Dajte priesvitný kryt späť na miesto, až kým nebudete počuť kliknutie.
· Opätovne neodstraňujte priesvitný kryt.
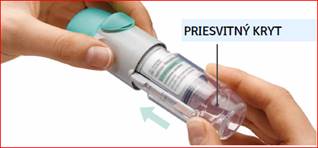
| 4. Otočte
· Ponechajte uzáver zatvorený.
· Otáčajte priesvitným krytom v smere šípok na obale, až kým nezapadne (polovičné otočenie).
|
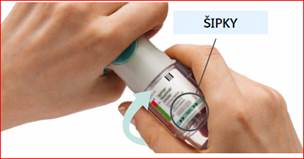
|
5. Otvorte
· Otvárajte uzáver, až kým nepovolí a nie je úplne otvorený.
|
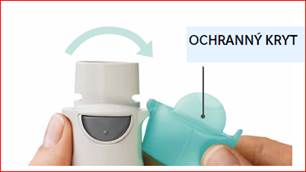
|
6. Stlačte
· Nasmerujte inhalátor smerom na zem.
· Stlačte tlačidlo na uvoľnenie dávky.
· Zatvorte uzáver.
· Zopakujte kroky 4-6, až kým sa neobjaví obláčik.
· Keď sa objaví obláčik, zopakujte kroky 4-6 ešte trikrát.
Teraz je váš inhalátor pripravený na použitie. Tieto kroky nebudú mať vplyv na počet dostupných dávok. Po príprave bude môcť váš inhalátor poskytnúť 60 vstrekov (30 dávok).
| 
|
Každodenné používanie OTOČTE
· Ponechajte uzáver zatvorený.
· OTÁČAJTE priesvitný kryt v smere šípok na obale, až kým nezapadne (polovičné otočenie).
|

|
OTVORTE
· OTVORTE uzáver, až kým nepovolí a nie je úplne otvorený.
|

|
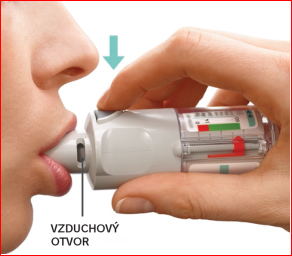
STLAČTE
· Pomaly a úplne vydýchnite.
· Zovrite pery okolo náustku bez toho, aby ste zakryli vzduchové otvory. Nasmerujte inhalátor na zadnú stranu vášho hrdla.
· Zatiaľ čo sa pomaly hlboko nadýchnete ústami, STLAČTE tlačidlo uvoľňujúce dávku a pokračujte pomaly v nádychu, kým vydržíte.
· Zadržte dych na 10 sekúnd alebo aj dlhšie, pokiaľ vydržíte.
· Pri obidvoch vstrekoch zopakujte: Otočte, Otvorte, Zatlačte.
· Zatvorte kryt, kým znovu použijete váš inhalátor.
| 
|
4.3 Kontraindikácie
Precitlivenosť na tiotropiumbromid alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 alebo na atropín alebo jeho deriváty, napr. ipratropium alebo oxitropium.
4.4 Osobitné upozornenia a opatrenia pri používaní
Tiotropiumbromid, ako raz denne dávkované udržiavacie bronchodilatans sa nemá používať ako iniciálna liečba akútnych záchvatov bronchospazmu alebo na úľavu od akútnych príznakov. V prípade akútneho záchvatu sa má použiť beta-2-agonista s rýchlym nástupom účinku.
Spiriva Respimat sa nemá používať ako monoterapia na liečbu astmy. Pacienti s astmou musia byť poučení, aby pokračovali v užívaní protizápalovej liečby, t. j. inhalačných kortikosteroidov, nezmenenej po zavedení Spirivy Respimat, aj keď sa ich príznaky zlepšia.
Po podaní inhalačného roztoku tiotropiumbromidu sa môžu vyskytnúť okamžité alergické reakcie.
Pre svoju anticholinergickú aktivitu sa má tiotropiumbromid podávať opatrne u pacientov s glaukómom so zatvoreným uhlom, hyperpláziou prostaty alebo obštrukciou hrdla močového mechúra.
Lieky na inhaláciu môžu spôsobiť inhaláciou indukovaný bronchospazmus.
Tiotropium sa má s opatrnosťou používať u pacientov s nedávno prekonaným infarktom myokardu < 6 mesiacov; s akoukoľvek nestabilnou alebo život ohrozujúcou srdcovou arytmiou alebo srdcovou arytmiou vyžadujúcou zákrok alebo zmenu liekov za posledný rok; s hospitalizáciou pre zlyhanie srdca (trieda II alebo IV podľa klasifikácie NYHA) v poslednom roku. Títo pacienti boli z klinických štúdií vylúčení a uvedené stavy môže ovplyvniť anticholinergický mechanizmus účinku.
Keďže pri znížených obličkových funkciách stúpa plazmatická koncentrácia, u pacientov so stredne ťažkou až ťažkou poruchou funkcie obličiek (klírens kreatinínu ≤ ako 50 ml/min) sa tiotropiumbromid podáva len ak očakávaný prínos prevyšuje potenciálne riziko. U pacientov s ťažkou poruchou funkcie obličiek nie sú dlhodobé skúsenosti (pozri časť 5.2).
Pacienti si majú dávať pozor, aby im roztok nevnikol do očí. Majú byť poučení, že to môže spôsobiť precipitáciu alebo zhoršenie glaukómu so zatvoreným uhlom, bolesť očí alebo diskomfort, prechodné neostré videnie, irizáciu videnia alebo farebné obrazce spojené s červenými očami z konjunktiválnej kongescie a korneálneho edému. Ak sa objaví ktorákoľvek z kombinácii týchto očných príznakov, pacienti majú prestať užívať tiotropiumbromid a okamžite to konzultovať so špecialistom.
Sucho v ústach, ktoré bolo zaznamenané pri anticholinergickej liečbe, môže pri dlhodobom používaní viesť k zubným kazom.
Tiotropiumbromid sa nemá používať častejšie než jedenkrát denne (pozri časť 4.9).
Spiriva Respimat sa neodporúča pri cystickej fibróze (CF). Ak sa použije u pacientov s CF, Spiriva Respimat môže spôsobiť nárast prejavov a príznakov CF (napr. závažné nežiaduce udalosti, pľúcne exacerbácie, infekcie dýchacích ciest).
4.5 Liekové a iné interakcie
Aj keď sa nevykonali žiadne formálne štúdie zamerané na liekové interakcie, tiotropiumbromid sa používa súbežne s inými liekmi všeobecne používanými pri liečbe CHOCHP a astmy vrátane sympatomimetických bronchodilatancií, metylxantínov, perorálnych a inhalačných steroidov, antihistaminík, mukolytík, modifikátorov leukotriénov, kromónov, liečby anti-IgE bez klinického záznamu liekových interakcií.
Nezistilo sa, že užívanie LABA alebo ICS zmenilo expozíciu tiotropia.
