
/u>
podávaniaSARCLISA je určená na intravenózne použitie. Pokyny na riedenie lieku pred podaním, pozri časť
6.6.
RýchlosťpodávaniainfúzieInfúziu SARCLISY je potrebné po riedení podať intravenózne s rýchlosťou podávania infúzie uvedenej v Tabuľke 2 nižšie (pozri časť 5.1). Postupné zvýšenie rýchlosti podávania infúzie je možné zvážiť len v prípade, ak sa nevyskytli žiadne reakcie na infúziu (pozri časť 4.8).
 Tabuľka 2: Rýchlosti podávania infúzie SARCLISY
Tabuľka 2: Rýchlosti podávania infúzie SARCLISY
| Nariedený objem
| Počiatočná rýchlosť
| Absencia reakcie na infúziu
| Prírastok rýchlosti infúzie
| Maximálna rýchlosť
|
Prvá infúzia
| 250 ml
| 25 ml/hodina
| Počas 60 minút
| 25 ml/hodina každých 30 minút
| 150 ml/hodina
|
Druhá infúzia
| 250 ml
| 50 ml/hodina
| Počas 30 minút
| 50 ml/hodina počas 30 minút, následne zvýšiť objem o
100 ml/hodina
každých 30 minút
| 200 ml/hodina
|
Ďalšie infúzie
| 250 ml
| 200 ml/hodina
|
|
| 200 ml/hodina
|
Ak sa u pacientov vyskytnú reakcie na infúziu, je potrebné upraviť podávanie (pozri časť 4.4).
• U pacientov, u ktorých sa vyskytli reakcie na infúziu 2. stupňa (stredne závažné), sa má zvážiť dočasné prerušenie podávania infúzie a je možné podať dodatočnú symptomatickú liečbu. Po zlepšení stavu na stupeň ≤ 1 (mierna reakcia), je možné opätovne podávať infúziu SARCLISY pri polovičnej počiatočnej rýchlosti podávania, pričom je podľa potreby nevyhnutné dôsledné monitorovanie a podporná liečba. Ak sa symptómy znovu neobjavia po 30 minútach, je možné zvýšiť rýchlosť podávania infúzie na počiatočnú rýchlosť a následne ju postupne zvyšovať
tak, ako je uvedené v Tabuľke 2.
• Ak po prerušení podávania infúzie SARCLISY symptómy rýchlo nevymiznú, ich stav sa nezlepší na stupeň ≤ 1, vyskytnú sa opakovane po úvodnom zmiernení pomocou vhodných liekov, vyžadujú si hospitalizáciu alebo ohrozujú život pacienta (stupeň ≥ 3), je nevyhnutné liečbu SARCLISOU natrvalo ukončiť a podľa potreby je nutné podávať dodatočnú podpornú liečbu.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníSledovateľnosťAby sa zlepšila sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo šarže podaného lieku.
R
ea
kcie
na
infúzie
U 38,2 % pacientov, ktorým bola podaná SARCLISA (pozri časť 4.8), boli zaznamenané mierne alebo stredne závažné reakcie na infúziu. Všetky reakcie na infúziu začali počas podávania prvej infúzie SARCLISY a v 98 % prípadov odzneli tieto reakcie v ten istý deň. Medzi najčastejšie symptómy reakcií na infúziu patrili dyspnoe, kašeľ, zimnica a nevoľnosť. Medzi najzávažnejšie prejavy a symptómy patrili hypertenzia a dyspnoe (pozri časť 4.8).
Aby sa znížilo riziko a závažnosť reakcií na infúziu, je potrebné pacientom pred podaním SARCLISY podať ako premedikáciu paracetamol, H2 antagonisty alebo inhibítory protónovej pumpy, difenhydramín alebo ekvivalent; dexametazón sa používa aj ako premedikácia a aj na liečbu myelómu (pozri časť 4.2). Počas celého trvania podávania infúzie SARCLISY je potrebné časté monitorovanie vitálnych funkcií. Ak je to potrebné, je potrebné prerušiť podávanie infúzie SARCLISY a vykonať vhodné medicínske a podporné opatrenia (pozri časť 4.2). Ak sa po prerušení podávania infúzie SARCLISY príznaky nezlepšia, vyskytnú sa opakovane po úvodnom zmiernení pomocou vhodných liekov, vyžadujú si hospitalizáciu, alebo ohrozujú život pacienta, je nevyhnutné liečbu SARCLISOU natrvalo ukončiť a zaviesť vhodnú liečbu.
Neutropénia
U pacientov liečených SARCLISOU boli zaznamenané prípady neutropénie 3. – 4. stupňa, nahlásené ako laboratórne abnormality (84,9 %) a neutropenické komplikácie (30,3 %) (pozri časť 4.8).
Počas liečby je potrebné pravidelne monitorovať kompletný krvný obraz. Pacienti s neutropéniou sa majú monitorovať, aby bolo možné odhaliť prejavy infekcie. Neodporúča sa znižovať dávku SARCLISY. Na zmiernenie rizika neutropénie (pozri časť 4.2) je možné odložiť podanie dávky
SARCLISY a využiť rastové faktory (napr. G-CSF).
Infekcie
V súvislosti so SARCLISOU sa zaznamenal vyšší výskyt infekcií vrátane infekcií stupňa ≥ 3, najmä
pneumónie, infekcií horných dýchacích ciest a bronchitídy (pozri časť 4.8). U pacientov dostávajúcich SARCLISU sa majú starostlivo monitorovať príznaky infekcie a má sa začať vhodná štandardná liečba. Počas liečby možno zvážiť profylaktickú liečbu antibiotikami, antimykotikami
a antivirotikami.
Ďalšieprimárnemalignity
V štúdii ICARIA-MM boli zaznamenané ďalšie primárne malignity (SPM, second primary malignancies) u 6 pacientov (3,9 %) liečených SARCLISOU a u 1 pacienta (0,7 %) liečeného pomadidomidom a dexametazónom a zahŕňali kožný skvamocelulárny karcinóm u 4 pacientov liečených SARCLISOU a u 1 pacienta liečeného pomadidomidom a dexametazónom (pozri časť 4.8). Po resekcii kožného skvamocelulárneho karcinómu pacienti pokračovali v liečbe. Celkový výskyt SPM u všetkých pacientov exponovaných SARCLISA je 3 %. Pred liečbou a počas liečby majú lekári v súlade s usmerneniami IMWG dôkladne zhodnotiť výskyt SPM a začať indikovanú liečbu.
