163 z 321 úmrtí).
d. Vykonávaná v ITT populácii s merateľným ochorením, ktorá mala objektívnu odpoveď. Podiel kompletných odpovedí bol u talazoparibu 5,5 % oproti ramenu s chemoterapiou, kde to bolo 0 %.
e. Podľa kritérií RECIST 1.1, nevyžadovalo sa potvrdenie CR/PR.
f. Stratifikovaný test CMH.
Obrázok 1 zobrazuje Kaplan-Meierovu krivku pre štúdiu EMBRACA.
Obrázok 1. Kaplan-Meierove krivky PFS – štúdia EMBRACA
|
)
% (
y t i l i
b a b o
r
P l a v
i
v r
u
S
e e r
F
-
n
o i
s s e
r
g o r
P
|
|


100
90
80
70
60
50
TalazoparibMedián PFS = 8,6 mesiacovChemotherapy
Median PFS=5.6 months
95% CI (4.2, 6.7)
Hazard Ratio=0.54
95% CI (0.41, 0.71)
p<0.0001
|
|
95% IS (7,2; 9,3)
Talazoparib
Median PFS=8.6 months
95% CI (7.2, 9.3)
|
|
 ChemoterapiaMedián PFS = 5,6 mesiacov95% IS (4,2; 6,7) Miera rizika = 0,5495% IS (0,41; 0,71)
ChemoterapiaMedián PFS = 5,6 mesiacov95% IS (4,2; 6,7) Miera rizika = 0,5495% IS (0,41; 0,71) p<0,0001
p<0,0001
40
30
20
10
0
Number of patients at risk
|
|

0 3 6 9 12 15 18 21 24 27 30 33 36 39
Počet rizikových pacientov
Čas (mesiace)
Talazoparib
Chemoterapia
287 229 148 91 55 42 29 23 16 12 5 3 1
144 68 34 22 9 8 4 2 2 1
Skratky: IS = interval spoľahlivosti, PFS = prežívanie bez progresie.
Vykonala sa séria vopred špecifikovaných podskupinových analýz PFS na základe prognostických faktorov a východiskových charakteristík, aby sa preskúmala vnútorná konzistencia liečebného účinku. V súlade s celkovými výsledkami sa pozorovalo zníženie rizika progresie ochorenia alebo úmrtia v prospech ramena talazoparibu vo všetkých samostatných podskupinách pacientov (obrázok 2).
Obrázok 2. „Forest plot“ pre analýzy PFS podľa kľúčových podskupín – štúdia EMBRACA
ITT a podskupina
Počet pacientov
n (%)
Miera rizika (95% IS)
Všetci randomizovaní pacienti (ITT) Stav hormonálnych receptorov
TNBC založené na najaktuálnejšej biopsii
HR+ založené na najaktuálnejšej biopsii
Anamnéza metastáz do CNS
Áno
Nie
Predchádzajúca liečba platinou
Áno
Nie
Predchádzajúce režimy cytotoxickej chemoterapie pre aBC
0
1
≥ 2
431 (100)
190 (44,1)
241 (55,9)
63 (14,6)
368 (85,4)
76 (17,6)
355 (82,4)
165 (38,3)
161 (37,4)
105 (24,4)
0,54 (0,41; 0,71)
0,60 (0,41; 0,87)
0,47 (0,32; 0,71)
0,32 (0,15; 0,67)
0,58 (0,43; 0,78)
0,76 (0,40; 1,45)
0,52 (0,39; 0,71)
0,57 (0,34;0,95 )
0,51 (0,33; 0,80)
0,56 (0,34; 0,95)
0,25 0,50 0,75 1,00 1,25 1,50
 ← v prospech talazoparibu ← → v prospech PCT →
← v prospech talazoparibu ← → v prospech PCT →
Skratky: aBC = pokročilý karcinóm prsníka, IS = interval spoľahlivosti, CNS = centrálna nervová sústava, HR+ = pozitivita na hormálne receptory, ITT = populácia „s úmyslom sa liečiť“, PCT = liečba podľa výberu lekára (chemoterapia), PFS = prežívanie bez progresie, TNBC = triple negatívny karcinóm prsníka.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s talazoparibom
vo všetkých podskupinách pediatrickej populácie pri karcinóme prsníka (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Expozícia talazoparibu sa vo všeobecnosti úmerne zvyšovala s dávkou v rozsahu 0,025 mg až 2 mg po dennom podaní viacerých dávok. Po opakovanom dennom podávaní 1 mg talazoparibu pacientom,
boli geometrický priemer (% variačného koeficientu [VK %]) plochy pod krivkou (AUC) pre závislosť plazmatickej koncentrácie od času a maximálna pozorovaná koncentrácia v plazme (Cmax) pre talazoparib v ustálenom stave v rozsahoch 126 (107) ng.h/ml až 208 (37) ng.h/ml a 11 (90) ng/ml až 19 (27) ng/ml. Po opakovanom dennom podávaní dosiahli koncentrácie talazoparibu v plazme ustálený stav po 2 až 3 týždňoch. Medián miery akumulácie talazoparibu po opakovanom perorálnom podaní 1 mg jedenkrát denne bol v rozsahu 2,3 až 5,2. Talazoparib je substrátom transportérov P-gp
a BCRP.
