
/>V prípade predávkovania sa odporúča, aby sa u pacienta sledovali prípadné prejavy alebo príznaky nežiaducich reakcií a aby sa okamžite začalo s vhodnou symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu, ATC kód: L04AC18
Mechanizmus účinku
Rizankizumab je humanizovaná monoklonálna imunoglobulínová protilátka (IgG1), ktorá sa
selektívne viaže s vysokou afinitou na podjednotku p19 humánneho cytokínu označovaného ako interleukín 23 (IL-23) bez väzby na IL-12 a inhibuje jeho interakciu s receptorovým komplexom
IL-23. IL-23 je cytokín, ktorý sa podieľa na zápalovej a imunitnej odpovedi. Tým, že rizankizumab blokuje IL-23 a bráni mu vo väzbe na jeho receptor, inhibuje bunkovú signalizáciu závislú od IL-23 a uvoľňovanie prozápalových cytokínov.
Farmakodynamické účinky
V štúdii u jedincov so psoriázou bola po jednotlivých dávkach rizankizumabu znížená expresia génov
súvisiacich s osou IL-23/IL-17 v koži. V psoriatických léziách bolo pozorované aj zníženie hrúbky epidermy, infiltrácie zápalovými bunkami a expresie markerov psoriázy.
V štúdii u jedincov so psoriatickou artritídou sa po liečbe rizankizumabom 150 mg podávaným subkutánne v 0. týždni, 4. týždni a potom každých 12 týždňov zaznamenalo v 24. týždni štatisticky
a klinicky významné zníženie IL-23 a biomarkerov súvisiacich s IL-17 vrátane IL-17A, IL-17F a IL-
22 v porovnaní s východiskovými hodnotami.
Klinická účinnosťabezpečnosť
Ložisková psoriáza
Účinnosť a bezpečnosť rizankizumabu boli hodnotené u 2109 jedincov so stredne ťažkou až ťažkou ložiskovou psoriázou v štyroch multicentrických, randomizovaných, dvojito zaslepených štúdiách (ULTIMMA-1, ULTIMMA-2, IMMHANCE a IMMVENT). Jedinci s ložiskovou psoriázou zaradení do štúdií boli vo veku 18 rokov a viac, ktorí mali postihnutie ≥ 10 % telesného povrchu (body surface area, BSA) a ktorí mali statické skóre celkového hodnotenia lekárom (static Physician Global Assessment, sPGA) ≥ 3 v celkovom hodnotení (hrúbka ložiska/indurácia, erytém a tvorba šupín) psoriázy na škále závažnosti od 0 do 4, index plochy postihnutia a závažnosti psoriázy (Psoriasis Area and Severity Index, PASI) ≥ 12 a ktorí boli kandidáti na systémovú liečbu alebo fototerapiu.
Celkovo mali jedinci medián východiskového skóre PASI 17,8, medián BSA 20,0 % a medián východiskového skóre DLQI 13, %. Východiskové skóre sPGA bolo závažné u 19,3 % jedincov a stredne závažné u 80,7 % jedincov. Celkovo 9,8 % jedincov v štúdiách malo diagnostikovanú psoriatickú artritídu v anamnéze.
Naprieč všetkými štúdiami 30,9 % jedincov bolo bez akejkoľvek predchádzajúcej systémovej liečby (vrátane nebiologickej a biologickej liečby); 38,1 % dostalo predchádzajúcu fototerapiu alebo fotochemoterapiu; 48,3 % dostalo predchádzajúcu nebiologickú systémovú liečbu; 42,1 % dostalo predchádzajúcu biologickú liečbu a 23,7 % dostalo najmenej jeden anti-TNF alfa liek na liečbu psoriázy.
ULTIMMA-1 a ULTIMMA-2
Do štúdií ULTIMMA-1 a ULTIMMA-2 bolo zaradených 997 jedincov (598 bolo randomizovaných na
rizankizumab 150 mg, 199 na ustekinumab 45 mg alebo 90 mg [podľa východiskovej telesnej hmotnosti] a 200 na placebo). Jedinci dostávali liečbu v 0. týždni, 4. týždni a následne každých
12 týždňov. Dva koprimárne cieľové ukazovatele v štúdiách ULTIMMA-1 a ULTIMMA-2 boli podiel jedincov, ktorí dosiahli 1) odpoveď PASI 90 a 2) skóre sPGA čistá alebo takmer čistá koža (sPGA 0 alebo 1) v 16. týždni v porovnaní s placebom. Výsledky pre koprimárne a ďalšie cieľové ukazovatele
sú uvedené v tabuľke 2 a na obrázku 1.
Tabuľka 2: Výsledky účinnosti a kvality života u dospelých s ložiskovou psoriázou v štúdiách
ULTIMMA 1 a ULTIMMA 2
|
ULTIMMA-1
|
ULTIMMA-2
|
|
Rizankizumab
(N = 304)
n (%)
|
Ustekinumab
(N = 100)
n (%)
|
Placebo (N = 102) n (%)
|
Rizankizumab
(N = 294)
n (%)
|
Ustekinumab
(N = 99)
n (%)
|
Placebo (N = 98) n (%)
|
sPGA čistá alebo takmer čistá koža (0 alebo 1)
|
16. týžde
ň
a
|
267 (87,8)
|
63 (63,0)
|
8 (7,8)
|
246 (83,7)
|
61 (61,6)
|
5 (5,1)
|
52. týždeň
|
262 (86,2)
|
54 (54,0)
|
--
|
245 (83,3)
|
54 (54,5)
|
--
|
sPG
A čistá koža (0)
|
16. týždeň
|
112 (36,8)
|
14 (14,0)
|
2 (2,0)
|
150 (51,0)
|
25 (25,3)
|
3 (3,1)
|
52. týždeň
|
175 (57,6)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
|
PASI 75
|
12. týždeň
|
264 (86,8)
|
70 (70,0)
|
10 (9,8)
|
261 (88,8)
|
69 (69,7)
|
8 (8,2)
|
52. týždeň
|
279 (91,8)
|
70 (70,0)
|
--
|
269 (91,5)
|
76 (76,8)
|
--
|
PASI 90
|
16. týžde
ň
a
|
229 (75,3)
|
42 (42,0)
|
5 (4,9)
|
220 (74,8)
|
47 (47,5)
|
2 (2,0)
|
52. týždeň
|
249 (81,9)
|
44 (44,0)
|
--
|
237 (80,6)
|
50 (50,5)
|
--
|
PASI 100
|
16. týždeň
|
109 (35,9)
|
12 (12,0)
|
0 (0,0)
|
149 (50,7)
|
24 (24,2)
|
2 (2,0)
|
52. týždeň
|
171 (56,3)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
|
DLQI 0 or 1
b
|
16. týždeň
|
200 (65,8)
|
43 (43,0)
|
8 (7,8)
|
196 (66,7)
|
46 (46,5)
|
4 (4,1)
|
52. týždeň
|
229 (75,3)
|
47 (47,0)
|
--
|
208 (70,7)
|
44 (44,4)
|
--
|
PSS 0 (bez symptómov)
c
|
16. týždeň
|
89 (29,3)
|
15(15,0)
|
2 (2,0)
|
92 (31,3)
|
15 (15,2)
|
0 (0,0)
|
52. týždeň
|
173 (56,9)
|
30 (30,0)
|
--
|
160 (54,4)
|
30 (30,3)
|
--
|
Všetky porovnania rizankizumabu oproti ustekinumabu a placebu dosiahli p < 0,001, okrem PASI 75 v 52. týždni v štúdii ULTIMMA-2, kde p = 0,001
a Koprimárne cieľové ukazovatele verzus placebo
b Bez dopadu na kvalitu života súvisiacu so zdravím
c Psoriasis Symptom Scale (PSS) 0 znamená žiadne príznaky bolesti, svrbenia, začervenania a pálenia počas posledných 24 hodín.
|
Obrázo
k 1: Časový priebeh priemernej percentuálnej zmeny od východiskového stavu PASI
v štúdii ULTIMMA-1 a ULTIMMA-2
0
-10
-20
-30
-40
-50
-60
RZB UST
PBO
-70
-80
-90

-100
0 4 8 12 16 22 28 34 40 46 52
RZB = rizankizumab UST = ustekinumab PBO = placebo
p < 0,001 v každom časovom bode
Týždne
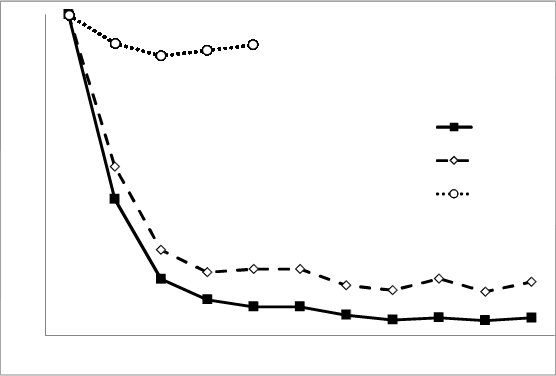
Nepreukázali sa rozdiely v odpovedi na rizankizumab medzi podskupinami podľa veku, pohlavia,
rasy, telesnej hmotnosti ≤ 130 kg, východiskového skóre PASI, súbežnej psoriatickej artritídy, predchádzajúcej nebiologickej systémovej liečby, predchádzajúcej biologickej liečby a predchádzajúceho zlyhania biologickej liečby.
Zlepšenia sa pozorovali pri psoriáze, ktorá postihovala kožu hlavy, nechty, dlane a chodidlá v 16. a 52. týždni u jedincov liečených rizankizumabom.
Tabuľka 3: Priemerné zmeny NAPSI, PPASI, and PSSI oproti východiskovým hodnotám
|
ULTIMMA-1
|
ULTIMMA-2
|
IMMHANCE
|
|
Rizankizumab
|
Placebo
|
Rizankizumab
|
Placebo
|
Rizankizumab
|
Placebo
|
NAPSI: Zmena
v 16. týždni
(SE)
|
N = 178;
-9,0 (1,17)
|
N = 56;
2,1
(1,86)
***
|
N = 177;
-7,5 (1,03)
|
N = 49;
3,0
(1,76)
***
|
N = 235;
-7,5 (0,89)
|
N = 58;
2,5
(1,70)
***
|
PPASI: Zmena
v 16. týždni
(SE)
|
N = 95;
-5,93 (0,324)
|
N = 34;
-3,17
(0,445)
***
|
N = 86;
-7,24 (0,558)
|
N = 23;
-3,74
(1,025)
**
|
N = 113;
-7,39 (0,654)
|
N = 26;
-0,27
(1,339)
***
|
PSSI: Zmena
v 16. týždni
(SE)
|
N = 267;
-17,6 (0,47)
|
N = 92;
-2,9
(0,69)
***
|
N = 252;
-18,4 (0,52)
|
N = 83;
-4,6 (0,82)
***
|
N = 357;
-20,1 (0,40)
|
N = 88;
-5,5 (0,77)
***
|
NAPSI: Zmena
v 52. týždni
(SE)
|
N = 178;
-15,7 (0,94)
|
-
|
N = 183;
-16,7 (0,85)
|
-
|
-
|
-
|
PPASI: Zmena
v 52. týždni
(SE)
|
N = 95;
-6,16 (0,296)
|
-
|
N = 89;
-8,35 (0,274)
|
-
|
-
|
-
|
PSSI: Zmena
v 52. týždni
(SE)
|
N = 269;
-17,9 (0,34)
|
-
|
N = 259;
-18,8 (0,24)
|
-
|
-
|
-
|
Nail Psoriasis Severity Index (NAPSI), Palmoplantar Psoriasis Severity Index (PPASI), Psoriasis
Scalp Severity Index (PSSI), Standard Error (SE)
** P < 0,01 v porovnaní s rizankizumabom
*** P < 0,001 v porovnaní s rizankizumabom
|
Podľa merania na stupnici Hospital Anxiety and Depression (HADS) sa skupina pacientov
s rizankizumabom zlepšila v 16. týždni v porovnaní so skupinou s placebom.
Udržanie odpovedeV integrovanej analýze jedincov, ktorí dostávali rizankizumab v štúdiách ULTIMMA-1 a ULTIMMA-
2, s odpoveďou PASI 100 v 16. týždni, si 79,8 % (206/258) jedincov, ktorí ďalej dostávali rizankizumab, udržalo odpoveď do 52. týždňa. V prípade jedincov s odpoveďou PASI 90 v 16. týždni si do 52. týždňa udržalo odpoveď 88,4 % (398/450) jedincov.
Bezpečnostný profil rizankizumabu až do 77 týždňov expozície bol konzistentný s profilom pozorovaným do 16 týždňov.
IMMHANCEDo štúdie IMMHANCE bolo zaradených 507 jedincov (407 bolo randomizovaných na rizankizumab
150 mg a 100 na placebo). Jedinci dostávali liečbu v 0. týždni, 4. týždni a následne každých
12 týždňov. Jedinci, ktorí dostávali pôvodne rizankizumab a mali odpoveď sPGA čistá alebo takmer čistá koža v 28. týždni, boli opakovane randomizovaní na pokračovanie v podávaní rizankizumabu každých 12 týždňov do 88. týždňa (so 16-týždňovým obdobím sledovania po poslednej dávke rizankizumabu) alebo bola liečba prerušená.
V 16. týždni bol rizankizumab lepší ako placebo v koprimárnych cieľových ukazovateľoch sPGA čistá alebo takmer čistá koža (83,5 % rizankizumab vs 7,0 % placebo) a PASI 90 (73,2 % rizankizumab vs
2,0 % placebo).
Z 31 jedincov zo štúdie IMMHANCE s latentnou tuberkulózou (TBC), ktorí nedostávali profylaxiu počas štúdie, sa u žiadneho neobjavila aktívna TBC počas priemerného obdobia sledovania 55 týždňov pri používaní rizankizumabu.
Z jedincov, ktorí mali sPGA čistá alebo takmer čistá koža v 28. týždni v štúdii IMMHANCE, si udržalo túto odpoveď 81,1 % (90/111) jedincov opakovane randomizovaných na pokračovanie liečby rizankizumabom do 104. týždňa v porovnaní so 7,1 % (16/225) jedincov, ktorí boli opakovane randomizovaní na vysadenie rizankizumabu. Z týchto jedincov dosiahlo sPGA čistá koža v 104. týždni
63,1 %(70/111) jedincov opakovane randomizovaných na pokračovanie liečby rizankizumabom
v porovnaní s 2,2 % (5/225) jedincov, ktorí boli opakovane randomizovaní na prerušenie liečby rizankizumabom.
Z jedincov, ktorí dosiahli sPGA čistá alebo takmer čistá koža v 28. týždni a po prerušení liečby rizankizumabom u nich došlo k relapsu na sPGA stredne ťažké alebo ťažké ochorenie, 83,7 % (128/153) znovu dosiahlo sPGA čistá alebo takmer čistá koža po 16 týždňoch opakovanej liečby. Strata sPGA čistá alebo takmer čistá koža sa pozorovala už po 12 týždňoch od vynechania dávky. Z jedincov, ktorí boli opakovane randomizovaní na prerušenie liečby, došlo u 80,9 % (182/225)
k relapsu a medián času do relapsu bol 295 dní. Nezistili sa žiadne charakteristiky, ktoré by umožňovali predvídať čas do straty odpovede alebo pravdepodobnosť opätovného dosiahnutia odpovede na úrovni jednotlivých pacientov.
IMMVENTDo štúdie IMMVENT bolo zaradených 605 jedincov (301 bolo randomizovaných na rizankizumab
a 304 na adalimumab). Jedinci randomizovaní na rizankizumab dostali 150 mg v 0. týždni, 4. týždni a následne každých 12 týždňov. Jedinci randomizovaní na adalimumab dostali 80 mg v 0. týždni,
40 mg v 1. týždni a 40 mg každý druhý týždeň do 15. týždňa. Od 16. týždňa jedinci, ktorí dostávali
adalimumab, pokračovali v liečbe alebo ich liečba bola zmenená v závislosti od odpovede:
· < PASI 50 liečba bola zmenená na rizankizumab
· PASI 50 až < PASI 90 jedinci boli opakovane randomizovaní buď na pokračovanie liečby adalimumabom, alebo ich liečba bola zmenená na rizankizumab
· PASI 90 jedinci dostávali naďalej adalimumab
Výsledky sú uvedené v tabuľke 4.