Interferenciasosérologickýmitestami(nepriamyantiglobulínovýtest)
SARCLISA sa viaže na CD38 na červené krvinky (red blood cells, RBC), čo môže spôsobiť falošný pozitívny výsledok nepriameho antiglobulínového testu (nepriamy Coombsov test). Aby sa predišlo potenciálnym problémom s transfúziou RBC, je potrebné, aby pacientom, ktorí podstúpia liečbu SARCLISOU, boli pred zavedením prvej infúzie vykonané testy na určenie krvnej skupiny a skríningové vyšetrenia. Pred začatím liečby SARCLISOU je možné zvážiť určiť fenotyp podľa lokálnej praxe. Ak sa s liečbou SARCLISOU už začalo, je potrebné informovať krvnú banku, že pacient podstupuje liečbu SARCLISOU. Pacienti majú byť informovaní o teoretickom riziku hemolýzy. Ak je potrebné podať pohotovostnú transfúziu, je možné podať krížovo nespárované ABO/RhD kompatibilné s RBC podľa bežnej praxe miestnej krvnej banky (pozri časť 4.5).
V súčasnosti nie sú k dispozícii informácie týkajúce sa toho, ako dlho po poslednej infúzii SARCLISY môže pretrvávať interferencia s nepriamym Coombsovým testom. Vychádzajúc z polčasu izatuximabu sa dá predpokladať, že izatuximabom sprostredkovaný pozitívny nepriamy Coombsov test môže pretrvávať približne počas 6 mesiacov od poslednej infúzie.
Interferenciasostanovenímkompletnejodpovede
Izatuximab je monoklonálna protilátka IgG kappa, ktorá môže byť rozpoznaná vo vzorkách elektroforézy sérových proteínov (SPE) aj vo vzorkách imunofixácie (IFE), ktoré sa používajú na klinické monitorovanie endogénneho M-proteínu (pozri časť 4.5). Táto interferencia môže
u niektorých pacientov s myelómovým proteínom IgG kappa ovplyvniť presnosť stanovenia kompletnej odpovede. Interferencia bola skúmaná u dvadsiatich dvoch pacientov zo skupiny, ktorí boli nastavení na režim podávania izatuximabu a zároveň splnili kritériá pre veľmi dobrú čiastočnú odpoveď (VGPR), pričom sa u nich vyskytla len zostatková imunofixačná pozitivita. Vzorky séra týchto pacientov boli podrobené skúške pomocou hmotnostnej spektrometrie, ktorá umožňuje oddeliť signál izatuximabu od signálu myelómu M-proteínu (pozri časť 4.5).
Staršíľudia
Údaje o populácii pacientov vo veku ≥ 85 rokov sú obmedzené (pozri časť 4.2).
4.5 Liekové a iné interakcie
Izatuximab neovplyvňuje farmakokinetiku pomalidomidu a naopak. Interferenciasosérologickýmitestami
Nakoľko proteín CD38 sa nachádza na povrchu červených krviniek, izatuximab, protilátka proti CD38, môže zasahovať do sérologického testovania krvnej banky a vyvolať tak falošne pozitívne reakcie pri nepriamych antiglobulínových testoch (nepriame Coombsove testy), pri testoch na rozpoznávanie protilátok (pri skríningu), na paneloch na identifikáciu protilátok a v prípade krížového párovania antihumánnych globulínov (AHG) u pacientov, ktorým je podávaný izatuximab (pozri časť
4.4). Interferenciu možno zmierniť ovplyvnením reagenčných RBCs (Red Blood Cells, červených
krviniek) s ditiotrietolom (DTT), čo bráni väzbe izatuximabu, alebo pomocou lokálne validovaných metód. Keďže Kellov systém krvných skupín je taktiež citlivý na ovplyvnenie s DTT, Kell negatívne jednotky musia byť dodané po vylúčení alebo identifikovaní allo-protilátok pri RBCs ovplyvnených
s DTT.
Zásahdoelektroforézysérovýchproteínovaimunofixačnýchtestov
Izatuximab môže byť detegovaný vo vzorkách elektroforézy sérových proteínov (SPE) aj vo vzorkách imunofixácie (IFE), ktoré sa používajú na klinické monitorovanie endogénneho M-proteínu a môže zasahovať do presnosti klasifikácie odpovede na základe kritérií medzinárodnej pracovnej skupiny pre myelóm (International Myelome Working Group, IMWG) (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/Antikoncepcia
Ženy vo fertilnom veku, ktoré podstupujú liečbu izatuximabom, musia používať účinnú antikoncepciu počas liečby, a minimálne 5 mesiacov po jej ukončení.
Gravidita
Nie sú k dispozícii údaje o používaní izatuximabu u gravidných žien. Neboli vykonané štúdie reprodukčnej toxicity u zvierat v súvislosti s používaním izatuximabu. Je známe, že imunoglobulínové monoklonálne protilátky G1 prechádzajú placentou po ukončení prvého trimestra tehotenstva.
Použitie izatuximabu sa u gravidných žien neodporúča.
Dojčenie
Nie je známe, či sa izatuximab vylučuje do ľudského materského mlieka. Je známe, že ľudské IgG sa vylučujú do materského mlieka počas prvých dní po pôrode, pričom sa ich koncentrácia krátko na to zníži. Riziko, ktorému je dojčené dieťa počas tohto krátkeho obdobia bezprostredne po pôrode vystavené, sa však nedá vylúčiť. Pre toto osobitné obdobie sa má rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu izatuximabom má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu. Následne, podľa klinickej potreby, možno izatuximab použiť počas dojčenia.
F
er
tilit
a
Nie sú k dispozícii žiadne údaje o ľuďoch ani o zvieratách, na základe ktorých by bolo možné stanoviť potenciálne účinky izatuximabu na mužskú a ženskú fertilitu (pozri časť 5.3).
V prípade iných liečiv podávaných spolu s izatuximabom sa riaďte podľa aktuálnych súhrnov charakteristických vlastností jednotlivých liekov.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeSARCLISA nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluNajčastejšie nežiaduce reakcie (
> 20 %) sú neutropénia (46,7 %), reakcie na infúziu (38,2 %), zápal pľúc (30,9 %), infekcie horných dýchacích ciest (28,3 %), hnačka (25,7 %) a bronchitída (23,7 %).
Najčastejšie sa vyskytujúce závažné nežiaduce reakcie sú zápal pľúc (9,9 %) a febrilná neutropénia
(6,6 %).
