
ej triedy orgánových systémov sú ADR zoradené podľa frekvencie, pričom najčastejšie reakcie sú prvé. V rámci každej skupiny frekvencií sa nežiaduce reakcie na lieky uvádzajú v poradí klesajúcej závažnosti. Okrem toho je zodpovedajúca kategória frekvencie každého ADR založená na tejto konvencii: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1000); veľmi zriedkavé (< 1/10 000).
V klinických skúšaniach vo vývoji zameranom na ložiskovú psoriázu bolo Taltzom liečených celkom
4 204 pacientov. Z týchto bolo 2 190 psoriatických pacientov vystavených pôsobeniu Taltzu najmenej
počas jedného roka, čo predstavuje 3 531 paciento-rokov expozície.
Tri placebom kontrolované štúdie fázy III s ložiskovou psoriázou sa spojili kvôli vyhodnoteniu
bezpečnosti Taltzu v porovnaní s placebom až do 12 týždňov od začiatku liečby. Hodnotilo sa celkom
3 119 pacientov (1 161 pacientov s dávkou 80 mg každé 4 týždne (Q4W), 1 167 pacientov s dávkou
80 mg každé 2 týždne (Q2W) a 791 pacientov s placebom).
Tabuľka č. 1. Prehľad nežiaducich reakcií v klinických skúšaniacha
Trieda orgánového systému Taltz Placebo
I
n
f
ekcie a nákazy
Q
4W
(
N = 1161)
n (%)
Q
2W
(
N = 1167)
n (%)
(
N = 791)
n (%)
veľmi časté infekcie horných dýchacích ciestb
155 (13,4) 163 (14,0) 101 (12,8)
časté tinea 10 (0,9) 17 (1,5) 1 (0,1)
menej časté chrípka 10 (0,9) 8 (0,7) 0 rinitída 10 (0,9) 9 (0,8) 0 orálna kandidózac 2 (0,2) 9 (0,8) 0 konjunktivitída 1 (0,1) 8 (0,7) 3 (0,4) celulitídad 10 (0,9) 9 (0,8) 2 (0,3)
Poruchy krvi a lymfatického systému
menej časté neutropéniaf 3 (0,3) 6 (0,5) 1 (0,1)
trombocytopéniaf 2 (0,2) 2 (0,2) 0
Poruchy dýchacej sústavy, hrudníka a mediastína
časté orofaryngálna
bolesť
Poruchy gastrointestinálneho traktu
20 (1,7) 16 (1,4) 4 (0,5)
časté nevoľnosť 15 (1,3) 23 (2,0) 5 (0,6)
Poruchy kože a podkožného tkaniva
menej časté žihľavka 6 (0,5) 10 (0,9) 0
Celkové poruchy a reakcie v mieste podania
veľmi časté reakcie v mieste
e
150 (12,9) 196 (16,8) 26 (3,3)
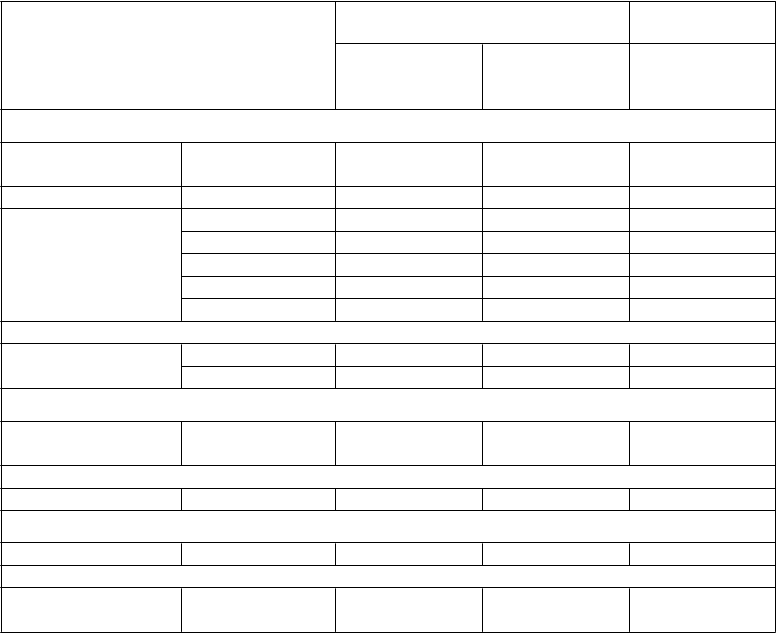
podania
a
Placebom kontrolované klinické skúšania (fázy III) u pacientov so stredne závažnou až závažnou
ložiskovou psoriázou s expozíciou ixekizumabu 80 mg Q2W, ixekizumabu 80 mg Q4W alebo placebu počas až 12 týždňov trvania liečby
b
Infekcia horných dýchacích ciest zahŕňa nazofaryngitídu a infekciu horných dýchacích ciest
c
Orálna kandidóza definovaná ako prípady s preferovanými termínmi orálna kandidóza
a orálna fungálna infekcia
d
Celulitída zahŕňa stafylokokovú celulitídu, celulitídu vonkajšieho ucha a eryzipel
e Reakcie v mieste podania boli častejšie u pacientov s telesnou hmotnosťou < 60 kg v porovnaní so skupinou s telesnou hmotnosťou ≥ 60 kg (25 % oproti 14 % u kombinovaných skupín Q2W a Q4W) f Na základe hlásených nežiaducich účinkov
Opis vybraných nežiaducich reakciíReakcie v mieste podania injekcieNajčastejšie pozorované reakcie v mieste podania injekcie boli erytém a bolesť. Tieto reakcie boli prevažne slabej až stredne závažnej intenzity a neviedli k ukončeniu podávania Taltzu.
InfekcieV placebom kontrolovanej časti klinických skúšaní fázy III s ložiskovou psoriázou boli infekcie hlásené u 27,2 % pacientov liečených Taltzom počas až 12 týždňov, v porovnaní s 22,9 % pacientov
liečených placebom.
Väčšina infekcií bola nezávažná a miernej až stredne závažnej intenzity a väčšina z nich nevyžadovala
prerušenie liečby. Závažné infekcie sa vyskytli u 13 (0,6 %) pacientov liečených Taltzom a u 3 (0,4 %) pacientov liečených placebom (pozri časť 4.4). Počas celej liečebnej doby boli infekcie
hlásené u 52,8 % pacientov liečených Taltzom (46,9 zo 100 paciento-rokov). Závažné infekcie boli
hlásené u 1,6 % pacientov liečených Taltzom (1,5 zo 100 paciento-rokov).
Laboratórne hodnotenie neutropénie a trombocytopénieU 9 % pacientov liečených Taltzom sa pozorovala neutropénia. Vo väčšine prípadov počet krvných neutrofilov bol ≥ 1 000 buniek/mm3. Takéto hladiny neutropénie môžu pretrvávať, fluktuovať alebo byť dočasné. U 0,1 % pacientov užívajúcich Taltz sa pozoroval počet neutrofilov <1 000 buniek/mm3. Vo všeobecnosti neutropénia nevyžadovala prerušenie podávania Taltzu.
U 3 % pacientov vystavených Taltzu sa zmenila hladina počtu krvných doštičiek z normálnej základnej hodnoty na hodnoty <150 000 doštičiek/mm3 a ≥75 000 doštičiek/mm3. Trombocytopénia môže pretrvávať, fluktuovať alebo byť dočasná.
ImunogenicitaPribližne u 9–17 % pacientov liečených Taltzom v odporúčanom dávkovacom režime sa vytvorili protilátky proti lieku, väčšina ktorých mala nízke titre a nesúviseli so zníženou klinickou odpoveďou
do 60. týždňa liečby. Avšak približne u 1 % pacientov liečených Taltzom sa potvrdili neutralizačné
protilátky súvisiace s nízkymi koncentráciami lieku a zníženou klinickou odpoveďou. Súvislosť medzi
imunogenicitou a liečbou vyvolanými nežiaducimi účinkami nebola jasne preukázaná.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieDávky do 180 mg sa podávali subkutánne v klinických skúšaniach bez dávku limitujúcej toxicity.
V klinických štúdiách boli hlásené predávkovania až do 240 mg v jednom subkutánnom podaní bez
akýchkoľvek závažných nežiaducich účinkov. V prípade predávkovania sa odporúča, aby bol pacient sledovaný na všetky prejavy a príznaky nežiaducich reakcií a aby sa okamžite začalo s príslušnou symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresíva, inhibítory interleukínu, ATC kód: L04AC13
Mechanizmus účinku
Ixekizumab je monoklonálna protilátka IgG4, ktorá sa s vysokou afinitou (< 3 pM) a špecificitou viaže
na interleukín 17A (ako IL-17A aj IL-17A/F). Zvýšené koncentrácie interleukínu IL-17A hrajú úlohu v patogenéze psoriázy prostredníctvom podpory proliferácie a aktivácie keratínocytov. Neutralizácia
IL-17A ixekizumabom túto aktivitu inhibuje. Ixekizumab sa neviaže na ligandy IL-17B, IL-17C,
IL-17D, IL-17E ani IL-17F.
Väzbové testy in vitro potvrdili, že ixekizumab sa neviaže na ľudské Fcγ receptory I, IIa a IIIa ani na zložku komplementu C1q.
Farmakodynamické účinky
Ixekizumab moduluje biologické odpovede, ktoré sú vyvolané alebo regulované interleukínom
IL-17A. Na základe údajov z biopsie psoriatickej kože z klinického skúšania fázy I bol zaznamenaný od dávky závislý trend znižovania hrúbky epidermy, množstva proliferujúcich keratinocytov, T-buniek a dendritických buniek, ako aj redukcie lokálnych zápalových markerov od počiatku po 43. deň. Priamym dôsledkom je, že liečba ixekizumabom redukuje erytém, induráciu a šupinatosť kože
v léziách ložiskovej psoriázy.
