
syndróm nádorového rozpadu a anafylaktické a hypersenzitívne reakcie, je opísaný nižšie.
Závažné reakcie súvisiace s infúziou so smrteľnými následkami boli hlásené po uvedení intravenóznej formy rituximabu na trh, s nástupom v rozsahu od 30 minút do 2 hodín od začiatku prvej intravenóznej infúzie rituximabu. Boli charakterizované pulmonálnymi udalosťami a v niektorých prípadoch zahŕňali rýchly rozpad nádoru s prejavmi syndrómu nádorového rozpadu spolu s horúčkou, zimnicou, triaškou, hypotenziou, žihľavkou, angioedémom a inými príznakmi (pozri časť 4.8).
Ťažký syndróm uvoľňovania cytokínov sa vyznačuje ťažkým dyspnoe, ktoré často sprevádzajú bronchospazmus a hypoxia, spolu s horúčkou, zimnicou, triaškou, žihľavkou a angioedémom. Tento syndróm sa môže spájať s niektorými prejavmi syndrómu nádorového rozpadu, ako sú hyperurikémia, hyperkaliémia, hypokalciémia, hyperfosfatémia, akútne zlyhávanie obličiek, zvýšená laktát dehydrogenáza (LDH), a môže sa spájať s akútnym respiračným zlyhávaním a smrťou. Akútne respiračné zlyhávanie môže byť sprevádzané udalosťami, akými sú pľúcna intersticiálna infiltrácia alebo edém, ktoré sú viditeľné na röntgenovej snímke hrudníka. Tento syndróm sa často prejavuje
do jednej alebo dvoch hodín od začiatku prvej infúzie. Pacienti s pľúcnou nedostatočnosťou
v anamnéze alebo s nádorovou infiltráciou pľúc, môžu mať zvýšené riziko zlých výsledkov a musia sa liečiť so zvýšenou opatrnosťou. Pacientom, u ktorých sa vyvinie závažný syndróm uvoľňovania
cytokínov, sa musí okamžite prerušiť podávanie infúzie (pozri časť 4.2) a majú dostať intenzívnu
symptomatickú liečbu. Keďže po počiatočnom zlepšení klinických príznakov môže nasledovať zhoršenie, títo pacienti musia byť dôkladne monitorovaní, pokiaľ syndróm nádorového rozpadu
a pľúcna infiltrácia nevymiznú alebo sa nevylúčia. Ďalšia liečba pacientov po úplnom vymiznutí
znakov a príznakov zriedka viedla k opakovanému závažnému syndrómu uvoľňovania cytokínov.
Pacienti s vysokou nádorovou záťažou alebo vysokým počtom (≥ 25 x 109/l) cirkulujúcich malígnych buniek, ako sú pacienti s CLL, ktorí môžu mať vyššie riziko hlavne závažného syndrómu uvoľňovania cytokínov, sa majú liečiť so zvýšenou opatrnosťou. Títo pacienti majú byť počas prvej infúzie veľmi dôkladne monitorovaní. U týchto pacientov sa má zvážiť použitie zníženej rýchlosti prvej infúzie alebo
rozdelenie dávkovania na dva dni počas prvého cyklu a v ktoromkoľvek nasledujúcom cykle, ak bude počet lymfocytov stále > 25 x 109/l.
Všetky druhy nežiaducich reakcií súvisiacich s infúziou sa pozorovali u 77 % pacientov liečených rituximabom (vrátane syndrómu uvoľňovania cytokínov sprevádzaného hypotenziou
a bronchospazmom u 10 % pacientov) (pozri časť 4.8). Tieto príznaky sú zvyčajne reverzibilné
po prerušení infúzie rituximabu a podaní antipyretika, antihistaminika a prípadne kyslíka, intravenózneho fyziologického roztoku alebo bronchodilatancií a glukokortikoidov, ak je to potrebné. Informácie o závažných reakciách sú uvedené vyššie v časti opisujúcej syndróm uvoľňovania cytokínov.
Po intravenóznom podávaní proteínov pacientom boli hlásené anafylaktické a iné hypersenzitívne reakcie. Na rozdiel od syndrómu uvoľňovania cytokínov, skutočné hypersenzitívne reakcie sa typicky objavujú do niekoľkých minút od začiatku infúzie. Počas podávania rituximabu musia byť k dispozícii lieky na liečbu hypersenzitívnych reakcií, napr. epinefrín (adrenalín), antihistaminiká
a glukokortikoidy na okamžité použitie v prípade alergickej reakcie. Klinické prejavy anafylaxie sa môžu podobať klinickým prejavom syndrómu uvoľňovania cytokínov (ako je opísané vyššie). Reakcie
spojené s hypersenzitivitou sa hlásili menej často ako reakcie spojené s uvoľňovaním cytokínov.
Ďalšie reakcie, hlásené v niekoľkých prípadoch, boli infarkt myokardu, atriálna fibrilácia, pľúcny edém a akútna reverzibilná trombocytopénia.
Keďže počas podávania rituximabu môže dochádzať k hypotenzii, má sa zvážiť prerušenie podávania antihypertenzív po dobu 12 hodín pred infúziou Ruxience.
Poruchy srdca a srdcovej činnosti
U pacientov liečených rituximabom sa vyskytli angina pectoris, srdcové arytmie, ako sú flutter
a fibrilácia predsiení, zlyhávanie srdca a/alebo infarkt myokardu. Pacienti s anamnézou ochorenia srdca a/alebo kardiotoxickej chemoterapie, majú byť preto dôkladne monitorovaní.
Hematologické toxicity
Hoci nie je rituximab v monoterapii myelosupresívny, pri zvažovaní liečby pacientov s počtom neutrofilov < 1,5 x 109/l a/alebo počtom trombocytov < 75 x 109/l sa musí postupovať opatrne, pretože klinické skúsenosti v tejto populácii sú obmedzené. Rituximab sa používal u 21 pacientov, ktorí podstúpili autológnu transplantáciu kostnej drene, a v iných rizikových skupinách, ktoré mali pravdepodobne redukovanú funkciu kostnej drene, bez navodenia myelotoxicity.
Počas liečby s Ruxience sa má pravidelne vyhodnocovať počet krviniek vrátane počtov neutrofilov a trombocytov.
Infekcie
Počas liečby rituximabom môže dôjsť k závažným infekciám vrátane úmrtí (pozri časť 4.8). Ruxience sa nesmie podávať pacientom s aktívnou, závažnou infekciou (napr. tuberkulózou, sepsou
a oportúnnymi infekciami, pozri časť 4.3).
Lekári musia postupovať opatrne, keď zvažujú použitie Ruxience u pacientov, ktorí majú v anamnéze opakované alebo chronické infekcie alebo sprievodné ochorenia, ktoré ich môžu ďalej predisponovať k závažnej infekcii (pozri časť 4.8).
U pacientov liečených rituximabom boli hlásené prípady reaktivácie hepatitídy B vrátane fulminantnej hepatitídy so smrteľnými následkami. Väčšina týchto pacientov podstúpila aj cytotoxickú chemoterapiu. Obmedzené informácie z jednej štúdie u pacientov s relapsovanou/refraktérnou CLL naznačujú, že liečba rituximabom môže zhoršovať aj výsledok primárnych infekcií hepatitídy B.
Pred začiatkom liečby Ruxience majú všetci pacienti podstúpiť skríning na vírus hepatitídy B (HBV). Skríning má minimálne zahŕňať stav HBsAg a stav HBcAb. Môže byť doplnený aj o ďalšie vhodné markery podľa lokálnych odporučených postupov. Pacienti s aktívnou hepatitídou B sa nesmú liečiť
byť konzultovaná s odborníkmi na choroby pečene pred začiatkom liečby a pacienti majú byť monitorovaní a liečení podľa lokálnych štandardných liečebných postupov na prevenciu reaktivácie hepatitídy B.
Po uvedení rituximabu na trh pri NHL a CLL boli hlásené veľmi zriedkavé prípady progresívnej multifokálnej leukoencefalopatie (PML) (pozri časť 4.8). Väčšina pacientov dostávala rituximab
v kombinácii s chemoterapiou alebo ako súčasť transplantátu hematopoetických kmeňových buniek.
Imunizácie
U pacientov s NHL a CLL sa neštudovala bezpečnosť imunizácie živými vírusovými vakcínami po liečbe rituximabom a očkovanie živými vírusovými vakcínami sa neodporúča. Pacienti liečení Ruxience sa môžu očkovať neživými vakcínami. Môžu ale byť redukované miery odpovedí na neživé vakcíny. V nerandomizovanej klinickej štúdii mali pacienti s relapsovaným NHL nízkeho stupňa, ktorí boli liečení rituximabom v monoterapii, nižšiu mieru odpovede na očkovanie antigénom (tzv. recall) proti tetanu (16 % vs. 81 %) a na očkovanie neoantigénom hemocyanínu zo svätojánskej mušky
(KLH) (4 % vs. 76 %, keď sa vyhodnocovali pre > 2-násobný nárast titrov protilátok) ako kontrolná skupina zdravých neliečených osôb. Pre pacientov s CLL sa dajú predpokladať podobné výsledky
berúc do úvahy podobnosti oboch ochorení, ale v klinických skúšaniach sa to neskúmalo.
Priemerné predterapeutické protilátkové titre proti panelu antigénov (Streptococcus pneumoniae, chrípka A, mumps, rubeola, kiahne) ostávali zachované najmenej 6 mesiacov po liečbe rituximabom.
Kožné reakcie
Hlásili sa ťažké kožné reakcie, ako sú toxická epidermálna nekrolýza (Lyellov syndróm)
a Stevensov-Johnsonov syndróm, niektoré so smrteľnými následkami (pozri časť 4.8). V prípade takejto udalosti s podozrením na vzťah s rituximabom sa liečba musí trvalo ukončiť.
Reumatoidnáartritída,granulomatózaspolyangiitídouamikroskopickápolyangiitídaapemphigusvulgaris
Pacienti s reumatoidnou artritídou bez predchádzajúcej liečby metotrexátom (MTX)
Použitie rituximabu sa neodporúča u pacientov predtým neliečených MTX, pretože priaznivý vzťah medzi prínosom a rizikom nebol stanovený.
Reakcie súvisiace s infúziou
Liečba rituximabom je spojená s reakciami súvisiacimi s infúziou (IRR), ktoré môžu súvisieť s uvoľňovaním cytokínov a/alebo iných chemických mediátorov.
Závažné IRR so smrteľnými následkami sa hlásili po uvedení lieku na trh u pacientov s reumatoidnou artritídou. Pri reumatoidnej artritíde bola väčšina udalostí súvisiacich s infúziou hlásených
v klinických skúšaniach mierna až stredne ťažká. Väčšinu častých príznakov tvorili alergické reakcie
ako bolesti hlavy, pruritus, dráždenie v hrdle, začervenanie, vyrážka, žihľavka, hypertenzia a pyrexia. Vo všeobecnosti bol podiel pacientov, u ktorých došlo k akejkoľvek reakcii na infúziu, vyšší po prvej infúzii ako po druhej infúzii v rámci ktoréhokoľvek liečebného cyklu. Incidencia IRR sa v priebehu nasledujúcich cyklov znižovala (pozri časť 4.8). Tieto hlásené reakcie boli zvyčajne reverzibilné po znížení rýchlosti alebo prerušení infúzie rituximabu a podaní antipyretika, antihistaminika a prípadne kyslíka, intravenózneho fyziologického roztoku alebo bronchodilatátorov, a glukokortikoidov
v prípade potreby. Dôkladne monitorujte pacientov s už prítomnými poruchami srdca alebo srdcovej činnosti a pacientov, u ktorých predtým došlo ku kardiopulmonálnym nežiaducim reakciám.
V závislosti od závažnosti IRR a potrebných intervencií prerušte alebo trvalo ukončite liečbu
Ruxience. Vo väčšine prípadov sa infúzia môže opäť začať podávať s o 50 % nižšou rýchlosťou (napr. zo 100 mg/hod na 50 mg/hod), keď príznaky úplne vymiznú.
Počas podávania Ruxience musia byť k dispozícii lieky na liečbu hypersenzitívnych reakcií, napr. epinefrín (adrenalín), antihistaminiká a glukokortikoidy na okamžité použitie v prípade alergickej reakcie.
Nie sú k dispozícii žiadne údaje o bezpečnosti rituximabu u pacientov so stredne ťažkým zlyhávaním srdca (III. trieda podľa NYHA) alebo ťažkým, nekontrolovaným kardiovaskulárnym ochorením.
U pacientov liečených rituximabom sa pozorovalo objavenie sa symptómov už existujúcich ischemických srdcových stavov, ako angina pectoris, ako aj fibrilácia a flutter predsiení. Preto sa
u pacientov so známym ochorením srdca v anamnéze a u pacientov, u ktorých došlo
k predchádzajúcim kardiopulmonálnym nežiaducim reakciám, má pred liečbou Ruxience zvážiť riziko kardiovaskulárnych komplikácií vyplývajúcich z reakcií na infúziu a títo pacienti sa majú
počas podávania dôkladne monitorovať. Keďže počas infúzie rituximabu môže dochádzať
k hypotenzii, má sa zvážiť prerušenie podávania antihypertenzív po dobu 12 hodín pred infúziou
Ruxience.
IRR pre pacientov s granulomatózou s polyangiitídou, mikroskopickou polyangiitídou a pemphigus vulgaris boli konzistentné s IRR pozorovanými u pacientov s reumatoidnou artritídou v klinických skúšaniach (pozri časť 4.8).
Poruchy srdca a srdcovej činnosti
U pacientov liečených rituximabom sa vyskytli angina pectoris, srdcové arytmie, ako sú flutter a fibrilácia predsiení, zlyhávanie srdca a/alebo infarkt myokardu. Preto majú byť pacienti s anamnézou ochorenia srdca dôkladne monitorovaní (pozri Reakcie súvisiace s infúziou, vyššie).
Infekcie
Vzhľadom na mechanizmus účinku rituximabu a poznatky o tom, že B-bunky hrajú dôležitú úlohu pri zachovávaní normálnej imunitnej odpovede, majú pacienti po liečbe rituximabom zvýšené riziko infekcie (pozri časť 5.1). Počas liečby rituximabom môže dôjsť k závažným infekciám vrátane smrteľných (pozri časť 4.8). Ruxience sa nesmie podávať pacientom s aktívnou, ťažkou infekciou (napr. tuberkulózou, sepsou a oportúnnymi infekciami, pozri časť 4.3) alebo ťažko imunokompromitovaným pacientom (napr. s veľmi nízkymi hladinami CD4 alebo CD8). Lekári musia postupovať opatrne, keď zvažujú použitie rituximabu u pacientov s anamnézou opakovaných alebo chronických infekcií alebo sprievodných ochorení, ktoré ich môžu ďalej predisponovať k závažnej infekcii, napr. hypogamaglobulinémie (pozri časť 4.8). Pred začiatkom liečby Ruxience sa odporúča stanoviť hladiny imunoglobulínov.
Pacienti hlásiaci znaky a príznaky infekcie po liečbe Ruxience majú byť okamžite vyšetrení a vhodne liečení. Pred podaním nasledujúceho cyklu liečby Ruxience sa u pacientov musí znova vyhodnotiť akékoľvek potenciálne riziko infekcií.
Veľmi zriedkavé prípady smrteľnej progresívnej multifokálnej leukoencefalopatie (PML) sa hlásili po používaní rituximabu na liečbu reumatoidnej artritídy a autoimunitných ochorení zahŕňajúcich systémový lupus erythematosus (SLE) a vaskulitídu.
Infekcie hepatitídy B
Prípady reaktivácie hepatitídy B vrátane prípadov so smrteľnými následkami boli hlásené u pacientov s reumatoidnou artritídou, granulomatózou s polyangiitídou a mikroskopickou polyangiitídou liečených rituximabom.
Pred začiatkom liečby Ruxience majú všetci pacienti podstúpiť skríning na vírus hepatitídy B (HBV). Skríning musí minimálne zahŕňať stav HBsAg a stav HBcAb. Môže byť doplnený aj o ďalšie vhodné markery podľa lokálnych odporučených postupov. Pacienti s aktívnou hepatitídou B sa nesmú liečiť rituximabom. Liečba pacientov so sérologiou pozitívnou na hepatitídu B (buď HBsAg, alebo HBcAb) sa má konzultovať s odborníkmi na choroby pečene pred začiatkom liečby a pacienti majú byť monitorovaní a liečení podľa štandardných liečebných postupov na prevenciu reaktivácie hepatitídy B.
Neskorá neutropénia
Počet neutrofilov v krvi stanovujte pred každým cyklom liečby Ruxience a pravidelne až do
6 mesiacov po ukončení liečby alebo do objavenia sa znakov alebo príznakov infekcie (pozri časť 4.8).
Kožné reakcie
Boli hlásené ťažké kožné reakcie, ako sú toxická epidermálna nekrolýza (Lyellov syndróm)
a Stevensov-Johnsonov syndróm, niektoré so smrteľnými následkami (pozri časť 4.8). V prípade takejto udalosti s podozrením na vzťah s Ruxience sa liečba musí trvalo ukončiť.
Imunizácia
Pred liečbou Ruxience lekári musia skontrolovať stav vakcinácie pacienta a dodržiavať platné imunizačné postupy. Vakcinácia sa musí ukončiť najmenej 4 týždne pred prvým podaním Ruxience.
Bezpečnosť imunizácie živými vírusovými vakcínami po liečbe rituximabom sa neštudovala. Očkovanie živými vírusovými vakcínami sa preto neodporúča pri liečbe Ruxience alebo pri úbytku periférnych B-buniek.
Pacienti liečení Ruxience môžu byť očkovaní neživými vakcínami. Môže však dôjsť k redukcii miery odpovede na neživé vakcíny. V randomizovanom klinickom skúšaní mali pacienti s reumatoidnou artritídou liečení rituximabom a metotrexátom porovnateľné miery odpovede na preočkovací antigén (tzv. recall) proti tetanu (39 % vs. 42 %), redukované miery odpovede na pneumokokovú polysacharidovú vakcínu (43 % vs. 82 % na najmenej 2 pneumokokové protilátkové sérotypy)
a na očkovací neoantigén KLH (47 % vs. 93 %), keď sa podávali 6 mesiacov po rituximabe,
v porovnaní s pacientami, ktorí dostávali len metotrexát. Ak sa vyžaduje očkovanie neživými vakcínami pri liečbe rituximabom, musí sa ukončiť najmenej 4 týždne pred začiatkom nasledujúceho
cyklu rituximabu.
V rámci celkových skúseností s liečbou rituximabom opakovanou počas jedného roka pri reumatoidnej artritíde boli podiely pacientov s pozitívnymi protilátkovými titrami proti S. pneumoniae, chrípke, mumpsu, rubeole, kiahňam a tetanovému toxoidu vo všeobecnosti podobné podielom
vo východiskovom stave.
Súbežné/následné používanie iných DMARD pri reumatoidnej artritíde
Neodporúča sa súbežné používanie Ruxience a iných antireumatických liekov ako sú tie, ktoré sú špecificky uvedené v časti týkajúcej sa indikácie a dávkovania pre reumatoidnú artritídu.