Súbežné podávanie tiotropiumbromidu s inými liekmi obsahujúcimi anticholinergiká nebolo sledované, preto sa neodporúča.
4.6 Fertilita, gravidita a laktácia
GraviditaO používaní tiotropia u tehotných žien je veľmi obmedzené množstvo údajov. Štúdie na zvieratách nenasvedčujú žiaden priamy ani nepriamy škodlivý účinok, pokiaľ ide o reprodukčnú toxicitu v klinicky relevantných dávkach (pozri časť 5.3). Ako preventívne opatrenie sa odporúča nepoužívať Spirivu Respimat počas tehotenstva.
DojčenieNie je známe, či sa tiotropiumbromid vylučuje do ľudského materského mlieka. Napriek štúdiám na hlodavcoch, ktoré dokázali, že tiotropiumbromid sa vylučuje do materského mlieka len v malých množstvách, použitie Spirivy Respimat sa neodporúča počas dojčenia. Tiotropiumbromid je dlhodobo účinkujúca látka. Rozhodnutie o tom, či pokračovať/ukončiť dojčenie alebo pokračovať/ukončiť liečbu Spirivou Respimat sa má urobiť po zohľadnení prínosu dojčenia pre dieťa a prínosu liečby Spirivou Respimat pre ženu.
FertilitaKlinické údaje týkajúce sa fertility nie sú pre tiotropium dostupné. Neklinická štúdia s tiotropiom nepreukázala žiadne príznaky nežiaducich účinkov na fertilitu (pozri 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Výskyt závratov alebo neostrého videnia môže ovplyvňovať schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnostiMnohé z uvedených nežiaducich účinkov môžu byť pripísané anticholinergickým vlastnostiam tiotropiumbromidu.
Tabuľkový zoznam nežiaducich účinkovFrekvencie prideľované nežiaducim účinkom uvedeným nižšie vychádzajú z približnej miery výskytu nežiaducich liekových reakcií (t.j. udalostí pripisovaných tiotropiu), pozorovaných v skupine s tiotropiom, získaných zo 7 placebom kontrolovaných klinických štúdií s CHOCHP (3 282 pacientov) a 12 placebom kontrolovaných klinických štúdií s dospelými a pediatrickými pacientmi s astmou
(1 930 pacientov) pri trvaní liečby od štyroch týždňov do jedného roka.
Frekvencia je uvedená použitím nasledujúcej konvencie:
Veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000), neznáme (nedajú sa odhadnúť z dostupných údajov).
Trieda orgánových systémov/MedDRA preferované termíny
| Frekvencia
pri CHOCHP
| Frekvencia
pri astme
|
|
|
|
Poruchy metabolizmu a výživy
|
|
|
Dehydratácia
| neznáme
| neznáme
|
|
|
|
Poruchy nervového systému
|
|
|
Závraty
| menej časté
| menej časté
|
Bolesti hlavy
| menej časté
| menej časté
|
Insomnia
| zriedkavé
| menej časté
|
|
|
|
Poruchy oka
|
|
|
Glaukóm
| zriedkavé
| neznáme
|
Zvýšený vnútroočný tlak
| zriedkavé
| neznáme
|
Neostré videnie
| zriedkavé
| neznáme
|
|
|
|
Poruchy srdca a srdcovej činnosti
|
|
|
Atriálna fibrilácia
| zriedkavé
| neznáme
|
Palpitácie
| zriedkavé
| menej časté
|
Supraventrikulárna tachykardia
| zriedkavé
| neznáme
|
Tachykardia
| zriedkavé
| neznáme
|
|
|
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
|
|
Kašeľ
| menej časté
| menej časté
|
Faryngitída
| menej časté
| menej časté
|
Dysfónia
| menej časté
| menej časté
|
Epistaxa
| zriedkavé
| zriedkavé
|
Bronchospazmus
| zriedkavé
| menej časté
|
Laryngitída
| zriedkavé
| neznáme
|
Sinusitída
| neznáme
| neznáme
|
|
|
|
Poruchy gastrointestinálneho traktu
|
|
|
Sucho v ústach
| časté
| menej časté
|
Zápcha
| menej časté
| zriedkavé
|
Orofaryngeálna kandidóza
| menej časté
| menej časté
|
Dysfágia
| zriedkavé
| neznáme
|
Porucha gastroezofageálneho refluxu
| zriedkavé
| neznáme
|
Zubný kaz
| zriedkavé
| neznáme
|
Gingivitída
| zriedkavé
| zriedkavé
|
Glositída
| zriedkavé
| neznáme
|
Stomatitída
| neznáme
| zriedkavé
|
Intestinálna obštrukcia, vrátane
paralytického ilea
|
neznáme
|
neznáme
|
Nauzea
| neznáme
| neznáme
|
|
|
|
Poruchy kože a podkožného tkaniva
Poruchy imunitného systému
|
|
|
Vyrážka
| menej časté
| menej časté
|
Svrbenie
| menej časté
| zriedkavé
|
Angioneurotický edém
| zriedkavé
| zriedkavé
|
Žihľavka
| zriedkavé
| zriedkavé
|
Infekcia kože/vred na koži
| zriedkavé
| neznáme
|
Suchá koža
| zriedkavé
| neznáme
|
Precitlivenosť (vrátane
okamžitých reakcií)
| neznáme
| zriedkavé
|
Anafylaktická reakcia
| neznáme
| neznáme
|
|
|
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
|
|
Opuch kĺbov
| neznáme
| neznáme
|
|
|
|
Poruchy obličiek a močových ciest
|
|
|
Retencia moču
| menej časté
| neznáme
|
Dyzúria
| menej časté
| neznáme
|
Infekcie močových ciest
| zriedkavé
| zriedkavé
|
Popis vybraných nežiaducich účinkovV kontrolovaných klinických štúdiách v CHOCHP boli častejšie pozorované anticholinergické nežiaduce účinky ako je sucho v ústach, ktoré sa vyskytlo približne u 2,9 % pacientov. Pri astme predstavoval výskyt sucha v ústach 0,83 %.
V 7 klinických štúdiách s CHOCHP, sucho v ústach viedlo k prerušeniu liečby u 3 z 3 282 pacientov liečených tiotropiom (0,1 %). V 12 klinických štúdiách zameraných na astmu (1 930 pacientov) nebolo hlásené žiadne prerušenie liečby v dôsledku sucha v ústach.
Závažné nežiaduce účinky súvisiace s anticholinergickým efektom, zahŕňajúce glaukóm, zápchu, intestinálnu obštrukciu vrátane paralytického ilea a retencie moču.
Pediatrická populáciaDatabáza bezpečnosti obsahuje 560 pediatrických pacientov (296 pacientov vo veku 1 až 11 rokov a 264 pacientov vo veku 12 až 17 rokov) z 5 placebom kontrolovaných klinických skúšaní pri trvaní liečby 12 týždňov až jeden rok. Frekvencia, druh a závažnosť nežiaducich reakcií v pediatrickej populácii sú podobné ako u dospelých.