Klinická účinnosť abezpečnosť
Účinnosť a bezpečnosť Taltzu sa hodnotili v troch randomizovaných, dvojito zaslepených, placebom
kontrolovaných klinických skúšaniach fázy III u dospelých pacientov so stredne závažnou až závažnou ložiskovou psoriázou, ktorí boli kandidátmi na fototerapiu alebo systémovú liečbu
(UNCOVER-1, UNCOVER-2 a UNCOVER-3). Účinnosť a bezpečnosť Taltzu sa tiež hodnotili
v porovnaní s etanerceptom (UNCOVER-2 a UNCOVER-3). Pacienti randomizovaní na Taltz, ktorí boli sPGA (0,1) respondéri v 12. týždni, boli re-randomizovaní na podávanie placeba alebo Taltzu
počas ďalších 48 týždňov (UNCOVER-1 a UNCOVER-2); pacienti randomizovaní na placebo,
etanercept alebo Taltz, ktorí boli sPGA (0,1) non-respondéri, dostávali Taltz počas ďalších až
48 týždňov.
Z 3 866 pacientov zaradených do týchto placebom kontrolovaných klinických skúšaní 64 % dostalo predtým systémovú liečbu (biologickú, konvenčnú systémovú alebo psoralen a liečbu ultrafialovým žiarením A (PUVA)), 43,5 % dostalo predtým fototerapiu, 49,3 % dostalo predtým konvenčnú systémovú liečbu a 26,4 % dostalo predtým biologickú liečbu psoriázy. Zo všetkých pacientov 14,9 % dostalo aspoň jeden anti-TNF alfa liek a 8,7 % dostalo anti-IL-12/IL-23 liek. 23,4 % pacientov malo pri vstupnom vyšetrení v anamnéze psoriatickú artritídu.
Vo všetkých troch klinických skúšaniach bolo koprimárnym koncovým ukazovateľom percento pacientov, ktorí dosiahli v porovnaní s placebom odpoveď PASI 75 a sPGA s hodnotou 0 („čisté“) alebo 1 („minimálne“) v 12. týždni. Pacienti vo všetkých liečebných skupinách mali medián vstupného PASI skóre v rozmedzí od 17,4 do 18,3; 48,3 % až 51,2 % pacientov malo závažné alebo veľmi závažné vstupné sPGA skóre a priemernú vstupnú hodnotu na numerickej škále svrbenia kože (Numeric Rating Scale, itch NRS) v rozmedzí od 6,3 do 7,1.
Klinická odpoveď v 12. týždni
Do UNCOVER-1 bolo zaradených 1 296 pacientov. Pacienti boli randomizovaní (1:1:1) na podávanie placeba alebo Taltzu (80 mg každé dva alebo štyri týždne [Q2W alebo Q4W] po podaní 160 mg počiatočnej dávky) po dobu 12 týždňov.
T
abuľka č. 2. Výsledky účinnosti v 12. týždni z UNCOVER-1
Koncové
Počet pacientov (%) Rozdiel oproti placebu v miere
odpovede (95% CI)
ukazovatele
sPGA
s hodnotou „0“
Placebo
(N = 431)
Taltz
80 mg Q4W (N = 432)

a
Taltz
80 mg Q2W (N = 433)
a
Taltz
80 mg Q4W
Taltz
80 mg Q2W
(čisté) alebo „1“ (minimálne)
14 (3,2) 330 (76,4)
354 (81,8)
73,1 (68,8; 77,5) 78,5 (74,5; 82,5)
sPGA s
hodnotou „0“
(čisté)
0 149 (34,5)a 160 (37,0)a 34,5 (30,0; 39,0) 37,0 (32,4; 41,5)
PASI 75 17 (3,9 357 (82,6)a 386 (89,1)a 78,7 (74,7; 82,7) 85,2 (81,7; 88,7)
PASI 90 2 (0,5) 279 (64,6)a 307 (70,9)a 64,1 (59,6; 68,7) 70,4 (66,1; 74,8) PASI 100 0 145 (33,6)a 153 (35,3)a 33,6 (29,1; 38,0) 35,3 (30,8; 39,8)
Pokles v škále
svrbenia
Itch NRS ≥ 4b
58 (15,5) 305 (80,5)a 336 (85,9)a 65,0 (59,5; 70,4) 70,4 (65,4; 75,5)
Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
Pacienti s itch NRS >= 4 pri vstupnom vyšetrení: placebo N = 374, Taltz 80 mg Q4W N = 379, Taltz
80 mg Q2W N = 391
Do UNCOVER-2 bolo zaradených 1 224 pacientov. Pacienti boli randomizovaní (1:2:2:2) na podávanie placeba alebo Taltzu (80 mg každé dva alebo štyri týždne [Q2W alebo Q4W] po 160 mg počiatočnej dávke) alebo etanerceptu 50 mg dvakrát týždenne počas 12 týždňov.
T
abuľka č. 3. Výsledky účinnosti v 12. týždni z UNCOVER-2
Počet pacientov (%) Rozdiel oproti placebu
v miere odpovede (95% CI)
Koncové ukazovatele
sPGA „0“ (čisté) alebo
Placebo
(N = 168)
Taltz
80 mg Q4W (N = 347)
a
Taltz
80 mg Q2W (N = 351)
a
Etanercept
50 mg dvakrát týždenne
(N = 358)
Taltz
80 mg Q4W
70,5 (65,3;
Taltz
80 mg Q2W
80,8 (76,3;
„1“ (minimálne)
4 (2,4) 253 (72,9)
292 (83,2)
129 (36,0)
75,7)
85,4)
sPGA „0“
a,b
a,b
c 31,7 (26,6;
41,3 (36,0;
(čisté) 1 (0,6) 112 (32,3)
147 (41,9)
21 (5,9)
36,7)
46,6)
PASI 75 4 (2,4) 269 (77,5)a 315 (89,7)a 149 (41,6)a 75,1 (70,2;
80,1)
PASI 90 1 (0,6) 207 (59,7)a,b 248 (70,7)a,b 67 (18,7)a 59,1 (53,8;
64,4)
PASI 100 1 (0,6) 107 (30,8)a,b 142 (40,5)a,b 19 (5,3)c 30,2 (25,2;
35,2)
87,4 (83,4;
91,3)
70,1 (65,2;
75,0)
39,9 (34,6;
45,1)
Pokles v škále svrbenia Itch
a,b
a,b
a 62,7 (55,1;
71,1 (64,0;
NRS ≥ 4d 19 (14,1) 225 (76,8)
258 (85,1)
177 (57,8)
70,3)
78,2)
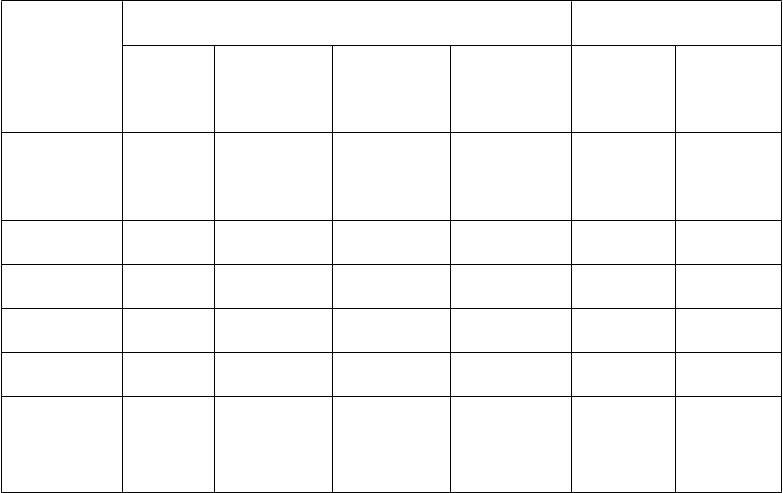 Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
p < 0,001 v porovnaní s etanerceptom
c
p < 0,01 v porovnaní s placebom
d
P
acienti s itch NRS > = 4 pri vstupnom vyšetrení: placebo N = 135, Taltz 80 mg Q4W N = 293, Taltz
80 mg Q2W N = 303, Etanercept N = 306
Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
p < 0,001 v porovnaní s etanerceptom
c
p < 0,01 v porovnaní s placebom
d
P
acienti s itch NRS > = 4 pri vstupnom vyšetrení: placebo N = 135, Taltz 80 mg Q4W N = 293, Taltz
80 mg Q2W N = 303, Etanercept N = 306
Do UNCOVER-3 bolo zaradených 1 346 pacientov. Pacienti boli randomizovaní (1:2:2:2) na podávanie placeba alebo Taltzu (80 mg každé dva alebo štyri týždne [Q2W alebo Q4W] po 160 mg počiatočnej dávke) alebo etanerceptu 50 mg dvakrát týždenne po dobu 12 týždňov.