K dispozícii je obmedzené množstvo údajov z klinických skúšaní na to, aby sa úplne vyhodnotila bezpečnosť následného používania iných DMARD (vrátane inhibítorov TNF a iných biologických liekov) po rituximabe (pozri časť 4.5). Z dostupných údajov vyplýva, že miera klinicky relevantnej infekcie ostáva nezmenená, keď sa takéto lieky používajú u pacientov predtým liečených rituximabom, avšak títo pacienti majú byť dôkladne sledovaní, či sa u nich nevyskytnú prejavy infekcie, keď sa biologické lieky a/alebo DMARD používajú po liečbe rituximabom.
Malignita
Imunomodulačné lieky môžu zvyšovať riziko malignity. Na základe obmedzených skúseností
s rituximabom u pacientov s reumatoidnou artritídou (pozri časť 4.8) súčasné údaje nenaznačujú žiadne zvýšené riziko malignity. V súčasnosti sa však nedá vylúčiť možné riziko vývoja solídnych nádorov.
Pomocnálátka: Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na jednu dávku, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
V súčasnosti sú údaje o možných liekových interakciách s rituximabom obmedzené.
U pacientov s CLL pri súbežnom podávaní s rituximabom sa nepozoroval vplyv na farmakokinetiku fludarabínu alebo cyklofosfamidu. Okrem toho nedochádzalo k žiadnemu zjavnému účinku fludarabínu a cyklofosfamidu na farmakokinetiku rituximabu.
Súbežné podávanie s metotrexátom nemalo žiadny vplyv na farmakokinetiku rituximabu u pacientov s reumatoidnou artritídou.
Pacienti s titrami ľudskej protilátky proti myším antigénom (HAMA) alebo protilátky proti liečivu (ADA) môžu mať alergické alebo hypersenzitívne reakcie, keď sa liečia inými diagnostickými alebo terapeutickými monoklonálnymi protilátkami.
U pacientov s reumatoidnou artritídou bolo 283 pacientov liečených biologickým DMARD po liečbe rituximabom. U týchto pacientov bola miera klinicky relevantnej infekcie počas liečby rituximabom
6,01 na 100 pacientorokov v porovnaní so 4,97 na 100 pacientorokov po liečbe biologickým DMARD.
4.6 Fertilita, gravidita a laktácia
Antikoncepcia umužovažien
Kvôli dlhému retenčnému času rituximabu u pacientov s úbytkom B-buniek musia ženy vo fertilnom
veku používať účinné antikoncepčné metódy počas liečby Ruxience a po dobu 12 mesiacov po jej ukončení.
Gravidita
O IgG imunoglobulínoch je známe, že prechádzajú cez placentovú bariéru.
Hladiny B-buniek u ľudských novorodencov po maternálnej expozícii rituximabu sa v klinických skúšaniach neštudovali. Nie sú dostupné žiadne adekvátne a dobre kontrolované údaje z klinických štúdií u tehotných žien, avšak prechodný úbytok B-buniek a lymfocytopénia sa hlásili u niektorých dojčiat narodených matkám, ktoré boli počas gravidity vystavené rituximabu. Podobné účinky sa pozorovali v štúdiách na zvieratách (pozri časť 5.3). Preto sa Ruxience nemá podávať gravidným ženám, pokiaľ možný prínos nepreváži potenciálne riziko.
Dojčenie
Nie je známe, či sa rituximab vylučuje do ľudského mlieka. Keďže sa však materský IgG vylučuje
do ľudského mlieka a rituximab sa detegoval v mlieku dojčiacich opíc, ženy nemajú dojčiť, pokiaľ sú liečené Ruxience a po dobu 12 mesiacov po ukončení liečby Ruxience.
Fertilita
Štúdie na zvieratách nepreukázali škodlivé účinky rituximabu na reprodukčné orgány.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie účinkov rituximabu na schopnosť viesť vozidlá a obsluhovať stroje,
hoci farmakologická aktivita a doteraz hlásené nežiaduce reakcie naznačujú, že rituximab nemá žiadny alebo len zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Skúsenosti znon-Hodgkinovholymfómuachronickejlymfocytovejleukémie
Súhrn bezpečnostného profilu
Celkový bezpečnostný profil rituximabu pri non-Hodgkinovom lymfóme a CLL je založený
na údajoch od pacientov z klinických skúšaní a zo sledovaní po uvedení lieku na trh. Títo pacienti sa liečili buď rituximabom v monoterapii (ako indukčná liečba alebo udržiavacia liečba po indukčnej liečbe) alebo v kombinácii s chemoterapiou.
Najčastejšie pozorovanými nežiaducimi reakciami na liek (adverse drug reactions, ADRs) u pacientov liečených rituximabom boli IRR, ktoré sa vyskytli u väčšiny pacientov počas podania prvej infúzie. Výskyt príznakov súvisiacich s infúziou sa podstatne znižuje pri následných infúziách a po ôsmich dávkach rituximabu je nižší ako 1 %.
K infekčným udalostiam (hlavne bakteriálnym a vírusovým) dochádzalo u približne 30 –
55 % pacientov počas klinických skúšaní u pacientov s NHL a u 30 – 50 % pacientov počas klinických skúšaní u pacientov s CLL.
Najčastejšie hlásenými alebo pozorovanými
závažnými nežiaducimi reakciami boli:
· IRR (vrátane syndrómu uvoľňovania cytokínov, syndrómu nádorového rozpadu), pozri časť 4.4,
· infekcie, pozri časť 4.4,
· kardiovaskulárne udalosti, pozri časť 4.4.
Iné závažné hlásené nežiaduce reakcie zahŕňali reaktiváciu hepatitídy B a PML (pozri časť 4.4.).
Tabuľkový zoznam nežiaducich reakciíFrekvencie nežiaducich reakcií hlásených pri liečbe rituximabom v monoterapii alebo pri liečbe
rituximabom v kombinácii s chemoterapiou sú zhrnuté v tabuľke 1. V rámci každej skupiny frekvencií sú nežiaduce účinky uvádzané v poradí klesajúcej závažnosti. Frekvencie sú definované ako veľmi
časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (nemožno odhadnúť z dostupných údajov).
Nežiaduce reakcie, ktoré sa identifikovali len počas sledovania po uvedení lieku na trh a pre ktoré nebolo možné odhadnúť frekvenciu, sa uvádzajú ako „neznáme“.
Tabuľka 1 Nežiaduce reakcie na liek hlásené v klinických skúšaniach alebo počas sledovaniaTrieda orgánových systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Veľmi zriedkavé
|
Neznáme
| Infekcie
a nákazy
| bakteriálne infekcie, vírusové infekcie,
+bronchitída
| sepsa,
+pneumónia,
+febrilná infekcia,
+herpes zoster,
+infekcia dýchacích ciest, hubové
infekcie, infekcie neznámeho pôvodu,
+akútna bronchitída,
+sinusitída, hepatitída B1
|
| závažná vírusová infekcia2, Pneumocystis jirovecii
| PML
|
| Poruchy krvi
a lymfatického systému
| neutropénia, leukopénia,
+febrilná neutropénia,
+trombocytopénia
| anémia,
+pancytopénia,
+granulocytopé- nia
| koagulačné poruchy, aplastická anémia, hemolytická anémia, lymfadenopatia
|
| prechodné zvýšenie sérových hladín IgM3
| neskorá neutropénia3
|
|
|
po uvedení lieku na trh u pacientov s NHL a CLL ochorením liečených rituximabom v monoterapii/udržiavacej liečbe alebo v kombinácii s chemoterapiou
Trieda orgánových
systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Veľmi zriedkavé
|
Neznáme
|
Poruchy imunitného
systému
|
reakcie súvisiace s infúziou4, angioedém
|
hypersenzitivita
|
|
anafylaxia
|
syndróm nádorového
rozpadu, syndróm uvoľňovania cytokínov4, sérová choroba
|
akútna reverzibilná
trombocytopénia súvisiaca
s infúziou4
|
Poruchy metabolizmu
a výživy
|
|
hyperglykémia, zníženie
hmotnosti,
periférny edém, edém tváre,
zvýšené LDH, hypokalciémia
|
|
|
|
|
Psychické poruchy
|
|
|
depresia, nervozita
|
|
|
|
Poruchy nervového systému
|
|
parestézia, hypestézia, agitácia,
insomnia, vazodilatácia, závrat, úzkosť
|
dysgeúzia
|
|
periférna neuropatia, paralýza
tvárového nervu5
|
kraniálna neuropatia, strata iných zmyslov5
|
Poruchy oka
|
|
porucha slzenia,
konjunktivitída
|
|
|
ťažkéá strata zraku5
|
|
Poruchy ucha a labyrintu
|
|
tinitus, bolesť ucha
|
|
|
|
strata sluchu5
|
Poruchy srdca a srdcovej
činnosti
|
|
+infarkt myokardu4 a 6,
arytmia,
+fibrilácia predsiení, tachykardia,
+porucha srdca
|
+zlyhávanie ľavej komory,
+supraventriku- lárna tachykardia,
+ventrikulárna tachykardia,
+angina pectoris,
+ischémia myokardu,
bradykardia
|
ťažké poruchy srdca 4 a 6
|
zlyhávanie srdca4 a 6
|
|
Poruchy ciev
|
|
hypertenzia, ortostatická
hypotenzia, hypotenzia
|
|
|
vaskulitída
(hlavne kožná),
leukocyto-
klastická vaskulitída
|
|
Poruchy dýchacej sústavy, hrudníka
a mediastína
|
|
bronchospaz- mus4, respiračné ochorenie,
bolesť
na hrudníku, dyspnoe, zhoršený kašeľ,
nádcha
|
astma, bronchiolitis obliterans, pľúcna
porucha, hypoxia
|
intersticiálna choroba pľúc7
|
respiračné zlyhávanie4
|
infiltrácia pľúc
|
Poruchy gastrointestiná
-lneho traktu
|
nevoľnosť
|
vracanie, hnačka, bolesť
brucha, dysfágia, stomatitída, zápcha,
dyspepsia, anorexia, dráždenie v hrdle
|
zväčšenie brucha
|
|
Gastrointes- tinálna perforácia7
|
|
Trieda orgánových
systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Veľmi zriedkavé
|
Neznáme
|
Poruchy kože a podkožného
t
k
aniva
|
pruritus, vyrážka,
+alopécia
|
žihľavka, potenie, nočné
potenie,
+porucha kože
|
|
|
závažné pľuzgierové
kožné reakcie, Stevensov- Johnsonov
syndróm, toxická epidermálna nekrolýza
(Lyellov syndróm)7
|
|
Poruchy kostrovej
a svalovej
sústavy
a spojivového tkaniva
|
|
hypertónia, myalgia,
artralgia, bolesť
chrbta, bolesť krku, bolesť
|
|
|
|
|
Poruchy obličiek
a močových
ciest
|
|
|
|
|
zlyhávanie obličiek4
|
|
Celkové poruchy
a reakcie v mieste podania
|
horúčka, zimnica, asténia, bolesť
hlavy
|
bolesť vyvolaná
nádorom, začervenanie, slabosť, syndróm
prechladnutia,
+únava,
+chvenie,
+multiorgánové zlyhanie4
|
bolesť v mieste infúzie
|
|
|
|
Laboratórne a funkčné vyšetrenia
|
znížené hladiny
IgG
|
|
|
|
|
|
Pre každý termín sa frekvencia vzťahovala na reakcie všetkých stupňov (od mierneho po ťažký) s výnimkou termínov označených s „+“, pri ktorých sa frekvencia vzťahuje len na ťažké reakcie (≥ 3. stupňa podľa NCI bežných kritérií toxicity). Uvádza sa len najvyššia frekvencia pozorovaná v skúšaniach.
1. zahŕňa reaktiváciu a primárne infekcie; frekvencia sa vzťahuje na R-FC režim pri relapsovanej/refraktérnej CLL
2. pozri aj časť Infekcie nižšie
3. pozri aj časť Hematologické nežiaduce reakcie nižšie
4. pozri aj časť Reakcie súvisiace s infúziou nižšie. Zriedkavo boli hlásené smrteľné prípady.
5. znaky a príznaky kraniálnej neuropatie. Dochádzalo k nim v rôznych časoch až do niekoľkých mesiacov po ukončení liečby rituximabom.
6. pozorované najmä u pacientov s predchádzajúcim kardiologickým ochorením a/alebo kardiotoxickou chemoterapiou
a väčšinou boli spojené s reakciami súvisiacimi s infúziou
7. zahŕňa smrteľné prípady
|
Nasledovné termíny sa hlásili ako nežiaduce udalosti počas klinických skúšaní, avšak hlásili sa
s podobnou alebo nižšou incidenciou v skupinách s rituximabom ako v kontrolných skupinách:
hematotoxicita, infekcia pri neutropénii, infekcia močovej sústavy, senzorická porucha, pyrexia.
Znaky a príznaky naznačujúce reakciu súvisiacu s infúziou sa hlásili u viac ako 50 % pacientov v klinických skúšaniach a boli pozorované najmä v priebehu podania prvej infúzie, zvyčajne
v priebehu prvej až dvoch hodín. Tieto príznaky zahŕňali najmä horúčku, zimnicu a triašku. Ďalšie
príznaky zahŕňali začervenanie, angioedém, bronchospazmus, vracanie, nevoľnosť, žihľavku/vyrážku, únavu, bolesť hlavy, dráždenie v hrdle, nádchu, pruritus, bolesť, tachykardiu, hypertenziu, hypotenziu, dyspnoe, dyspepsiu, asténiu a prejavy syndrómu nádorového rozpadu. K závažným reakciám súvisiacim s infúziou (ako je bronchospazmus, hypotenzia) došlo až v 12 % prípadov. Ďalšie reakcie, ktoré boli v niektorých prípadoch hlásené, boli infarkt myokardu, atriálna fibrilácia, pľúcny edém
a akútna reverzibilná trombocytopénia. S nižšími alebo neznámymi frekvenciami sa hlásili zhoršenia už prítomných srdcových ochorení, ako sú angina pectoris alebo kongestívne zlyhávanie srdca alebo
závažné poruchy srdca (zlyhávanie srdca, infarkt myokardu, atriálna fibrilácia), pľúcny edém, multiorgánové zlyhávanie, syndróm nádorového rozpadu, syndróm uvoľňovania cytokínov, zlyhávanie obličiek a respiračné zlyhávanie. Incidencia príznakov súvisiacich s infúziou sa podstatne znížila
pri následných infúziách a po ôsmich cykloch liečby rituximabom (liečby obsahujúcej rituximab)
zodpovedá < 1 % pacientov.
Opis vybraných nežiaducich reakcií
Infekcie
Rituximab vedie k úbytku B-buniek u približne 70 – 80 % pacientov, ale len u menšej časti pacientov sa spájal so znížením sérových imunoglobulínov.
Lokalizované kvasinkové infekcie ako aj herpes zoster boli hlásené v randomizovaných klinických štúdiách s vyššou incidenciou v skupine obsahujúcej rituximab. Závažné infekcie boli hlásené
u približne 4 % pacientov liečených rituximabom v monoterapii. Vyššie frekvencie celkových infekcií vrátane infekcií 3. alebo 4. stupňa sa pozorovali počas udržiavacej liečby rituximabom po dobu
2 rokov pri porovnávaní s pozorovanou skupinou. Nedochádzalo k žiadnej kumulatívnej toxicite, pokiaľ ide o infekcie hlásené v priebehu 2-ročného liečebného obdobia. Okrem toho sa pri liečbe
rituximabom hlásili iné závažné vírusové infekcie, buď nové, reaktivované alebo zhoršené, pričom niektoré z nich so smrteľnými následkami. Väčšina pacientov bola liečená rituximabom v kombinácii s chemoterapiou alebo ako súčasť transplantátu hematopoetických kmeňových buniek. Príkladmi
týchto závažných vírusových infekcií sú infekcie spôsobené herpesovými vírusmi
(cytomegalovírusom, vírusom Varicella zoster a vírusom Herpes simplex), JC vírusom (vírusom progresívnej multifokálnej leukoencefalopatie (PML)) a vírusom hepatitídy C. V klinických
skúšaniach sa hlásili aj prípady PML so smrteľnými následkami, ku ktorým došlo po progresii
ochorenia a opakovanej liečbe. Boli hlásené prípady reaktivácie hepatitídy B, z ktorých väčšina sa vyskytla u pacientov liečených rituximabom v kombinácii s cytotoxickou chemoterapiou. U pacientov
s relapsovanou/refraktérnou CLL bola incidencia infekcie hepatitídy B 3./4. stupňa (reaktivácia
a primárna infekcia) 2 % v R-FC vs. 0 % v FC. Progresia Kaposiho sarkómu sa pozorovala
u pacientov vystavených rituximabu, u ktorých už bol Kaposiho sarkóm prítomný. K týmto prípadom dochádzalo pri neschválených indikáciách a väčšina pacientov bola HIV-pozitívna.
Hematologické nežiaduce reakcie
V klinických skúšaniach s rituximabom v monoterapii podávaným po dobu 4 týždne sa hematologické abnormality vyskytli u malého počtu pacientov a zvyčajne boli mierne a reverzibilné. Závažná
(3./4. stupňa) neutropénia bola hlásená u 4,2 %, anémia u 1,1 % a trombocytopénia u 1,7 % pacientov.
V priebehu udržiavacej liečby rituximabom trvajúcej až 2 roky sa hlásili leukopénia (5 % vs. 2 %,
3./4. stupňa) a neutropénia (10 % vs. 4 %, 3./4. stupňa) s vyššou incidenciou v porovnaní
s pozorovanou skupinou. Incidencia trombocytopénie bola nízka (< 1 %, 3./4. stupňa) a medzi liečebnými skupinami sa nelíšila. Počas liečebného cyklu v štúdiách s rituximabom v kombinácii
s chemoterapiou sa hlásili leukopénia 3./4. stupňa (R-CHOP 88 % vs. CHOP 79 %, R-FC 23 % vs. FC
12 %), neutropénia (R-CVP 24 % vs. CVP 14 %; R-CHOP 97 % vs. CHOP 88 %, R-FC 30 % vs. FC
19 % u predtým neliečenej CLL), pancytopénia (R-FC 3 % vs. FC 1 % u predtým neliečenej CLL)
zvyčajne s vyššími frekvenciami v porovnaní len s chemoterapiou. Vyššia incidencia neutropénie
u pacientov liečených rituximabom a chemoterapiou sa však nespájala s vyššou incidenciou infekcií a nákaz v porovnaní s pacientami liečenými len chemoterapiou. Klinické štúdie v prípade predtým
neliečenej a relapsovanej/refraktérnej CLL stanovili, že až u 25 % pacientov liečených s R-FC sa neutropénia predĺžila (definované ako počet neutrofilov nižší ako 1 x 109/l od 24 do 42 dní
po poslednej dávke) alebo mala neskorý nástup (definovaný ako počet neutrofilov nižší ako 1 x 109/l neskôr ako 42 dní po poslednej dávke u pacientov bez predchádzajúcej predĺženej neutropénie alebo
u pacientov, ktorí sa zotavili pred 42. dňom) po liečbe rituximabom plus FC. Nehlásili sa žiadne rozdiely v incidencii anémie. Hlásilo sa niekoľko prípadov neskorej neutropénie, ku ktorým došlo viac ako štyri týždne po poslednej infúzii rituximabu. V klinickej štúdii v prvej línii liečby CLL u pacientov
v štádiu C podľa Bineta došlo k viacerým nežiaducim udalostiam v skupine R-FC v porovnaní
so skupinou FC (R-FC 83 % vs. FC 71 %). V klinickej štúdii s relapsovanou/refraktérnou CLL sa trombocytopénia 3./4. stupňa hlásila u 11 % pacientov v skupine R-FC v porovnaní s 9 % pacientov
v skupine FC.