Tabuľka 4: Výsledky účinnosti a kvality života v 16. týždni u dospelých s ložiskovou psoriázou v štúdii IMMVENT
| Rizankizumab
(N = 301)
n (%)
| Adalimumab
(N = 304)
n (%)
|
sPGA čistá alebo takmer čistá kožaa
|
252 (83,7)
|
183 (60,2)
|
PASI 75
| 273 (90,7)
| 218 (71,7)
|
PASI 90a
| 218 (72,4)
| 144 (47,4)
|
PASI 100
| 120 (39,9)
| 70 (23,0)
|
DLQI 0 alebo 1b
| 198 (65,8)
| 148 (48,7)
|
Všetky porovnania dosiahli p < 0,001
a Koprimárne cieľové ukazovatele
b Bez dopadu na kvalitu života súvisiacu so zdravím
|
U jedincov, ktorí mali pri používaní adalimumabu v 16. týždni PASI 50 až < PASI 90 a boli opakovane randomizovaní, boli zaznamenané rozdiely v miere odpovede PASI 90 medzi zmenou liečby na rizankizumab a pokračovaním v podávaní adalimumabu 4 týždne po opakovanej randomizácii (49,1 % vs 26,8 %, v uvedenom poradí).
Výsledky po opakovanej randomizácii v 28. týždni sú prezentované v tabuľke 5 a na obrázku 2.
Tabuľka 5: Výsledky účinnosti 28 týždňov po opakovanej randomizácii v štúdii IMMVENT
| Zmena na rizankizumab (N = 53)
n (%)
| Pokračovanie
s adalimumabom
(N = 56)
n (%)
|
PASI 90
| 35 (66,0)
| 12 (21,4)
|
PASI 100
| 21 (39,6)
| 4 (7,1)
|
Všetky porovnania dosiahli p < 0,001
|
Obrázok 2: Časový priebeh PASI 90 po opakovanej randomizácii v štúdii IMMVENT
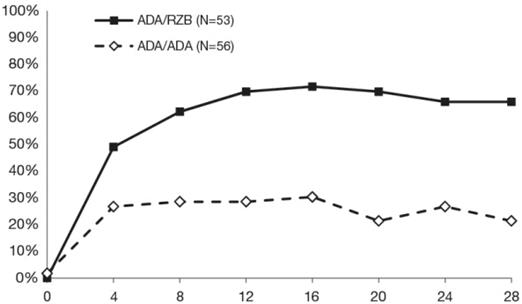 Týždne po opakovanej randomizácii
Týždne po opakovanej randomizáciiADA/ADA: Jedinci, ktorí boli randomizovaní na adalimumab a pokračovali v liečbe adalimumabom
ADA/RZB: Jedinci, ktorí boli randomizovaní na adalimumab a ich liečba bola zmenená na rizankizumab
p < 0,05 v 4. týždni a p < 0,001 v každom časovom bode počínajúc 8. týždňom
U 270 jedincov, ktorí prešli z adalimumabu na rizankizumab bez obdobia bez liečby (wash-out), bol bezpečnostný profil rizankizumabu podobný ako u jedincov, ktorí začali liečbu rizankizumabom po období bez liečby po akejkoľvek predchádzajúcej systémovej liečbe.
Psoriatická artritídaUkázalo sa, že rizankizumab zlepšuje u dospelých s aktívnou psoriatickou artritídou (PsA) prejavy
a príznaky, fyzickú funkciu, kvalitu života súvisiacu so zdravím a podiel jedincov bez rádiografickej progresie.
Bezpečnosť a účinnosť rizankizumabu sa hodnotila u 1407 jedincov s aktívnou PsA v 2 randomizovaných dvojito zaslepených, placebom kontrolovaných štúdiách (964 v KEEPSAKE1 a 443 v KEEPSAKE2).
Jedinci v týchto štúdiách mali najmenej 6 mesiacov diagnostikovanú PsA na základe klasifikačných kritérií pre psoriatickú artritídu (Classification Criteria for Psoriatic Arthritis, CASPAR), medián trvania PsA 4,9 roka na začiatku štúdie, ≥ 5 citlivých kĺbov a ≥ 5 opuchnutých kĺbov a aktívnu ložiskovú psoriázu alebo psoriázu nechtov na začiatku štúdie. 55,9 % jedincov malo ≥ 3 % BSA postihnutých ložiskovou psoriázou. Entezitídu malo 63,4 % a daktylitídu 27,9 % jedincov. V štúdii KEEPSAKE1, kde sa ďalej hodnotila psoriáza nechtov, malo 67,3 % jedincov psoriázu nechtov.
V oboch štúdiách boli jedinci randomizovaní na podávanie 150 mg rizankizumabu alebo placeba v 0.,
4. a 16. týždni. Od 28. týždňa dostávali všetci jedinci rizankizumab každých 12 týždňov.
V štúdii KEEPSAKE1 mali všetci jedinci predchádzajúcu nedostatočnú odpoveď alebo intoleranciu na nebiologickú liečbu DMARD a zatiaľ neabsolvovali biologickú liečbu. V štúdii KEEPSAKE2 malo
53,5 % jedincov predchádzajúcu nedostatočnú odpoveď alebo intoleranciu na nebiologickú liečbu
DMARD a 46,5 % jedincov malo predchádzajúcu nedostatočnú odpoveď alebo intoleranciu na biologickú liečbu.
V oboch štúdiách 59,6 % jedincov dostávalo súbežne metotrexát (MTX); 11,6 % dostávalo súbežne nebiologické DMARD iné ako MTX a 28,9 % dostávalo monoterapiu rizankizumabom.
Klinická odpoveďLiečba rizankizumabom viedla k významnému zlepšeniu ukazovateľov aktivity ochorenia v porovnaní
s placebom v 24. týždni. V oboch štúdiách bol primárnym cieľovým ukazovateľom podiel jedincov,
ktorí v 24. týždni dosiahli odpoveď ACR20 podľa Amerického kolégia reumatológie (American
College of Rheumatology, ACR). Kľúčové výsledky účinnosti sú uvedené v tabuľke 6.
Tabuľka 6: Výsledky účinnosti v štúdiách KEEPSAKE1 a KEEPSAKE2
| KEEPSAKE1
| KEEPSAKE2
|
Cieľový
Ukazovateľ
| Placebo N = 481 n (%)
| Rizankizumab
N = 483
n (%)
| Placebo N = 219 n (%)
| Rizankizumab
N = 224
n (%)
|
Odpoveď ACR20
|
16. týždeň
| 161 (33,4)
| 272 (56,3)a
| 55 (25,3)
| 108 (48,3)a
|
24. týždeň
| 161 (33,5)
| 277 (57,3)a
| 58 (26,5)
| 115 (51,3)a
|
52. týždeň*
| -
| 338/433 (78,1)
| -
| 131/191 (68,6)
|
Odpoveď ACR50
|
24. týždeň
| 54 (11,3)
| 162 (33,4)b
| 20 (9,3)
| 59 (26,3)b
|
52. týždeň*
| -
| 209/435 (48,0)
| -
| 72/192 (37,5)
|
Odpoveď ACR70
|
24. týždeň
| 23 (4,7)
| 74 (15,3)b
| 13 (5,9)
| 27 (12,0)c
|
52. týždeň*
| -
| 125/437 (28,6)
| -
| 37/192 (19,3)
|
Riešenie entezitídy (LEI = 0)
|
24. týždeň*
| 156/448 (34,8)d
| 215/444 (48,4)a, d
| -
| -
|
52. týždeň*
| -
| 244/393 (62,1)d
| -
| -
|
Riešenie daktylitídy (LDI = 0)
|
24. týždeň*
| 104/204 (51,0)e
| 128/188 (68,1)a, e
| -
| -
|
52. týždeň*
|
-
|
143/171 (83,6)e
|
-
|
-
|
Odpoveď MDA (Minimal Disease Activity, minimálna aktivita ochorenia)
|
24. týždeň
|
49 (10,2)
|
121 (25,0)a
|
25 (11,4)
|
57 (25,6)a
|
52. týždeň*
|
-
|
183/444 (41,2)
|
-
|
61/197 (31,0)
|
* Údaje sú zobrazené pre dostupných jedincov vo formáte n/N pozorovaných (%).
a) Multiplicitou kontrolované porovnanie p ≤ 0,001 rizankizumab vs placebo.
b) Nominálne porovnanie p ≤ 0,001 rizankizumab vs placebo.
c) Nominálne porovnanie p ≤ 0,05 rizankizumab vs placebo.
d) Sumarizované zo združených údajov zo štúdií KEEPSAKE1 a KEEPSAKE2 pre jedincov s východiskovým LEI > 0.
e) Sumarizované zo združených údajov zo štúdií KEEPSAKE1 a KEEPSAKE2 pre jedincov
s východiskovým LDI > 0.
|
Odpoveď v čase
V štúdii KEEPSAKE1 sa už v 4. týždni pozorovala väčšia odpoveď ACR20 v skupine
s rizankizumabom v porovnaní s placebom (25,7 %) a rozdiel v liečbe pretrvával až do 24. týždňa
(obrázok 3).
Obrázok 3: Percentuálny podiel pacientov dosahujúcich odpovede ACR20 v štúdii KEEPSAKE1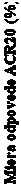 do 24. týždňa
do 24. týždňaZačiatok štúdie 4. týždeň 8. týždeň 12. týždeň 16. týždeň 24. týždeň
Návšteva
○
Placebo (N = 481) ──■── Rizankizumab (N = 483)U 19,6 % jedincov v štúdii KEEPSAKE2 sa už v 4. týždni pozorovala väčšia odpoveď ACR20 na
rizankizumab v porovnaní s placebom.
Odpovede pozorované v skupinách s rizankizumabom boli podobné bez ohľadu na súbežné používanie nebiologického DMARD, počet predchádzajúcich nebiologických DMARD, vek, pohlavie, rasu
a BMI. V štúdii KEEPSAKE2 sa odpovede pozorovali bez ohľadu na predchádzajúcu biologickú
liečbu.
Bezpečnostný profil rizankizumabu s expozíciou do 52 týždňov bol konzistentný s profilom pozorovaným do 24 týždňov.
V oboch štúdiách bol podiel jedincov, ktorí dosiahli modifikované kritériá odpovede PsA (PsARC) v 24. týždni vyšší u jedincov, ktorí dostávali rizankizumab v porovnaní s placebom. Okrem toho jedinci, ktorí dostávali rizankizumab dosiahli v 24. týždni väčšie zlepšenie skóre aktivity ochorenia (28 kĺbov) hodnoteného pomocou CRP (DAS28-CRP) v porovnaní s placebom. V prípade PsARC
a DAS28-CRP sa zlepšenia udržali do 52. týždňa.
Liečba rizankizumabom viedla v porovnaní s placebom k zlepšeniu jednotlivých zložiek ACR, dotazníka hodnotenia zdravia – index postihnutia (Health Assessment Questionnaire-Disability Index, HAQ-DI), hodnotenia bolesti a vysoko citlivého C-reaktívneho proteínu (hsCRP).
Liečba rizankizumabom viedla u jedincov s PsA k štatisticky významnému zlepšeniu kožných prejavov psoriázy.
Liečba rizankizumabom viedla v štúdii KEEPSAKE1 k štatisticky významnému zlepšeniu modifikovaného indexu závažnosti psoriázy nechtov (modified Nail Psoriasis Severity Index, mNAPSI) a 5-bodového skóre celkového hodnotenia psoriázy nechtov lekárom (Physician's Global Assessment of Fingerail Psoriasis, PGA-F) u jedincov so psoriázou nechtov na začiatku štúdie
(67,3 %). Toto zlepšenie sa udržalo až do 52. týždňa (pozri tabuľku 7).
Tabuľka 7: Výsledky účinnosti pri psoriáze nechtov v štúdii KEEPSAKE1
| Placebo
N = 338
| Rizankizumab
N = 309
|
Zmena mNAPSI od začiatku štúdiea
|
24. týždeň
| -5,57
| -9,76b
|
52. týždeň
| -
| -13,64
|
Zmena PGA-F od začiatku štúdiea
|
24. týždeň
| -0,4
| -0,8b
|
52. týždeň
| -
| -1,2
|
PGA-F čistá koža/minimálne prejavy a zlepšenie o ≥ 2 stupnec
|
24. týždeň n (%)
| 30 (15,9)
| 71 (37,8)d
|
52. týždeň n (%)
| -
| 105 (58,0)
|
a) Sumarizované pre jedincov so psoriázou nechtov na začiatku štúdie (placebo
N = 338; rizankizumab N = 309; v 52. týždni, pre mNAPSI pozorovaný rizankizumab N = 290, pre PGA-F pozorovaný rizankizumab N = 291).
b) Multiplicitou kontrolované porovnanie p ≤ 0,001 rizankizumab vs placebo.
c) Sumarizované pre jedincov so psoriázou nechtov a celkovým skóre globálneho hodnotenia PGA-F „mierne“, „stredne závažné“ alebo „závažné“ na začiatku štúdie (placebo N = 190; rizankizumab N = 188, v 52. týždni pozorovaný rizankizumab
N = 181).
d) Nominálne porovnanie p ≤ 0,001 rizankizumab vs placebo.
|
RádiografickáodpoveďV štúdii KEEPSAKE1 sa inhibícia progresie štrukturálneho poškodenia hodnotila rádiograficky
a vyjadrila sa ako zmena modifikovaného celkového Sharpovho skóre (mTSS) v 24. týždni
v porovnaní s východiskovou hodnotou. Skóre mTSS bolo pre PsA modifikované pridaním distálnych interfalangeálnych kĺbov (DIP) ruky. V 24. týždni nebola priemerná progresia štrukturálneho poškodenia pri rizankizumabe (priemer mTSS 0,23) v porovnaní s placebom (priemer mTSS 0,32) štatisticky významná. V 24. týždni bol podiel jedincov bez rádiografickej progresie (definovanej ako
zmena oproti východiskovej hodnote mTSS ≤ 0) vyšší pri rizankizumabe (92,4 %) v porovnaní s placebom (87,7 %). Táto odpoveď sa udržala až do 52. týždňa.
Fyzická funkciaakvalitaživotasúvisiacasozdravím
V oboch štúdiách sa u jedincov liečených rizankizumabom preukázalo štatisticky významné zlepšenie
fyzickej funkcie od začiatku štúdie hodnotené pomocou HAQ-DI v 24. týždni (KEEPSAKE1 (-0,31) v porovnaní s placebom (-0,11) (p ≤ 0,001)), (KEEPSAKE2 (-0,22) v porovnaní s placebom (-0,05) (p ≤ 0,001)). V 24. týždni dosiahol väčší podiel jedincov v skupine s rizankizumabom klinicky významné zníženie skóre HAQ-DI aspoň o 0,35 bodu oproti východiskovej hodnote v porovnaní so skupinou s placebom. Zlepšenia fyzickej funkcie sa udržali do 52. týždňa.
V oboch štúdiách sa u jedincov liečených rizankizumabom preukázalo v 24. týždni významné zlepšenie súhrnného skóre fyzickej zložky SF-36 V2 a skóre FACIT-únava v porovnaní s placebom, pričom zlepšenie sa udržalo do 52. týždňa.
Na začiatku štúdie bola psoriatická spondylitída hlásená u 19,6 % (7,9 % diagnostikovaných na základe RTG alebo MRI vyšetrenia) jedincov v štúdii KEEPSAKE1 a u 19,6 % (5 % diagnostikovaných na základe RTG alebo MRI vyšetrenia) jedincov v štúdii KEEPSAKE2.
U jedincov s klinicky hodnotenou psoriatickou spondylitídou, ktorí boli liečení rizankizumabom, sa v 24. týždni preukázalo zlepšenie skóre Bathovho indexu aktivity ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Disease Activity Index, BASDAI) od začiatku štúdie v porovnaní s placebom. Zlepšenia sa udržali do 52. týždňa. Vzhľadom na malý počet skúmaných jedincov nie sú k dispozícii dostatočné dôkazy o účinnosti rizankizumabu u jedincov so psoriatickou artropatiou podobnou ankylozujúcej spondylitíde potvrdenou RTG alebo MRI vyšetrením.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre rizankizumab
v jednej alebo viacerých podskupinách pediatrickej populácie v liečbe ložiskovej psoriázy a psoriatickej artritídy (pozri časť 4.2 pre informácie o použití u pediatrickej populácie).
