
/>aPoznámka: Intenzita klinických nežiaducich udalostí je odstupňovaná podľa National Cancer Institute Common
Terminology Criteria for Adverse Events, verzia 4.0 (NCI-CTCAE).
EKG: Elektrokardiogram; QTc: interval QT korigovaný na srdcovú frekvenciu
Osobitné populácie
Nie je potrebná úprava dávkovania v súvislosti s vekom, telesnou hmotnosťou, pohlavím, etnickou príslušnosťou a fajčením (pozri časť 5.2).
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene (celkový bilirubín ≤ horná hranica normálu (upper limit of normal, ULN) a aspartátaminotransferáza (AST) > ULN, alebo celkový bilirubín > 1,0 až
1,5 x ULN a akákoľvek AST) sa úprava dávky neodporúča, ale pri podávaní TAGRISSA týmto pacientom je potrebná opatrnosť. Bezpečnosť a účinnosť tohto lieku u pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene neboli stanovené. Použitie u pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene sa až do získania ďalších údajov neodporúča (pozri časť 5.2).
Porucha funkcie obličiek
U pacientov s miernou a stredne ťažkou poruchou funkcie obličiek sa úprava dávky neodporúča. U pacientov s ťažkou poruchou funkcie obličiek je dostupné obmedzené množstvo údajov. Bezpečnosť a účinnosť tohto lieku u pacientov s terminálnym štádiom ochorenia obličiek [klírens kreatinínu (ClCr) < 15 ml/min, vypočítaný podľa Cockcroftovho a Gaultovho vzorca], alebo
u pacientov na dialýze neboli doteraz stanovené. Pri liečbe pacientov s ťažkou poruchou funkcie obličiek alebo terminálnym štádiom ochorenia obličiek je potrebná opatrnosť (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť u detí a dospievajúcich vo veku menej ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Tento liek je na perorálne použitie. Tableta sa má prehltnúť vcelku a zapiť vodou, nemá sa drviť, deliť alebo žuvať.
Ak pacient nevie tabletu prehltnúť, tabletu je možné najprv rozpustiť v 50 ml neperlivej vody. Bez lámania sa má hodiť do vody, miešať kým sa nerozpustí a potom okamžite vypiť. Aby sa zabezpečilo že v pohári neostanú zvyšky lieku, pohár sa má do polovice naplniť ďalšou vodou a obsah ihneď vypiť. Nesmie sa pridávať žiadna iná tekutina.
Ak je potrebné na podávanie použiť nazogastrickú sondu, má sa postupovať rovnako, ako je uvedené vyššie s použitím objemu 15 ml na úvodné rozpustenie a 15 ml na vypláchnutie zvyškov lieku. Výsledných 30 ml roztoku sa má podať podľa pokynov výrobcu nazogastrickej sondy s primeraným množstvom preplachov. Disperzia a zvyšky sa majú podať do 30 minút od vhodenia tablety do vody.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Ľubovník bodkovaný by sa nemal používať spoločne s TAGRISSOM (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
HodnoteniestavumutácieEGRFT790M
Pri zvažovaní použitia TAGRISSA na liečbu lokálne pokročilého alebo metastatického NSCLC je dôležité u všetkých pacientov určiť stav mutácie EGFR T790M. Validovaný test sa má uskutočniť
s použitím nádorovej DNA získanej zo vzorky tkaniva alebo cirkulujúcej nádorovej DNA (ctDNA)
získanej zo vzorky plazmy.
Na určenie stavu mutácie T790M v nádorovej DNA (zo vzorky tkaniva alebo plazmy) sa majú použiť len robustné, spoľahlivé a citlivé testy s preukázaným prínosom.
Pozitívny výsledok určenia stavu mutácie T790M s použitím testovania tkanív alebo plazmy indikuje spôsobilosť na liečbu TAGRISSOM. Ak sa však testuje ctDNA získaná z plazmy a výsledok je negatívny odporúča sa, ak je to možné, následne otestovať aj tkanivo z dôvodu možných falošne negatívnych výsledkov pri testovaní plazmy.
Intersticiálnachorobapľúc(ILD)
U pacientov liečených TAGRISSOM v klinických štúdiách sa pozorovala ťažká život ohrozujúca alebo fatálna intersticiálna choroba pľúc (ILD) alebo nežiaduce reakcie podobné ILD. Vo väčšine prípadov sa po prerušení liečby zmiernili alebo ustúpili. Pacienti s anamnézou ILD, liekmi navodenou ILD, radiačnou pneumonitídou vyžadujúcu si liečbu steroidmi, alebo akýmkoľvek znakom klinicky aktívnej ILD boli z klinických štúdií vyradení (pozri časť 4.8).
Intersticiálna pľúcna choroba (ILD) alebo nežiaduce reakcie podobné ILD (napr. pneumonitída) sa hlásili u 2,9% a boli fatálne u 0,3% z 1221 pacientov, ktorí dostávali TAGRISSO vo všetkých klinických štúdiách. ILD alebo ILD podobné nežiaduce reakcie sa hlásili u 11/411 (2,7%) pacientov, ktorí dostávali TAGRISSO v dvoch štúdiách fázy II, z toho 0,7% boli 3.stupňa alebo 4.stupňa a 1% boli fatálne. Výskyt ILD bol 6,2% u pacientov japonského etnického pôvodu, 1,2% u pacientov ázijskeho pôvodu a 2,4% u non-ázijských pacientov (pozri časť 4.8).