V klinických štúdiách s rituximabom u pacientov s Waldenstromovou makroglobulinémiou sa pozorovali prechodné zvýšenia sérových hladín IgM po začiatku liečby, ktoré sa môžu spájať
s hyperviskozitou a súvisiacimi príznakmi. Toto prechodné zvýšenie IgM sa zvyčajne vrátilo aspoň na
východiskovú úroveň do 4 mesiacov.
Kardiovaskulárne nežiaduce reakcie
Kardiovaskulárne reakcie v priebehu klinických skúšaní s rituximabom v monoterapii boli hlásené u 18,8 % pacientov, pričom najčastejšie hlásenými udalosťami boli hypotenzia a hypertenzia.
V priebehu infúzie sa hlásili prípady arytmie 3. alebo 4. stupňa (vrátane ventrikulárnej
a supraventrikulárnej tachykardie) a anginy pectoris. V priebehu udržiavacej liečby bola incidencia porúch srdca 3./4. stupňa porovnateľná medzi pacientami liečenými rituximabom a pacientami
v pozorovanej skupine. Poruchy srdca sa hlásili ako závažné nežiaduce udalosti (vrátane atriálnej
fibrilácie, infarktu myokardu, zlyhávania ľavej komory, ischémie myokardu) u 3 % pacientov liečených rituximabom v porovnaní s < 1 % pacientov v pozorovanej skupine. V štúdiách
vyhodnocujúcich rituximab v kombinácii s chemoterapiou bola incidencia srdcových arytmií
3. a 4. stupňa, hlavne supraventrikulárnych arytmií, ako sú tachykardia a flutter/fibrilácia predsiení, vyššia v R-CHOP skupine (14 pacienti, 6,9 %) ako v CHOP skupine (3 pacienti, 1,5 %). Ku všetkým týmto arytmiám buď dochádzalo v súvislosti s infúziou rituximabu alebo sa spájali s predisponujúcimi stavmi, ako sú horúčka, infekcia, akútny infarkt myokardu alebo už prítomné respiračné
a kardiovaskulárne ochorenie. Nepozoroval sa žiadny rozdiel medzi skupinami R-CHOP a CHOP
v incidencii iných srdcových udalostí 3. a 4. stupňa vrátane srdcového zlyhávania, ochorenia
myokardu a prejavov koronárnej choroby srdca. Pri CLL bola celková incidencia porúch srdca 3. alebo
4. stupňa nižšia aj v klinickej štúdii v prvej línii liečby (4 % R-FC, 3 % FC), aj v klinickej štúdii u relapsovaných/refraktérnych stavov (4 % R-FC, 4 % FC).
Respiračný systém
Boli hlásené prípady intersticiálneho ochorenia pľúc, niektoré so smrteľnými následkami.
Neurologické poruchy
V priebehu liečebného obdobia (indukčná fáza liečby zahŕňajúca skupinu R-CHOP po dobu maximálne ôsmich cyklov) štyria pacienti (2 %) liečení v skupine R-CHOP, pričom všetci pacienti
mali kardiovaskulárne rizikové faktory, zaznamenali tromboembolické cerebrovaskulárne príhody
v priebehu prvého liečebného cyklu. Medzi liečebnými skupinami nebol žiadny rozdiel v incidencii iných tromboembolických príhod. Naopak, u troch pacientov (1,5 %) došlo k cerebrovaskulárnym udalostiam v skupine CHOP, pričom ku všetkým došlo v priebehu obdobia následného sledovania.
Pri CLL bola celková incidencia porúch nervového systému 3. alebo 4. stupňa nižšia v klinickej štúdii v prvej línii liečby (4 % R-FC, 4% FC) aj v klinickej štúdii u relapsovaných/refraktérnych stavov
(3 % R-FC, 3 % FC).
Boli hlásené prípady posteriórneho reverzibilného encefalopatického syndrómu (PRES)/reverzibilného posteriórneho leukoencefalopatického syndrómu (RPLS). Znaky a príznaky zahŕňali poruchu videnia, bolesť hlavy, záchvaty a zmenený mentálny stav s asociovanou hypertenziou alebo bez nej. Diagnóza PRES/RPLS sa musí potvrdiť na základe zobrazovacieho vyšetrenia mozgu. V hlásených prípadoch boli potvrdené rizikové faktory pre PRES/RPLS zahŕňajúce základné ochorenie pacienta, hypertenziu, imunosupresívnu liečbu a/alebo chemoterapiu.
Poruchy gastrointestinálneho traktu
Gastrointestinálna perforácia, ktorá v niektorých prípadoch viedla k úmrtiu, sa pozorovala u pacientov s non-Hodgkinovým lymfómom liečených rituximabom. Vo väčšine týchto prípadov sa rituximab podával v kombinácii s chemoterapiou.
Hladiny IgG
V klinickom skúšaní hodnotiacom udržiavaciu liečbu rituximabom pri relapsovanom/refraktérnom folikulárnom lymfóme boli mediány hladín IgG pod dolnou hranicou normy (LLN) (< 7 g/l)
po indukčnej liečbe v pozorovanej skupine aj v skupine s rituximabom. V pozorovanej skupine sa medián hladiny IgG následne zvýšil nad LLN, ale zostal konštantný v skupine s rituximabom. Podiel
pacientov s hladinami IgG nižšími ako LLN bol približne 60 % v skupine s rituximabom počas 2- ročného liečebného obdobia, zatiaľ čo v pozorovanej skupine sa znížil (36 % po 2 rokoch).
Malý počet spontánnych hlásení a v literatúre opísaných prípadov hypogamaglobulinémie sa pozoroval u pediatrických pacientov liečených rituximabom, pričom niektoré prípady boli závažné
a vyžadovali dlhodobú imunoglobulínovú substitučnú liečbu. Následky dlhodobého úbytku B-buniek
u pediatrických pacientov nie sú známe.
Poruchy kože a podkožného tkaniva
Toxická epidermálna nekrolýza (Lyellov syndróm) a Stevensov-Johnsonov syndróm, niektoré so smrteľnými následkami, boli hlásené veľmi zriedkavo.
Subpopulácie pacientov – rituximab v monoterapii
Starší pacienti (vo veku ≥ 65 rokov):
Incidencia nežiaducich reakcií všetkých stupňov a nežiaducich reakcií 3./4. stupňa bola u starších pacientov podobná ako incidencia u mladších pacientov (vo veku < 65 rokov).
Pacienti s rozsiahlou nádorovou masou (tzv. bulky disease)
Vyššia incidencia nežiaducich reakcií 3./4. stupňa bola u pacientov s rozsiahlou nádorovou masou ako u pacientov bez rozsiahlej nádorovej masy (25,6 % vs. 15,4 %). Incidencia nežiaducich reakcií akéhokoľvek stupňa bola v týchto dvoch skupinách podobná.
Opakovaná liečba
Percento pacientov hlásiacich nežiaduce reakcie po opätovnej liečbe ďalšími cyklami rituximabu bolo podobné ako percento pacientov hlásiacich nežiaduce reakcie po prvej expozícii (nežiaduce reakcie akéhokoľvek stupňa a 3./4. stupňa).
Subpopulácie pacientov – kombinovaná liečba s rituximabom
Starší pacienti (vo veku ≥ 65 rokov)
Incidencia krvných a lymfatických nežiaducich udalostí 3./4. stupňa bola vyššia u starších pacientov ako u mladších pacientov (vo veku < 65 rokov) s predtým neliečenou alebo relapsovanou/refraktérnou
CLL.
Skúsenosti zreumatoidnejartritídy
Súhrn bezpečnostného profilu
Celkový bezpečnostný profil rituximabu pri reumatoidnej artritíde je založený na údajoch od pacientov
z klinických skúšaní a zo sledovaní po uvedení lieku na trh.
Bezpečnostný profil rituximabu u pacientov so stredne ťažkou až ťažkou reumatoidnou artritídou (RA) je zhrnutý v nižšie uvedených častiach. V klinických skúšaniach bolo viac ako 3 100 pacientov liečenáých najmenej jedným cyklom liečby a títo pacienti boli sledovaní po dobu od 6 mesiacov do viac ako 5 rokov. Približne 2 400 pacientov bolo liečených dvomi alebo viac cyklami liečby, pričom
1 000 bolo liečených 5 alebo viac cyklami. Bezpečnostné informácie získané po uvedení lieku na trh odrážajú očakávaný profil nežiaducich reakcií ako sa pozoroval v klinických skúšaniach pre rituximab
(pozri časť 4.4).
Pacienti boli liečení 2 x 1 000 mg rituximabu s odstupom dvoch týždňov v kombinácii s a metotrexátom (10 – 25 mg/týždeň). Infúzie Rituximabu sa podávali po intravenóznej infúzii
100 mg metylprednizolónu. Pacienti tiež 15 dní používali perorálnu liečbu prednizónom.
Tabuľkový zoznam nežiaducich reakcií
Zoznam nežiaducich reakcií je uvedený v tabuľke 2. Frekvencie sú definované ako veľmi časté
(≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), a veľmi zriedkavé (< 1/10 000). V rámci každej skupiny frekvencií sú nežiaduce účinky
uvádzané v poradí klesajúcej závažnosti.
Najčastejšími hlásenými nežiaducimi reakciami, ktorých výskyt sa dáva do súvislosti s rituximabom boli IRR. Celková incidencia IRR v klinických skúšaniach bola 23 % pri podaní prvej infúzie
a pri následných infúziách klesala. Závažné IRR boli menej časté (0,5 % pacientov) a pozorovali sa hlavne počas úvodného cyklu. Okrem nežiaducich reakcií zaznamenaných v klinických skúšaniach pri
RA s rituximabom sa po uvedení lieku na trh hlásili aj progresívna multifokálna leukoencefalopatia
(PML) (pozri časť 4.4) a reakcia podobná sérovej chorobe.
Trieda orgánových systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Veľmi zriedkavé
| Infekcie a nákazy
| infekcia horných dýchacích
ciest, infekcie močovej sústavy
| bronchitída, sinusitída, gastroenteritída,
tinea pedis
|
|
| PML, reaktivácia hepatitídy B
| Poruchy krvi
a lymfatického systému
|
| neutropénia1
|
| neskorá neutropénia2
| reakcia podobná sérovej chorobe
| Poruchy srdca
a srdcovej činnosti
|
|
|
| angina pectoris,
atriálna
fibrilácia, zlyhávanie
srdca, infarkt myokardu
| Flutter predsiení
| Poruchy
imunitného systému
| 3reakcie súvisiace
s infúziou
(hypertenzia, nevoľnosť, vyrážka,
pyrexia, pruritus, žihľavka, dráždenie v
hrdle, návaly tepla, hypotenzia, nádcha,
triaška, tachykardia, únava, orofaryngeálna
bolesť, periférny edém, erytém)
|
| 3reakcie súvisiace
s infúziou
(generalizovaný edém, bronchospazmus,
sipot, laryngeálny edém, angioneurotický
edém, generalizovaný pruritus, anafylaxia,
anafylaktoidná reakcia)
|
|
| Celkové poruchy
a reakcie v mieste podania
| Poruchy metabolizmu a výživy
|
| hypercholesterolémia
|
|
|
| Poruchy nervového systému
| bolesť hlavy
| parestézia, migréna, závraty, ischias
|
|
|
| Poruchy kože a podkožného
tkaniva
|
| alopécia
|
|
| toxická epidermálna nekrolýza (Lyellov
syndróm),
Stevensov-Johnsonov syndróm5
| Psychické poruchy
|
| depresia, úzkosť
|
|
|
| Poruchy gastrointestinálneho traktu
|
| dyspepsia, hnačka, gastroezofageálny reflux, vredy
v ústach, bolesť
v hornej časti brucha
|
|
|
|
|
|
Tabuľka 2 Súhrn nežiaducich reakcií na liek hlásených v klinických skúšaniach alebo počas sledovania po uvedení lieku na trh u pacientov s reumatoidnou artritídou liečených rituximabom
Trieda orgánových systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Veľmi zriedkavé
|
Poruchy kostrovej a svalovej sústavy
|
|
artralgia/bolesť kostí a svalov, osteoartritída,
burzitída
|
|
|
|
Laboratórne a funkčné
vyšetrenia
|
znížené hladiny IgM4
|
znížené hladiny IgG4
|
|
|
|
1. Kategória frekvencie odvodená od laboratórnych hodnôt získaných ako súčasť rutinného laboratórneho monitorovania v klinických skúšaniach.
2. Kategória frekvencie odvodená na základe údajov získaných po uvedení lieku na trh.
3. Reakcie, ktoré sa vyskytnú počas podania infúzie alebo do 24 hodín od podania infúzie. Pozri aj Reakcie súvisiace s infúziou nižšie. K IRR môže dochádzať v dôsledku hypersenzitivity a/alebo v dôsledku mechanizmu účinku.
4. Zahŕňa pozorovania získané ako súčasť rutinného laboratórneho monitorovania.
5. Zahŕňa smrteľné prípady.
|
Opakované liečebné cykly
Opakované liečebné cykly sa spájajú s podobným profilom nežiaducich reakcií na liek ako sa pozoroval po prvej expozícii. Miera výskytu všetkých nežiaducich reakcií na liek po prvej expozícii
rituximabu bola najvyššia v priebehu prvých 6 mesiacov a potom klesala. Hlavný podiel na
nežiaducich reakciách na liek mali IRR (najčastejšie v priebehu prvého liečebného cyklu), zhoršenie
RA a infekcie, pričom všetky boli častejšie v priebehu prvých 6 mesiacov liečby.
Opis vybraných nežiaducich reakciíReakcie súvisiace s infúziouNajčastejšími nežiaducimi reakciami, ktoré sa vyskytli po podaní rituximabu boli v klinických skúšaniach IRR (pozri tabuľku 2). Spomedzi 3 189 pacientov liečených rituximabom sa u 1 135
(36 %) vyskytla najmenej jedna IRR, pričom u 733/3 189 (23 %) pacientov došlo k IRR po prvej infúzii pri prvej expozícii rituximabu. Pri následných infúziách incidencia IRR klesala. V klinických
skúšaniach došlo k závažnej IRR u menej ako 1 % (17/3 189) pacientov. V klinických skúšaniach nedošlo k žiadnej IRR 4. stupňa CTC a IRR nespôsobili žiadne úmrtie. Podiel udalostí 3. stupňa CTC a IRR vedúcich k ukončeniu účasti v skúšaní sa znižoval s každým cyklom a od 3. cyklu boli
zriedkavé. Premedikácia s intravenóznym glukokortikoidom signifikantne redukovala incidenciu a závažnosť IRR (pozri časti 4.2 a 4.4). Závažné IRR so smrteľnými následkami boli hlásené po
uvedení lieku na trh.
V skúšaní zameranom na hodnotenie bezpečnosti rýchlejšej infúzie rituximabu u pacientov
s reumatoidnou artritídou mohli pacienti so stredne ťažkou až ťažkou aktívnou RA, u ktorých sa nevyskytla závažná IRR v priebehu prvej infúzie alebo do 24 hodín po nej, dostať 2-hodinovú intravenóznu infúziu rituximabu. Pacienti, ktorí mali v anamnéze závažnú reakciu na infúziu
pri biologickej liečbe RA boli zo skúšania vylúčení. Incidencia, typy a závažnosť IRR boli konzistentné s dosiaľ pozorovanými údajmi. Neboli pozorované žiadne závažné IRR.
InfekcieCelková miera výskytu infekcie bola približne 94 na 100 pacientorokov u pacientov liečených rituximabom. Infekcie boli hlavne mierne až stredne ťažké a pozostávali najmä z infekcií horných dýchacích ciest a infekcií močovej sústavy. Incidencia infekcií, ktoré boli závažné alebo vyžadovali intravenózne antibiotiká, bola približne 4 na 100 pacientorokov. Miera výskytu závažných infekcií nevykazovala žiadne signifikantné zvýšenie po opakovaných cykloch rituximabu. Infekcie dolných dýchacích ciest (vrátane pneumónie) boli v priebehu klinických skúšaní hlásené ako podobné
v skupinách s rituximabom a v kontrolných skupinách.
Prípady progresívnej multifokálnej leukoencefalopatie so smrteľnými následkami sa hlásili po použití rituximabu na liečbu autoimunitných ochorení. Tie zahŕňali reumatoidnú artritídu a autoimunitné ochorenia mimo schválenej indikácie vrátane systémového lupus erythematosus (SLE) a vaskulitídy.
U pacientov s non-Hodgkinovým lymfómom, ktorí boli liečení rituximabom v kombinácii
s cytotoxickou chemoterapiou, sa hlásili prípady reaktivácie hepatitídy B (pozri non-Hodgkinov
lymfóm). Reaktivácia infekcie hepatitídy B sa veľmi zriedkavo hlásila aj u pacientov s reumatoidnou artritídou, ktorí boli liečení rituximabom (pozri časť 4.4).
Kardiovaskulárne nežiaduce reakcie
Závažné kardiologické reakcie boli hlásené v miere 1,3 na 100 pacientorokov u pacientov liečených rituximabom v porovnaní s mierou 1,3 na 100 pacientorokov u pacientov liečených placebom. Podiel pacientov, u ktorých došlo ku kardiologickým reakciám (všetkým alebo závažným) sa s opakovanými cyklami nezvyšoval.
Neurologické udalosti
Boli hlásené prípady posteriórneho reverzibilného encefalopatického syndrómu (PRES)/reverzibilného posteriórneho leukoencefalopatického syndrómu (RPLS). Znaky a príznaky zahŕňali poruchu videnia,
bolesť hlavy, záchvaty a zmenený mentálny stav s asociovanou hypertenziou alebo bez nej. Diagnóza
PRES/RPLS sa musí potvrdiť na základe zobrazovacieho vyšetrenia mozgu. V hlásených prípadoch boli potvrdené rizikové faktory pre PRES/RPLS zahŕňajúce základné ochorenie pacienta, hypertenziu,
imunosupresívnu liečbu a/alebo chemoterapiu.
Neutropénia
Pri liečbe rituximabom boli pozorované prípady neutropénie, z ktorých väčšina bola prechodná
a mierna alebo stredne ťažká. K neutropénii môže dôjsť niekoľko mesiacov po podaní rituximabu
(pozri časť 4.4).
V placebom kontrolovaných obdobiach klinických skúšaní sa ťažká neutropénia vyvinula u 0,94 % (13/1 382) pacientov liečených rituximabom a u 0,27 % (2/731) pacientov na placebe.
Po uvedení lieku na trh sa zriedkavo hlásili prípady neutropénie vrátane ťažkej neutropénie s neskorým nástupom a perzistentnej neutropénie, pričom niektoré z nich so smrteľnými infekciami.
Poruchy kože a podkožného tkaniva
Toxická epidermálna nekrolýza (Lyellov syndróm) a Stevensov-Johnsonov syndróm, niektoré so smrteľnými následkami, boli hlásené veľmi zriedkavo.