Ďalšie osobitné skupiny pacientovVo vyššom veku sa môže vyskytnúť zvýšenie anticholinergických účinkov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Vysoké dávky tiotropiumbromidu môžu viesť k anticholinergickým prejavom a príznakom.Avšak u zdravých dobrovoľníkov sa nevyskytol žiadny nežiaduci systémový anticholinergický účinok po jednej inhalačnej dávke do 340 mikrogramov tiotropiumbromidu. Navyše u zdravých dobrovoľníkov neboli pozorované žiadne relevantné nežiaduce účinky, okrem sucha v ústach/krku a suchej sliznice nosa, po 14-dňovej liečbe do 40 mikrogramov inhalačného roztoku tiotropia, s výnimkou zjavne zníženej produkcie slín od 7. dňa.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné inhalačné antiastmatiká, anticholinergiká
ATC kód: R03B B04
MechanizmusúčinkuTiotropiumbromid je dlhodobo účinkujúci, špecifický antagonista muskarínových receptorov. Má podobnú afinitu k subtypom M
1 – M
5 . V dýchacích cestách sa tiotropiumbromid kompetitívne a reverzibilne viaže na M
3 receptory hladkého svalstva bronchov, antagonizuje cholinergický (bronchokonstrikčný) účinok acetylcholínu, čím spôsobuje relaxáciu hladkého svalstva bronchov. Účinok je závislý od dávky a trvá viac než 24 hodín. Tiotropiumbromid, ako N-kvartérne anticholinergikum je topicky (broncho-)selektívny pri inhalačnom podávaní, čím je daný akceptovateľný terapeutický rozsah predtým, ako sa prejavia systémové anticholinergické účinky.
Farmakodynamické účinkyUvoľňovanie tiotropia najmä z M
3 receptorov je veľmi pomalé, má preukázateľne dlhší polčas disociácie než ipratropium. Uvoľňovanie z receptorov M
2 je rýchlejšie ako z receptorov M
3, čo vo funkčných
in vitro štúdiách viedlo ku (kineticky kontrolovanej) M
3-receptorovej selektivite oproti M
2 receptorom. Vysoká potencia, veľmi pomalé uvoľňovanie z receptorov a lokálna selektivita po inhalačnej aplikácii majú klinickú koreláciu v signifikantnej a dlhodobej bronchodilatácii u pacientov s CHOCHP a astmou.
Klinická účinnosť a bezpečnosť pri CHOCHP:Program klinických vývojových štúdií fázy III pozostával z dvoch 1-ročných, dvoch 4-týždňových a dvoch 12-týždňových randomizovaných, dvojito zaslepených štúdii u 2 901 pacientov s CHOCHP
(1 308 užívalo dávku 5 µg tiotropiumbromidu).
Jednoročný program pozostával z dvoch placebom kontrolovaných štúdií. Obidve 12-týždňové štúdie boli s kontrolou aktívnou (ipratropium) a placebom. Všetkých šesť štúdií zahŕňalo hodnotenie pľúcnych funkcií. Navyše dve 1-ročné štúdie zahŕňali sledovanie miery dyspnoe, posúdenie kvality života a vplyv na exacerbácie.
Placebom kontrolované štúdiePľúcne funkciePľúcne funkcie (forsírovaný expiračný objem počas prvej sekundy - FEV
1 a forsírovaná vitálna kapacita FVC) sa signifikantne zlepšili pri podávaní inhalačného roztoku tiotropia jedenkrát denne, pričom zlepšenie nastalo do 30 minút po podaní prvej dávky v porovnaní s placebom (priemerné zlepšenie FEV
1 do 30 minút: 0,113 litrov; 95 % konfidenčný interval (CI): 0,102 až 0,125 litrov, p<0,0001). Zlepšenie pľúcnych funkcií sa udržalo v rovnovážnom stave počas 24 hodín v porovnaní s placebom (priemerné zlepšenie FEV
1: 0,122 litrov; 95 % CI: 0,106 až 0,138 litrov, p<0,0001). Farmakodynamický rovnovážny stav bol dosiahnutý počas prvého týždňa.
Denne vykonávané vyšetrenia dokázali, že Spiriva Respimat signifikantne zlepšuje ranný a večerný PEFR (maximálna výdychová rýchlosť) v porovnaní s placebom (priemerné zlepšenie PEFR: priemerné zlepšenie ráno 22 l/min; 95 % CI: 18 až 55 l/min, p<0,0001; večer 26 l/min; 95 % CI: 23 až 30 l/min, p<0,0001). Používanie Spirivy Respimat znížilo potrebu záchrannej bronchodilatačnej liečby v porovnaní s placebom (priemerná redukcia použitia pri záchrane 0,66 prípadov na deň, 95 % CI: 0,51 až 0,81 prípadov na deň, p<0,0001).
Bronchodilatačný efekt pri používaní Spirivy Respimat sa pozoroval počas 1 roku podávania bez znakov tolerancie.
Dyspnoe, kvalita života ovplyvnená zdravotným stavom, exacerbácie CHOCHP v dlhodobých 1-ročných štúdiáchDyspnoe
Spiriva Respimat signifikantne zlepšuje dyspnoe (hodnotené použitím Transition Dyspnea Index) v porovnaní s placebom (priemerné zlepšenie 1,05 jednotiek; 95 % CI: 0,73 až 1,38 jednotiek, p<0,0001). Toto zlepšenie sa udržalo počas celého obdobia liečby.
Kvalita života ovplyvnená zdravotným stavom
Priemerné zlepšenie v celkovom hodnotení kvality života pacientov (sledované dotazníkom St. George’s Respiratory Questionnaire) medzi Spirivou Respimat oproti placebu na konci dvoch 1-ročných štúdií bolo 3,5 jednotiek (95 % CI: 2,1 až 4,9; p<0,0001). 4-jednotkové zníženie je považované za klinicky relevantné.
CHOCHP exacerbácie
V troch jednoročných, randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických štúdiách liečba Spirivou Respimat, v porovnaní s placebom, viedla ku signifikantnému zníženiu rizika CHOCHP exacerbácie. CHOCHP exacerbácie sa definovali ako „súbor minimálne dvoch respiračných príhod/príznakov s dobou trvania tri dni alebo viac vyžadujúcich zmenu liečby (predpísanie antibiotík a/alebo systémových kortikosteroidov a/alebo signifikantnú zmenu v predpísaných respiračných liekoch)“. Liečba Spirivou Respimat viedla ku zníženiu rizika hospitalizácie kvôli CHOCHP exacerbácii (signifikantne v primerane rozsiahlej exacerbačnej štúdii).
Súbor analýz dvoch štúdií fázy III a individuálnej analýzy dodatočnej exacerbačnej štúdie je znázornený v tabuľke 1. Všetky respiračné lieky okrem anticholinergík a dlhodobo pôsobiacich beta-agonistov boli povolené ako súbežná liečba t.j. rýchlo pôsobiace beta-agonisty, inhalačné kortikosteroidy a xantíny. Dlhodobo pôsobiace beta-agonisty boli dodatočne povolené v exacerbačnej štúdii.
