
makokinetiky granisetronu je v tejto populácii nevyhnutná určitá miera opatrnosti.
Deti a dospievajúci
Bezpečnosť a účinnosť SANCUSA u detí vo veku 0 až 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podania
Transdermálna náplasť sa má aplikovať na čistú, suchú, neporušenú zdravú kožu na vonkajšiu časť
nadlaktia. Ak nie je možné aplikovať transdermálnu náplasť na rameno, môže sa aplikovať na brucho. Transdermálna náplasť sa nemá umiestniť na kožu, ktorá je červená, podráždená alebo poškodená.
Každá transdermálna náplasť je balená vo vrecku a má sa aplikovať bezprostredne po otvorení vrecka. Pred aplikáciou sa odstraňuje odlepovacia vrstva.
Transdermálna náplasť sa nemá strihať na kúsky.
V prípade úplného alebo čiastočného odlepenia náplasti sa má pôvodná náplasť prelepiť na rovnakom mieste leukoplastom (ak je to nevyhnutné). Ak prelepenie nie je možné alebo je transdermálna náplasť poškodená, nová transdermálna náplasť sa má aplikovať na rovnaké miesto ako pôvodná
transdermálna náplasť. Ak to nie je možné, nová transdermálna náplasť sa má aplikovať na druhé
rameno. Novo aplikovaná transdermálna náplasť sa má odstrániť po uplynutí času odporúčaného vyššie.
4.3 Kontraindikácie
Precitlivenosť na liečivo, na iné antagonisty receptora 5-HT3 alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Reakcie v mieste aplikácie
V klinických skúšaniach so SANCUSOM boli hlásené reakcie v mieste aplikácie, ktoré boli zvyčajne
miernej intenzity a neviedli k ukončeniu používania. Ak sa vyskytnú závažné reakcie alebo generalizovaná kožná reakcia (napr. alergická vyrážka, vrátane erytematóznej, makulóznej, papulóznej vyrážky alebo pruritu), transdermálna náplasť sa musí odstrániť.
Gastrointestinálne poruchy
Pretože granisetron môže znižovať motilitu v hrubom čreve, po jeho podaní sa majú sledovať pacienti
s prejavmi subakútnej intestinálnej obštrukcie.
Srdcové poruchy
Antagonisty 5-HT3, ako je granisetron, môžu súvisieť s arytmiami alebo anomáliami EKG. To môže
mať potenciálne klinický význam u pacientov s preexistujúcimi arytmiami alebo poruchami srdcového
prevodu alebo u pacientov, ktorí sú liečení antiarytmikami alebo beta-blokátormi. V klinických štúdiách so SANCUSOM sa nepozorovali žiadne klinicky významné účinky.
Expozícia slnečnému žiareniu
Priame prírodné alebo umelé slnečné žiarenie môže ovplyvňovať granisetron. Je potrebné pacientom
odporučiť, aby si miesto aplikácie transdermálnej náplasti zakryli, napr. odevom, ak existuje riziko expozície slnečnému žiareniu počas nosenia náplasti a počas 10 dní po jej odstránení.
Sprchovanie alebo umývanie
Počas nosenia SANCUSA sa môže aj naďalej normálne pokračovať v sprchovaní alebo umývaní.
Činnostiam, ako je plávanie, namáhavé cvičenie alebo používanie sauny, sa treba vyhýbať.
Externé teplo
V oblasti transdermálnej náplasti sa treba vyhýbať aplikácii externého tepla (napríklad termoforu
alebo vyhrievacej podložky).
Osobitné skupiny pacientov
U starších pacientov alebo pacientov s poruchou funkcie obličiek alebo pečene nie je potrebná žiadna
úprava dávky. Hoci sa u pacientov s poruchou funkcie obličiek alebo pečene, ktorí užívali granisetron perorálne a intravenózne, nepozoroval žiadny dôkaz zvýšeného výskytu nežiaducich reakcií , na základe farmakokinetiky granisetronu je v tejto populácii nevyhnutná určitá miera opatrnosti.
4.5 Liekové a iné interakcie
In vitro štúdie použitím ľudských mikrozómov naznačujú, že granisetron ani nestimuluje ani neinhibuje enzýmový systém cytochrómu P450.