Abnormality v laboratórnych vyšetreniach
Hypogamaglobulinémia (hladina IgG alebo IgM nižšia ako dolná hranica normálneho rozmedzia) sa pozorovala u pacientov s RA liečených rituximabom. Vplyvom nízkych hladín IgG alebo IgM nedošlo k zvýšeniu miery výskytu celkových infekcií alebo závažných infekcií (pozri časť 4.4).
Malý počet spontánnych hlásení a v literatúre opísaných prípadov hypogamaglobulinémie sa pozoroval u pediatrických pacientov liečených rituximabom, pričom niektoré prípady boli závažné
a vyžadovali dlhodobú imunoglobulínovú substitučnú liečbu. Následky dlhodobého úbytku B-buniek u pediatrických pacientov nie sú známe.
Skúsenosti zgranulomatózyspolyangiitídouamikroskopickejpolyangiitídy
Indukcia remisie
99 pacientov sa liečilo s cieľom indukcie remisie GPA a MPA v klinickom skúšaní s rituximabom
(375 mg/m2, raz týždenne počas 4 týždňov) a s glukokortikoidmi (pozri časť 5.1).
Nežiaduce reakcie na liek uvedené v tabuľke 3 boli všetky nežiaduce reakcie, ku ktorým došlo
s incidenciou ≥ 5 % v skupine s rituximabom a to s vyššou frekvenciou ako v porovnávacej skupine.
Trieda orgánových systémov
Nežiaduce reakcie
|
Rituximab
(
n = 99)
|
Infekcie a nákazy
|
Infekcia močovej sústavy
|
7 %
|
Bronchitída
|
5 %
|
Herpes zoster
|
5 %
|
Nazofaryngitída
|
5 %
|
Poruchy krvi a lymfatického systému
|
Trombocytopénia
|
7 %
|
Poruchy imunitného systému
|
Syndróm uvoľňovania cytokínov
|
5 %
|
Poruchy metabolizmu a výživy
|
Hyperkaliémia
|
5 %
|
Psychické poruchy
|
Nespavosť
|
14 %
|
Poruchy nervového systému
|
Závraty
|
10 %
|
Tras
|
10 %
|
Poruchy ciev
|
Hypertenzia
|
12 %
|
Návaly tepla
|
5 %
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Kašeľ
|
12 %
|
Dyspnoe
|
11 %
|
Epistaxa
|
11 %
|
Upchatý nos
|
6 %
|
Poruchy gastrointestinálneho traktu
|
Hnačka
|
18 %
|
Dyspepsia
|
6 %
|
Zápcha
|
5 %
|
Poruchy kože a podkožného tkaniva
|
Akné
|
7 %
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Svalové kŕče
|
18 %
|
Artralgia
|
15 %
|
Bolesť chrbta
|
10 %
|
Svalová slabosť
|
5 %
|
Bolesti kostí a svalov
|
5 %
|
Bolesť končatín
|
5 %
|
Celkové poruchy a reakcie v mieste podania
|
Periférny edém
|
16 %
|
Laboratórne a funkčné vyšetrenia
|
Znížený hemoglobín
|
6 %
|
|
|
Tabuľka 3 Nežiaduce reakcie na liek, ku ktorým došlo po 6 mesiacoch u ≥ 5 % pacientov liečených rituximabom na indukciu remisie GPA a MPA a to s vyššou frekvenciou ako v porovnávacej skupine.
Udržiavacia liečba
V ďalšej klinickej štúdii sa celkovo 57 pacientov s ťažkými aktívnymi GPA a MPA s ochorením v remisii liečilo rituximabom na udržanie remisie (pozri časť 5.1).
Tried
a orgánových systémov
Nežiaduca reakcia na liek1
|
Rituximab
(
n = 57)
|
Infekci
e a nákazy
|
Bronchitída
|
14 %
|
Nádcha
|
5 %
|
Celkové poruchy a reakcie v mieste podania
|
Pyrexia
|
9 %
|
Ochorenie podobné chrípke
|
5 %
|
Periférny edém
|
5 %
|
Poruch
y gastrointestinálneho traktu
|
Hnačka
|
7 %
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Dyspnoe
|
9 %
|
Úrazy
, otravy a komplikácie liečebného postupu
|
Reakcie súvisiace s infúziou2
|
12 %
|
1 Udalosti sa považovali za nežiaduce reakcie na liek len po dôkladnom vyhodnotení a ak sa dal príčinný vzťah medzi liekom a danou nežiaducou udalosťou aspoň rozumne predpokladať.
2 Podrobnosti o reakciách súvisiacich s infúziou sú uvedené v časti popisujúcej vybrané nežiaduce reakcie
na liek.
|
|
|
Tabuľka 4 Nežiaduce reakcie na liek, ku ktorým došlo u ≥ 5 % pacientov liečených rituximabom v udržiavacej liečbe GPA a MPA a to s vyššou frekvenciou ako v porovnávacej skupine
Celkový bezpečnostný profil bol konzistentný s dobre stanoveným bezpečnostným profilom
rituximabu v schválených autoimunitných indikáciách vrátane GPA/MPA. Celkovo došlo
u 4 % pacientov v skupine s rituximabom k nežiaducim udalostiam vedúcim k ukončeniu liečby. Väčšina nežiaducich udalostí v skupine s rituximabom mala miernu alebo strednú intenzitu.
U žiadneho z pacientov v skupine s rituximabom nedošlo k smrteľným nežiaducim účinkom.
Najčastejšie hlásenými udalosťami považovanými za nežiaduce reakcie na liek boli reakcie súvisiace s infúziou a infekcie.
V dlhodobej pozorovacej bezpečnostnej klinickej štúdii bolo 97 pacientov s GPA/MPA liečených rituximabom (priemerne 8 infúzií [rozsah 1 – 28]) až 4 roky podľa štandardnej praxe a posúdenia lekára. Celkový bezpečnostný profil bol konzistentný s dobre stanoveným bezpečnostným profilom rituximabu pri RA a GPA/MPA a nehlásili sa žiadne nové nežiaduce reakcie na liek.
Opis vybraných nežiaducich reakcií na liekReakcie súvisiace s infúziouV klinickom skúšaní hodnotiacom indukciu remisie pri ťažkej aktívnej GPA a MPA sa IRR definovali ako akákoľvek nežiaduca udalosť, ku ktorej dôjde do 24 hodín po infúzii a skúšajúci ju považuje za
súvisiacu s infúziou v bezpečnostnej populácii. Z 99 pacientov liečených rituximabom, u 12 (12 %)
došlo k najmenej jednej IRR. Všetky IRR boli CTC 1. alebo 2. stupňa. Najčastejšie IRR zahŕňali syndróm uvoľňovania cytokínov, začervenanie, podráždenie hrdla a tras. Rituximab sa podával
v kombinácii s intravenóznymi glukokortikoidmi, ktoré môžu redukovať incidenciu a závažnosť týchto
udalostí.
V klinickom skúšaní s udržiavacou liečbou došlo u 7/57 (12 %) pacientov v rituximabovej skupine k najmenej jednej reakcii súvisiacej s infúziou. Incidencia príznakov IRR bola najvyššia v priebehu podanie prvej infúzie alebo po prvej infúzii (9 %) a znižovala sa s následnými infúziami (< 4 %). Všetky príznaky IRR boli mierne alebo stredne ťažké a väčšina z nich sa patrila do tried orgánových systémov „Poruchy dýchacej sústavy, hrudníka a mediastína“ a „Poruchy kože a podkožného tkaniva“.
InfekcieV klinickom skúšaní s cieľom dosiahnuť indukcie remisie, ktoré zahŕňalo 99 pacientov liečených rituximabom, bola celková miera výskytu infekcie približne 237 na 100 pacientorokov (95 % IS 197 –
285) v prípade 6-mesačného primárneho koncového ukazovateľa. Infekcie boli hlavne mierne až stredne ťažké a pozostávali najmä z infekcií horných dýchacích ciest, herpes zoster a infekcií močovej sústavy. Miera výskytu závažných infekcií bola približne 25 na 100 pacientorokov. Najčastejšie hlásenou závažnou infekciou v rituximabovej skupine bola pneumónia s frekvenciou 4 %.
V klinickom skúšaní s udržiavacou liečbou zaznamenalo 30/57 (53 %) pacientov v rituximabovej skupine infekcie. Incidencia infekcií všetkých stupňov bola v skupinách podobná. Infekcie boli hlavne mierne až stredne ťažké. Najčastejšie infekcie v rituximabovej skupine zahŕňali infekcie horných dýchacích ciest, gastroenteritídu, infekcie močovej sústavy a herpes zoster. Incidencia závažných infekcií bola podobná v oboch skupinách (približne 12 %). Najčastejšie hlásenou závažnou infekciou
v rituximabovej skupine bola mierna alebo stredne ťažká bronchitída.
Malignity
V klinickom skúšaní s cieľom dosiahnuť indukcie remisie bola incidencia malignity u pacientov s granulomatózou s polyangiitídou a mikroskopickou polyangiitídou liečených rituximabom
2,00 na 100 pacientorokov pri uzavretí zberu dát tohto skúšania podľa bežného postupu (keď posledný
pacient dokončil obdobie následného sledovania). Na základe štandardizovaných mier incidencie sa incidencia malignít javí ako podobná s incidenciou, ktorá bola predtým hlásená u pacientov s
s ANCA-asociovanými vaskulitídami.
Kardiovaskulárne nežiaduce reakcie
V klinickom skúšaní s cieľom dosiahnuť indukciu remisie dochádzalo ku kardiovaskulárnym udalostiam v miere približne 273 na 100 pacientorokov (95 % IS 149 – 470) v prípade 6-mesačného primárneho koncového ukazovateľa. Miera výskytu závažných kardiologických udalostí bola 2,1 na
100 pacientorokov (95 % IS 3 – 15). Najčastejšie hlásenými udalosťami boli tachykardia (4 %)
a atriálna fibrilácia (3 %) (pozri časť 4.4).
Neurologické udalosti
Boli hlásené prípady posteriórneho, reverzibilného encefalopatického syndrómu (PRES)/reverzibilného, posteriórneho leukoencefalopatického syndrómu (RPLS) pri autoimunitných stavoch. Znaky a príznaky zahŕňali poruchu videnia, bolesť hlavy, záchvaty a zmenený mentálny stav s asociovanou hypertenziou alebo bez nej. Diagnóza PRES/RPLS sa musí potvrdiť na základe zobrazovacieho vyšetrenia mozgu. V hlásených prípadoch boli potvrdené rizikové faktory
pre PRES/RPLS zahŕňajúce základné ochorenie pacienta, hypertenziu, imunosupresívnu liečbu a/alebo chemoterapiu.
Reaktivácia hepatitídy-B
Malý počet prípadov reaktivácie hepatitídy B, niektoré so smrteľnými následkami, bol hlásený
u pacientov s granulomatózou s polyangiitídou a mikroskopickou polyangiitídou, ktorí boli liečení rituximabom po uvedení lieku na trh.
Hypogamaglobulinémia
Hypogamaglobulinémia (hladina IgA, IgG alebo IgM nižšia ako dolná hranica normálneho rozmedzia)
sa pozorovala u pacientov s granulomatózou s polyangiitídou a mikroskopickou polyangiitídou liečených rituximabom. Miera výskytu celkových infekcií a závažných infekcií sa vplyvom nízkych hladín IgA, IgG alebo IgM nezvyšovala.
V klinickom skúšaní na dosiahnutie indukcie remisie malo v 6. mesiaci 27 %, 58 % a 51 % pacientov v rituximabovej skupine s normálnymi imunoglobulínovými hladinami v úvode znížené hladiny IgA, IgG a IgM, v príslušnom poradí, v porovnaní s 25 %, 50 % a 46 % v cyklofosfamidovej skupine.
V klinickom skúšaní s udržiavacou liečbou sa nepozorovali žiadne klinicky významné rozdiely medzi týmito dvoma liečebnými skupinami alebo pokles celkových hladín imunoglobulínov IgG, IgM alebo IgA v priebehu celého skúšania.
Neutropénia
V klinickom skúšaní s cieľom dosiahnuť indukciu remisie sa u 24 % pacientov v rituximabovej
skupine (jediný cyklus) a u 23 % pacientov v cyklofosfamidovej skupine vyvinula neutropénia 3. alebo vyššieho stupňa CTC. Neutropénia sa nespájala s pozorovaným zvýšením závažných infekcií
u pacientov liečených rituximabom.
V klinickom skúšaní s udržiavacou liečbou bola incidencia neutropénie všetkých stupňov 0 %
u pacientov liečených rituximabom vs 5 % u pacientov liečených azatioprínom.
Poruchy kože a podkožného tkanivaToxická epidermálna nekrolýza (Lyellov syndróm) a Stevensov-Johnsonov syndróm, niektoré so smrteľnými následkami, boli hlásené veľmi zriedkavo.
Skúsenosti zpemphigusvulgarisSúhrn bezpečnostného profiluBezpečnostný profil rituximabu v kombinácii s krátkodobými, nízkodávkovými glukokortikoidmi
v liečbe pacientov s pemphigus vulgaris bol skúmaný v 3. fáze randomizovanej, kontrolovanej, multicentrickej, otvorenej klinickej štúdie u pacientov s pemphigus, ktorá zahŕňala 38 pacientov
s pemphigus vulgaris (PV) randomizovaných do rituximabovej skupiny. Pacienti randomizovaní do rituximabovej skupiny boli najprv liečení 1 000 mg intravenózne v 1. deň a potom ďalšími 1 000 mg intravenózne v 15. deň klinickej štúdie. V 12. a 18. mesiaci sa podávali udržiavacie dávky 500 mg
intravenózne. V čase relapsu pacienti mohli byť liečení 1 000 mg intravenózne (pozri časť 5.1).
Bezpečnostný profil rituximabu u pacientov s PV bol konzistentný s bezpečnostným profilom pozorovaným u pacientov s RA a GPA/MPA.
Tabuľkový zoznam nežiaducich reakciíNežiaduce reakcie na liek uvedené v tabuľke 5 boli nežiaduce udalosti, ktoré sa vyskytli v miere ≥ 5 %
u pacientov s PV liečených rituximabom, s ≥ 2 % absolútneho rozdielu v incidencii medzi skupinou liečenou rituximabom a skupinou liečenou štandardnou dávkou prednizónu až do 24. mesiaca. Žiadny
pacient neprerušil liečbu kvôli nežiaducim reakciám na liek.
Trieda orgánových systémov
Nežiaduca reakcia na liek
| Rituximab + prednizón v nízkej dávke
(n = 38)
| Infekcie a nákazy
| Infekcia herpetickým vírusom
| 8 %
| Herpes zoster
| 5 %
| Orálny herpes
| 5 %
| Konjunktivitída
| 5 %
| Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
| Kožný papilóm
| 5 %
| Psychické poruchy
| Perzistentná depresívna porucha
| 13 %
| Veľká depresívna porucha
| 5 %
| Podráždenosť
| 5 %
| Poruchy nervového systému
| Bolesť hlavy
| 5 %
| Závraty
| 5 %
|
|
|
Tabuľka 5 Nežiaduce reakcie na liek u pacientov s pemphigus vulgaris liečených rituximabom v klinickej štúdii až do 24. mesiaca
Tried
a orgánových systémov
Nežiaduca reakcia na liek
|
Rituxima
b + prednizón v nízkej dávke
(
n = 38)
|
Poruch
y srdca a srdcovej činnosti
|
Tachykardia
|
5 %
|
Poruch
y gastrointestinálneho traktu
|
Bolesť v hornej časti brucha
|
5 %
|
Poruch
y kože a podkožného tkaniva
|
Alopécia
|
13 %
|
Pruritus
|
5 %
|
Žihľavka
|
5 %
|
Porucha kože
|
5 %
|
Poruch
y kostrovej a svalovej sústavy a spojivového tkaniva
|
Bolesti kostí a svalov
|
5 %
|
Celkov
é poruchy a reakcie v mieste podania
|
Únava
|
8 %
|
Pyrexia
|
5 %
|
Úrazy
, otravy a komplikácie liečebného postupu
|
Reakcie súvisiace s infúziou*
|
58 %
|
* Reakcie súvisiace s infúziou zahŕňali príznaky zaznamenané pri ďalšej plánovanej návšteve po každej infúzii a nežiaduce udalosti, ku ktorým došlo v deň podania infúzie alebo jeden deň
po infúzii. Najčastejšie príznaky reakcie súvisiacej s infúziou/preferované výrazy zahŕňali bolesti hlavy, zimnicu, vysoký krvný tlak, nevoľnosť, asténiu a bolesť.
|
Opis vybraných nežiaducich reakcií
Reakcie súvisiace s infúziou
Reakcie súvisiace s infúziou v klinickej štúdii u pacientov s pemphigus vulgaris boli časté (58 %). Takmer všetky reakcie súvisiace s infúziou boli mierne až stredne ťažké. Podiel pacientov, u ktorých došlo k reakcii súvisiacej s infúziou, bol 29 % (11 pacientov) po prvej infúzii, 40 % (15 pacientov) po druhej infúzii, 13 % (5 pacientov) po tretej infúzii a 10 % (4 pacienti) po štvrtej infúzii. Žiaden pacient neprerušil liečbu kvôli reakciám súvisiacim s infúziou. Príznaky reakcií súvisiacich s infúziou boli podobného typu a závažnosti ako príznaky u pacientov s RA a GPA/MPA.
InfekcieU 14 pacientov (37 %) v rituximabovej skupine došlo k infekciám súvisiacim s liečbou v porovnaní s 15 pacientami (42 %) v skupine, ktorej sa podávala štandardná dávka prednizónu. Najčastejšími infekciami v rituximabovej skupine boli infekcie vírusom herpes simplex a herpes zoster, bronchitída, infekcia močovej sústavy, hubová infekcia a konjunktivitída. U troch pacientov (8 %) v rituximabovej skupine došlo celkovo k 5 závažným infekciám (pneumónia vyvolaná
Pneumocystis jirovecii, infekčnej trombóze, medzistavcovej discitíde, pľúcnej infekcii, stafylokokovej sepse) a u jedného pacienta (3 %) v skupine so štandardnou dávkou prednizónu došlo k závažnej infekcii (pneumónia vyvolaná
Pneumocystis jirovecii).
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Z klinických skúšaní u ľudí sú k dispozícii obmedzené skúsenosti s dávkami, ktoré boli vyššie ako schválená dávka pre intravenóznu formu obsahujúcu rituximab. Najvyššia doteraz testovaná intravenózna dávka rituximabu je 5 000 mg (2 250 mg/m2), testovaná v klinickej štúdii so zvyšovaním dávky u pacientov s CLL. Neidentifikovali sa žiadne ďalšie bezpečnostné signály.
U pacientov, ktorí sa predávkovali, sa musí okamžite prerušiť podávanie infúzie a musia sa dôkladne monitorovať.