Tabuľka 1: Štatistická analýza CHOCHP exacerbácií a hospitalizovaných CHOCHP exacerbácií u pacientov so strednou až veľmi závažnou CHOCHP
Štúdia
(NSpiriva, Nplacebo)
| Koncový ukazovateľ
| Spiriva Respimat
| Placebo
| % zníženie rizika
(95 % CI)a
| p-hodnota
|
1-ročná fáza III
súhrn analýzd
(670, 653)
| Počet dní do prvej CHOCHP exacerbácie
| 160a
| 86a
| 29
(16 až 40)b
| <0,0001b
|
Stredná hodnota výskytu exacerbácie na pacientorok
| 0,78c
| 1,00c
| 22
(8 až 33)c
| 0,002c
|
Čas do prvej hospitalizácie pacienta s CHOCHP exacerbáciou
|
|
| 25
(-16 až 51)b
| 0,20b
|
Stredná hodnota výskytu hospitalizovaných pacientov s exacerbáciou na pacientorok
| 0,09 c
| 0,11 c
| 20
(-4 až 38) c
| 0,096 c
|
1-ročná fáza IIIb exacerbačná štúdia
(1939, 1953)
| Počet dní do prvej CHOCHP exacerbácii
| 169a
| 119a
| 31
(23 až 37)b
| <0,0001b
|
Stredná hodnota výskytu exacerbácií na pacientorok
| 0,69c
| 0,87c
| 21
(13 až 28)c
| <0,0001c
|
Čas do prvej hospitalizácie pacienta s CHOCHP exacerbáciou
|
|
| 27
(10 až 41)b
| 0,003b
|
Stredná hodnota výskytu hospitalizovaných pacientov s exacerbáciou na pacientorok
| 0,12c
| 0,15c
| 19
(7 až 30)c
| 0,004c
|
a Čas do výskytu prvej príhody: počet dní na liečbe, pričom 25 % pacientov malo minimálne jednu exacerbáciu CHOCHP /bolo hospitalizovaných kvôli CHOCHP exacerbácii.
V štúdii A 25 % pacientov užívajúcich placebo malo exacerbáciu do dňa 112, pričom pri Spirive Respimat 25 % pacientov malo exacerbáciu do dňa 173 ( p=0.09); v štúdii B 25 % pacientov užívajúcich placebo malo exacerbáciu do dňa 74, pričom pri Spirive Respimat 25 % pacientov malo exacerbáciu do dňa 149 (p<0.0001).b Miera rizika sa určila na základe Coxovho proporčného modelu rizika. Percentuálne zníženie rizika je 100(1 - miera rizika).
c Regresívne Poissonovo rozdelenie pravdepodobnosti. Zníženie rizika je 100(1 - miera rizika).
d Súbor analýz sa určil pri návrhu štúdií. Exacerbačné koncové ukazovatele sa významne zlepšili v individuálnych analýzach dvoch jednoročných štúdií.
Dlhodobá štúdia s tiotropiom kontrolovaná aktívnym liekomBola vykonaná dlhodobá, rozsiahla, randomizovaná, dvojito zaslepená, aktívnym liekom kontrolovaná klinická štúdia s obdobím pozorovania až 3 roky, aby porovnala účinnosť a bezpečnosť Spirivy HandiHaler a Spirivy Respimat (5 694 pacientov užívalo Spirivu HandiHaler; 5 711 pacientov užívalo Spirivu Respimat). Primárnymi koncovými ukazovateli boli čas do prvej CHOCHP exacerbácie, čas do mortality zo všetkých príčin a v podskupine (906 pacientov) minimálny FEV
1 (pred podaním dávky).
Čas do prvej CHOCHP exacerbácie bol numericky podobný v štúdii so Spirivou Respimat a Spirivou HandiHaler (pomer rizika (Spiriva Respimat/Spiriva HandiHaler) 0,98 s 95 % CI 0,93 až 1,03). Medián počtu dní do prvej CHOCHP exacerbácie bol 756 dní pri Spirive Respimat a 719 dní pri Spirive HandiHaler.
Bronchodilatačný účinok Spirivy Respimat sa udržal vyše 120 týždňov a bol podobný ako pri Spirive HandiHaler. Priemerný rozdiel v minimálnom FEV
1 pri Spirive Respimat oproti Spirive HandiHaler bol -0,010 l (95 % CI −0,038 až 0,018 l).
V postmarketingovej štúdii TIOSPIR porovnávajúcej Spirivu Respimat a Spirivu HandiHaler bola mortalita zo všetkých príčin (vrátane ďalšieho sledovania vitálneho statusu) podobná s pomerom rizika (Spiriva Respimat/Spiriva HandiHaler) = 0,96 , 95 % CI 0,84 - 1,09). Príslušná expozícia liečbe bola 13 135 a 13 050 pacientorokov.
V placebom kontrolovaných štúdiách s ďalším sledovaním vitálneho statusu po koniec zamýšľaného obdobia liečby, Spiriva Respimat preukázala numerické zvýšenie v mortalite zo všetkých príčin v porovnaní s placebom (pomer podielov (95 % interval spoľahlivosti) 1,33 (0,93, 1,92)) s liečebnou expozíciou Spirivou Respimat 2 574 pacientorokov; nárast mortality sa pozoroval u pacientov so známymi poruchami srdcového rytmu. Spiriva HandiHaler preukázala 13 % zníženie rizika úmrtia (pomer rizika vrátane ďalšieho sledovania vitálneho statusu (tiotropium/placebo) = 0,87; 95% CI, 0,76 až 0,99). Expozícia liečbe Spirivou HandiHaler bola 10 927 pacientorokov. V podskupine pacientov so známymi poruchami srdcového rytmu v štúdii so Spirivou HandiHaler kontrolovanou placebom ani v štúdii TIOSPIR porovnávajúcej Spirivu Respimat so Spirivou HandiHaler nebol pozorovaný žiadny nárast rizika mortality.
Klinická účinnosť a bezpečnosť v astmeProgram klinických štúdií fázy III, zameraný na perzistujúcu astmu dospelých, pozostával z dvoch 1-ročných randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdií celkovo u 907 pacientov s astmou (453 užívalo Spiriva Respimat), liečených kombináciou IKS (≥ 800 µg budezonidu/deň alebo ekvivalent) a LABA. Štúdie zahŕňali merania pľúcnych funkcií a závažné exacerbácie ako primárne koncové ukazovatele.
Štúdie PrimoTinA-asthmaVo dvoch 1-ročných štúdiách u pacientov, ktorí boli symptomatickí na udržiavacej liečbe aspoň s IKS (≥ 800 µg budezonidu/deň alebo ekvivalent)
plus LABA, Spiriva Respimat preukázala klinicky relevantné zlepšenia pľúcnych funkcií v porovnaní s placebom pri používaní ako prídavnej liečby k liečbe základnej.