Na vylúčenie ILD sa má vykonať dôkladné zhodnotenie všetkých pacientov s akútnym nástupom a/alebo nevysvetliteľným zhoršením pľúcnych príznakov (dyspnoe, kašeľ, horúčka). Liečba týmto
liekom sa má až do preskúmania týchto symptómov prerušiť. Ak sa diagnostikuje ILD, liečba
TAGRISSOM sa musí natrvalo ukončiť a podľa potreby sa má začať vhodná liečba.
PredĺženieQTcintervalu
U pacientov liečených TAGRISSOM sa vyskytuje predĺženie QTc intervalu. Predĺženie QTc intervalu môže viesť k zvýšenému riziku ventrikulárnych tachyarytmií (napr. torsade de pointes) alebo
k náhlemu úmrtiu. V štúdiách AURAex alebo AURA2 sa nehlásili žiadne prípady arytmií (pozri časť
4.8). Pacienti s klinicky významnými abnormalitami srdcového rytmu a vedenia vzruchu, merané použitím elektrokardiogramu (EKG) v kľudovom stave (napr. QTc interval viac ako 470 ms), boli
z týchto štúdií vyradení (pozri časť 4.8).
Ak je to možné, vyhnite sa použitiu osimertinibu u pacientov s vrodeným syndrómom dlhého intervalu QT. U pacientov s kongestívnym zlyhávaním srdca, abnormalitami elektrolytov, alebo u tých, ktorí užívajú lieky, o ktorých je známe že predlžujú QTc interval pravidelne sledujte EKG a elektrolyty. Podávanie prerušte u pacientov, u ktorých sa predĺžil QTc interval na viac ako 500 ms na minimálne 2 separátnych EKG, kým nie je QTc interval menej ako 481 ms alebo do obnovenia východiskových hodnôt, ak východisková hodnota QTc je viac alebo sa rovná 481 ms, potom pokračujete v podávaní TAGRISSA v znížených dávkach tak, ako je uvedené v tabuľke 1. Podávanie osimertinibu natrvalo ukončite u pacientov, u ktorých sa vyvinulo predĺženie QTc intervalu spolu s ktorýmkoľvek
s nasledujúcich: Torsades de pointes, polymorfná ventrikulárna tachykardia, prejavy/príznaky závažnej arytmie.
4.5 Liekové a iné interakcie
Farmakokinetickéinterakcie
Silné induktory CYP3A4 môžu znížiť expozíciu osimertinibu. Osimertinib môže zvýšiť expozíciu substrátov BCRP.
Liečivá, ktoré môžu zvýšiť koncentrácie osimertinibu v plazme
Štúdie in vitro preukázali že metabolizmus osimertinibu fázy I je prednostne sprostredkovaný CYP3A4 a CYP3A5. V klinickej farmakokinetickej štúdií u pacientov súbežné podávanie s 200 mg itrakonazolu (silný inhibítor CYP3A4) dvakrát denne nemalo klinicky významný účinok na expozíciu osimertinibu (plocha pod časovou krivkou koncentrácie (AUC) zvýšená o 24 % a Cmax znížený
o 20 %). Pri inhibítoroch CYP3A4 sa preto nepredpokladá, že ovplyvnia expozíciu osimertinibu.
Ďalšie katalyzujúce enzýmy neboli identifikované.
Liečivá, ktoré môžu znížiť koncentrácie osimertinibu v plazme
V klinickej farmakokinetickej štúdii u pacientov sa pri súbežnom podávaní s rifampicínom (600 mg denne počas 21 dní) znížil rovnovážny stav AUC osimertinibu o 78 %. Podobne aj expozícia
metabolitu AZ5104 znížila AUC o 82 % a Cmax o 78 %. Odporúča sa vyhnúť súčasnému použitiu silných induktorov CYP3A (napr. fenytoín, rifampicín, karbamazepín) s TAGRISSOM. Mierne induktory CYP3A4 (napr. bosentan, efavirenz, etravirin, modafinil) môžu tiež znížiť expozíciu
osimertinibu a mali by sa používať opatrne alebo pokiaľ je to možné sa im vyhnúť. Na odporučenie úpravy dávky TAGRISSA nie je k dispozícii dostatok klinických údajov. Súbežné užívanie ľubovníka bodkovaného je kontraindikované (pozri časť 4.3).
Účinok liečiv znižujúcich hladinu žalúdočnej kyseliny na osimertinib
V klinickej farmakokinetickej štúdii neviedlo súbežné podávanie omeprazolu ku klinicky významným zmenám v expozíciách osimertinibu. Látky upravujúce gastrické pH sa môžu používať súčasne
s TAGRISSOM bez akýchkoľvek obmedzení.
Liečivá, ktorých plazmatické koncentrácie môžu byť zmenené TAGRISSOM
Na základe štúdií in vitro je osimertinib kompetitívnym inhibítorom transportérov BCRP.
V klinickej FK štúdii súbežné podávanie TAGRISSA s rosuvastatínom (citlivý substrát BCRP) zvýšilo AUC rosuvastatínu o 35 % a Cmax o 72 %. Pacientov súčasne užívajúcich liečivá s dispozíciou závislou na BCRP a s úzkym terapeutickým indexom majú byť starostlivo sledovaní na prejavy zmenenej
znášanlivosti súčasne podávaného liečiva, ako výsledku zvýšenej expozície počas užívania
TAGRISSA (pozri časť 5.2).