Po uvedení lieku na trh bolo hlásených päť prípadov predávkovania rituximabom. Tri prípady nehlásili žiadnu nežiaducu udalosť. Dvoma hlásenými nežiaducimi udalosťami boli príznaky podobné chrípke pri dávke 1,8 g rituximabu a respiračné zlyhanie so smrteľnými následkami pri dávke 2 g rituximabu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, monoklonálne protilátky
, ATC kód: L01XC02
Ruxience je biologicky podobný liek. Podrobné informácie sú dostupné na internetovej stránke
Európskej agentúry pre lieky
http://www.ema.europa.eu.Rituximab sa špecificky viaže na transmembránový antigén CD20, neglykozylovaný fosfoproteín,
ktorý sa nachádza na pre-B lymfocytoch a zrelých B lymfocytoch. Tento antigén sa exprimuje na > 95 % všetkých B-buniek non-Hodgkinových lymfómov.
CD20 sa nachádza na normálnych aj na malígnych B-bunkách, ale nie na hematopoetických kmeňových bunkách, pro-B-bunkách, normálnych plazmatických bunkách alebo v iných normálnych tkanivách. Po naviazaní sa protilátky sa tento antigén neinternalizuje a neuvoľnuje sa z bunkového povrchu. CD20 necirkuluje v plazme vo forme voľného antigénu a nedochádza ku kompetícii
o naviazanie protilátky.
Fab doména rituximabu sa viaže na CD20 antigén na povrchu B-lymfocytov a prostredníctvom Fc domény môže byť spustená imunitná efektorová funkcia, ktorá vedie k lýze B-buniek. Možné mechanizmy takto sprostredkovanej bunkovej lýzy zahŕňajú cytotoxicitu závislú od komplementu (CDC), ktorá je výsledkom naviazania sa C1q, a bunkovú cytotoxicitu závislú od protilátky (ADCC) sprostredkovanú jedným alebo viacerými Fcg receptormi na povrchu granulocytov, makrofágov a NK buniek. Preukázalo sa tiež, že naviazanie sa rituximabu na CD20 antigén na B lymfocytoch indukuje bunkovú smrť prostredníctvom apoptózy.
Po podaní prvej dávky rituximabu počty periférnych B-buniek poklesli pod normálnu hodnotu. U pacientov liečených na hematologické malignity obnovovanie B-buniek začalo v priebehu
6 mesiacov po liečbe a vo všeobecnosti dosiahli opäť normálne hodnoty do 12 mesiacov po ukončení liečby, hoci u niektorých pacientov to mohlo trvať dlhšie (až do mediánu času obnovovania
23 mesiacov po indukčnej liečbe). U pacientov s reumatoidnou artritídou sa pozoroval okamžitý úbytok B-buniek v periférnej krvi po dvoch infúziách v dávke 1 000 mg rituximabu s odstupom
14 dní. Počty B-buniek v periférnej krvi sa začínajú zvyšovať od 24. týždňa a dôkazy o repopulácii sa u väčšiny pacientov pozorujú do 40. týždňa, či už sa rituximab podáva ako monoterapia alebo
v kombinácii s metotrexátom. U malého počtu pacientov došlo k dlhšiemu úbytku periférnych B-
buniek trvajúcemu 2 roky alebo dlhšie po poslednej dávke rituximabu. U pacientov s granulomatózou s polyangiitídou alebo mikroskopickou polyangiitídou klesol počet B-buniek v periférnej krvi na
< 10 buniek/μl po dvoch týždňových infúziách rituximabu v dávke 375 mg/m2 a u väčšiny pacientov zostal na tejto úrovni až do 6. mesiaca. Väčšina pacientov (81 %) vykazovala prejavy obnovy B- buniek, s počtom > 10 buniek/μl v 12. mesiaci, a s nárastom na 87 % pacientov v 18. mesiaci.
Klinické skúsenostipri non-Hodgkinovomlymfómea chronickejlymfocytovejleukémie
Folikulárny lymfóm
Monoterapia
Úvodná liečba, 4 dávky raz týždenne
V pivotnom skúšaní bolo 166 pacientov s relapsovaným alebo chemorezistentným B-bunkovým NHL nízkeho stupňa alebo folikulárnym B-bunkovým NHL liečených dávkou 375 mg/m2 rituximabu vo forme intravenóznej infúzie raz týždenne počas štyroch týždňov. Celková miera odpovede (ORR)
v populácii so zámerom liečiť (intent-to-treat, ITT) bola 48 % (IS95% 41 % – 56 %) so 6 % mierou úplnej odpovede (CR) a 42 % mierou čiastočnej odpovede (PR). Predpokladaný medián času
do progresie (TTP) u pacientov s odpoveďou bol 13,0 mesiaca. V analýze podskupín bola ORR vyššia u pacientov s IWF B, C a D histologickými podtypmi ako s IWF A podtypom (58 % vs. 12 %), vyššia
u pacientov, ktorých najväčšia lézia mala v najväčšom priemere < 5 cm vs. > 7 cm (53 % vs. 38 %), a vyššia u pacientov s chemosenzitívnym relapsom v porovnaní s pacientmi s chemorezistentným
relapsom (definovaným ako trvanie odpovede < 3 mesiace) (50 % vs. 22 %). ORR u pacientov, ktorí podstúpili autológnu transplantáciu kostnej drene (ABMT) bola 78 % verzus 43 % u pacientov bez
ABMT. Vek, pohlavie, stupeň lymfómu, začiatočná diagnóza, prítomnosť alebo neprítomnosť rozsiahlej nádorovej masy, normálna alebo vysoká LDH alebo prítomnosť extranodálneho ochorenia nemali štatisticky signifikantný vplyv (Fisherov exaktný test) na odpoveď na rituximab. Štatisticky
signifikantná korelácia sa zaznamenala v miere odpovede u pacientov s infiltráciou kostnej drene. Odpovedalo 40 % pacientov s infiltráciou kostnej drene v porovnaní s 59 % pacientov bez infiltrácie
kostnej drene (p = 0,0186). Toto zistenie nebolo podporené postupnou logistickou regresnou analýzou, pri ktorej sa identifikovali nasledovné faktory ako prognostické faktory: histologický typ, východisková bcl-2 pozitivita, rezistencia voči poslednej chemoterapii a rozsiahla nádorová masa.
Úvodná liečba, 8 dávok raz týždenne
V multicentrickom klinickom skúšaní s jedným ramenom bolo 37 pacientov s relapsovaným alebo chemorezistentným B-bunkovým NHL nízkeho stupňa alebo folikulárnym B-bunkovým NHL liečených 8 dávkami 375 mg/m2 rituximabu vo forme intravenóznej infúzie raz týždenne. ORR bola
57 % (95 % interval spoľahlivosti (IS); 41 % – 73 %; CR 14 %, PR 43 %) s predpokladaným mediánom TTP 19,4 mesiaca u pacientov s odpoveďou (rozmedzie 5,3 až 38,9 mesiaca).
Úvodná liečba pri rozsiahlej nádorovej mase, 4 dávky raz týždenne
Na základe súhrnných údajov z troch skúšaní, 39 pacientov s relapsovanou alebo chemorezistentnou rozsiahlou nádorovou masou (jedna lézia s priemerom ≥ 10 cm), B-bunkovým NHL nízkeho stupňa alebo folikulárnym B-bunkovým NHL dostalo štyri dávky 375 mg/m2 rituximabu vo forme intravenóznej infúzie raz týždenne. ORR bola 36 % (IS95% 21 % – 51 %; CR 3 %, PR 33 %)
s mediánom TTP 9,6 mesiaca u pacientov s odpoveďou (rozmedzie 4,5 až 26,8 mesiaca).
Opakovaná liečba, 4 dávky raz týždenne
V multicentrickom klinickom skúšaní s jedným ramenom sa 58 pacientov s relapsovaným alebo chemorezistentným B-bunkovým NHL nízkeho stupňa alebo folikulárnym B-bunkovým NHL,
u ktorých sa dosiahla objektívna klinická odpoveď na predchádzajúci cyklus rituximabu, znova liečilo
štyrmi dávkami 375 mg/m2 rituximabu vo forme intravenóznej infúzie raz týždenne. Traja z nich dostali dva cykly rituximabu pred vstupom do klinickej štúdie a v klinickej štúdii dostali tretí cyklus. Dvaja pacienti boli v klinickej štúdii opakovane liečení dvakrát. Pre 60 opakovaných terapií v klinickej
štúdii bola ORR 38 % (IS95% 26 % – 51 %; 10 % CR, 28 % PR) s predpokladaným mediánom TTP
17,8 mesiaca u pacientov s odpoveďou (rozsah 5,4 až 26,6). To je priaznivé v porovnaní s TTP
dosiahnutým po predchádzajúcom cykle rituximabu (12,4 mesiaca).
Úvodná liečba v kombinácii s chemoterapiou
V otvorenom randomizovanom klinickom skúšaní bolo celkovo 322 predtým neliečených pacientov s folikulárnym lymfómom randomizovaných buď na CVP chemoterapiu (cyklofosfamid 750 mg/m2,
každé 3 týždne počas 8 cyklov alebo na rituximab 375 mg/m2 v kombinácii s CVP (R-CVP). Rituximab sa podával v prvý deň každého liečebného cyklu. Celkovo sa liečilo a analyzovalo
z hľadiska účinnosti 321 pacientov (162 R-CVP, 159 CVP). Medián následného sledovania pacientov bol 53 mesiacov. R-CVP viedla k signifikantnému prínosu vo vzťahu k CVP pre primárny koncový
ukazovateľ, t.j. čas do zlyhania liečby (27 mesiacov vs. 6,6 mesiaca, p < 0,0001, log-rank test). Podiel pacientov s odpoveďou nádoru na liečbu (CR, CRu, PR) bol signifikantne vyšší (p < 0,0001
Chi-kvadrát test) v R-CVP skupine (80,9 %) ako v CVP skupine (57,2 %). Liečba s R-CVP signifikantne predlžovala čas do progresie ochorenia alebo do úmrtia v porovnaní s CVP, 33,6 mesiaca v porovnaní s 14,7 mesiaca (p < 0,0001, log-rank test). Medián trvania odpovede bol 37,7 mesiaca
v R-CVP skupine a 13,5 mesiaca v CVP skupine (p < 0,0001, log-rank test).
Rozdiel medzi liečebnými skupinami z hľadiska celkového prežívania preukázal signifikantný klinický rozdiel (p = 0,029, log-rank test stratifikovaný podľa centra): miery prežívania v 53. mesiaci boli
80,9 % u pacientov v R-CVP skupine v porovnaní so 71,1 % u pacientov v CVP skupine.
Aj výsledky z troch ďalších randomizovaných klinických skúšaní s použitím rituximabu v kombinácii s chemoterapeutickým režimom iným ako CVP (CHOP, MCP, CHVP/interferón-α) preukázali signifikantné zlepšenia podielov odpovedí, parametrov závislých od času, ako aj celkového prežívania. Kľúčové výsledky zo všetkých štyroch klinických štúdií sú zhrnuté v tabuľke 6.
Tabuľka 6 Súhrn kľúčových výsledkov zo štyroch randomizovaných klinických štúdií III. fázy hodnotiacich prínos rituximabu v kombinácii s rôznymi chemoterapeutickými režimami pri folikulárnom lymfóme
Klinické štúdia
|
Liečba, N
|
Medián FU, mesiace
|
ORR,
%
|
CR,
%
| Medián
TTF/PFS/EFS
mesiace
| OS
miery,
%
|
M39021
|
CVP, 159
R-CVP, 162
|
53
|
57
81
|
10
41
| Medián TTP:
14,7
33,6
p < 0,0001
| 53-mesiacov
71,1
80,9
p = 0,029
|
GLSG’00
|
CHOP, 205
R-CHOP, 223
|
18
|
90
96
|
17
20
| Medián TTF:
2,6 roka Nedosiahnuté p < 0,001
| 18-mesiacov
90
95
p = 0,016
|
OSHO-39
|
MCP, 96
R-MCP, 105
|
47
|
75
92
|
25
50
|
Medián PFS: 28,8
Nedosiahnuté p < 0,0001
| 48-mesiacov
74
87
p = 0,0096
|
FL2000
| CHVP-IFN,
183
R-CHVP-IFN,
175
|
42
|
85
94
|
49
76
|
Medián EFS: 36
Nedosiahnuté p < 0,0001
| 42-mesiacov
84
91
p = 0,029
|
EFS – Prežívanie bez udalosti
TTP – Čas do progresie ochorenia alebo úmrtia
PFS – Prežívanie bez progresie ochorenia
TTF – Čas do zlyhania liečby
Miery OS – miery celkového prežívania v čase analýz
Udržiavacia liečbaPredtým neliečený folikulárny lymfóm
V prospektívnom, otvorenom, medzinárodnom, multicentrickom klinickom skúšaní III. fázy dostalo
1 193 pacientov s predtým neliečeným pokročilým folikulárnym lymfómom indukčnú liečbu
s R-CHOP (n = 881), R-CVP (n = 268) alebo R-FCM (n = 44), podľa rozhodnutia skúšajúcich. Celkovo na indukčnú liečbu odpovedalo 1 078 pacientov, z ktorých 1 018 bolo randomizovaných
na udržiavaciu liečbu rituximabom (n = 505) alebo na pozorovanie (n = 513). Tieto dve liečebné skupiny boli dobre vyvážené z hľadiska východiskových charakteristík a stavu ochorenia. Udržiavacia
liečba rituximabom pozostávala z jednej infúzie rituximabu v dávke 375 mg/m2 plochy povrchu tela, ktorá sa podávala každé 2 mesiace až do progresie ochorenia alebo maximálne po dobu dva roky.
Vopred špecifikovaná primárna analýza sa uskutočnila s mediánom času pozorovania 25 mesiacov od randomizácie, pričom udržiavacia liečba rituximabom viedla ku klinicky relevantnému a štatisticky signifikantnému zlepšeniu primárneho koncového ukazovateľa, ktorým bolo skúšajúcim hodnotené prežívanie bez progresie (PFS), pri porovnaní s pozorovanou skupinou u pacientov s predtým neliečeným folikulárnym lymfómom (tabuľka 7).
Signifikantný prínos udržiavacej liečby rituximabom sa pozoroval aj pre sekundárne koncové ukazovatele, ktorými boli prežívanie bez udalosti (EFS), čas do nasledujúcej anti-lymfómovej liečby (TNLT), čas do nasledujúcej chemoterapie (TNCT) a celková miera odpovede (ORR) v čase primárnej analýzy (tabuľka 7).
Údaje z predĺženého následného sledovania pacientov v klinickej štúdii (medián následného sledovania 9 rokov) potvrdili dlhodobý prínos udržiavacej liečby rituximabom vo vzťahu k PFS, EFS, TNLT a TNCT (tabuľka 7).
| Primárna analýza
(medián FU: 25 mesiacov)
| Finálna analýza
(medián FU: 9,0 roka)
| Pozorovanie Rituximab n = 513 n = 505
| Pozorovanie Rituximab n = 513 n = 505
| Primárna účinnosť
Prežívanie bez progresie (medián)
log-rank p-hodnota pomer rizík (95 % IS)
redukcia rizika
|
NR NR
< 0,0001
0,50 (0,39; 0,64)
50 %
|
4,06 roka 10,49 roka
< 0,0001
0,61 (0,52; 0,73)
39 %
| Sekundárna účinnosť Celkové prežívanie (medián) log-rank p-hodnota
pomer rizík (95 % IS)
redukcia rizika
|
NR NR
0,7246
0,89 (0,45; 1,74)
11 %
|
NR NR
0,7948
1,04 (0,77; 1,40)
-6 %
| Prežívanie bez udalosti (medián)
log-rank p-hodnota pomer rizík (95 % IS)
redukcia rizika
| 38 mesiacov NR
< 0,0001
0,54 (0,43; 0,69)
46 %
| 4,04 roka 9,25 roka
< 0,0001
0,64 (0,54; 0,76)
36 %
| TNLT (medián)
log-rank p-hodnota pomer rizík (95 % IS)
redukcia rizika
| NR NR
0,0003
0,61 (0,46; 0,80)
39 %
| 6,11 roka NR
< 0,0001
0,66 (0,55; 0,78)
34 %
| TNCT (medián)
log-rank p-hodnota pomer rizík (95 % IS)
redukcia rizika
| NR NR
0,0011
0,60 (0,44; 0,82)
40 %
| 9,32 roka NR
0,0004
0,71 (0,59; 0,86)
39 %
| Celková miera odpovede*
p-hodnota chi-kvadrát testu pomer šancí (95 % IS)
| 55 % 74 %
< 0,0001
2,33 (1,73; 3,15)
| 61 % 79 %
< 0,0001
2,43 (1,84; 3,22)
| Miera úplnej odpovede (CR/CRu)*
p-hodnota chi-kvadrát testu pomer šancí (95 % IS)
| 48 % 67 %
< 0,0001
2,21 (1,65; 2,94)
| 53 % 67 %
< 0,0001
2,34 (1,80; 3,03)
|
|
|
Tabuľka 7 Prehľad výsledkov účinnosti s udržiavacou liečbou rituximabom vs. pozorovanie pri primárnej analýze definovanej v protokole a po 9-ročnom mediáne následného sledovania (finálna analýza)* na konci udržiavacej liečby/pozorovania; výsledok finálnej analýzy je založený na mediáne následného sledovania
73 mesiacov.
FU: následné sledovanie; NR: nedosiahnuté v dobe ukončenia zberu klinických údajov, TNCT: čas do nasledujúcej chemoterapeutickej liečby; TNLT: čas do nasledujúcej anti-lymfómovej liečby.
Udržiavacia liečba rituximabom poskytla konzistentný prínos vo všetkých hodnotených
vopred definovaných podskupinách: pohlavie (mužské, ženské), vek (< 60 rokov, >= 60 rokov), skóre
FLIPI (<= 1, 2 alebo >= 3), indukčná liečba (R-CHOP, R-CVP alebo R-FCM) a bez ohľadu na kvalitu odpovede na indukčnú liečbu (CR, CRu alebo PR). Exploratívne analýzy prínosu udržiavacej liečby
preukázali menej výrazný účinok u starších pacientov (vo veku > 70 rokov), avšak hodnotená vzorka
bola malá.
Relapsovaný/refraktérny folikulárny lymfómV prospektívnom, otvorenom, medzinárodnom, multicentrickom klinickom skúšaní III. fázy bolo
465 pacientov s relapsovaným/refraktérnym folikulárnym lymfómom randomizovaných v prvom kroku na indukčnú liečbu buď CHOP (cyklofosfamid, doxorubícín, vinkristín, prednizolón; n = 231) alebo rituximabom plus CHOP (R-CHOP, n = 234). Tieto dve liečebné skupiny boli dobre vyvážené
z hľadiska východiskových charakteristík a stavu ochorenia. Celkovo bolo 334 pacientov, u ktorých sa dosiahla úplná alebo čiastočná remisia po indukčnej liečbe, randomizovaných v druhom kroku
na udržiavaciu liečbu rituximabom (n = 167) alebo na pozorovanie (n = 167). Udržiavacia liečba rituximabom pozostávala z jednej infúzie rituximabu 375 mg/m2 plochy povrchu tela, ktorá sa podávala každé 3 mesiace až do progresie ochorenia alebo maximálne po dobu dva roky.
Finálna analýza účinnosti zahŕňala všetkých pacientov randomizovaných do oboch častí štúdie.