5.2 Farmakokinetické vlastnosti
U jedincov s ložiskovou psoriázou a jedincov so psoriatickou artritídou bola farmakokinetika rizankizumabu podobná.
Absorpcia
Rizankizumab vykazoval lineárnu farmakokinetiku so zvýšením expozície úmerným dávke
v rozmedzí dávok 18 až 300 mg a 0,25 až 1 mg/kg podávaných subkutánne a 200 až 1200 mg a 0,01
až 5 mg/kg podávaných intravenózne.
Po subkutánnom podaní rizankizumabu boli maximálne plazmatické koncentrácie dosiahnuté
v rozmedzí 3 až 14 dní od podania s odhadovanou absolútnou biologickou dostupnosťou 89 %. Pri podaní dávky 150 mg v 0. týždni, 4. týždni a následne každých 12 týždňov sú predpokladané maximálne resp. minimálne plazmatické koncentrácie v rovnovážnom stave 12 resp. 2 µg/ml.
Bola preukázaná bioekvivalencia jednej injekcie rizankizumabu 150 mg a dvoch injekcií rizankizumabu 75 mg v naplnenej injekčnej striekačke. Bola preukázaná aj bioekvivalencia naplnenej injekčnej striekačky s rizankizumabom 150 mg a naplneného pera.
Distribúcia
Priemerný (± smerodajná odchýlka) distribučný objem v rovnovážnom stave (Vss) rizankizumabu bol v štúdiách fázy 3 u jedincov so psoriázou 11,4 (± 2,7) l, čo naznačuje, že distribúcia rizankizumabu je
primárne obmedzená na vaskulárne a intersticiálne priestory.
Biotransformácia
Terapeutické monoklonálne protilátky IgG sú typicky degradované na malé peptidy a aminokyseliny
katabolickými procesmi rovnakým spôsobom ako endogénne IgG. Nepredpokladá sa, že sa
rizankizumab metabolizuje enzýmami P450.
Eliminácia
Priemerný (± smerodajná odchýlka) systémový klírens (CL) rizankizumabu bol v štúdiách fázy 3
u jedincov so psoriázou 0,3 (± 0,1) l/deň. Priemerný terminálny polčas eliminácie rizankizumabu sa v štúdiách fázy 3 u jedincov so psoriázou pohyboval od 28 do 29 dní.
Nepredpokladá sa, že rizankizumab ako monoklonálna protilátka IgG1 by sa filtroval glomerulárnou filtráciou v obličkách alebo bol vylučovaný ako intaktná molekula v moči.
Linearita/nelinearita
Rizankizumab vykazoval lineárnu farmakokinetiku s približnými zvýšeniami systémovej expozície
úmernými dávke (Cmax a AUC) v hodnotených rozmedziach dávky 18 až 300 mg alebo 0,25 až
1 mg/kg pri subkutánnom podaní u zdravých jedincov alebo jedincov so psoriázou.
Interakcie
U jedincov s ložiskovou psoriázou bola vykonaná štúdia interakcií na zhodnotenie účinku
opakovaného podávania rizankizumabu na farmakokinetiku substrátov citlivých na cytochróm P450 (CYP). Expozícia kofeínu (substrát CYP1A2), warfarínu (substrát CYP2C9), omeprazolu (substrát CYP2C19), metoprololu (substrát CYP2D6) a midazolamu (substrát CYP3A) po liečbe rizankizumabom bola porovnateľná s ich expozíciami pred liečbou rizankizumabom, čo svedčí o tom, že nedochádza k žiadnym významným interakciám prostredníctvom týchto enzýmov.
Populačné farmakokinetické analýzy ukazujú, že expozícia rizankizumabu nebola ovplyvnená súbežnou liečbou, ktorú používali niektorí jedinci s ložiskovou psoriázou alebo psoriatickou artritídou počas klinických štúdií.
Osobitné skupinypacientov
Pediatrická populácia
Farmakokinetické vlastnosti rizankizumabu u pediatrických jedincov neboli stanovené.
Staršie osoby
Z 2234 jedincov s ložiskovou psoriázou vystavených rizankizumabu bolo 243 vo veku 65 rokov alebo
starších a 24 bolo veku 75 rokov alebo starších. Z 1542 jedincov so psoriatickou artritídou
vystavených rizankizumabu bolo 246 vo veku 65 rokov alebo starších a 34 jedincov bolo vo veku
75 rokov alebo starších. Medzi staršími a mladšími jedincami, ktorí dostávali rizankizumab, neboli pozorované žiadne celkové rozdiely v expozícii rizankizumabu.
Pacienti s poruchou funkcie pečene a obličiek
Neuskutočnili sa žiadne špecifické štúdie na stanovenie vplyvu poruchy funkcie obličiek alebo pečene
na farmakokinetické vlastnosti rizankizumabu. Podľa populačnej farmakokinetickej analýzy nemali sérové hladiny kreatinínu, klírens kreatinínu alebo markery pečeňových funkcií (ALT/AST/bilirubín) významný vplyv na klírens rizankizumabu u jedincov s ložiskovou psoriázou alebo psoriatickou artritídou.
Rizankizumab ako monoklonálna protilátka IgG1 je eliminovaný hlavne intracelulárnym katabolizmom a nepredpokladá sa, že sa metabolizuje enzýmami hepatálneho cytochrómu P450 alebo vylučuje obličkami.
Telesná hmotnosť
Klírens a distribučný objem rizankizumabu sa zvyšujú s rastom telesnej hmotnosti, čo môže mať za
následok zníženú účinnosť u jedincov s vysokou telesnou hmotnosťou (> 130 kg). Toto pozorovanie je
však založené na obmedzenom počte jedincov. V súčasnosti sa neodporúča úprava dávky na základe telesnej hmotnosti.
Pohlavie alebo rasa
Klírens rizankizumabu nebol významne ovplyvnený pohlavím alebo rasou u dospelých jedincov
s ložiskovou psoriázou alebo psoriatickou artritídou. V klinickej farmakokinetickej štúdii neboli
pozorované žiadne klinicky významné rozdiely v expozícii rizankizumabu u čínskych alebo japonských jedincov v porovnaní s belošskými jedincami.
5.3 Predklinické údaje o bezpečnosti
Neklinické údaje neodhalili žiadne osobitné riziko pre ľudí na základe štúdií toxicity po opakovanom podaní, vrátane farmakologických hodnotení bezpečnosti, a na základe štúdie reprodukčnej
a vývojovej toxicity na makakoch dlhochvostých v dávkach až do 50 mg/kg/týždeň (vedúcej
k expozíciám zodpovedajúcim približne 70-násobku klinickej expozície pri maximálnej odporúčanej dávke u ľudí [MRHD]).
S rizankizumabom neboli vykonané štúdie mutagenity a karcinogenity. V 26-týždňovej štúdii chronickej toxicity u opíc rodu Cynomolgus v dávkach až do 50 mg/kg/týždeň (približne 70-násobok klinickej expozície pri MRHD) neboli pozorované žiadne preneoplastické alebo neoplastické lézie
a nebola zaznamenaná žiadna nežiaduca imunotoxicita alebo kardiovaskulárne účinky.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Skyrizi 150 mginjekčnýroztokvnaplnenompereanaplnenejinjekčnejstriekačke
Trihydrát octanu sodného
Kyselina octová Dihydrát trehalózy Polysorbát 20
Voda na injekcie
Skyrizi 75 mginjekčnýroztokvnaplnenejinjekčnejstriekačke
Jantaran disodný, hexahydrát
Kyselina jantárová
Sorbitol
Polysorbát 20
Voda na injekcie
6.2 Inkompatibility
Štúdie kompatibility nie sú k dispozícii, a preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Naplnené pero alebo naplnenú(é) injekčnú(é) striekačku(y) uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Skyrizi 150 mg naplnené pero alebo naplnenú injekčnú striekačku možno uchovávať mimo chladničky
(max. do teploty 25 °C) počas 24 hodín v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Skyrizi 150 mg injekčnýroztokvnaplnenompere
Naplnená sklenená injekčná striekačka umiestnená v naplnenom pere s automatickým puzdrom ihly.
Skyrizi 150 mginjekčnýroztokvnaplnenejinjekčnejstriekačke
Naplnená sklenená injekčná striekačka s fixnou ihlou a krytom ihly, umiestnenými v automatickom
chrániči ihly.
Skyrizi 150 ml je k dispozícii v baleniach, ktoré obsahujú 1 naplnené pero alebo 1 naplnenú injekčnú striekačku.
Skyrizi 75 mginjekčnýroztokvnaplnenejinjekčnejstriekačke
Naplnená sklenená injekčná striekačka s fixnou ihlou a krytom ihly, umiestnenými v automatickom
chrániči ihly.
Skyrizi 75 mg je k dispozícii v baleniach, ktoré obsahujú 2 naplnené injekčné striekačky a 2 tampóny napustené alkoholom.
Na trh nemusia byť uvedené všetky formy.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Skyrizi 150 mginjekčnýroztokvnaplnenompere
Pred podaním injekcie majú pacienti vybrať škatuľku z chladničky a nechať ju pri izbovej teplote
mimo priameho slnečného žiarenia (30 až 90 minút) bez toho, aby naplnené pero vybrali zo škatuľky.
Roztok má byť bezfarebný až žltý a číry až mierne opaleskujúci.
Skyriz
i 150 mginjekčnýroztokvnaplnenejinjekčnejstriekačke
Pred podaním injekcie môžu pacienti vybrať škatuľku z chladničky a nechať ju pri izbovej teplote
mimo priameho slnečného žiarenia (15 až 30 minút) bez toho, aby naplnenú striekačku vybrali zo škatuľky.
Roztok má byť bezfarebný až žltý a číry až mierne opaleskujúci. Skyrizi75 mginjekčnýroztokvnaplnenejinjekčnej striekačke
Pred podaním injekcie môžu pacienti vybrať škatuľku z chladničky a nechať ju pri izbovej teplote
mimo priameho slnečného žiarenia (15 až 30 minút) bez toho, aby naplnené striekačky vybrali zo
škatuľky.
Roztok má byť bezfarebný až svetložltý a číry až mierne opaleskujúci.
Na podanie celej dávky 150 mg je potrebné podať dve naplnené injekčné striekačky. Všeobecnéosobitnéupozornenia
Pred použitím sa odporúča vizuálna kontrola každého naplneného pera alebo naplnenej injekčnej
striekačky. Roztok môže obsahovať zopár priehľadných až bielych čiastočiek, ktoré sú súčasťou lieku.
Skyrizi sa nemá použiť, ak je roztok zakalený alebo má zmenenú farbu alebo ak obsahuje veľké čiastočky. Naplneným perom ani naplnenou injekčnou striekačkou netraste.
Úplný návod na použitie je uvedený v písomnej informácii pre používateľa.
Každé naplnené pero alebo naplnená injekčná striekačka sú určené len na jedno použitie. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AbbVie Deutschland GmbH & Co. KG Knollstrasse
67061 Ludwigshafen
Nemecko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
Skyrizi150 mginjekčnýroztokvnaplnenompere
EU/1/19/1361/002
Skyrizi 150 mginjekčnýroztok vnaplnenejinjekčnejstriekačke
EU/1/19/1361/003
Skyrizi 75 mginjekčnýroztokvnaplnenejinjekčnejstriekačke
EU/1/19/1361/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 26. apríl 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUSkyrizi 600 mg koncentrát na infúzny roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEJedna injekčná liekovka obsahuje 600 mg rizankizumabu (risankizumabum) v 10,0 ml roztoku. Rizankizumab je humanizovaná monoklonálna imunoglobulínová protilátka G1 (IgG1) produkovaná
v ovariálnych bunkách čínskeho škrečka pomocou technológie rekombinantnej DNA. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAKoncentrát na infúzny roztok (sterilný koncentrát).
Roztok je bezfarebný až svetložltý a číry až mierne opaleskujúci.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiek Skyrizi je indikovaný na liečbu dospelých pacientov so stredne ťažkou až ťažkou aktívnou Crohnovou chorobou, ktorí mali nedostatočnú odpoveď na konvenčnú liečbu alebo biologickú liečbu, alebo na ňu prestali reagovať, prípadne ju netolerovali.
4.2 Dávkovanie a spôsob podávaniaTento liek je určený na použitie pod vedením a dohľadom lekára, ktorý má skúsenosti s diagnostikou a liečbou stavov, na ktoré je liek Skyrizi indikovaný.
DávkovanieOdporúčaná dávka je 600 mg podávaná intravenóznou infúziou v 0. týždni, 4. týždni a 8. týždni, po
ktorej nasleduje 360 mg subkutánnou injekciou v 12. týždni a potom každých 8 týždňov. V prípade pacientov, u ktorých sa nepreukázal žiadny terapeutický prínos po 24 týždňoch liečby, sa má zvážiť ukončenie liečby.
Dávkovanie následného subkutánneho dávkovacieho režimu, pozri časť 4.2 súhrnu charakteristických vlastností lieku Skyrizi 360 mg injekčný roztok v náplni.
Vynechanie dávkyAk dôjde k vynechaniu dávky, dávka má byť podaná čo najskôr. Následne má byť dávkovanie
obnovené podľa pôvodného plánu.
Osobitné skupinypacientov
Starší pacienti (vo veku 65 rokov a viac)
Nie je potrebná úprava dávkovania (pozri časť5.2).
K dispozícii sú obmedzené údaje u pacientov vo veku ³ 65 rokov.
Porucha funkcie obličiek alebo pečene
Neuskutočnili sa žiadne špecifické štúdie na hodnotenie vplyvu poruchy funkcie pečene alebo obličiek na farmakokinetické vlastnosti lieku Skyrizi. Všeobecne sa nepredpokladá, že tieto stavy budú mať významný vplyv na farmakokinetické vlastnosti monoklonálnych protilátok a nepovažuje sa za potrebné upravovať dávkovanie (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Skyrizi pri liečbe Crohnovej choroby u pacientov mladších ako 18 rokov neboli doteraz stanovené.
Pacienti s nadváhou
Nie je potrebná úprava dávkovania (pozri časť 5.2). Spôsob podávania
Na intravenóznu infúziu.
Skyrizi 600 mg koncentrát na infúzny roztok je určený len na intravenózne použitie. Má sa podávať najmenej jednu hodinu. Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Klinicky významné aktívne infekcie (napr. aktívna tuberkulóza, pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Infekcie
Rizankizumab môže zvýšiť riziko infekcie.
U pacientov s chronickou infekciou, recidivujúcou infekciou v anamnéze alebo známymi rizikovými faktormi pre infekciu sa má rizankizumab používať s opatrnosťou. Liečba rizankizumabom sa nemá začať u pacientov s akoukoľvek klinicky významnou aktívnou infekciou, pokým infekcia neodznie alebo nie je adekvátne liečená.
Pacienti liečení rizankizumabom majú byť poučení, aby vyhľadali lekársku pomoc, ak sa u nich
objavia prejavy alebo príznaky klinicky významnej chronickej alebo akútnej infekcie. Ak sa u pacienta objaví takáto infekcia alebo ak nereaguje na štandardnú liečbu infekcie, pacient má byť starostlivo sledovaný a rizankizumab sa nemá podávať, pokým neodznie infekcia.
Tuberkulóza
Pacienti majú byť pred začatím liečby rizankizumabom vyšetrení na tuberkulózu (TBC). Pacienti,
ktorí dostávajú rizankizumab, majú byť monitorovaní na prejavy a symptómy aktívnej TBC. Pred začatím liečby rizankizumabom má byť zvážená liečba TBC u pacientov s latentnou alebo aktívnou TBC v anamnéze, u ktorých nie je možné potvrdiť adekvátnu liečbu.
