
ch reakcií v tabuľkách
Nižšie uvedené údaje o bezpečnosti odzrkadľujú expozíciu ustekinumabu u dospelých v 12 štúdiách fázy II a III s účasťou 5 884 pacientov (4 135 so psoriázou a/alebo psoriatickou artritídou a 1 749
s Crohnovou chorobou). Toto zahŕňa expozíciu STELARE v kontrolovaných a nekontrolovaných obdobiach klinických štúdií počas najmenej 6 mesiacov alebo 1 roka (4 105 a 2 846 pacientov v tomto poradí so psoriázou, psoriatickou artritídou alebo Crohnovou chorobou) a expozíciu počas najmenej 4 alebo 5 rokov (1 482 a 838 pacientov so psoriázou, v tomto poradí).
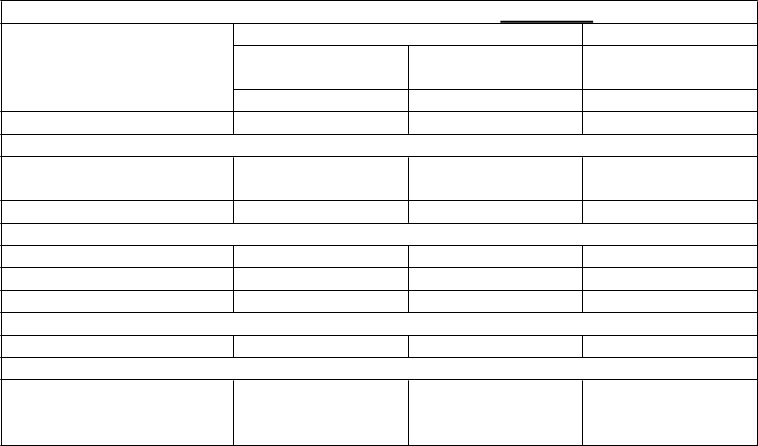
Tabuľka č. 2 obsahuje zoznam nežiaducich reakcií z klinických štúdií zameraných na psoriázu, psoriatickú artritídu a Crohnovu chorobu u dospelých ako aj nežiaducich účinkov hlásených z postmarketingových skúseností. Nežiaduce účinky sú usporiadané podľa triedy orgánových systémov a frekvencie s použitím nasledujúcej konvencie: Veľmi časté (≥ 1/10), Časté (≥ 1/100 až < 1/10), Menej časté (≥ 1/1 000 až < 1/100), Zriedkavé (≥ 1/10 000 až < 1/1 000), Veľmi zriedkavé
(< 1/10 000), neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka č. 2 Zoznam nežiaducich reakcií
Trieda orgánových systémov Frekvencia: nežiaduci účinok
Infekcie a nákazy Časté: infekcia horných dýchacích ciest, nazofaryngitída
Menej časté: celulitída, infekcia zubov, herpes zoster, vírusová infekcia horných dýchacích ciest, vulvovaginálna mykotická infekcia
Poruchy imunitného systému Menej časté: reakcie z precitlivenosti (vrátane vyrážky, žihľavky)
Zriedkavé: závažné reakcie z precitlivenosti (vrátane anafylaxie, angioedému)
Psychické poruchy Menej časté: depresia
Poruchy nervového systému Časté: závraty, bolesti hlavy
Menej časté: ochrnutie tváre
Poruchy dýchacej sústavy,
hrudníka a mediastína
Časté: orofaryngeálna bolesť

Menej časté: upchaný nos
Poruchy gastrointestinálneho
traktu
Poruchy kože a podkožného tkaniva
Časté: hnačka, nauzea, vracanie
Časté: svrbenie
Menej časté: pustulárna psoriáza, odlupovanie kože, akné
Zriedkavé: exfoliatívna dermatitída
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
Celkové poruchy a reakcie v mieste podania
Časté: bolesti chrbta, myalgia, artralgia
Časté: únava, erytém v mieste vpichu injekcie, bolesť v mieste podania injekcie
Menej časté: reakcie v mieste vpichu injekcie (vrátane hemorágie, hematómu, zatvrdnutia, opuchu a svrbenia), asténia
Opis vybraných nežiaducich reakcií
Infekcie
V placebom kontrolovaných štúdiách u pacientov so psoriázou, psoriatickou artritídou a Crohnovou chorobou bola miera prípadov infekcie alebo závažnej infekcie podobná medzi pacientmi liečenými ustekinumabom a pacientmi, ktorí dostávali placebo. V placebom kontrolovanej časti klinických štúdií u pacientov so psoriázou, pacientov so psoriatickou artritídou a pacientov s Crohnovou chorobou bola miera prípadov infekcie 1,38 na pacientorok sledovania u chorých liečených ustekinumabom a 1,35 u pacientov dostávajúcich placebo. Závažné infekcie sa vyskytli v miere 0,03 na pacientorok sledovania v skupine chorých, ktorým sa podával ustekinumab (27 závažných infekcií z 829 pacientorokov sledovania), a 0,03 v skupine chorých dostávajúcich placebo (11 závažných infekcií z 385 pacientorokov sledovania) (pozri časť 4.4).
V kontrolovaných a nekontrolovaných obdobiach klinických štúdií so psoriázou, psoriatickou artritídou a Crohnovou chorobou, ktoré predstavujú 10 953 pacientorokov expozície
u 5 884 pacientov, bol medián sledovania 0,99 roka; 3,2 rokov pre štúdie so psoriázou, 1,0 roka pre štúdie so psoriatickou artritídou a 0,6 roka pre štúdie s Crohnovou chorobou. Miera prípadov infekcií bola 0,91 na pacientorok sledovania u chorých liečených ustekinumabom a miera závažných infekcií bola 0,02 na pacientorok sledovania u chorých liečených ustekinumabom (178 závažných infekcií
z 10 953 pacientorokov sledovania) a k hláseným závažným infekciám patrili análny absces, celulitída, pneumónia, divertikulitída, gastroenteritída a vírusové infekcie.
V klinických štúdiách u pacientov s latentnou tuberkulózou, ktorí boli súčasne liečení izoniazidom, sa tuberkulóza nerozvinula.
Malignity
V placebom kontrolovanej časti klinických štúdií so psoriázou, psoriatickou artritídou a Crohnovou chorobou bol výskyt malignít s výnimkou nemelanómových kožných nádorov 0,12 na 100 pacientorokov sledovania u chorých liečených ustekinumabom (1 pacient z 829 pacientorokov sledovania) v porovnaní s 0,26 u pacientov, ktorí dostávali placebo (1 pacient z 385 pacientorokov sledovania). Výskyt nemelanómových kožných nádorov bol 0,48 na 100 pacientorokov sledovania
u chorých liečených ustekinumabom (4 pacienti z 829 pacientorokov sledovania) v porovnaní s 0,52
u pacientov dostávajúcich placebo (2 pacienti z 385 pacientorokov sledovania).
V kontrolovaných a nekontrolovaných obdobiach klinických štúdií so psoriázou, psoriatickou artritídou a Crohnovou chorobou, ktoré predstavujú 10 935 pacientorokov expozície
u 5 884 pacientov, bol medián sledovania 1,0 roka; 3,2 rokov pre štúdie so psoriázou, 1,0 roka pre štúdie so psoriatickou artritídou a 0,6 roka pre štúdie s Crohnovou chorobou. Malignity s výnimkou nemelanómovej rakoviny kože boli hlásené u 58 pacientov z 10 935 pacientorokov sledovania (incidencia 0,53 na 100 pacientorokov sledovania u pacientov liečených ustekinumabom). Výskyt malignít hlásených u pacientov liečených ustekinumabom bol porovnateľný s výskytom predpokladaným v bežnej populácii (štandardizovaná miera incidencie = 0,87 [95 % interval spoľahlivosti: 0,66; 1,14], upravené podľa veku, pohlavia a rasy). Najčastejšie pozorované malignity, iné ako nemelanómová rakovina kože, boli rakovina prostaty, melanóm, rakovina kolorekta a prsníka. Incidencia nemelanómovej rakoviny kože bola 0,49 na 100 pacientorokov sledovania u pacientov liečených ustekinumabom (53 pacientov z 10 919 pacientorokov sledovania). Pomer pacientov
s bazocelulárnym verzus skvamocelulárnym karcinómom kože (4:1) je porovnateľný s pomerom predpokladaným vo všeobecnej populácii (pozri časť 4.4).
Reakcie z precitlivenosti a reakcie na infúziu
V úvodných (indukčných) štúdiách s Crohnovou chorobou neboli po jednorazovej intravenóznej dávke hlásené žiadne prípady anafylaxie ani iných závažných reakcií súvisiacich s infúziou. V týchto štúdiách 2,4 % zo 466 placebom liečených pacientov a 2,6 % zo 470 pacientov liečených odporúčanou dávkou ustekinumabu hlásilo nežiaduce udalosti, ktoré sa vyskytli počas infúzie alebo do jednej hodiny od nej.
Imunogenicita
V klinických štúdiách so psoriázou a psoriatickou artritídou sa u menej ako 8 % pacientov liečených ustekinumabom vyvinuli protilátky proti ustekinumabu. V klinických štúdiách s Crohnovou chorobou sa u menej ako 3 % pacientov liečených ustekinumabom vyvinuli protilátky proti ustekinumabu. Nepozorovala sa nijaká zjavná súvislosť medzi vznikom protilátok na ustekinumab a rozvojom reakcií v mieste vpichu injekcie. Väčšina pacientov, ktorí boli pozitívni na protilátky proti ustekinumabu,
mala neutralizujúce protilátky. Účinnosť mala klesajúcu tendenciu u pacientov s pozitívnym testom na protilátky voči ustekinumabu, avšak pozitivita na protilátky nevylúčila klinickú odpoveď.
Pediatrická populáciaNežiaduce účinky u pediatrických pacientov vo veku 12 rokov a starších s ložiskovou psoriázou
Bezpečnosť ustekinumabu bola sledovaná v štúdii fázy III u 110 pacientov od 12 do 17 rokov počas
60 týždňov. Nežiaduce udalosti hlásené v tejto štúdii boli podobné ako udalosti pozorované v predchádzajúcich štúdiách u dospelých s ložiskovou psoriázou.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách sa podávali intravenózne jednotlivé dávky v množstve do 6 mg/kg bez dávkového obmedzenia vzhľadom na toxicitu. V prípade predávkovania sa odporúča u pacienta sledovať akékoľvek známky alebo symptómy nežiaducich účinkov a bezodkladne začať náležitú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory interleukínov, ATC kód: L04AC05.
Mechanizmus účinkuUstekinumab je plne humánna monoklonová protilátka IgG1κ, ktorá sa viaže s špecificitou na
spoločnú p40 podjednotku proteínu humánnych cytokínov interleukín (IL)-12 a IL-23. Ustekinumab inhibuje bioaktivitu humánnych IL-12 a IL-23 tak, že zabraňuje p40 naviazať sa na receptor proteínu IL-12Rb1 s expresiou na povrchu imunitných buniek. Ustekinumab sa nemôže viazať na IL-12 alebo IL-23, ktoré sa už naviazali na bunkové povrchové receptory IL-12Rb1. Je preto nepravdepodobné, že by ustekinumab mohol prispievať ku komplementácii alebo cytotoxicite buniek s receptormi IL-12 a/alebo IL-23. IL-12 a IL-23 sú heterodimerické cytokíny vylučované bunkami aktivovanými antigénmi, ako sú makrofágy a dendritické bunky, a oba cytokíny sa podieľajú na imunitnej funkcii;
IL-12 aktivuje „prirodzených zabíjačov“ (NK, z angl. natural killer) bunky a vedie k diferenciácii CD4+ T buniek smerom k fenotypu Th1 (T helper 1), IL-23 aktivuje dráhu Th17 (T helper 17). Neprimerané regulovanie IL 12 a IL 23 sa však spájalo s imunitou sprostredkovanými ochoreniami, ako napríklad psoriáza, psoriatická artritída a Crohnova choroba.
Naviazaním na spoločnú p40 podjednotku IL-12 a IL-23 môže ustekinumab vyvíjať svoj klinický účinok na psoriázu, psoriatickú artritídu a Crohnovu chorobu prostredníctvom prerušenia cytokínových dráh Th1 a Th17, ktoré sú dôležité pre patológiu týchto ochorení.
U pacientov s Crohnovou chorobou viedla liečba ustekinumabom k zníženiu hodnôt zápalových markerov vrátane C-reaktívneho proteínu (CRP) a fekálneho kalprotektínu počas indukčnej fázy. Toto zníženie sa počas udržiavacej fázy zachovalo.
Imuni
z
ácia
V priebehu dlhodobého predĺženia druhej štúdie psoriázy (PHOENIX 2) došlo u dospelých pacientov liečených STELAROU najmenej 3,5 roka k podobným protilátkovým odpovediam na obe, pneumokokovú polysacharidovú aj tetanovú vakcínu, ako u sledovanej skupiny s nesystematicky liečenou psoriázou. Podobné podiely dospelých pacientov rozvinuli ochranné hladiny anti- pneumokokových a anti-tetanových protilátok a titre protilátok boli u pacientov liečených STELAROU a u sledovaných pacientov podobné.
Klinická účinnosť
Crohnova choroba
Bezpečnosť a účinnosť ustekinumabu sa hodnotila v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných multicentrických štúdiách u dospelých pacientov so stredne ťažkou až ťažkou aktívnou Crohnovou chorobou (skóre CDAI [Crohn’s Disease Activity Index] ≥ 220 a ≤ 450). Klinický vývoj predstavovali dve 8-týždňové indukčné štúdie s intravenóznym podaním (UNITI-1 a UNITI-2), po ktorých nasledovala 44 týždňov trvajúca randomizovaná štúdia so subkutánnym podávaním (IM-UNITI), sledujúca udržanie účinku u pacientov, ktorí dosiahli klinickú odpoveď v indukčných štúdiách, čo celkovo predstavovalo 52 týždňov liečby.
Indukčné štúdie zahŕňali 1 409 (UNITI-1, n = 769; UNITI-2 n = 640) pacientov. Primárny koncový ukazovateľ v oboch indukčných štúdiách bol podiel jedincov s klinickou odpoveďou (definovanou
ako zníženie skóre CDAI o ≥ 100 bodov) v 6. týždni. V oboch štúdiách boli údaje o účinnosti zbierané a analyzované až do 8. týždňa. Súbežné dávky perorálnych kortikosteroidov,
imunomodulátorov, aminosalicylátov a antibiotík boli povolené a 75 % pacientov naďalej dostávalo najmenej jeden z týchto liekov. V oboch štúdiách boli pacienti randomizovaní na jednorazové intravenózne podanie buď odporúčanej odstupňovanej dávky približne 6 mg/kg (pozri Tabuľku 1, časť
4.2), fixnej dávky 130 mg ustekinumabu alebo placeba v 0. týždni.
Pacienti v UNITI-1 na predchádzajúcej anti-TNFα terapii zlyhali alebo ju netolerovali. Približne 48 % pacientov zlyhalo na 1 predchádzajúcej anti-TNFa terapii a 52 % zlyhalo na 2 alebo 3 predchádzajúcich anti-TNFα terapiách. V tejto štúdii 29,1 % pacientov nedosiahlo dostačujúcu
úvodnú odpoveď (primárni non-respondéri), 69,4 % odpovedalo, ale odpoveď neudržalo (sekundárni non-respondéri) a 36,4 % netolerovalo anti-TNFα terapie.
Pacienti v UNITI-2 zlyhali aspoň na jednej konvenčnej terapii, vrátane kortikosteroidov alebo imunomodulátorov, a buď predtým nedostali anti-TNF-α terapiu (68,6 %) alebo anti-TNFα terapiu predtým dostali, ale na nej nezlyhali (31,4 %).
V oboch štúdiách UNITI-1 a UNITI-2 bol významne vyšší podiel pacientov s klinickou odpoveďou
a remisiou v skupine liečenej ustekinumabom v porovnaní s placebom (Tabuľka 3). Klinická odpoveď a remisia boli významné už od 3. týždňa u pacientov liečených ustekinumabom a ďalej sa zlepšovala do 8. týždňa. V týchto indukčných štúdiách bola účinnosť vyššia a lepšie udržateľná v skupine
s odstupňovanou dávkou v porovnaní so skupinou so 130 mg dávkou, a preto sa odstupňované dávkovanie odporúča pre intravenóznu indukčnú dávku.