V klinickej FK štúdii súbežné podávanie TAGRISSA so simvastatínom (citlivým substrátom CYP3A4) znížilo AUC simvastatínu o 9 % a Cmax o 23 %. Tieto zmeny sú malé a pravdepodobne bez klinického významu. Klinicky významné FK interakcie so substrátmi CYP3A4 nie sú pravdepodobné. Neskúmal sa Pregnanový X receptor (PXR) regulujúci enzýmové interakcie iné ako CYP3A4. Nie je možné vylúčiť riziko zníženej expozície hormonálnej antikoncepcie.
4.6 Fertilita, gravidita a laktácia
Antikoncepciaumužovažien
Ženy vo fertilnom veku majú byť poučené, aby počas užívania TAGRISSA zabránili otehotneniu. Pacienti majú byť poučení, aby používali účinnú antikoncepciu počas nasledujúceho obdobia po
ukončení liečby týmto liekom: minimálne 2 mesiace u žien a 4 mesiace u mužov. Nie je možné
vylúčiť riziko zníženej expozície hormonálnej antikoncepcie.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití osimertinibu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (embryoletalitu, spomalený rast plodu a smrť čerstvo narodených mláďat, pozri časť 5.3). Na základe mechanizmu účinku a predklinických údajov môže osimertinib spôsobiť poškodenie plodu ak sa podáva gravidným ženám. TAGRISSO sa nemá používať počas gravidity pokiaľ klinický stav ženy nevyžaduje liečbu osimertinibom.
Dojčenie
Nie je známe, či sa osimertinib alebo jeho metabolity vylučujú do ľudského mlieka. Nie sú dostatočné informácie o vylučovaní osimertinibu a jeho metabolitov do mlieka u zvierat. Osimertinib a jeho metabolity sa však zistili u neodstavených mláďat a pozorovali sa nežiaduce účinky na rast
a prežívanie mláďat (pozri časť 5.3). Riziko u dojčiat nemôže byť vylúčené. Dojčenie sa má počas liečby TAGRISSOM ukončiť.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku TAGRISSA na fertilitu u ľudí. Výsledky zo štúdií na zvieratách ukázali, že osimertinib má účinky na mužské a ženské reprodukčné orgány a môže zhoršiť fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
TAGRISSO nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Bezpečnostné údaje lieku TAGRISSO odrážajú údaje od 411 predtým liečených pacientov s NSCLC
s pozitívnou mutáciou T790M dostávajúcich dávku 80 mg denne. Porovnávacie bezpečnostné údaje
z randomizovaných klinických skúšaní nie sú zatiaľ k dispozícii. Väčšina nežiaducich reakcií bola 1. alebo 2. stupňa závažnosti. Najčastejšie hlásenými nežiaducimi reakciami (adverse drug reactions,
ADRs) boli hnačka (42 %) a vyrážka (24 %). Nežiaducich udalostí 3. stupňa bolo 26 % a 4. stupňa
1,2 % v oboch štúdiách. U pacientov liečených TAGRISSOM 80 mg jedenkrát denne sa znížila dávka z dôvodu ADR u 2,2 % pacientov. Podávanie z dôvodu nežiaducich udalostí alebo abnormalít
laboratórnych parametrov sa ukončilo u 3,2 % pacientov.
Tabuľkovýzoznamnežiaducichreakcií
V tabuľke 2 sú uvedené často hlásené nežiaduce reakcie u pacientov dostávajúcich TAGRISSO.
Nežiaduce liekové reakcie sú uvedené podľa tried orgánových systémov s použitím MedDRA terminológie. V každej triede orgánových systémov sú nežiaduce reakcie usporiadané podľa frekvencie, najčastejšie reakcie sú uvedené ako prvé. V rámci jednotlivých skupín frekvencie sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. Okrem toho, súvisiaca kategória frekvencie pre každú ADR je podľa konvencie CIOMS III a je definovaná ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (nie je možné odhadnúť z dostupných údajov). Táto časť zahŕňa len údaje získané z ukončených štúdií so známou expozíciou pacienta. Údaje v tabuľke 2 sú
kumulatívnymi údajmi z predĺženia štúdie AURA (fáza II) a zo štúdií AURA 2; zhrnuté sú len udalosti u pacientov, ktorí dostali minimálne jednu dávku TAGRISSA.
Tabuľka 2. Nežiaduce liekové reakcie hlásené v štúdiách AURAa
T
rieda
orgánových systémov podľa
MedDRA
Terminológia
MedDRA
Podľa konvencie
C
IOM
S
/celková frekvencia
(všetky stupne CTCAE)b
Frekvencia 3. – 4.