Imunizácia
Pred začatím liečby rizankizumabom sa má zvážiť absolvovanie všetkých príslušných imunizácií
podľa aktuálnych odporúčaní pre imunizáciu. Ak bola pacientovi podaná živá vakcína (vírusová alebo bakteriálna), odporúča sa so začatím liečby rizankizumabom počkať najmenej 4 týždne. Pacientom liečeným rizankizumabom sa počas liečby a najmenej 21 týždňov po ukončení liečby nesmú podávať živé vakcíny (pozri časť 5.2).
Precitlivenosť
Ak sa vyskytne závažná reakcia z precitlivenosti, podávanie rizankizumabu sa má okamžite ukončiť
a má sa začať príslušná liečba.
Pomocné látkysoznámymúčinkom
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v injekčnej liekovke, t. j. v podstate
zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Nepredpokladá sa, že rizankizumab podlieha metabolizácii pečeňovými enzýmami alebo vylučovaniu obličkami. Nepredpokladajú sa interakcie medzi rizankizumabom a inhibítormi, induktormi alebo substrátmi enzýmov metabolizujúcich lieky, a nie je potrebná úprava dávky (pozri časť 5.2).
Súbežná imunosupresívnaliečba
Bezpečnosť a účinnosť rizankizumabu v kombinácii s imunosupresívami, vrátane biologických liekov,
neboli hodnotené.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženy vo fertilnom veku majú používať účinnú metódu antikoncepcie počas liečby a po dobu najmenej
21 týždňov po liečbe.
Gravidita
Údaje o použití rizankizumabu u tehotných žien sú obmedzené alebo nie sú k dispozícii (menej ako
300 výsledkov tehotenstva). Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky s ohľadom na reprodukčnú toxicitu. Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu rizankizumabu v gravidite.
Dojčenie
Nie je známe, či sa rizankizumab vylučuje do ľudského materského mlieka. Je známe, že humánne
IgG sa počas prvých niekoľkých dní po pôrode vylučujú do materského mlieka, čo sa čoskoro po tomto období znižuje na nízke koncentrácie; preto počas tohto krátkeho obdobia nemožno riziko pre dojčené dieťa vylúčiť. Je potrebné vziať do úvahy prínos dojčenia pre dieťa a prínos liečby rizankizumabom pre matku pri zvážení rozhodnutia o ukončení/nezačatí liečby rizankizumabom.
Fertilita
Účinok rizankizumabu na ľudskú fertilitu nebol hodnotený. Štúdie na zvieratách nepreukázali priame
alebo nepriame škodlivé účinky s ohľadom na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Rizankizumab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie boli infekcie horných dýchacích ciest (od 13,0 % pri psoriáze
do 15,6 % pri Crohnovej chorobe).
Tabuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie rizankizumabu pozorované v klinických štúdiách (tabuľka 1) sú uvedené podľa
triedy orgánových systémov (MedDRA) a vychádzajú z nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (nie je možné odhadnúť z dostupných údajov).
Tabuľk
a 1: Zoznam nežiaducich reakcií
Tried
a orgánových systémov
|
Frekvencia
|
Nežiaduc
e reakcie
|
Infekcie a nákazy
|
Veľmi časté
|
Infekcie horných dýchacích ciesta
|
Časté
|
Infekcie tineab
|
Menej časté
|
Folikulitída
|
Poruchy nervového systému
|
Časté
|
Bolesť hlavyc
|
Poruchy kože
a podkožného tkaniva
|
Časté
|
Pruritus
|
Neznáme
|
Vyrážka
Žihľavka
|
Celkové poruchy
a reakcie v mieste podania
|
Časté
|
Únavad
Reakcie v mieste vpichue
|
a Patrí sem: infekcia dýchacích ciest (vírusová, bakteriálna alebo nešpecifikovaná), sinusitída (vrátane akútnej), rinitída, nazofaryngitída, faryngitída (vrátane
vírusovej), tonzilitída, laryngitída, tracheitída
b Patrí sem: tinea pedis, tinea cruris, tinea corporis, tinea versicolor, tinea manuum, onychomykóza, mykotická infekcia kože
c Patrí sem: bolesť hlavy, tenzná bolesť hlavy, bolesť hlavy pri sínusitíde
d Patrí sem: únava, asténia
e Patrí sem: modrina v mieste vpichu, erytém, hematóm, hemorágia, podráždenie, bolesť, pruritus, reakcia, opuch, indurácia, vyrážka, precitlivenosť, uzlík, vyrážka, žihľavka, vezikuly, pocit tepla
|
Opis
vybraných
nežiaducich
reakcií
Infekcie
Psoriáza
Počas celého programu zameraného na psoriázu vrátane dlhodobej expozície rizankizumabu bol
výskyt infekcií 75,5 prípadov na 100 pacientorokov. Väčšina prípadov bola nezávažná a bola mierna až stredne závažná a neviedla k prerušeniu liečby rizankizumabom. Výskyt závažných infekcií bol 1,7 prípadu na 100 pacientorokov (pozri časť 4.4).
Crohnova chorobaCelkovo bol bezpečnostný profil pozorovaný u pacientov s Crohnovou chorobou liečených
rizankizumabom v súlade s bezpečnostným profilom pozorovaným u pacientov s ložiskovou psoriázou.
Miera výskytu infekcií v súhrnných údajoch z 12-týždňových indukčných štúdií bola 83,3 prípadov na
100 pacientorokov u jedincov liečených rizankizumabom 600 mg intravenózne v porovnaní so
117,7 prípadmi na 100 pacientorokov pri placebe. Výskyt závažných infekcií bol 3,4 prípadu na 100
pacientorokov (pozri časť 4.4) u jedincov liečených intravenózne podávaným rizankizumabom
600 mg v porovnaní so 16,7 prípady na 100 pacientorokov pri placebe (pozri časť 4.4).
Výskyt infekcií v 52-týždňovej udržiavacej štúdii bol 57,7 prípadu na 100 pacientorokov (pozri
časť 4.4) u jedincov liečených subkutánne podávaným rizankizumabom 360 mg v porovnaní so 76,0 prípadov na 100 pacientorokov u jedincov, ktorým bolo podané placebo po indukcii rizankizumabom. Výskyt závažných infekcií bol 6,0 prípadu na 100 pacientorokov u jedincov liečených subkutánne podávaným rizankizumabom 360 mg po indukcii rizankizumabom v porovnaní s 5,0 prípadmi na
100 pacientorokov u jedincov, ktorým bolo podané placebo po indukcii rizankizumabom (pozri
časť 4.4).
Imunogenita
Tak ako pri všetkých terapeutických proteínoch, aj pri rizankizumabe existuje možnosť imunogenity. Detekcia tvorby protilátok vysoko závisí od citlivosti a špecifickosti testu.
U jedincov s Crohnovou chorobou liečených odporúčanou indukčnou intravenóznou dávkou rizankizumabu a subkutánnou udržiavacou dávkou rizankizumabu počas obdobia až 64 týždňov
v klinických skúšaniach zameraných na liečbu Crohnovej choroby boli detegované protilátky proti lieku a neutralizujúce protilátky u 3,4 % (2/58), resp. u 0 % (0/58) hodnotených jedincov.
Protilátky proti rizankizumabu, vrátane neutralizujúcich protilátok, neboli spojené so zmenami klinickej odpovede alebo bezpečnosti.
Starší pacientiK dispozícii sú obmedzené údaje o bezpečnosti u pacientov vo veku ³ 65 rokov.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania sa odporúča, aby sa u pacienta sledovali prípadné prejavy alebo príznaky nežiaducich reakcií a aby sa okamžite začalo s vhodnou symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu, ATC kód: L04AC18
Mechanizmus účinkuRizankizumab je humanizovaná monoklonálna imunoglobulínová protilátka G1 (IgG1), ktorá sa
selektívne viaže s vysokou afinitou na podjednotku p19 humánneho cytokínu označovaného ako interleukín 23 (IL-23) bez väzby na IL-12 a inhibuje jeho interakciu s receptorovým komplexom IL-
23. IL-23 je cytokín, ktorý sa podieľa na zápalovej a imunitnej odpovedi. Tým, že rizankizumab blokuje IL-23 a bráni mu vo väzbe na jeho receptor, inhibuje bunkovú signalizáciu závislú od IL-23 a uvoľňovanie prozápalových cytokínov.
Farmakodynamické účinkyV štúdii u jedincov so psoriázou bola po jednotlivých dávkach rizankizumabu znížená expresia génov
súvisiacich s osou IL-23/IL-17 v koži. V psoriatických léziách bolo pozorované aj zníženie hrúbky epidermy, infiltrácie zápalovými bunkami a expresie markerov psoriázy.
V štúdii fázy 2 e u jedincov s Crohnovou chorobou bola po viacnásobnom podaní rizankizumabu znížená expresia génov súvisiacich s osou IL-23/IL-17 v tkanivách čreva. Po viacerých dávkach
v indukčných štúdiách fázy 3 u pacientov s Crohnovou chorobou sa pozorovalo zníženie hladiny fekálneho kalprotektínu (faecal calprotectin, FCP), sérového C-reaktívneho proteínu (CRP) a IL-22. Zníženia FCP, CRP a IL-22 v sére sa udržali až do 52. týždňa udržiavacej štúdie.
Klinick
á účinnosťÚčinnosť a bezpečnosť rizankizumabu boli hodnotené u 1419 jedincov so stredne ťažkou až ťažkou
aktívnou Crohnovou chorobou v troch multicentrických, randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách. Do štúdie boli zaradené jedinci vo veku 16 rokov alebo starší s indexom aktivity Crohnovej choroby (Crohn’s Disease Activity Index, CDAI) 220 až 450, priemernou dennou frekvenciou stolice (stool frequency, SF) ≥ 4 a/alebo priemerným denným skóre bolesti brucha (abdominal pain score, APS) ≥ 2 a zjednodušeným endoskopickým skóre pre CD (Simple Endoscopic Score for CD, SES-CD) ≥ 6 alebo ≥ 4 v prípade izolovaného ileálneho ochorenia, s vylúčením zužujúcej sa zložky a s potvrdením centrálneho hodnotiteľa.
Uskutočnili sa dve 12-týždňové indukčné štúdie (ADVANCE a MOTIVATE) s intravenóznym podávaním, ktoré zahŕňali 12-týždňové predĺženie pre jedincov, ktorí nedosiahli klinickú odpoveď v hodnotení SF/APS (≥ 30 % pokles SF a/alebo ≥ 30 % pokles APS a obidve hodnoty neboli horšie ako východiskové hodnoty) v 12. týždni. Po štúdiách ADVANCE a MOTIVATE nasledovala 52-
týždňová randomizovaná štúdia vysadenia pri subkutánnej udržiavacej liečbe (FORTIFY), do ktorej boli zaradení jedinci s klinickou odpoveďou v hodnotení SF/APS na intravenózne podávanú indukčnú liečbu, čo predstavuje najmenej 64 týždňov liečby.
ADVANCE a MOTIVATEV štúdiách ADVANCE a MOTIVATE boli jedinci randomizovaní na používanie rizankizumabu buď
v dávke 600 mg (odporúčaná dávka), 1200 mg alebo placeba, v 0. týždni, 4. týždni a 8. týždni.
V štúdii ADVANCE u 58 % (491/850) jedincov zlyhala liečba jednou alebo viacerými biologickými terapiami alebo túto liečbu netolerovali (predchádzajúce zlyhanie biologickej liečby) a u 42 % (359/850) jedincov zlyhala liečba konvenčnými terapiami alebo ju netolerovali, ale nezlyhala biologická terapia (bez predchádzajúceho zlyhania biologickej liečby). V štúdii ADVANCE bolo spomedzi jedincov bez predchádzajúceho zlyhania biologickej liečby 87 % (314/359) jedincov bez predchádzajúcej biologickej liečby a zvyšných 13 % jedincov už používalo biologickú liečbu, ale nikdy nedošlo k jej zlyhaniu alebo sa u nich neprejavila intolerancia. U všetkých pacientov v štúdii MOTIVATE došlo k predchádzajúcemu biologickému zlyhaniu liečby.
V oboch štúdiách väčší podiel jedincov liečených rizankizumabom dosiahol koprimárne cieľové ukazovatele: klinickú remisiu v 12. týždni a endoskopickú odpoveď v porovnaní s placebom
v 12. týždni. Zvýšená klinická odpoveď v hodnotení SF/APS a klinická remisia boli u jedincov liečených rizankizumabom významné už v 4. týždni a naďalej sa zlepšovali až do 12. týždňa (tabuľka 2).
Tabuľka 2. Výsledky účinnosti v štúdiách ADVANCE a MOTIVATE
| ADVANCE
| MOTIVATE
|
| Placebo
(N=175)
%
| Rizankizumab
600 mg i.v. (N=336)
%
| Liečebný rozdiel
(95 % CI)
| Placebo i. v. (N=187)
%
| Rizankizumab
600 mg i.v. (N=191)
%
| Liečebný rozdield (95 % CI)
|
Koprimárne cieľové ukazovatele
|
Klinická remisia
v 12. týždni
|
22 %
|
43 %
| 22 %
[14 %,
30 %]a
|
19 %
|
35 %
|
15 %
[6 %, 24 %]b
|
Endoskopická odpoveď
v 12. týždni
|
12 %
|
40 %
| 28 %
[21 %,
35 %]a
|
11 %
|
29 %
| 18 %
[10 %,
25 %]a
|
Dodatočné cieľové ukazovatele
|
Zvýšená klinická odpoveď
v hodnotení
SF/APS vo
4. týždni
g
|
31 %
|
46 %
|
15 %
[6 %, 23 %]b
|
32 %
|
45 %
|
14 %
[4 %, 23 %]c
|
Zvýšená klinická odpoveď
v hodnotení
SF/AP
S v
12. týždni
g
|
42 %
|
63 %
|
21 %
[12 %,
30 %]a
|
39 %
|
62 %
|
23 %
[13 %,
33 %]a
|
CDAI < 150
v
o 4. týždni
|
10 %
|
18 %
|
8 %
[1 %, 14 %]c
|
11 %
|
21 %
|
10 %
[2 %, 17 %]c
|
CDAI < 150
vo 12. týždni
|
25 %
|
45 %
|
21 %
[12 %,
29 %]a
|
20 %
|
42 %
|
22 %
[13 %,
31 %]a
|
Za
h
ojenie sliznice
v 12. týždni
h
|
(N=173)
8 %
|
(N=336)
21 %
|
14 %
a
|
(N=186)
4 %
|
(N=190)
14 %
|
9 %
b
|
Endoskopická odpoveď
v 12. týždni
|
9 %
|
24 %
|
15 %
a
|
4 %
|
19 %
|
15 %
a
[9 %, 21 %]
|
a Štatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p < 0,001).
b Štatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p ≤ 0,01).
c Nominálne porovnanie p ≤ 0,05 rizankizumab vs placebo.
d Upravený liečebný rozdiel.
e Klinická remisia na základe SF/APS: priemerná denná SF ≤ 2,8 a nie horšia ako východisková hodnota a priemerné denné skóre AP ≤ 1 a nie horšie ako východisková hodnota.
f Endoskopická odpoveď: viac ako 50 % pokles SES-CD oproti východiskovej hodnote alebo pokles aspoň o 2 body u jedincov s východiskovým skóre 4 a izolovaným ileálnym ochorením.
g Zvýšená klinická odpoveď v hodnotení SF/APS: ≥ 60 % pokles priemerného denného skóre SF a/alebo ≥ 35 % pokles priemerného denného skóre AP a obe nie horšie ako východiskové skóre, a/alebo klinická remisia.
h Zahojenie sliznice: Podskóre SES-CD ulcerovaného povrchu 0 u jedincov s podskóre ≥ 1 na
začiatku liečby.
i Endoskopická remisia SES-CD ≤ 4 a zníženie aspoň o 2 body v porovnaní s východiskovou hodnotou a žiadne čiastkové skóre väčšie ako 1 v žiadnej jednotlivej premennej.
|
|
|
[8%, 19%]
[4 %, 15 %]
[9 %, 21 %]
V 12. týždni dosiahol vyšší podiel jedincov liečených rizankizumabom pokles aspoň o 100 bodov od
východiskovej hodnoty CDAI v porovnaní s placebom (ADVANCE, rizankizumab = 60 %, placebo37 %, p < 0,001; MOTIVATE, rizankizumab =60 %, placebo=30 %, p < 0,001).
