
ti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Konestat alfa je odvodený z mlieka transgenických králikov a obsahuje stopy králičieho proteínu.
Pred začatím liečby Ruconestom je potrebné opýtať sa pacientov na predchádzajúci kontakt s králikmi a na prejavy a príznaky, ktoré by naznačovali alergickú reakciu.
Hypersenzitívne reakcie sa nedajú vylúčiť.
Pacienti musia byť počas celej doby podávania lieku dôkladne monitorovaní a pozorovaní na prítomnosť akýchkoľvek príznakov hypersenzitivity. Pacienti musia byť informovaní o včasných príznakoch hypersenzitívnych reakcií vrátane žihľavky, generalizovanej urtikárie, zvierania v hrudi, sipotu, hypotenzie a anafylaxie. Ak sa tieto príznaky vyskytnú po podaní, pacienti musia upozorniť svojho lekára.
V prípade anafylaktických reakcií alebo šoku je potrebné podať pohotovostnú liečbu.
Hoci sa skrížená reaktivita medzi kravským a králičím mliekom považuje za nepravdepodobnú, možnosť takejto skríženej reaktivity u pacienta s preukázanou klinickou alergiou na kravské mlieko sa nedá vylúčiť a pacient sa má kvôli prejavom a príznakom hypersenzitivity po podaní Ruconestu
sledovať. Pacienti s alergiou na kravské mlieko majú byť informovaní o tom, že môžu mať reakciu na Ruconest. Na vylúčenie skríženej reaktivity medzi kravským a králičím mliekom sa môže vykonať kožný vpichový (prick) test.
Sodík
Každá injekčná liekovka obsahuje približne 19,5 mg sodíka. Má sa to vziať do úvahy u pacientov na diéte s kontrolovaným obsahom sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Vedecká literatúra uvádza interakciu tkanivového aktivátora plazminogénu(tPA) a C1INH
obsahujúceho lieky. Liek Ruconest sa nesmie podávať zároveň s tPA.
4.6 Fertilita, gravidita a laktácia
Gravidita a dojčenie
S použitím lieku Ruconest v prípade gravidných a dojčiacich žien nie sú žiadne skúsenosti.
V jednej štúdii na zvieratách sa preukázala reprodukčná toxicita (pozri časť 5.3). Liek Ruconest sa neodporúča používať počas gravidity alebo laktácie s výnimkou prípadu, keď ošetrujúci lekár vyhodnotí, že prínosy prevažujú nad možnými rizikami.
Fertilita
Nie sú k dispozícii žiadne údaje o účinkoch lieku Ruconest na mužskú ani ženskú fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Na základe známej farmakológie a profilu vedľajších účinkov lieku Ruconest sa neočakávajú žiadne nežiaduce účinky na schopnosť viesť vozidlá a obsluhovať stroje. Počas užívania lieku Ruconest však boli nahlásené bolesti hlavy alebo vertigo, tie sa však môžu vyskytnúť aj v dôsledku záchvatu HAE. Pacientom sa musí odporučiť, aby neriadili vozidlá a neobsluhovali stroje, ak sa u nich vyskytne bolesť hlavy alebo vertigo.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
V klinických štúdiách s Ruconestom sa pozoroval jeden prípad hypersenzitivity. Najčastejším nežiaducim účinkom pozorovaným po podaní Ruconestu je bolesť hlavy.
Tabuľkovýsúhrnnežiaducichreakcií
Klinické skúsenosti podporujúce bezpečnosť lieku Ruconest obsahujú 300 podaní (83 podaní
zdravým osobám alebo pacientom asymptomatickým na HAE a 217 podaní 119 pacientom s HAE). V nasledujúcej tabuľke sa uvádza zoznam všetkých nežiaducich účinkov, ktoré sa vyskytli počas 7 dní po liečbe liekom Ruconest, ako boli hlásené v šiestich liečebných štúdiách.
Nežiaduce účinky boli zvyčajne miernej až strednej závažnosti. Výskyt nežiaducich účinkov bol podobný pre všetky skupiny s dávkou a po opakovaných podaniach sa nezvýšil.