Absorpcia
Po perorálnom podaní talazoparibu bol medián času do Cmax (Tmax) vo všeobecnosti medzi 1 až
2 hodinami po podaní dávky. Štúdia absolútnej biologickej dostupnosti nebola u ľudí vykonaná. Na
základe údajov vylučovania močom je však absolútna biologická dostupnosť minimálne 41 %
s absorbovanou frakciou minimálne 69 % (pozri časť Eliminácia). Neočakáva sa žiaden významný účinok látok redukujúcich kyseliny na expozíciu talazoparibu, keďže je rozpustnosť talazoparibu dostatočná pri všetkých pH medzi 1 a 6,8. Dvadsaťosem percent (28 %) pacientov v pivotnej štúdii užívalo látky redukujúce kyseliny, najmä inhibítory protónovej pumpy.
Účinok jedla
Požitie jedla znížilo rýchlosť, ale nie rozsah absorpcie talazoparibu. Po jednorazovej perorálnej dávke talazoparibu s vysokokalorickým jedlom s vysokým obsahom tukov (približne 827 kalórií, 57 % tuku)
sa priemerná Cmax talazoparibu znížila o približne 46 %, medián Tmax sa predĺžil z 1 hodiny na
4 hodiny a AUCinf nebola ovplyvnená. Na základe týchto výsledkov sa môže Talzenna podávať
s jedlom alebo bez neho (pozri časť 4.2).
Distribúcia
Priemerný zdanlivý distribučný objem (Vss/F) talazoparibu v populácii bol 420 l. In vitro sa približne
74 % talazoparibu viaže na plazmatické bielkoviny bez koncentračnej závislosti v rozsahu koncentrácií
0,01 µM až 1 µM.
Biotransformácia
Talazoparib u ľudí podlieha minimálnemu metabolizmu v pečeni. Po perorálnom podaní jednorazovej
1 mg dávky [14C]talazoparibu ľuďom neboli v plazme identifikované žiadne hlavné cirkulujúce metabolity a talazoparib bol jedinou identifikovanou cirkulujúcou od lieku odvodenou entitou. Z moču ani stolice sa nezískali žiadne metabolity, ktoré by individuálne zastupovali viac ako 10 % podanej
dávky.
In vitro talazoparib v klinicky relevantných koncentráciách neinhibuje cytochrómy (CYP)1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ani CYP3A4/5 a neindukuje CYP1A2, CYP2B6 ani CYP3A4.
In vitro talazoparib v klinicky relevantných koncentráciách neinhiboval žiadne hlavné črevné, pečeňové ani obličkové membránové prenášače (P-gp, BCRP, polypeptid prenášajúci organické anióny [OATP]1B1, OATP1B3, prenášač organických katiónov [OCT]1, OCT2, prenášač
organických aniónov [OAT]1, OAT3, exportnú pumpu žlčových solí [BSEP], multiliekové a toxínové extrúzie [MATE]1 a MATE2-K).