Tabuľka 3: Indukcia klinickej odpovede a remisie v UNITI-1 a UNITI 2
UNITI-1* UNITI-2**
Placebo
 N = 247
Odporúčaná dávka ustekinumabu N = 249
Placebo
N = 209
Odporúčaná dávka ustekinumabu N = 209
N = 247
Odporúčaná dávka ustekinumabu N = 249
Placebo
N = 209
Odporúčaná dávka ustekinumabu N = 209
Klinická remisia, 8. týždeň 18 (7,3 %) 52 (20,9 %)a 41 (19,6 %) 84 (40,2 %)a
Klinická odpoveď (100 bodov),
6. týždeň
53 (21,5 %) 84 (33,7 %)b 60 (28,7 %) 116 (55,5 %)a
Klinická odpoveď (100 bodov),
8. týždeň
50 (20,2 %) 94 (37,8 %)a 67 (32,1 %) 121 (57,9 %)a
Odpoveď 70 bodov, 3. týždeň 67 (27,1 %) 101 (40,6 %)b 66 (31,6 %) 106 (50,7 %)a
Odpoveď 70 bodov, 6. týždeň 75 (30,4 %) 109 (43,8 %)b 81 (38,8 %) 135 (64,6 %)a
Klinická remisia je definovaná ako skóre CDAI < 150; klinická odpoveď je definovaná ako zníženie skóre CDAI minimálne o 100 bodov alebo ako stav klinickej remisie
Odpoveď 70 bodov je definovaná ako zníženie skóre CDAI minimálne o 70 bodov
* Zlyhania na anti-TNFα
** Zlyhania na konvenčných terapiách
a p < 0,001
b p < 0,01
Udržiavacia štúdia (IM-UNITI) hodnotila 388 pacientov, ktorí dosiahli klinickú odpoveď 100 bodov
v 8. týždni indukcie s ustekinumabom v štúdiách UNITI-1 a UNITI-2. Pacienti boli randomizovaní na subkutánny udržiavací režim buď 90 mg ustekinumabu každých 8 týždňov, 90 mg ustekinumabu každých 12 týždňov alebo placebo počas 44 týždňov (odporúčané udržiavacie dávkovanie, pozri časť
4.2 v SPC STELARA injekčný roztok (injekčná liekovka) a injekčný roztok naplnený v injekčnej striekačke).
Významne vyššie podiely pacientov udržali klinickú remisiu a odpoveď v skupine liečenej ustekinumabom v porovnaní so skupinou s placebom v 44. týždni (pozri Tabuľku 4).
 Tabuľka 4: Udržanie klinickej odpovede a remisie v IM-UNITI (44. týždeň; 52 týždňov od podania indukčnej dávky)
Tabuľka 4: Udržanie klinickej odpovede a remisie v IM-UNITI (44. týždeň; 52 týždňov od podania indukčnej dávky)
P
lacebo*
N = 131
†
90 mg ustekinumabu každých
8 týždňov
N = 128
†
90 mg ustekinumabu každých
12 týždňov
N = 129
†
Klinická remisia 36 % 53 %a 49 %b Klinická odpoveď 44 % 59 %b 58 %b Klinická remisia bez kortikosteroidov 30 % 47 %a 43 %c Klinická remisia u pacientov:
v remisii na začiatku udržiavacej liečby 46 % (36/79) 67 % (52/78)a 56 % (44/78) ktorí boli zaradení zo štúdie CRD3002‡ 44 % (31/70) 63 % (45/72)c 57 % (41/72) ktorí neboli doteraz liečení anti-TNFα 49 % (25/51) 65 % (34/52)c 57 % (30/53) ktorí pristúpili zo štúdie CRD3001§ 26 % (16/61) 41 % (23/56) 39 % (22/57)
Klinická remisia je definovaná ako skóre CDAI < 150; klinická odpoveď je definovaná ako zníženie CDAI minimálne o 100 bodov alebo ako stav klinickej remisie
* Skupina s placebom sa skladala z pacientov, ktorí odpovedali na ustekinumab a boli randomizovaní na placebo na začiatku udržiavacej liečby.
† Pacienti so 100 bodovou klinickou odpoveďou na ustekinumab na začiatku udržiavacej liečby
‡ Pacienti, ktorí zlyhali na konvenčnej terapii, ale nie na anti-TNFα terapii
§ Pacienti, ktorí sú anti-TNFα refraktórni/intolerantní
a p < 0,01
b p < 0,05
c nominálne významné (p < 0,05)
V IM-UNITI, 29 zo 129 pacientov si neudržali odpoveď na ustekinumab, keď boli liečení každých 12
týždňov a mali povolenú úpravu dávky tak, aby dostávali ustekinumab každých 8 týždňov. Strata odpovede bola definovaná ako skóre CDAI ≥ 220 bodov a zvýšenie o 100 bodov oproti skóre CDAI pri vstupe do štúdie. U týchto pacientov bola klinická remisia dosiahnutá v prípade 41,4 % pacientov
16 týždňov po úprave dávky.
Pacienti, ktorí nezaznamenali klinickú odpoveď na indukciu ustekinumabu v 8. týždni indukčných štúdií UNITI-1 a UNITI-2 (476 pacientov), boli zaradení do nerandomizovanej časti udržiavacej štúdie (IM-UNITI) a dostávali celý čas 90 mg subkutánnu injekciu ustekinumabu. O osem týždňov
neskôr dosiahlo 50,5 % pacientov klinickú odpoveď a pokračovalo v užívaní udržiavacieho dávkovania každých 8 týždňov; spomedzi týchto pacientov s pokračujúcim udržiavacím dávkovaním si väčšina udržala odpoveď (68,1 %) a dosiahla remisiu (50,2 %) v 44. týždni, t.j. v podieloch, ktoré boli podobné ako u pacientov, ktorí pôvodne odpovedali na indukciu ustekinumabu.
Zo 131 pacientov, ktorí odpovedali na indukciu ustekinumabu a boli randomizovaní do skupiny s placebom na začiatku udržiavacej štúdie, 51 následne stratilo odpoveď a dostávalo 90 mg ustekinumabu subkutánne každých 8 týždňov. Väčšina pacientov, ktorá stratila odpoveď a vrátila sa k ustekinumabu tak urobila do 24 týždňov od indukčnej infúzie. Z týchto 51 pacientov, 70,6 % dosiahlo klinickú odpoveď a 39,2 % dosiahlo klinickú remisiu 16 týždňov po podaní prvej subkutánnej dávky ustekinumabu.
Endoskopia
V podštúdii sa hodnotil endoskopický vzhľad sliznice u 252 pacientov s vhodnou východiskovou endoskopickou aktivitou ochorenia. Primárny koncový ukazovateľ bola zmena oproti
východiskovému skóre SES-CD (Simplified Endoscopic Disease Severity Score for Crohn’s Disease), zloženému skóre hodnotiacemu 5 ileo-kolonických segmentov na prítomnosť/veľkosť vredov, podiel povrchu sliznice pokrytej vredmi, podiel povrchu sliznice postihnutej akýmikoľvek inými léziami
a prítomnosť/typ zúženia/striktúr. V 8. týždni bola po jednorazovej intravenóznej indukčnej dávke zmena v SES-CD skóre väčšia v skupine s ustekinumabom (n = 155, priemerná zmena = -2,8) ako v skupine s placebom (n = 97, priemerná zmena = -0,7, p = 0,012).
Odpoveď fistuly
V podskupine pacientov s drénovanou fistulou v úvode (8,8 %; n = 26), 12/15 (80 %) ustekinumabom liečených pacientov dosiahlo odpoveď fistuly v priebehu 44 týždňov (definovaná ako ≥ 50 % zníženie oproti východiskovému stavu v indukčnej štúdii v počte drénovaných fistúl) v porovnaní s 5/11
(45,5 %) dostávajúcimi placebo.
Kvalita života súvisiaca so zdravím
Kvalita života súvisiaca so zdravím bola posúdená na základe dotazníkov IBDQ a SF-36. V 8. týždni prejavovali pacienti dostávajúci ustekinumab štatisticky významne väčšie a klinicky významnejšie zlepšenia na celkovom skóre IBDQ a SF-36 Mental Component Summary Score v oboch štúdiách UNITI-1 a UNITI-2, a SF-36 Physical Component Summary Score v UNITI-2, pri porovnaní
s placebom. Tieto zlepšenia boli všeobecne lepšie udržateľné u pacientov liečených ustekinumabom v štúdii IM-UNITI až do 44. týždňa pri porovnaní s placebom.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s ustekinumabom v jednej alebo viacerých podskupinách pediatrickej populácie pre Crohnovu chorobu (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po podaní odporúčanej intravenóznej indukčnej dávky bol u pacientov s Crohnovou chorobou medián maximálnej koncentrácie ustekinumabu v sére, pozorovaný 1 hodinu po infúzii, 126,1 μg/ml.
Distribúcia
Medián distribučného objemu počas terminálnej fázy (Vz) po jednorazovom intravenóznom podaní pacientom so psoriázou sa pohyboval v rozsahu od 57 do 83 ml/kg.
Biotransformácia
Presná metabolická dráha ustekinumabu nie je známa.
Eliminácia
Medián systémového klírensu (CL) po jednorazovom intravenóznom podaní sa u pacientov so psoriázou pohyboval v rozsahu od 1,99 do 2,34 ml/deň/kg. Priemerný polčas (t1/2) ustekinumabu bol
približne 3 týždne u pacientov s Crohnovou chorobou, psoriázou a/alebo psoriatickou artritídou, v rozsahu od 15 do 32 dní počas všetkých štúdií zameraných na psoriázu a psoriatickú artritídu.
Linearita dávky
U pacientov so psoriázou sa systémová expozícia ustekinumabu (Cmax a AUC) zvyšovala proporcionálne v závislosti na dávke po jednorazovom intravenóznom podaní v rozsahu dávok od
0,09 mg/kg do 4,5 mg/kg.
Osobitné skupiny pacientov
Farmakokinetické údaje týkajúce sa pacientov s poruchou funkcie obličiek alebo pečene nie sú k dispozícii.
Neuskutočnili sa žiadne špeciálne štúdie s intravenóznym ustekinumabom so staršími ľuďmi alebo pediatrickými pacientmi.
U pacientov s Crohnovou chorobou bola variabilita CL ustekinumabu ovplyvnená telesnou hmotnosťou, hladinou albumínu v sére, CRP, úrovňou zlyhania TNF antagonistu, pohlavím, rasou (ázijská rasa oproti neázijskej rase) a úrovňou protilátok proti ustekinumabu, pričom telesná hmotnosť bol hlavný kovariát ovplyvňujúci distribučný objem. Súbežné používanie imunomodulátorov nemalo významný vplyv na dispozíciu ustekinumabu. Vplyv týchto štatisticky významných kovariátov na príslušné PK parametre bol v rozmedzí ± 20 % pri hodnotení naprieč reprezentatívnou škálou hodnôt kovariátov alebo kategórií údajov, ktoré je v rámci celkovej variability pozorované v PK ustekinumabu.
RegulovanieenzýmovCYP450
Účinky IL-12 alebo IL-23 na reguláciu enzýmov CYP450 boli hodnotené v in vitro štúdii na ľudských hepatocytoch, čo preukázalo, že IL-12 a/alebo IL-23 v sérových koncentráciách 10 ng/ml nemenia aktivitu ľudských enzýmov CYP450 (CYP1A2, 2B6, 2C9, 2C19, 2D6 alebo 3A4; pozri časť 4.5).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií toxicity po opakovanom podávaní a vývojovej a reprodukčnej toxicity, vrátane farmakologického hodnotenia bezpečnosti, neodhalili žiadne osobitné riziko (napr. orgánovej toxicity) pre ľudí. V štúdiách vývojovej a reprodukčnej toxicity na makakoch sa nepozorovali ani nežiaduce účinky na fertilitu samcov ani vrodené chyby alebo vývojová toxicita. Takisto sa nepozorovali žiadne nežiaduce účinky na fertilitu samíc pri použití analogickej protilátky voči IL-12/23 na myšiach.
Hladiny dávok v štúdiách na zvieratách boli približne až 45-násobne vyššie než je najvyššia ekvivalentná dávka určená na podanie pacientom so psoriázou a výsledné vrcholové sérové koncentrácie u opíc boli viac než 100-násobne vyššie než koncentrácia pozorovaná u ľudí.
Štúdie karcinogenity sa s ustekinumabom neuskutočnili vzhľadom na absenciu vhodných modelov na protilátku s neskríženou reaktivitou voči IL-12/23 p40 u hlodavcov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Disodná soľ kyseliny etyléndiamín-tetraoctovej (EDTA), dihydrát
Histidín
Monohydrát histidíniummonohydrochloridu
Metionín
Polysorbát 80
Sacharóza
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi. Stelara sa má riediť len s 0,9 % roztokom chloridu sodného. STELARA sa nemá podávať v tej istej intravenóznej linke súbežne s inými liekmi.
6.3 Čas použiteľnosti
2 roky
Neuchovávajte v mrazničke.
Chemická a fyzikálna stabilita počas používania (in-use) bola preukázaná počas 4 hodín pri 15-25 °C. Z mikrobiologického hľadiska, ak spôsob riedenia nevylučuje riziko mikrobiologickej kontaminácie,
sa má liek použiť ihneď. Ak sa nepodá ihneď, čas a podmienky uchovávania sú v zodpovednosti užívateľa.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C – 8°C). Neuchovávajte v mrazničke. Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
26 ml sterilného roztoku v 30 ml injekčnej liekovke zo skla typu I uzavretej gumovou zátkou potiahnutou butylom. V jednom balení je jedna injekčná liekovka s liekom STELARA.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Roztok v injekčnej liekovke so STELAROU sa nemá pretrepávať. Pred podaním treba roztok vizuálne skontrolovať, či nevidno pevné prachové častice alebo či nedošlo k zmene sfarbenia. Roztok je číry, bezfarebný až svetložltý. Liek sa nemá použiť, ak roztok zmenil farbu alebo je mútny, alebo ak sú prítomné cudzorodé pevné častice.
Riedenie
STELARA koncentrát na infúzny roztok musí byť zriedená a pripravená zdravotníckym pracovníkom použitím aseptickej techniky.
1. Vypočítajte dávku a potrebný počet injekčných liekoviek STELARY na základe hmotnosti pacienta (pozri časť 4.2, Tabuľka 1). Jedna 26 ml injekčná liekovka STELARY obsahuje
130 mg ustekinumabu. Použite len kompletné injekčné liekovky Stelary.
2. Z 250 ml infúzneho vaku odoberte a potom zlikvidujte objem 0,9 % roztoku chloridu sodného rovnajúci sa objemu STELARY, ktorý sa má pridať (odstráňte 26 ml chloridu sodného na každú potrebnú injekčnú liekovku STELARY, pri 2 injekčných liekovkách - odstráňte 52 ml, pri 3 injekčných liekovkách - odstráňte 78 ml, pri 4 injekčných liekovkách – odstráňte 104 ml).
3. Odoberte 26 ml STELARY z každej potrebnej injekčnej liekovky a pridajte ich do 250 ml infúzneho vaku. Konečný objem v infúznom vaku má byť 250 ml. Jemne pomiešajte.
4. Pred podaním vizuálne skontrolujte zriedený roztok. Nepoužite ho, ak spozorujete viditeľné nepriehľadné častice, sfarbenie alebo cudzie častice.
5. Zriedený roztok podávajte počas najmenej jednej hodiny. Po zriedení možno infúzny roztok uchovávať do štyroch hodín pred infúziou.
6. Použite len infúzny set s in-line sterilným nepyrogénnym filtorm s nízkou väzbou bielkovín
(veľkosť pórov 0,2 mikrometra).
7. Každá injekčná liekovka je len na jednorazové použitie a akýkoľvek nepoužitý liek sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen-Cilag International NV Turnhoutseweg 30
2340 Beerse
Belgicko
8. REGISTRAČNÉ ČÍSLOEU/1/08/494/005
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. január 2009
Dátum posledného predĺženia registrácie: 19. september 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
STELARA 45 mg injekčný roztok
STELARA 90 mg injekčný roztok
STELARA 45 mg injekčný roztok naplnený v injekčnej striekačke
STELARA 90 mg injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
STELARA45 mg injekčný roztok
Jedna injekčná liekovka obsahuje 45 mg ustekinumabu v 0,5 ml.
STELARA 90 mg injekčný roztok
Jedna injekčná liekovka obsahuje 90 mg ustekinumabu v 1 ml.
STELARA 45 mg roztok naplnený v injekčnej striekačke.