T
abuľka č. 4. Výsledky účinnosti v 12. týždni z UNCOVER 3
Koncové
Počet pacientov (%)
Etanercept
Rozdiel oproti placebu v miere
odpovede (95% CI)
ukazovatele
sPGA
s hodnotou
Placebo
(N = 193)
Taltz
80 mg Q4W (N = 386)
Taltz
80 mg Q2W (N = 385)
50 mg dvakrát týždenne (N = 382)
Taltz
80 mg Q4W
Taltz
80 mg Q2W
„0“ (čisté) alebo „1“ (minimálne) sPGA s hodnotou „0“ (čisté)
13 (6,7) 291 (75,4)a,b 310 (80,5)a,b 159 (41,6)a 68,7 (63,1;
74,2)
0 139 (36,0)a,b 155 (40,3)a,b 33 (8,6)a 36,0 (31,2;
40,8)
73,8 (68,5;
79,1)
40,3 (35,4;
45,2)
PASI 75 14 (7,3) 325 (84,2)a,b 336 (87,3)a,b 204 (53,4)a 76,9 (71,8;
82,1)
PASI 90 6 (3,1) 252 (65,3)a,b 262 (68,1)a,b 98 (25,7)a 62,2 (56,8;
67,5)
80,0 (75,1;
85,0)
64,9 (59,7;
70,2)
PASI 100 0 135 (35,0)a,b 145 (37,7)a,b 28 (7,3)a 35 (30,2; 39,7) 37,7 (32,8;
42,5)
Pokles v škále svrbenia Itch NRS ≥ 4c
33 (20,9) 250 (79,9)a,b 264 (82,5)a,b 200 (64,1)a 59,0 (51,2;
66,7)
61,6 (54,0;
69,2)
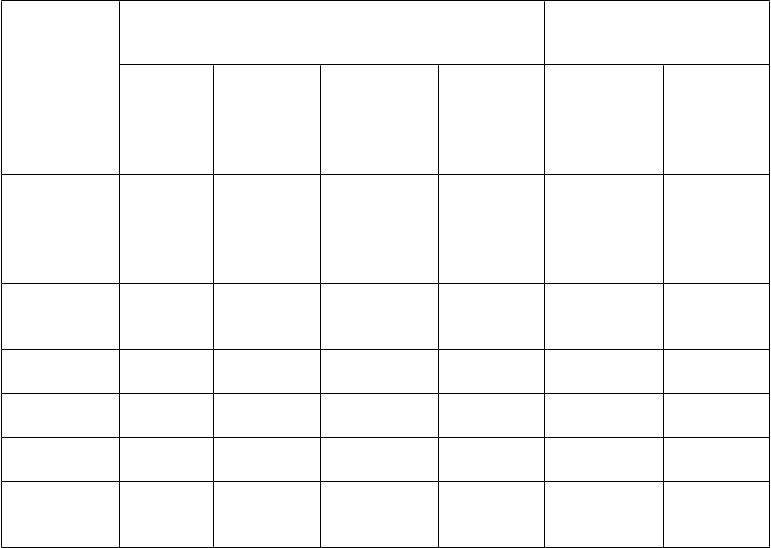 Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
p < 0,001 v porovnaní s etanerceptom
c
P
acienti s itch NRS >= 4 pri vstupnom vyšetrení: placebo N = 158, Taltz 80 mg Q4W N = 313, Taltz
80 mg Q2W N = 320, Etanercept N = 312
Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
p < 0,001 v porovnaní s etanerceptom
c
P
acienti s itch NRS >= 4 pri vstupnom vyšetrení: placebo N = 158, Taltz 80 mg Q4W N = 313, Taltz
80 mg Q2W N = 320, Etanercept N = 312
Taltz bol spojený s rýchlym nástupom účinnosti s > 50 % znížením priemerného PASI do 2. týždňa
(obrázok č. 1). Percento pacientov, ktorí dosiahli PASI 75, bolo signifikantne vyššie u Taltzu
v porovnaní s placebom a etanerceptom už v 1. týždni. Približne 25 % pacientov liečených Taltzom dosiahlo PASI skóre < 5 do 2. týždňa, viac ako 55 % dosiahlo PASI skóre < 5 do 4. týždňa a ich počet
sa zvýšil na 85 % do 12. týždňa (v porovnaní s 3 %, 14 % a 50 % u etanerceptu). Významné zlepšenie
závažnosti svrbenia bolo pozorované u pacientov liečených Taltzom v 1. týždni.
O
brázok č. 1. PASI skóre, percentuálne zlepšenie pri každej návšteve od počiatku (mBOCF)) v populácii so zámerom liečiť (ITT populácii) v priebehu fázy indukčného dávkovania - UNCOVER-2 a UNCOVER-3

Účinnosť a bezpečnosť Taltzu bola preukázaná bez ohľadu na vek, pohlavie, rasu, telesnú hmotnosť, iniciálnu závažnosť PASI, umiestnenie ložiska, konkurentnú psoriatickú artritídu a predchádzajúcu biologickú liečbu. Taltz bol účinný u pacientov doteraz neliečených systémovou liečbou, doteraz neliečených biologickou liečbou, u pacientov, ktorí podstúpili biologickú/anti-TNF liečbu
aj u pacientov, u ktorých biologická/anti-TNF liečba zlyhala.
Účinnosť u non-respondérov na etanercept: z pacientov, ktorí boli sPGA (0,1) non-respondéri na etanercept v 12. týždni v UNCOVER-2 (N = 200) a ktorí boli prestavení na Taltz 80 mg Q4W po 4- týždňovom období bez podávania liečiva („washout“ obdobie), sa po 12 týždňoch liečby Taltzom
73 % pacientov podarilo dosiahnuť sPGA (0,1) a 83,5 % pacientov PASI 75.
V 2 klinických skúšaniach, ktoré zahŕňali aktívny komparátor (UNCOVER-2 a UNCOVER-3), bola miera výskytu závažných nežiaducich účinkov u etanerceptu aj u Taltzu 1,9 % a miera ukončenia liečby kvôli nežiaducim účinkom bola 1,2 % u etanerceptu a 2,0 % u Taltzu. Miera výskytu infekcií bola 21,5 % u etanerceptu a 26,0 % u Taltzu, pričom väčšina prípadov bola miernej až stredne závažnej závažnosti. Miera výskytu závažných infekcií bola 0,4 % u etanerceptu a 0,5 % u Taltzu.
Udržanie odpovede v 60. týždniPacienti pôvodne randomizovaní na Taltz, ktorí reagovali na liečbu v 12. týždni (t.j. sPGA skóre
s hodnotou 0,1) v UNCOVER-1 aj UNCOVER-2, boli re-randomizovaní na dobu ďalších 48 týždňov
na jeden z týchto liečebných režimov: placebo alebo Taltz (80 mg každé štyri alebo dvanásť týždňov
[Q4W alebo Q12W]).
T
abuľka č. 5. Udržanie odpovede a účinnosti v 60. týždni
(št
údie UNCOVER-1 a UNCOVER-2)
Počet pacientov (%)
Rozdiel oproti placebu v miere
odpovede (95% CI)
Koncové
ukazovatele
Udržané sPGA „0“
80 mg Q4W (indukcia) / Placebo (udržanie) (N
= 191)
80 mg Q2W (indukcia) / Placebo (udržanie) (N
= 211)
80 mg Q4W (indukcia) /
80 mg Q4W (udržanie) (N

= 195)
80 mg Q2W (indukcia) /
80 mg Q4W (udržanie) (N
= 221)
80 mg Q4W (indukcia) /
80 mg Q4W (udržanie)
80 mg Q2W (indukcia) /
80 mg Q4W (udržanie)
(čisté) alebo
„1“ (minimálne)
Udržané alebo dosiahnuté sPGA „0“ (čisté) Udržané alebo dosiahnuté PASI 75
Udržané alebo dosiahnuté PASI 90
Udržané alebo dosiahnuté PASI 100
12 (6,3) 16 (7,6) 134 (68.7)a 173 (78,3)a 62,4 (55,1;
69,8)
3 (1,6) 6 (2,8) 96 (49.2)a 130 (58,8)a 47,7 (40,4;
54,9)
15 (7,9) 19 (9,0) 145 (74.4)a 184 (83,3)a 66,5 (59,3;
73,7)
9 (4,7) 10 (4,7) 130 (66.7)a 169 (76,5)a 62,0 (54,7;
69,2)
3 (1,6) 6 (2,8) 97 (49,7)a 127 (57,5)a 48.2 (40,9;
55,4)
70,7 (64,2;
77,2)
56,0 (49,1;
62,8)
74,3 (68,0;
80,5)
71,7 (65,4;
78,0)
54.6 (47,7;
61,5)
Skratky: N = počet pacientov v analyzovanej populácii
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítajú ako non-respondéri
a
p < 0,001 v porovnaní s placebom
Taltz bol účinný pri udržaní odpovede u pacientov bez predchádzajúcej systémovej liečby, bez predchádzajúcej biologickej liečby, u pacientov s anamnézou biologickej/anti-TNF liečby
aj u pacientov, u ktorých biologická/anti-TNF liečba zlyhala.
U respondérov s sPGA (0,1) v 12. týždni, re-randomizovaných na vysadenie liečby (t.j. na placebo), bol medián času do relapsu (sPGA ≥ 3) 164 dní v integrovaných štúdiách UNCOVER-1 a UNCOVER-2. Z týchto pacientov 71,5 % znovu dosiahlo odpoveď aspoň sPGA (0,1) v priebehu
12 týždňov po opakovanom nastavení na Taltz 80 mg Q4W
V porovnaní s placebom a etanerceptom sa prejavilo signifikantne väčšie zlepšenie od vstupného vyšetrenia do 12. týždňa u nechtovej psoriázy (pri meraní podľa Indexu závažnosti nechtovej psoriázy
[Nail Psoriasis Severity Index, NAPSI]), u psoriázy pokožky hlavy (pri meraní podľa Indexu závažnosti psoriázy pokožky hlavy [Psoriasis Scalp Severity Index, PSSI]) a u palmoplantárnej psoriázy (pri meraní podľa Indexu závažnosti palmoplantárnej psoriázy [Psoriasis Palmoplantar
Severity Index, PPASI]). Tieto zlepšenia nechtovej psoriázy, psoriázy pokožky hlavy a palmoplantárnej psoriázy sa u pacientov liečených Taltzom, ktorí boli v 12. týždni respondéri na
liečbu s sPGA (0,1), udržali aj v 60. týždni.