V 12. týždni dosiahol vyšší podiel jedincov liečených rizankizumabom zvýšenú klinickú odpoveď
v hodnotení SF/APS aj endoskopickú odpoveď v porovnaní s placebom (ADVANCE, rizankizumab =
31 %, placebo = 8 %, p < 0,001; MOTIVATE, rizankizumab = 21 %, placebo = 7 %, p < 0,001).
Výsledky pre koprimárne cieľové ukazovatele v podskupinách jedincov so zlyhaním predchádzajúcej biologickej liečby a bez tohto zlyhania (bez pripustenia multiplicity) sú uvedené v tabuľke 3.
Tabuľk
a 3. Výsledky účinnosti v 12. týždni v podskupine jedincov s predchádzajúcim zlyhaním biologickej liečby a jedincov bez predchádzajúceho zlyhania biologickej liečby v štúdii ADVANCE
|
ADVANCE
|
Placeb
o i.v.
|
Rizankizuma
b 600 mg
|
Liečebný rozdiel (95 % IS)
|
Klinická remisia podľa skóre SF/AP
|
Predchádzajúce zlyhanie biologickej liečby
|
23 % (N=97)
|
41 % (N=195)
|
18 % [7 %, 29 %]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
21 % (N=78)
|
48 % (N=141)
|
27 % [15 %, 39 %]
|
Endoskopick
á odpoveď
|
Predchádzajúce zlyhanie biologickej liečby
|
11 % (N=97)
|
33 % (N=195)
|
21 % [12 %, 31 %]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
13 % (N=78)
|
50 % (N=141)
|
38 % [27 %, 49 %]
|
V štúdii ADVANCE dosiahol vyšší podiel jedincov liečených rizankizumabom s predchádzajúcim
biologickým zlyhaním aj bez neho CDAI < 150 v porovnaní s placebom (s predchádzajúcim biologickým zlyhaním, rizankizumab = 42 %, placebo = 26 %; bez predchádzajúceho biologického zlyhania, rizankizumab = 49 %, placebo = 23 %).
Hospitalizácie súvisiace s Crohnovou chorobouMiera hospitalizácií súvisiacich s Crohnovou chorobou v 12. týždni bola nižšia u osôb liečených rizankizumabom v porovnaní s placebom (ADVANCE, rizankizumab = 3 %, placebo = 12 %,
p < 0,001; MOTIVATE, rizankizumab = 3 %, placebo = 11 %, p ≤ 0,01).
FORTIFYUdržiavacia štúdia FORTIFY hodnotila 462 jedincov s klinickou odpoveďou v hodnotení SF/APS na
12 týždňov intravenóznej indukčnej liečby rizankizumabom (i.v.) v štúdiách ADVANCE a MOTIVATE. Jedinci boli randomizovaní na pokračovanie v udržiavacom režime subkutánne (s.c.) podávaného rizankizumabu 360 mg (odporúčaná dávka) alebo rizankizumabu 180 mg s.c. každých
8 týždňov, alebo na ukončenie indukcie rizankizumabom a podávanie placeba s.c. každých 8 týždňov až do 52 týždňov.
Koprimárne cieľové ukazovatele boli klinická remisia v 52. týždni a endoskopická odpoveď
v 52. týždni. U jedincov s predchádzajúcim biologickým zlyhaním a bez neho sa merali aj koprimárne cieľové ukazovatele (pozri tabuľku 4).
Tabuľka 4. Výsledky účinnosti v štúdii FORTIFY v 52. týždni (64 týždňov od začatia liečby indukčnou dávkou).
| FORTIFY
|
| Rizankizumab i.v. indukcia/s.c. placebof (N=164) %
| Rizankizumab i.v. indukcia/rizankizumab
360 mg s.c.
(N=141) %
| Liečebný rozdiel
(95 % IS)
|
Koprimárne cieľové ukazovatele
|
Klinická remisia
|
40 %
|
52 %
|
15% [5%, 25%]a,g
|
Predchádzajúce zlyhanie biologickej liečby
|
34 % (N=123)
|
48 % (N=102)
|
14 % [1 %,27 %]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
56 % (N=41)
|
62 % (N=39)
|
5 % [-16 %,27 %]
|
Endoskopick
á odpoveď
|
22 %
|
47 %
|
28% [19%, 37%]b,g
|
Predchádzajúce zlyhanie biologickej liečby
|
20 % (N=123)
|
44 % (N=102)
|
23% [11%, 35%]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
27 % (N=41)
|
54 % (N=39)
|
27 % [6 %, 48 %]
|
Dodatočn
é cieľové ukazovatele
|
Zvýšená klinická odpoveď v hodnotení SF/APS
|
49 %
|
59 %
|
13% [2%, 23%]e,g
|
Udržanie klinickej remisie
h
|
(N = 91)
51 %
|
(N = 72)
69 %
|
21% [6%, 35%]d,g
|
Endoskopická remisia
|
13 %
|
39 %
|
28% [20%, 37%]c,g
|
Hojenie sliznice
|
(N = 162)
10 %
|
(N = 141)
31 %
|
22% [14%, 30%]c,g
|
a Štatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p < 0,001). b Štatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p < 0,001). c Nominálne porovnanie p < 0,001 rizankizumab vs placebo bez celkovej kontroly chýb typu I.
d Nominálne porovnanie p ≤ 0,01 rizankizumab vs placebo bez celkovej kontroly chýb typu I.
e Nominálne porovnanie p ≤ 0,05 rizankizumab vs placebo bez celkovej kontroly chýb typu I.
f Skupinu len s indukčnou liečbou tvorili jedinci, u ktorých sa dosiahla klinická odpoveď na indukčnú liečbu rizankizumabom a ktorí boli randomizovaní na podávanie placeba v udržiavacej
štúdii (FORTIFY).
g Upravený liečebný rozdiel.
h Udržanie klinickej remisie: klinická remisia v 52. týždni u jedincov s klinickou remisiou v 0. týždni
|
Hlboká remisia (klinická remisia a endoskopická remisia) v 52. týždni sa pozorovala vo vyššej miere
u osôb liečených rizankizumabom i.v./rizankizumabom s.c. v porovnaní s jedincami, ktorí dostávali rizankizumab i.v./placebo s.c.(28 % vs 10 %, nominálna hodnota p < 0,001).
V 52. týždni dosiahol vyšší podiel jedincov liečených rizankizumabom i.v./rizankizumabom s.c. CDAI < 150 v porovnaní s jedincami, ktorí dostávali rizankizumab i.v./placebo s.c. (52 % vs 41 %, nominálna hodnota p < 0,01). Vyšší podiel jedincov liečených rizankizumabom i.v./rizankizumabom s.c zaznamenal zníženie aspoň o 100 bodov v skóre východiskovej CDAI v porovnaní s jedincami, ktorí dostávali rizankizumab i.v./placebo s.c. (62 % vs 48 %, nominálna hodnota p < 0,001).
91 jedincom, u ktorých sa nepreukázala klinická odpoveď v hodnotení SF/APS 12 týždňov po indukcii rizankizumabom v štúdiách ADVANCE a MOTIVATE, sa podávala subkutánna 360 mg dávka rizankizumabu v 12. a 20. týždni. Z týchto jedincov 64 % (58/91) dosiahlo klinickú odpoveď
v hodnotení SF/APS v 24. týždni; 33 jedincov, ktorí dosiahli klinickú odpoveď v hodnotení SF/APS, sa zaradilo do štúdie FORTIFY a pokračovalo v s.c. používaní rizankizumabu 360 mg každých
8 týždňov až do 52 týždňov. Spomedzi týchto jedincov dosiahlo 55 % (18/33) klinickú remisiu a 45 % (15/33) endoskopickú odpoveď v 52. týždni.
Počas štúdie FORTIFY 30 jedincov stratilo odpoveď na liečbu rizankizumabom 360 mg s.c. a dostalo záchrannú liečbu rizankizumabom (jednorazová i.v. dávka 1200 mg, po ktorej nasledovalo 360 mg s.c. každých 8 týždňov). Spomedzi týchto jedincov dosiahlo 57 % (17/30) klinickú odpoveď v hodnotení SF/APS v 52. týždni. Navyše spomedzi týchto jedincov dosiahlo 20 % (6/30) klinickú remisiu a 34 % (10/29) endoskopickú odpoveď v 52. týždni.
Výsledky týkajúce sa zdravia a kvality života
Kvalita života súvisiaca so zdravím sa hodnotila pomocou Dotazníka o zápalových črevných
ochoreniach (Inflammatory Bowel Disease Questionnaire, IBDQ) a Krátkeho dotazníka kvality života SF-36 (36-Item Short Form Health Survey, SF-36). Zmiernenie únavy sa hodnotilo pomocou dotazníka k Funkčnému hodnoteniu liečby chronických ochorení - únava (Functional Assessment of Chronic Illness Therapy-Fatigue, FACIT-Fatigue). Produktivita práce bola hodnotená pomocou Dotazníka o zhoršení produktivity práce a aktivity (Work Productivity and Activity Impairment CD, WPAI-CD).
V 12. týždni štúdií ADVANCE a MOTIVATE dosiahli jedinci liečení rizankizumabom klinicky významné zlepšenie oproti východiskovému stavu v celkovom skóre IBDQ, vo všetkých doménových skóre IBDQ (črevné príznaky, systémová funkcia, emocionálna funkcia a sociálna funkcia),
v súhrnnom skóre fyzických a psychických komponentov SF-36, a v skóre FACIT-Fatigue a WPAI- CD v porovnaní s placebom. Pre WPAI-CD sa v štúdii ADVANCE preukázalo nižšie ovplyvnenie pri práci, celkové ovplyvnenie práce a menší ovplyvnenie aktivity a v štúdii MOTIVATE bolo zaznamenane nižšie ovplyvnenie aktivity. Tieto zlepšenia sa udržali u osôb liečených
rizankizumabom i.v./rizankizumabom s.c. v štúdii FORTIFY do 52. týždňa. Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre rizankizumab
v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe Crohnovej choroby (pre informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Rizankizumab vykazoval lineárnu farmakokinetiku so zvýšením expozície úmerným dávke
v rozmedzí dávok 18 až 360 mg a 0,25 až 1 mg/kg podávaných subkutánne a 200 až 1800 mg a 0,01
až 5 mg/kg podávaných intravenózne.
Po subkutánnom podaní rizankizumabu boli maximálne plazmatické koncentrácie dosiahnuté
v rozmedzí 3 až 14 dní od podania s odhadovanou absolútnou biologickou dostupnosťou 74 - 89 %. Pri podaní dávky 150 mg v 0. týždni, 4. týždni a následne každých 12 týždňov sú predpokladané maximálne resp. minimálne plazmatické koncentrácie v rovnovážnom stave 12 resp. 2 µg/ml.
U jedincov s Crohnovou chorobou liečených indukčnou dávkou 600 mg intravenózne (i.v.) v 0., 4. a 8. týždni, po ktorej nasleduje udržiavacia dávka 360 mg subkutánne (s.c.) v 12. týždni a potom každých
8 týždňov, maximálny medián vrcholovej a minimálnej (trough) koncentrácie pred podaním ďalšej
dávky sa odhaduje na 156 a 38, 8 µg/ml počas indukčného obdobia (8. – 12. týždeň) a medián ustálených vrcholových a minimálnych (trough) koncentrácií pred podaním ďalšej dávky sa odhaduje na 28,0 a 8,13 µg/ml počas udržiavacieho obdobia (40. – 48. týždeň).
Distribúcia
Priemerný (± smerodajná odchýlka) distribučný objem v rovnovážnom stave (Vss) rizankizumabu bol v štúdiách fázy 3 u jedincov so psoriázou 11,4 (± 2,7) l, čo naznačuje, že distribúcia rizankizumabu je
primárne obmedzená na vaskulárne a intersticiálne priestory. Typický 70 kg jedinec s Crohnovou chorobou mal Vss 7,68 l.
Biotransformácia
Terapeutické monoklonálne protilátky IgG sú typicky degradované na malé peptidy a aminokyseliny
katabolickými procesmi rovnakým spôsobom ako endogénne IgG. Nepredpokladá sa, že sa rizankizumab metabolizuje enzýmami P450.
Eliminácia
Priemerný (± smerodajná odchýlka) systémový klírens (CL) rizankizumabu bol v štúdiách fázy 3
u jedincov so psoriázou 0,3 (± 0,1) l/deň. Priemerný terminálny polčas eliminácie rizankizumabu sa
v štúdiách fázy 3 u jedincov so psoriázou pohyboval od 28 do 29 dní. Typický 70 kg jedinec
s Crohnovou chorobou mal systémový klírens 0,30 l/deň a terminálny polčas eliminácie 21 dní.
Nepredpokladá sa, že rizankizumab ako monoklonálna protilátka IgG1 by sa filtroval glomerulárnou filtráciou v obličkách alebo bol vylučovaný ako intaktná molekula v moči.
Linearita/nelinearita
Rizankizumab vykazoval lineárnu farmakokinetiku so zvýšeniami systémovej expozície približne
úmernými dávke (Cmax a AUC) v hodnotených rozmedziach dávky 18 až 360 mg alebo 0,25 až
1 mg/kg pri subkutánnom podaní a 200 až 1800 mg a 0,01 až 5 mg/kg pri intravenóznom podaní u zdravých jedincov alebo jedincov so psoriázou alebo Crohnovou chorobou.
Interakcie
U jedincov s ložiskovou psoriázou bola vykonaná štúdia interakcií na zhodnotenie účinku
opakovaného podávania rizankizumabu na farmakokinetiku substrátov citlivých na cytochróm P450
(CYP). Expozícia kofeínu (substrát CYP1A2), warfarínu (substrát CYP2C9), omeprazolu (substrát CYP2C19), metoprololu (substrát CYP2D6) a midazolamu (substrát CYP3A) po liečbe rizankizumabom bola porovnateľná s ich expozíciami pred liečbou rizankizumabom, čo svedčí o tom, že nedochádza k žiadnym významným interakciám prostredníctvom týchto enzýmov.
Populačné farmakokinetické analýzy ukazujú, že expozícia rizankizumabu nebola ovplyvnená súbežne podávanými liekmi, ktorú používali niektorí jedinci s ložiskovou psoriázou počas klinických štúdií. Podobne nebol nepozorovaný vplyv súbežne podávaných liekov na základe populačných farmakokinetických analýz pri Crohnovej chorobe.
Osobitné skupinypacientov
Pediatrická populácia
Farmakokinetické vlastnosti rizankizumabu u pediatrických jedincov mladších ako 16 rokov neboli stanovené. Z 1574 jedincov s Crohnovou chorobou vystavených rizankizumabu bolo 12 vo veku
16 - 17 rokov. Expozície rizankizumabu u 16- až 17-ročných osôb s Crohnovou chorobou boli
podobné ako u dospelých. Na základe populačných farmakokinetických analýz sa zistilo, že vek nemá významný vplyv na expozíciu rizankizumabu.
Starší pacienti
Z 2234 jedincov s ložiskovou psoriázou vystavených rizankizumabu bolo 243 vo veku 65 rokov alebo starších a 24 bolo veku 75 rokov alebo starších. Z 1574 jedincov s Crohnovou chorobou vystavených rizankizumabu bolo 72 vo veku 65 rokov alebo starších a 5 vo veku 75 rokov alebo starších. Medzi staršími a mladšími jedincami, ktorí dostávali rizankizumab, neboli pozorované žiadne celkové rozdiely v expozícii rizankizumabu.
Pacienti s poruchou funkcie obličiek alebo pečene
Neuskutočnili sa žiadne špecifické štúdie na stanovenie vplyvu poruchy funkcie obličiek alebo pečene na farmakokinetické vlastnosti rizankizumabu. Podľa populačných farmakokinetických analýz nemali sérové hladiny kreatinínu, klírens kreatinínu ani markery pečeňových funkcií (ALT/AST/bilirubín) významný vplyv na klírens rizankizumabu u jedincov s psoriázou alebo Crohnovou chorobou.