Jedna naplnená injekčná striekačka obsahuje 45 mg ustekinumabu v 0,5 ml.
STELARA 90 mg roztok naplnený v injekčnej striekačke
Jedna naplnená injekčná striekačka obsahuje 90 mg ustekinumabu v 1 ml.
Ustekinumab je plne humánna monoklonová protilátka IgG1κ proti interleukínu (IL)-12/23, ktorá vzniká v myších myelómových bunkových líniách použitím rekombinantnej DNA technológie.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
STELARA45 mg injekčný roztok
Injekčný roztok.
STELARA90 mg injekčný roztok
Injekčný roztok.
STELARA45 mg injekčný roztok naplnený v injekčnej striekačke
Injekčný roztok.
STELARA90 mg injekčný roztok naplnený v injekčnej striekačke
Injekčný roztok.
Roztok je číry až slabo opalescentný, bezfarebný až svetložltý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Ložisková psoriáza
STELARA je indikovaná na liečbu stredne závažnej až závažnej ložiskovej formy psoriázy
u dospelých pacientov, u ktorých sa nedostavila klinická odpoveď na iné systémové terapie, vrátane cyklosporínu, metotrexátu (MTX) alebo PUVA (psolaren a ultrafialové žiarenie pásma A), prípadne sú im takéto terapie kontraindikované alebo ich netolerujú (pozri časť 5.1).
Ložisková psoriáza u pediatrických pacientov
STELARA je indikovaná na liečbu stredne závažnej až závažnej ložiskovej psoriázy u adolescentných pacientov od 12 rokov a starších, ktorí nie sú dostatočne kontrolovaní alebo netolerujú iné systémové terapie alebo fototerapie (pozri časť 5.1).
Psoriatická artritída (PsA)
STELARA, v monoterapii alebo v kombinácii s MTX, je indikovaná na liečbu aktívnej psoriatickej artritídy u dospelých pacientov, ak nebola odpoveď na predchádzajúcu nebiologickú liečbu antireumatickým liekom modifikujúcim chorobu (DMARD, z angl. disease-modifying anti-rheumatic drug) dostatočná (pozri časť 5.1).
Crohnova choroba
STELARA je indikovaná na liečbu dospelých pacientov so stredne ťažkou až ťažkou aktívnou Crohnovou chorobou, u ktorých odpoveď buď na konvenčnú terapiu alebo na terapiu antagonistom tumor nekrotizujúceho faktoru alfa - TNFα bola neadekvátna alebo došlo k strate odpovede alebo takúto terapiu netolerujú alebo je im kontraindikovaná.
4.2 Dávkovanie a spôsob podávania
STELARA je určená na podávanie pod vedením a dozorom lekárov, ktorí majú skúsenosti s diagnostikovaním a liečbou ochorení, na ktoré je STELARA indikovaná.
Dávkovanie
Ložisková psoriáza
Odporúčané dávkovanie STELARY je úvodná dávka 45 mg podaná subkutánne, po čom nasleduje dávka 45 mg o 4 týždne neskôr, a potom sa liek podáva každých 12 týždňov.
U pacientov, u ktorých sa nedostavila klinická odpoveď ani po 28 týždňoch liečby, treba uvažovať o prerušení terapie.
Pacienti s hmotnosťou > 100 kg
Pacientom s hmotnosťou > 100 kg sa podáva úvodná dávka 90 mg subkutánne, po čom nasleduje dávka 90 mg o 4 týždne neskôr, a potom sa liek podáva každých 12 týždňov. U týchto pacientov sa zistilo, že je účinná aj dávka 45 mg. Napriek tomu dávka 90 mg bola účinnejšia (pozri časť 5.1, Tabuľka č. 4).
Psoriatická artritída (PsA)
Odporúčané dávkovanie STELARY je úvodná dávka 45 mg podaná subkutánne, po ktorej nasleduje
45 mg dávka o 4 týždne neskôr, a potom každých 12 týždňov. U pacientov s hmotnosťou > 100 kg sa môže prípadne použiť dávka 90 mg.
U pacientov, ktorí neodpovedali na liečbu do 28. týždňa, treba zvážiť prerušenie liečby.
Starší ľudia (vo veku ≥ 65 rokov)
Starším pacientom nie je potrebné upravovať dávkovanie (pozri časť 4.4).
Porucha funkcie obličiek a pečene
Použitie STELARY sa neskúmalo v tejto populácii pacientov. Nie je možné poskytnúť žiadne odporúčania ohľadne dávkovania.
Pediatrická populácia
Bezpečnosť a účinnosť STELARY u detí so psoriázou mladších ako 12 rokov alebo u detí so psoriatickou artritídou mladších ako 18 rokov neboli doteraz stanovené.
 Ložisková
psoriáza u pediatrických pacientov (12 rokov a viac)
Ložisková
psoriáza u pediatrických pacientov (12 rokov a viac)
Odporúčaná dávka STELARY odvodená od telesnej hmotnosti sa uvádza nižšie (Tabuľky 1 a 2). STELARA sa má podať v 0. a 4. týždni, a potom každých 12 týždňov.
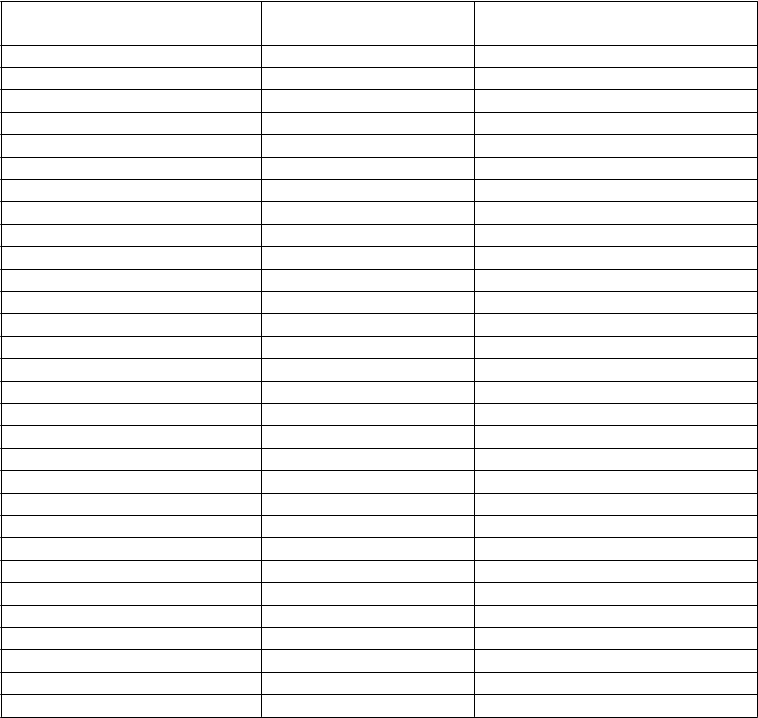
Tabuľka 1: Odporúčaná dávka STELARY pre psoriázu u pediatrických pacientovTelesná hmotnosť v čase dávkovania Odporúčaná dávka< 60 kg 0,75 mg/kga
≥ 60-≤ 100 kg 45 mg
> 100 kg 90 mg
a Pre výpočet objemu injekcie (ml) pre pacientov < 60 kg, použite nasledujúci vzorec:
telesná hmotnosť (kg) x
0,0083(ml/kg) alebo pozrite Tabuľku 2. Vypočítaný objem sa má zaokrúhliť na najbližšiu 0,01 ml a podať pomocou 1 ml kalibrovanej injekčnej striekačky. Pre pediatrických pacientov, ktorí potrebujú dostať menšiu dávku ako 45 mg, je dostupná 45 mg injekčná liekovka.
Tabuľka 2: Objemy injekcie STELARY pre pediatrických psoriatických pacientov < 60 kgTelesná hmotnosť v časedávkovania (kg) Dávka (mg) Objem injekcie (ml)30 22,5 0,25
31 23,3 0,26
32 24,0 0,27
33 24,8 0,27
34 25,5 0,28
35 26,3 0,29
36 27,0 0,30
37 27,8 0,31
38 28,5 0,32
39 29,3 0,32
40 30,0 0,33
41 30,8 0,34
42 31,5 0,35
43 32,3 0,36
44 33,0 0,37
45 33,8 0,37
46 34,5 0,38
47 35,3 0,39
48 36,0 0,40
49 36,8 0,41
50 37,5 0,42
51 38,3 0,42
52 39,0 0,43
53 39,8 0,44
54 40,5 0,45
55 41,3 0,46
56 42,0 0,46
57 42,8 0,47
58 43,5 0,48
59 44,3 0,49
U pacientov, u ktorých sa nedostavila klinická odpoveď ani po 28 týždňoch liečby, treba uvažovať
o prerušení terapie.
Crohnova chorobaV liečebnom režime sa prvá dávka STELARY podáva intravenózne. Dávkovanie pri režime intravenózneho podávania, pozri časť 4.2 v SPC STELARA 130 mg koncentrát na infúzny roztok.
Prvé subkutánne podanie 90 mg STELARY sa má uskutočniť v týždni 8 po intravenóznej dávke. Po tomto sa odporúča dávkovanie každých 12 týždňov.
Pacienti, ktorí nemali primeranú odpoveď v 8. týždni po prvej subkutánnej dávke, môžu v tom čase dostať druhú subkutánnu dávku (pozri časť 5.1).
Pacienti, ktorí stratia odpoveď pri dávkovaní každých 12 týždňov, môžu mať prínos zo zvýšenia frekvencie dávkovania na každých 8 týždňov (pozri časť 5.1).
U pacientov môže byť dávkovanie následne každých 8 týždňov alebo každých 12 týždňov, podľa klinického posúdenia (pozri časť 5.1).
Ukončenie liečby sa má zvážiť u pacientov, u ktorých sa nezaznamená žiadny terapeutický prínos do 16. týždňa alebo 16 týždňov po zmene na 8 týždňové dávkovanie.
Počas liečby STELAROU možno pokračovať podávaním imunomodulátorov a/alebo kortikosteroidov. U pacientov, ktorí odpovedali na liečbu STELAROU, sa v súlade so štandardmi liečby má znížiť
alebo ukončiť používanie kortikosteroidov.
Ak sa terapia preruší, obnovenie liečby so subkutánnym dávkovaním každých 8 týždňov je bezpečné a účinné.
Starší ľudia (vo veku ≥ 65 rokov)
Starším pacientom nie je potrebné upravovať dávkovanie (pozri časť 4.4).
Porucha funkcie obličiek a pečene
Použitie STELARY sa neskúmalo v tejto populácii pacientov. Nie je možné poskytnúť žiadne odporúčania ohľadne dávkovania.
Pediatrická populácia
Bezpečnosť a účinnosť STELARY v liečbe Crohnovej choroby u detí mladších ako 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
STELARA 45 mg a 90 mg injekčné liekovky alebo naplnené injekčné striekačky sú určené iba na subkutánnu injekciu. Ak je to možné, miestami aplikácie nemajú byť plochy kože postihnuté psoriázou.
Po náležitom zaškolení, ako sa podáva subkutánna injekcia, môžu STELARU podávať pacienti alebo ich ošetrovatelia, ak to lekár uzná za vhodné. Lekár má však pacienta naďalej starostlivo sledovať. Pacientov alebo ich ošetrovateľov treba poučiť, aby podávali predpísané množstvo STELARY podľa návodu uvedenom v písomnej informácii pre používateľa. Zrozumiteľné pokyny o podávaní lieku sú v písomnej informácii pre používateľa.
Pozri časť 6.6, kde sú podrobnejšie informácie o príprave a zaobchádzaní s liekom.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Klinicky závažná aktívna infekcia (napr. aktívna tuberkulóza, pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Infekcie
Ustekinumab môže mať potenciál zvyšovať riziko vzniku infekcií a reaktivovať latentné infekcie. V klinických štúdiách sa pozorovali závažné bakteriálne, plesňové a vírusové infekcie u pacientov liečených STELAROU (pozri časť 4.8).
Pri zvažovaní použitia STELARY u pacientov s chronickou infekciou alebo s anamnézou rekurentnej infekcie je potrebná opatrnosť (pozri časť 4.3).
Pred začatím terapie STELAROU je potrebné pacienta vyšetriť na prítomnosť infekcie tuberkulózy. STELARA sa nesmie podávať chorým s aktívnou tuberkulózou (pozri časť 4.3). K liečbe latentnej infekcie tuberkulózy treba pristúpiť pred podaním STELARY. Antituberkulóznu terapiu treba takisto zvážiť pred začatím liečby STELAROU u pacientov s latentnou alebo aktívnou tuberkulózou
v minulosti, u ktorých nie je možné zaručiť adekvátny priebeh liečby. U pacientov užívajúcich
STELARU treba počas liečby a po nej starostlivo sledovať známky a symptómy aktívnej tuberkulózy.
Pacientov treba poučiť, aby vyhľadali lekársku pomoc v prípade, ak sa vyskytnú známky alebo symptómy poukazujúce na infekciu. Ak sa u pacienta rozvinie závažná infekcia, je potrebné pacienta starostlivo sledovať a nesmie sa mu STELARA podávať, kým nie je infekcia vyliečená.
Malignity
Imunosupresíva ako ustekinumab majú potenciál zvyšovať riziko vzniku malignity. U niektorých pacientov, ktorí dostávali v klinických skúšaniach STELARU, sa rozvinuli kutánne a nekutánne malignity (pozri časť 4.8).
Neuskutočnili sa žiadne klinické štúdie, do ktorých by boli zaradení pacienti s anamnézou malignity, alebo pacienti, u ktorých by liečba pokračovala, ak u nich počas terapie STELAROU došlo k vzniku malignity. Preto je potrebné starostlivo zvažovať použitie STELARY u týchto pacientov.
U všetkých pacientov, predovšetkým u pacientov starších ako 60 rokov, u pacientov s predĺženou imunosupresívnou liečbou v anamnéze alebo u pacientov s liečbou PUVA v anamnéze, je potrebné sledovať objavenie nemelanómovej rakoviny kože (pozri časť 4.8).
Reakcie z precitlivenosti
V postmarketingovom sledovaní boli hlásené závažné reakcie z precitlivenosti, v niektorých prípadoch niekoľko dní po liečbe. Vyskytla sa anafylaxia a angioedém. Ak sa vyskytne anafylaktická alebo iná závažná reakcia z precitlivenosti, je potrebné pristúpiť k náležitej liečbe a STELARU vysadiť (pozri časť 4.8).
Citlivosť na latex
Kryt ihly na striekačke v prípade STELARY naplnených striekačiek je vyrobený zo suchej prírodnej gumy (derivát latexu), čo môže u jedincov citlivých na latex vyvolať alergickú reakciu.
Očkovania
Odporúča sa nepodávať živé vírusové alebo živé bakteriálne vakcíny (ako Bacillus Calmette-Guérin (BCG)) súčasne so STELAROU. Špeciálne klinické štúdie s účasťou pacientov, ktorí nedávno dostali živé vírusové alebo živé bakteriálne vakcíny, sa neuskutočnili. K dispozícii nie sú žiadne údaje
o sekundárnom prenose infekcie živými vakcínami u pacientov liečených STELAROU. Pred očkovaním živými vírusovými alebo živými bakteriálnymi vakcínami sa terapia STELAROU nemá podávať najmenej 15 týždňov od poslednej dávky a k terapii sa možno vrátiť najskôr 2 týždne po očkovaní. Indikujúci lekári sa majú oboznámiť so Súhrnmi charakteristických vlastností lieku pre špecifické vakcíny kvôli podrobnejším informáciám a pokynom o súčasnom podávaní imunosupresív po očkovaní.
Pacienti liečení STELAROU môžu súbežne dostať inaktivované alebo mŕtve vakcíny.
Dlhodobá liečba STELAROU nepotláča humorálnu imunitnú odpoveď na pneumokokovú polysacharidovú alebo tetanovú vakcínu (pozri časť 5.1).
Súbežná imunosupresívna liečba
V štúdiách so psoriázou sa nehodnotila bezpečnosť a účinnosť STELARY v kombinácii
s imunosupresívami, vrátane biologických látok alebo fototerapie. V štúdiách so psoriatickou artritídou sa nepreukázalo, že by súbežné podávanie MTX malo vplyv na bezpečnosť alebo účinnosť STELARY. V štúdiách s Crohnovou chorobou sa nepreukázalo, že by súbežné podávanie imunosupresív alebo kortikosteroidov malo vplyv na bezpečnosť alebo účinnosť STELARY. Je potrebné starostlivo zvážiť súčasné podávanie iných imunosupresív a STELARY alebo prechod na túto terapiu z iného imunosupresívneho biologického liečiva (pozri časť 4.5).