Frekvencie nežiaducich účinkov uvedených nižšie sú definované nasledujúcim spôsobom:
veľmi časté (≥ 1/10),
časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
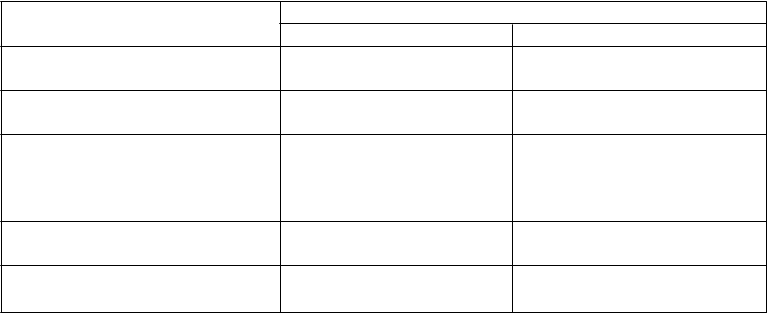
Nežiaduce účinky
Časté Menej časté
Poruchy nervového systému bolesť hlavy vertigo parestézia
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Poruchy kože a podkožného
tkaniva
Celkové poruchy a reakcie v mieste podania
podráždenie hrdla
hnačka
nauzea
žalúdočná nevoľnosť orálna parestézia urtikária
opuch
 H
l
ásenie
podozrení
na
nežiaduce
reakcie
H
l
ásenie
podozrení
na
nežiaduce
reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii nie sú žiadne klinické informácie o predávkovaní.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné hematologické liečivá, liečivá používané pri dedičnom angioedéme, ATC kód: B06AC04.
Plazmový proteín C1INH je hlavným regulátorom aktivácie kontaktných a komplementárnych systémov
in vivo. Pacienti s HAE majú heterozygótnu deficienciu plazmového proteínu C1INH. Preto môžu trpieť nekontrolovanou aktiváciou kontaktných a komplementárnych systémov s tvorbou zápalových mediátorov, ktoré sa klinicky prejavujú ako výskyt akútnych záchvatov angioedému.
Konestat alfa, rekombinantný ľudský komplementárny komponent 1 (C1) inhibítora esterázy (rhC1INH), je analógom ľudského C1INH a získava sa z mlieka králikov, vyjadruje kódovanie génov pre ľudské C1INH. Sekvencia aminokyselín konestatu alfa je rovnaká ako sekvencia endogénneho C1INH.
C1INH využíva inhibičný účinok na niekoľkých proteázach (cieľových proteázach) kontaktných a komplementárnych systémov. Účinok konestatu alfa na nasledujúcich cieľových proteázach sa hodnotil
in vitro: aktivované C1s, kalikreín, faktor XIIa a faktor XIa. Inhibičná kinetika je porovnateľná s kinetikou pozorovanou pri ľudskom C1INH odvodenom z plazmy.
Komplementárny komponent (proteín) C4 je substrátom pre aktivované C1. Pacienti s HAE majú nízke hladiny C4 v obehu. Čo sa týka C1INH odvodeného z plazmy, farmakodynamické účinky konestatu alfa na C4 vykazujú zotavenie komplementárnej homeostázy závislé od dávky v prípade pacientov s HAE na úrovni aktivity plazmového C1INH väčšie než 0,7 jednotiek/ml, čo je najnižší limit normálneho rozsahu. V prípade pacientov s HAE zvyšuje Ruconest v dávke 50 jednotiek/kg
úroveň aktivity plazmového C1INH na úroveň vyššiu ako 0,7 jednotiek/ml na približne 2 hodiny
(pozri časť 5.2).