stupňa podľa
CTCAE
Poruchy dýchacej sústavy, hrudníka a mediastína
intersticiálna choroba pľúcc
časté (2,7 %) d 0,7 %
Poruchy gastrointestinálne ho traktu
Poruchy kože a podkožného tkaniva
hnačka veľmi časté (42 %) 1 % stomatitída veľmi časté (12 %) 0 % vyrážkae veľmi časté (41 %) 0,5 %
suchá kožaf veľmi časté (31 %) 0 %
paronychiag veľmi časté (25 %) 0 %
pruritus veľmi časté (14 %) 0 %
Laboratórne
a funkčné vyšetrenia (nálezy
na základe výsledkov
vyšetrení uvádzané ako stupne CTCAE)
znížený počet
trombocytovh
znížený počet leukocytovh
znížený počet neutrofilovh
veľmi časté (54 %) 1,2 %
veľmi časté (67 %) 1,2 %
veľmi časté (33 %) 3,4 %
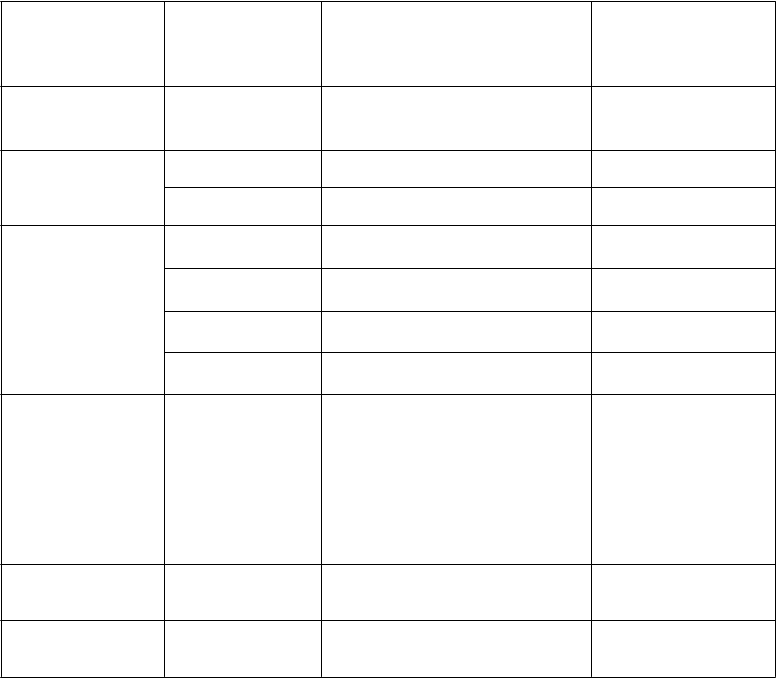
a Údaje sú kumulatívnymi údajmi z predĺženia štúdie AURA (fáza II) a zo štúdií AURA 2; zhrnuté sú len udalosti u pacientov, ktorí dostali minimálne jednu dávku TAGRISSA.
b National Cancer Institute Common Terminology Criteria for Adverse Events, verzia 4.0.
c Zahŕňa prípady hlásené v rámci zlúčených pojmov: intersticiálna choroba pľúc a pneumonitída.
d Hlásené udalosti stupňa 5 (fatálne) podľa CTCAE verzia 4.
e Zahŕňa prípady hlásené v rámci zlúčených pojmov pre nežiaduce udalosti vyrážky: vyrážka, generalizovaná vyrážka, erytematózna vyrážka, makulárna vyrážka, makulo-papulárna vyrážka, papulárna vyrážka, pustulárna vyrážka, erytém, folikulitída, akné, dermatitída a akneiformná dermatitída.
f Zahŕňa prípady hlásené v rámci zlúčených pojmov: suchá koža, kožné fisúry, xeróza, ekzém.
g Zahŕňa prípady hlásené v rámci zlúčených pojmov: poruchy nechtového lôžka, zápal nechtového lôžka, citlivosť nechtového lôžka, zmena farby nechtu, porucha nechtu, dystrofia nechtu, infekcia nechtu, zmena štruktúry nechtu, lámavosť nechtov, onycholýza, onychomadéza, paronychia.
h Predstavuje výskyt laboratórnych nálezov, ktoré neboli súčasťou hlásených nežiaducich udalostí.
Popis vybranýchnežiaducichreakcií
Intersticiálna choroba pľúc (ILD)
V štúdiách fázy 2 bol výskyt ILD u pacientov japonského etnika 6,2 %, 1,2 % u pacientov nejaponského ázijského etnika a 2,4 % u pacientov iného ako ázijského etnika. Medián času do nástupu ILD alebo ILD podobných nežiaducich reakcií bol 2,7 mesiacov (pozri časť 4.4).
Predĺženie QTc intervalu
Zo 411 pacientov v štúdiách AURAex a AURA2 sa u jedného pacienta (menej ako 1 %) zistil QTc viac ako 500 ms a 11 pacientov (2,7 %) malo predĺženie QTc oproti východiskovej hodnote viac ako
60 ms. Farmakokinetická analýza s TAGRISSOM predikovala od koncentrácie závislé zvýšenie predĺženia QTc intervalu. V štúdiách AURAex a AURA2 sa nehlásili žiadne prípady arytmií (pozri časti 4.4 a 5.1).
Staršie osoby
Z celkového počtu pacientov v klinických štúdiách s osimertinibom (N=411) bolo 46 % vo veku 65 rokov alebo starších, z ktorých 13 % bolo vo veku 75 rokov a starších. V porovnaní s mladšími osobami (< 65) malo viac osôb vo veku ≥ 65 rokov nežiaduce reakcie, ktoré viedli k úprave dávky skúšaného lieku (prerušenia alebo zníženia) (23 % oproti 17 %). Nežiaduce udalosti boli podobné bez ohľadu na vek. U starších pacientov bolo viac nežiaducich reakcií stupňa 3 alebo vyššieho v porovnaní s mladšími pacientmi (32 % oproti 28 %). Medzi týmito a mladšími osobami sa nepozorovali žiadne celkové rozdiely v účinnosti.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v PríloheV.