Imunoterapia
STELARA sa nehodnotila u pacientov, ktorí podstúpili alergénovú imunoterapiu. Nie je známe, či
STELARA môže mať vplyv na alergénovú imunoterapiu.
Vážne ochorenia kože
U pacientov so psoriázou bola po liečbe ustekinumabom hlásená exfoliatívna dermatitída (pozri
časť 4.8). U pacientov s ložiskovou psoriázou sa môže v rámci prirodzeného priebehu ich ochorenia vyskytnúť erytrodermálna psoriáza s príznakmi, ktoré môžu byť klinicky na nerozoznanie od exfoliatívnej dermatitídy. V rámci sledovania psoriázy u pacienta si majú lekári pozorne všímať príznaky erytrodermálnej psoriázy alebo exfoliatívnej dermatitídy. Ak sa tieto príznaky objavia, má sa začať vhodná liečba. Liečba STELAROU sa má ukončiť, ak existuje podozrenie na reakciu na liek.
Osobitné skupiny pacientov
Starší ľudia (≥ 65 rokov)
U pacientov starších ako 65 rokov, ktorí dostávali STELARU, sa nepozorovali žiadne rozdiely v účinnosti alebo bezpečnosti v porovnaní s mladšími pacientmi, počet pacientov starších ako
65 rokov však nie je dostatočný na to, aby sa dalo určiť, či odpovedajú na liečbu odlišne ako mladší pacienti. Keďže sa vo všeobecnosti u starších pacientov vyskytujú infekcie vo vyššej miere, liečbe tejto skupiny pacientov treba venovať zvýšenú pozornosť.
4.5 Liekové a iné interakcie
Živé vakcíny sa nesmú podávať súčasne so STELAROU (pozri časť 4.4).
Neuskutočnili sa žiadne interakčné štúdie u ľudí. V populačných farmakokinetických analýzach klinických štúdií v III. fáze sa skúmal účinok najčastejšie používaných súčasne podávaných liekov
u pacientov so psoriázou (vrátane paracetamolu, ibuprofénu, kyseliny acetylsalicylovej, metformínu, atorvastatínu, levotyroxínu) na farmakokinetiku ustekinumabu. Nezistili sa žiadne náznaky interakcie
s týmito súčasne podávanými liekmi. Daná analýza vychádzala zo základu, že najmenej 100 pacientov
(> 5 % sledovanej populácie) dostávalo súbežnú liečbu týmito liekmi po obdobie najmenej 90 % z času trvania štúdie. U pacientov so psoriatickou artritídou alebo Crohnovou chorobou nebola farmakokinetika ustekinumabu ovplyvnená súbežným užívaním MTX, NSAID, 6-merkaptopurínu, azatioprínu a perorálnych kortikosteroidov alebo predchádzajúcou expozíciou anti-TNFα látkam.
Výsledky štúdie in vitro nenaznačujú potrebu úpravy dávky u pacientov, ktorí užívajú zároveň substráty CYP450 (pozri časť 5.2).
V štúdiách so psoriázou sa nehodnotila bezpečnosť a účinnosť STELARY v kombinácii
s imunosupresívami, vrátane biologických látok alebo fototerapie. V štúdiách so psoriatickou artritídou sa nepreukázalo, že by súbežné podávanie MTX malo vplyv na bezpečnosť alebo účinnosť STELARY. V štúdiách s Crohnovou chorobou sa nepreukázalo, že by súbežné podávanie imunosupresív alebo kortikosteroidov malo vplyv na bezpečnosť alebo účinnosť STELARY(pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a až do 15 týždňov po liečbe.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití ustekinumabu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri 5.3). Z dôvodu bezpečnosti sa neodporúča užívať STELARU počas gravidity.
Laktácia
Nie je známe, či sa ustekinumab vylučuje do ľudského materského mlieka. V štúdiách na zvieratách sa zistilo vylučovanie do materského mlieka v malom množstve. Nie je známe, či sa ustekinumab po požití systémovo absorbuje. Vzhľadom na potenciál pre vznik nežiaducich reakcií spôsobených ustekinumabom u dojčiat, je potrebné rozhodnúť, či prerušiť laktáciu počas liečby a ešte 15 týždňov po liečbe alebo či prerušiť terapiu STELAROU so zreteľom na prínos laktácie pre dieťa a prínos terapie pre pacientku.
Fertilita
Vplyv ustekinumabu na fertilitu u ľudí sa neskúmal (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Stelara nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšie nežiaduce reakcie (> 5 %) v kontrolovaných obdobiach klinických štúdií psoriázy, psoriatickej artritídy a Crohnovej choroby s ustekinumabom u dospelých boli nazofaryngitída a bolesť hlavy. Väčšinou sa považovali za ľahké a nevyžadovali si prerušenie liečby v štúdii. Najzávažnejšia nežiaduca reakcia, ktorá bola hlásená pre STELARU, je závažná reakcia z precitlivenosti vrátane anafylaxie (pozri časť 4.4). Celkový bezpečnostný profil bol u pacientov so psoriázou, psoriatickou artritídou a Crohnovou chorobou podobný.
Zoznam nežiaducich reakcií v tabuľkách
Nižšie uvedené údaje o bezpečnosti odzrkadľujú expozíciu ustekinumabu u dospelých v 12 štúdiách fázy II a III s účasťou 5 884 pacientov (4 135 so psoriázou a/alebo psoriatickou artritídou a 1 749
s Crohnovou chorobou). Toto zahŕňa expozíciu STELARE v kontrolovaných a nekontrolovaných obdobiach klinických štúdií počas najmenej 6 mesiacov alebo 1 roka (4 105 a 2 846 pacientov v tomto poradí so psoriázou, psoriatickou artritídou alebo Crohnovou chorobou) a expozíciu počas najmenej 4 alebo 5 rokov (1 482 a 838 pacientov so psoriázou, v tomto poradí).
Tabuľka č. 3 obsahuje zoznam nežiaducich reakcií z klinických štúdií zameraných na
psoriázu, psoriatickú artritídu a Crohnovu chorobu u dospelých ako aj nežiaducich účinkov hlásených z postmarketingových skúseností. Nežiaduce účinky sú usporiadané podľa triedy orgánových systémov a frekvencie s použitím nasledujúcej konvencie: Veľmi časté (≥ 1/10), Časté (≥ 1/100 až
< 1/10), Menej časté (≥ 1/1 000 až < 1/100), Zriedkavé (≥ 1/10 000 až < 1/1 000), Veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka č. 3 Zoznam nežiaducich reakcií
Trieda orgánových systémov Frekvencia: nežiaduci účinok
Infekcie a nákazy Časté: infekcia horných dýchacích ciest, nazofaryngitída
Menej časté: celulitída, infekcia zubov, herpes zoster, vírusová infekcia horných dýchacích ciest, vulvovaginálna mykotická infekcia
Poruchy imunitného systému Menej časté: reakcie z precitlivenosti (vrátane vyrážky, žihľavky)
Zriedkavé: závažné reakcie z precitlivenosti (vrátane anafylaxie, angioedému)
Psychické poruchy Menej časté: depresia
Poruchy nervového systému Časté: závraty, bolesti hlavy
Menej časté: ochrnutie tváre
Poruchy dýchacej sústavy,
hrudníka a mediastína
Časté: orofaryngeálna bolesť
Menej časté: upchaný nos
Poruchy gastrointestinálneho
traktu
Poruchy kože a podkožného tkaniva
Časté: hnačka, nauzea, vracanie
Časté: svrbenie
Menej časté: pustulárna psoriáza, odlupovanie kože, akné
Zriedkavé: exfoliatívna dermatitída
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
Celkové poruchy a reakcie v mieste podania
Časté: bolesti chrbta, myalgia, artralgia
Časté: únava, erytém v mieste vpichu injekcie, bolesť v mieste podania injekcie
Menej časté: reakcie v mieste vpichu injekcie (vrátane hemorágie, hematómu, zatvrdnutia, opuchu a svrbenia), asténia
 Opis vybraných nežiaducich reakcií
Infekcie
Opis vybraných nežiaducich reakcií
Infekcie
V placebom kontrolovaných štúdiách u pacientov so psoriázou, psoriatickou artritídou a Crohnovou chorobou bola miera prípadov infekcie alebo závažnej infekcie podobná medzi pacientmi liečenými ustekinumabom a pacientmi, ktorí dostávali placebo. V placebom kontrolovanej časti klinických štúdií u pacientov so psoriázou, pacientov so psoriatickou artritídou a pacientov s Crohnovou chorobou bola miera prípadov infekcie 1,38 na pacientorok sledovania u chorých liečených ustekinumabom a 1,35 u pacientov dostávajúcich placebo. Závažné infekcie sa vyskytli v miere 0,03 na pacientorok sledovania v skupine chorých, ktorým sa podával ustekinumab (27 závažných infekcií z 829 pacientorokov sledovania), a 0,03 v skupine chorých dostávajúcich placebo (11 závažných infekcií z 385 pacientorokov sledovania) (pozri časť 4.4).
V kontrolovaných a nekontrolovaných obdobiach klinických štúdií so psoriázou, psoriatickou artritídou a Crohnovou chorobou, ktoré predstavujú 10 953 pacientorokov expozície
u 5 884 pacientov, bol medián sledovania 0,99 roka; 3,2 rokov pre štúdie so psoriázou, 1,0 roka pre štúdie so psoriatickou artritídou a 0,6 roka pre štúdie s Crohnovou chorobou. Miera prípadov infekcií bola 0,91 na pacientorok sledovania u chorých liečených ustekinumabom a miera závažných infekcií bola 0,02 na pacientorok sledovania u chorých liečených ustekinumabom (178 závažných infekcií
z 10 953 pacientorokov sledovania) a k hláseným závažným infekciám patrili análny absces, celulitída, pneumónia, divertikulitída, gastroenteritída a vírusové infekcie.
V klinických štúdiách u pacientov s latentnou tuberkulózou, ktorí boli súčasne liečení izoniazidom, sa tuberkulóza nerozvinula.
MalignityV placebom kontrolovanej časti klinických štúdií so psoriázou, psoriatickou artritídou a Crohnovou chorobou bol výskyt malignít s výnimkou nemelanómových kožných nádorov 0,12 na 100 pacientorokov sledovania u chorých liečených ustekinumabom (1 pacient z 829 pacientorokov sledovania) v porovnaní s 0,26 u pacientov, ktorí dostávali placebo (1 pacient z 385 pacientorokov sledovania). Výskyt nemelanómových kožných nádorov bol 0,48 na 100 pacientorokov sledovania
u chorých liečených ustekinumabom (4 pacienti z 829 pacientorokov sledovania) v porovnaní s 0,52
u pacientov dostávajúcich placebo (2 pacienti z 385 pacientorokov sledovania).
V kontrolovaných a nekontrolovaných obdobiach klinických štúdií so psoriázou, psoriatickou artritídou a Crohnovou chorobou, ktoré predstavujú 10 935 pacientorokov expozície
u 5 884 pacientov, bol medián sledovania 1,0 roka; 3,2 rokov pre štúdie so psoriázou, 1,0 roka pre štúdie so psoriatickou artritídou a 0,6 roka pre štúdie s Crohnovou chorobou. Malignity s výnimkou nemelanómovej rakoviny kože boli hlásené u 58 pacientov z 10 935 pacientorokov sledovania (incidencia 0,53 na 100 pacientorokov sledovania u pacientov liečených ustekinumabom). Výskyt malignít hlásených u pacientov liečených ustekinumabom bol porovnateľný s výskytom predpokladaným v bežnej populácii (štandardizovaná miera incidencie = 0,87 [95 % interval spoľahlivosti: 0,66; 1,14], upravené podľa veku, pohlavia a rasy). Najčastejšie pozorované malignity, iné ako nemelanómová rakovina kože, boli rakovina prostaty, melanóm, rakovina kolorekta a prsníka. Incidencia nemelanómovej rakoviny kože bola 0,49 na 100 pacientorokov sledovania u pacientov liečených ustekinumabom (53 pacientov z 10 919 pacientorokov sledovania). Pomer pacientov
s bazocelulárnym verzus skvamocelulárnym karcinómom kože (4:1) je porovnateľný s pomerom predpokladaným vo všeobecnej populácii (pozri časť 4.4).
Reakcie z precitlivenostiV priebehu kontrolovaných období klinických štúdií psoriázy a psoriatickej artritídy s ustekinumabom sa pozorovali vyrážky a žihľavka, pričom každá z týchto reakcií sa vyskytla u < 1 % pacientov (pozri časť 4.4).
ImunogenicitaV klinických štúdiách psoriázou a psoriatickou artritídou sa u menej ako 8 % pacientov liečených ustekinumabom vyvinuli protilátky proti ustekinumabu. V klinických štúdiách s Crohnovou chorobou sa u menej ako 3 % pacientov liečených ustekinumabom vyvinuli protilátky proti ustekinumabu. Nepozorovala sa nijaká zjavná súvislosť medzi vznikom protilátok na ustekinumab a rozvojom reakcií v mieste vpichu injekcie. Väčšina pacientov, ktorí boli pozitívni na protilátky proti ustekinumabu,
mala neutralizujúce protilátky. Účinnosť mala klesajúcu tendenciu u pacientov s pozitívnym testom na protilátky voči ustekinumabu, avšak pozitivita na protilátky nevylúčila klinickú odpoveď.
Pediatrická populáciaNežiaduce účinky u pediatrických pacientov vo veku 12 rokov a starších s ložiskovou psoriázou
Bezpečnosť ustekinumabu bola sledovaná v štúdii fázy III u 110 pacientov od 12 do 17 rokov počas
60 týždňov. Nežiaduce udalosti hlásené v tejto štúdii boli podobné ako udalosti pozorované v predchádzajúcich štúdiách u dospelých s ložiskovou psoriázou.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách sa podávali intravenózne jednotlivé dávky v množstve do 6 mg/kg bez dávkového obmedzenia vzhľadom na toxicitu. V prípade predávkovania sa odporúča u pacienta
sledovať akékoľvek známky alebo symptómy nežiaducich účinkov a bezodkladne začať náležitú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresíva, inhibítory interleukínov, ATC kód: L04AC05. Mechanizmus účinku
Ustekinumab je plne humánna monoklonová protilátka IgG1κ, ktorá sa viaže s špecificitou na
spoločnú p40 podjednotku proteínu humánnych cytokínov interleukín (IL)-12 a IL-23. Ustekinumab inhibuje bioaktivitu humánnych IL-12 a IL-23 tak, že zabraňuje p40 naviazať sa na receptor proteínu IL-12Rb1 s expresiou na povrchu imunitných buniek. Ustekinumab sa nemôže viazať na IL-12 alebo IL-23, ktoré sa už naviazali na bunkové povrchové receptory IL-12Rb1. Je preto nepravdepodobné, že by ustekinumab mohol prispievať ku komplementácii alebo cytotoxicite buniek s receptormi IL-12 a/alebo IL-23. IL-12 a IL-23 sú heterodimerické cytokíny vylučované bunkami aktivovanými antigénmi, ako sú makrofágy a dendritické bunky, a oba cytokíny sa podieľajú na imunitnej funkcii;
IL-12 aktivuje „prirodzených zabíjačov“ (NK, z angl. natural killer) bunky a vedie k diferenciácii CD4+ T buniek smerom k fenotypu Th1 (T helper 1), IL-23 aktivuje dráhu Th17 (T helper 17). Neprimerané regulovanie IL 12 a IL 23 sa však spájalo s imunitou sprostredkovanými ochoreniami, ako napríklad psoriáza, psoriatická artritída a Crohnova choroba.
Naviazaním na spoločnú p40 podjednotku IL-12 a IL-23 môže ustekinumab vyvíjať svoj klinický účinok na psoriázu, psoriatickú artritídu a Crohnovu chorobu prostredníctvom prerušenia cytokínových dráh Th1 a Th17, ktoré sú dôležité pre patológiu týchto ochorení.
U pacientov s Crohnovou chorobou viedla liečba ustekinumabom k zníženiu hodnôt zápalových markerov vrátane C-reaktívneho proteínu (CRP) a fekálneho kalprotektínu počas indukčnej fázy. Toto zníženie sa počas udržiavacej fázy zachovalo.