Pretože granisetron je metabolizovaný pečeňovými enzýmami cytochrómu P450 metabolizujúcimi liečivo (CYP1A1 a CYP3A4), induktory alebo inhibítory týchto enzýmov môžu zmeniť klírens a tak polčas granisetronu.
Po intravenóznom podaní granisetronu viedla indukcia pečeňových enzýmov fenobarbitalom u ľudí k zvýšeniu celkového plazmatického klírensu (približne o 25 %).
Zaznamenalo sa, že súbežné podávanie intravenóznych antagonistov receptora 5-HT3 s perorálnym paracetamolom u ľudí malo za následok blokádu analgetického účinku prostredníctvom farmakodynamického mechanizmu.
In vitro štúdie preukázali, že ketokonazol môže inhibovať metabolizmus granisetronu prostredníctvom skupiny izoenzýmov cytochrómu P450 3A. Klinický význam nie je známy.
V štúdiách u zdravých osôb sa nezistil žiadny dôkaz o interakcii medzi granisetronom
a benzodiazepínmi (lorazepam), neuroleptikami (haloperidol) alebo antiulcerózami (cimetidín).
Medzi SANCUSOM a emetogénnymi protirakovinovými chemoterapiami sa nepozorovali žiadne klinicky významné liekové interakcie. Okrem toho sa nepozorovali žiadne interakcie medzi granisetronom a emetogénnymi protirakovinovými chemoterapiami. V súlade s týmito údajmi sa
v klinických štúdiách so SANCUSOM nezaznamenali žiadne klinicky významné liekové interakcie.
V klinických interakčných štúdiách nemal aprepitant klinicky významné účinky na farmakokinetiku granisetronu.
Deti a dospievajúci
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití granisetronu u gravidných žien. Štúdie na zvieratách nepreukázali
priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu SANCUSA počas gravidity.
Laktácia
Nie je známe, či sa granisetron alebo jeho metabolity vylučujú do ľudského mlieka. Laktácia má byť
počas liečby SANCUSOM ukončená.
Fertilita
Nie sú k dispozícii údaje o vplyve granisetronu na fertilitu ľudí. Po liečbe granisetronom u potkanov
nebola ovplyvnená fertilita.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
SANCUSO nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať
stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnostný profil SANCUSA je odvodený z kontrolovaných klinických skúšaní a zo skúseností po
uvedení lieku na trh. Najčastejšie hlásená nežiaduca reakcia v klinických štúdiách bola zápcha, ktorá sa vyskytovala približne u 8,7 % pacientov. Väčšina nežiaducich reakcií bola mierna alebo stredne závažná.
Zoznam nežiaducich reakcií zoradených do tabuľky
Nežiaduce reakcie z klinických štúdií a spontánnych hlásení so SANCUSOM sú uvedené nižšie
v tabuľke:
V rámci triedy orgánových systémov sú nežiaduce reakcie usporiadané podľa frekvencie pomocou nasledujúcej konvencie: veľmi časté (³ 1/10); časté (³ 1/100 až < 1/10); menej časté (³ 1/1 000 až
< 1/100); zriedkavé (³ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie hlásené pre SANCUSO
Trieda orgánových systémov
|
N
e
ž
i
a
du
c
a reakcia
|
F
rekvencia
|
Poruchy metabolizmu a výživy
|
Znížená chuť do jedla
|
Menej časté
|
Poruchy nervového systému
|
Bolesť hlavy
|
Menej časté
|
Dystónia
|
Zriedkavé
|
Dyskinéza
|
Zriedkavé
|
Poruchy ucha a labyrintu
|
Vertigo
|
Menej časté
|
Poruchy ciev
|
Sčervenanie
|
Menej časté
|
Poruchy gastrointestinálneho traktu
|
Zápcha
|
Časté
|
Sucho v ústach, nauzea, dvíhanie žalúdka
|
Menej časté
|
Poruchy pečene a žlčových ciest
|
Zvýšenie alanínaminotransferázy, zvýšenie aspartátaminotransferázy, zvýšenie gamaglutamyltransferázy
|
Menej časté
|
Poruchy kože a podkožného tkaniva
|
Podráždenie v mieste aplikácie
|
Menej časté
|
Reakcie v mieste aplikácie (bolesť v mieste aplikácie, pruritus v mieste aplikácie, erytém v mieste aplikácie, vyrážka v mieste aplikácie, podráždenie v mieste aplikácie)*
|
Neznáme
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Artralgia
|
Menej časté
|
Celkové poruchy a reakcie v mieste podania
|
Generalizovaný edém
|
Menej časté
|
*Spontánne hlásenia
Popis vybraných nežiaducich reakciíU pacientov liečených stredne alebo vysoko emetogénnou chemoterapiou sa môže aj tak objaviť
vracanie napriek liečbe s antiemetikami, vrátane SANCUSA.