4.9 Predávkovanie
Vo fáze I/II klinických skúšaní bolo obmedzené množstvo pacientov liečených dennou dávkou TAGRISSA do 240 mg bez toxicít limitujúcich dávku. V týchto štúdiách mali pacienti, ktorí dostávali TAGRISSO v denných dávkach 160 mg a 240 mg zvýšenú frekvenciu výskytu a závažnosť typických EGFR TKI navodených nežiaducich účinkov (predovšetkým hnačka a kožná vyrážka) v porovnaní
s pacientmi dostávajúcimi 80 mg dávku. Skúsenosti s náhodným predávkovaním u ľudí sú obmedzené. Všetky prípady boli izolovanými incidentmi pacientov, ktorí omylom užívali dodatočnú dennú dávku TAGRISSA bez akýchkoľvek klinických dôsledkov.
Neexistuje žiadna špecifická liečba predávkovania TAGRISSOM. Pri podozrení na predávkovanie sa má podávanie TAGRISSA prerušiť a začať so symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE35. Mechanizmusúčinku
Osimertinib je inhibítor tyrozínkinázy (TKI). Je to ireverzibilný inhibítor receptora epidermálneho
rastového faktora (EGFR) so senzitizujúcimi mutáciami (EGFRm) a mutáciou T790M rezistentnou na
TKI.
Farmakodynamickéúčinky
Štúdie in vitro preukázali, že osimertinib má veľmi silnú a inhibičnú aktivitu proti EGFR v rámci všetkých klinicky významných senzitizujúcich mutácií EGFR a mutácií T790M bunkových línií
(zdanlivé IC50 od 6 nM do 54 nM oproti fosfo-EGFR) nemalobunkového karcinómu pľúc (NSCLC). Toto vedie k inhibícii bunkového rastu so signifikantne nižšou aktivitou proti EGFR v bunkových líniách wild-type (zdanlivé IC50 od 480 nM do 1,8 μM oproti fosfo-EGFR). Perorálne podávanie osimertinibu in vivo viedlo k zmenšeniu nádoru pri oboch EGFRm a T790M xenotransplantátu NSCLC a transgénnych myšacích modeloch nádoru pľúc.
Elektrofyziologickévyšetreniesrdca
Potenciál TAGRISSA na predĺženie QTc intervalu sa hodnotil u 210 pacientov, ktorí dostávali denne
80 mg osimertinibu v štúdii AURA2. Po podaní jednej dávky a v rovnovážnom stave boli zaznamenané série EKG na vyhodnotenie účinku osimertinibu na QTc interval.
Farmakokinetická analýza predikovala s liekom súvisiace predĺženie QTc intervalu pri dávke 80 mg
14 ms s hornou hranicou 16 ms (90% IS).
Klinickáúčinnosťabezpečnosť
Dve jednoramenné otvorené klinické štúdie AURAex (fázy II s predĺženou kohortou, (n=201))
a AURA2 (n=210) sa vykonali u pacientov s karcinómom pľúc s pozitívnou mutáciou EGFR T790M, s progresiou na predchádzajúcej systémovej liečbe vrátane EGFR liečených TKI. U všetkých pacientov sa vyžadovalo, aby mali NSCLC s pozitívnou mutáciou EGFR T790M zistenou použitím
„cobas EGFR mutation test“ urobeným pred liečbou v centrálnom laboratóriu. Všetci pacienti dostali
80 mg dávku TAGRISSA jedenkrát denne. V týchto dvoch skúšaniach bol primárnym ukazovateľom účinnosti objektívna miera odpovede (objective response rate, ORR) podľa kritérií RECIST verzia 1.1
hodnotená zaslepenou nezávislou centrálnou revíziou (Blinded Independent Central Review, BICR).
Sekundárne ukazovatele účinnosti zahŕňali trvanie odpovede (Duration of Response, DoR), mieru kontroly ochorenia (Disease Control Rate, DCR) a prežívanie bez progresie (Progression-Free
Survival, PFS).
Vstupné charakteristiky celkovej populácie štúdie (AURAex a AURA2) boli nasledovné: medián veku
63 rokov, 13 % pacientov bolo vo veku ≥75 rokov, ženy (68 %), biela rasa (36 %), ázijská rasa (60 %).Všetci pacienti dostali minimálne jednu predchádzajúcu liečbu. 31 % (N=129) dostalo 1.- líniovú liečbu (len liečba EGFR-TKI), 69 % (N=282) dostalo 2. alebo viac líniovú liečbu. 72 % pacientov nikdy nefajčilo, 99 % malo výkonnostný stav podľa WHO 0 alebo 1 a 39 % pacientov malo metastázy v mozgu (stabilné počas minimálne 4 týždňov, ktoré si nevyžadovali liečbu kortikosteroidmi). Väčšina pacientov (83 %) mala na začiatku viscerálne metastázy. Medián následného sledovania pre AURAex bol 6,9 mesiacov a pre AURA2 6,7 mesiacov.
AURA (fáza I) bola otvorené jednoramenné skúšanie so zvyšujúcou sa dávkou a rozšírením fázy I vrátane 271 predtým liečených pacientov s lokálne pokročilým alebo metastatickým NSCLC vo všetkých rozšírených kohortách s viacerými dávkami. Bezpečnosť a účinnosť 80 mg dávky TAGRISSA jedenkrát denne sa pozorovala v rozšírenej kohorte 63 predtým liečených pacientov
s centrálne potvrdenou pozitívnou mutáciou T790M NSCLC. Predchádzajúce liečby zahŕňali EGFR- TKI a chemoterapiu. Demografické charakteristiky populácie štúdie (n=63) s pozitívnou T790M boli:
medián veku 60 rokov, ženy (62 %), biela rasa (35 %), ázijská rasa (59 %), výkonnostný stav podľa
WHO 0 alebo 1 (100 %) a nikdy nefajčili (67 %). Počet predchádzajúcich terapií bol v rozmedzí 1 až
9. Medián trvania nasledujúceho sledovania bol 8,2 mesiacov. Výsledky účinnosti zo štúdií AURA
ako aj združená analýza (AURA ex a AURA2) sú zhrnuté v tabuľke 3.