Imunizácia
V priebehu dlhodobého predĺženia druhej štúdie psoriázy (PHOENIX 2) došlo u dospelých pacientov liečených STELAROU najmenej 3,5 roka k podobným protilátkovým odpovediam na obe, pneumokokovú polysacharidovú aj tetanovú vakcínu, ako u sledovanej skupiny s nesystematicky liečenou psoriázou. Podobné podiely dospelých pacientov rozvinuli ochranné hladiny anti- pneumokokových a anti-tetanových protilátok a titre protilátok boli u pacientov liečených STELAROU a u sledovaných pacientov podobné.
Klinická účinnosť
Ložisková psoriáza (dospelí)
Bezpečnosť a účinnosť ustekinumabu sa hodnotila u 1 996 pacientov v dvoch randomizovaných dvojito zaslepených placebom kontrolovaných klinických štúdiách u pacientov so stredne závažnou až závažnou ložiskovou psoriázou, ktorí boli kandidátmi na fototerapiu alebo systémovú terapiu. Ďalšia randomizovaná, zaslepená, aktívne kontrolovaná štúdia porovnávala ustekinumab a etanercept
u pacientov so stredne závažnou až závažnou ložiskovou psoriázou, ktorí nedostatočne odpovedali, netolerovali alebo im bola kontraindikovaná liečba cyklosporínom, MTX alebo PUVA.
Prvá štúdia psoriázy (PHOENIX 1) hodnotila 766 pacientov. 53 % z nich neodpovedalo na liečbu, netolerovali ju alebo mali kontraindikovanú inú systémovú terapiu. Pacienti randomizovaní do skupiny s ustekinumabom dostávali dávky 45 mg alebo 90 mg v 0. týždni a v 4. týždni, v liečbe sa potom pokračovalo v tej istej dávke každých 12 týždňov. Pacienti randomizovaní do skupiny
s placebom v 0. týždni a 4. týždni boli v skríženej fáze preradení na ustekinumab (dávka buď 45 mg alebo 90 mg) v 12. týždni a 16. týždni, po čom nasledovalo podávanie lieku každých 12 týždňov.
Pacienti pôvodne randomizovaní na ustekinumab, ktorí dosiahli 75 %-ný index klinickej odpovede podľa Psoriasis Area and Severity Index 75 (zlepšenie PASI najmenej o 75 % oproti východiskovej hodnote), v 28. aj 40. týždni boli podľa opakovanej randomizácie vybraní na ustekinumab každých 12 týždňov alebo placebo (t. j. liečba bola vysadená). Pacienti, ktorí boli pri ďalšej randomizácii určení
na liečbu placebom, v 40. týždni znova začali dostávať ustekinumab v pôvodnom dávkovacom režime, ak sa u nich v 40. týždni zistila najmenej 50 %-ná strata zlepšenia PASI. Všetci pacienti boli
sledovaní v priebehu 76 týždňov od prvého podania skúmanej liečby.
V druhej štúdii psoriázy (PHOENIX 2) sa hodnotilo 1 230 pacientov. 61 % z nich buď neodpovedalo na liečbu, netolerovali ju alebo mali kontraindikovanú inú systémovú terapiu. Pacienti randomizovaní do skupiny s ustekinumabom dostávali dávky 45 mg alebo 90 mg v 0. týždni a v 4. týždni, po čom nasledovala ďalšia dávka v 16. týždni. Pacienti randomizovaní do skupiny s placebom v 0. týždni
a 4. týždni boli v skríženej fáze preradení na ustekinumab (buď 45 mg alebo 90 mg) v 12. týždni
a 16. týždni. Všetci pacienti boli sledovaní v priebehu 52 týždňov od prvého podania skúmanej liečby.
3. štúdia psoriázy (ACCEPT) hodnotila 903 pacientov so stredne závažnou až závažnou psoriázou, ktorí nedostatočne odpovedali, netolerovali alebo im bola kontraindikovaná iná systémová liečba, porovnala účinnosť ustekinumabu a etanerceptu a vyhodnotila bezpečnosť ustekinumabu a etanerceptu. Počas 12-týdňovej aktívne kontrolovanej časti štúdie boli pacienti randomizovaní na etanercept (50 mg dvakrát týždenne), ustekinumab 45 mg v 0. a 4. týždni, alebo ustekinumab 90 mg v 0. a 4. týždni.
Východisková charakteristika ochorenia bola vo všeobecnosti konzistentná vo všetkých liečených skupinách v 1. a 2. štúdii psoriázy s mediánom východiskového skóre PASI od 17 do 18, východiskovým mediánom plochy telesného povrchu (BSA, z angl. Body Surface Area) ≥ 20 a indexom mediánu Dermatology Life Quality Index (DLQI, dermatologický index kvality života)
v rozsahu od 10 do 12. Približne jedna tretina (v 1. štúdii psoriázy) a jedna štvrtina (v 2. štúdii psoriázy) subjektov mala psoriatickú artritídu (PsA). Podobná závažnosť ochorenia sa pozorovala aj v 3. štúdii psoriázy.
Primárne kritérium v týchto štúdiách bol podiel pacientov, u ktorých sa dosiahla odpoveď PASI 75
v 12. týždni od východiskového stavu (pozri tabuľky č. 4 a 5).
Tabuľka č. 4 Súhrn klinických odpovedí v 1. štúdii psoriázy (PHOENIX 1) a v 2. štúdii psoriázy
(PHOENIX 2)
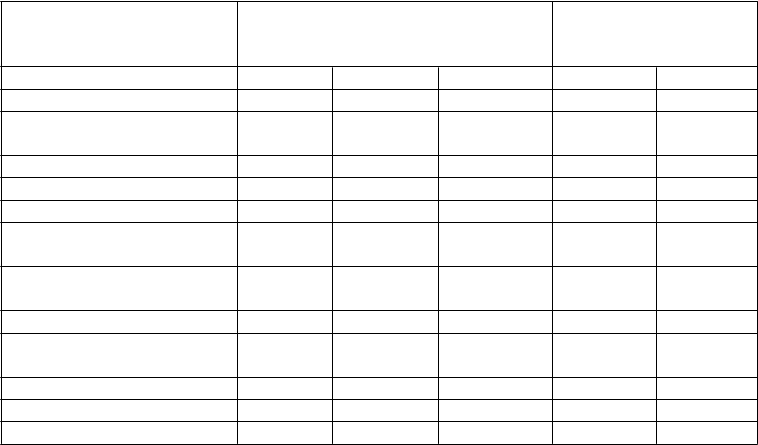
12. týždeň
2 dávky (0. týždeň a 4. týždeň)
28. týždeň
3 dávky (0. týždeň,
4. týždeň a 16. týždeň)
1. štúdia psoriázy
Počet randomizovaných
PBO 45 mg 90 mg 45 mg 90 mg
pacientov 255 255 256 250 243
Odpoveď PASI 50 N (%) 26 (10 %) 213 (84 %)a 220 (86 %)a 228 (91 %) 234 (96 %) Odpoveď PASI 75 N (%) 8 (3 %) 171 (67 %)a 170 (66 %)a 178 (71 %) 191 (79 %) Odpoveď PASI 90 N (%) 5 (2 %) 106 (42 %)a 94 (37 %)a 123 (49 %) 135 (56 %)
PGAb vyčisteného alebo a
minimálneho N (%) 10 (4 %) 151 (59 %)
Počet pacientov
156 (61 %)a
146 (58 %) 160 (66 %)
s hmotnosťou ≤ 100 kg 166 168 164 164 153
Odpoveď PASI 75 N (%) 6 (4 %) 124 (74 %) 107 (65 %) 130 (79 %) 124 (81 %) Počet pacientov
s hmotnosťou > 100 kg 89 87 92 86 90
Odpoveď PASI 75 N (%) 2 (2 %) 47 (54 %) 63 (68 %) 48 (56 %) 67 (74 %)
2. štúdia psoriázy
Počet randomizovaných
pacientov 410 409 411 397 400
Odpoveď PASI 50 N (%) 41 (10 %) 342 (84 %)a 367 (89 %)a 369 (93 %) 380 (95 %) Odpoveď PASI 75 N (%) 15 (4 %) 273 (67 %)a 311 (76 %)a 276 (70 %) 314 (79 %) Odpoveď PASI 90 N (%) 3 (1 %) 173 (42 %)a 209 (51 %)a 178 (45 %) 217 (54 %)
PGAb vyčisteného alebo a
minimálneho N (%) 18 (4 %) 277 (68 %)
Počet pacientov
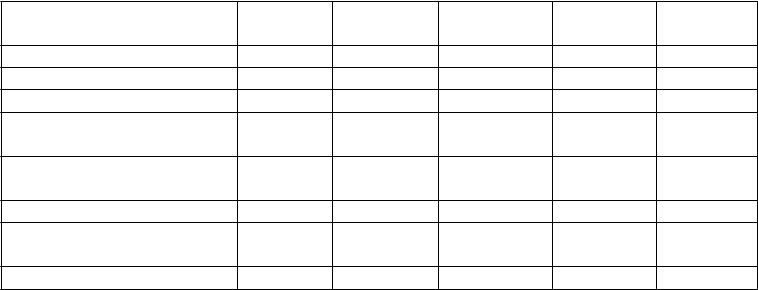
300 (73 %)a
241 (61 %) 279 (70 %)
s hmotnosťou ≤ 100 kg 290 297 289 287 280
Odpoveď PASI 75 N (%) 12 (4 %) 218 (73 %) 225 (78 %) 217 (76 %) 226 (81 %) Počet pacientov
s hmotnosťou > 100 kg 120 112 121 110 119
Odpoveď PASI 75 N (%) 3 (3 %) 55 (49 %) 86 (71 %) 59 (54 %) 88 (74 %)
a p < 0,001 pre 45 mg alebo 90 mg ustekinumabu v porovnaní s placebom (PBO).
b PGA = celkové hodnotenie lekára (z angl. Physician Global Assessment)
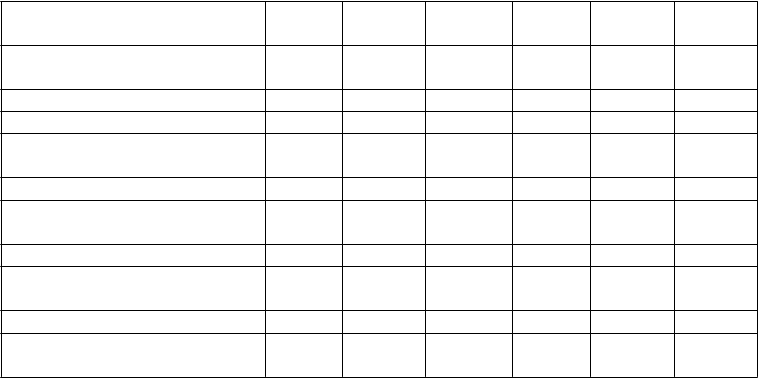
Tabuľka č. 5 Súhrn klinických odpovedí v 12. týždni v 3. štúdii psoriázy (ACCEPT)
3. štúdia psoriázy
Počet randomizovaných
Etanercept
24 dávok
(50 mg dvakrát týždenne)
Ustekinumab
2 dávky (0. týždeň a 4. týždeň)
45 mg 90 mg
pacientov 347 209 347
Odpoveď PASI 50 N (%) 286 (82 %) 181 (87 %) 320 (92 %)a Odpoveď PASI 75 N (%) 197 (57 %) 141 (67 %)b 256 (74 %)a Odpoveď PASI 90 N (%) 80 (23 %) 76 (36 %)a 155 (45 %)a
PGA vyčisteného alebo a
minimálneho N (%) 170 (49 %) 136 (65 %)
Počet pacientov s hmotnosťou
245 (71 %)a
≤ 100 kg 251 151 244
Odpoveď PASI 75 N (%) 154 (61 %) 109 (72 %) 189 (77 %) Počet pacientov s hmotnosťou
> 100 kg 96 58 103
Odpoveď PASI 75 N (%) 43 (45 %) 32 (55 %) 67 (65 %)
a p < 0,001 pre 45 mg alebo 90 mg ustekinumabu v porovnaní s etanerceptom.
b p = 0,012 pre 45 mg ustekinumabu v porovnaní s etanerceptom.
V 1. štúdii psoriázy bolo pretrvávanie PASI 75 signifikantne lepšie pri kontinuálnej liečbe
v porovnaní s tým, keď sa liečba vysadila (p < 0,001). Podobné výsledky sa zistili pri každej dávke ustekinumabu. V 1. roku (52. týždeň) u 89 % pacientov, ktorým bola pri ďalšej randomizácii určená udržiavacia liečba, sa dostavila klinická odpoveď (respondenti) PASI 75 v porovnaní so 63 % pacientov, ktorým bolo pri ďalšej randomizácii pridelené placebo (vysadenie liečby) (p < 0,001). Po
18 mesiacoch (76. týždeň) 84 % pacientov vybraných pri ďalšej randomizácii na udržiavaciu liečbu boli respondenti PASI 75 – v porovnaní s 19 % pacientov, ktorým po opakovanej randomizácii bola pridelená liečba placebom (vysadenie liečby). Po 3 rokoch (148. týždeň), 82 % pacientov po opakovanej randomizácii na udržiavaciu liečbu, boli respondenti PASI 75. V 5. roku (244. týždeň),
80 % pacientov, ktorí boli znovu randomizovaní na udržiavaciu liečbu, boli respondenti PASI 75.
U 85 % pacientov, ktorým bolo pri opakovanej randomizácii pridelené placebo a ktorí znova nastúpili na pôvodný liečebný režim ustekinumabom po strate ≥ 50 % zlepšenia PASI, sa opäť dostavila odpoveď PASI 75 do 12 týždňov od opakovaného začatia terapie.
V 1. štúdii psoriázy, v 2. a v 12. týždni, sa v porovnaní s placebom signifikantne zlepšil DLQI
v každej skupine liečenej ustekinumabom oproti východiskovému stavu. Zlepšenie pretrvalo až do 28. týždňa. Podobné signifikantné zlepšenie sa zistilo v 2. štúdii psoriázy v 4. a 12. týždni, ktoré pretrvalo
až do 24. týždňa. V 1. štúdii psoriázy zlepšenia v psoriáze nechtov (tzv. Nail Psoriasis Severity Index), vo fyzickej a duševnej zložke sumárnych skóre SF-36 a v pocite svrbenia podľa vizuálnej analógovej škály (VAS, z angl. Visual Analogue Scale) boli takisto významné v každej skupine liečenej ustekinumabom v porovnaní s placebom. V 2. štúdii psoriázy sa škála anxiozity a depresie hospitalizovaných pacientov (HADS, z angl. Hospital Anxiety and Depression Scale) a dotazník pracovného obmedzenia (WLQ, z angl. Work Limitations Questionnaire) takisto významne zlepšili v každej skupine liečenej ustekinumabom pri porovnaní s placebom.
Psoriatická artritída (PsA) (dospelí)Preukázalo sa, že ustekinumab zlepšuje známky a príznaky, fyzickú kondíciu a kvalitu života súvisiacu so zdravím a znižuje mieru progresie poškodenia periférnych kĺbov u dospelých pacientov s aktívnou PsA.
Bezpečnosť a účinnosť ustekinumabu bola hodnotená u 927 pacientov v dvoch randomizovaných dvojito zaslepených placebom kontrolovaných štúdiách na pacientoch s aktívnou PsA
(≥ 5 opuchnutých kĺbov a ≥ 5 citlivých kĺbov) aj napriek liečbe nesteroidnými antiflogistikami (NSAID) alebo antireumatickými liekmi modifikujúcimi chorobu (DMARD). U pacientov v týchto štúdiách bola diagnostikovaná PsA aspoň 6 mesiacov. Zaradení boli pacienti s každým podtypom PsA, vrátane polyartikulárnej artritídy bez zjavných reumatoidných uzlíkov (39 %), spondylitídy
s periférnou artritídou (28 %), asymetrickou periférnou artritídou (21 %), distálneho interfalangeálneho postihnutia (12 %) a mutilujúcej artritídy (0,5 %). V oboch štúdiách malo v úvode viac ako 70 % pacientov entezitídu a viac ako 40 % pacientov daktylitídu. Pacienti boli randomizovaní na liečbu ustekinumabom 45 mg, 90 mg alebo placebo subkutánne v 0. a 4. týždni
s nasledujúcim dávkovaním každých 12 týždňov (q12w). Približne 50 % pacientov pokračovalo na ustálených dávkach MTX (≤ 25 mg/týždeň).