Triedne účinkyTriedne účinky granisetronu pozorované pri iných formách (perorálnej a intravenóznej) zahŕňajú
nasledovné:
- hypersenzitívne reakcie, napr. anafylaxia, urtikária
- insomnia
- bolesť hlavy
- extrapyramídové reakcie
- ospalosť
- závrat
- predĺženie QT
- zápcha
- hnačka
- zvýšenie pečeňových transamináz
- vyrážka
- asténia
4.9 Predávkovanie
Pre granisetron neexistuje žiadne špecifické antidotum. V prípade predávkovania sa má transdermálna náplasť odstrániť. Je potrebné podať symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiemetiká a lieky proti nevoľnosti, antagonisty serotonínu (5HT3) ATC kód: A04AA02
Granisetron je silné antiemetikum a vysoko selektívny antagonista 5-hydroxytryptamínu (5-HT3 receptora). Farmakologické štúdie preukázali, že granisetron je účinný proti nauzey a vracaniu, čo sú dôsledky cytostatickej liečby. Štúdie väzby rádioligandu preukázali, že granisetron má zanedbateľnú afinitu k iným typom receptora, vrátane 5HT1, 5HT2, 5HT4 a väzobných miest dopamínu D2.
Pivotná, randomizovaná, dvojito zaslepená, double-dummy, medzinárodná štúdia fázy III porovnávala účinnosť, znášanlivosť a bezpečnosť SANCUSA s 2 mg perorálneho granisetronu jedenkrát denne
v prevencii nauzey a vracania u celkovo 641 pacientov užívajúcich viacdňovú chemoterapiu. Štúdia bola navrhnutá na preukázanie neinferiority SANCUSA oproti perorálnemu granisetronu.
Populácia randomizovaná do skúšania zahŕňala 48 % mužov a 52 % žien vo veku 16 až 86 rokov užívajúcich stredne emetogénnu (moderately emetogenic – ME) alebo vysoko emetogénnu (highly emetogenic – HE) viacdňovú chemoterapiu. 78 % pacientov bolo belochov, pričom 12 % bolo Ázijcov a 10 % Hispáncov/Latinská Amerika.
Transdermálna náplasť granisetronu bola aplikovaná 24 až 48 hodín pred prvou dávkou chemoterapie a bola nalepená počas 7 dní. Perorálny granisetron sa podával denne po dobu trvania chemoterapeutického režimu, jednu hodinu pred každou dávkou chemoterapie. Antiemetická aktivita bola hodnotená od prvého podania až do 24 hodín po začatí posledného dňa podávania ME alebo HE chemoterapeutického režimu.
Neinferiorita SANCUSA v porovnaní s perorálnym granisetronom sa potvrdila, pričom úplná kontrola
(complete control – CC) sa dosiahla u 60,2 % pacientov v skupine so SANCUSOM a 64,8 %
pacientov užívalo perorálny granisetron podľa protokolu (rozdiel -4,89 %; 95 % interval spoľahlivosti
–12,91 % až +3,13 %; n = 284 transdermálna náplasť, n = 298 perorálna forma). CC bola definovaná ako žiadne vracanie a/alebo dvíhanie žalúdka, maximálne mierna nauzea a žiadny záchranný liek od prvého podania až do 24 hodín po začatí posledného dňa podávania viacdňovej chemoterapie.
Vzhľadom na postupné zvyšovanie plazmatických hladín granisetronu po aplikácii transdermálnej náplasti môžu byť úvodné plazmatické hladiny na začiatku chemoterapie nižšie ako pri 2 mg perorálneho granisetronu a preto sa môže pozorovať pomalší nástup účinnosti. Preto je SANCUSO indikované na používanie u pacientov, kde podanie perorálnych antiemetík je skomplikované faktormi, ktoré sťažujú prehĺtanie.