Tabuľka 3. Výsledky účinnosti zo štúdií AURA
Fáza I Fáza II
Parameter účinnosti
1
Miera objektívnej
AURA (rozšírenie fázy I) (N=63)
AURAex (fáza II) (N=201)
AURA2
(N=210)
Celkovo
(N=411)
odpovede
2,3
% (95% IS)
Trvanie odpovede (DoR)3
Medián, Mesiace
(95% IS)
DoR vyššia ako 6 mesiacov v % (95% IS)
Miera kontroly odpovede (DCR)3
% (95% IS)
Prežívanie bez progresie Medián, Mesiace (95% IS)
62 (48, 74) 61(54, 68) 71 (64, 77) 66 (61, 71)
9,7 (8,3, NE) NE (NE, NE) 7,8 (7.1, NE) NE (8,3, NE)
72 (54, 84) 83 (74, 89) 75 (65, 82) 78 (72, 84)
95 (86, 99) 90 (85, 94) 91 (87, 95) 91 (88, 94)
11 (7, 15) NE (8,1; NE) 8,6 (8,3; 9,7) 9,7 (8,3; NE)

1 Na základe BICR, Blinded Independent Central Review, sledovanie PFS.
2 Miera objektívnej odpovede stanovená podľa RECIST v1.1 podľa BICR v hodnotiteľnej populácii s odpoveďou
(merateľné ochorenie na začiatku podľa BICR) n=60 pre AURA,199 pre AURAex,199 pre AURA2 a 398 celkovo; NE, nemožno odhadnúť, vrátane 2 úplných odpovedí.
3 Len u pacientov s odpoveďou; DoR definované ako čas od dátumu prvej zadokumentovanej odpovede (potvrdená úplná
alebo čiastočná odpoveď alebo stabilizované ochorenie ≥ 6 týždňov).
Miery objektívnej odpovede nad 50 % sa pozorovali vo všetkých predefinovaných analyzovaných podskupinách, vrátane liečby, etnickej príslušnosti, veku a regiónu.
V celkovej populácii malo zadokumentovanú odpoveď v čase prvého skenu (6 týždňov) 86 % (227/263) pacientov a v čase druhého skenu (12 týždňov) 96 % pacientov (253/263).
Klinické štúdie sa neuskutočnili u pacientov s NSCLC s pozitívnou mutáciou EGFR T790M de novo.
PediatrickápopuláciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s TAGRISSOM'
vo všetkých podskupinách pediatrickej populácie s NSCLC (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnostiFarmakokinetické parametre osimertinibu boli charakterizované u zdravých osôb a pacientov s NSCLC. Na základe farmakokinetickej analýzy populácie je zdanlivý plazmatický klírens osimertinibu 14,2 l/h, zdanlivý distribučný objem je 986 l a terminálny polčas približne 48 hodín. AUC a Cmax sa zvýšili úmerne s dávkou pri rozmedzí dávky od 20 až 240 mg. Podávanie osimertinibu jedenkrát denne vedie ku približne 3-násobnej kumulácii s dosiahnutím rovnovážneho stavu do 15.
dňa dávkovania. Pri rovnovážnom stave sú cirkulujúce plazmatické koncentrácie zvyčajne udržiavané v rámci 1,6-násobného rozmedzia počas 24 hodinového dávkovacieho intervalu.
Absorpcia
Po perorálnom podávaní TAGRISSA sa maximálne plazmatické koncentrácie osimertinibu dosiahli s mediánom (min-max) tmax 6 (3 - 24) hodín s niekoľkými maximálnymi hodnotami pozorovanými
u niektorých pacientov počas prvých 24 hodín. Absolútna biologická dostupnosť TAGRISSA sa
nestanovila. Na základe klinickej farmakokinetickej štúdie u pacientov užívajúcich 80 mg dávku, jedlo nemenilo biologickú dostupnosť osimertinibu v klinicky významnej miere (zvýšenie AUC o 6 % (90%
IS: -5; 19) a zníženie Cmax o 7 % (90% IS: -19; 6)). U zdravých dobrovoľníkov, ktorí dostávali 80 mg tabletu, sa pri podávaní omeprazolu počas 5 dní zvýšilo gastrické pH a pri 90% IS pre mieru expozície obsiahnutej v rámci limitu 80 – 125 % nebola expozícia osimertinibu ovplyvnená (zvýšenie AUC
o 7 % a Cmax o 2 %).
Distribúcia
Priemerný distribučný objem osimertinibu v rovnovážnom stave (Vss/F) je podľa populačných odhadov 986 l a indikuje rozsiahlu distribúciu do tkaniva. Väzbu na plazmatické bielkoviny nie je možné zmerať z dôvodu nestability, ale na základe fyzikálno-chemických vlastností osimertinibu sa predpokladá silná väzba na plazmatické bielkoviny. Osimertinib preukázal kovalentnú väzbu na plazmatické bielkoviny potkanov a ľudí, ľudský sérový albumín a hepatocyty potkanov a ľudí.