Rizankizumab ako monoklonálna protilátka IgG1 je eliminovaný hlavne intracelulárnym katabolizmom a nepredpokladá sa, že sa metabolizuje enzýmami hepatálneho cytochrómu P450 alebo vylučuje obličkami.
Telesná hmotnosť
Klírens a distribučný objem rizankizumabu sa zvyšujú s rastom telesnej hmotnosti, čo môže mať za následok zníženú účinnosť u jedincov s vysokou telesnou hmotnosťou (> 130 kg). Toto pozorovanie je však založené na obmedzenom počte jedincov s ložiskovou psoriázou. V súčasnosti sa neodporúča úprava dávky na základe telesnej hmotnosti.
Pohlavie alebo rasa
Klírens rizankizumabu nebol významne ovplyvnený pohlavím alebo rasou u dospelých jedincov
s ložiskovou psoriázou alebo Crohnovou chorobou. V klinickej farmakokinetickej štúdii so zdravými dobrovoľníkmi neboli pozorované žiadne klinicky významné rozdiely v expozícii rizankizumabu
u čínskych alebo japonských jedincov v porovnaní s belošskými jedincami.
5.3 Predklinické údaje o bezpečnosti
Neklinické údaje neodhalili žiadne osobitné riziko pre ľudí na základe štúdií toxicity po opakovanom podaní, vrátane farmakologických hodnotení bezpečnosti, a rozšírenej štúdie pre- a postnatálnej vývojovej toxicity na opiciach rodu Cynomolgus v dávkach do 50 mg/kg/týždeň, ktoré predstavujú 10- násobok klinických expozícií počas indukcie pri dávke 600 mg i.v. každé 4 týždne a 39-násobok klinických expozícií pri udržiavacej dávke 360 mg s.c. každých 8 týždňov.
S rizankizumabom neboli vykonané štúdie mutagenity a karcinogenity. V 26-týždňovej štúdii chronickej toxicity u opíc rodu Cynomolgus v dávkach až do 50 mg/kg/týždeň (približne 7-násobok klinickej expozície počas indukcie pri dávkovaní 600 mg i.v. každé 4 týždne a 28-násobok klinickej expozície počas udržiavacej fázy pri podávaní 360 mg s.c každých 8 týždňov) neboli pozorované žiadne preneoplastické alebo neoplastické lézie a nebola zaznamenaná žiadna nežiaduca imunotoxicita alebo kardiovaskulárne účinky.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Trihydrát octanu sodného
Kyselina octová
Dihydrát trehalózy
Polysorbát 20
Voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.
3 Čas použiteľnosti
24 mesiacov
Zriedený roztoknaintravenóznuinfúziu
Chemická a fyzikálna stabilita pri používaní bola preukázaná počas 20 hodín pri teplote 2 °C až 8 °C
alebo do 8 hodín pri izbovej teplote (kumulatívny čas po príprave vrátane doby uchovávania a infúzie), ak je zriedený roztok chránený pred priamym a nepriamym slnečným žiarením.
Z mikrobiologického hľadiska sa má pripravená infúzia použiť ihneď. Ak sa nepoužije ihneď, za dĺžku a podmienky uchovávania zriedeného lieku pred použitím je zodpovedný používateľ a doba uchovávania nemá byť dlhšia ako 20 hodín pri teplote 2 °C až 8 °C.
Neuchovávajte v mrazničke.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom. Podmienky na uchovávanie po nariedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
10,0 ml koncentrátu na infúzny roztok v sklenenej injekčnej liekovke uzavretej potiahnutou gumovou zátkou.
Skyrizi je k dispozícii v baleniach, ktoré obsahujú 1 injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Roztokom v injekčnej liekovke ani roztokom po zriedení sa nemá triasť. Pred podaním sa má roztok vizuálne skontrolovať; nemal by obsahovať žiadne častice ani by nemal mať neobvyklú farbu. Roztok má byť bezfarebný až svetložltý a číry až mierne opaleskujúci. Tekutina môže obsahovať drobné biele alebo číre čiastočky. Liek alebo zriedený liek sa nemajú používať, ak majú zmenenú farbu alebo ak je roztok zakalený alebo ak obsahuje cudzorodé častice.
Pokyny nariedenie
Tento liek má na podávanie pripravovať kvalifikovaný zdravotnícky pracovník dodržiavajúci
aseptický postup. Pred podaním sa musí zriediť.
Roztok na infúziu sa pripravuje zriedením koncentrátu do intravenózneho infúzneho vaku alebo sklenenej fľaše obsahujúcej 5% dextrózu vo vode (D5W) (600 mg/10 ml v 100 ml, 250 ml alebo
500 ml) na konečnú koncentráciu približne 1,2 mg/ml až 6 mg/ml.
Pred začiatkom intravenóznej infúzie má mať obsah intravenózneho infúzneho vaku alebo sklenenej fľaše izbovú teplotu.
Zriedený roztok podávajte počas najmenej jednej hodiny. Po zriedení v infúznom vaku sa má infúzia úplne podať do 8 hodín od zriedenia.
Každá injekčná liekovka je určená na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AbbVie Deutschland GmbH & Co. KG Knollstrasse
67061 Ludwigshafen
Nemecko
8. REGISTRAČNÉ ČÍSLO/ČÍSLAEU/1/19/1361/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 26. apríl 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.'
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUSkyrizi 360 mg injekčný roztok v náplni
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEJedna náplň obsahuje 360 mg rizankizumabu v 2,4 ml roztoku.
Rizankizumab je humanizovaná monoklonálna imunoglobulínová protilátka G1 (IgG1) produkovaná v ovariálnych bunkách čínskeho škrečka pomocou technológie rekombinantnej DNA.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný roztok (injekcia)
Roztok je bezfarebný až žltý a číry až mierne opaleskujúci.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLiek Skyrizi je indikovaný na liečbu dospelých pacientov so stredne ťažkou až ťažkou aktívnou Crohnovou chorobou, ktorí mali nedostatočnú odpoveď na konvenčnú liečbu alebo biologickú liečbu, alebo na ňu prestali reagovať, prípadne ju netolerovali.
4.2 Dávkovanie a spôsob podávaniaTento liek je určený na použitie pod vedením a dohľadom lekára, ktorý má skúsenosti s diagnostikou a liečbou stavov, na ktoré je liek Skyrizi indikovaný.
DávkovanieOdporúčaná dávka je 600 mg podávaná intravenóznou infúziou v 0. týždni, 4. týždni a 8. týždni,
následne 360 mg subkutánnou injekciou v 12. týždni a potom každých 8 týždňov. V prípade pacientov, u ktorých sa nepreukázal žiadny terapeutický prínos po 24 týždňoch liečby, sa má zvážiť ukončenie liečby.
Dávkovanie úvodného intravenózneho dávkovacieho režimu, pozri časť 4.2 súhrnu charakteristických vlastností lieku Skyrizi 600 mg koncentrát na infúzny roztok.
Vynechanie dávkyAk dôjde k vynechaniu dávky, dávka má byť podaná čo najskôr. Následne má byť dávkovanie
obnovené podľa pôvodného plánu.
Osobitné skupinypacientov
Starší pacienti (vo veku 65 rokov a viac)
Nie je potrebná úprava dávkovania (pozri časť 5.2).
K dispozícii sú obmedzené údaje u pacientov vo veku ≥ 65 rokov.
Porucha funkcie obličiek alebo pečene
Neuskutočnili sa žiadne špecifické štúdie na hodnotenie vplyvu poruchy funkcie pečene alebo obličiek na farmakokinetické vlastnosti Skyrizi. Všeobecne sa nepredpokladá, že tieto stavy budú mať významný vplyv na farmakokinetické vlastnosti monoklonálnych protilátok a nepovažuje sa za potrebné upravovať dávkovanie (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Skyrizi pri liečbe Crohnovej choroby u pacientov mladších ako 18 rokov neboli doteraz stanovené.
Pacienti s nadváhou
Nie je potrebná úprava dávkovania (pozri časť 5.2). Spôsob podávania
Skyrizi sa podáva subkutánnou injekciou.
Injekcia sa má podávať do stehna alebo brucha. Pacienti si nemajú podávať injekciu do oblastí, kde je koža citlivá, s podliatinami, erytematózna, zatvrdnutá alebo porušená.
Pacienti si môžu aplikovať liek Skyrizi sami po vyškolení v technike podávania subkutánnej injekcie
„on-body“ injektorom. Pacienti majú byť informovaní, aby si pred podaním prečítali „Návod na použitie“, ktorý je uvedený v písomnej informáci pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Klinicky významné aktívne infekcie (napr. Aktívna tuberkulóza, pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Infekcie
Rizankizumab môže zvýšiť riziko infekcie.
U pacientov s chronickou infekciou, recidivujúcou infekciou v anamnéze alebo známymi rizikovými faktormi pre infekciu sa má rizankizumab používať s opatrnosťou. Liečba rizankizumabom sa nemá začať u pacientov s akoukoľvek klinicky významnou aktívnou infekciou, pokým infekcia neodznie alebo nie je adekvátne liečená.
Pacienti liečení rizankizumabom majú byť poučení, aby vyhľadali lekársku pomoc, ak sa u nich
objavia prejavy alebo príznaky klinicky významnej chronickej alebo akútnej infekcie. Ak sa u pacienta objaví takáto infekcia alebo ak nereaguje na štandardnú liečbu infekcie, pacient má byť starostlivo sledovaný a rizankizumab sa nemá podávať, pokým neodznie infekcia.
Tuberkulóza
Pacienti majú byť pred začatím liečby rizankizumabom vyšetrení na tuberkulózu (TBC). Pacienti,
ktorí dostávajú rizankizumab, majú byť monitorovaní na prejavy a symptómy aktívnej TBC. Pred
začatím liečby rizankizumabom má byť zvážená liečba TBC u pacientov s latentnou alebo aktívnou
TBC v anamnéze, u ktorých nie je možné potvrdiť adekvátnu liečbu.
Imunizácia
Pred začatím liečby rizankizumabom sa má zvážiť absolvovanie všetkých príslušných imunizácií
podľa aktuálnych odporúčaní pre imunizáciu. Ak bola pacientovi podaná živá vakcína (vírusová alebo bakteriálna), odporúča sa so začatím liečby rizankizumabom počkať najmenej 4 týždne. Pacientom liečeným rizankizumabom sa počas liečby a najmenej 21 týždňov po ukončení liečby nesmú podávať živé vakcíny (pozri časť 5.2).
Precitlivenosť
Ak sa vyskytne závažná reakcia z precitlivenosti, podávanie rizankizumabu sa má okamžite ukončiť
a má sa začať príslušná liečba.
Pomocné látkysoznámymúčinkom
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v náplni, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Nepredpokladá sa, že rizankizumab podlieha metabolizácii pečeňovými enzýmami alebo vylučovaniu obličkami. Nepredpokladajú sa interakcie medzi rizankizumabom a inhibítormi, induktormi alebo substrátmi enzýmov metabolizujúcich lieky, a nie je potrebná úprava dávky (pozri časť 5.2).
Súbežná imunosupresívna liečba
Bezpečnosť a účinnosť rizankizumabu v kombinácii s imunosupresívami, vrátane biologických liekov,
neboli hodnotené.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženy vo fertilnom veku majú používať účinnú metódu antikoncepcie počas liečby a po dobu najmenej
21 týždňov po liečbe.
Gravidita
Údaje o použití rizankizumabu u tehotných žien sú obmedzené alebo nie sú k dispozícii (menej ako
300 výsledkov tehotenstva). Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky s ohľadom na reprodukčnú toxicitu. Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu rizankizumabu v gravidite.
Dojčenie
Nie je známe, či sa rizankizumab vylučuje do ľudského materského mlieka. Je známe, že humánne
IgG sa počas prvých niekoľkých dní po pôrode vylučujú do materského mlieka, čo sa čoskoro po tomto období znižuje na nízke koncentrácie; preto počas tohto krátkeho obdobia nemožno riziko pre dojčené dieťa vylúčiť. Je potrebné vziať do úvahy prínos dojčenia pre dieťa a prínos liečby rizankizumabom pre matku pri zvážení rozhodnutia o ukončení/nezačatí liečby rizankizumabom.
Fertilita
Účinok rizankizumabu na ľudskú fertilitu nebol hodnotený. Štúdie na zvieratách nepreukázali priame
alebo nepriame škodlivé účinky s ohľadom na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Rizankizumab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie boli infekcie horných dýchacích ciest (od 13,0 % pri psoriáze
do 15,6 % pri Crohnovej chorobe).
Tabuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie rizankizumabu pozorované v klinických štúdiách (tabuľka 1) sú uvedené podľa
triedy orgánových systémov (MedDRA) a vychádzajú z nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (nie je možné odhadnúť z dostupných údajov).
Tabuľk
a 1: Zoznam nežiaducich reakcií
Tried
a orgánových systémov
|
Frekvencia
|
Nežiaduc
e reakcie
|
Infekcie a nákazy
|
Veľmi časté
|
Infekcie horných dýchacích ciesta
|
Časté
|
Infekcie tineab
|
Menej časté
|
Folikulitída
|
Poruchy nervového systému
|
Časté
|
Bolesť hlavyc
|
Poruchy kože
a podkožného tkaniva
|
Časté
|
Pruritus
|
Neznáme
|
Vyrážka
Žihľavka
|
Celkové poruchy
a reakcie v mieste podania
|
Časté
|
Únavad
Reakcie v mieste vpichue
|
a Patrí sem: infekcia dýchacích ciest (vírusová, bakteriálna alebo nešpecifikovaná), sinusitída (vrátane akútnej), rinitída, nazofaryngitída, faryngitída (vrátane
vírusovej), tonzilitída, laryngitída, tracheitída
b Patrí sem: tinea pedis, tinea cruris, tinea corporis, tinea versicolor, tinea manuum, onychomykóza, mykotická infekcia kože
c Patrí sem: bolesť hlavy, tenzná bolesť hlavy, bolesť hlavy pri sínusitíde
d Patrí sem: únava, asténia
e Patrí sem: modrina v mieste vpichu, erytém, hematóm, hemorágia, podráždenie, bolesť, pruritus, reakcia, opuch, indurácia, vyrážka, precitlivenosť, uzlík, vyrážka, žihľavka, vezikuly, pocit tepla
|
Opis
vybraných
nežiaducich
reakcií
Infekcie
Psoriáza
Počas celého programu zameraného na psoriázu vrátane dlhodobej expozície rizankizumabu bol
výskyt infekcií 75,5 prípadov na 100 pacientorokov. Väčšina prípadov bola nezávažná a bola mierna až stredne závažná a neviedla k prerušeniu liečby rizankizumabom. Výskyt závažných infekcií bol 1,7 prípadu na 100 pacientorokov (pozri časť 4.4).
Crohnova chorobaCelkovo bol bezpečnostný profil pozorovaný u pacientov s Crohnovou chorobou liečených
rizankizumabom v súlade s bezpečnostným profilom pozorovaným u pacientov s ložiskovou psoriázou.
Miera výskytu infekcií v súhrnných údajoch z 12-týždňových indukčných štúdií bola 83,3 prípadov na
100 pacientorokov u jedincov liečených rizankizumabom 600 mg intravenózne v porovnaní so
117,7 prípadmi na 100 pacientorokov pri placebe. Výskyt závažných infekcií bol 3,4 prípadu na
100 pacientorokov (pozri časť 4.4) u jedincov liečených intravenózne podávaným rizankizumabom
600 mg v porovnaní so 16,7 prípady na 100 pacientorokov pri placebe (pozri časť 4.4).
Výskyt infekcií v 52-týždňovej udržiavacej štúdii bol 57,7 prípadu na 100 pacientorokov u jedincov liečených subkutánne podávaným rizankizumabom 360 mg po indukcii rizankizumabom v porovnaní so 76,0 prípadov na 100 pacientorokov u jedincov, ktorým bolo podané placebo po indukcii rizankizumabom. Výskyt závažných infekcií bol 6,0 prípadu na 100 pacientorokov u jedincov liečených subkutánne podávaným rizankizumabom 360 mg po indukcii rizankizumabom v porovnaní
s 5,0 prípadmi na 100 pacientorokov u jedincov, ktorým bolo podané placebo po indukcii rizankizumabom (pozri časť 4.4).