Po mediáne času pozorovania 31 mesiacov pre pacientov randomizovaných na indukčnú fázu liečby, R-CHOP signifikantne zlepšila výsledky pacientov s relapsovaným/refraktérnym folikulárnym lymfómom, keď sa porovnávali s výsledkami CHOP (pozri tabuľku 8).
| CHOP
| R-CHOP
| p-hodnota
| Redukcia rizika1)
| Primárna účinnosť
ORR2)
CR2)
PR2)
|
74 %
16 %
58 %
|
87 %
29 %
58 %
|
0,0003
0,0005
0,9449
|
Na Na Na
|
|
|
Tabuľka 8 Indukčná fáza: prehľad výsledkov účinnosti pre CHOP vs. R-
CHOP (medián času pozorovania 31 mesiacov)1) Odhady sa vypočítavali podľa pomeru rizík
2) Posledná odpoveď nádoru na liečbu vyhodnocovaná skúšajúcim. „Primárnym“ štatistickým testom pre „odpoveď“ bol použitý test trendu CR verzus PR verzus žiadna odpoveď (p < 0,0001)
Skratky: NA, nedostupné; ORR: celková miera odpovede; CR: úplná odpoveď; PR: čiastočná odpoveď
U pacientov randomizovaných na udržiavaciu liečbu bol medián času pozorovania 28 mesiacov od randomizácie do udržiavacej liečby. Udržiavacia liečba rituximabom viedla ku klinicky relevantnému a štatisticky signifikantnému zlepšeniu primárneho koncového ukazovateľa, PFS, (čas od randomizácie na udržiavaciu liečbu do relapsu, progresia ochorenia alebo smrti) v porovnaní len
s pozorovaním (p < 0,0001 log-rank test). Medián PFS bol 42,2 mesiaca v rituximabovej udržiavacej skupine v porovnaní so 14,3 mesiaca v pozorovacej skupine. Použitím cox modelu regresnej analýzy
sa riziko progresie ochorenia alebo úmrtia redukovalo o 61 % udržiavacou liečbou s rituximab
v porovnaní s pozorovaním (95 % IS; 45 % – 72 %). Odhadované miery odpovedí bez progresie na základe výpočtu podľa Kaplanovej-Meierovej metódy v 12. mesiaci boli 78 % v skupine
s udržiavacou liečbou rituximabom vs. 57 % v pozorovacej skupine. Analýza celkového prežívania
potvrdila signifikantný prínos udržiavacej liečby rituximabom v porovnaní s pozorovaním (p = 0,0039
log-rank test). Udržiavacia liečba rituximabom redukovala riziko úmrtia o 56 % (95 % IS; 22 % –
75 %).
Tabuľka 9 Udržiavacia fáza: prehľad výsledkov účinnosti pre rituximab vs. pozorovanie
(medián času pozorovania 28 mesiacov)
Parameter účinnosti
|
Medián času do udalosti na základe odhadu
podľa Kaplanovej-Meierovej metódy (mesiace)
|
Redukcia
rizika
|
Pozorovanie
(
n = 167)
|
Rituximab
(
n = 167)
|
Log-rank
p-hodnota
|
Prežívanie bez progresie
(PFS)
|
14,3
|
42,2
|
< 0,0001
|
61 %
|
Celkové prežívanie
|
NR
|
NR
|
0,0039
|
56 %
|
Čas do novej liečby lymfómu
|
20,1
|
38,8
|
< 0,0001
|
50 %
|
Prežívanie bez ochoreniaa
|
16,5
|
53,7
|
0,0003
|
67 %
|
Analýza podskupín
PFS
CHOP R-CHOP CR
PR OS
CHOP R-CHOP
|
11,6
22,1
14,3
14,3
NR NR
|
37,5
51,9
52,8
37,8
NR NR
|
< 0,0001
0,0071
0,0008
< 0,0001
0,0348
0,0482
|
71 %
46 %
64 %
54 %
55 %
56 %
|
NR: nedosiahnuté; a: aplikovateľné len u pacientov, ktorí dosiahli CR
Prínos udržiavacej liečby rituximabom sa potvrdil vo všetkých analyzovaných podskupinách,
bez ohľadu na indukčný režim (CHOP alebo R-CHOP) alebo kvalitu odpovede na indukčnú liečbu
(CR alebo PR) (tabuľka 9). Udržiavacia liečba rituximabom signifikantne predlžovala medián PFS
u pacientov odpovedajúcich na CHOP indukčnú liečbu (medián PFS 37,5 mesiaca vs. 11,6 mesiaca, p < 0,0001), ako aj u pacientov, ktorí odpovedali na R-CHOP indukčnú liečbu (medián PFS
51,9 mesiaca vs. 22,1 mesiaca, p = 0,0071). Hoci boli podskupiny malé, rituximabová udržiavacia liečba poskytovala signifikatný prínos vo vzťahu k celkovému prežívaniu u pacientov odpovedajúcich na CHOP, aj u pacientov odpovedajúcich na R-CHOP, hoci na potvrdenie tohto pozorovania je
potrebné dlhšie obdobie následného sledovania.
Difúzny veľkobunkový Non-Hodgkinov B-lymfómV randomizovanom, otvorenom klinickom skúšaní bolo celkovo 399 predtým neliečených starších
pacientov (vo veku 60 až 80 rokov) s difúznym veľkobunkovým lymfómom z B-buniek liečených štandardnou CHOP chemoterapiu (cyklofosfamid 750 mg/m2, doxorubicín 50 mg/m2, vinkristín
1,4 mg/m2 až do maximálnej dávky 2 mg v 1. deň a prednizolón 40 mg/m2/deň v 1. – 5. deň) každé
3 týždne počas ôsmich cyklov alebo rituximabom 375 mg/m2 plus CHOP (R-CHOP). Rituximab sa podával v prvý deň tohto liečebného cyklu.'
Finálna analýza účinnosti zahŕňala všetkých randomizovaných pacientov (197 CHOP, 202 R-CHOP) a mala medián trvania následného sledovania približne 31 mesiacov. Tieto dve liečebné skupiny boli dobre vyvážené z hľadiska východiskových charakteristík ochorenia a stavu ochorenia. Finálna analýza potvrdila, že R-CHOP liečba sa spájala s klinicky relevantným a štatisticky signifikantným zlepšením trvania prežívania bez udalosti (parameter primárnej účinnosti, pričom udalosťami boli úmrtie, relaps alebo progresia lymfómu alebo zavedenie novej anti-lymfómovej liečby) (p = 0,0001). Medián trvania prežívania bez udalosti na základe odhadu podľa Kaplanovej-Meierovej metódy bol
35 mesiacov v R-CHOP skupine v porovnaní s 13 mesiacmi v CHOP skupine, čo predstavuje redukciu rizika o 41 %. V 24. mesiaci boli odhady pre celkové prežívanie 68,2 % v R-CHOP skupine
v porovnaní s 57,4 % v CHOP skupine. Následná analýza trvania celkového prežívania sa uskutočnila s mediánom trvania následného sledovania 60 mesiacov, pričom potvrdila prínos R-CHOP liečby
oproti CHOP liečbe (p = 0,0071) vyjadrený znížením rizika o 32 %.
Analýza všetkých sekundárnych parametrov (miery odpovede, prežívanie bez progresie, prežívanie bez ochorenia, trvanie odpovede) verifikovala liečebný účinok R-CHOP v porovnaní s CHOP. Miera
kompletnej (úplnej) odpovede na liečbu po 8. cykle bola 76,2 % v R-CHOP skupine a 62,4 %
v CHOP skupine (p = 0,0028). Riziko progresie ochorenia sa zredukovalo o 46 % a riziko relapsu
o 51 %. Vo všetkých podskupinách pacientov (pohlavie, vek, IPI upravené podľa veku, Ann Arbor stupeň, ECOG, β2 mikroglobulín, LDH, albumín, B príznaky, rozsiahla nádorová masa, extranodálne
postihnutie, postihnutie kostnej drene) boli miery rizika pre prežívanie bez udalosti a celkové prežívanie (R-CHOP v porovnaní s CHOP) nižšie ako 0,83, respektíve 0,95. R-CHOP sa spájala so
zlepšením výsledkov liečby u vysokorizikových aj u nízkorizikových pacientov podľa IPI upraveného podľa veku.
Klinické laboratórne nálezyZo 67 pacientov hodnotených na prítomnosť ľudskej protilátky proti myším antigénom (HAMA) sa
u žiadneho nezaznamenala reakcia. Z 356 pacientov hodnotených na prítomnosť protilátok proti lieku
(ADA) bolo pozitívnych 1,1 % (4 pacienti).
Chronická lymfocytová leukémiaV dvoch otvorených randomizovaných klinických skúšaniach bolo celkovo 817 predtým neliečených
pacientov a 552 pacientov s relapsovanou/refraktérnou CLL randomizovaných na liečbu buď FC
chemoterapiu (fludarabín 25 mg/m2, cyklofosfamid 250 mg/m2, 1. – 3. deň) každé 4 týždne počas 6 cyklov alebo rituximabom v kombinácii s FC (R-FC). Rituximab bol podávaný v dávke
375 mg/m2 v priebehu prvého cyklu jeden deň pred chemoterapiou a v dávke 500 mg/m2 v 1. deň každého nasledujúceho liečebného cyklu. Pacienti s relapsovanou/refraktérnou CLL boli vylúčení zo
štúdie, ak boli predtým liečení monoklonálnymi protilátkami alebo boli refraktérni (definované ako zlyhanie dosiahnutia čiastočnej remisie na dobu najmenej 6 mesiacov) na fludarabín alebo akýkoľvek nukleozidový analóg. Účinnosť sa analyzovala celkovo u 810 pacientov (403 R-FC, 407 FC) v prvej
línii liečby (tabuľka 10a a tabuľka 10b) a u 552 pacientov (276 R-FC, 276 FC) u pacientov s relapsovanou/refraktérnou CLL (tabuľka 11).
V klinickej štúdii v prvej línii liečby pri mediáne času pozorovania 48,1 mesiaca bol medián PFS
55 mesiacov v R-FC skupine a 33 mesiacov v FC skupine (p < 0,0001, log-rank test). Analýza celkového prežívania preukázala signifikantný prínos R-FC liečby v porovnaní so samotnou FC
chemoterapiou (p = 0,0319, log-rank test) (tabuľka 10a). Prínos vo vzťahu k PFS sa konzistentne
pozoroval vo väčšine podskupín pacientov analyzovaných podľa rizika ochorenia v úvode (tzn. Binet štádiá A – C) (tabuľka 10b).
Tabuľka 10a Prvá línia liečby chronickej lymfocytovej leukémieParameter účinnosti
| Medián času do udalosti na základe odhadu
podľa Kaplanovej-Meierovej metódy
(mesiace)
| Redukcia
rizika
| FC
(n = 409)
| R-FC
(n = 408)
| Log-rank
p-hodnota
| Prežívanie bez progresie (PFS)
| 32,8
| 55,3
| < 0,0001
| 45 %
| Celkové prežívanie
| NR
| NR
| 0,0319
| 27 %
| Prežívanie bez udalosti
| 31,3
| 51,8
| < 0,0001
| 44 %
| Miera odpovede (CR, nPR alebo
PR)
Miery CR
| 72,6 %
16,9 %
| 85,8 %
36,0 %
| < 0,0001
< 0,0001
| n.a. n.a.
| Trvanie odpovede*
| 36,2
| 57,3
| < 0,0001
| 44 %
| Prežívanie bez ochorenia (DFS)**
| 48,9
| 60,3
| 0,0520
| 31 %
| Čas do novej liečby
| 47,2
| 69,7
| < 0,0001
| 42 %
|
|
|
Prehľad výsledkov účinnosti rituximabu plus FC vs. FC v monoterapii -
medián času pozorovania 48,1 mesiacaMiera odpovede na liečbu a miery CR sa analyzovali s použitím Chi-kvadrát testu. NR: nedosiahnuté; n.a.: neaplikovateľné
*: aplikovateľné len u pacientov, ktorí dosiahli CR, nPR, PR
**: aplikovateľné len u pacientov, ktorí dosiahli CR
Tabuľka 10b Prvá línia liečby chronickej lymfocytovej leukémie
 Pomer rizík prežívania bez progresie podľa Binet štádia (ITT) – medián času pozorovania 48,1 mesiaca
Pomer rizík prežívania bez progresie podľa Binet štádia (ITT) – medián času pozorovania 48,1 mesiaca
Prežívanie bez progresie (PFS) Počet pacientov Pomer rizík
(95 % IS)
p-hodnota
(Waldov test,
|
FC
|
R-FC
|
|
neupravené)
|
Binet štádium A
|
22
|
18
|
0,39 (0,15; 0,98)
|
0,0442
|
Binet štádium B
|
259
|
263
|
0,52 (0,41; 0,66)
|
< 0,0001
|
Binet štádium C
|
126
|
126
|
0,68 (0,49; 0,95)
|
0,0224
|
IS: Interval spoľahlivosti
|
|
|
|
|
V klinickej štúdii u pacientov s relapsovanou/refraktérnou CLL bol medián prežívania bez progresie
(primárny koncový ukazovateľ) 30,6 mesiaca v R-FC skupine a 20,6 mesiaca v FC skupine
(p = 0,0002, log-rank test). Prínos vo vzťahu k PFS sa pozoroval v takmer všetkých podskupinách pacientov analyzovaných podľa rizika ochorenia v úvode. Mierne, ale nie signifikantné zlepšenie celkového prežívania sa hlásilo v R-FC skupine v porovnaní s FC skupinou.
Tabuľka 11 Prehľad výsledkov účinnosti pri relapsovanej/refraktérnej chronickej lymfocytovej leukémii pre rituximab plus FC vs. FC v monoterapii (medián času pozorovania 25,3 mesiaca)Parameter účinnosti
| Medián času do udalosti na základe odhadu podľa Kaplanovej-Meierovej metódy (mesiace)
| Redukcia rizika
|
FC
(n = 276)
| R-FC (n = 276)
| Log-rank p-hodnota
|
Prežívanie bez progresie (PFS)
| 20,6
| 30,6
| 0,0002
| 35 %
|
Celkové prežívanie
| 51,9
| NR
| 0,2874
| 17 %
|
Prežívanie bez udalosti
| 19,3
| 28,7
| 0,0002
| 36 %
|
Miera odpovede (CR, nPR alebo
PR)
Miery CR Trvanie odpovede*
Prežívanie bez ochorenia (DFS)**
Čas do novej liečby CLL
| 58,0 %
13,0 %
27,6
42,2
34,2
| 69,9 %
24,3 %
39,6
39,6
NR
| 0,0034
0,0007
0,0252
0,8842
0,0024
| n.a.
n.a.
31 %
-6 %
35 %
|
Miera odpovede na liečbu a miery CR sa analyzovali s použitím Chi-
kvadrát testu.*: aplikovateľné len u pacientov, ktorí dosiahli CR, nPR, PR; NR: nedosiahnuté n.a. neaplikovateľné
**: aplikovateľné len u pacientov, ktorí dosiahli CR;
Výsledky z ďalších podporných klinických štúdií používajúcich rituximab v kombinácii s inými chemoterapeutickými režimami (vrátane CHOP, FCM, PC, PCM, bendamustínu a kladribínu)
na liečbu predtým neliečených pacientov a/alebo pacientov s relapsovanou/refraktérnou CLL tiež preukázali vysoké celkové miery odpovede s prínosom vo vzťahu k mieram PFS, hoci s mierne
zvýšenou toxicitou (najmä myelotoxicitou). Tieto štúdie podporujú použitie rituximabu s akoukoľvek chemoterapiou.
Údaje od približne 180 pacientov predtým liečených rituximabom preukázali klinický prínos (vrátane
CR) a podporujú opakovanú liečbu rituximabom.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s rituximabom vo
všetkých podskupinách pediatrickej populácie s folikulárnym lymfómom a CLL. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
Klinické skúsenostipri reumatoidnejartritídeÚčinnosť a bezpečnosť rituximabu na zmiernenie znakov a príznakov reumatoidnej artritídy
u pacientov s nedostatočnou odpoveďou na inhibítory TNF sa preukázala v pivotnom, randomizovanom, kontrolovanom, dvojito zaslepenom, multicentrickom klinickom skúšaní
(skúšanie 1).
V skúšaní 1 bolo hodnotených 517 pacientov, ktorí nedostatočne odpovedali alebo netolerovali jeden alebo viaceré inhibítory TNF. Vhodní pacienti mali aktívnu reumatoidnú artritídu diagnostikovanú podľa kritérií Americkej reumatologickej akadémie (American College of Rheumatology, ACR). Rituximab sa podával vo forme dvoch intravenóznych infúzií s odstupom 15 dní. Pacienti boli liečení
2 x 1 000 mg intravenóznymi infúziami rituximabu alebo placeba v kombinácii s MTX. Všetci pacienti súbežne dostávali 60 mg perorálneho prednizónu v 2. – 7. deň a 30 mg v 8. – 14. deň po prvej infúzii. Primárnym koncovým ukazovateľom bol podiel pacientov, ktorí dosiahli odpoveď ACR20
v 24. týždni. U pacientov sa sledovali dlhodobé koncové ukazovatele viac ako 24 týždňov, pričom zahŕňali rádiografické hodnotenie po 56 týždňoch a 104 týždňoch. V tomto čase bolo 81 % pacientov
z pôvodnej placebo skupiny liečených rituximabom v 24. až 56. týždni v rámci protokolu nezaslepeného predĺženia klinickej štúdie.
Klinické skúšania s rituximabom u pacientov s včasnou reumatoidnou artritídou (pacienti bez predchádzajúcej liečby metotrexátom a pacienti s nedostatočnou odpoveďou na metotrexát, ale ešte neliečení inhibítormi TNF-alfa) dosiahli primárne koncové ukazovatele. Rituximab nie je indikovaný na liečbu týchto pacientov, pretože bezpečnostné údaje o dlhodobej liečbe rituximabom nie sú dostatočné, najmä pokiaľ ide o riziko vývoja malignít a PML.
Výsledky z hľadiska aktivity ochoreniaRituximab v kombinácii s metotrexátom signifikantne zvyšoval podiel pacientov, ktorí dosiahli najmenej 20 % zlepšenie ACR skóre v porovnaní s pacientmi liečenými len metotrexátom (tabuľka 12). Vo všetkých klinických štúdiách v rámci klinického vývoja lieku bol prínos liečby
u pacientov podobný nezávisle od veku, pohlavia, plochy povrchu tela, rasy, počtu predchádzajúcich terapií alebo stavu ochorenia.
Klinicky a štatisticky signifikantné zlepšenie sa zaznamenalo aj pre všetky individuálne zložky odpovede ACR (počet citlivých a opuchnutých kĺbov, celkové hodnotenie stavu ochorenia pacientom a lekárom, skóre poškodenia kĺbových funkcií (HAQ), hodnotenie stupňa bolesti a C-reaktívny proteín (mg/dl).
| Výsledok†
| Placebo + MTX
| Rituximab + MTX (2 x 1 000 mg)
| 1. skúšanie
|
| n = 201
| n = 298
|
| ACR20
ACR50
ACR70
| 36 (18 %)
11 (5 %)
3 (1 %)
| 153 (51 %)***
80 (27 %)***
37 (12 %)***
|
| EULAR odpoveď
(dobrá/stredná)
|
44 (22 %)
|
193 (65 %)***
|
| Priemerná zmena v DAS
| -0,34
| -1,83***
|
|
|
Tabuľka 12 Výsledky klinických odpovedí v čase hodnotenia primárneho koncového ukazovateľa v skúšaní 1 (populácia ITT)† Výsledok v 24. týždni
Signifikantný rozdiel oproti placebu + MTX v čase hodnotenia primárneho koncového ukazovateľa: ***p ≤ 0,0001
Pacienti liečení rituximabom v kombinácii s metotrexátom mali signifikantne väčšiu redukciu skóre aktivity ochorenia (DAS28) ako pacienti liečení len metotrexátom (tabuľka 12). Podobne, signifikantne viac pacientov liečených rituximabom v kombinácii s metotrexátom ako pacientov liečených metotrexátom v monoterapii dosiahlo dobrú až strednú odpoveď EULAR (Európska liga proti reumatizmu, European League Against Rheumatism) (tabuľka 12).