In vitro talazoparib v klinicky relevantných koncentráciách neinhiboval žiadne hlavné izoformy uridíndifosfátglukuronosyltransferázy (UGT) (1A1, 1A4, 1A6, 1A9, 2B7 a 2B15).
Eliminácia
Hlavnou cestou eliminácie talazoparibu je renálna eliminácia nezmeneného lieku (pasívna filtrácia
a aktívna sekrécia). Na aktívnej renálnej sekrécii talazoparibu sa pravdepodobne podieľa P-gp. Priemer (± smerodajná odchýlka) koncového polčasu eliminácie talazoparibu v plazme bol 90
(±58) hodín a priemer zdanlivého perorálneho klírensu (CL/F) v populácii (variabilita medzi
účastníkmi) bol 6,5 (31 %) l/h u pacientov s rakovinou. U 6 pacientok, ktorým bola podaná jedna perorálna dávka [14C]talazoparibu,sa priemerne z moču a stolice získalo 69 % (±8,6 %) a 20 % (±5,5 %), z celkovej podanej rádioa ktívnej dávky. Vylučovanie nezmeneného talazoparibu v moči
bolo hlavnou cestou eliminácie tvoriac 55 % podanej dávky, pričom nezmenený talazoparib zachytený v stolici tvoril 14 %.
Vek, pohlavieatelesnáhmotnosť
Vykonala sa populačná analýza PK s použitím údajov od 490 pacientov s rakovinou za účelom
hodnotenia vplyvu veku (od 18 do 88 rokov), pohlavia (53 mužov a 437 žien) a telesnej hmotnosti (od
35,7 kg do 162 kg) na PK talazoparibu. Výsledky ukázali, že vek, pohlavie ani telesná hmotnosť nemajú žiaden klinicky relevantný účinok na PK talazoparibu.
Rasa
Na základe populačnej analýzy PK zahŕňajúcej 490 pacientov, z čoho bolo 41 pacientov aziatov
a 449 pacientov neaziatov (361 belochov, 16 černochov, 9 iných a 63 nehlásených), bol CL/F
talazoparibu vyšší u aziatov ako u neaziatov, čo vedie k o 19 % nižšej expozícii (AUC) u aziatov.
Pediatrická populácia
Farmakokinetika talazoparibu nebola hodnotená u pacientov vo veku < 18 rokov.
Porucha funkcie obličiek
Na základe populačnej analýzy PK vykonanej u 490 pacientov, kde malo 132 pacientov miernu
poruchu funkcie obličiek (60 ml/min ≤ CrCl < 90 ml/min), 33 pacientov malo stredne závažnú poruchu funkcie obličiek (30 ml/min ≤ CrCl < 60 ml/min) a 1 pacient mal závažnú poruchu funkcie
obličiek (CrCl < 30 ml/min) sa CL/F talazoparibu znížilo o 14 %, u pacientov s miernym resp. o 37 %
so stredne závažnou poruchou funkcie obličiek v porovnaní s pacientmi s normálnou funkciou obličiek
(CrCl ≥ 90 ml/min). Na odhadnutie vplyvu závažného poškodenia obličiek na CL/F talazoparibu
v tejto populácii pacientov nie je dostupný dostatok údajov. U pacientov vyžadujúcich hemodialýzu sa neskúmala PK talazoparibu (pozri časť 4.2).
Porucha funkcie pečene
Na základe populačnej analýzy PK, ktorá zahŕňala 490 pacientov, kde malo 118 pacientov miernu
poruchu funkcie pečene (celkový bilirubín ≤ 1,0 x ULN a AST > ULN alebo celkový bilirubín > 1,0
až 1,5 × ULN a akékoľvek AST), nemala mierna porucha funkcie pečene žiadny vplyv na PK
talazoparibu. PK talazoparibu nebola skúmaná u pacientov so stredne závažnou (celkový
bilirubín > 1,5 až 3,0 × ULN a akákoľvek hladina AST) alebo závažnou poruchou funkcie pečene
(celkový bilirubín > 3,0 × ULN a akákoľvek hladina AST) (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Karcinogenita
Štúdie karcinogenity s talazoparibom neboli vykonané.