Úplná kontrola podľa dní je zobrazená nižšie.
Úplná kontrola podľa dní
Podľa protokolu
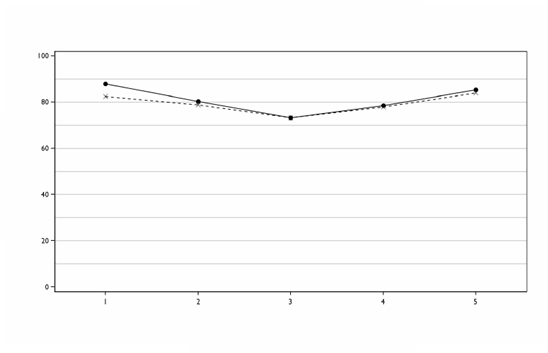
Deň štúdie
Perorálna forma _ _ _ Náplasť
V klinických skúšaniach so SANCUSOM sa nezistili žiadne účinky na srdcovú frekvenciu alebo
krvný tlak súvisiace s liečbou. Hodnotenie sériových EKG záznamov u pacientov nepreukázalo žiadne predĺženie QT ani žiadnu zmenu v EKG morfológii. Vplyv SANCUSA na QTc interval sa špecificky hodnotil v zaslepenom, randomizovanom, paralelnom, placebom a pozitívne (moxifloxacín) kontrolovanom QTc skúšaní so SANCUSOM u 240 dospelých mužov a žien. Pri SANCUSE sa nepozoroval žiadny významný účinok na predĺženie QTc.
Hodnotenie adhézie transdermálnej náplasti u 621 pacientov užívajúcich buď transdermálne náplasti s účinnou látkou alebo placebom preukázalo, že menej ako 1 % transdermálnych náplastí sa odlepilo
v priebehu 7 dní od aplikácie transdermálnej náplasti.
Nie sú k dispozícii žiadne skúsenosti z klinických skúšaní so SANCUSOM a u pacientov na chemoterapii menej ako 3 po sebe nasledujúce dni alebo na viacerých cykloch chemoterapie alebo na vysoko dávkovej chemoterapii pred transplantáciou kmeňových buniek.
5.2 Farmakokinetické vlastnostiAbsorpciaGranisetron prechádza neporušenou kožou do systémovej cirkulácie procesom pasívnej difúzie.
Po aplikácii SANCUSA sa granisetron absorbuje pomaly, pričom maximálne koncentrácie sa dosahujú v rozmedzí 24 až 48 hodín.
Na základe merania reziduálneho obsahu transdermálnej náplasti po jej odstránení je podaných približne 65 % granisetronu, čo zabezpečuje priemernú dennú dávku 3,1 mg na deň.
Súbežné podávanie jednorazového intravenózneho bolusu 0,01 mg/kg (maximálne 1 mg) granisetronu v rovnakom čase ako bola aplikovaná transdermálna náplasť SANCUSO sa hodnotilo u zdravých jedincov. Úvodné maximum plazmatických koncentrácií granisetronu pripísateľné intravenóznej
dávke sa dosiahlo 10 minút po podaní dávky. Známy farmakokinetický profil transdermálnej náplasti
počas nosenia náplasti (7 dní) nebol ovplyvnený.
Po následnej aplikácii dvoch transdermálnych náplastí SANCUSO zdravým jedincom, každej na sedem dní, sa hladiny granisetronu udržiavali počas obdobia štúdie s dôkazom minimálnej kumulácie.
V štúdii navrhnutej na hodnotenie účinku tepla na transdermálne dodanie granisetronu zo SANCUSA u zdravých jedincov bola na 4 hodiny na transdermálnu náplasť aplikovaná vyhrievacia podložka vyžarujúca teplotu v priemere 42°C každý deň počas 5 dní nosenia. Zatiaľ čo aplikácia vyhrievacej podložky bola spojená s miernym a prechodným zvýšením toku v transdermálnej náplasti počas obdobia aplikácie vyhrievacej podložky, žiadne celkové zvýšenie expozície granisetronu sa nepozorovalo v porovnaní s kontrolnou skupinou.'