Rádiografická odpoveď
Štrukturálne poškodenie kĺbov sa hodnotilo na základe rádiografického vyšetrenia a bolo vyjadrené ako zmena v modifikovanom celkovom Sharpovom skóre (mTSS) a v jeho zložkách, skóre erózií
a skóre zúženia kĺbovej štrbiny.
V skúšaní 1 u pacientov s nedostatočnou odpoveďou na jeden alebo viac TNF inhibítorov alebo u pacientov, ktorí netolerovali liečbu jedným alebo viacerými TNF inhibítormi, ktorí boli liečení rituximabom v kombinácii s metotrexátom, sa v 56. týždni preukázala signifikantne nižšia rádiografická progresia ako u pacientov s metotrexátom. Z pacientov, ktorí boli pôvodne liečení len metotrexátom do 56. týždňa, bolo 81 % liečených rituximabom buď ako záchrannou liečbou v 16. –
24. týždni alebo v predĺžení skúšania. Väčší podielu pacientov liečených pôvodnou liečbou rituximabom a MTX nemal žiadnu erozívnu progresiu v priebehu 56 týždňov (tabuľka 13).
Tabuľka 13 Rádiografické výsledky po 1 roku (populácia mITT)
| Placebo + MTX
| Rituximab + MTX
2 x 1 000 mg
|
Skúšanie 1
| (n = 184)
| (n = 273)
|
Priemerná zmena oproti východiskovej hodnote: Modifikované celkové Sharpovo skóre
Skóre erózií
Skóre zúženia kĺbovej štrbiny
Podiel pacientov bez rádiografickej progresie
Podiel pacientov bez erozívnej zmeny
|
2,30
1,32
0,98
46 %
52 %
|
1,01*
0,60*
0,41**
53 %, NS
60 %, NS
|
150 pacientov pôvodne randomizovaných na placebo + MTX v skúšaní 1 bolo liečených najmenej jedným cyklom
RTX + MTX do jedného roka
* p < 0,05, ** p < 0,001. Skratka: NS, nesignifikantné
Dlhodobo sa pozorovala aj inhibícia rýchlosti progresie poškodenia kĺbov. Rádiografická analýza
v 2. roku v skúšaní 1 preukázala signifikantne redukovanú progresiu štrukturálneho poškodenia kĺbov u pacientov liečených rituximabom v kombinácii s metotrexátom v porovnaní s metotrexátom, ako aj signifikantne vyšší podiel pacientov bez progresie štrukturálneho poškodenia kĺbov počas 2-ročného obdobia.
Výsledky z hľadiska fyzických funkcií a kvality životaSignifikantné redukcie indexu invalidity podľa dotazníka na hodnotenie poškodenia kĺbovej funkcie
(HAQ-DI) a skóre únavy (FACIT-Fatigue) sa pozorovali u pacientov liečených rituximabom
v kombinácii s metotrexátom v porovnaní s pacientmi liečenými metotrexátom. Podiely pacientov liečených rituximabom vykazujúcich minimálny klinicky významný rozdiel (MCID) v HAQ-DI
(definovaný ako pokles individuálneho celkového skóre o > 0,22) bol tiež vyšší ako u pacientov liečených metotrexátom (tabuľka 14).
Preukázalo sa aj signifikantné zlepšenie kvality života súvisiacej so zdravím aj so signifikantným zlepšením skóre fyzického zdravia (PHS) askóre mentálneho zdravia (MHS) na základe SF-36. Okrem toho signifikantne vyšší podiel pacientov dosiahol MCID pre tieto skóre (tabuľka 14).
Tabuľka 14 Výsledky pre funkčné schopnosti a kvalitu života v 24. týždni skúšania 1
Výsledok†
|
Placebo + MTX
|
Rituximab + MTX
(
2 x 1 000 mg)
|
Priemerná zmena HAQ-DI
% MCID pre HAQ-DI Priemerná zmena FACIT-T
Priemerná zmena PHS na základe SF-36
% MCID pre PHS na základe SF-36
Priemerná zmena MHS na základe SF-36
% MCID pre PHS na základe SF-36
|
n = 201
0,1
20 %
-0,5
n = 197
0,9
13 %
1,3
20 %
|
n = 298
-0,4***
51 %
-9,1***
n = 294
5,8***
48 %***
4,7**
38 %*
|
† Výsledok v 24. týždni
Signifikantný rozdiel oproti placebu v dobe hodnotenia primárneho koncového ukazovateľa: * p < 0,05, **p < 0,001,
***p ≤ 0,0001
MCID pre HAQ-DI ≥ 0,22, MCID pre PHS na základe SF-36 > 5,42, MCID pre MHS na základe SF-36 > 6,33
Účinnosť u pacientov séropozitívnych na autoprotilátku (RF a/alebo anti-
CCP)Pacienti séropozitívni na reumatoidný faktor (RF) a/alebo protilátky proti cyklickému citrulinovanému peptidu (anti-CCP), ktorí sa liečili rituximabom v kombinácii s metotrexátom, vykazovali lepšiu
odpoveď v porovnaní s pacientmi negatívnymi pre obidve autoprotilátky.
Výsledky účinnosti u pacientov liečených rituximabom sa analyzovali na základe hodnoty autoprotilátok pred začiatkom liečby. V 24. týždni pacienti séropozitívni na RF a/alebo anti-CCP v úvode mali signifikantne vyššiu pravdepodobnosť dosiahnutia ACR20 a ACR50 odpovedí v porovnaní so séronegatívnymi pacientmi (p = 0,0312 a p = 0,0096) (tabuľka 15). Tieto zistenia sa replikovali
v 48. týždni, keď séropozitivita na autoprotilátku tiež signifikantne zvyšovala pravdepodobnosť dosiahnutia odpovede ACR70. V 48. týždni mali séropozitívni pacienti 2 – 3-krát vyššiu
pravdepodobnosť dosiahnutia ACR odpovedí ako séronegatívni pacienti. U séropozitívnych pacientov
tiež došlo k signifikantne výraznejšiemu poklesu DAS28-FW ako u séronegatívnych pacientov
(obrázok 1).
Tabuľka 15 Súhrn účinnosti podľa stavu autoprotilátok v úvode
| 24. týždeň
| 48. týždeň
|
| Séropozitívni
(n = 514)
| Séronegatívni
(n = 106)
| Séropozitívni
(n = 506)
| Séronegatívni
(n = 101)
|
ACR20 (%)
| 62,3*
| 50,9
| 71,1*
| 51,5
|
ACR50 (%)
| 32,7*
| 19,8
| 44,9**
| 22,8
|
ACR70 (%)
| 12,1
| 5,7
| 20,9*
| 6,9
|
EULAR odpoveď (%)
| 74,8*
| 62,9
| 84,3*
| 72,3
|
Priemerná zmena
DAS28-FW
| -1,97**
| -1,50
| -2,48***
| -1,72
|
Úrovne signifikantnosti sú definované ako * p < 0,05, **p < 0,001, ***p < 0,0001.

 Obrázok 1: Zmena DAS28
Obrázok 1: Zmena DAS28-
FW oproti východiskovej hodnote podľa východiskového stavu autoprotilátok0,0-0,2-0,4-0,6 -0,8-1,0-1,2-1,4-1,6-1,8-2,0-2,2-2,4-2,6-2,8-3,0
-0,8-1,0-1,2-1,4-1,6-1,8-2,0-2,2-2,4-2,6-2,8-3,0Týždeň
| Týždeň
| Týždeň
| Týždeň
| Týždeň
| Týždeň
| Týždeň
|
4
| 8
| 16
| 24
| 32
| 40
| 48
|
Anti-CCP +ve a/alebo RF +ve (n = 562) Anti-CCP -ve a RF -ve (n = 116)Dlhodobá účinnosť pri liečbe s opakovanými cyklamiLiečba rituximabom v kombinácii s metotrexátom pri opakovaných cykloch viedla k pretrvávaniu zlepšení klinických znakov a príznakov RA ako vyplýva z odpovedí ACR, DAS28-FW a EULAR,
ktoré bolo preukázané vo všetkých hodnotených populáciách pacientov (obrázok 2). Pozorovalo sa
pretrvávanie zlepšenia fyzických funkcií na základe HAQ-DI skóre a podielov pacientov dosahujúcich MCID pre HAQ-DI.
Obrázok 2: ACR odpovede pre 4 liečebné cykly (24 týždňov po každom cykle (na pacienta, a návštevu)) u pacientov s nedostatočnou odpoveďou na inhibítory TNF-
(n = 146)ACR20ACR50ACR701.
| 2.
| 3.
| 4.
|
cyklus
| cyklus
| cyklus
| cyklus
|
 Klinické laboratórne nálezy
Klinické laboratórne nálezyCelkovo 392/3 095 (12,7 %) pacientov s reumatoidnou artritídou malo pozitívne testy na prítomnosť
ADA v klinických štúdiách po liečbe rituximabom. Objavenie sa ADA sa u väčšiny pacientov nespájalo s klinickým zhoršením alebo zvýšením rizika reakcií na následné infúzie. Prítomnosť ADA sa môže spájať so zhoršením reakcií na infúziu alebo alergických reakcií po druhej infúzii
v následných cykloch.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s rituximabom
vo všetkých podskupinách pediatrickej populácie s autoimunitnou artritídou. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
Klinickéskúsenostiprigranulomatózespolyangiitídou(Wegenerovou)amikroskopickoupolyangiitídouIndukcia remisieCelkovo 197 pacientov vo veku 15 rokov alebo starších s ťažkou aktívnou granulomatózou
s polyangiitídou (75 %) a mikroskopickou polyangiitídou (24 %) bolo zaradených a liečených
v randomizovanom, dvojito zaslepenom, multicentrickom, klinickom skúšaní na preukázanie non- inferiority, s aktívnym komparátorom.
Pacienti boli randomizovaní v pomere 1:1 buď na liečbu perorálnym cyklofosfamidom raz denne
(2 mg/kg/deň) počas 3 – 6 mesiacov alebo na liečbu rituximabom (375 mg/m2) raz týždenne počas
4 týždňov. Všetci pacienti v skupine s cyklofosfamidom boli liečení azatioprinom v udržiavacej liečbe počas následného sledovania. Pacienti v oboch skupinách používali 1 000 mg pulzy intravenózneho (IV) metylprednizolónu (alebo ekvivalentnej dávky iného glukokortikoidu) denne po dobu 1 až 3 dní a potom nasledoval perorálny prednizón (1 mg/kg/deň, maximálne 80 mg/deň). Postupné znižovanie dávky prednizónu sa ukončilo po 6 mesiacoch od začiatku liečby v klinickom skúšaní.
Primárnym koncovým ukazovateľom bolo dosiahnutie úplnej remisie v 6. mesiaci definované ako Birminghamove skóre aktivity vaskulitídy pre Wegenerovu granulomatózu (BVAS/WG) s hodnotou 0 a bez liečby glukokortikoidmi. Vopred špecifikovaná hranica non-inferiority pre rozdiel medzi terapiami bola 20 %. Toto skúšanie preukázalo non-inferioritu rituximabu oproti cyklofosfamidu pri dosiahnutí úplnej remisie po 6 mesiacoch (tabuľka 16).
Účinnosť sa pozorovala u pacientov s novodiagnostikovaným ochorením aj u pacientov s relapsom ochorenia (tabuľka 17).
| Rituximab
(n = 99)
| Cyklofosfamid
(n = 98)
| Rozdiel medzi liečbami
(Rituximab-
cyklofosfamid)
| Miera
| 63,6 %
| 53,1 %
| 10,6 %
95,1 %b IS (-3,2 %, 24,3 %) a
| - IS = interval spoľahlivosti.
- * podľa metódy „worst case imputation“
a Preukázala sa non-inferiorita, pretože dolná hranica (-3,2 %) bola vyššia ako vopred stanovená hranica non-inferiority
(-20 %).
b V 95,1 % intervale spoľahlivosti je naviac zohľadnená hodnota alfa 0,001 pre predbežnú analýzu účinnosti.
|
|
|
Tabuľka 16 Percento pacientov, u ktorých sa dosiahla úplná remisia v 6. mesiaci (populácia so zámerom liečiť*)Tabuľka 17 Úplná remisia v 6. mesiaci podľa stavu ochorenia
| Rituximab
| Cyklofosfamid
| Rozdiel (95 % IS)
|
Všetci pacienti Novodiagnostikovaní S relapsom
| n = 99 n = 48 n = 51
| n = 98 n = 48 n = 50
|
|
Úplná remisia
|
Všetci pacienti
| 63,6 %
| 53,1 %
| 10,6 % (-3,2; 24,3)
|
Novodiagnostikovaní
| 60,4 %
| 64,6 %
| −4,2 % (−23,6; 15,3)
|
S relapsom
| 66,7 %
| 42,0 %
| 24,7 % (5,8; 43,6)
|
V prípade pacientov s chýbajúcimi údajmi sa aplikovala metóda „worst case imputation“.
Úplná remisia v 12. a 18. mesiaciV skupine s rituximabom 48 % pacientov dosiahlo úplnú remisiu v 12. mesiaci a 39 % pacientov
dosiahlo úplnú remisiu v 18. mesiaci. U pacientov liečených cyklofosfamidom (po ktorom nasledoval azatioprín na udržanie úplnej remisie) 39 % pacientov dosiahlo úplnú remisiu v 12. mesiaci
a 33 % pacientov dosiahlo úplnú remisiu v 18. mesiaci. Od 12. mesiaca do 18. mesiaca sa pozorovalo
8 relapsov v rituximabovej skupine v porovnaní so štyrmi v cyklofosfamidovej skupine.
Laboratórne vyšetrenia
Celkovo bolo 23/99 (23 %) pacientov liečených rituximabom zo skúšania s cieľom dosiahnuť indukciu remisie pozitívnych na prítomnosť ADA v 18. mesiaci. Žiadny z 99 pacientov liečených rituximabom nebol pozitívny na prítomnosť ADA pri skríningu. Prítomnosť ADA nemala žiadny zjavný negatívny dopad na bezpečnosť alebo účinnosť v klinickom skúšaní s cieľom dosiahnuť indukciu remisie.
Udržiavacia liečba
Celkovo 117 pacientov (88 s GPA, 24 s MPA a 5 s renálnou vaskulitídou súvisiacou s prítomnosťou autoprotilátok proti antigénom cytoplazmatických granúl neutrofilných granulocytov (ANCA))
s ochorením v remisii bolo randomizovaných na liečbu azatioprínom (59 pacientov) alebo rituximabom (58 pacientov) v prospektívnej, multicentrickej, kontrolovanej, nezaslepenej klinickej
štúdii. Zaradení pacienti mali 21 až 75 rokov a mali novodiagnostikované alebo relapsované ochorenie v úplnej remisii po kombinovanej liečbe glukokortikoidmi a cyklofosfamidom podávaným v pulzoch.
Väčšina pacientov bola ANCA-pozitívna pri diagnostikovaní alebo v priebehu ochorenia, a mali histologicky potvrdenú nekrotizujúcu vaskulitídu malých ciev s klinickým fenotypom GPA/MPA alebo renálnu ANCA asociovanú vaskulitídu, alebo obe.
Liečba na indukciu remisie zahŕňala intravenózny prednizón, podávaný podľa posúdenia skúšajúceho, ktorému u niektorých pacientov predchádzali pulzy metylprednizolónu a pulzy cyklofosfamidu, pokiaľ sa nedosiahla remisia po 4 až 6 mesiacoch. V tom čase a maximálne do 1 mesiaca po poslednom
podaní pulzu cyklofosfamidu boli pacienti náhodne zaradení buď na liečbu rituximabom (dve 500 mg intravenózne infúzie s odstupom dvoch týždňov (v 1. deň a 15. deň), po ktorých nasledovalo 500 mg
intravenózne každých 6 mesiacov počas 18 mesiacov) alebo azatioprínom (podávaný perorálne
v dávke 2 mg/kg/deň počas 12 mesiacov, potom 1,5 mg/kg/deň počas 6 mesiacov a nakoniec
1 mg/kg/deň počas 4 mesiacov (po týchto 22 mesiacoch sa liečba vysadila)). Liečba prednizónom sa postupne znižovala a potom sa udržiavala na nízkej dávke (približne 5 mg denne) najmenej
18 mesiacov po randomizácii. Postupné znižovanie dávok prednizónu a rozhodnutie o ukončení liečby prednizónom po 18 mesiacoch boli ponechané na posúdenie skúšajúceho.
Všetci pacienti boli sledovaní do 28. mesiaca (10 mesiacov po poslednej infúzii rituximabu alebo
6 mesiacov po poslednej dávke azatioprínu). Prevencia pneumónie spôsobenej Pneumocystis jirovecii
sa vyžadovala pre všetkých pacientov s počtom CD4+ T-lymfocytov nižším ako 250/mm3.
Primárnym koncovým ukazovateľom bol výskyt závažného relapsu v 28. mesiaci.
Výsledky
V 28. mesiaci došlo k závažnému relapsu (definovanému ako znovuobjavenie sa klinických a/alebo laboratórnych prejavov aktivity vaskulitídy ([BVAS] > 0), ktorý môže viesť k zlyhávaniu alebo poškodeniu orgánov alebo môže byť život ohrozujúci) u 3 pacientov (5 %) v rituximabovej skupine
a u 17 pacientov (29 %) v azatioprínovej skupine (p = 0,0007). Menej závažné relapsy (neohrozujúce život a bez závažného orgánového poškodenia) sa vyskytli u siedmich pacientov v rituximabovej
skupine (12 %) a u ôsmich pacientov v azatioprínovej skupine (14 %).
Krivky kumulatívnej miery incidencie ukázali, že čas do prvého závažného relapsu bol dlhší u pacientov s rituximabom od 2. mesiaca, a to trvalo až do 28. mesiaca (obrázok 3).
Obrázok 3: Kumulatívna miera incidencie v čase prvého závažného relapsu
Percent o pacient ov
s prvý m veľkým

relapso m
Azatioprín
Rituximab
Cenzuro
Čas prežívania (mesiace)
Počet účastníkov so závažným relapsom
|
Azatioprín
| 0
| 0
| 3
| 3
| 5
| 5
| 8
| 8
| 9
| 9
| 9
| 10
| 13
| 15
| 17
|
Rituximab
| 0
| 0
| 0
| 0
| 1
| 1
| 1
| 1
| 1
| 1
| 1
| 1
| 3
| 3
| 3
|
Počet účastníkov s rizikom
|
Azatioprín
| 59
| 56
| 52
| 50
| 47
| 47
| 44
| 44
| 42
| 41
| 40
| 39
| 36
| 34
| 0
|
Rituximab
| 58
| 56
| 56
| 56
| 55
| 54
| 54
| 54
| 54
| 54
| 54
| 54
| 52
| 50
| 0
|
Poznámka: Pacienti sa cenzurovali v 28. mesiaci, keď u nich nedošlo k žiadnej udalosti.