V 24. týždni boli priemerné zlepšenia v „peak“ a „trough“ FEV
1 0,110 litra (95 % CI: 0,063 až 0,158 litra, p < 0,0001) a 0,093 litra (95 % CI: 0,050 až 0,137 litra, p < 0,0001), v uvedenom poradí. Zlepšenie pľúcnych funkcií v porovnaní s placebom sa udržalo 24 hodín.
V štúdiách PrimoTinA-asthma znížila liečba symptomatických pacientov (N = 453) s IKS plus LABA plus tiotropium riziko závažných exacerbácií astmy o 21 % v porovnaní s liečbou symptomatických pacientov (N = 454) s IKS plus LABA plus placebo. Zníženie rizika v priemernej hodnote počtu závažných exacerbácií astmy/pacientorok bolo 20 %.
Toto bolo podporené 31 % znížením rizika zhoršenia astmy a 24 % znížením rizika v priemernej hodnote počtu zhoršení astmy/pacientorok (pozri tabuľku 2).
Tabuľka 2: Exacerbácie u pacientov symptomatických pri užívaní IKS (≥ 800 µg budezonidu/deň alebo ekvivalent) plus LABA (štúdie PrimoTinA-asthma)
Štúdia
| Koncový ukazovateľ
| Spiriva Respimat,
pridaná aspoň k IKSa/LABA
(N = 453)
| Placebo,
pridané aspoň k IKSa/LABA
(N = 454)
| % zníženie rizika
(95 % CI)
| p-hodnota
|
Dve 1-ročné štúdie fázy III,
súhrn analýz
| Dni do 1. závažnej exacerbácie astmyt
| 282c
| 226c
| 21b
(0, 38)
| 0,0343
|
Priemerný počet závažných exacerbácií astmy/pacientorok
| 0,530
| 0,663
| 20d
(0, 36)
| 0,0458
|
Dni do 1. zhoršenia astmy
| 315c
| 181c
| 31b
(18, 42)
| < 0,0001
|
Priemerný počet zhoršení astmy/pacientorok
| 2,145
| 2,835
| 24d
(9, 37)
| 0,0031
|
a ≥ 8 00 µg budezonidu/deň alebo ekvivalentu
b Pomer rizík, interval spoľahlivosti a p-hodnota získané z Coxovho modelu proporcionálnych rizík iba s liečbou ako efektom. Percentuálne zníženie rizika je 100 (1 - pomer rizík).
c Čas do prvej udalosti: dni na liečbe dovtedy, kým sa u 25 %/50 % pacientov nevyskytne najmenej jedna závažná exacerbácia astmy/zhoršenie astmy
d Pomer podielov bol získaný z regresívne Poissonovho rozdelenia pravdepodobnosti s log expozíciou (v rokoch) ako ofset. Percentuálne zníženie rizika je 100 (1 - pomer podielov).
Pediatrická populáciaCHOCHP
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Spirivou Respimat vo všetkých podskupinách pediatrickej populácie s CHOCHP (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Astma
Všetky klinické štúdie fázy III v programe zameranom na perzistujúcu astmu u pediatrických pacientov (1 - 17 rokov) boli randomizované, dvojito zaslepené a placebom kontrolované. Všetci pacienti používali základnú liečbu, ktorá obsahoval IKS.
Závažná astmaDospievajúci (12 - 17 rokov) V 12-týždňovej štúdii PensieTinA-asthma bolo zaradených celkovo 392 pacientov (130 užívalo Spiriva Respimat), ktorí boli symptomatickí pri užívaní vysokej dávky IKS s jedným kontrolným liekom alebo stredne vysokej dávky IKS s dvoma kontrolnými liekmi.
U pacientov vo veku 12
- 17 rokov bola vysoká dávka IKS definovaná ako dávka > 800
- 1600 µg budezonidu/deň alebo ekvivalent; stredne vysoká dávka IKS ako 400
- 800 µg budezonidu/deň alebo ekvivalent. Okrem toho pacienti vo veku 12
- 14 rokov mohli dostať dávku IKS > 400 µg budezonidu/deň alebo ekvivalent a aspoň jeden kontrolný liek alebo ≥ 200 µg budezonidu/deň alebo ekvivalent a aspoň dva kontrolné lieky.
V tejto štúdii Spiriva Respimatpreukázala zlepšenia pľúcnych funkcií v porovnaní s placebom pri používaní ako prídavnej liečby k liečbe základnej, rozdiely v „peak“ a „trough“ FEV
1 však nebolo štatisticky signifikantné.
- V 12. týždni boli priemerné zlepšenia v „peak“ a „trough“ FEV1 0,090 litra (95 % CI:
-0.019 až 0,198 litra, p = 0,1039) a 0,054 litra (95 % CI: -0,061 to 0,168 litra, p = 0,3605), v uvedenom poradí.
- V 12. týždni Spiriva Respimat signifikantne zlepšila ranný a večerný PEF (ráno 17,4 l/min; 95 % CI: 5,1 až 29,6 l/min; večer 17,6 l/min; 95 % CI: 5,9 až 29,6 l/min).
Deti (6 – 11 rokov)V 12-týždňovej štúdii VivaTinA-asthma bolo zaradených celkovo 400 pacientov (130 užívalo Spirivu Respimat), ktorí boli symptomatickí pri užívaní vysokej dávky IKS s jedným kontrolným liekom alebo stredne vysokej dávky IKS s dvoma kontrolnými liekmi. Vysoká dávka IKS bola definovaná ako dávka > 400 µg budezonidu/deň alebo ekvivalent, stredne vysoká dávka ako 200
- 400 µg budezonidu/deň alebo ekvivalent.
V tejto štúdii Spiriva Respimatpreukázala významné zlepšenia pľúcnych funkcií v porovnaní s placebom pri používaní ako prídavnej liečby k liečbe základnej.
- V 12. týždni boli priemerné zlepšenia v „peak“ a „trough“ FEV1 0,139 litra (95 % CI: 0,075 až 0,203 litra, p < 0,0001) a 0,087 litra (95 % CI: 0,019 až 0,154 litra, p = 0,0117), v uvedenom poradí.
Stredne závažná astmaDospievajúci (12 - 17 rokov)V 1-týždňovej štúdii RubaTinA-asthma celkovo u 397 pacientov (134 užívalo Spirivu Respimat), ktorí boli symptomatickí pri užívaní stredne vysokej dávky IKS (200
- 800 µg budezonidu/deň alebo ekvivalent u pacientov vo veku 12
- 14 rokov alebo 400
- 800 µg budezonidu/deň alebo ekvivalent u pacientov vo veku 15
- 17 rokov), Spiriva Respimatpreukázala významné zlepšenia pľúcnych funkcií v porovnaní s placebom pri používaní ako prídavnej liečby k liečbe základnej.
Deti (6 - 11 rokov) V 1-týždňovej štúdii CanoTinA-asthma celkovo u 401 pacientov (135 užívalo Spirivu Respimat), ktorí boli symptomatickí pri užívaní stredne vysokej dávky IKS (200
- 400 µg budezonidu/deň alebo ekvivalent), Spiriva Respimatpreukázala významné zlepšenia pľúcnych funkcií v porovnaní s placebom pri používaní ako prídavnej liečby k liečbe základnej.