ZoznamnežiaducichreakciíuvedenývtabuľkeNežiaduce reakcie sú popísané pomocou Všeobecných kritérií toxicity NCI, terminológie COSTART a MedDRA. Frekvencie sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé
(< 1/10 000); neznáme (z dostupných údajov).
V rámci každej skupiny zostavenej na základe frekvencie výskytu sú nežiaduce reakcie uvedené v
poradí podľa klesajúcej miery závažnosti.
Nežiaduce reakcie boli roztriedené podľa výskytu ≥ 5 % (všetky stupne) alebo ≥ 2 % (stupne
≥ 3)
u pacientov liečených v skupine s izatuximabom a na základe miery výskytu ≥ 5 % väčšej v skupine
s režimom izatuximabu v porovnaní so skupinou s porovnávacím režimom (pomalidomid a nízka dávka dexametazónu). Atriálna fibrilácia a kožný skvamocelulárny karcinóm boli pridané z dôvodu ich klinického významu.
Tabuľka 3a: Nežiaduce reakcie zaznamenané u pacientov s mnohopočetným myelómom, liečených izatuximabom v kombinácii s pomalidomidom a nízkou dávkou dexametazónu (štúdia ICARIA-MM)Systém triedy orgánov
Preferovaný termín
| Nežiaduca reakcia
| Frekvencia
| Výskyt (%) (N = 152)
|
Akýkoľvek stupeň
| Stupeň ≥ 3
|
Infekcie a nákazy
| Zápal pľúc b
| Veľmi časté
|
47 (30,9)
|
40 (26,3)
|
Infekcia horných dýchacích ciest*
| Veľmi časté
|
43 (28,3)
|
5 (3,3)
|
Bronchitída*
| Veľmi časté
|
36 (23,7)
|
5 (3,3)
|
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
| Kožný skvamocelulárny karcinóm
| Časté
|
4 (2,6)
|
2 (1,3)
|
Poruchy krvného
| Neutropéniac
| Veľmi časté
|
71 (46,7)
|
70 (46,1)
|
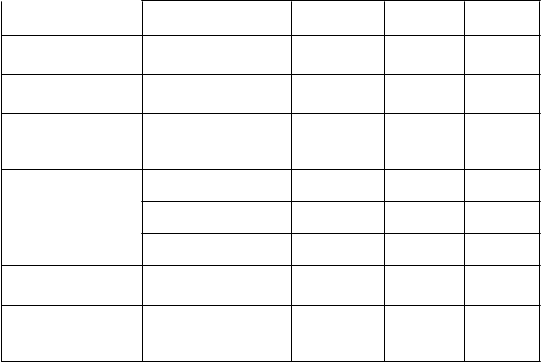
obehu a lymfatického systému
|
Febrilná neutropénia
|
Veľmi časté
|
18 (11,8)
|
18 (11,8)
|
Poruchy
m
et
abolizmu a výživy
|
Znížená chuť do jedla*
|
Časté
|
15 (9,9)
|
2 (1,3)
|
Poruchy srdca
a srdcovej činnosti
|
Atriálna fibrilácia
|
Časté
|
7 (4,6)
|
3 (2,0)
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Dyspnoe*
|
Veľmi časté
|
23 (15,1)
|
6 (3,9)
|
Poruchy gastrointestinálneho
|
Hnačka*
|
Veľmi časté
Veľmi časté
|
39 (25,7)
|
3 (2,0)
|
tr
aktu
|
|
|
23 (15,1)
|
0
|
|
Vracanie*
|
Veľmi časté
|
18 (11,8)
|
2 (1,3)
|
L
aboratórne
a funkčné vyšetrenia
|
Úbytok hmotnosti*
|
Časté
|
10 (6,6)
|
0
|
Ú
r
azy, otravy a komplikácie liečebného postupu
|
Reakcia na infúziu
|
Veľmi časté
|
58 (38,2)
|
4 (2,6)
|
|
|
Nevoľnosť*
a V Tabuľke 3 sú uvedené iba TEAE. Hematologické laboratórne hodnoty sú zaznamenané v Tabuľke 4.
b Termín zápal pľúc zahŕňa nasledujúce pojmy: atypický zápal pľúc, bronchopulmonálna aspergilóza, zápal pľúc, zápal pľúc spôsobený baktériou Haemophilus, zápal pľúc spôsobený chrípkou, zápal pľúc spôsobený pneumokokmi, zápal pľúc spôsobený streptokokmi, zápal pľúc spôsobený vírusmi, zápal pľúc spôsobený kvasinkou Candida, zápal pľúc spôsobený baktériami, infekcia baktériou hemophilus, pľúcna infekcia, zápal pľúc spôsobený hubovou infekciou a zápal pľúc spôsobený parazitom Pneumocystis jirovecii.
c Hematologické laboratórne hodnoty sú zaznamenané ako TEAE iba vtedy, ak viedli k prerušeniu liečby a/alebo úprave dávky a/alebo splnili závažné kritérium a/alebo boli definované ako AESI.
*Žiaden stupeň 4
Popis vybraných nežiaducich reakciíReakcienainfúziePočas klinického skúšania ICARIA-MM boli reakcie na infúzie zaznamenané u 58 pacientov (38,2 %) liečených SARCLISOU. V prípade všetkých pacientov, u ktorých sa vyskytli reakcie na infúzie, sa tieto reakcie vyskytli počas podávania prvej infúzie SARCLISY, u 3 pacientov (2,0 %) sa vyskytli reakcie na infúziu aj počas podávania druhej infúzie a u 2 pacientov (1,3 %) aj počas podávania 4.
infúzie. U 3,9 % pacientov sa vyskytli reakcie na infúziu 1. stupňa, u 31,6 % reakcie 2. stupňa, u 1,3 %
reakcie 3. stupňa a u 1,3 % pacientov sa vyskytli reakcie 4. stupňa. Všetky reakcie na infúziu boli reverzibilné a u 98 % pacientov vymizli v deň podania infúzie. Medzi prejavy a symptómy minimálne
3. stupňa reakcií na infúziu patrili dyspnoe, hypertenzia a bronchospazmus.
Miera prerušenia podávania infúzie z dôvodu reakcií na infúziu dosiahla úroveň 28,9 %. Priemerná dĺžka podávania infúzie pred jej prerušením bola 55 minút.
Prerušenie liečby z dôvodu reakcie na infúziu bolo zaznamenané u 2,6 % pacientov v skupine nastavenej na režim podávania izatuximabu.