V 1. štúdii PsA (PSUMMIT I) a v 2. štúdii PsA (PSUMMIT II) bolo 80 % resp. 86 % pacientov predtým liečených DMARD. V 1. štúdii nebola povolená predchádzajúca liečba tumor nekrotizujúcim faktorom-alfa (TNF)α. V 2. štúdii bola väčšina pacientov (58 %, n = 180) predtým liečená jednou alebo viacerými anti-TNFα látkami, z ktorých viac ako 70 % ukončilo liečbu anti-TNFα z dôvodu nedostatočnej účinnosti alebo intolerancie v ktoromkoľvek čase.
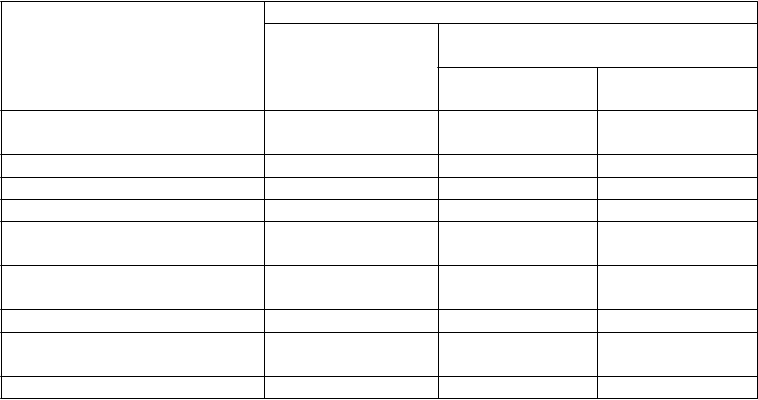
Prejavy a príznakyV 24. týždni mala liečba ustekinumabom za následok významné zlepšenie miery aktivity ochorenia v porovnaní s placebom. Primárny cieľ bol podiel pacientov, ktorí dosiahli v 24. týždni odpoveď podľa ACR (z angl. American College of Rheumatology) 20. Hlavné výsledky účinnosti sú uvedené nižšie v tabuľke č. 6.
 Tabuľka č. 6 Počet pacientov, ktorí dosiahli klinickú odpoveď v 24. týždni 1. (PSUMMIT I) a 2. (PSUMMIT II) štúdie psoriatickej artritídy
Tabuľka č. 6 Počet pacientov, ktorí dosiahli klinickú odpoveď v 24. týždni 1. (PSUMMIT I) a 2. (PSUMMIT II) štúdie psoriatickej artritídy
1. štúdia psoriatickej artritídy
2. štúdia psoriatickej artritídy
Počet randomizovaných
PB
O 45 mg 90 mg PBO 45 mg 90 mg
pacientov
206 205 204 104 103 105
Odpoveď ACR 20, N (%) 47 (23 %)
87 (42 %)a
101 (50 %)a
21 (20 %)
45 (44 %)a
46 (44 %)a
Odpoveď ACR 50, N (%) 18
(9 %)
51 (25 %)a
57 (28 %)a
7 (7 %) 18 (17 %)b
24 (23 %)a
Odpoveď ACR 70, N (%) 5 (2 %) 25 (12 %)a
29 (14 %)a
3 (3 %) 7 (7 %)c 9 (9 %)c
Počet pacientov s ≥ 3 % BSA
d 146 145 149 80 80 81
Odpoveď PASI 75, N (%) 16 (11 %)
83 (57 %)a
93 (62 %)a
4 (5 %) 41 (51 %)a
45 (56 %)a
Odpoveď PASI 90, N (%) 4 (3 %) 60 (41 %)a
65 (44 %)a
3 (4 %) 24 (30 %)a
36 (44 %)a
Kombinovaná odpoveď
PASI 75 a ACR 20, N (%) 8 (5 %)
40 (28 %)a
62
(42 %)a 2 (3 %)
24 (30 %)a
31 (38 %)a
Počet pacientov ≤ 100 kg 154 153 154 74 74 73
Odpoveď ACR 20, N (%) 39 (25 %)
67 (44 %)
78 (51 %)
17 (23 %)
32 (43 %)
34 (47 %)
Počet pacientov s ≥ 3 % BSA
d 105 105 111 54 58 57
Odpoveď PASI 75, N (%) 14 (13 %)
64 (61 %)
73 (66 %)
4 (7 %) 31 (53 %)
32 (56 %)
Počet pacientov > 100 kg 52 52 50 30 29 31
Odpoveď ACR 20, N (%) 8
(15 %)
20 (38 %)
23 (46 %)
4 (13 %)
13 (45 %)
12 (39 %)
Počet pacientov s ≥ 3 % BSA
d 41 40 38 26 22 24
Odpoveď PASI 75, N (%) 2 (5 %) 19 (48 %)
20 (53 %)
0 10 (45 %)
13 (54 %)
a p < 0,001
b p < 0,05
c p = NS
d Počet pacientov s kožou postihnutou psoriázou na ≥ 3 % povrchu tela v úvode liečby.
Odpovede ACR 20, 50 a 70 sa naďalej zlepšovali alebo boli udržané do 52. týždňa (1. a 2. štúdia PsA)'
a 100. týždňa (1. štúdia PsA). V 1. štúdii PsA dosiahlo v 100. týždni odpoveď ACR 20 57 % so 45 mg a 64 % s 90 mg. V 2. štúdii PsA dosiahlo v 52. týždni odpoveď ACR 20 47 % so 45 mg a 48 %
s 90 mg.
V 24. týždni bol podiel pacientov dosahujúcich zmenenú odpoveď podľa PsARC (z angl. PsA response criteria) tiež výrazne vyšší v skupine s ustekinumabom v porovnaní s placebom. Odpovede podľa PsARC boli udržané do 52. a 100. týždňa. V 24. týždni vykazoval vyšší podiel pacientov liečených ustekinumabom, ktorí mali spondylitídu s periférnou artritídou pri prvej návšteve, 50 a 70 percentné zlepšenie skóre BASDAI (z angl. Bath Ankylosing Spondylitis Disease Activity Index)
v porovnaní s placebom.
Odpovede pozorované v skupinách liečených ustekinumabom boli podobné u pacientov súbežne dostávajúcich a nedostávajúcich MTX a boli udržané do 52. a 100. týždňa. Pacienti predtým liečení s anti-TNFα látkami, ktorí dostávali ustekinumab, dosiahli v 24. týždni vyššiu odpoveď ako pacienti dostávajúci placebo (odpoveď ACR 20 v 24. týždni pre 45 mg a 90 mg bola 37 % resp. 34 %,
v porovnaní s placebom 15 %; p < 0,05) a odpovede boli udržané do 52. týždňa.
V 24. týždni 1. štúdie PsA bolo u pacientov s entezitídou a/alebo daktylitídou v úvode liečby pozorované výrazné zlepšenie skóre entezitídy a daktylitídy v skupinách s ustekinumabom
v porovnaní s placebom, V 2. štúdii PsA bolo v 24. týždni výrazné zlepšenie skóre entezitídy a numerické zlepšenie (nie štatisticky významné) v skóre daktylitídy pozorované v skupine
s ustekinumabom 90 mg v porovnaní s placebom. Zlepšenia v skóre entezitídy a daktylitídy boli udržané do 52. a 100. týždňa.
Rádiografická odpoveď
Štrukturálne poškodenie na rukách aj nohách bolo vyjadrené ako zmena v celkovom skóre podľa Sharpa modifikované van der Heijdemovou (vdH-S skóre), upravené pre PsA pridaním distálnych medzičlánkových kĺbov ruky, oproti východiskovému stavu. Bola vykonaná vopred určená zjednotená analýza kombinujúca údaje od 927 pacientov z 1. a 2. štúdie PsA. Ustekinumab preukázal štatisticky významný pokles v miere progresie štrukturálneho poškodenia v porovnaní s placebom, keď sa meral zmenou celkového upraveného vdH-S skóre od východiskového stavu do 24. týždňa (stredná hodnota
± SD skóre bolo 0,97 ± 3,85 v skupine s placebom v porovnaní s 0,4 ± 2,11 v skupine
s ustekinumabom 45 mg (p < 0,05) a 0,39 ± 2,40 v skupine s ustekinumabom 90 mg (p < 0,001). Tento účinok bol podporený 1. štúdiou PsA. Tento účinok sa považuje za preukázaný bez ohľadu na súbežné použitie MTX a bol udržaný do 52. (zjednotená analýza) a 100. týždňa (1. štúdia PsA).
Fyzická kondícia a kvalita života súvisiaca so zdravím
Pacienti liečení ustekinumabom vykazovali v 24. týždni výrazné zlepšenie fyzickej kondície podľa skóre HAQ-DI (z angl. Disability Index of the Health Assessment Questionnaire). Podiel pacientov dosahujúcich klinicky významné ≥ 0,3 zlepšenie skóre HAQ-DI oproti východiskovej hodnote bol výrazne vyšší v skupinách s ustekinumabom v porovnaní s placebom. Zlepšenie skóre HAQ-DI oproti východiskovej hodnote bolo udržané do 52. a 100. týždňa.
V 24. týždni sa zaznamenalo výrazné zlepšenie skóre DLQI v skupinách s ustekinumabom
v porovnaní splacebom, ktoré bolo udržané do 52. a 100. týždňa. V 24. týždni bolo v 2. štúdii PsA zaznamenané výrazné zlepšenie skóre FACIT-F (z angl. Functional Assessment of Chronic Illness Therapy – Fatigue) v skupinách s ustekinumabom v porovnaní s placebom. Podiel pacientov dosahujúcich klinicky výrazné zlepšenie únavy (4 body v FACIT-F) bol tiež výrazne vyšší
v skupinách s ustekinumabom v porovnaní s placebom. Zlepšenie skóre FACIT bolo udržané do 52. týždňa.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre ustekinumab
v jednej alebo viacerých podskupinách pediatrickej populácie vo veku 6 až 11 rokov v stredne závažnej až závažnej ložiskovej forme psoriázy a v juvenilnej idiopatickej artritíde (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Ložisková psoriáza u pediatrických pacientov
Preukázalo sa, že ustekinumab zlepšuje prejavy a príznaky a kvalitu života súvisiacu so zdravím u pediatrických pacientov vo veku 12 rokov a starších s ložiskovou psoriázou.
Účinnosť ustekinumabu sa hodnotila u 110 pediatrických pacientov vo veku 12 až 17 rokov so stredne závažnou až závažnou ložiskovou psoriázou v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii fázy III (CADMUS). Pacienti boli randomizovaní buď na placebo
(n = 37) alebo na odporúčanú dávku ustekinumabu (pozri časť 4.2; n = 36) alebo na polovicu odporúčanej dávky ustekinumabu (n = 37) v subkutánnej injekcii v 0. a 4. týždni, po čom nasledovalo podávanie lieku každých 12 týždňov (q12w). V 12. týždni boli pacienti dostávajúci placebo preradení na ustekinumab.
Do tejto štúdie boli zaradení pacienti s PASI ≥ 12, PGA ≥ 3 a postihnutím BSA aspoň 10 %, ktorí boli kandidátmi na systémovú terapiu alebo fototerapiu. Približne 60 % pacientov bolo predtým liečených konvenčnou systémovou terapiou alebo fototerapiou. Približne 11 % pacientov bolo predtým
liečených biologickými látkami.
Primárny koncový ukazovateľ bol podiel pacientov, ktorí v 12. týždni dosiahli PGA čisté (0) alebo minimálne (1). Sekundárne koncové ukazovatele zahŕňali PASI 75, PASI 90, zmenu oproti východiskovej hodnote CDLQI (Children’s Dermatology Life Quality Index), zmenu oproti východiskovej hodnote celkového skóre PedsQL (Paediatric Quality of Life Inventory) škály v 12. týždni. V 12. týždni dosiahli jedinci liečení ustekinumabom významne väčšie zlepšenie psoriázy
a kvality života súvisiacej so zdravím v porovnaní s placebom (Tabuľka 7).
U všetkých pacientov bola sledovaná účinnosť v priebehu 52 týždňov od prvého podania skúšaného lieku. Podiel pacientov s PGA čisté (0) alebo minimálne (1) a s PASI 75 preukázal rozdiel medzi skupinou liečenou ustekinumabom a placebom pri prvej návšteve po úvode liečby, t.j. v 4. týždni, pričom dosiahol vrchol v 12. týždni. Zlepšenia PGA, PASI, CDLQI a PedsQL sa zachovali do 52. týždňa (Tabuľka 7).
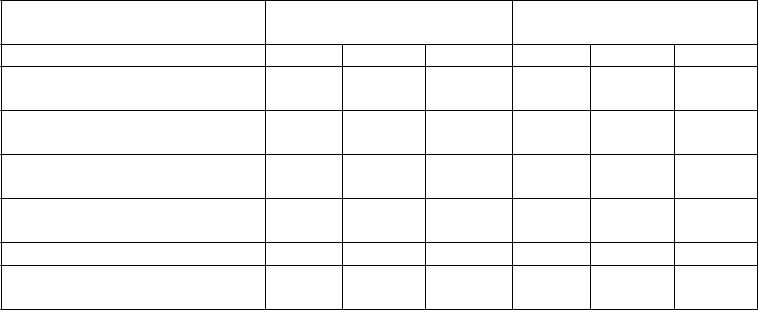
Tabuľka 7: Súhrn primárnych a sekundárnych koncových ukazovateľov v 12. a 52. týždni
Štúdia psoriázy u pediatrických pacientov (CADMUS)
12. týždeň 52. týždeň
Placebo Odporúčaná dávka ustekinumabu
Odporúčaná dávka ustekinumabu
N (%) N (%) N (%) Randomizovaní pacienti 37 36 35
PGA
PGA čisté (0) alebo a
minimálne (1) 2 (5,4 %) 25 (69,4 %)
20 (57,1 %)
PGA čisté (0) 1 (2,7 %) 17 (47,2 %)a 13 (37,1 %)
 PASI
PASIPASI 75 4 (10,8 %) 29 (80,6 %)a 28 (80,0 %) PASI 90 2 (5,4 %) 22 (61,1 %)a 23 (65,7 %) PASI 100 1 (2,7 %) 14 (38,9 %)a 13 (37,1 %)
CDLQICDLQI 0 alebo 1b 6 (16,2 %) 18 (50,0 %)c 20 (57,1 %)
PedsQLzmena oproti východiskovej
hodnote Priemer (SD)d a p < 0,001
3,35 (10,04) 8,03 (10,44)e 7,26 (10,92)
b CDLQI: CDLQI je dermatologický nástroj na posúdenie vplyvu kožných problémov na kvalitu života súvisiacu so zdravím v pediatrickej populácii. CDLQI s hodnotou 0 alebo 1 znamená žiadny vplyv na kvalitu života dieťaťa.
c p = 0,002
d PedsQL: Celkové skóre PedsQL škály je všeobecné meradlo kvality života súvisiacej so zdravím, ktoré bolo vyvinuté pre použitie v detskej a adolescentnej populácii. Pre skupinu s placebom v 12. týždni, N = 36.
e p = 0,028
Počas placebom kontrolovaného obdobia do 12. týždňa bola účinnosť odporúčanej dávky aj polovice
odporúčanej dávky všeobecne porovnateľná pri primárnom koncovom ukazovateli (69,4 % a 67,6 % v tomto poradí), existoval však dôkaz o odpovedi na dávku pri kritériách účinnosti vyššieho stupňa (napr. PGA čisté (0), PASI 90). Po 12. týždni bola účinnosť všeobecne vyššia a lepšie sa udržala
v skupine s odporúčanou dávkou v porovnaní so skupinou s polovicou odporúčanej dávky, v ktorej sa častejšie pozorovalo mierne zníženie účinnosti ku koncu každého 12-týždňového dávkovacieho intervalu. Profily bezpečnosti odporúčanej dávky a polovice odporúčanej dávky boli porovnateľné.
Crohnova choroba
Bezpečnosť a účinnosť ustekinumabu sa hodnotila v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných multicentrických štúdiách u dospelých pacientov so stredným až ťažkým stupňom aktivity Crohnovej choroby (skóre CDAI [Crohn’s Disease Activity Index] ≥ 220 a ≤ 450). Klinický vývoj predstavovali dve 8-týždňové indukčné štúdie s intravenóznym podávaním (UNITI-1 a UNITI-2), po ktorých nasledovala 44 týždňov trvajúca randomizovaná štúdia so subkutánnym podávaním (IM-UNITI), sledujúca udržanie účinku u pacientov, ktorí dosiahli klinickú odpoveď v indukčných štúdiách, čo celkovo predstavovalo 52 týždňov liečby.