Biotransformácia
Štúdie in vitro naznačujú, že osimertinib je prednostne metabolizovaný CYP3A4 a CYP3A5. Metabolizmus sprostredkovaný CYP3A4 môže byť menej dôležitá cesta. Môžu existovať alternatívne
metabolické cesty, ktoré neboli úplne charakterizované. Na základe štúdií in vitro sa po perorálnom
podaní osimertinibu v plazme predklinických druhov a ľudí zistili 2 farmakologicky aktívne metabolity (AZ7550 a AZ5104); AZ7550 preukázal farmakologicky podobný profil ako TAGRISSO zatiaľ čo AZ5104 preukázal väčší potenciál u oboch mutovaného a wild-type (divokého typu) EGFR. Po perorálnom podaní TAGRISSA pacientom sa oba metabolity v plazme objavujú pomaly,
s mediánom (min-max) tmax 24 (4-72) a 24 (6-72) hodín v uvedenom poradí. V ľudskej plazme je pôvodný osimertinib zastúpený v 0,8% s 2 metabolitmi podieľajúcimi sa 0,08% a 0,07% z celkovej rádioaktivity, s prevahou rádioaktivity kovalentne viazanej na plazmatické bielkoviny. Geometrický priemer expozície oboch AZ5104 aj AZ7550, na základe AUC, bol pre každý metabolit približne 10 % z expozície osimertinibu v rovnovážnom stave.
Hlavnou metabolickou dráhou osimertinibu bola oxidácia a dealkylácia. Minimálne 12 zložiek sa pozorovalo v združených vzorkách ľudského moču a stolice a 5 zložiek predstavovalo > 1 % dávky
z toho nezmenený osimertinib predstavoval 1,9 %, AZ5104 6,6% a AZ7550 2,7% z dávky, zatiaľ čo adičná molekulárna zlúčenina cysteinylu (M21) predstavovala 1,5 % a neznámy metabolit (M25)
1,9 % z dávky.
Na základe štúdií in vitro je osimertinib kompetitívnym inhibítorom CYP 3A4/5, ale nie CYP1A2,
2A6, 2B6, 2C8, 2C9, 2C19, 2D6 a 2E1 v klinicky významných koncentráciách. Na základe štúdií in vitro osimertinib nie je inhibítorom UGT1A1 a UGT2B7 v klinicky významných koncentráciách v pečeni. Intestinálna inhibícia UGT1A1 je možná, ale klinický význam nie je známy.
Eliminácia
Po jednorazovej perorálnej dávke 20 mg osimertinibu sa 67,8 % zistilo v stolici (1,2 % ako pôvodné liečivo), zatiaľ čo 14,2 % podanej dávky sa zistilo v moči o 84 dní od odberu vzoriek (0,8 % ako pôvodné liečivo). Nezmenený osimertinib sa podieľal na približne 2 % eliminácii so zastúpením 0,8 % v moči a 1,2 % v stolici.
Interakciestransportnýmiproteínmi
Štúdie in vitro ukázali, že osimertinib nie je substrátom OATP1B1 a OATP1B3. Osimertinib in vitro
neinhibuje OAT1, OAT3, OATP1B1, OATP1B3 a MATE2K v klinicky významných koncentráciách. Avšak interakcie so substrátom MATE1 a OCT2 nie je možné vylúčiť.
Účinky osimertinibu na P-gp a BCRP
Na základe štúdií in vitro je osimertinib substrátom P-glykoproteínu a proteínu zodpovedného za rezistenciu pri rakovine prsníka (breast cancer resistance protein, BCRP), ale pri klinických dávkach osimertinibu nie je pravdepodobné, že dôjde ku klinicky významným liekovým interakciám. Na základe in vitro údajov je osimertinib inhibítorom BCRP a P-gp. Neskúmal sa PXR regulujúci enzýmové interakcie iné ako CYP3A4 (pozri časť 4.5).
Osobitnépopulácie
Vo farmakokinetických analýzach populácie (n=778) neboli identifikované žiadne klinicky významné vzťahy medzi predikovanou expozíciou v rovnovážnom stave (AUCss) a vekom pacienta (rozmedzie
21 až 89 rokov), pohlavím, etnickou príslušnosťou (vrátene bielych, ázijských, japonských, čínskych pacientov a pacientov inej ako ázijskej a bielej rasy) a stavu fajčenia (n=24 súčasní fajčiari,
n=232 bývalí fajčiari). FK analýza populácie naznačila že telesná hmotnosť bola významným
kovariátom s -20 % až +30 % zmenou AUCss osimertinibu očakávanou pri rozmedzí telesných hmotností 90 kg až 43 kg v uvedenom poradí (kvantily 95 % až 5 %) v porovnaní s AUCss pri mediáne telesnej hmotnosti 62 kg. Pri zohľadnení extrémnych telesných hmotností od < 43 kg do
> 90 kg boli miery metabolitu AZ5104 v rozmedzí od 11,8 % do 9,6 %, zatiaľ čo miery AZ7550 boli v rozmedzí od 12,8 % do 9,9 % v uvedenom poradí. Tieto zmeny expozície z dôvodu rozdielnosti telesných hmotností sa nepovažujú za klinicky významné.