InfekciePočas klinického skúšania ICARIA-MM bola miera výskytu infekcií minimálne 3. stupňa 42,8 %. Najčastejšie zaznamenanou závažnou infekciou bol zápal pľúc, pričom infekcia 3. stupňa sa vyskytla u
21,7 % pacientov v skupine s izatuximabovým režimom, v porovnaní so 16,1 % pacientov v skupine s porovnávacím režimom (pomalidomid a nízke dávky dexametazónu). Infekcia 4. stupňa sa vyskytla u
3,3 % pacientov v skupine s izatuximabovým režimom, v porovnaní s 2,7 % v skupine s porovnávacím režimom. Predčasné ukončenie liečby z dôvodu infekcie bolo zaznamenané u 2,6 %
pacientov v skupine s izatuximabovým režimom, v porovnaní s 5,4 % v skupine s porovnávacím
režimom. Fatálne infekcie boli zaznamenané u 3,3 % pacientov v skupine s režimom podávania izatuximabu a u 4,0 % v skupine s porovnávacím režimom.
Hematologické laboratórnehodnotyTabuľka 4: Hematologické laboratórne anomálie u pacientov, ktorým bol podávaný izatuximabv kombinácii s pomalidomidom a nízkymi dávkami dexametazónom, v porovnaní s podávaním pomalidomidu a nízkych dávok dexametazónu (ICARIA-MM)Laboratórna hodnota
| SARCLISA + pomalidomid + nízke dávky dexametazónu
n (%)
(N = 152)
| pomalidomid + nízke dávky dexametazónu
n (%)
(N = 147)
|
| Všetky stupne
| Stupeň 3
| Stupeň 4
| Všetky stupne
| Stupeň 3
| Stupeň 4
|
Anémia
| 151 (99,3)
| 48 (31,6)
| 0
| 145 (98,6)
| 41 (27,9)
| 0
|
Neutropénia
| 146 (96,1)
| 37 (24,3)
| 92 (60,5)
| 137 (93,2)
| 57 (38,8)
| 46 (31,3)
|
Lymfopénia
| 140 (92,1)
| 64 (42,1)
| 19 (12,5)
| 137 (93,2)
| 52 (35,4)
| 12 (8,2)
|
Trombocytopénia
| 127 (83,6)
| 22 (14,5)
| 25 (16,4)
| 118 (80,3)
| 14 (9,5)
| 22 (15,0)
|
Spoločným menovateľom použitým na výpočet percentuálneho zastúpenia je počet pacientov, u
ktorých bol počas zohľadneného pozorovacieho obdobia vyhodnotený aspoň 1 laboratórny test.
ImunogenicitaNaprieč 6 klinickými štúdiami zameranými na podávanie izatuximabu, ako samostatného liečiva aj
v kombinácii s inými liečivami, pacientom s mnohopočetným myelómom (MM), vrátane klinického
skúšania ICARIA-MM (N = 564), bol výskyt protilátok proti liečivu (ADA) súvisiaci s liečbou 2,3 %. Vplyv ADA na farmakokinetiku, bezpečnosť ani účinnosť izatuximabu nebol zaznamenaný.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePrejavyasymptómyPočas klinických skúšaní sa nevyskytli žiadne prípady predávkovania. Počas klinických štúdií boli podávané intravenózne dávky s obsahom najviac 20 mg/kg izatuximabu.
LiečbaV prípade predávkovania SARCLISOU neexistuje špeciálna protilátka. Pri predávkovaní je potrebné monitorovať pacienta kvôli prejavom alebo symptómom nežiaducich reakcií a okamžite prijať všetky vhodné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakologické vlastnostiFarmakoterapeutická skupina: cytostatiká, monoklonálne protilátky, ATC kód: L01XC38 izatuximab.
MechanizmusúčinkuIzatuximab je monoklonálna protilátka odvodená od IgG1, ktorá sa viaže k špecifickému mimobunkovému epitopu receptoru CD38. CD38 je transmembránový glykoproteín, ktorý je jasne definovaný na bunkách mnohopočetných myelómov.
I
n vitro, izatuximab pôsobí na základe IgG mechanizmu závislom od Fc vrátane: cytotoxicity sprostredkovanej bunkami závislej od protilátok (antibody dependent cell mediated, ADCC), bunkovej fagocytózy závislej od protilátok (antibody dependent cellular phagocytosis, ADCP) a cytotoxicity závislej od doplnkov (complement dependent cytotoxicity, CDC). Izatuximab dokáže tiež usmrtiť nádorové bunky pomocou indukcie apoptózy prostredníctvom mechanizmu nezávislého na Fc.
In vitro izatuximab blokuje enzymatickú aktivitu CD38, čo katalyzuje syntézu a hydrolýzu cyklickej
ADP-ribózy (cADPR), činidla, ktoré mobilizuje vápnik. Izatuximab inhibuje produkciu cADPR
z mimobunkových NAD (nicotinamide adenine dinucleotide) v bunkách mnohopočetného myelómu.
In vitro môže izatuximab aktivovať bunky NK v prípade chýbajúceho pozitívneho cieľa nádorových buniek CD38.
In vivo pokles absolútneho počtu celkových CD16+ a CD56+ buniek NK, CD19+ B-bunky, CD4+ T- bunky a TREG (CD3+, CD4+, CD25+, CD127-) bol zaznamenaný v periférnej krvi pacientov, ktorí boli liečení izatuximabom v monoterapii.
U pacientov s mnohopočetným myelómom vyvolala monoterapia SARCLISOU klonálnu expanziu repertoáru receptoru T-bunky, ktorá preukazuje adaptívnu imunitnú odpoveď.
In vitro kombinácia izatuximabu a pomalidomidu v porovnaní len so samotným izatuximabom zlepšuje rozpad bunky CD38, v dôsledku čoho sú pomocou efektorových buniek (ADCC) a priameho usmrtenia nádorových buniek bunky mnohopočetného myelómu vytláčané. In vivo pokusy za použitia modelu štepu z cudzieho tkaniva ľudského mnohopočetného myelómu preukázali, že kombinácia izatuximabu a pomalidomidu prispieva k lepšej protinádorovej aktivite, v porovnaní s aktivitou izatuximabu podaného samostatne alebo pomalidomidu podaného samostatne.