Indukčné štúdie zahŕňali 1 409 (UNITI-1, n = 769; UNITI-2 n = 640) pacientov. Primárny koncový ukazovateľ v oboch indukčných štúdiách bol podiel jedincov s klinickou odpoveďou (definovanou
ako zníženie skóre CDAI o ≥ 100 bodov) v 6. týždni. V oboch štúdiách boli údaje o účinnosti zbierané a analyzované až do 8. týždňa. Súbežné dávky perorálnych kortikosteroidov,
imunomodulátorov, aminosalycilátov a antibiotík boli povolené a 75 % pacientov naďalej dostávalo najmenej jeden z týchto liekov. V oboch štúdiách boli pacienti randomizovaní na jednorazové intravenózne podanie buď odporúčanej odstupňovanej dávky približne 6 mg/kg (pozri časť 4.2 v SPC STELARA 130 mg koncentrát na infúzny roztok), fixnú dávku 130 mg ustekinumabu alebo placebo
v 0. týždni.
Pacienti v UNITI-1 na predchádzajúcej anti-TNFα terapii zlyhali alebo ju netolerovali. Približne 48 % pacientov zlyhalo na 1 predchádzajúcej anti-TNFa terapii a 52 % zlyhalo na 2 alebo 3 predchádzajúcich anti-TNFα terapiách. V tejto štúdii 29,1 % pacientov nedosiahlo dostačujúcu
úvodnú odpoveď (primárni non-respondéri), 69,4 % odpovedalo, ale odpoveď neudržalo (sekundárni non-respondéri) a 36,4 % netolerovalo anti-TNFα terapie.
Pacienti v UNITI-2 zlyhali aspoň na jednej konvenčnej terapii, vrátane kortikosteroidov alebo imunomodulátorov, a buď predtým nedostali anti-TNF-α terapiu (68,6 %) alebo anti-TNFα terapiu predtým dostali, ale na nej nezlyhali (31,4 %).
V oboch štúdiách UNITI-1 a UNITI-2 bol významne vyšší podiel pacientov s klinickou odpoveďou
a remisiou v skupine liečenej ustekinumabom v porovnaní s placebom (Tabuľka 3). Klinická odpoveď a remisia boli významné už od 3. týždňa u pacientov liečených ustekinumabom a ďalej sa zlepšovala do 8. týždňa. V týchto indukčných štúdiách bola účinnosť vyššia a lepšie udržateľná v skupine
s odstupňovanou dávkou v porovnaní so skupinou so 130 mg dávkou, a preto sa odstupňované dávkovanie odporúča pre intravenóznu indukčnú dávku.
Tabuľka 8: Indukcia klinickej odpovede a remisie v UNITI-1 a UNITI 2
UNITI-1* UNITI-2**
Placebo
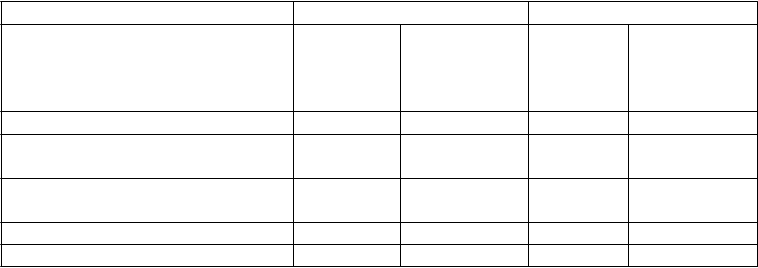 N = 247
Odporúčaná dávka ustekinumabu N = 249
Placebo
N = 209
Odporúčaná dávka ustekinumabu N = 209
N = 247
Odporúčaná dávka ustekinumabu N = 249
Placebo
N = 209
Odporúčaná dávka ustekinumabu N = 209
Klinická remisia, 8. týždeň 18 (7,3 %) 52 (20,9 %)a 41 (19,6 %) 84 (40,2 %)a
Klinická odpoveď (100 bodov),
6. týždeň
Klinická odpoveď (100 bodov),
8. týždeň
53 (21,5 %) 84 (33,7 %)b 60 (28,7 %) 116 (55,5 %)a
50 (20,2 %) 94 (37,8 %)a 67 (32,1 %) 121 (57,9 %)a
Odpoveď 70 bodov, 3. týždeň 67 (27,1 %) 101 (40,6 %)b 66 (31,6 %) 106 (50,7 %)a
Odpoveď 70 bodov, 6. týždeň 75 (30,4 %) 109 (43,8 %)b 81 (38,8 %) 135 (64,6 %)a
Klinická remisia je definovaná ako skóre CDAI < 150; klinická odpoveď je definovaná ako zníženie skóre CDAI minimálne o 100 bodov alebo ako stav klinickej remisie
Odpoveď 70 bodov je definovaná ako zníženie skóre CDAI minimálne o 70 bodov
* Zlyhania na anti-TNFα
** Zlyhania na konvenčných terapiách
a p < 0,001
b p < 0,01
Udržiavacia štúdia (IM-UNITI) hodnotila 388 pacientov, ktorí dosiahli klinickú odpoveď 100 bodov
v 8. týždni indukcie s ustekinumabom v štúdiách UNITI-1 a UNITI-2. Pacienti boli randomizovaní na subkutánny udržiavací režim buď 90 mg ustekinumabu každých 8 týždňov, 90 mg ustekinumabu každých 12 týždňov alebo placebo po dobu 44 týždňov (odporúčané udržiavacie dávkovanie, pozri časť 4.2).
Významne vyššie podiely pacientov udržali klinickú remisiu a odpoveď v skupine liečenej ustekinumabom v porovnaní so skupinou s placebom v 44. týždni (pozri Tabuľku 9).
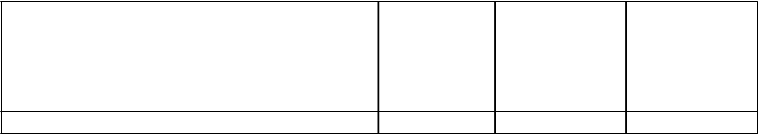 Tabuľka 9: Udržanie klinickej odpovede a remisie v IM-UNITI (44. týždeň; 52 týždňov od podania indukčnej dávky)
Tabuľka 9: Udržanie klinickej odpovede a remisie v IM-UNITI (44. týždeň; 52 týždňov od podania indukčnej dávky)
Placebo*
N = 131
†
90 mg ustekinumabu každých
8 týždňov
N = 128
†
90 mg ustekinumabu každých
12 týždňov
N = 129
†
Klinická remisia 36 % 53 %a 49 %b
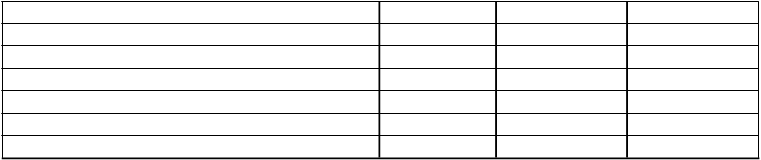
Klinická odpoveď 44 % 59 %b 58 %b Klinická remisia bez kortikosteroidov 30 % 47 %a 43 %c Klinická remisia u pacientov:
v remisii na začiatku udržiavacej liečby 46 % (36/79) 67 % (52/78)a 56 % (44/78) ktorí boli zaradení zo štúdie CRD3002‡ 44 % (31/70) 63 % (45/72)c 57 % (41/72) ktorí neboli doteraz liečení anti-TNFα 49 % (25/51) 65 % (34/52)c 57 % (30/53) ktorí pristúpili zo štúdie CRD3001§ 26 % (16/61) 41 % (23/56) 39 % (22/57)
Klinická remisia je definovaná ako skóre CDAI < 150; klinická odpoveď je definovaná ako zníženie CDAI minimálne o 100 bodov alebo ako stav klinickej remisie
* Skupina s placebom sa skladala z pacientov, ktorí odpovedali na ustekinumab a boli randomizovaní na placebo na začiatku udržiavacej liečby.
† Pacienti so 100 bodovou klinickou odpoveďou na ustekinumab na začiatku udržiavacej liečby
‡ Pacienti, ktorí zlyhali na konvenčnej terapii, ale nie na anti-TNFα terapii
§ Pacienti, ktorí sú anti-TNFα refraktórni/intolerantní
a p < 0,01
b p < 0,05
c nominálne významné (p < 0,05)
V IM-UNITI, 29 zo 129 pacientov si neudržali odpoveď na ustekinumab, keď boli liečení každých 12
týždňov a mali povolenú úpravu dávky tak, aby dostávali ustekinumab každých 8 týždňov. Strata odpovede bola definovaná ako skóre CDAI ≥ 220 bodov a zvýšenie o 100 bodov oproti skóre CDAI pri vstupe do štúdie. U týchto pacientov bola klinická remisia dosiahnutá v prípade 41,4 % pacientov
16 týždňov po úprave dávky.
Pacienti, ktorí nezaznamenali klinickú odpoveď na indukciu ustekinumabu v 8. týždni indukčných štúdií UNITI-1 a UNITI-2 (476 pacientov), boli zaradení do nerandomizovanej časti udržiavacej štúdie (IM-UNITI) a dostávali celý čas 90 mg subkutánnu injekciu ustekinumabu. O osem týždňov neskôr dosiahlo 50,5 % pacientov klinickú odpoveď a pokračovalo v užívaní udržiavacieho dávkovania každých 8 týždňov; spomedzi týchto pacientov s pokračujúcim udržiavacím dávkovaním si väčšina udržala odpoveď (68,1 %) a dosiahla remisiu (50,2 %) v 44. týždni, t.j. v podieloch, ktoré boli podobné ako u pacientov, ktorí pôvodne odpovedali na indukciu ustekinumabu.
Zo 131 pacientov, ktorí odpovedali na indukciu ustekinumabu a boli randomizovaní do skupiny s placebom na začiatku udržiavacej štúdie, 51 následne stratilo odpoveď a dostávalo 90 mg ustekinumabu subkutánne každých 8 týždňov. Väčšina pacientov, ktorá stratila odpoveď a vrátila sa k ustekinumabu, tak urobila do 24 týždňov od indukčnej infúzie. Z týchto 51 pacientov, 70,6 % dosiahlo klinickú odpoveď a 39,2 % dosiahlo klinickú remisiu 16 týždňov po podaní prvej subkutánnej dávky ustekinumabu.
EndoskopiaV podštúdii sa hodnotil endoskopický vzhľad sliznice u 252 pacientov s vhodnou východiskovou endoskopickou aktivitou ochorenia. Primárny koncový ukazovateľ bola zmena oproti
východiskovému skóre SES-CD (Simplified Endoscopic Disease Severity Score for Crohn’s Disease), zloženému skóre hodnotiacemu 5 ileo-kolonických segmentov na prítomnosť/veľkosť vredov, podiel povrchu sliznice pokrytej vredmi, podiel povrchu sliznice postihnutej akýmikoľvek inými léziami
a prítomnosť/typ zúženia/striktúr. V 8. týždni bola po jednorazovej intravenóznej indukčnej dávke zmena v SES-CD skóre väčšia v skupine s ustekinumabom (n = 155, priemerná zmena = -2,8) ako v skupine s placebom (n = 97, priemerná zmena = -0,7, p = 0,012).
Odpoveď fistulyV podskupine pacientov s drénovanou fistulou v úvode (8,8 %; n = 26), 12/15 (80 %) ustekinumabom liečených pacientov dosiahlo odpoveď fistuly v priebehu 44 týždňov (definovaná ako ≥ 50 % zníženie oproti východiskovému stavu v indukčnej štúdii v počte drénovaných fistúl) v porovnaní s 5/11
(45,5 %) dostávajúcimi placebo.
Kvalita života súvisiaca so zdravím
Kvalita života súvisiaca so zdravím bola posúdená na základe dotazníkov IBDQ a SF-36. V 8. týždni prejavovali pacienti dostávajúci ustekinumab štatisticky významne väčšie a klinicky významnejšie zlepšenia na celkovom skóre IBDQ a SF-36 Mental Component Summary Score v oboch štúdiách UNITI-1 a UNITI-2, a SF-36 Physical Component Summary Score v UNITI-2, pri porovnaní
s placebom. Tieto zlepšenia boli všeobecne lepšie udržateľné u pacientov liečených ustekinumabom v štúdii IM-UNITI až do 44. týždňa pri porovnaní s placebom.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s ustekinumabom v jednej alebo viacerých podskupinách pediatrickej populácie pre Crohnovu chorobu (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Priemerný čas na dosiahnutie maximálnej koncentrácie v sére (tmax) bol 8,5 dňa po jednorazovom subkutánnom podaní dávky 90 mg zdravým jedincom. Priemerné hodnoty tmax ustekinumabu po jednorazovom subkutánnom podaní dávky buď 45 mg alebo 90 mg pacientom so psoriázou boli porovnateľné s hodnotami pozorovanými u zdravých jedincov.
Absolútna biologická dostupnosť ustekinumabu po jednorazovom subkutánnom podaní u pacientov so psoriázou bola stanovená na 57,2 %.
Distribúcia
Medián distribučného objemu počas terminálnej fázy (Vz) po jednorazovom intravenóznom podaní pacientom so psoriázou sa pohyboval v rozsahu od 57 do 83 ml/kg.
Biotransformácia
Presná metabolická dráha ustekinumabu nie je známa.
Eliminácia
Medián systémového klírensu (CL) po jednorazovom intravenóznom podaní sa u pacientov so psoriázou pohyboval v rozsahu od 1,99 do 2,34 ml/deň/kg. Priemerný polčas (t1/2) ustekinumabu bol približne 3 týždne u pacientov so psoriázou, psoriatickou artritídou alebo Crohnovou chorobou,
v rozsahu od 15 do 32 dní počas všetkých štúdií zameraných na psoriázu a psoriatickú artritídu. V populačnej farmakokinetickej analýze zdanlivý klírens (CL/F) bol 0,465 l/deň a zdanlivý distribučný objem (V/F) 15,7 l u pacientov so psoriázou. Pohlavie pacientov nemalo vplyv na CL/F ustekinumabu. V populačnej farmakokinetickej analýze sa zistila tendencia k vyššiemu klírensu ustekinumabu u pacientov s pozitívnym testom na protilátky voči ustekinumabu.
Linearita dávky
U pacientov so psoriázou sa systémová expozícia ustekinumabu (Cmax a AUC) zvyšovala proporcionálne v závislosti na dávke po jednorazovom intravenóznom podaní v rozsahu dávok od
0,09 mg/kg do 4,5 mg/kg alebo po jednorazovom subkutánnom podaní v rozsahu dávok od približne
24 mg do 240 mg.
Jednorazová dávka verzus opakované dávky
Časové profily ustekinumabu v sére boli zvyčajne po jednorazovom alebo opakovanom podaní subkutánnej dávky predikovateľné. U pacientov so psoriázou rovnovážna koncentrácia ustekinumabu v sére sa dosiahla na 28. týždeň po počiatočných subkutánnych dávkach v 0. a 4. týždni, po čom sa dávky podávali každých 12 týždňov. Priemerná rovnovážna spodná hodnota koncentrácie sa pohybovala od 0,21 μg/ml do 0,26 μg/ml (45 mg) a od 0,47 μg/ml do 0,49 μg/ml (90 mg). Nedochádzalo k zjavnej kumulácii sérovej koncentrácie ustekinumabu v priebehu času, keď sa podával subkutánne každých 12 týždňov.
Pacientom s Crohnovou chorobou sa po intravenóznej dávke ~6 mg/kg podávala počnúc 8. týždňom subkutánna udržiavacia dávka 90 mg ustekinumabu každých 8 alebo 12 týždňov. Rovnovážna koncentrácia ustekinumabu sa dosiahla na začiatku druhej udržiavacej dávky. Priemerná rovnovážna priebežná koncentrácia sa pohybovala od 1,97 μg/ml do 2,24 μg/ml a od 0,61 μg/ml do 0,76 μg/ml pre
90 mg ustekinumabu každých 8 týždňov alebo každých 12 týždňov, v tomto poradí. Rovnovážne priebežné hladiny ustekinumabu po podaní 90 mg ustekinumabu každých 8 týždňov súviseli s vyššími mierami klinickej remisie v porovnaní s rovnovážnymi priebežnými hladinami po 90 mg každých 12 týždňov.