Genotoxicita
Talazoparib nebol mutagénny v bakteriálnom teste reverzných mutácií (Amesov test). Talazoparib bol
klastogénny v in vitro teste chromozómových aberácií v lymfocytoch z ľudskej periférnej krvi
a in vivo mikrojadrovom teste u potkanov pri expozíciách podobných klinicky relevantným dávkam. Táto klastogenita je konzistentná s genómovou nestabilitou vyplývajúcou z primárnej farmakológie talazoparibu, čo naznačuje možnú genotoxicitu pre ľudí.
Toxicita po opakovanom podaní
V štúdiách toxicity po opakovanom podaní u potkanov a psov zahŕňali hlavné nálezy pri
subterapeutických expozíciách hypocelularitu kostnej drene s poklesom hematopoetických buniek závislým od dávky, spotrebovanie lymfoidného tkaniva vo viacerých orgánoch a atrofiu a/alebo
degeneratívne zmeny v semenníkoch, nadsemenníkoch a semenotvorných tubuloch. Ďalšie nálezy pri
vyšších expozíciách zahŕňali nárast v apoptóze/nekróze závislý od dávky v gastrointestinálnom trakte (GI), pečeni a vaječníkoch. Väčšina histopatologických nálezov bola vo všeobecnosti reverzibilná, pričom nálezy na semenníkoch boli čiastočne reverzibilné po 4 týždňoch od prerušenia dávkovania. Tieto nálezy týkajúce sa toxicity sú konzistentné s farmakológiou talazoparibu a jeho spôsobom distribúcie v tkanivách.
Vývojová toxikológia
V štúdii embryo-fetálneho vývoja u potkanov viedol talazoparib k embryo-fetálnemu úmrtiu,
malformácii plodu (zasunutým očiam, malým očiam, oddeleniu častí hrudnej kosti, spojeniu oblúka cervikálnych stavcov) a štruktúrnym variáciám kostí pri systémovej expozícii samíc AUC24 približne
0,09-násobnej z relevantnej expozície u ľudí pri odporúčanej dávke.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly
Silicifikovaná mikrokryštalická celulóza (SMCC) (mikrokryštalická celulóza a oxid kremičitý)
Obal0,25 mg kapsuly
Hypromelóza (HPMC)
Žltý oxid železa (E172) Oxid titaničitý (E171)
Obal1mg kapsuly
Hypromelóza (HPMC)
Červený oxid železa (E172) Žltý oxid železa (E172)
Oxid titaničitý (E171)
Tlačiarenský atrament
Šelak (E904)
Propylénglykol (E1520) Hydroxid amónny (E527) Čierny oxid železa (E172) Hydroxid draselný (E525)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Talzenna 0,25mgtvrdékapsuly
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým (PP) uzáverom s tesniacou
vložkou. Veľkosť balenia: škatule s 30 kapsulami vo fľaši z HDPE.
Polyvinylchlorid/polyvinylidénchloridový (PVC/PVdC) perforovaný blister s jednotlivými dávkami prekrytý hliníkovou odlupovacou fóliou. Veľkosti balenia: škatule 30 x 1 kapsula, 60 x 1 kapsula alebo 90 x 1 kapsula v blistroch s jednotlivými dávkami.
Talzenna 1mgtvrdékapsuly
Fľaša z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým (PP) uzáverom s tesniacou
vložkou. Veľkosť balenia: škatule s 30 kapsulami vo fľaši z HDPE.
Polyvinylchlorid/polyvinylidénchloridový (PVC/PVdC) perforovaný blister s jednotlivými dávkami prekrytý hliníkovou odlupovacou fóliou. Veľkosť balenia: škatule 30 x 1 kapsula v blistroch
s jednotlivými dávkami.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
Talzenna
0,25
m
g
tvrdékapsuly
EU/1/19/1377/001
EU/1/19/1377/002
EU/1/19/1377/003
EU/1/19/1377/004
Talzenna1mgtvrdékapsulyEU/1/19/1377/005
EU/1/19/1377/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.