Klinickáúčinnosťabezpečnosť
ICARIA-MM (EFC14335)
Účinnosť a bezpečnosť SARCLISY v kombinácii s pomalidomidom a nízkodávkovým dexametazónom boli vyhodnotené na základe ICARIA-MM (EFC14335), multicentrickej medzinárodnej, randomizovanej, otvorenej, dvojramennej štúdie fázy III u pacientov s relapsujúcim a refraktérnym mnohopočetným myelómom. Pacienti absolvovali aspoň dve predchádzajúce liečby, vrátane podávania lenalidomidu a inhibítoru proteáz (PI), a došlo u nich počas poslednej liečby alebo do 60 dní po jej ukončení k progresii ochorenia. Pacienti so základným refraktérnym ochorením boli vylúčení.
Celkovo bolo randomizovaných 307 pacientov v pomere 1:1 na liečbu buď SARCLISOU
v kombinácii s pomalidomidom a nízkodávkovým dexametazónom (režim podávania izatuximabu,
154 pacientov) alebo pomalidomidom a nízkymi dávkami dexametazónu (porovnávací režim, 153 pacientov). V oboch skupinách boli lieky podávané v 28-dňových cykloch, až kým nedošlo k progresii ochorenia alebo k nežiaducej toxicite. SARCLISA 10 mg/kg bola podávaná vo forme intravenóznej infúzie týždenne počas prvého cyklu a následne každé dva týždne.
Jedenkrát denne sa podávala dávka 4 mg pomalidomidu v rozmedzí od 1. do 21. dňa každého 28- dňového cyklu. Nízka dávka dexametazónu 40 mg (20 mg u pacientov vo veku ≥ 75 rokov) (perorálne/intravenózne) sa podávala v 1., 8., 15. a 22. deň každého 28-dňového cyklu.
Celkovo bola východisková demografická charakteristika a východisková charakteristika ochorení
v oboch skupinách podobná, vyskytli sa len mierne asymetrie. Priemerný vek pacientov bol 67 rokov
(v rozmedzí od 36 – 86 rokov), 19,9 % pacientov bolo vo veku ≥ 75 rokov. ECOG PS bol 0 u 35,7 %
pacientov v ramene s izatuximabom a u 45,1 % v porovnávacom ramene, 1 u 53,9 % v ramene
s izatuximabom a u 44,4 % v porovnávacej skupine a 2 u 10,4 % v ramene s izatuximabom a 10,5 %
v porovnávacom ramene, 10,4 % pacientov v ramene s izatuximabom v porovnaní s 10,5 % pacientov
v porovnávacom ramene, ktorí sa zapojili do klinického skúšania, sa v minulosti liečili na COPD alebo astmu a 38,6 % pacientov s poruchou funkcie obličiek (klírens kreatinínu < 60 ml/min/1,73 m²) bolo zaradených do skupiny užívajúcej izatuximab, v porovnaní s 33,3 % pacientov, ktorí boli zaradení do porovnávacej skupiny. Na základe medzinárodného systému na určovanie štádia ochorenia
(International Staging System, ISS) na začiatku klinického skúšania bolo štádium I stanovené u
37,5 % (41,6 % v ramene s izatuximabom a 33,3 % v porovnávacom ramene), štádium II u 35,5 % (34,4 % v ramene s izatuximabom a 36,6 % v porovnávacom ramene) a štádium III u 25,1 % (22,1 % v ramene s izatuximabom a 28,1 % v porovnávacom ramene) pacientov. Celkovo u 19,5 % pacientov (15,6 % v ramene s izatuximabom a 23,5 % v porovnávacom ramene) bolo na začiatku klinického skúšania vysoké riziko chromozomálnych anomálií; del (17 p) sa vyskytovalo u 12,1 % (9,1 %
v ramene s izatuximabom a 15,0 % v porovnávacom ramene), t (4; 14) sa vyskytovalo u 8,5 % (7,8 %
v ramene s izatuximabom a 9,2 % v porovnávacom ramene) a t (14; 16) sa vyskytovalo u 1,6 % (0,6 %
v ramene užívajúcom izatuximab a 2,6 % v porovnávacom ramene) pacientov.
Priemerný počet absolvovaných línií liečby bol 3 (v rozmedzí od 2 – 11). Všetkým pacientom boli v minulosti podávané inhibítor proteazómu, lenalidomid a 56,4 % pacientov v minulosti podstúpilo transplantáciu kmeňových buniek. Väčšina pacientov (92,5 %) bola refraktérna voči lenalidomidu,
75,9 % voči inhibítoru proteazómu a 72,6 % bolo refraktérnych aj voči imunomodulačnému inhibítoru aj voči inhibítoru proteazómu a 59 % pacientov bolo refraktérnych voči lenalidomidu v poslednej línii liečby.
Priemerná dĺžka trvania liečby bola 41,0 týždňov v skupine s izatuximabom, v porovnaní s 24,0
týždňami v porovnávacej skupine.
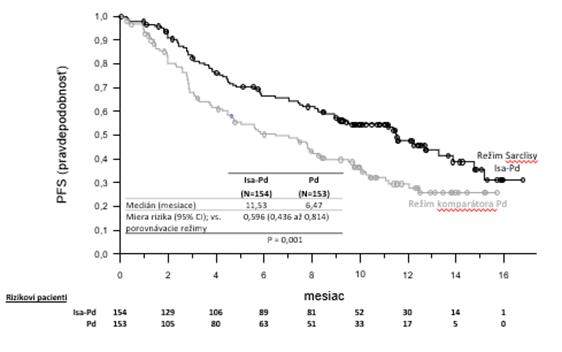
Primárnym cieľom účinnosti klinického skúšania ICARIA-MM bolo prežitie bez progresie (PFS). Zlepšenie PFS preukazovalo 40,4 % zníženie rizika progresie ochorenia alebo úmrtia pacientov, ktorí dodržiavali režim podávania izatuximabu.