Kvalita života/výsledky hlásené pacientom
V 12. týždni a počas klinických skúšaní sa Taltz spájal so štatisticky signifikantným zlepšením
v kvalite života spojenej so zdravím meraným priemerným poklesom oproti východiskovému stavu
podľa Dermatologického indexu kvality života (Dermatology Life Quality Index, DLQI) (Taltz 80 mg
Q2W od -10,2 do -11,1, Taltz 80 mg Q4W od -9,4 do -10,7, etanercept od -7,7 do -8,0 a placebo od -
1,0 do -2,0). Signifikantne väčší podiel pacientov liečených Taltzom dosiahlo DLQI 0 alebo 1. Naprieč štúdiami sa Taltz spájal so štatisticky signifikantným zlepšením závažnosti svrbenia
hodnoteného podľa Itch NRS skóre. Signifikantne väčší podiel pacientov liečených Taltzom
dosiahol redukciu Itch NRS ≥ 4 body v 12. týždni (84,6 % pre Taltz Q2W, 79,2 % pre Taltz Q4W
a 16,5 % pre placebo) a benefit bol udržaný počas celého času až do 60. týždňa u pacientov liečených
Taltzom, ktorí boli sPGA (0 alebo 1) respondéri v 12. týždni. Neboli žiadne dôkazy zhoršenia depresie
počas 60 týždňov liečby Taltzom podľa hodnotenia Stručného súpisu sebahodnotiacej depresívnej
symptomatológie (Quick Inventory of Depressive Symptomatology Self Report).
Imunizácia
V štúdii so zdravými jedincami neboli identifikované žiadne bezpečnostné riziká s dvoma
inaktivovanými vakcínami (proti tetanu a pneumokokom), po prijatí dvoch dávok ixekizumabu (160 mg nasledovaná druhou dávkou 80 mg o dva týždne neskôr ). Avšak údaje týkajúce sa imunizácie boli nedostatočné, aby sa dospelo k záveru o primeranej imunitnej reakcii na tieto vakcíny po podaní Taltzu.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Taltzom v jednej
alebo vo viacerých podskupinách pediatrickej populácie pri liečbe ložiskovej psoriázy (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po jednom subkutánnom podaní dávky ixekizumabu pacientom so psoriázou boli priemerné
maximálne koncentrácie dosiahnuté v priebehu 4 až 7 dní v rozpätí všetkých dávok od 5 do 160 mg. Priemerná (SD) maximálna plazmatická koncentrácia (Cmax) ixekizumabu po počiatočnej dávke
160 mg bola 19,9 (8,15) µg/ml.
Po počiatočnej dávke 160 mg bol rovnovážny stav dosiahnutý do 8. týždňa s dávkovacím režimom
80 mg Q2W. Odhad priemernej (SD) Cmax, ss, je 21,5 (9,16) µg/ml a odhad priemernej Ctrough, ss je
5,23 (3,19) µg/ml.
Po prestavení dávkovacieho režimu z 80 mg Q2W na dávkovací režim 80 mg Q4W v 12. týždni by sa
mal rovnovážny stav dosiahnuť po približne 10 týždňoch. Odhad priemernej (SD) Cmax, ss, je
14,6 (6,04) µg/ml a odhad priemernej Ctrough, ss je 1,87 (1,30) µg/ml.
Priemerná biodostupnosť ixekizumabu po subkutánnom podaní bola vo všetkých analýzach 54 % až
90 %.
Distribúcia
Z populačných farmakokinetických analýz vyplýva, že priemerný celkový distribučný objem
v rovnovážnom stave bol 7,11 l.
Biotransformácia
Ixekizumab je monoklonálna protilátka a očakáva sa, že sa bude rozkladať na malé peptidy a
aminokyseliny katabolickými dráhami takým istým spôsobom ako endogénne imunoglobulíny.
E
li
m
i
nácia
V populačnej PK analýze bol priemerný sérový klírens 0,0161 l/h. Klírens nezávisí od dávky.
Priemerný eliminačný polčas podľa odhadu z populačnej farmakokinetickej analýzy je u pacientov s ložiskovou psoriázou 13 dní.
Linearita / nelinearita
Expozícia (AUC) sa pri podaní vo forme subkutánnej injekcie úmerne zvyšovala v rozmedzí dávok od
5 do 160 mg.
Starší pacienti
Z 4204 pacientov s ložiskovou psoriázou vystavených pôsobeniu Taltzu v klinických skúšaniach bolo
celkom 301 pacientov 65-ročných alebo starších a 36 pacientov bolo 75-ročných alebo starších. Na základe populačnej farmakokinetickej analýzy obmedzeného počtu starších pacientov (n = 94 pre vek
≥ 65 rokov a n = 12 pre vek ≥ 75 rokov) bol klírens u starších pacientov a pacientov mladších ako 65-
ročných podobný.
Porucha funkcie obličiek a pečene
Špecifické štúdie klinickej farmakológie na vyhodnotenie účinkov poruchy funkcie obličiek a poruchy
funkcie pečene na PK ixekizumabu sa neuskutočnili. Očakáva sa, že renálna eliminácia intaktného ixekizumabu, monoklonálnej IgG bude nízka a menšieho významu; a takisto monoklonálne protilátky
IgG sa hlavne eliminujú intracelulárnym katabolizmom a neočakáva sa, že porucha funkcie pečene
bude mať vplyv na klírens ixekizumabu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje od opíc makak cynomolgus získané na základe farmakologických štúdií toxicity po opakovanom podávaní, vyhodnotení farmakologickej bezpečnosti, reprodukčnej a vývinovej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Podávanie ixekizumabu opiciam makak cynomolgus v subkutánnych dávkach do 50 mg/kg týždenne po dobu 39 týždňov nevyvolalo žiadnu orgánovú toxicitu ani nežiaduce účinky na imunitné funkcie (napr. protilátková odpoveď závislá od T-buniek a aktivita NK buniek). Týždenná subkutánna dávka pre opice v hodnote 50 mg/kg je približne 19-násobok 160 mg počiatočnej dávky Taltzu a u opíc má za následok expozíciu (AUC), ktorá je najmenej 61-násobne vyššia ako predpovedaná priemerná rovnovážna expozícia u ľudí pri odporúčanom dávkovacom režime.
Predklinické štúdie na vyhodnotenie karcinogénneho alebo mutagénneho potenciálu ixekizumabu sa
neuskutočnili.
U pohlavne dospelých opíc makak cynomolgus, ktorým bol podávaný ixekizumab po dobu 13 týždňov v týždennej subkutánne podávanej dávke 50 mg/kg, neboli pozorované žiadne účinky na reprodukčné orgány, menštruačný cyklus ani spermie.
V štúdiách vývojovej toxicity bolo preukázané, že ixekizumab prechádza placentou a bol prítomný v krvi potomstva až do veku 6 mesiacov. Vyšší výskyt postnatálnej mortality v porovnaní so súbežnými kontrolami bol zaznamenaný u potomstva opíc, ktorým bol podávaný ixekizumab. Toto sa týkalo predovšetkým predčasného pôrodu alebo zanedbávania potomstva matkou, čo sú bežné nálezy v štúdiách s nehumánnymi primátmi a boli považované za nesúvisiace s ixekizumabom.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
citrát sodný
kyselina citrónová, bezvodá chlorid sodný
polysorbát 80
voda na injekcie
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 ºC – 8 ºC).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
1 ml roztoku v injekčnej striekačke z čistého skla typu I. Veľkosti balenia s 1, 2 alebo 3 naplnenými
injekčnými striekačkami. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Návod na použitie
Pokyny na používanie injekčnej striekačky, ktoré sú súčasťou písomnej informácie pre používateľa, sa musia dôsledne dodržiavať.
Naplnená injekčná striekačka je iba na jedno použitie.
Taltz sa nemá používať, ak sa v ňom objavia častice alebo ak je roztok zakalený a/alebo zreteľne
hnedý.
Taltz, ktorý bol uchovávaný v mrazničke, sa nesmie používať.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Holandsko.
8. REGISTRAČNÉ ČÍSLO(ČÍSLA)
EU/1/15/1085/004
EU/1/15/1085/005
EU/1/15/1085/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUTaltz 80 mg injekčný roztok naplnený v pere.
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEJedno naplnené pero obsahuje v 1 ml 80 mg ixekizumabu*.
*Ixekizumab je rekombinantná humanizovaná monoklonálna protilátka, ktorá sa tvorí v CHO
bunkách.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA Injekčný roztok naplnený v pere. Roztok je číry a bezfarebný až žltkastý.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieTaltz je určený na liečbu stredne závažnej až závažnej ložiskovej psoriázy u dospelých, ktorí sú kandidátmi na systémovú liečbu.
4.2 Dávkovanie a spôsob podávaniaTaltz je určený na použitie pod vedením a dohľadom lekára so skúsenosťami v diagnostike a liečbe
psoriázy.
DávkovanieOdporúčaná dávka je 160 mg v subkutánnej injekcii (dvoch 80 mg injekciách) v 0. týždni, po ktorej
nasleduje 80 mg (jedna injekcia) v 2., 4., 6., 8., 10. a 12. týždni, potom udržiavacie dávky s 80 mg
(jedna injekcia) každé 4 týždne.
Pozornosť sa má venovať prerušeniu liečby u pacientov, u ktorých sa neobjavila žiadna odpoveď ani po 16 až 20 týždňoch liečby. U niektorých pacientov s počiatočnou čiastočnou odpoveďou môže nastať zlepšenie počas pokračovania liečby dlhšie ako po 20 týždňov.