Vo farmakokinetickej štúdii so zdravými dobrovoľníkmi, kde sa SANCUSO aplikovalo na dobu 7 dní, bola priemerná celková expozícia (AUC0-nekonečno) 416 ng•h/ml (rozmedzie 55–1 192 ng•h/ml),
s variabilitou medzi jednotlivcami 89 %. Priemerné Cmax bolo 3,9 ng/ml (rozmedzie 0,7–9,5 ng/ml),
s variabilitou medzi jednotlivcami 77 %. Táto variabilita je rovnaká ako známa vysoká variabilita
farmakokinetiky granisetronu po perorálnom alebo intravenóznom podaní.
Distribúcia
Granisetron je distribuovaný s priemerným distribučným objemom približne 3 l/kg. Väzba na
plazmatické proteíny je približne 65 %. Granisetron sa voľne distribuuje medzi plazmou a červenými krvinkami.
Biotransformácia
Medzi perorálnym a transdermálnym použitím sa nepozorovali žiadne rozdiely v metabolických
profiloch granisetronu.
Granisetron je metabolizovaný predovšetkým na 7-hydroxygranisetron a 9´N-dezmetylgranisetron. In vitro štúdie použitím ľudských pečeňových mikrozómov naznačujú, že CYP1A1 je hlavný enzým zodpovedný za 7-hydroxyláciu granisetronu, zatiaľ čo CYP3A4 prispieva k 9´dezmetylácii.
Eliminácia
Granisetron sa vylučuje predovšetkým pečeňovým metabolizmom. Po intravenóznom podaní dávky sa
priemerný plazmatický klírens pohyboval od 33,4 do 75,7 l/h u zdravých osôb a od 14,7 do 33,6 l/h
u pacientov so širokou interindividuálnou variabilitou. Priemerný plazmatický polčas u zdravých osôb je 4-6 hodín a u pacientov je 9-12 hodín. Po aplikácii transdermálnej náplasti je zdanlivý plazmatický polčas granisetronu u zdravých osôb predĺžený na približne 36 hodín z dôvodu pomalej rýchlosti absorpcie granisetronu cez kožu.
V klinických štúdiách uskutočnených so SANCUSOM sa preukázalo, že klírens u pacientov s rakovinou je približne polovicou klírensu u zdravých osôb.
Po intravenóznej injekcii sa u zdravých osôb približne 12 % dávky vylučuje v nezmenenej forme močom v priebehu 48 hodín. Zvyšok dávky sa vylúči vo forme metabolitov, pričom 49 % močom a 34 % stolicou.
Farmakokinetika u osobitných skupín pacientov
Vplyv pohlavia na farmakokinetiku SANCUSA sa špeciálne neskúmal. V klinických štúdiách so
SANCUSOM sa nepozorovali žiadne zodpovedajúce účinky pohlavia na farmakokinetiku, pričom u oboch pohlaví sa zaznamenala široká interindividuálna variabilita. Model FK populácie potvrdil chýbajúci vplyv pohlavia na farmakokinetiku SANCUSA.
Starší pacienti
V klinickej štúdii sa nepozoroval žiadny rozdiel v plazmatickej farmakokinetike SANCUSA u mužov a žien v pokročilom veku (≥ 65 rokov) v porovnaní s mladšími osobami (vo veku 18-45 rokov vrátane).
Porucha funkcie obličiek alebo pečene
Neuskutočnili sa žiadne klinické štúdie, ktoré by špecificky skúmali farmakokinetiku SANCUSA
u pacientov s poruchou funkcie obličiek alebo pečene. Medzi renálnou funkciou (meranou klírensom kreatinínu) a klírensom granisetronu sa v modeli FK populácie neidentifikoval žiadny jednoznačný vzťah. U pacientov so zlyhaním obličiek alebo poruchou funkcie pečene sa určovala farmakokinetika granisetronu po jednorazovej intravenóznej dávke 40 mg/kg granisetronhydrochloridu.
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene z dôvodu neoplastického postihnutia pečene bol celkový plazmatický klírens približne polovičný v porovnaní s pacientmi bez poruchy funkcie pečene. Vzhľadom na širokú variabilitu vo farmakokinetických parametroch granisetronu a dobrú znášanlivosť oveľa vyššej dávky ako je odporúčaná dávka nie je potrebné u pacientov s funkčnou poruchou pečene upravovať dávku.
Porucha funkcie obličiek
U pacientov s rakovinou sa nepozoroval žiadny vzťah medzi klírensom kreatinínu a celkovým klírensom, čo svedčí o tom, že porucha funkcie obličiek nemá žiadny vplyv na farmakokinetiku granisetronu.