Výsledky účinnosti sú uvedené v tabuľke 5 a Kaplanove-Meierove krivky pre PFS a OS sú znázornené na obrázkoch 1 a 2:
Tabuľka 5: Účinnosť SARCLISY v kombinácii s pomalidomidom a nízkymi dávkami dexametazónu, v porovnaní s pomalidomidom a nízkymi dávkami dexametazónu, pri liečbe mnohopočetného myelómu (analýza zámeru liečiť (intent to treat))Záver
| SARCLISA + pomalidomid + nízke dávky dexametazónu
N = 154
| Pomalidomid + nízke dávky dexametazónu
N = 153
|
Prežívanie bez progresiea b
|
|
|
Priemerná dĺžka (v mesiacoch)
[95 % CI]
| 11,53
[8,936 – 13,897]
| 6,47
[4,468 – 8,279]
|
Miera rizikac [95 % CI]
| 0,596 [0,436 – 0,814]
|
hodnota-p (stratifikovanýlog-ranktest)c
| 0,0010
|
Celková miera odpoveded
Počet pacientov, u ktorých sa vyskytla odpoveď (sCR + CR + VGPR + PR) n (%)
[95 % CI]e
|
93 (60,4)
[0,5220 – 0,6817]
|
54 (35,3)
[0,2775 – 0,4342]
|
Miera pravdepodobnosti vs. komparátor [95 % presný CI]
| 2,795 [1,715 – 4,562]
|
hodnota-p (stratifikovaný Cochranov-Mantelov-Haenszelov test)c
| < 0,0001
|
Z
áver
'
|
S
ARC
L
ISA + pomalidomid + nízke dávky dexametazónu
N = 154
|
Pomalidomid + nízke dávky dexametazónu
N = 153
|
Striktne úplná odpoveď (sCR)
+ úplná odpoveď (CR) n (%)
|
7 (4,5)
|
3 (2,0)
|
Veľmi dobrá čiastočná odpoveď (VGPR) n (%)
|
42 (27,3)
|
10 (6,5)
|
Čiastočná odpoveď (PR) n
(%)
|
44 (28,6)
|
41 (26,8)
|
V
G
PR alebo lepšia n (%)
[95 % CI]e
|
49 (31,8)
[0,2455 – 0,3980]
|
13 (8,5)
[0,0460 – 0,1409]
|
Miera pravdepodobnosti vs. komparátor [95 % presný CI]
|
5,026 [2,514 – 10,586]
|
hodnota-p (stratifikovaný Cochranov-Mantelov-Haenszelov test)c
|
< 0,0001
|
T
r
vanie odpovede f *
Medián v mesiacoch [95% CI]g
|
13,27 [10,612-NR]
|
11,07 [8,542-NR]
|
|
|
|
|
|
a Výsledky PFS posudzovala na základe centrálnych laboratórnych údajov o M-proteíne a revízií centrálnych rádiologických zobrazení nezávislá komisia pre odpoveď (Independent Response Committee, IRC) pomocou kritérií medzinárodnej pracovnej skupiny pre myelóm (International Myeloma Working Group, IMWG).
b Pacienti s prežitím alebo bez progresie ochorenia pred ukončením analýzy alebo pred dátumom iniciácie ďalšej anti-myelómovej liečby boli kontrolovaní k dátumu posledného platného posúdenia ochorenia nepreukazujúceho
progresiu ochorenia pred začatím ďalšej anti-myelómovej liečby (ak nejaká bola) alebo pred dátumom ukončenia, podľa toho, čo nastane skôr.
c Rozdelenie podľa veku (< 75 rokov versus
> 75 rokov) a počtu predchádzajúcich absolvovaných línií liečby
(2 alebo 3 verzus > 3) podľa IRT.
d sCR, CR, VGPR a PR vyhodnotila IRC na základe kritérií pre odpoveď IMWG.
e Odhad na základe Clopper-Pearsonovej metódy.
f Trvanie odpovede bolo stanovené pre pacientov, ktorí dosiahli odpoveď ≥PR (93 pacienti v ramene
s izatuximabom a 54 pacientov porovnávacom ramene). Kaplanove-Meierove odhady trvania odpovede. g CI pre Kaplanove-Meierove odhady sa počíta pomocou transformácie log-log funkcie prežitia a Brookmeyerovych a Crowleho metód.
* Dátum ukončenia bol 11. október 2018. Priemerné trvanie následného kontrolného obdobia = 11,60 mesiacov.
HR < 1 v prospech skupiny s režimom podávania izatuximabu. NR: nedosiahnutý
V prípade pacientov s vysokorizikovou cytogenetikou (centrálne laboratórne posúdenie) dosahovalo priemerné PFS 7,49 (95 % CI: 2,628 až NC) v skupine s režimom izatuximabu a 3,745 (95 % CI:
2,793 až 7,885) v skupine s porovnávacím režimom (HR = 0,655; 95 % CI: 0,334 až 1,283). Zlepšenie
PFS bolo v skupine s režimom podávania izatuximabu zaznamenané aj u pacientov vo veku
> 75 rokov (HR = 0,479; 95 % CI: 0,242 až 0,946) v štádiu III podľa ISS na začiatku klinického skúšania (HR = 0,635; 95 % CI: 0,363 až 1,110) so základným klírensom kreatinínu < 60 ml/min/1,73 m² (HR
= 0,502; 95 % CI: 0,297 až 0,847) s > 3 predchádzajúcimi líniami liečby (HR = 0,590; 95 % CI: 0,356
až 0,977) v prípade pacientov refraktérnych voči predchádzajúcej liečbe lenalidomidom (HR = 0,593;
95 % CI: 0,431 až 0,816) alebo inhibítorom proteáz (HR = 0,578; 95 % CI: 0,405 až 0,824) a v prípade pacientov refraktérnych voči lenalidomidu podávanému počas poslednej línie liečby pred začiatkom klinického skúšania (HR = 0,601; 95 % CI: 0,436 až 0,828).
Nie sú dostupné dostatočné údaje pre posúdenie účinnosti izatuximabového režimu u pacientov, ktorí boli v minulosti liečení daratumumabom (1 pacient v ramene s izatuximabom a žiaden pacient
v porovnávacom ramene).
Priemerný počet dní do prvej odpovede u pacientov, u ktorých sa odpoveď vyskytla, bol 35 dní v izatuximabovej skupine, v porovnaní s 58 dňami v porovnávacej skupine. S mediánom dĺžky sledovania 11,56 mesiacov v skupine s izatuximabom a 11,73 mesiacov v porovnávacej skupine, celkový medián prežívania nebol dosiahnutý ani pre jednu liečebnú skupinu. Miera rizika v prípade OS bola 0,687 (95 % CI: 0,461 – 1,023, p-hodnota = 0,0631).