Účinnosť a bezpečnosť lieku Ruconest pri liečbe akútnych záchvatov angioedému v prípade pacientov s HAE bola vyhodnotená v dvoch dvojito zaslepených randomizovaných placebom kontrolovaných
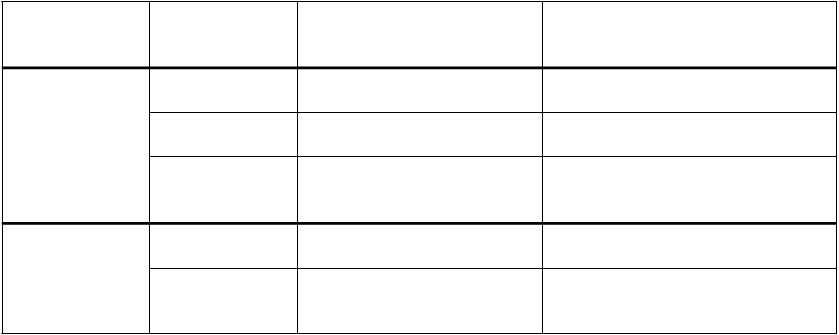
a štyroch otvorených klinických štúdiách. Dávky vyhodnotené v klinických štúdiách boli v rozsahu jednej liekovky 2 100 jednotiek (zodpovedajúcej 18 - 40 jednotiek/kg) až 50 a 100 jednotiek/kg. Účinnosť lieku Ruconest pri liečbe akútnych záchvatov angioedému bola preukázaná výrazne kratším časom do začiatku úľavy od príznakov a času nastúpenia minimálnych príznakov, ako aj malým počtom zlyhaní liečby. V nasledujúcej tabuľke sa uvádzajú výsledky (primárne a sekundárne parametre) dvoch randomizovaných kontrolovaných skúšok:
Štúdia Liečba
C1-1205 RCT 100 jednotiek/kg n = 13
50 jednotiek/kg n = 12
Čas (v min.) do
začiatku úľavy,
priemerná hodnota (95 % IS)
68 (62, 132)
p = 0,001
122 (72, 136)
p < 0,001
Čas (v min.) do
minimálnych príznakov, priemerná hodnota (95 % IS)
245 (125, 270)
p = 0,04
247 (243, 484)
Fyziologický roztok
n = 13
258 (240, 720) 1101 (970, 1494)
C1-1304 RCT 100 jednotiek/kg n = 16
62 (40, 75)
p = 0,003
480 (243, 723)
p = 0,005
Fyziologický roztok
n = 16
508 (70, 720) 1440 (720, 2885)
Výsledky otvorených štúdií boli konzistentné s uvedenými zisteniami a podporujú opakované
užívanie lieku Ruconest pri liečbe následných záchvatov angioedému.
Pri randomizovaných kontrolovaných skúškach sa pri 39/41 (95 %) pacientov liečených liekom Ruconest dosiahol čas do začiatku úľavy do 4 hodín. Pri otvorenej štúdii sa pri 114/119 (95 %) pacientov liečených jednou dávkou 50 jednotiek/kg dosiahol čas do začiatku úľavy do 4 hodín. Ďalšia dávka 50 jednotiek/kg sa podávala v prípade 13/133 (10 %) záchvatov.
Pediatrická populáciaDeväť dospievajúcich pacientov s HAE (vo veku od 13 do 17 rokov) sa liečilo dávkou 50 jednotiek/kg pri 26 akútnych záchvatoch angioedému a 7 pacientov (vo veku od 16 do 17 rokov) dávkou 2 100 jednotiek pri 24 akútnych záchvatoch angioedému. Účinnosť a bezpečnosť u dospievajúcich pacientov boli konzistentné s výsledkami u dospelých.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Ruconestom v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe akútnych záchvatov angioedému (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiDistribúciaNeuskutočnili sa žiadne štúdie o formálnej distribúcii. Objem distribúcie konestatu alfa bol približne
3 l, čo je porovnateľné s objemom plazmy.
Biotransformácia a elimináciaNa základe údajov o zvieratách sa konestat alfa dostane z obehu pomocou pečene prostredníctvom
receptorom sprostredkovanej endocytózy, po ktorej nasleduje úplná hydrolýza/degradácia.
Po podaní lieku Ruconest (50 jednotiek/kg) asymptomatickým pacientom s HAE sa pozorovalo Cmax =
1,36 jednotiek/ml. Eliminačný polčas konestatu alfa bol približne 2 hodiny.