Laboratórne vyšetreniaU celkovo 6/34 (18 %) pacientov liečených rituximabom v klinickom skúšaní s udržiavacou liečbou sa vytvorili ADA. Prítomnosť ADA nemala žiadny zjavný negatívny dopad na bezpečnosť alebo
účinnosť v klinickom skúšaní s udržiavacou liečbou.
Klinické skúsenostipripemphigusvulgarisÚčinnosť a bezpečnosť rituximabu v kombinácii s krátkodobou liečbou nízkymi dávkami
glukokortikoidov (prednizónu) sa hodnotila u novodiagnostikovaných pacientov so stredne ťažkým až ťažkým pemphigom (74 pacientov s pemphigus vulgaris [PV] a 16 pacientov s pemphigus foliaceus
[PF]) v tejto randomizovanej, nezaslepenej, kontrolovanej, multicentrickej klinickej štúdii. Pacienti
boli vo veku od 19 do 79 rokov a predtým sa neliečili na pemphigus. V populácii PV malo
5 (13 %) pacientov v rituximabovej skupine a 3 (8 %) pacienti v skupine so štandardnou dávkou prednizónu stredne ťažké ochorenie a 33 (87 %) pacientov v rituximabovej skupine
a 33 (92 %) pacientov v skupine so štandardnou dávkou prednizónu malo ťažké ochorenie podľa závažnosti ochorenia definovanej Harmanovými kritériami.
Pacienti boli stratifikovaní podľa východiskovej závažnosti ochorenia (stredne ťažké alebo ťažké)
a randomizovaní sa v pomere 1 : 1 buď na podávanie rituximabu a nízkodávkového prednizónu alebo na štandardnú dávku prednizónu. Pacienti randomizovaní do rituximabovej skupiny dostávali
začiatočnú intravenóznu infúziu 1 000 mg rituximabu v 1. deň klinickej štúdie v kombinácii
s 0,5 mg/kg/deň perorálneho prednizónu postupne znižovaného v priebehu 3 mesiacov, ak mali stredne ťažké ochorenie alebo 1 mg/kg/deň perorálneho prednizónu postupne znižovaného v priebehu
6 mesiacov, ak mali ťažké ochorenie, a druhú intravenóznu infúziu 1 000 mg v 15. deň štúdie.
V 12. a 18. mesiaci sa podávali udržiavacie infúzie rituximabu v dávke 500 mg. Pacienti randomizovaní do skupiny so štandardnou dávkou prednizónu dostávali najskôr 1 mg/kg/deň perorálneho prednizónu postupne znižovaného v priebehu 12 mesiacov, ak mali stredne ťažké ochorenie alebo 1,5 mg/kg/deň perorálneho prednizónu postupne znižovaného v priebehu
18 mesiacov, ak mali ťažké ochorenie. Pacienti v rituximabovej skupine, u ktorých došlo k relapsu, mohli dostať ďalšiu infúziu rituximabu v dávke 1 000 mg v kombinácii so znovu zavedenou alebo zvýšenou dávkou prednizónu. Infúzie na udržiavaciu liečbu a infúzie v prípade relapsu sa nepodávali skôr ako 16 týždňov po predchádzajúcej infúzii.
Primárnym koncovým ukazovateľom tejto štúdie bola úplná remisia (úplná epitelizácia a absencia nových a/alebo pôvodných lézií) v 24. mesiaci bez používania prednizónovej liečby počas dvoch mesiacov alebo dlhšieho obdobia (CRoff počas ≥ 2 mesiacov).
VýsledkyTáto štúdia dokázala štatisticky signifikantne lepšie výsledky liečby rituximabom v kombinácii s nízkodávkovým prednizónom v porovnaní so štandardnou dávkou prednizónu pri dosahovaní CRoff
≥ 2 mesiace v 24. mesiaci u pacientov s PV (pozri tabuľku 18).
| Rituximab +
Prednizón n = 38
| Prednizón
n = 36
| p-hodnotaa
| 95 % ISb
| Počet pacientov s odpoveďou (miera odpovede [%])
| 34 (89,5 %)
| 10 (27,8 %)
| < 0,0001
| 61,7 % (38,4; 76,5)
|
|
|
Tabuľka 18 Percento PV pacientov, u ktorých došlo k úplnej remisii bez kortikosteroidovej liečby počas dvoch mesiacov alebo dlhšieho obdobia v 24. mesiaci (populácia-PV Intent-to-Treat)ap-hodnota je odvodená z Fisherovho exaktného testu so strednou korekciou p-hodnoty
b95 % interval spoľahlivosti je korigovaný Newcombov interval
Počet pacientov liečených rituximabom plus nízkodavkovým prednizónom, ktorým bol prednizón
vysadený alebo ostali na minimálnej dávke (denná dávka prednizónu 10 mg alebo menej) v porovnaní s pacientmi na štandardnej dávke prednizónu počas 24-mesačného liečebného obdobia preukazuje
steroidy šetriaci účinok rituximabu (obrázok 4).
Obrázok 4: Počet pacientov, ktorí nedostávali kortikosteroidnú liečbu alebo dostávali minimálnu kortikosteroidnú dávku (≤ 10 mg/deň) v priebehu daného času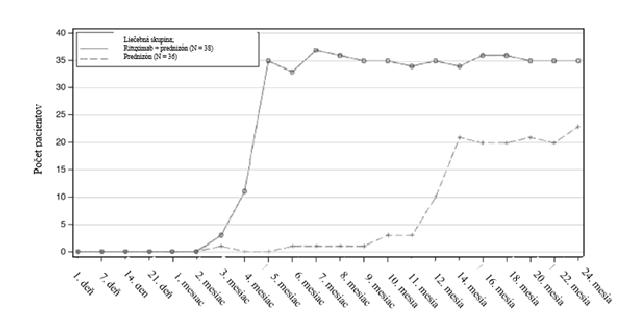
Návšteva
Retrospektívne post-hoc hodnotenie laboratórnych výsledkovCelkovo malo 19/34 (56 %) pacientov s PV, ktorí sa liečili rituximabom, pozitívny test na prítomnosť
ADA protilátok v 18. mesiaci. Klinická relevancia vytvárania ADA u pacientov s PV liečených rituximabom nie je jasná.
5.2 Farmakokinetické vlastnosti
Non-Hodgkinov lymfóm
Na základe populačnej farmakokinetickej analýzy 298 pacientov s NHL, ktorí dostali jednu alebo viac
infúzií rituximabu v monoterapii alebo v kombinácii s CHOP liečbou (použité rituximabové dávky boli v rozsahu od 100 do 500 mg/m2), odhad hodnoty nešpecifického klírensu (CL1) pre typickú populáciu bol 0,14 l/deň, odhad hodnoty špecifického klírensu (CL2) pre typickú populáciu bol
0,59 l/deň, na ktorých sa pravdepodobne podieľajú B-bunky alebo nádorová záťaž, a odhad hodnoty distribučného objemu centrálneho kompartmentu (V1) pre typickú populáciu bol 2,7 l. Odhadovaný medián terminálneho eliminačného polčasu rituximabu bol 22 dní (rozsah 6,1 až 52 dní). Východiskové počty CD19-pozitívnych buniek a veľkosť merateľných nádorových lézií sa podieľali na určitej variabilite v hodnote CL2 rituximabu v prípade údajov získaných od 161 pacientov, ktorým sa podávalo 375 mg/m2 vo forme intravenóznej infúzie v dávkach počas 4 týždňov. Pacienti s vyššími počtami CD19-pozitívnych buniek alebo nádorových lézií mali vyššie hodnoty CL2. Veľká zložka variability medzi jednotlivcami však pretrvávala pre hodnoty CL2 po korekcii pre počty
CD19-pozitívnych buniek a veľkosť nádorovej lézie. V1 sa menilo podľa plochy povrchu tela (BSA) a liečby CHOP. Táto variabilita v hodnote V1 (27,1 % a 19,0 %), na ktorej sa podieľali rozsah BSA (1,53 až 2,32 m2) a súčasná liečba CHOP, bola relatívne malá. Vek, pohlavie a výkonnostný stav
podľa WHO nemali žiadny vplyv na farmakokinetiku rituximabu. Z tejto analýzy vyplýva, že od úpravy dávkovania rituximabu podľa ktoréhokoľvek z testovaných kovariantov sa nedá očakávať zmysluplná redukcia jeho farmakokinetickej variability.
Rituximab, podávaný vo forme intravenóznej infúzie v dávke 375 mg/m2 v týždňových intervaloch, celkovo 4 dávky 203 pacientom s NHL, ktorí ešte neboli liečení rituximabom, viedol k priemernému Cmax po štvrtej infúzii v dávke 486 µg/ml (v rozsahu 77,5 až 996,6 µg/ml). Rituximab bol detegovateľný v sére pacientov 3 – 6 mesiacov po ukončení poslednej liečby.
Po podávaní rituximabu v dávke 375 mg/m2 vo forme intravenóznej infúzie v týždňových intervaloch, celkovo 8 dávok, 37 pacientom s NHL, sa priemerné Cmax zvýšilo s každou ďalšou infúziou, v škále
od priemerne 243 µg/ml (v rozsahu 16 až 582 µg/ml) po prvej infúzii až do 550 µg/ml (v rozsahu 171
až 1 177 µg/ml) po ôsmej infúzii.
Farmakokinetický profil rituximabu, keď sa podával vo forme 6 infúzií v dávke 375 mg/m2
v kombinácii so 6 cyklami chemoterapie CHOP, bol podobný ako farmakokinetický profil pozorovaný pri samostatnom rituximabe.
Chronická lymfocytováleukémia
Rituximab sa podával ako intravenózna infúzia v prvom cykle v dávke 375 mg/m2, ktorá sa v každom
cykle zvýšila na 500 mg/m2, celkovo 5 dávok v kombinácii s fludarabínom a cyklofosfamidom
u pacientov s CLL. Priemerné Cmax (n = 15) bolo 408 µg/ml (v rozsahu 97 – 764 µg/ml) po piatej
500 mg/m2 infúzii a priemerný terminálny polčas bol 32 dní (v rozsahu 14 – 62 dní).
Reumatoidná artritída
Po dvoch intravenóznych infúziách rituximabu v dávke 1 000 mg s odstupom dvoch týždňov bol
priemerný terminálny polčas 20,8 dňa (v rozsahu 8,58 až 35,9 dní), priemerný systémový klírens bol
0,23 l/deň (v rozsahu 0,091 až 0,67 l/deň) a priemerný rovnovážny distribučný objem bol 4,6 l
(v rozsahu 1,7 až 7,51 l). Populačná farmakokinetická analýza rovnakých údajov poskytla podobné priemerné hodnoty pre systémový klírens 0,26 l/deň a pre polčas 20,4 dňa. Populačná farmakokinetická analýza odhalila, že BSA a pohlavie sú najsignifikantnejšími kovariantmi
na vysvetlenie variability medzi jednotlivcami vo farmakokinetických parametroch. Po úprave hodnôt v závislosti na BSA mali muži vyšší distribučný objem a rýchlejší klírens ako ženy. Farmakokinetické
rozdiely súvisiace s pohlavím sa nepovažujú za klinicky relevantné a nie je potrebné upravovať dávku. Pre pacientov s poškodenou funkciou pečene alebo obličiek nie sú dostupné žiadne farmakokinetické údaje.
Farmakokinetika rituximabu sa vyhodnocovala po dvoch intravenóznych (IV) dávkach 500 mg
a 1 000 mg v 1. a 15. deň v štyroch štúdiách. Vo všetkých štúdiách bola farmakokinetika rituximabu
priamo úmerná dávke v rámci študovaného limitovaného rozsahu dávok. Priemerné Cmax rituximabu v sére po prvej infúzii bolo v rozsahu od 157 do 171 µg/ml pre dávku 2 x 500 mg a v rozsahu od 298 do 341 µg/ml pre dávku 2 x 1 000 mg. Po druhej infúzii bolo priemerné Cmax v rozsahu od 183
do 198 µg/ml pre dávku 2 x 500 mg a v rozsahu od 355 do 404 µg/ml pre dávku 2 x 1 000 mg. Priemerný terminálny eliminačný polčas bol v rozsahu 15 až 16 dní pre skupinu s dávkou 2 x 500 mg a 17 až 21 dní pre skupinu s dávkou 2 x 1 000 mg. Priemerné Cmax bolo o 16 až 19 % vyššie po druhej infúzii v porovnaní s prvou infúziou pre obe dávky.
Farmakokinetika rituximabu sa vyhodnocovala po dvoch intravenóznych dávkach 500 mg a 1 000 mg pri opakovanej liečbe v druhom cykle. Priemerné Cmax rituximabu v sére po prvej infúzii bolo 170 až
175 µg/ml pre dávku 2 x 500 mg a 317 až 370 µg/ml pre dávku 2 x 1 000 mg. Cmax po druhej infúzii bolo 207 µg/ml pre dávku 2 x 500 mg a v rozsahu od 377 do 386 µg/ml pre dávku 2 x 1 000 mg.
Priemerný terminálny eliminačný polčas po druhej infúzii po druhom cykle bol 19 dní pre dávku
2 x 500 mg a bol v rozsahu 21 až 22 dní pre dávku 2 x 1 000 mg. Farmakokinetické parametre pre rituximab boli v týchto dvoch liečebných cykloch porovnateľné.
Farmakokinetické (PK) parametre v populácii pacientov, ktorí neadekvátne odpovedali na anti-TNF po rovnakom dávkovacom režime (2 x 1 000 mg intravenózne s 2-týždňovým odstupom) boli podobné, s priemernou maximálnou sérovou koncentráciou 369 µg/ml a priemerným terminálnym polčasom 19,2 dňa.
Granulomatóza spolyangiitídouamikroskopickoupolyangiitídou
Na základe populačnej farmakokinetickej analýzy údajov od 97 pacientov s granulomatózou
s polyangiitídou a mikroskopickou polyangiitídou dostávajúcich 375 mg/m2 rituximabu raz týždenne, celkovo štyri dávky, bol odhadovaný medián terminálneho eliminačného polčasu 23 dní (v rozsahu 9
až 49 dní).
Priemerný klírens rituximabu bol 0,313 l/deň (v rozsahu 0,116 až 0,726 l/deň) a distribučný objem rituximabu bol 4,50 l (v rozsahu 2,25 až 7,39 l). Farmakokinetické parametre rituximabu u týchto pacientov sa javili podobné ako parametre pozorované u pacientov s reumatoidnou artritídou.
5.3 Predklinické údaje o bezpečnosti
Rituximab sa ukázal byť vysoko špecifický voči CD20 antigénu na B-bunkách. Štúdie toxicity na makakoch nepreukázali žiadny iný účinok ako očakávaný farmakologický úbytok B-buniek v periférnej krvi a lymfatickom tkanive.
Štúdie vývojovej toxicity sa uskutočňovali na makakoch v dávkach do 100 mg/kg (liečba v gestačných dňoch 20 – 50) a neodhalili žiadny dôkaz o toxicite pre plod spôsobenej rituximabom. Pozoroval sa však od dávky závislý farmakologický úbytok B-buniek v lymfatických orgánoch plodu, ktorý pretrvával aj postnatálne a bol sprevádzaný poklesom hladín IgG u postihnutých novonarodených zvierat. Počet B-buniek sa u týchto zvierat vrátil na normálnu úroveň do 6 mesiacov po narodení
a nemal negatívny vplyv na odpoveď na imunizáciu.
Neuskutočňovali sa štandardné testy na preskúmanie mutagenity, pretože takéto testy pre túto molekulu nie sú relevantné. Na stanovenie karcinogénneho potenciálu rituximabu sa neuskutočňovali žiadne dlhodobé štúdie na zvieratách.
Neuskutočňovali sa špecifické štúdie na stanovenie účinkov rituximabu na fertilitu. V štúdiách všeobecnej toxicity na makakoch sa nepozorovali žiadne škodlivé účinky na reprodukčné orgány u samčekov alebo samičiek.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
Histidínium-chlorid monohydrát
Edetan disodný
Polysorbát 80
Sacharóza
Voda, na injekcie
6.2 Inkompatibility
Nepozorovali sa žiadne inkompatibility medzi Ruxience a polyvinylchloridovými alebo polyetylénovými vakmi alebo infúznymi súpravami.
6.3 Čas použiteľnosti
Neotvorená injekčnáliekovka
24 mesiacov
Nariedený liek
· Po aseptickom nariedení v roztoku chloridu sodného
Infúzny roztok Ruxience pripravený v 0,9 % roztoku chloridu sodného je fyzikálne a chemicky stabilný 24 hodín pri teplote 2 °C – 8 °C plus ďalších 24 hodín pri teplote ≤ 30 °C.
· Po aseptickom nariedení v D-glukózovom roztoku
Infúzny roztok Ruxience pripravený v 5 % roztoku D-glukózy je fyzikálne a chemicky stabilný
24 hodín pri teplote 2 °C – 8 °C plus ďalších 24 hodín pri teplote ≤ 30 °C.
Z mikrobiologického hľadiska sa má pripravený infúzny roztok použiť okamžite. Ak sa nepoužije okamžite, za dobu uchovávania a podmienky uchovávania pred použitím je zodpovedný používateľ
a normálne nemajú prekročiť 24 hodín pri teplote 2 °C – 8 °C, pokiaľ sa nariedenie neuskutočňovalo
v kontrolovaných a overených aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Nádobu uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Ruxience 100mginfúznykoncentrát
Číre sklenené injekčné liekovky I. typu s chlórbutylovou gumenou zátkou obsahujúce
100 mg rituximabu v 10 ml.
Balenie obsahujúce 1 injekčnú liekovku.
Ruxience 500mginfúznykoncentrát
Číre sklenené injekčné liekovky I. typu s chlórbutylovou gumenou zátkou obsahujúce
500 mg rituximabu v 50 ml.
Balenie obsahujúce 1 injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomRuxience sa dodáva v sterilných, jednorazových, nepyrogénnych injekčných liekovkách bez prítomnosti konzervačných látok.
Asepticky vytiahnite potrebné množstvo Ruxience a narieďte ho na vypočítanú koncentráciu
1 až 4 mg/ml rituximabu do infúzneho vaku obsahujúceho sterilný, nepyrogénny injekčný roztok chloridu sodného 9 mg/ml (0,9 %) alebo 5 % D-glukózu vo vode. Na premiešanie roztoku jemne prevráťte vak, aby sa nevytvárala pena. Pri príprave roztokov sa musí dbať na dôkladne dodržiavanie sterility. Keďže tento liek neobsahuje žiadne antimikrobiálne konzervačné látky alebo bakteriostatiká, musí sa používať aseptická metóda. Pred podaním sa parenterálne lieky musia vizuálne skontrolovať, či neobsahujú častice alebo nemajú zmenenú farbu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLARuxience 100mginfúznykoncentrátEU/1/20/1431/001
Ruxience 500mginfúznykoncentrátEU/1/20/1431/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.