Porucha funkcie pečene
Osimertinib je eliminovaný hlavne pečeňou a preto môžu mať pacienti s poruchou funkcie pečene zvýšenú expozíciu. Farmakokinetické skúšanie u osôb s poruchou funkcie pečene sa nevykonalo. Na
základe FK analýzy populácie nebol vzťah medzi markermi funkcie pečene (ALT, AST, bilirubín)
a expozíciou osimertinibu. Marker poruchy funkcie pečene, sérový albumín ukázal účinok na FK
osimertinibu. Z klinických štúdií boli vyradení pacienti s AST alebo ALT > 2,5 x horná hranica normálu (ULN), alebo z dôvodu pôvodného malígneho ochorenia > 5,0 x ULN, alebo s celkovým bilirubínom > 1,5 x ULN. Na základe farmakokinetickej analýzy 44 pacientov s miernou poruchou funkcie pečene a 330 pacientov s normálnou funkciou pečene boli expozície osimertinibu podobné. U pacientov s poruchou funkcie pečene sú k dispozícii obmedzené údaje (pozri časť 4.2).
Porucha funkcie obličiek
Farmakokinetická štúdia u pacientov s poruchou funkcie obličiek sa nevykonala. Na základe farmakokinetickej analýzy populácie boli expozície osimertinibu podobné u 330 pacientov s miernou poruchou funkcie obličiek (ClCr 60 až menej ako 90 ml/min), u 149 pacientov so stredne ťažkou poruchou funkcie obličiek (ClCr 30 až < ako 60 ml/min), u 3 pacientov s ťažkou poruchou funkcie obličiek (ClCr 15 až < ako 30 ml/min) a 295 pacientov s normálnou funkciou obličiek(≥ 90 ml/min). Ťažká porucha funkcie obličiek môže ovplyvniť elimináciu liekov eliminovaných v pečeni. Pacienti
s ClCr menej ako 15 ml/min neboli zaradení do klinických skúšaní.
5.3 Predklinické údaje o bezpečnosti
Hlavné zistenia pozorované v štúdiách toxicity po opakovanom podávaní u potkanov a psov pozostávali z atrofických, zápalových a degeneratívnych zmien postihujúcich rohovku (sprevádzané korneálnymi translucenciami a opacitami u psov pri vyšetrení oftalmoskopom), gastrointestinálny trakt (vrátane jazyka), kožu a samčie a samičie reprodukčné orgány so sekundárnymi zmenami sleziny. Tieto nálezy sa vyskytli pri plazmatických koncentráciách, ktoré boli nižšie ako tie pozorované u pacientov dostávajúcich 80 mg terapeutickú dávku. Nálezy prítomné po 1 mesiaci dávkovania boli vo veľkej miere reverzibilné počas 1 mesiaca ukončenia dávkovania, s výnimkou čiastočného upravenia niektorých zmien rohovky.
Predklinické údaje naznačujú, že osimertinib a jeho metabolit (AZ5104) inhibujú kanál h-ERG
a účinok na predĺženie QTc intervalu nie je možné vylúčiť.
Karcinogenitaamutagenita
Nevykonali sa štúdie karcinogenity s osimertinibom. V in vitro a in vivo skúškach osimertinib nespôsoboval genetické poškodenie.
Reprodukčná
toxicita
Degeneratívne zmeny sa pozorovali v semenníkoch potkanov a psov vystavených osimertinibu počas
≥ 1 mesiaca a po expozícii osimertinibu počas 3 mesiacov sa u samcov potkanov pozorovala znížená fertilita. Tieto nálezy sa pozorovali pri klinicky relevantných plazmatických koncentráciách. Patologické nálezy v semenníkoch pozorované po 1 mesiaci dávkovania boli u potkanov reverzibilné, nie je však možné poskytnúť definitívne stanovisko k reverzibilite týchto lézií u psov.
Nevykonali sa štúdie samičej fertility. V štúdiách toxicity po opakovanom podávaní potkanom vystaveným osimertinibu počas ≥ 1 mesiaca pri klinicky relevantných plazmatických koncentráciách sa pozoroval zvýšený výskyt anoestrus, degenerácia corpora lutea v ováriách a stenčenie epitelu maternice a vagíny. Nálezy v ováriách pozorované po 1 mesiaci dávkovania boli reverzibilné.
V modifikovanej štúdii embryofetálneho vývoja u potkanov osimertinib podávaný potkanom pred implantáciou embrya spôsobil embryoletalitu. Tieto účinky sa pozorovali pri samicou tolerovanej dávke 20 mg/kg, kedy bola expozícia rovnaká ako expozícia u ľudí pri odporúčanej dávke 80 mg denne (na základe celkovej AUC). Expozícia pri dávkach 20 mg/kg a vyšších spôsobila počas organogenézy znížené hmotnosti plodu, ale žiadne nežiaduce účinky na externú alebo viscerálnu morfológiu plodu. Pri podávaní osimertinibu gravidným samiciam potkanov počas gestácie a potom počas včasnej laktácie sa preukázala expozícia osimertinibu a jeho metabolitov u neodstavených mláďat a zníženie prežívania a slabý rast mláďat (pri dávkach 20 mg/kg a vyšších).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety manitol
mikrokryštalická celulóza
čiastočne substituovaná hydroxypropylcelulóza nátrium-stearylfumarát
Obaltablety polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastenec
žltý oxid železitý (E172)
červený oxid železitý (E172)
čierny oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Al/Al blistre s perforáciou umožňujúcou oddelenie jednotlivej dávky. Škatuľky s obsahom 30 x 1
tableta (3 blistre).
Al/Al blistre s perforáciou umožňujúcou oddelenie jednotlivej dávky. Škatuľky s obsahom 28 x 1
tableta (4 blistre).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAstraZeneca AB Gärtunavägen
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLAEU/1/16/1086/001
EU/1/16/1086/002
EU/1/16/1086/003
EU/1/16/1086/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 2. február 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.