Deti (1 - 5 rokov)Uskutočnila sa jedna 12-týždňová randomizovaná, dvojito zaslepená, placebom kontrolovaná klinická štúdia fázy II/III (NinoTinA-asthma) celkovo so 101 pacientmi (31 užívalo Spirivu Respimat) s astmou používajúcich základnú liečbu, ktorá obsahovala IKS. Na podanie skúšaného lieku sa u 98 pacientov použil inhalačný nadstavec s ventilom Aerochamber Plus Flow-Vu
® s tvárovou maskou.
Primárnym cieľom štúdie bola bezpečnosť; hodnotenia účinnosti boli exploratívne.
Počet a percento pacientov, ktorí hlásili nežiaduce udalosti bez ohľadu na súvislosť, sú uvedené v Tabuľke 3. Počet nežiaducich udalostí súvisiacich s astmou bol nižší pri Spirive Respimat v porovnaní s placebom. Exploratívne hodnotenia účinnosti nepreukázali rozdiely medzi Spirivou Respimat a placebom.
Tabuľka 3: Frekvencia pacientov s nežiaducimi udalosťami hlásená u ≥ 5 pacientov v štúdii NinoTinA-asthma (deti vo veku 1 až 5 rokov)'
| Placebo N (%)
| Spiriva Respimat N (%)
|
Počet pacientov
| 34 (100,0)
| 31 (100,0)
|
Pacienti s akýmkoľvek nežiaducim účinkom
| 25 (73,5)
| 18 (58,1)
|
Nazofaryngitída
| 5 (14,7)
| 2 (6,5)
|
Infekcia horných dýchacích ciest
| 1 (2,9)
| 5 (16,1)
|
Astma*
| 10 (29,4)
| 2 (6,5)
|
Horúčka
| 6 (17,6)
| 3 (9,7)
|
* MedDRA termíny najnižšej úrovne pod preferovaným termínom „Astma“ boli buď „Zhoršenie astmy“, alebo „Exacerbácia astmy“.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Spirivou Respimat v podskupine pediatrických pacientov mladších ako 1 rok (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Klinická účinnosť a bezpečnosť pri cystickej fibróze (CF):Klinický vývojový program s CF zahŕňal 3 multicentrické štúdie s 959 pacientmi vo veku 5 mesiacov a viac. Pacienti mladší ako 5 rokov používali „spacer“ (AeroChamber Plus) s tvárovou maskou a boli zaradení len do hodnotenia bezpečnosti. Dve pivotné štúdie (štúdia fázy II so zisťovaním dávky a potvrdzujúca štúdia fázy III) porovnávali účinky na funkciu pľúc (percento predpovedajúce FEV
1 AUC
0-4h a „trough“ (minimálny) FEV
1) po podaní Spirivy Respimat (5 µg tiotropia: 469 pacientov) oproti placebu (315 pacientov) v 12-týždňových randomizovaných dvojito zaslepených periódach; štúdia fázy III zahŕňala aj predĺženie dlhodobej nezaslepenej štúdie až do 12 mesiacov. V týchto štúdiách boli ako súbežná liečba povolené všetky lieky na respiračné ochorenia okrem anticholinergík, napr. dlhodobo pôsobiace beta-agonisty, mukolytiká a antibiotiká.
Účinky na funkciu pľúc sú uvedené v Tabuľke 4. Nepozorovalo sa žiadne signifikantné zlepšenie príznakov ani zdravotného stavu (exacerbácie pomocou Dotazníka respiračných a systémových príznakov a Dotazníka kvality života pri cystickej fibróze).
Tabuľka 4: Upravený priemerný odklon absolútnych zmien východiskových hodnôt po 12 týždňoch od hodnôt placeba
| Fáza II
| Fáza III
|
Všetci pacienti
(NSpiriva = 176,
Nplacebo = 168)
| Všetci pacienti
(NSpiriva = 293,
Nplacebo = 147)
| ≤11 rokov
| ≥12 rokov
|
(NSpiriva = 95, Nplacebo = 47)
| (NSpiriva = 198, Nplacebo = 100)
|
priemer
(95 % CI)
| p-hodnota
| priemer
(95 % CI)
| p- hodnota
| priemer
(95 % CI)
| priemer
(95 % CI)
|
FEV1AUC0-4h (% odhadovaného) a
absolútne zmeny
| 3,39
(1,67; 5,12)
| <0,001
| 1,64
(-0,27; 3,55)
| 0,092
| -0,63
(-4,58; 3,32)
| 2,58
(0,50; 4,65)
|
FEV1AUC0-4h
(litre)
absolútne zmeny
| 0,09
(0,05; 0,14)
| <0,001
| 0,07
(0,02; 0,12)
| 0,010
| 0,01
(-0,07; 0,08)
| 0,10
(0,03; 0,17)
|
„Trough“ FEV1 (% odhadovaného) a
absolútne zmeny
| 2,22
(0,38; 4,06)
| 0,018
| 1,40
-0,50; 3,30
| 0,150
| -1,24
(-5,20; - 271)
| 2,56
(0,49; 4,62)
|
„Trough“ FEV1
(litre)
absolútne zmeny
| 0,06
(0,01, 0,11)
| 0,028
| 0,07
(0,02, 0,12)
| 0,012
| -0,01
(-0,08, 0,06)
| 0,10
(0,03, 0,17)
|
a Súbežné primárne koncové ukazovatele
Všetky nežiaduce rekcie lieku pozorované v štúdiách s CF sú známe nežiaduce účinky tiotropia (pozri časť 4.8). Najčastejšie pozorovanými nežiaducimi udalosťami považovanými za súvisiace počas 12-týždňovej dvojito zaslepenej periódy boli kašeľ (4,1 %) a sucho v ústach (2,8 %).
Počet a percento pacientov, ktorí hlásili nežiaduce udalosti osobitného záujmu pri cystickej fibróze bez ohľadu na súvislosť sú uvedené v Tabuľke 5. Prejavy a príznaky, ktoré sa považujú za prejavy cystickej fibrózy, sa zvýšili po podaní tiotropia numericky, no nie štatisticky významne, osobitne u pacientov ≤11 rokov.
Tabuľka 5: Percento pacientov s nežiaducimi udalosťami osobitného záujmu pri cystickej fibróze podľa vekových skupín počas 12-týždňov liečby bez ohľadu na súvislosť (súhrn fázy II a fázy III)
| ≤11 rokov
| ≥12 rokov
|
| Nplacebo = 96
| NSpiriva = 158
| Nplacebo = 215
| NSpiriva = 307
|
Bolesť brucha
| 7,3
| 7,0
| 5,1
| 6,2
|
Zápcha
| 1,0
| 1,9
| 2,3
| 2,6
|
Syndróm distálnej intestinálnej obštrukcie
| 0,0
| 0,0
| 1,4
| 1,3
|
Infekcie dýchacích ciest
| 34,4
| 36,7
| 28,4
| 28,3
|
Zvýšené spútum
| 1,0
| 5,1
| 5,6
| 6,2
|
Exacerbácie
| 10,4
| 14,6
| 18,6
| 17,9
|
„Syndróm distálnej intestinálnej obštrukcie“ a „zvýšené spútum" sú preferované termíny MedDRA. „Infekcie dýchacích ciest“ je termín najvyššej úrovne skupiny MedDRA. „Bolesť brucha“, „zápcha“ a „exacerbácie“ sú súborom preferovaných termínov MedDRA.