O
b
r
ázok 1: Kaplanove-Meierove krivky PFS – Populácia ITT – ICARIA-MM (posúdila IRC)
 O
b
r
ázok 2 – Kaplanove-Meierove krivky OS – Populácia ITT – ICARIA-MM
O
b
r
ázok 2 – Kaplanove-Meierove krivky OS – Populácia ITT – ICARIA-MM
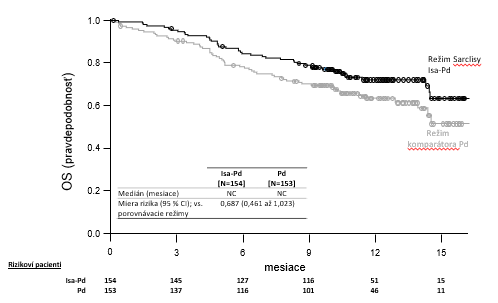
Dátum ukončenia = 11. október 2018
V štúdii ICARIA-MM (EFC14335) bol použitý objem infúzie izatuximabu podľa hmotnosti. Metóda
s pevne stanoveným objemom, ako je popísaná v časti 4.2, bola hodnotená v štúdii TCD14079 časť B a farmakokinetické simulácie potvrdili minimálne rozdiely medzi farmakokinetikou po aplikácii objemu injekcie podľa hmotnosti pacienta a pevného objemu 250 ml (pozri časť 5.2). V štúdii TCD14079 časť B neboli žiadne nové bezpečnostné signály alebo rozdiely v účinnosti v porovnaní so štúdiou ICARIA-MM.
Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so SARCLISOU pri liečbe malígnych neoplaziem hematopoetického a lymfatického tkaniva vo všetkých podskupinách pediatrickej populácie. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmakokinetické vlastnosti
Farmakokinetika izatuximabu sa posudzovala u 476 pacientov s mnohopočetným myelómom, ktorým boli intravenózne podávané infúzie izatuximabu, buď samostatne alebo v kombinácii
s pomalidomidom a dexametazónom, pričom podávané dávky sa pohybovali v rozmedzí od 1 do
20 mg/kg a boli podávané buď raz týždenne, každé 2 týždne, alebo každé 2 týždne počas 8 týždňov a následne každé 4 týždne alebo každý týždeň počas 4 týždňov a následne každé 2 týždne.
Izatuximab preukazuje nelineárnu farmakokinetiku s ovplyvnením základných farmakokinetických dejov v dôsledku väzby na receptor CD38.
Expozícia izatuximabu (plocha pod krivkou závislosti plazmatickej koncentrácie od času pri dávkovacom intervale AUC) sa zvyšuje rýchlejšie než úmerne s dávkou z 1 na 20 mg/kg podľa harmonogramu podávania každé 2 týždne, pričom v prípade harmonogramu podávania každý týždeň počas 4 týždňov a následne každé 2 týždne nebola zaznamenaná žiadna odchýlka od proporcionality dávok v rozmedzí od 5 do 20 mg/kg. Je to spôsobené tým, že pri dávkach menších ako 5 mg/kg nelineárny klírens v dôsledku naviazania na cieľovú štruktúru výrazne prispieva k celkovému klírensu, čo je však zanedbateľné pri vyšších dávkach. Po podávaní 10 mg/kg izatuximabu každý týždeň počas
4 týždňov a následne každé 2 týždne bola priemerná doba potrebná na dosiahnutie stabilného stavu 8
týždňov s 3,1-násobným nárastom. Priemerné (CV %) predpokladané maximálne koncentrácia plazmy Cmax v stabilnom stave dosahovali 351 µg/ml (36,0 %) a priemerný (CV %) predpokladaný maximálny AUC v stabilnom stave dosiahol 72 600 µg.h/ml (51,7 %). Aj keď, zmena spôsobu podávania infúzie izatuximabu z objemu v závislosti od hmotnosti na pevne stanovený objem vyústila do zmien v tmax, zmena mala malý vplyv na farmakokinetiku s porovnateľným simulovaným Cmax v rovnovážnom stave (283 µg/ml oproti 284 µg/ml) a Ctrough po 4 týždňoch (119 µg/ml oproti 119 µg/ml) u pacientov s priemernou hmotnosťou (76 kg). Tiež pre iné hmotnostné skupiny pacientov boli Cmax a Ctrough porovnateľné.
Súbežné podávanie izatuximabu a pomalidomidu nemá vplyv na ich farmakokinetiku. Distribúcia
Odhadovaný celkový objem distribúcie izatuximabu je 8,75 l.
Metabolizmus
Predpokladá sa, že izatuximab sa ako veľký proteín metabolizuje pri nenasýtených proteolytických
katabolických procesoch.
Eliminácia
Izatuximab sa eliminuje dvomi paralelnými cestami. Pri nízkych koncentráciách dominuje nelineárna cesta ovplyvnená cieľovou štruktúrou a pri vyšších koncentráciách dominuje nešpecifická lineárna cesta. V prípade terapeutického rozmedzia koncentrácií plazmy dominuje lineárna cesta, ktorá sa
s postupom času znižuje o 50 % na hodnotu 9,55 ml/h (0,229 l/deň) v rovnovážnom stave. Súvisí to
s konečným polčasom rozpadu v trvaní 28 dní.
Osobitné skupinypacientov
Vek
Farmakokinetické analýzy populácie 476 pacientov vo veku od 36 do 85 rokov preukázali
porovnateľnú expozíciu izatuximabu u pacientov vo veku < 75 rokov (n = 406), v porovnaní s pacientmi vo veku ≥ 75 rokov (n = 70).
Pohlavie
Farmakokinetická analýza populácie 207 žien (43,5 %) a 269 mužov (56,5 %) nepreukázala žiadny klinicky významný vplyv pohlavia na farmakokinetiku izatuximabu.
Rasa
Farmakokinetická analýza populácie 377 belochov (79 %), 25 aziatov (5 %), 18 afroameričanov (4 %)
a 33 pacientov inej rasy (7 %) nepreukázala žiadny klinicky významný vplyv rasy na farmakokinetiku izatuximabu.
Telesnáhmotnosť
Expozícia izatuximabu (AUC) v rovnovážnom stave so zvyšujúcou sa telesnou hmotnosťou klesá.
Poruchyfunkciepečene
Neboli vykonané žiadne oficiálne klinické skúšania u pacientov s poruchou funkcie pečene. Zo 476 pacientov v populácii zohľadnenej vo farmakokinetickej analýze bola mierna porucha funkcie pečene prítomná u 65 pacientov [celková hodnota bilirubínu dosahovala 1- až 1,5-násobok hornej hranice normálu (ULN) alebo aspartát-aminotransferáza (AST) > ULN] a stredne závažná porucha funkcie pečene bola prítomná u 1 pacienta (celkový bilirubín > 1,5- až 3-násobok ULN a akékoľvek AST). Mierna porucha funkcie pečene nemala žiadny klinicky významný vplyv na farmakokinetiku izatuximabu. Vplyv stredne závažnej (celkový bilirubín > 1,5- až 3-násobok ULN a akékoľvek AST) a závažnej poruchy funkcie pečene (celkový bilirubín > 3-násobok ULN a akékoľvek AST) na farmakokinetiku izatuximabu nie je známy. Avšak, keďže je izatuximab monoklonálnou protilátkou, nie je možné očakávať, že by bol eliminovaný prostredníctvom metabolizmu sprostredkovaného enzýmami pečene, a teda sa neočakáva, že by odchýlky funkcie pečene ovplyvňovali vylučovanie izatuximabu (pozri časť 4.2).