ImunogenitaTak ako pri všetkých terapeutických proteínoch, aj pri rizankizumabe existuje možnosť imunogenity. Detekcia tvorby protilátok vysoko závisí od citlivosti a špecifickosti testu.
U jedincov s Crohnovou chorobou liečených odporúčanou indukčnou intravenóznou dávkou rizankizumabu a subkutánnou udržiavacou dávkou rizankizumabu počas obdobia až 64 týždňov
v klinických skúšaniach zameraných na liečbu Crohnovej choroby boli detegované protilátky proti lieku a neutralizujúce protilátky u 3,4 % (2/58), resp. u 0 % (0/58) hodnotených jedincov.
Protilátky proti rizankizumabu, vrátane neutralizujúcich protilátok, neboli spojené so zmenami klinickej odpovede alebo bezpečnosti.
Starší pacientiK dispozícii sú obmedzené údaje o bezpečnosti u pacientov vo veku ≥ 65 rokov.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania sa odporúča, aby sa u pacienta sledovali prípadné prejavy alebo príznaky nežiaducich reakcií a aby sa okamžite začalo s vhodnou symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu, ATC kód: L04AC18
Mechanizmus účinkuRizankizumab je humanizovaná monoklonálna protilátka imunoglobulín G1 (IgG1), ktorá sa
selektívne viaže s vysokou afinitou na podjednotku p19 humánneho cytokínu označovaného ako interleukín 23 (IL-23) bez väzby na IL-12 a inhibuje jeho interakciu s receptorovým komplexom
IL-23. IL-23 je cytokín, ktorý sa podieľa na zápalovej a imunitnej odpovedi. Tým, že rizankizumab blokuje IL-23 a bráni mu vo väzbe na jeho receptor, inhibuje bunkovú signalizáciu závislú od IL-23 a uvoľňovanie prozápalových cytokínov.
Farmakodynamické účinkyV štúdii u jedincov so psoriázou bola po jednotlivých dávkach rizankizumabu znížená expresia génov
súvisiacich s osou IL-23/IL-17 v koži. V psoriatických léziách bolo pozorované aj zníženie hrúbky epidermy, infiltrácie zápalovými bunkami a expresie markerov psoriázy.
V štúdii fázy 2 u jedincov s Crohnovou chorobou bola po viacnásobnom podaní rizankizumabu znížená expresia génov súvisiacich s osou IL-23/IL-17 v tkanivách čreva. Po viacerých dávkach v indukčných štúdiách fázy 3 u pacientov s Crohnovou chorobou sa pozorovalo zníženie hladiny
fekálneho kalprotektínu (
faecal calprotectin, FCP), sérového C-reaktívneho proteínu (CRP) a IL-22. Zníženia FCP, CRP a IL-22 v sére sa udržali až do 52. týždňa udržiavacej štúdie.
Klinická účinnosťÚčinnosť a bezpečnosť rizankizumabu boli hodnotené u 1419 jedincov so stredne ťažkou až ťažkou
aktívnou Crohnovou chorobou v troch multicentrických, randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách. Do štúdie boli zaradené jedinci vo veku 16 rokov alebo starší s indexom aktivity Crohnovej choroby (Crohn’s Disease Activity Index, CDAI) 220 až 450,
priemernou dennou frekvenciou stolice (stool frequency, SF) ≥ 4 a/alebo priemerným denným skóre bolesti brucha (abdominal pain score, APS) ≥ 2 a zjednodušeným endoskopickým skóre pre CD (Simple Endoscopic Score for CD, SES-CD) ≥ 6 alebo ≥ 4 v prípade izolovaného ileálneho ochorenia, s vylúčením zužujúcej sa zložky a s potvrdením centrálneho hodnotiteľa.
Uskutočnili sa dve 12-týždňové indukčné štúdie (ADVANCE a MOTIVATE) s intravenóznym podávaním, ktoré zahŕňali 12-týždňové predĺženie pre jedincov, ktorí nedosiahli klinickú odpoveď v hodnotení SF/APS (≥ 30 % pokles SF a/alebo ≥ 30 % pokles APS a obidve hodnoty neboli horšie ako východiskové hodnoty) v 12. týždni. Po štúdiách ADVANCE a MOTIVATE nasledovala 52-
týždňová randomizovaná štúdia vysadenia pri subkutánnej udržiavacej liečbe (FORTIFY), do ktorej
boli zaradení jedinci s klinickou odpoveďou v hodnotení SF/APS na intravenózne podávanú indukčnú liečbu, čo predstavuje najmenej 64 týždňov liečby.
ADVANCE a MOTIVATEV štúdiách ADVANCE a MOTIVATE boli jedinci randomizovaní na používanie rizankizumabu buď
v dávke 600 mg (odporúčaná dávka), 1200 mg alebo placeba, v 0. týždni, 4. týždni a 8. týždni.
V štúdii ADVANCE u 58 % (491/850) jedincov zlyhala liečba jednou alebo viacerými biologickými terapiami alebo túto liečbu netolerovali (predchádzajúce zlyhanie biologickej liečby) a u 42 % (359/850) jedincov zlyhala liečba konvenčnými terapiami alebo ju netolerovali, ale nezlyhala biologická terapia (bez predchádzajúceho zlyhania biologickej liečby). V štúdii ADVANCE bolo spomedzi jedincov bez predchádzajúceho zlyhania biologickej liečby 87 % (314/359) jedincov bez predchádzajúcej biologickej liečby a zvyšných 13 % jedincov už používalo biologickú liečbu, ale nikdy nedošlo k jej zlyhaniu alebo sa u nich neprejavila intolerancia. U všetkých pacientov v štúdii MOTIVATE došlo k predchádzajúcemu biologickému zlyhaniu liečby.
V oboch štúdiách väčší podiel jedincov liečených rizankizumabom dosiahol koprimárne cieľové ukazovatele: klinickú remisiu v 12. týždni a endoskopickú odpoveď v porovnaní s placebom
v 12. týždni. Zvýšená klinická odpoveď v hodnotení SF/APS a klinická remisia boli u jedincov liečených rizankizumabom významné už v 4. týždni a naďalej sa zlepšovali až do 12. týždňa (tabuľka 2).
Tabuľka 2. Výsledky účinnosti v štúdiách ADVANCE a MOTIVATE
| ADVANCE
| MOTIVATE
|
| Placebo
(N=175)
%
| Rizankizumab
600 mg i.v. (N=336)
%
| Liečebný rozdiel
(95 % CI)
| Placebo i. v. (N=187)
%
| Rizankizumab
600 mg i.v. (N=191)
%
| Liečebný rozdield (95 % CI)
|
Koprimárne cieľové ukazovatele
|
Klinická remisia
v 12. týždni
|
22 %
|
43 %
| 22 %
[14 %,
30 %]a
|
19 %
|
35 %
|
15 %
[6 %, 24 %]b
|
Endoskopi
c
- ká odpoveď v 12. týždni
|
12 %
|
40 %
|
28 %
[21 %,
35 %]a
|
11 %
|
29 %
|
18 %
[10 %, 25 %]a
|
Dodatočn
é cieľové ukazovatele
|
Zvýšená klinická odpoveď
v hodnotení
SF/APS vo
4. týždni
g
|
31 %
|
46 %
|
15 %
[6 %,
23 %]b
|
32 %
|
45 %
|
14 %
[4 %, 23 %]c
|
Zvýšená klinická odpoveď
v hodnotení
SF/APS
v 12. týždni
g
|
42 %
|
63 %
|
21 %
[12 %,
30 %]a
|
39 %
|
62 %
|
23 %
[13 %, 33 %]a
|
CDAI < 150
v
o 4. týždni
|
10 %
|
18 %
|
8 %
[1 %,
14 %]c
|
11 %
|
21 %
|
10 %
[2 %, 17 %]c
|
CDAI < 150
vo 12. týždni
|
25 %
|
45 %
|
21 %
[12 %,
29 %]a
|
20 %
|
42 %
|
22 %
[13 %, 31 %]a
|
Za
h
ojenie sliznice
v 12. týždni
h
|
(N=173)
8 %
|
(N=336)
21 %
|
14 %
a
|
(N=186)
4 %
|
(N=190)
14 %
|
9 %
b
|
Endoskopi
c
- ká odpoveď v 12. týždni
|
9 %
|
24 %
|
15 %
[9 %,
21 %]a
|
4 %
|
19 %
|
15 %
[9 %, 21 %]a
|
aŠtatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p < 0,001).
bŠtatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p ≤ 0,01).
c Nominálne porovnanie p ≤ 0,05 rizankizumab vs placebo.
dUpravený liečebný rozdiel.
e Klinická remisia na základe SF/APS: priemerná denná SF ≤ 2,8 a nie horšia ako východisková hodnota a priemerné denné skóre AP ≤ 1 a nie horšie ako východisková hodnota.
f Endoskopická odpoveď: viac ako 50 % pokles SES-CD oproti východiskovej hodnote alebo pokles aspoň o 2 body u jedincov s východiskovým skóre 4 a izolovaným ileálnym ochorením.
g Zvýšená klinická odpoveď v hodnotení SF/APS: ≥ 60 % pokles priemerného denného skóre SF
a/alebo ≥ 35 % pokles priemerného denného skóre AP a obe nie horšie ako východiskové skóre, a/alebo klinická remisia.
h Zahojenie sliznice: Podskóre SES-CD ulcerovaného povrchu 0 u jedincov s podskóre ≥ 1 na
začiatku liečby.
i Endoskopická remisia SES-CD ≤ 4 a zníženie aspoň o 2 body v porovnaní s východiskovou hodnotou a žiadne čiastkové skóre väčšie ako 1 v žiadnej jednotlivej premennej.
|
|
|
[8%, 19%]
[4 %, 15 %]
V 12. týždni dosiahol vyšší podiel jedincov liečených rizankizumabom pokles aspoň o 100 bodov od
východiskovej hodnoty CDAI v porovnaní s placebom (ADVANCE, rizankizumab = 60 %, placebo37 %, p < 0,001; MOTIVATE, rizankizumab = 60 %, placebo=30 %, p < 0,001).
V 12. týždni dosiahol vyšší podiel jedincov liečených rizankizumabom zvýšenú klinickú odpoveď
v hodnotení SF/APS aj endoskopickú odpoveď v porovnaní s placebom (ADVANCE, rizankizumab
=31 %, placebo = 8 %, p < 0,001; MOTIVATE, rizankizumab = 21 %, placebo = 7 %, p < 0,001).
Výsledky pre koprimárne cieľové ukazovatele v podskupinách jedincov so zlyhaním predchádzajúcej biologickej liečby a bez tohto zlyhania (bez pripustenia multiplicity) sú uvedené v tabuľke 3.
Tabuľk
a 3. Výsledky účinnosti v 12. týždni u jedincov s predchádzajúcim zlyhaním biologickej liečby a jedincov bez predchádzajúceho zlyhania biologickej liečby v štúdii ADVANCE
|
ADVANCE
|
Placeb
o i.v.
|
Rizankizuma
b 600 mg
|
Liečebný rozdiel (95 % IS)
|
Klinická remisia podľa skóre SF/AP
|
Predchádzajúce zlyhanie biologickej liečby
|
23 % (N=97)
|
41 % (N=195)
|
18 % [7 %, 29 %]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
21 % (N=78)
|
48 % (N=141)
|
27 % [15 %, 39 %]
|
Endoskopick
á odpoveď
|
Predchádzajúce zlyhanie biologickej liečby
|
11 % (N=97)
|
33 % (N=195)
|
21 % [12 %, 31 %]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
13 % (N=78)
|
50 % (N=141)
|
38 % [27 %, 49 %]
|
V štúdii ADVANCE dosiahol vyšší podiel jedincov liečených rizankizumabom s predchádzajúcim
biologickým zlyhaním aj bez neho CDAI < 150 v porovnaní s placebom (s predchádzajúcim biologickým zlyhaním, rizankizumab = 42 %, placebo = 26 %; bez predchádzajúceho biologického zlyhania, rizankizumab = 49 %, placebo = 23 %).
Hospitalizácie súvisiace s Crohnovou chorobouMiera hospitalizácií súvisiacich s Crohnovou chorobou v 12. týždni bola nižšia u osôb liečených rizankizumabom v porovnaní s placebom (ADVANCE, rizankizumab = 3 %, placebo = 12 %,
p < 0,001; MOTIVATE, rizankizumab = 3 %, placebo = 11 %, p ≤ 0,01).
FORTIFYUdržiavacia štúdia FORTIFY hodnotila 462 jedincov s klinickou odpoveďou v hodnotení SF/APS na
12 týždňov intravenóznej indukčnej liečby rizankizumabom (i.v.) v štúdiách ADVANCE
a MOTIVATE. Jedinci boli randomizovaní na pokračovanie v udržiavacom režime subkutánne (s.c.)
podávaného rizankizumabu 360 mg (odporúčaná dávka) alebo rizankizumabu 180 mg s.c. každých
8 týždňov, alebo na ukončenie indukcie rizankizumabom a podávanie placeba s.c. každých 8 týždňov až do 52 týždňov.
Koprimárne cieľové ukazovatele boli klinická remisia v 52. týždni a endoskopická odpoveď
v 52. týždni. U jedincov s predchádzajúcim biologickým zlyhaním a bez neho sa merali aj koprimárne cieľové ukazovatele (pozri tabuľku 4).
Tabuľka 4. Výsledky účinnosti v štúdii FORTIFY v 52. týždni (64 týždňov od začatia liečby indukčnou dávkou).
| FORTIFY
|
| Rizankizumab i.v. indukcia/s.c. placebof (N=164) %
| Rizankizumab i.v. indukcia/rizankizumab
360 mg s.c. (N=141) %
| Liečebný rozdiel
(95 % IS)
|
Koprimárne cieľové ukazovatele
|
Klinická remisia
|
40 %
|
52 %
|
15% [5%, 25%]a,g
|
Predchádzajúce zlyhanie biologickej liečby
|
34 % (N=123)
|
48 % (N=102)
|
14 % [1 %,27 %]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
56 % (N=41)
|
62 % (N=39)
|
5 % [-16 %,27 %]
|
Endoskopick
á odpoveď
|
22 %
|
47 %
|
28% [19%, 37%]b,g
|
Predchádzajúce zlyhanie biologickej liečby
|
20 % (N=123)
|
44 % (N=102)
|
23% [11%, 35%]
|
Bez predchádzajúceho zlyhania biologickej liečby
|
27 % (N=41)
|
54 % (N=39)
|
27 % [6 %, 48 %]
|
Dodatočn
é cieľové ukazovatele
|
Zvýšená klinická odpoveď v hodnotení SF/APS
|
49 %
|
59 %
|
13% [2%, 23%]e,g
|
Udržanie klinickej remisie
h
|
(N = 91)
51 %
|
(N = 72)
69 %
|
21% [6%, 35%]d,g
|
Endoskopická remisia
|
13 %
|
39 %
|
28% [20%, 37%]c,g
|
Hojenie sliznice
|
(N = 162)
10 %
|
(N = 141)
31 %
|
22% [14%, 30%]c,g
|
a Štatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p < 0,001). b Štatisticky významné pri kontrole multiplicity pri porovnaní rizankizumabu a placeba (p < 0,001). c Nominálne porovnanie p < 0,001 rizankizumab vs placebo bez celkovej kontroly chýb typu I.
d Nominálne porovnanie p ≤ 0,01 rizankizumab vs placebo bez celkovej kontroly chýb typu I.
e Nominálne porovnanie p ≤ 0,05 rizankizumab vs placebo bez celkovej kontroly chýb typu I.
f Skupinu len s indukčnou liečbou tvorili jedinci, u ktorých sa dosiahla klinická odpoveď na indukčnú liečbu rizankizumabom a ktorí boli randomizovaní na podávanie placeba v udržiavacej
štúdii (FORTIFY).
g Upravený liečebný rozdiel.
h Udržanie klinickej remisie: klinická remisia v 52. týždni u jedincov s klinickou remisiou v 0. Týždni
|
Hlboká remisia (klinická remisia a endoskopická remisia) v 52. týždni sa pozorovala vo vyššej miere
u osôb liečených rizankizumabom i.v./rizankizumabom s.c. v porovnaní s jedincami, ktorí dostávali rizankizumab i.v./placebo s.c.(28 % vs 10 %, nominálna hodnota p < 0,001).