U tridsiatich štyroch (10,9 %) pacientov randomizovaných na placebo a 56 (12,0 %) pacientov randomizovaných na Spirivu Respimat sa vyskytla závažná nežiaduca udalosť.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Spirivou Respimat vo všetkých podskupinách pediatrickej populácie do veku 1 roka.
5.2 Farmakokinetické vlastnosti
a) Všeobecný úvodTiotropiumbromid je nechirálna kvartérna amónna látka a je slabo rozpustná vo vode. Tiotropiumbromid je dostupný ako inhalačný roztok podávaný pomocou Respimat inhalátora. Približne 40 % inhalovanej dávky sa ukladá v cieľovom orgáne, v pľúcach, zvyšná časť sa ukladá v gastrointestinálnom trakte. Niektoré nižšie popísané farmakokinetické údaje boli získané s použitím vyšších dávok ako sú dávky odporúčané na liečbu.
b) Všeobecná charakteristika liečiva po podaní liekuAbsorpcia: Údaje o vylučovaní do moču dokazujú, že po inhalácii u mladých zdravých dobrovoľníkov sa približne 33 % inhalovanej dávky dostane do cirkulácie. Perorálny roztok tiotropiumbromidu má absolútnu biologickú dostupnosť 2 - 3 %. Nepredpokladá sa, že jedlo má vplyv na absorpciu tejto kvartérnej amónnej zlúčeniny.
Maximálne plazmatické koncentrácie tiotropia boli pozorované 5 - 7 minút po inhalácii.
Vrcholové plazmatické hladiny tiotropia boli v rovnovážnom stave u pacientov s CHOCHP 10,5 pg/ml a rýchlo klesali multikompartmentovým spôsobom. Rovnovážny stav hladiny plazmatických koncentrácií bol 1,60 pg/ml.
Vrcholová plazmatická koncentrácia tiotropia v rovnovážnom stave 5,15 pg/ml bola dosiahnutá 5 minút po podaní rovnakej dávky pacientom s astmou.
Systémová expozícia tiotropia po inhalovaní tiotropia cez inhalátor Respimat bola podobná tiotropiu inhalovanému cez pomôcku HandiHaler.
Distribúcia: Väzba liečiva na plazmatické bielkoviny je 72 % a distribučný objem je 32 l/kg. Lokálna koncentrácia v pľúcach nie je známa, ale spôsob podávania poukazuje na podstatne vyššie koncentrácie v pľúcach. Štúdie na potkanoch ukazujú, že tiotropium vo významnom rozsahu neprechádza hematoencefalickou bariérou.
Biotransformácia: Rozsah biotransformácie je malý. Je to zrejmé z renálnej exkrécie 74 % nezmenenej látky po intravenóznom podaní u mladých zdravých dobrovoľníkov. Ester tiotropiumbromidu sa neenzymaticky štiepi na alkohol (N-metylskupín) a kyselinu (kyselinu dietylénglykolovú), ktoré sú na muskarínových receptoroch inaktívne. In vitro experimenty na mikrozómoch ľudskej pečene a ľudských hepatocytoch naznačujú, že určité množstvo látky (<20 % dávky po intravenóznom podaní) sa metabolizuje oxidáciou závislou na cytochróme P450 (CYP) a následne konjugáciou s glutatiónom na rôzne metabolity fázy II.
V in vitro štúdiách na pečeňových mikrozómoch sa zistilo, že enzymatická cesta môže byť inhibovaná CYP 2D6 (a 3A4) inhibítormi, chinidínom, ketokonazolom a gestodénom. CYP 2D6 a 3A4 sa zúčastňujú pri eliminácii len malého množstva dávky. Tiotropiumbromid aj pri veľmi vysokých terapeutických koncentráciách neinhibuje CYP 1A1, 1A2, 2B6, 2C9, 2D6, 2E1 alebo 3A v mikrozómoch ľudskej pečene.
Eliminácia: Účinný polčas tiotropia po inhalácii zdravými dobrovoľníkmi a pacientmi s CHOCHP sa pohybuje medzi 27 - 45 hodinami. Účinný polčas u pacientov s astmou bol 34 hodín. Celkový klírens kreatinínu po intravenóznej dávke u zdravých mladých dobrovoľníkov bol 880 ml/min. Intravenózne podané tiotropium je väčšinou nezmenené vylúčené močom (74 %).
Po inhalácii roztoku pacientmi s CHOCHP do dosiahnutia rovnovážneho stavu tvorí renálne vylučovanie 18,6 % (0,3 µg dávky, zvyšok predstavuje neabsorbovanú látku v čreve, ktorá je vylúčená stolicou.
Po inhalácii roztoku zdravými dobrovoľníkmi renálne vylučovanie tvorí 20,1 - 29,4 % dávky, zvyšok predstavuje neabsorbovanú látku v čreve, ktorá je vylúčená stolicou.
U pacientov s astmou sa 11,9 % (0,595 µg) dávky vylučuje nezmenenej močom v priebehu 24 hodín od podania dávky v rovnovážnom stave. Renálny klírens tiotropia prevyšuje klírens kreatinínu, čo naznačuje sekréciu látky do moču.
Po dlhodobej inhalácii jedenkrát denne pacientmi s CHOCHP sa farmakokinetický rovnovážny stav dosiahol do 7 dní bez ďalšej kumulácie liečiva.
Linearita/nelinearita: Tiotropium vykazoval lineárnu farmakokinetiku v terapeutických dávkach nezávisle od formy.
c) Osobitné skupiny pacientovStarší pacienti: Tak ako sa očakáva pri všetkých prevažne renálne vylučovaných liekoch, zvyšujúci vek sa spája so znížením renálneho klírensu tiotropia (347 ml/min u pacientov s CHOCHP < ako 65-ročných oproti 275 ml/min u pacientov s CHOCHP > ako 65-ročných). Toto neviedlo k zodpovedajúcemu zvýšeniu AUC
0-6, ss ani hodnôt C
max,ss. Nezistilo sa, že by sa u pacientov s astmou expozícia tiotropia odlišovala v závislosti od veku.
Pacienti s poruchou funkcie obličiek:
Po inhalačnom podaní tiotropia raz denne do dosiahnutia rovnovážneho stavu u pacientov s CHOCHP viedla mierna porucha funkcie obličiek (CL
CR 50 - 80 ml/min) k mierne vyššej AUC
0-6,ss (o 1,8 – 30 % vyššej) a podobným hodnotám C
max,ss v porovnaní s pacientmi s normálnou funkciou obličiek (CL
CR > 80 ml/min).