Poruchafunkcieobličiek
Neboli vykonané žiadne oficiálne klinické skúšania u pacientov s poruchou funkcie obličiek. Farmakokinetické analýzy populácie u 476 pacientov zahŕňali 192 pacientov s miernou poruchou funkcie obličiek (60 ml/min/1,73 m2 ≤ odhadovaná miera glomerulárnej filtrácie (e-GFR)
< 90 ml/min/1,73 m2), 163 pacientov so stredne závažnou poruchou funkcie obličiek (30 ml/min/1,73
m2 ≤ e-GFR < 60 ml/min/1,73 m2) a 12 pacientov so závažnou poruchou funkcie obličiek (e-GFR
< 30 ml/min/1,73 m2). Analýzy nepreukázali žiadny klinicky významný vplyv miernej až stredne
závažnej poruchy funkcie obličiek na farmakokinetiku izatuximabu, v porovnaní s normálnou funkciou obličiek.
Pediatrickápopulácia
Izatuximabu nebol hodnotený u pacientov mladších ako 18 rokov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií toxicity po opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí, aj keď sa u zvoleného druhu farmakologické odpovede nevyskytujú a z tohto dôvodu nie je závažnosť u ľudí známa. Štúdie genotoxicity, karcinogénneho potenciálu a reprodukčnej toxicity a vývinu neboli vykonané.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
sacharóza
histidínium-chlorid, monohydrát histidín
polysorbát 80
voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
N
e
otvorená
i
njekčná
li
e
kovka
3 roky
Poriedení
Chemická a fyzikálna stabilita pri používaní infúzneho roztoku SARCLISY bola preukázaná počas 48
hodín pri teplote od 2 °C – 8 °C a následne počas 8 hodín pri izbovej teplote (vrátane času podávania infúzie).
Z mikrobiologického hľadiska sa má liek použiť ihneď. Ak sa nepoužije okamžite, čas skladovania
počas používania a podmienky uchovávania pred použitím sú na zodpovednosti používateľa a za normálnych okolností takéto uchovávanie nemá prekročiť 24 hodín pri 2 °C až 8 °C, ak sa riedenie nevykonalo v kontrolovaných a validovaných aseptických podmienkach.
Ak sa liek uchováva v infúznom vaku, nie je potrebné ho chrániť pred svetlom.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale, na ochranu pred svetlom.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
5 ml koncentrátu obsahuje 100 mg izatuximabu v 6 ml injekčnej liekovke z bezfarebného číreho skla typu I uzatvorenej zátkou z brómbutylu potiahnutou ETFE (kopolymérom etylénu a tetrafluóretylénu). Injekčné liekovky sú olemované hliníkovou fóliou so sivým ľahko otvárateľným tlačidlom. Plniaci objem bol stanovený tak, aby bolo umožnené odobrať 5 ml (t.j. 5,4 ml). Balenie obsahuje jednu alebo tri injekčné liekovky.
25 ml koncentrátu obsahuje 500 mg izatuximabu v 30 ml injekčnej liekovke z bezfarebného číreho skla typu I uzatvorenej zátkou z brómbutylu potiahnutou ETFE (kopolymérom etylénu a tetrafluóretylénu). Injekčné liekovky sú olemované hliníkovou fóliou s modrým ľahko otvárateľným tlačidlom. Plniaci objem bol stanovený tak, aby bolo umožnené odobrať 25 ml (t.j. 26 ml). Balenie obsahuje jednu injekčnú liekovku.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Prípravanaintravenóznepodanie
Príprava infúzneho roztoku musí prebiehať za aseptických podmienok.
• Dávku (mg) koncentrátu SARCLISY je nutné vypočítať podľa telesnej hmotnosti pacienta (váženie je potrebné opakovať pred začiatkom každého cyklu, aby bolo možné dávku primerane upraviť, pozri časť 4.2). Na získanie požadovanej dávky pre pacienta môže byť potrebné použiť viac ako jednu injekčnú liekovku.
• Injekčné liekovky s koncentrátom SARCLISY je nutné pred riedením vizuálne skontrolovať, aby bolo možné sa uistiť, že neobsahuje žiadne častice a nedošlo k zmene zafarbenia.
• Injekčnými liekovkami netraste.
• Z 250 ml vaku s injekčným roztokom chloridu sodného 9 mg/ml (0,9 %) alebo 5 % roztokom glukózy sa odoberie objem riedidla rovnajúci sa požadovanému objemu koncentrátu SARCLISY.
• Odoberie sa primeraný objem koncentrátu SARCLISY a rozriedi sa v 250 ml infúznom vaku s injekčným roztokom chloridu sodného 9 mg/ml (0,9 %) alebo s 5 % roztokom glukózy.
• Infúzny vak musí byť vyrobený z polyolefínov (PO), polyetylénu (PE), polypropylénu (PP), polyvinylchloridu (PVC) s di (2-etylhexyl) ftalátom (DEHP) alebo s etylvinylacetátom (EVA).
• Jemne prevráťte vak naopak, aby sa rozriedený roztok zhomogenizoval. Netraste.
Podávanie• Infúzny roztok sa podáva intravenóznou infúziou, pričom sa používa infúzna súprava
s intravenóznymi hadičkami (vyrobenými z PE, PVC s DEHP alebo bez DEHP, polybutadiénu
(PBD) alebo polyuretánu (PU)) s líniovým filtrom (z polyétersulfónu (PES), polysulfónu alebo nylonu).
• Infúzny roztok sa má podávať počas doby závislej od rýchlosti podávania infúzie (pozri časť
4.2).
• Ak sa liek skladuje v infúznom vaku, pri bežnom umelom osvetlení nie je potrebné ho chrániť pred svetlom.
• Nezavádzajte infúziu s roztokom SARCLISY do tej istej intravenóznej línie súbežne s inými liečivami.
LikvidáciaVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIsanofi-aventis groupe
54 rue La Boétie
75008 Paríž
Francúzsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/20/1435/001
EU/1/20/1435/002
EU/1/20/1435/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: {DD Mesiac RRRR}
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eua na internetovej stránke Štátneho ústavu pre kontrolu liečiv.