Starší pacienti (≥ 65 rokov)Úprava dávky nie je potrebná (pozri časť 5.2).
U pacientov vo veku ≥ 75 rokov sú k dispozícii len obmedzené údaje.
Porucha funkcie obličiek a pečeneV týchto skupinách pacientov nebol Taltz skúmaný. Odporúčanú dávku nemožno stanoviť.
P
ediatrická populácia
Bezpečnosť a účinnosť Taltzu u detí a dospievajúcich vo veku 6 až 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Neexistuje žiadne relevantné použitie Taltzu u detí mladších ako 6-ročných v liečbe stredne závažnej
až závažnej ložiskovej psoriázy.
Spôsob podávania
Subkutánne použitie.
Taltz je určený na podávanie v subkutánnej injekcii. Miesta podania injekcie sa môžu striedať. Ak je to možné, oblasti kože s výskytom psoriázy sa nemajú používať ako miesta vpichu. Roztok/striekačka sa nemá triasť.
Ak lekár rozhodne, že je to vhodné, môže si pacient po riadnom zácviku v technike podávania subkutánnej injekcie podávať injekciu Taltzu sám. Lekár však má zabezpečiť vhodné sledovanie pacientov. Všeobecné pokyny na podávanie lieku sú uvedené v písomnej informácii pre používateľa.
4.3 Kontraindikácie
Závažná precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Klinicky významné aktívne infekcie (napr. aktívna tuberkulóza, pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Infekcie
Liečba Taltzom sa spája so zvýšeným výskytom infekcií, ako sú napríklad infekcie horných dýchacích
ciest, kandidóza ústnej dutiny, konjunktivitída a tinea (pozri časť 4.8).
Taltz sa má používať s opatrnosťou u pacientov s klinicky významnou chronickou infekciou. Ak takáto infekcia vznikne, dôsledne liečbu sledujte a ak pacient nereaguje na štandardnú liečbu alebo ak sa infekcia stane závažnou, podávanie Taltzu ukončite. Kým sa infekcia nevylieči, v liečbe Taltzom sa nemá pokračovať.
Taltz sa nesmie podávať pacientom s aktívnou tuberkulózou (TBC). Pred začiatkom podávania Taltzu
pacientom s latentnou TBC zvážte protituberkulóznu liečbu.
Precitlivenosť
Boli hlásené závažné reakcie z precitlivenosti, vrátane niekoľkých prípadov angioedému a žihľavky
a zriedkavo neskoré (10-14 dní po podaní injekcie) závažné reakcie z precitlivenosti, vrátane celkovej žihľavky, dyspnoe a vysokých titrov protilátok. Ak sa objaví závažná reakcia z precitlivenosti, podávanie Taltzu sa má okamžite ukončiť a má sa začať s vhodnou liečbou.
Zápalové črevné ochorenie
Boli hlásené nové prípady alebo zhoršenia Crohnovej choroby a ulceróznej kolitídy. Opatrnosť je
potrebná pri predpisovaní Taltzu pacientom so zápalovým črevným ochorením vrátane Crohnovej choroby a ulceróznej kolitídy a pacienti majú byť dôkladne sledovaní.
Imunizácia
Taltz sa nemá používať so živými vakcínami. O reakcii na živé vakcíny nie sú k dispozícii žiadne
údaje; údaje o reakciách na inaktivované vakcíny sú obmedzené (pozri časť 5.1).
Pomocné látky
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v 80 mg dávke, t.j. je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Bezpečnosť Taltzu v kombinácii s inými imuno-modulačnými liekmi alebo s fototerapiou nebola
skúmaná.
Neuskutočnili sa žiadne formálne štúdie liekových interakcií in vivo. Nebola hlásená úloha IL-17 pri regulácii enzýmov CYP450. Tvorba niektorých enzýmov CYP450 je však počas chronických zápalov potláčaná zvýšenou hladinou cytokínov. Protizápalová liečba, ako napríklad liečba inhibítorom IL-17A ixekizumabom, preto môže mať za následok normalizáciu hladiny CYP450 so sprievodnou nižšou expozíciou súbežne podávaných liekov metabolizovaných enzýmom CYP450. Preto sa nedá vylúčiť klinicky významný účinok na substráty CYP450 s úzkym terapeutickým indexom, u ktorých sa dávka individuálne upravuje (napr. warfarín). Na začiatku liečby ixekizumabom u pacientov liečených týmito druhmi liekov sa má zvážiť terapeutické monitorovanie.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku majú počas liečby a najmenej 10 týždňov po liečbe používať účinnú metódu
antikoncepcie.
Gravidita
K dispozícii je len obmedzené množstvo údajov o používaní ixekizumabu u gravidných žien. Štúdie
na zvieratách nepreukázali priame ani nepriame škodlivé účinky týkajúce sa gravidity,
embryonálneho/fetálneho vývoja, pôrodu alebo postnatálneho vývoja (pozri časť 5.3). Ako preventívne opatrenie je počas gravidity vhodnejšie vyhnúť sa používaniu Taltzu.
Dojčenie
Nie je známe, či sa ixekizumab vylučuje do ľudského mlieka alebo či sa po použití systémovo
vstrebáva. Avšak ixekizumab sa vylučuje v nízkych hladinách do mlieka opíc makak cynomolgus. Je potrebné vziať do úvahy prínos dojčenia pre dieťa a prínos liečby pre ženu a potom sa rozhodnúť, či ukončiť dojčenie alebo ukončiť podávanie Taltzu.
Fertilita
Účinok ixekizumabu na ľudskú fertilitu ešte nebol skúmaný. Štúdie na zvieratách nepreukazujú
priame ani nepriame škodlivé účinky týkajúce sa fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Taltz nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Najčastejšie hlásenými nežiaducimi reakciami na lieky (adverse drug reactions, ADR) boli reakcie v
mieste podania injekcie a infekcie horných dýchacích ciest (najčastejšie nazofaryngitída).
T
abuľka s prehľadom nežiaducich reakcií
ADR z klinických skúšaní (tabuľka č.1) sú zoradené podľa triedy orgánových systémov MedDRA.
V rámci každej triedy orgánových systémov sú ADR zoradené podľa frekvencie, pričom najčastejšie reakcie sú prvé. V rámci každej skupiny frekvencií sa nežiaduce reakcie na lieky uvádzajú v poradí klesajúcej závažnosti. Okrem toho je zodpovedajúca kategória frekvencie každého ADR založená na tejto konvencii: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1000); veľmi zriedkavé (< 1/10 000).
V klinických skúšaniach vo vývoji zameranom na ložiskovú psoriázu bolo Taltzom liečených celkom
4 204 pacientov. Z týchto bolo 2 190 psoriatických pacientov vystavených pôsobeniu Taltzu najmenej
počas jedného roka, čo predstavuje 3 531 paciento-rokov expozície.
Tri placebom kontrolované štúdie fázy III s ložiskovou psoriázou sa spojili kvôli vyhodnoteniu
bezpečnosti Taltzu v porovnaní s placebom až do 12 týždňov od začiatku liečby. Hodnotilo sa celkom
3 119 pacientov (1 161 pacientov s dávkou 80 mg každé 4 týždne (Q4W), 1 167 pacientov s dávkou
80 mg každé 2 týždne (Q2W) a 791 pacientov s placebom).
Tabuľka č. 1. Prehľad nežiaducich reakcií v klinických skúšaniacha
Trieda orgánového systému Taltz Placebo
I
n
f
ekcie a nákazy
Q
4W
(
N = 1161)
n (%)
Q
2W
(
N = 1167)
n (%)
(
N = 791)
n (%)
veľmi časté infekcie horných dýchacích ciestb
155 (13,4) 163 (14,0) 101 (12,8)
časté tinea 10 (0,9) 17 (1,5) 1 (0,1)
menej časté chrípka 10 (0,9) 8 (0,7) 0 rinitída 10 (0,9) 9 (0,8) 0 orálna kandidózac 2 (0,2) 9 (0,8) 0 konjunktivitída 1 (0,1) 8 (0,7) 3 (0,4) celulitídad 10 (0,9) 9 (0,8) 2 (0,3)
Poruchy krvi a lymfatického systému
menej časté neutropéniaf 3 (0,3) 6 (0,5) 1 (0,1)
trombocytopéniaf 2 (0,2) 2 (0,2) 0
Poruchy dýchacej sústavy, hrudníka a mediastína
časté orofaryngálna
bolesť
Poruchy gastrointestinálneho traktu
20 (1,7) 16 (1,4) 4 (0,5)
časté nevoľnosť 15 (1,3) 23 (2,0) 5 (0,6)
Poruchy kože a podkožného tkaniva
menej časté žihľavka 6 (0,5) 10 (0,9) 0
Celkové poruchy a reakcie v mieste podania
veľmi časté reakcie v mieste
e
150 (12,9) 196 (16,8) 26 (3,3)
podania
a
Placebom kontrolované klinické skúšania (fázy III) u pacientov so stredne závažnou až závažnou
ložiskovou psoriázou s expozíciou ixekizumabu 80 mg Q2W, ixekizumabu 80 mg Q4W alebo placebu
počas až 12 týždňov trvania liečby
b
Infekcia horných dýchacích ciest zahŕňa nazofaryngitídu a infekciu horných dýchacích ciest
c
Orálna kandidóza definovaná ako prípady s preferovanými termínmi orálna kandidóza
a orálna fungálna infekcia
d
Celulitída zahŕňa stafylokokovú celulitídu, celulitídu vonkajšieho ucha a eryzipel
e Reakcie v mieste podania boli častejšie u pacientov s telesnou hmotnosťou < 60 kg v porovnaní so skupinou s telesnou hmotnosťou ≥ 60 kg (25 % oproti 14 % u kombinovaných skupín Q2W a Q4W) f Na základe hlásených nežiaducich účinkov
Opis vybraných nežiaducich reakciíReakcie v mieste podania injekcieNajčastejšie pozorované reakcie v mieste podania injekcie boli erytém a bolesť. Tieto reakcie boli
prevažne slabej až stredne závažnej intenzity a neviedli k ukončeniu podávania Taltzu.