U pacientov s CHOCHP, ktorí majú stredne ťažkú až ťažkú poruchu funkcie obličiek (CL
CR < 50 ml/min), intravenózne podávanie jednej dávky tiotropia zdvojnásobuje celkovú expozíciu (o 82 % vyššia AUC
0-4h a o 52 % vyššia C
max) v porovnaní s pacientmi s CHOCHP s normálnou funkciou obličiek, čo sa potvrdilo aj pri plazmatických koncentráciách po inhalácii suchého prášku. U astmatických pacientov s miernou poruchou funkcie obličiek (CL
CR 50 - 80 ml/min) inhalačne podávané tiotropium neviedlo k relevantným nárastom v expozícii v porovnaní s pacientmi s normálnou funkciou obličiek.
Pacienti s poruchou funkcie pečene: Neočakáva sa, že by pečeňová nedostatočnosť mala relevantný vplyv na farmakokinetiku tiotropia. Tiotropium je predominantne eliminovaný renálnou exkréciou (74 % u mladých zdravých dobrovoľníkov) a jednoduchý ester sa neenzymaticky štiepi na farmakokineticky neaktívne látky.
Japonskí pacienti s CHOCHP: Pri vzájomnom porovnaní štúdií boli priemerné vrcholové plazmatické koncentrácie tiotropia 10 minút po podaní dávky v rovnovážnom stave o 20 % až 70 % vyššie u japonských pacientov oproti pacientom bielej rasy s CHOCHP po inhalovaní tiotropia, ale u japonských pacientov v porovnaní s pacientmi bielej rasy nebol náznak vyššej mortality ani vyššieho rizika pre srdce. Za ďalšie etnicity alebo rasy nie sú k dispozícii dostačujúce farmakokinetické údaje.
Pediatrickí pacienti:AstmaVrcholová a celková (AUC a renálne vylučovanie) expozícia tiotropiu sú porovnateľné medzi pacientmi s astmou, ktorí boli vo veku 6 - 11 rokov, 12 - 17 rokov a ≥18 rokov. Na základe renálneho vylučovania bola celková expozícia tiotropiu u pacientov vo veku 1 až 5 rokov o 52 až 60 % nižšia ako v iných starších vekových skupinách. Zistilo sa, že údaje o celkovej expozícii boli po upravení na plochu povrchu tela porovnateľné vo všetkých vekových skupinách. Spiriva Respimat sa pacientom vo veku 1 až 5 rokov podávala s inhalačným nadstavcom s ventilom a s tvárovou maskou.
CHOCHP
V programe CHOCHP neboli žiadni pediatrickí pacienti (pozri 4.2).
Cystická fibrózaPo inhalácii 5 µg tiotropia bola plazmatická hladina tiotropia u pacientov s CF ≥ 5 rokov 10,1 pg/ml 5 minút po podaní dávky pri rovnovážnom stave a potom sa rapídne znížila. Frakcia dávky dostupnej u pacientov s CF vo veku < 5 rokov, ktorí používali spacer a masku bola približne 3- až 4-násobne nižšia než dávka, ktorá sa pozorovala u 5-ročných a starších pacientov s CF. Expozícia tiotropia súvisela s telesnou hmotnosťou pacientov s CF < 5 rokov.
d) Vzťah farmakokinetika/farmakodynamikaNie je priamy vzťah medzi farmakokinetikou a farmakodynamikou.
5.3 Predklinické údaje o bezpečnosti
Mnohé účinky pozorované v konvenčných štúdiách, týkajúce sa bezpečnosti farmakológie, toxicity opakovanej dávky a reprodukčnej toxicity, sa dajú vysvetliť anticholinergickými vlastnosťami tiotropiumbromidu. U zvierat sa pozorovala znížená spotreba potravy, nižší váhový prírastok, sucho slizníc úst a nosa, znížená lakrimácia a salivácia, mydriáza a zvýšená srdcová frekvencia. Ostatné relevantné účinky sledované pri štúdiách toxicity opakovanej dávky boli: mierne dráždenie dýchacích ciest u potkanov a myší v zmysle rinitídy a zmien epitelu v nosovej dutine a v hrtane, prostatitída s proteínovými depozitmi, litiáza močového mechúra u potkanov.
U juvenilných potkanov vystavených účinku od 7. dňa po narodení do pohlavnej zrelosti sa pozorovali rovnaké priame a nepriame farmakologické zmeny ako v štúdiách toxicity s opakovaným podávaním dávky ako aj rinitídou. Nezaznamenala sa žiadna systémová toxicita a nepozorovali sa žiadne účinky súvisiace s toxicitou na kľúčové vývojové parametre, vývoj priedušnice ani na vývoj dôležitých orgánov.
Škodlivý vplyv na tehotenstvo, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj sa zistil len pri maternálnych toxických dávkach. Tiotropiumbromid nie je teratogénny u potkanov alebo králikov. Vo všeobecnosti, štúdie reprodukcie a fertility u potkanov nepreukázali, pri akomkoľvek dávkovaní, žiadny nežiaduci účinok na fertilitu alebo schopnosť párenia sa oboch liečených rodičov alebo ich potomkov.
Zmeny na respiračnom (iritácia) a urogenitálnom (prostatitída) systéme a reprodukčná toxicita sa zistili pri lokálnom a systémovom podávaní dávok vyšších ako päťnásobok terapeutickej dávky. Štúdie zamerané na genotoxicitu a karcinogénny potenciál nepreukázali u ľudí žiadne výnimočné riziko.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
benzalkóniumchlorid
dinátriumedetát
čistená voda
kyselina chlorovodíková 3,6% (na úpravu pH)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
Čas použiteľnosti po prvom otvorení: 3 mesiace
6.4 Špeciálne upozornenia na uchovávanie
Nezmrazujte.
6.5 Druh obalu a obsah balenia
Druh a materiál kontajnera, ktorý je v kontakte s liekom:
Roztok je naplnený do polyetylén/polypropylénových náplní s polypropylénovým ochranným uzáverom s integrovaným silikónovým tesniacim krúžkom. Náplň je uložená v hliníkovom valci.
Veľkosť balenia a priložená pomôcka:
Jednotlivé balenie: 1 Respimat inhalátor a 1 náplň, obsahujúca 60 vstrekov (30 liečebných dávok)
Dvojité balenie: 2 samostatné balenia, každé obsahuje 1 Respimat inhalátor a 1 náplň, obsahujúcu 60 vstrekov (30 liečebných dávok)
Trojité balenie: 3 samostatné balenia, každé obsahuje 1 Respimat inhalátor a 1 náplň, obsahujúcu 60 vstrekov (30 liečebných dávok)
Osem-balenie: 8 samostatných balení, každé obsahuje 1 Respimat inhalátor a 1 náplň, obsahujúcu 60 vstrekov (30 liečebných dávok)
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Boehringer Ingelheim International GmbH
Binger Strasse 173
D-55216 Ingelheim nad Rýnom
Nemecko
8. REGISTRAČNÉ ČÍSLO
14/0315/07-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 28. 08. 2007
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTU
Apríl 2018