V 52. týždni dosiahol vyšší podiel jedincov liečených rizankizumabom i.v./rizankizumabom s.c. CDAI
< 150 v porovnaní s jedincami, ktorí dostávali rizankizumab i.v./placebo s.c. (52 % vs 41 %, nominálna hodnota p < 0,01). Vyšší podiel jedincov liečených rizankizumabom i.v./rizankizumabom s.c zaznamenal zníženie aspoň o 100 bodov v skóre východiskovej CDAI v porovnaní s jedincami, ktorí dostávali rizankizumab i.v./placebo s.c. (62 % vs 48 %, nominálna hodnota p < 0,001).
91 jedincom, u ktorých sa nepreukázala klinická odpoveď v hodnotení SF/APS 12 týždňov po indukcii rizankizumabom v štúdiách ADVANCE a MOTIVATE, sa podávala subkutánna 360 mg dávka rizankizumabu v 12. a 20. týždni. Z týchto jedincov 64 % (58/91) dosiahlo klinickú odpoveď
v hodnotení SF/APS v 24. týždni; 33 jedincov, ktorí dosiahli klinickú odpoveď v hodnotení SF/APS, sa zaradilo do štúdie FORTIFY a pokračovalo v s.c. používaní rizankizumabu 360 mg každých
8 týždňov až do 52 týždňov. Spomedzi týchto jedincov dosiahlo 55 % (18/33) klinickú remisiu a 45 %
(15/33) endoskopickú odpoveď v 52. týždni.
Počas štúdie FORTIFY 30 jedincov stratilo odpoveď na liečbu rizankizumabom 360 mg s.c. a dostalo záchrannú liečbu rizankizumabom (jednorazová i.v. dávka 1200 mg, po ktorej nasledovalo 360 mg s.c. každých 8 týždňov). Spomedzi týchto jedincov dosiahlo 57 % (17/30) klinickú odpoveď v hodnotení SF/APS v 52. týždni. Navyše spomedzi týchto jedincov dosiahlo 20 % (6/30) klinickú remisiu a 34 % (10/29) endoskopickú odpoveď v 52. týždni.
Výsledky týkajúce sa zdravia a kvality života
Kvalita života súvisiaca so zdravím sa hodnotila pomocou Dotazníka o zápalových črevných
ochoreniach (Inflammatory Bowel Disease Questionnaire, IBDQ) a Krátkeho dotazníka kvality života SF-36 (36-Item Short Form Health Survey, SF-36). Zmiernenie únavy sa hodnotilo pomocou dotazníka k Funkčnému hodnoteniu liečby chronických ochorení - únava (Functional Assessment of Chronic Illness Therapy-Fatigue, FACIT-Fatigue). Produktivita práce bola hodnotená pomocou Dotazníka o zhoršení produktivity práce a aktivity (Work Productivity and Activity Impairment CD, WPAI-CD).
V 12. týždni štúdií ADVANCE a MOTIVATE dosiahli jedinci liečení rizankizumabom klinicky významné zlepšenie oproti východiskovému stavu v celkovom skóre IBDQ, vo všetkých doménových skóre IBDQ (črevné príznaky, systémové funkcie, emocionálne funkcie a sociálne funkcie),
v súhrnnom skóre fyzických a psychických komponentov SF-36, a v skóre FACIT-Fatigue a WPAI- CD v porovnaní s placebom. Pre WPAI-CD sa v štúdii ADVANCE preukázalo nižšie ovplyvnenie pri práci, celkové ovplyvnenie práce a menší ovplyvnenie aktivity a v štúdii MOTIVATE bolo zaznamenane nižšie ovplyvnenie aktivity. Tieto zlepšenia sa udržali u osôb liečených
rizankizumabom i.v./rizankizumabom s.c. v štúdii FORTIFY do 52. týždňa.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre rizankizumab
v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe Crohnovej choroby (pre informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Rizankizumab vykazoval lineárnu farmakokinetiku so zvýšením expozície úmerným dávke
v rozmedzí dávok 18 až 360 mg a 0,25 až 1 mg/kg podávaných subkutánne a 200 až 1800 mg a 0,01
až 5 mg/kg podávaných intravenózne.
Po subkutánnom podaní rizankizumabu boli maximálne plazmatické koncentrácie dosiahnuté
v rozmedzí 3 až 14 dní od podania s odhadovanou absolútnou biologickou dostupnosťou 74 – 89 %. Pri podaní dávky 150 mg v 0. týždni, 4. týždni a následne každých 12 týždňov sú predpokladané maximálne resp. Minimálne plazmatické koncentrácie v rovnovážnom stave 12 resp. 2 µg/ml.
U jedincov s Crohnovou chorobou liečených indukčnou dávkou 600 mg intravenózne (i.v.) v 0., 4. a 8. týždni, po ktorej nasleduje udržiavacia dávka 360 mg subkutánne (s.c.) v 12. týždni a potom každých
8 týždňov, maximálny medián vrcholovej a minimálnej (trough) koncentrácie pred podaním ďalšej dávky sa odhaduje na 156 a 38, 8 µg/ml počas indukčného obdobia (8. – 12. týždeň) a medián ustálených vrcholových a minimálnych (trough) koncentrácií pred podaním ďalšej dávky sa odhaduje na 28,0 a 8,13 µg/ml počas udržiavacieho obdobia (40. – 48. týždeň).
Distribúcia
Priemerný (± smerodajná odchýlka) distribučný objem v rovnovážnom stave (Vss) rizankizumabu bol v štúdiách fázy 3 u jedincov so psoriázou 11,4 (± 2,7) l, čo naznačuje, že distribúcia rizankizumabu je
primárne obmedzená na vaskulárne a intersticiálne priestory. Typický 70 kg jedinec s Crohnovou chorobou mal Vss 7,68 l.
Biotransformácia
Terapeutické monoklonálne protilátky IgG sú typicky degradované na malé peptidy a aminokyseliny
katabolickými procesmi rovnakým spôsobom ako endogénne IgG. Nepredpokladá sa, že sa rizankizumab metabolizuje enzýmami P450.
Eliminácia
Priemerný (± smerodajná odchýlka) systémový klírens (CL) rizankizumabu bol v štúdiách fázy 3
u jedincov so psoriázou 0,3 (± 0,1) l/deň. Priemerný terminálny polčas eliminácie rizankizumabu sa
v štúdiách fázy 3 u jedincov so psoriázou pohyboval od 28 do 29 dní. Typický 70 kg jedinec
s Crohnovou chorobou mal systémový klírens 0,30 l/deň a terminálny polčas eliminácie 21 dní.
Nepredpokladá sa, že rizankizumab ako monoklonálna protilátka IgG1 by sa filtroval glomerulárnou filtráciou v obličkách alebo bol vylučovaný ako intaktná molekula v moči.
Linearita/nelinearita
Rizankizumab vykazoval lineárnu farmakokinetiku so zvýšeniami systémovej expozície približne
úmernými dávke (Cmax a AUC) v hodnotených rozmedziach dávky 18 až 360 mg alebo 0,25 až
1 mg/kg pri subkutánnom podaní a 200 až 1800 mg a 0,01 až 5 mg/kg pri intravenóznom podaní u zdravých jedincov alebo jedincov so psoriázou alebo Crohnovou chorobou.
Interakcie
U jedincov s ložiskovou psoriázou bola vykonaná štúdia interakcií na zhodnotenie účinku
opakovaného podávania rizankizumabu na farmakokinetiku substrátov citlivých na cytochróm P450
(CYP). Expozícia kofeínu (substrát CYP1A2), warfarínu (substrát CYP2C9), omeprazolu (substrát CYP2C19), metoprololu (substrát CYP2D6) a midazolamu (substrát CYP3A) po liečbe rizankizumabom bola porovnateľná s ich expozíciami pred liečbou rizankizumabom, čo svedčí o tom, že nedochádza k žiadnym významným interakciám prostredníctvom týchto enzýmov.
Populačné farmakokinetické analýzy ukazujú, že expozícia rizankizumabu nebola ovplyvnená súbežne podávanými liekmi, ktorú používali niektorí jedinci s ložiskovou psoriázou počas klinických štúdií. Podobne nebol nepozorovaný vplyv súbežne podávaných liekov na základe populačných farmakokinetických analýz pri Crohnovej chorobe.
Osobitné skupinypacientov
Pediatrická populácia
Farmakokinetické vlastnosti rizankizumabu u pediatrických jedincov mladších ako 16 rokov neboli stanovené. Z 1574 jedincov s Crohnovou chorobou vystavených rizankizumabu bolo 12 vo veku
16 – 17 rokov. Expozícia rizankizumabu u 16- až 17-ročných osôb s Crohnovou chorobou bola
podobná ako u dospelých. Na základe populačných farmakokinetických analýz sa zistilo, že vek nemá významný vplyv na expozíciu rizankizumabu.
Starší pacienti
Z 2234 jedincov s ložiskovou psoriázou vystavených rizankizumabu bolo 243 vo veku 65 rokov alebo starších a 24 bolo veku 75 rokov alebo starších. Z 1574 jedincov s Crohnovou chorobou vystavených rizankizumabu bolo 72 vo veku 65 rokov alebo starších a 5 vo veku 75 rokov alebo starších. Medzi staršími a mladšími jedincami, ktorí dostávali rizankizumab, neboli pozorované žiadne celkové rozdiely v expozícii rizankizumabu.
Pacienti s poruchou funkcie obličiek alebo pečene
Neuskutočnili sa žiadne špecifické štúdie na stanovenie vplyvu poruchy funkcie obličiek alebo pečene na farmakokinetické vlastnosti rizankizumabu. Podľa populačných farmakokinetických analýz nemali sérové hladiny kreatinínu, klírens kreatinínu ani markery pečeňových funkcií (ALT/AST/bilirubín) významný vplyv na klírens rizankizumabu u jedincov s psoriázou alebo Crohnovou chorobou.
Rizankizumab ako monoklonálna protilátka IgG1 je eliminovaný hlavne intracelulárnym katabolizmom a nepredpokladá sa, že sa metabolizuje enzýmami hepatálneho cytochrómu P450 alebo vylučuje obličkami.
Telesná hmotnosť
Klírens a distribučný objem rizankizumabu sa zvyšujú s rastom telesnej hmotnosti, čo môže mať za následok zníženú účinnosť u jedincov s vysokou telesnou hmotnosťou (> 130 kg). Toto pozorovanie je však založené na obmedzenom počte jedincov s ložiskovou psoriázou. V súčasnosti sa neodporúča úprava dávky na základe telesnej hmotnosti.
Pohlavie alebo rasa
Klírens rizankizumabu nebol významne ovplyvnený pohlavím alebo rasou u dospelých jedincov
s ložiskovou psoriázou alebo Crohnovou chorobou. V klinickej farmakokinetickej štúdii so zdravými dobrovoľníkmi neboli pozorované žiadne klinicky významné rozdiely v expozícii rizankizumabu
u čínskych alebo japonských jedincov v porovnaní s belošskými jedincami.
5.3 Predklinické údaje o bezpečnosti
Neklinické údaje neodhalili žiadne osobitné riziko pre ľudí na základe štúdií toxicity po opakovanom podaní, vrátane farmakologických hodnotení bezpečnosti, a rozšírenej štúdie pre- a postnatálnej vývojovej toxicity na opiciach rodu Cynomolgus v dávkach do 50 mg/kg/týždeň, ktoré predstavujú
10-násobok klinických expozícií počas indukcie pri dávke 600 mg i.v. každé 4 týždne a 39-násobok klinických expozícií pri udržiavacej dávke 360 mg s.c. každých 8 týždňov.
S rizankizumabom neboli vykonané štúdie mutagenity a karcinogenity. V 26-týždňovej štúdii chronickej toxicity u opíc rodu Cynomolgus v dávkach až do 50 mg/kg/týždeň (približne 7-násobok klinickej expozície počas indukcie pri dávkovaní 600 mg i.v. každé 4 týždne a 28-násobok klinickej expozície počas udržiavacej fázy pri podávaní 360 mg s.c každých 8 týždňov) neboli pozorované žiadne preneoplastické alebo neoplastické lézie a nebola zaznamenaná žiadna nežiaduca imunotoxicita alebo kardiovaskulárne účinky.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Trihydrát octanu sodného
Kyselina octová
Dihydrát trehalózy
Polysorbát 20
Voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi.
6.
3 Čas použiteľnosti
24 mesiacov
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Náplň uchovávajte vo vonkajšom obale
Pred použitím sa odporúča vizuálna kontrola každej náplni na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
360 mg roztok v jednorazovej náplni vyrobenej z cyklickej olefínovej živice s gumovou prepážkou a gumovým piestom ako kontaktnými materiálmi s produktom a živicovým uzáverom. Náplň je zostavená pomocou teleskopickej skrutkovej zostavy. Náplň je balená spolu s „on-body“ injektorom (zariadenie na podávanie). Kvapalina v „on-body“ injektore prechádza polyvinylchloridovou hadičkou a 29-gauge ihlou z nehrdzavejúcej ocele. „On-body“ injektor obsahuje batérie s oxidom strieborným
a zinkom a lepiacu náplasť na kožu vyrobenú z polyesteru s akrylovým lepidlom. Zariadenie na podávanie je určené na použitie s dodanou 360 mg náplňou.
Skyrizi 360 mg je k dispozícii v baleniach, ktoré obsahujú 1 náplň a 1 „on-body“ injektor.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Liek Skyrizi je určený na použitie pod vedením a dohľadom zdravotníckeho pracovníka.
Pred použitím sa odporúča vizuálna kontrola každej náplni. Roztok neonsahuje cudzorodé častice a je prakticky bez častíc súvisiacich s liekom. Skyrizi sa nemá použiť, ak je roztok zakalený alebo má zmenenú farbu alebo ak obsahuje veľké čiastočky.
Roztok má byť bezfarebný až žltý a číry až mierne opaleskujúci.
Pred podaním injekcie majú pacienti vybrať škatuľku z chladničky a nechať ju pri izbovej teplote mimo priameho slnečného žiarenia po dobu 45 až 90 minút bez toho, aby náplň vybrali zo škatuľky.
Úplný návod na použitie je uvedený v písomnej informácii pre používateľa. Každý „on-body“ injektor s náplňou je určený len na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AbbVie Deutschland GmbH & Co. KG Knollstrasse
67061 Ludwigshafen
Nemecko
8. REGISTRAČNÉ ČÍSLO/ČÍSLA
EU/1/19/1361/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 26. apríl 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.PRÍLOHA IVVEDECKÉ ZÁVERY A DÔVODY ZMENY PODMIENOK ROZHODNUTIA O REGISTRÁCIIVedecké záveryVzhľadom na hodnotiacu správu Výboru pre hodnotenie rizík liekov (PRAC) o periodicky aktualizovaných správach o bezpečnosti lieku (PSURs) pre rizankizumab dospel Výbor pre humánne lieky (CHMP) k týmto vedeckým záverom:
Vzhľadom na dostupné údaje zo spontánnych hlásení o vyrážke a žihľavke, zahŕňajúcich v 30, resp.
118 prípadoch úzku časovú súvislosť a vzhľadom na pravdepodobný mechanizmus účinku, výbor PRAC považuje kauzálny vzťah medzi rizankizumabom a vyrážkou a žihľavkou za prinajmenšom odôvodnenú možnosť. PRAC dospel k záveru, že informácie o liekoch obsahujúcich rizankizumab sa majú zodpovedajúcim spôsobom zmeniť a doplniť.
Výbor pre humánne lieky (CHMP) súhlasí s vedeckými závermi PRAC.
Dôvody zmeny podmienok rozhodnutia o registráciiNa základe vedeckých záverov pre rizankizumab je CHMP toho názoru, že pomer prínosu a rizika lieku (liekov) obsahujúceho rizankizumab je nezmenený za predpokladu, že budú prijaté navrhované zmeny v informáciách o lieku.
CHMP odporúča zmenu podmienok rozhodnutia o registrácii.