InfekcieV placebom kontrolovanej časti klinických skúšaní fázy III s ložiskovou psoriázou boli infekcie hlásené u 27,2 % pacientov liečených Taltzom počas až 12 týždňov, v porovnaní s 22,9 % pacientov
liečených placebom.
Väčšina infekcií bola nezávažná a miernej až stredne závažnej intenzity a väčšina z nich nevyžadovala
prerušenie liečby. Závažné infekcie sa vyskytli u 13 (0,6 %) pacientov liečených Taltzom a u 3 (0,4 %) pacientov liečených placebom (pozri časť 4.4). Počas celej liečebnej doby boli infekcie
hlásené u 52,8 % pacientov liečených Taltzom (46,9 zo 100 paciento-rokov). Závažné infekcie boli
hlásené u 1,6 % pacientov liečených Taltzom (1,5 zo 100 paciento-rokov).
Laboratórne hodnotenie neutropénie a trombocytopénieU 9 % pacientov liečených Taltzom sa pozorovala neutropénia. Vo väčšine prípadov počet krvných neutrofilov bol ≥ 1 000 buniek/mm3. Takéto hladiny neutropénie môžu pretrvávať, fluktuovať alebo byť dočasné. U 0,1 % pacientov užívajúcich Taltz sa pozoroval počet neutrofilov <1 000 buniek/mm3. Vo všeobecnosti neutropénia nevyžadovala prerušenie podávania Taltzu.
U 3 % pacientov vystavených Taltzu sa zmenila hladina počtu krvných doštičiek z normálnej základnej hodnoty na hodnoty <150 000 doštičiek/mm3 a ≥75 000 doštičiek/mm3. Trombocytopénia môže pretrvávať, fluktuovať alebo byť dočasná.
ImunogenicitaPribližne u 9–17 % pacientov liečených Taltzom v odporúčanom dávkovacom režime sa vytvorili protilátky proti lieku, väčšina ktorých mala nízke titre a nesúviseli so zníženou klinickou odpoveďou
do 60. týždňa liečby. Avšak približne u 1 % pacientov liečených Taltzom sa potvrdili neutralizačné
protilátky súvisiace s nízkymi koncentráciami lieku a zníženou klinickou odpoveďou. Súvislosť medzi
imunogenicitou a liečbou vyvolanými nežiaducimi účinkami nebola jasne preukázaná.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieDávky do 180 mg sa podávali subkutánne v klinických skúšaniach bez dávku limitujúcej toxicity.
V klinických štúdiách boli hlásené predávkovania až do 240 mg v jednom subkutánnom podaní bez
akýchkoľvek závažných nežiaducich účinkov. V prípade predávkovania sa odporúča, aby bol pacient sledovaný na všetky prejavy a príznaky nežiaducich reakcií a aby sa okamžite začalo s príslušnou symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresíva, inhibítory interleukínu, ATC kód: L04AC13
Mechanizmus účinku
Ixekizumab je monoklonálna protilátka IgG4, ktorá sa s vysokou afinitou (< 3 pM) a špecificitou viaže
na interleukín 17A (ako IL-17A aj IL-17A/F). Zvýšené koncentrácie interleukínu IL-17A hrajú úlohu v patogenéze psoriázy prostredníctvom podpory proliferácie a aktivácie keratínocytov. Neutralizácia
IL-17A ixekizumabom túto aktivitu inhibuje. Ixekizumab sa neviaže na ligandy IL-17B, IL-17C,
IL-17D, IL-17E ani IL-17F.
Väzbové testy in vitro potvrdili, že ixekizumab sa neviaže na ľudské Fcγ receptory I, IIa a IIIa ani na zložku komplementu C1q.
Farmakodynamické účinky
Ixekizumab moduluje biologické odpovede, ktoré sú vyvolané alebo regulované interleukínom
IL-17A. Na základe údajov z biopsie psoriatickej kože z klinického skúšania fázy I bol zaznamenaný od dávky závislý trend znižovania hrúbky epidermy, množstva proliferujúcich keratinocytov, T-buniek a dendritických buniek, ako aj redukcie lokálnych zápalových markerov od počiatku po 43. deň. Priamym dôsledkom je, že liečba ixekizumabom redukuje erytém, induráciu a šupinatosť kože'
v léziách ložiskovej psoriázy.
Klinická účinnosť abezpečnosť
Účinnosť a bezpečnosť Taltzu sa hodnotili v troch randomizovaných, dvojito zaslepených, placebom
kontrolovaných klinických skúšaniach fázy III u dospelých pacientov so stredne závažnou až
závažnou ložiskovou psoriázou, ktorí boli kandidátmi na fototerapiu alebo systémovú liečbu
(UNCOVER-1, UNCOVER-2 a UNCOVER-3). Účinnosť a bezpečnosť Taltzu sa tiež hodnotili
v porovnaní s etanerceptom (UNCOVER-2 a UNCOVER-3). Pacienti randomizovaní na Taltz, ktorí boli sPGA (0,1) respondéri v 12. týždni, boli re-randomizovaní na podávanie placeba alebo Taltzu
počas ďalších 48 týždňov (UNCOVER-1 a UNCOVER-2); pacienti randomizovaní na placebo,
etanercept alebo Taltz, ktorí boli sPGA (0,1) non-respondéri, dostávali Taltz počas ďalších až
48 týždňov.
Z 3 866 pacientov zaradených do týchto placebom kontrolovaných klinických skúšaní 64 % dostalo predtým systémovú liečbu (biologickú, konvenčnú systémovú alebo psoralen a liečbu ultrafialovým žiarením A (PUVA)), 43,5 % dostalo predtým fototerapiu, 49,3 % dostalo predtým konvenčnú systémovú liečbu a 26,4 % dostalo predtým biologickú liečbu psoriázy. Zo všetkých pacientov 14,9 % dostalo aspoň jeden anti-TNF alfa liek a 8,7 % dostalo anti-IL-12/IL-23 liek. 23,4 % pacientov malo pri vstupnom vyšetrení v anamnéze psoriatickú artritídu.
Vo všetkých troch klinických skúšaniach bolo koprimárnym koncovým ukazovateľom percento pacientov, ktorí dosiahli v porovnaní s placebom odpoveď PASI 75 a sPGA s hodnotou 0 („čisté“) alebo 1 („minimálne“) v 12. týždni. Pacienti vo všetkých liečebných skupinách mali medián vstupného PASI skóre v rozmedzí od 17,4 do 18,3; 48,3 % až 51,2 % pacientov malo závažné alebo veľmi závažné vstupné sPGA skóre a priemernú vstupnú hodnotu na numerickej škále svrbenia kože (Numeric Rating Scale, itch NRS) v rozmedzí od 6,3 do 7,1.
Klinická odpoveď v 12. týždni
Do UNCOVER-1 bolo zaradených 1 296 pacientov. Pacienti boli randomizovaní (1:1:1) na podávanie placeba alebo Taltzu (80 mg každé dva alebo štyri týždne [Q2W alebo Q4W] po podaní 160 mg
počiatočnej dávky) po dobu 12 týždňov.
T
abuľka č. 2. Výsledky účinnosti v 12. týždni z UNCOVER-1
Koncové
Počet pacientov (%) Rozdiel oproti placebu v miere
odpovede (95% CI)
ukazovatele
sPGA
s hodnotou „0“
Placebo
(N = 431)
Taltz
80 mg Q4W (N = 432)
a
Taltz
80 mg Q2W (N = 433)
a
Taltz
80 mg Q4W
Taltz
80 mg Q2W
(čisté) alebo „1“ (minimálne)
14 (3,2) 330 (76,4)
354 (81,8)
73,1 (68,8; 77,5) 78,5 (74,5; 82,5)
sPGA s
hodnotou „0“
(čisté)
0 149 (34,5)a 160 (37,0)a 34,5 (30,0; 39,0) 37,0 (32,4; 41,5)
PASI 75 17 (3,9 357 (82,6)a 386 (89,1)a 78,7 (74,7; 82,7) 85,2 (81,7; 88,7)
PASI 90 2 (0,5) 279 (64,6)a 307 (70,9)a 64,1 (59,6; 68,7) 70,4 (66,1; 74,8) PASI 100 0 145 (33,6)a 153 (35,3)a 33,6 (29,1; 38,0) 35,3 (30,8; 39,8)
Pokles v škále
svrbenia
Itch NRS ≥ 4b
58 (15,5) 305 (80,5)a 336 (85,9)a 65,0 (59,5; 70,4) 70,4 (65,4; 75,5)
Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
P
acienti s itch NRS >= 4 pri vstupnom vyšetrení: placebo N = 374, Taltz 80 mg Q4W N = 379, Taltz
80 mg Q2W N = 391
Do UNCOVER-2 bolo zaradených 1 224 pacientov. Pacienti boli randomizovaní (1:2:2:2) na podávanie placeba alebo Taltzu (80 mg každé dva alebo štyri týždne [Q2W alebo Q4W] po 160 mg počiatočnej dávke) alebo etanerceptu 50 mg dvakrát týždenne počas 12 týždňov.