Index telesnej hmotnosti (Body mass index - BMI)
V klinickej štúdii navrhnutej na hodnotenie expozície granisetronu zo SANCUSA u osôb s rozličnými hladinami telesných tukov, pomocou BMI ako náhradného meradla telesného tuku, sa nepozorovali žiadne rozdiely v plazmatickej farmakokinetike SANCUSA u mužov a žien s nízkym BMI
[< 19,5 kg/m2 (muži), < 18,5 kg/m2 (ženy)] a s vysokým BMI (30,0 až 39,9 kg/m2 vrátane)
v porovnaní s kontrolnou skupinou (BMI 20,0 až 24,9 kg/m2 vrátane).
Deti a dospievajúci
Neuskutočnili sa žiadne štúdie na preskúmanie farmakokinetiky SANCUSA u detí a dospievajúcich.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, reprodukčnej toxicity a genotoxicity neodhalili žiadne osobitné riziko pre ľudí. Štúdie karcinogenity nepreukázali žiadne osobitné riziko pre ľudí pri používaní v odporúčanej dávke. Avšak pri podávaní vyšších dávok a dlhšiu dobu nie je možné vylúčiť riziko karcinogenity, ale pri krátkodobej aplikácii odporúčanej pre transdermálny prenosový systém sa riziko karcinogenity pre
ľudí nepredpokladá.
SANCUSO transdermálne náplasti nepreukázal žiadny potenciál fotopodráždenia alebo fotosenzitivity pri testovaní in vivo u morčiat. Granisetron nebol fototoxický pri testovaní in vitro na myších bunkových líniách fibroblastov. Pri testovaní potenciálu fotogenotoxicity in vitro na bunkových
líniách ovárií čínskych škrečkov zvýšil granisetron percento buniek s poškodením chromozómov po fotopodráždení. Hoci klinický význam tohto zistenia nie je úplne objasnený, pacientom sa odporúča zakryť miesto aplikácie transdermálnej náplasti, ak existuje riziko expozície slnečnému žiareniu počas obdobia nosenia náplasti a počas 10 dní po jej odstránení (pozri časť 4.4).
Pri testovaní potenciálu kožnej senzibility u morčiat preukázalo SANCUSO nízky potenciál pre dráždenie.
Štúdia s klonovanými ľudskými srdcovými iónovými kanálmi preukázala, že granisetron má potenciál ovplyvňovať srdcovú repolarizáciu prostredníctvom blokády draslíkových hERG kanálov. Preukázalo sa, že granisetron blokuje sodíkové aj draslíkové kanály, čo by mohlo ovplyvňovať depolarizáciu
a repolarizáciu srdca a tým PR, QRS a QT intervaly. Tieto údaje pomáhajú objasniť mechanizmus,
v dôsledku ktorého sa môžu vyskytovať niektoré z EKG zmien (predovšetkým predĺženie QT a QRS) súvisiace s touto triedou látok. Žiadne klinicky významné účinky na EKG sa však v klinických štúdiách so SANCUSOM nepozorovali, vrátane QT štúdie u 240 zdravých osôb (pozri časť 5.1).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Podkladová vrstva
Polyester
Matricová vrstva
Kopolymér akrylát-vinylacetátu
Odlepovacia vrstva
Silikonizovaný polyester
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Každá transdermálna náplasť je zabalená v zatavenom vrecku, ktoré je vyrobené z papiera potiahnutého polyesterom/hliníka/LLDPE a ochrannej vrstvy z polyetyléntereftalátu pokrytého silikónom.
Každá škatuľka obsahuje 1 transdermálnu náplasť.
6.6 Špeciálne opatrenia na likvidáciu
Po použití transdermálna náplasť stále obsahuje liečivo. Po jej odstránení sa má použitá transdermálna náplasť pevne preložiť na polovicu, lepiacou stranou dovnútra a potom zlikvidovať mimo dosahu detí.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
ProStrakan Limited Galabank Business Park Galashiels
TD1 1QH
Veľká Británia
Tel: +44 (0)1896 664000
Fax: +44 (0)1896 664001
medinfo@prostrakan.com8. REGISTRAČNÉ ČÍSLO (ČÍSLA)9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.