Vplyv hmotnosti na farmakokinetiku
V populačnej farmakokinetickej analýze údajov od pacientov so psoriázou sa ukázalo, že telesná hmotnosť je najvýznamnejším činiteľom ovplyvňujúcim klírens ustekinumabu. Priemerný CL/F
u pacientov s hmotnosťou > 100 kg bol približne o 55 % vyšší v porovnaní s pacientmi s hmotnosťou
≤ 100 kg. Priemerný V/F u pacientov s hmotnosťou > 100 kg bol približne o 37 % vyšší v porovnaní s pacientmi s hmotnosťou ≤ 100 kg. Priemerná spodná hladina sérovej koncentrácie ustekinumabu
u pacientov s vyššou telesnou hmotnosťou (> 100 kg) v skupine dostávajúcej dávku 90 mg bola porovnateľná s koncentráciou u pacientov s nižšou telesnou hmotnosťou (≤ 100 kg) v skupine dostávajúcej dávku 45 mg. Podobné výsledky sa dosiahli v potvrdzujúcej populačnej farmakokinetickej analýze použitím údajov od pacientov so psoriatickou artritídou.
Osobitné skupiny pacientov
Farmakokinetické údaje týkajúce sa pacientov s poškodenou funkciou obličiek alebo pečene nie sú k dispozícii.
Neuskutočnili sa žiadne špeciálne štúdie so staršími pacientmi.
Farmakokinetika ustekinumabu bola vo všeobecnosti porovnateľná medzi pacientmi ázijského a neázijského pôvodu so psoriázou.
U pacientov s Crohnovou chorobou bola variabilita CL ustekinumabu ovplyvnená telesnou hmotnosťou, hladinou albumínu v sére, CRP, úrovňou zlyhania TNF antagonistu, pohlavím, rasou (ázijská rasa oproti neázijskej rase) a úrovňou protilátok proti ustekinumabu, pričom telesná hmotnosť bol hlavný kovariát ovplyvňujúci distribučný objem. Súbežné užívanie imunomodulátorov nemalo významný vplyv na dispozíciu ustekinumabu. Vplyv týchto štatisticky významných kovariátov na príslušné PK parametre bol v rozmedzí ± 20 % pri hodnotení naprieč reprezentatívnou škálou hodnôt kovariátov alebo kategórií údajov, ktoré je v rámci celkovej variability pozorované v PK ustekinumabu.
V populačnej farmakokinetickej analýze sa nezistil žiadny vplyv tabaku alebo alkoholu na farmakokinetiku ustekinumabu.
U pediatrických pacientov so psoriázou vo veku 12 až 17 rokov liečených odporúčanou dávkou odvodenou od hmotnosti boli koncentrácie ustekinumabu v sére zvyčajne porovnateľné
s koncentráciami u dospelej psoriatickej populácie liečenej dávkou pre dospelých, zatiaľ čo
u pediatrických psoriatických pacientov liečených polovicou odporúčanej dávky odvodenej od hmotnosti boli koncentrácie ustekinumabu v sére zvyčajne nižšie ako koncentrácie u dospelých.
RegulovanieenzýmovCYP450
Účinky IL-12 alebo IL-23 na reguláciu enzýmov CYP450 boli hodnotené v in vitro štúdii na ľudských hepatocytoch, čo preukázalo, že IL-12 a/alebo IL-23 v sérových koncentráciách 10 ng/ml nemenia aktivitu ľudských enzýmov CYP450 (CYP1A2, 2B6, 2C9, 2C19, 2D6 alebo 3A4; pozri časť 4.5).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií toxicity po opakovanom podávaní a vývojovej a reprodukčnej toxicity, vrátane farmakologického hodnotenia bezpečnosti, neodhalili žiadne osobitné
sa nepozorovali ani nežiaduce účinky na fertilitu samcov ani vrodené chyby alebo vývojová toxicita. Takisto sa nepozorovali žiadne nežiaduce účinky na fertilitu samíc pri použití analogickej protilátky voči IL-12/23 na myšiach.
Hladiny dávok v štúdiách na zvieratách boli približne až 45-násobne vyššie než je najvyššia ekvivalentná dávka určená na podanie pacientom so psoriázou a výsledné vrcholové sérové koncentrácie u opíc boli viac než 100-násobne vyššie než koncentrácia pozorovaná u ľudí.
Štúdie karcinogenity sa s ustekinumabom neuskutočnili vzhľadom na absenciu vhodných modelov na protilátku s neskríženou reaktivitou voči IL-12/23 p40 u hlodavcov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Histidín
Monohydrát histidíniumchloridu
Polysorbát 80
Sacharóza
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke pri teplote (2°C – 8°C). Neuchovávajte v mrazničke.
Uchovávajte injekčnú liekovku alebo naplnenú injekčnú striekačku vo vonkajšom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
STELARA45 mg injekčný roztok
0,5 ml sterilného roztoku v 2 ml injekčnej liekovke zo skla typu I uzavretej gumovou zátkou potiahnutou butylom.
STELARA90 mg injekčný roztok
1 ml sterilného roztoku v 2 ml injekčnej liekovke zo skla typu I uzavretej gumovou zátkou potiahnutou butylom.
STELARA45 mg injekčný roztok naplnený v injekčnej striekačke
0,5 ml roztoku v 1 ml striekačke zo skla typu I s pripevnenou ihlou z nehrdzavejúcej ocele a krytom ihly, ktorý obsahuje suchú prírodnú gumu (derivát latexu). Na striekačke je pripevnený bezpečnostný kryt.
STELARA90 mg injekčný roztok naplnený v injekčnej striekačke
1 ml roztoku v 1 ml striekačke zo skla typu I s pripevnenou ihlou z nehrdzavejúcej ocele a krytom ihly, ktorý obsahuje suchú prírodnú gumu (derivát latexu). Na striekačke je pripevnený bezpečnostný kryt.
V jednom balení je 1 injekčná liekovka alebo 1 naplnená striekačka s liekom STELARA.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomRoztok v injekčnej liekovke alebo naplnenej striekačke so STELAROU sa nemá pretrepávať. Pred subkutánnym podaním treba roztok vizuálne skontrolovať, či nevidno pevné prachové častice alebo či nedošlo k zmene sfarbenia. Roztok je číry až slabo opalescentný, bezfarebný až svetložltý a môže obsahovať drobné priesvitné alebo biele častice proteínu. Takýto vzhľad nie je pre proteínové roztoky nezvyčajný. Liek sa nemá použiť, ak roztok zmenil farbu alebo je mútny, alebo ak sú prítomné cudzorodé pevné častice. Pred podaním sa má umožniť, aby STELARA dosiahla izbovú teplotu (približne pol hodiny). Podrobné informácie o použití sú uvedené v písomnej informácii pre používateľa.
STELARA neobsahuje konzervačné látky; z toho dôvodu nespotrebovaný liek, ktorý ostal v injekčnej liekovke a v striekačke, sa už nemá použiť. STELARA sa dodáva vo forme sterilnej injekčnej liekovky na jedno použitie alebo naplnenej striekačke na jedno použitie. Striekačka, ihla a injekčná liekovka sa nesmú nikdy opakovane použiť. Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen-Cilag International NV Turnhoutseweg 30
2340 Beerse
Belgicko
8. REGISTRAČNÉ ČÍSLOSTELARA45 mg injekčný roztokEU/1/08/494/001
STELARA90 mg injekčný roztokEU/1/08/494/002
STELARA45 mg injekčný roztok naplnený v injekčnej striekačkeEU/1/08/494/003
STELARA90 mg injekčný roztok naplnený v injekčnej striekačkeEU/1/08/494/004
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. január 2009
Dátum posledného predĺženia registrácie: 19. september 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.Nasledujúca informácia je určená len pre zdravotníckych pracovníkov:
Návod na riedenie:STELARA koncentrát na infúzny roztok musí byť zriedený, pripravený a podaný infúzne
zdravotníckym pracovníkom použitím aseptickej techniky.
1. Vypočítajte dávku a potrebný počet injekčných liekoviek STELARY na základe hmotnosti pacienta (pozri časť 3, Tabuľka 1). Jedna 26 ml injekčná liekovka STELARY obsahuje 130 mg ustekinumabu.
2. Z 250 ml infúzneho vaku odoberte a potom zlikvidujte objem 0,9 % roztoku chloridu sodného rovnajúci sa objemu STELARY, ktorý sa má pridať (odstráňte 26 ml chloridu sodného na každú potrebnú injekčnú liekovku STELARY, pri 2 injekčných liekovkách - odstráňte 52 ml, pri 3 injekčných liekovkách odstráňte 78 ml, pri 4 injekčných liekovkách – odstráňte 104 ml).
3. Odoberte 26 ml STELARY z každej potrebnej injekčnej liekovky a pridajte ich do 250 ml infúzneho vaku. Konečný objem v infúznom vaku má byť 250 ml. Jemne pomiešajte.
4. Pred infúziou vizuálne skontrolujte zriedený roztok. Nepoužite ho, ak spozorujete viditeľné nepriehľadné častice, sfarbenie alebo cudzie častice.
5. Zriedený roztok podávajte infúzne po dobu najmenej jednej hodiny. Po zriedení možno infúzny roztok uchovávať do štyroch hodín pred infúziou.
6. Použite len infúzny set s in-line sterilným nepyrogénnym filtorm s nízkou väzbou bielkovín
(veľkosť pórov 0,2 mikrometra).
7. Každá injekčná liekovka je len na jednorazové použitie a akýkoľvek nepoužitý liek sa má zlikvidovať v súlade s národnými požiadavkami.
UchovávanieAk je to potrebné, infúzny roztok sa môže uchovávať až do štyroch hodín pri izbovej teplote. Neuchovávajte v mrazničke.
Pokyny na podávanie liekuNa začiatku liečby vám prvú injekciu podá lekársky personál. Vy a váš lekár sa však môžete rozhodnúť, že si budete liek podávať sami. V tomto prípade vás poučia, ako sa injekcia podáva. Ak máte nejaké otázky o tom, ako si injikovať liek, prediskutujte ich s lekárom.
· Nemiešajte Stelaru s inými injekčnými tekutinami
· Injekčnými liekovkami netraste. Je to preto, že prudké trasenie môže liek znehodnotiť.
Nepoužívajte liek, ak sa ním silno triaslo.
1. Skontrolujte počet injekčných liekoviek a pripravte si potrebný materiál:Vyberte injekčnú liekovku/liekovky z chladničky. Nechajte injekčnú liekovku postáť približne pol hodinu. Tekutina tým získa vhodnú teplotu na podanie injekcie (izbová teplota).
Skontrolujte injekčnú liekovku/liekovky, aby ste si boli istí, že:
· počet injekčných liekoviek a sila je správna
o Ak je pre vás určená dávka 45 mg alebo menšia, dostanete jednu injekčnú liekovku s obsahom 45 mg lieku Stelara
o Ak je pre vás určená dávka 90 mg, dostanete dve injekčné liekovky s obsahom 45 mg lieku Stelara a bude potrebné, aby ste si podali dve injekcie. Vyberte si dve rôzne miesta na podanie injekcií (napr. jednu injekciu do pravého stehna a druhú injekciu do ľavého stehna) a injekcie podajte jednu za druhou. Pre každú injekciu použite novú ihlu
a striekačku.
· máte správny liek
· neuplynula doba použiteľnosti lieku
· injekčná liekovka nie je poškodená a plomba nie je zlomená
· roztok v injekčnej liekovke je číry až slabo opalescentný (má perleťový lesk) a bezfarebný až svetložltý
· roztok nemá zmenenú farbu ani nie je mútny a neobsahuje žiadne cudzorodé častice
· roztok nie je zamrazený.
Deti s hmotnosťou menej ako 60 kg potrebujú dávku nižšiu ako 45 mg. Uistite sa, že poznáte správne množstvo (objem), ktoré sa má vytiahnuť z injekčnej liekovky a typ striekačky potrebnej na podanie dávky. Ak neviete potrebné množstvo alebo typ striekačky, obráťte sa na svojho lekára pre ďalšie inštrukcie.
Prineste si všetko potrebné a položte to na čistý povrch. Patrí sem striekačka, ihla, antiseptické utierky, vata alebo gáza a kontajner na ostré predmety (pozri obrázok 1).
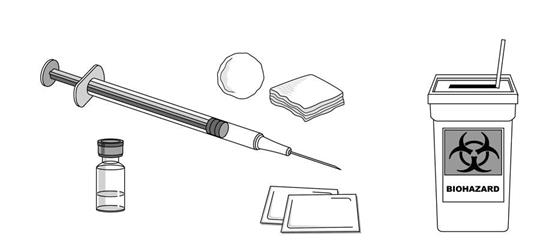
Obrázok 1
2. Zvoľte a pripravte si miesto vpichu injekcie:Zvoľte miesto vpichu injekcie (pozri obrázok 2)
· Stelara sa podáva injekčne pod kožu (subkutánne)
· Vhodné miesta na injekciu sú horná časť stehna alebo okolie brucha najmenej 5 cm od pupka
· Podľa možnosti si nevyberte miesta so známkami psoriázy na koži
· Ak vám niekto pomáha pri podávaní injekcie, potom možno zvoliť ako miesto na pichnutie injekcie aj hornú časť ramena
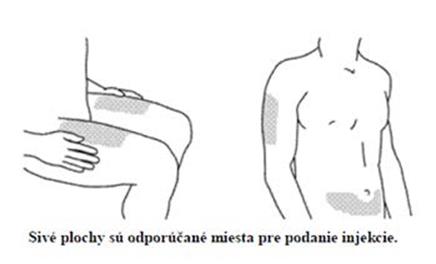
Obrázok 2
Pripravte si miesto vpichu injekcie
· Dôkladne si umyte ruky mydlom a teplou vodou
· Antiseptickou utierkou vyčistite miesto vpichu injekcie do kože
· Pred podaním injekcie sa už tohto miesta nedotýkajte
3. Pripravte si dávku:· Zložte uzáver injekčnej liekovky (pozri obrázok 3)

Obrázok 3
· Neodstraňujte zátku
· Vyčistite zátku antiseptickým tampónom
· Položte injekčnú liekovku na rovný plochý povrch
· Zdvihnite striekačku a odstráňte kryt ihly
· Nedotýkajte sa ihly a dbajte na to, aby sa ničoho nedotýkala
· Pretlačte ihlu cez gumenú zátku
· Otočte injekčnú liekovku a striekačku hore dnom
· Vyťahujte piest striekačky, aby ste ju naplnili množstvom tekutiny predpísaným lekárom
· Je dôležité, aby ihla zostávala vždy ponorená v tekutine. Tým sa zabráni vzniku vzduchových bublín v striekačke (pozri obrázok 4)

Obrázok 4
· Vyberte ihlu z injekčnej liekovky
· Podržte striekačku s ihlou smerujúcou nahor, aby ste videli, či vo vnútri nie sú vzduchové bubliny
· Ak sú prítomné vzduchové bubliny, jemne zboku poklopávajte prstom na striekačku, kým bubliny nevyplávajú nahor (pozri obrázok 5)
Obrázok 5
· Potom tlačte piest dovtedy, kým všetok vzduch neodstránite (tekutina nesmie vytiecť)
· Neotáčajte striekačku nadol a nedovoľte, aby sa ihla niečoho dotkla.
4. Injikovanie dávky:· Jemne stlačte vyčistenú kožu medzi palec a ukazovák. Nestláčajte ju tuho
· Zapichnite ihlu do stlačenej kože
· Palcom tlačte na piest dovtedy, kým sa neinjikuje všetka tekutina. Tlačte pomaly a rovnomerne, pričom stále jemne pridŕžajte kožu stlačenú
· Keď je piest stlačený až na doraz, vyberte ihlu z kože
5. Po injekcii:· Pritlačte antiseptickú utierku na miesto vpichu injekcie a podržte ju tam pár sekúnd
· V mieste podania injekcie sa môže objaviť trochu krvi alebo tekutina. Je to normálne
· Miesto podania injekcie môžete pritlačiť vatou alebo gázou a 10 sekúnd podržať
· Kožu v mieste podania injekcie nešúchajte. Ak je potrebné, miesto podania injekcie môžete prekryť náplasťou.
6. Likvidácia:· Použité striekačky a ihly treba odložiť do obalu odolnému proti prepichnutiu ako je nepriepustný kontajner na ostré predmety. Nikdy nepoužívajte ihly a striekačky opakovane kvôli svojej bezpečnosti a svojmu zdraviu a kvôli bezpečnosti ostatných. Kontajner zlikvidujte v súlade s národnými požiadavkami
· Prázdne injekčné liekovky, antiseptické utierky a iné pomôcky možno likvidovať domovým odpadom.