T
abuľka č. 3. Výsledky účinnosti v 12. týždni z UNCOVER-2
Počet pacientov (%) Rozdiel oproti placebu
v miere odpovede (95% CI)
Koncové ukazovatele
sPGA „0“ (čisté) alebo
Placebo
(N = 168)
Taltz
80 mg Q4W (N = 347)
a
Taltz
80 mg Q2W (N = 351)
a
Etanercept
50 mg dvakrát týždenne
(N = 358)
Taltz
80 mg Q4W
70,5 (65,3;
Taltz
80 mg Q2W
80,8 (76,3;
„1“ (minimálne)
4 (2,4) 253 (72,9)
292 (83,2)
129 (36,0)
75,7)
85,4)
sPGA „0“
a,b
a,b
c 31,7 (26,6;
41,3 (36,0;
(čisté) 1 (0,6) 112 (32,3)
147 (41,9)
21 (5,9)
36,7)
46,6)
PASI 75 4 (2,4) 269 (77,5)a 315 (89,7)a 149 (41,6)a 75,1 (70,2;
80,1)
PASI 90 1 (0,6) 207 (59,7)a,b 248 (70,7)a,b 67 (18,7)a 59,1 (53,8;
64,4)
PASI 100 1 (0,6) 107 (30,8)a,b 142 (40,5)a,b 19 (5,3)c 30,2 (25,2;
35,2)
87,4 (83,4;
91,3)
70,1 (65,2;
75,0)
39,9 (34,6;
45,1)
Pokles v škále svrbenia Itch
a,b
a,b
a 62,7 (55,1;
71,1 (64,0;
NRS ≥ 4d 19 (14,1) 225 (76,8)
258 (85,1)
177 (57,8)
70,3)
78,2)
Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
p < 0,001 v porovnaní s etanerceptom
c
p < 0,01 v porovnaní s placebom
d
P
acienti s itch NRS > = 4 pri vstupnom vyšetrení: placebo N = 135, Taltz 80 mg Q4W N = 293, Taltz
80 mg Q2W N = 303, Etanercept N = 306
Do UNCOVER-3 bolo zaradených 1 346 pacientov. Pacienti boli randomizovaní (1:2:2:2) na podávanie placeba alebo Taltzu (80 mg každé dva alebo štyri týždne [Q2W alebo Q4W] po 160 mg počiatočnej dávke) alebo etanerceptu 50 mg dvakrát týždenne po dobu 12 týždňov.
T
abuľka č. 4. Výsledky účinnosti v 12. týždni z UNCOVER 3
Koncové
Počet pacientov (%)
Etanercept
Rozdiel oproti placebu v miere
odpovede (95% CI)
ukazovatele
sPGA
s hodnotou
Placebo
(N = 193)
Taltz
80 mg Q4W (N = 386)
Taltz
80 mg Q2W (N = 385)
50 mg dvakrát týždenne (N = 382)
Taltz
80 mg Q4W
Taltz
80 mg Q2W
„0“ (čisté) alebo „1“ (minimálne) sPGA s hodnotou „0“ (čisté)
13 (6,7) 291 (75,4)a,b 310 (80,5)a,b 159 (41,6)a 68,7 (63,1;
74,2)
0 139 (36,0)a,b 155 (40,3)a,b 33 (8,6)a 36,0 (31,2;
40,8)
73,8 (68,5;
79,1)
40,3 (35,4;
45,2)
PASI 75 14 (7,3) 325 (84,2)a,b 336 (87,3)a,b 204 (53,4)a 76,9 (71,8;
82,1)
PASI 90 6 (3,1) 252 (65,3)a,b 262 (68,1)a,b 98 (25,7)a 62,2 (56,8;
67,5)
80,0 (75,1;
85,0)
64,9 (59,7;
70,2)
PASI 100 0 135 (35,0)a,b 145 (37,7)a,b 28 (7,3)a 35 (30,2; 39,7) 37,7 (32,8;
42,5)
Pokles v škále svrbenia Itch NRS ≥ 4c
33 (20,9) 250 (79,9)a,b 264 (82,5)a,b 200 (64,1)a 59,0 (51,2;
66,7)
61,6 (54,0;
69,2)
Skratky: N = počet pacientov v populácii so zámerom liečiť (intent-to-treat population)
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítali ako non-respondéri
a
p < 0,001 v porovnaní s placebom
b
p < 0,001 v porovnaní s etanerceptom
c
P
acienti s itch NRS >= 4 pri vstupnom vyšetrení: placebo N = 158, Taltz 80 mg Q4W N = 313, Taltz
80 mg Q2W N = 320, Etanercept N = 312
Taltz bol spojený s rýchlym nástupom účinnosti s > 50 % znížením priemerného PASI do 2. týždňa
(obrázok č. 1). Percento pacientov, ktorí dosiahli PASI 75, bolo signifikantne vyššie u Taltzu
v porovnaní s placebom a etanerceptom už v 1. týždni. Približne 25 % pacientov liečených Taltzom dosiahlo PASI skóre < 5 do 2. týždňa, viac ako 55 % dosiahlo PASI skóre < 5 do 4. týždňa a ich počet
sa zvýšil na 85 % do 12. týždňa (v porovnaní s 3 %, 14 % a 50 % u etanerceptu). Významné zlepšenie
závažnosti svrbenia bolo pozorované u pacientov liečených Taltzom v 1. týždni.
O
brázok č. 1. PASI skóre, percentuálne zlepšenie pri každej návšteve od počiatku (mBOCF)) v populácii so zámerom liečiť (ITT populácii) v priebehu fázy indukčného dávkovania - UNCOVER-2 a UNCOVER-3
Účinnosť a bezpečnosť Taltzu bola preukázaná bez ohľadu na vek, pohlavie, rasu, telesnú hmotnosť, iniciálnu závažnosť PASI, umiestnenie ložiska, konkurentnú psoriatickú artritídu a predchádzajúcu biologickú liečbu. Taltz bol účinný u pacientov doteraz neliečených systémovou liečbou, doteraz neliečených biologickou liečbou, u pacientov, ktorí podstúpili biologickú/anti-TNF liečbu
aj u pacientov, u ktorých biologická/anti-TNF liečba zlyhala.
Účinnosť u non-respondérov na etanercept: z pacientov, ktorí boli sPGA (0,1) non-respondéri na etanercept v 12. týždni v UNCOVER-2 (N = 200) a ktorí boli prestavení na Taltz 80 mg Q4W po 4- týždňovom období bez podávania liečiva („washout“ obdobie), sa po 12 týždňoch liečby Taltzom
73 % pacientov podarilo dosiahnuť sPGA (0,1) a 83,5 % pacientov PASI 75.
V 2 klinických skúšaniach, ktoré zahŕňali aktívny komparátor (UNCOVER-2 a UNCOVER-3), bola miera výskytu závažných nežiaducich účinkov u etanerceptu aj u Taltzu 1,9 % a miera ukončenia liečby kvôli nežiaducim účinkom bola 1,2 % u etanerceptu a 2,0 % u Taltzu. Miera výskytu infekcií bola 21,5 % u etanerceptu a 26,0 % u Taltzu, pričom väčšina prípadov bola miernej až stredne závažnej závažnosti. Miera výskytu závažných infekcií bola 0,4 % u etanerceptu a 0,5 % u Taltzu.
Udržanie odpovede v 60. týždniPacienti pôvodne randomizovaní na Taltz, ktorí reagovali na liečbu v 12. týždni (t.j. sPGA skóre
s hodnotou 0,1) v UNCOVER-1 aj UNCOVER-2, boli re-randomizovaní na dobu ďalších 48 týždňov na jeden z týchto liečebných režimov: placebo alebo Taltz (80 mg každé štyri alebo dvanásť týždňov
[Q4W alebo Q12W]).
T
abuľka č. 5. Udržanie odpovede a účinnosti v 60. týždni
(št
údie UNCOVER-1 a UNCOVER-2)
Počet pacientov (%)
Rozdiel oproti placebu v miere
odpovede (95% CI)
Koncové
ukazovatele
Udržané sPGA „0“
80 mg Q4W (indukcia) / Placebo (udržanie) (N
= 191)
80 mg Q2W (indukcia) / Placebo (udržanie) (N
= 211)
80 mg Q4W (indukcia) /
80 mg Q4W (udržanie) (N
= 195)
80 mg Q2W (indukcia) /
80 mg Q4W (udržanie) (N
= 221)
80 mg Q4W (indukcia) /
80 mg Q4W (udržanie)
80 mg Q2W (indukcia) /
80 mg Q4W (udržanie)
(čisté) alebo
„1“ (minimálne)
Udržané alebo dosiahnuté sPGA „0“ (čisté) Udržané alebo dosiahnuté PASI 75
Udržané alebo dosiahnuté PASI 90
Udržané alebo dosiahnuté PASI 100
12 (6,3) 16 (7,6) 134 (68.7)a 173 (78,3)a 62,4 (55,1;
69,8)
3 (1,6) 6 (2,8) 96 (49.2)a 130 (58,8)a 47,7 (40,4;
54,9)
15 (7,9) 19 (9,0) 145 (74.4)a 184 (83,3)a 66,5 (59,3;
73,7)
9 (4,7) 10 (4,7) 130 (66.7)a 169 (76,5)a 62,0 (54,7;
69,2)
3 (1,6) 6 (2,8) 97 (49,7)a 127 (57,5)a 48.2 (40,9;
55,4)
70,7 (64,2;
77,2)
56,0 (49,1;
62,8)
74,3 (68,0;
80,5)
71,7 (65,4;
78,0)
54.6 (47,7;
61,5)
Skratky: N = počet pacientov v analyzovanej populácii
U
pozornenie: pacienti s chýbajúcimi údajmi sa počítajú ako non-respondéri
a
p < 0,001 v porovnaní s placebom
Taltz bol účinný pri udržaní odpovede u pacientov bez predchádzajúcej systémovej liečby, bez predchádzajúcej biologickej liečby, u pacientov s anamnézou biologickej/anti-TNF liečby
aj u pacientov, u ktorých biologická/anti-TNF liečba zlyhala.
U respondérov s sPGA (0,1) v 12. týždni, re-randomizovaných na vysadenie liečby (t.j. na placebo), bol medián času do relapsu (sPGA ≥ 3) 164 dní v integrovaných štúdiách UNCOVER-1 a UNCOVER-2. Z týchto pacientov 71,5 % znovu dosiahlo odpoveď aspoň sPGA (0,1) v priebehu
12 týždňov po opakovanom nastavení na Taltz 80 mg Q4W
V porovnaní s placebom a etanerceptom sa prejavilo signifikantne väčšie zlepšenie od vstupného
vyšetrenia do 12. týždňa u nechtovej psoriázy (pri meraní podľa Indexu závažnosti nechtovej psoriázy
[Nail Psoriasis Severity Index, NAPSI]), u psoriázy pokožky hlavy (pri meraní podľa Indexu závažnosti psoriázy pokožky hlavy [Psoriasis Scalp Severity Index, PSSI]) a u palmoplantárnej psoriázy (pri meraní podľa Indexu závažnosti palmoplantárnej psoriázy [Psoriasis Palmoplantar Severity Index, PPASI]). Tieto zlepšenia nechtovej psoriázy, psoriázy pokožky hlavy a palmoplantárnej psoriázy sa u pacientov liečených Taltzom, ktorí boli v 12. týždni respondéri na liečbu s sPGA (0,1), udržali aj v 60. týždni.
Kvalita života/výsledky hlásené pacientom
V 12. týždni a počas klinických skúšaní sa Taltz spájal so štatisticky signifikantným zlepšením
v kvalite života spojenej so zdravím meraným priemerným poklesom oproti východiskovému stavu
podľa Dermatologického indexu kvality života (Dermatology Life Quality Index, DLQI) (Taltz 80 mg
Q2W od -10,2 do -11,1, Taltz 80 mg Q4W od -9,4 do -10,7, etanercept od -7,7 do -8,0 a placebo od -
1,0 do -2,0). Signifikantne väčší podiel pacientov liečených Taltzom dosiahlo DLQI 0 alebo 1. Naprieč štúdiami sa Taltz spájal so štatisticky signifikantným zlepšením závažnosti svrbenia
hodnoteného podľa Itch NRS skóre. Signifikantne väčší podiel pacientov liečených Taltzom
dosiahol redukciu Itch NRS ≥ 4 body v 12. týždni (84,6 % pre Taltz Q2W, 79,2 % pre Taltz Q4W
a 16,5 % pre placebo) a benefit bol udržaný počas celého času až do 60. týždňa u pacientov liečených
Taltzom, ktorí boli sPGA (0 alebo 1) respondéri v 12. týždni. Neboli žiadne dôkazy zhoršenia depresie
počas 60 týždňov liečby Taltzom podľa hodnotenia Stručného súpisu sebahodnotiacej depresívnej
symptomatológie (Quick Inventory of Depressive Symptomatology Self Report).
Imunizácia
V štúdii so zdravými jedincami neboli identifikované žiadne bezpečnostné riziká s dvoma
inaktivovanými vakcínami (proti tetanu a pneumokokom), po prijatí dvoch dávok ixekizumabu (160 mg nasledovaná druhou dávkou 80 mg o dva týždne neskôr ). Avšak údaje týkajúce sa imunizácie boli nedostatočné, aby sa dospelo k záveru o primeranej imunitnej reakcii na tieto vakcíny po podaní Taltzu.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Taltzom v jednej
alebo vo viacerých podskupinách pediatrickej populácie pri liečbe ložiskovej psoriázy (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po jednom subkutánnom podaní dávky ixekizumabu pacientom so psoriázou boli priemerné
maximálne koncentrácie dosiahnuté v priebehu 4 až 7 dní v rozpätí všetkých dávok od 5 do 160 mg. Priemerná (SD) maximálna plazmatická koncentrácia (Cmax) ixekizumabu po počiatočnej dávke
160 mg bola 19,9 (8,15) µg/ml.
Po počiatočnej dávke 160 mg bol rovnovážny stav dosiahnutý do 8. týždňa s dávkovacím režimom
80 mg Q2W. Odhad priemernej (SD) Cmax, ss, je 21,5 (9,16) µg/ml a odhad priemernej Ctrough, ss je
5,23 (3,19) µg/ml.
Po prestavení dávkovacieho režimu z 80 mg Q2W na dávkovací režim 80 mg Q4W v 12. týždni by sa
mal rovnovážny stav dosiahnuť po približne 10 týždňoch. Odhad priemernej (SD) Cmax, ss, je
14,6 (6,04) µg/ml a odhad priemernej Ctrough, ss je 1,87 (1,30) µg/ml.
Priemerná biodostupnosť ixekizumabu po subkutánnom podaní bola vo všetkých analýzach 54 % až
90 %.
Distribúcia
Z populačných farmakokinetických analýz vyplýva, že priemerný celkový distribučný objem
v rovnovážnom stave bol 7,11 l.
Biotransformácia
Ixekizumab je monoklonálna protilátka a očakáva sa, že sa bude rozkladať na malé peptidy a
aminokyseliny katabolickými dráhami takým istým spôsobom ako endogénne imunoglobulíny.
E
li
m
i
nácia
V populačnej PK analýze bol priemerný sérový klírens 0,0161 l/h. Klírens nezávisí od dávky.
Priemerný eliminačný polčas podľa odhadu z populačnej farmakokinetickej analýzy je u pacientov s ložiskovou psoriázou 13 dní.
Linearita / nelinearita
Expozícia (AUC) sa pri podaní vo forme subkutánnej injekcie úmerne zvyšovala v rozmedzí dávok od
5 do 160 mg.
Starší pacienti
Z 4204 pacientov s ložiskovou psoriázou vystavených pôsobeniu Taltzu v klinických skúšaniach bolo
celkom 301 pacientov 65-ročných alebo starších a 36 pacientov bolo 75-ročných alebo starších. Na
základe populačnej farmakokinetickej analýzy obmedzeného počtu starších pacientov (n = 94 pre vek
≥ 65 rokov a n = 12 pre vek ≥ 75 rokov) bol klírens u starších pacientov a pacientov mladších ako 65-
ročných podobný.
Porucha funkcie obličiek a pečene
Špecifické štúdie klinickej farmakológie na vyhodnotenie účinkov poruchy funkcie obličiek a poruchy
funkcie pečene na PK ixekizumabu sa neuskutočnili. Očakáva sa, že renálna eliminácia intaktného
ixekizumabu, monoklonálnej IgG bude nízka a menšieho významu; a takisto monoklonálne protilátky IgG sa hlavne eliminujú intracelulárnym katabolizmom a neočakáva sa, že porucha funkcie pečene bude mať vplyv na klírens ixekizumabu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje od opíc makak cynomolgus získané na základe farmakologických štúdií toxicity po opakovanom podávaní, vyhodnotení farmakologickej bezpečnosti, reprodukčnej a vývinovej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Podávanie ixekizumabu opiciam makak cynomolgus v subkutánnych dávkach do 50 mg/kg týždenne po dobu 39 týždňov nevyvolalo žiadnu orgánovú toxicitu ani nežiaduce účinky na imunitné funkcie (napr. protilátková odpoveď závislá od T-buniek a aktivita NK buniek). Týždenná subkutánna dávka pre opice v hodnote 50 mg/kg je približne 19-násobok 160 mg počiatočnej dávky Taltzu a u opíc má za následok expozíciu (AUC), ktorá je najmenej 61-násobne vyššia ako predpovedaná priemerná rovnovážna expozícia u ľudí pri odporúčanom dávkovacom režime.
Predklinické štúdie na vyhodnotenie karcinogénneho alebo mutagénneho potenciálu ixekizumabu sa neuskutočnili.
U pohlavne dospelých opíc makak cynomolgus, ktorým bol podávaný ixekizumab po dobu 13 týždňov v týždennej subkutánne podávanej dávke 50 mg/kg, neboli pozorované žiadne účinky na reprodukčné orgány, menštruačný cyklus ani spermie.
V štúdiách vývojovej toxicity bolo preukázané, že ixekizumab prechádza placentou a bol prítomný v krvi potomstva až do veku 6 mesiacov. Vyšší výskyt postnatálnej mortality v porovnaní so súbežnými kontrolami bol zaznamenaný u potomstva opíc, ktorým bol podávaný ixekizumab. Toto sa týkalo predovšetkým predčasného pôrodu alebo zanedbávania potomstva matkou, čo sú bežné nálezy v štúdiách s nehumánnymi primátmi a boli považované za nesúvisiace s ixekizumabom.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
citrát sodný
kyselina citrónová, bezvodá chlorid sodný
polysorbát 80
voda na injekcie
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 ºC – 8 ºC). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
1 ml roztoku v injekčnej striekačke z čistého skla typu I. Injekčná striekačka uložená v pere na jedno použitie s jednorazovou dávkou. Balenia s 1, 2 alebo 3 naplnenými perami. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Návod na použitie
Pokyny na používanie pera, ktoré sú súčasťou písomnej informácie pre používateľa, sa musia dôsledne
dodržiavať.
Naplnené pero je iba na jedno použitie.
Taltz sa nemá používať, ak sa v ňom objavia častice alebo ak je roztok zakalený a/alebo zreteľne
hnedý.
Taltz, ktorý bol uchovávaný v mrazničke, sa nesmie používať.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Holandsko.
8. REGISTRAČNÉ ČÍSLO(ČÍSLA)
EU/1/15/1085/001
EU/1/15/1085/002
EU/1/15/1085/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.