Vylučovanie
Ku vylučovaniu nedochádza, keďže konestat alfa sa z obehu dostáva prostredníctvom receptorom sprostredkovanej endocytózy, po ktorej nasleduje úplná hydrolýza/degradácia v pečeni.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje nenaznačujú žiadne bezpečnostné riziko týkajúce sa používania konestatu alfa
u ľudí, a to na základe farmakologických štúdií bezpečnosti, toxicity po jednej dávke, dvojtýždňovej subchronickej toxicity a lokálnej tolerancie u rôznych druhov zvierat, vrátane potkanov, psov, králikov a makakov. Genotoxický a karcinogénny potenciál sa neočakáva.
Embryofetálne štúdie na potkanoch a králikoch; denné jednorazové dávky nosiča alebo
625 jednotiek/kg/podanie rhC1INH boli podávané intravenózne spáreným potkanom a králikom.
V štúdii na potkanoch neexistovali žiadne malformácie plodov, a to ani v skupine, ktorej sa podával konestat alfa, ani v kontrolnej skupine. V štúdii embryotoxicity králikov sa pozorovalo zvýšenie výskytu defektov srdcových ciev plodu (1,12 % v liečebnej skupine oproti 0,03 % v kontrolnej skupine pozorovanej v minulosti) u zvierat, ktorým sa podal konestat alfa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sacharóza
Citrónan sodný (E331) Kyselina citrónová
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
4 roky.
Rekonštituovanýroztok
Chemická a fyzikálna stabilita pri používaní bola preukázaná počas 48 hodín pri teplote 5 ˚C a 25 ˚C. Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa nepodáva hneď, potom čas uchovávania pri používaní a podmienky pred používaním sú zodpovednosťou používateľa a normálne by nemal prekročiť 24 hodín pri teplote 2 °C až 8 °C, ak sa rekonštitúcia lieku nevykonala
v kontrolovaných a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 25 °C. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
2 100 jednotiek konestatu alfa, prášku, v 25 ml liekovke (sklo typu 1) so zátkou (silikonizovaná chlorobutylová guma) a odnímateľným viečkom (hliník a farebný plast).
Veľkosť balenia: 1.
6.6 Špeciálne opatrenia na likvidáciuKaždá liekovka lieku Ruconest je určená len na jedno použitie.
Rekonštitúciu, kombinovanie a zmiešavanie roztokov je potrebné vykonávať za aseptických
podmienok.
RekonštitúciaKaždá injekčná liekovka Ruconestu (2 100 jednotiek) sa rekonštituuje so 14 ml vody pre injekcie. Voda na injekciu sa pridáva pomaly, aby nedošlo k prudkému nárazu na prášok a opatrne sa mieša, aby sa minimalizovalo spenenie roztoku. Rekonštituovaný roztok obsahuje 150 jednotiek/ml konestatu alfa a javí sa ako číry, bezfarebný roztok.
Rekonštituovaný roztok v každej liekovke sa má vizuálne skontrolovať na prítomnosť častíc a zmenu farby. Roztok obsahujúci častice a so zmeneným sfarbením sa nesmie použiť. Liek sa musí použiť okamžite (pozri časť 6.3).
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPharming Group N.V. Darwinweg 24
NL-2333 CR LEIDEN Holandsko
8. REGISTRAČNÉ ČÍSLOEU/1/10/641/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. október 2010
Dátum posledného predĺženia registrácie: 18. september 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Ruconest 2100 U, prášok a rozpúšťadlo na injekčný roztok.
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Injekčnáliekovka s práškom
Jedna injekčná liekovka obsahuje 2 100 jednotiek konestatu alfa, zodpovedajúcich 2 100 jednotkám
na 14 ml po rekonštitúcii alebo zodpovedajúcich koncentrácii 150 jednotiek/ml.
Konestat alfa je rekombinantný analóg ľudského inhibítora C1 esterázy (rhC1INH) vyrobený technológiou rekombinantnej DNA v mlieku transgenických králikov.
Jedna jednotka aktivity konestatu alfa sa definuje ako ekvivalent inhibičnej aktivity C1 esterázy
prítomnej v 1 ml zmesnej normálnej plazmy.
Pomocnálátkasoznámymúčinkom:
Každá injekčná liekovka s práškom obsahuje približne 19,5 mg sodíka. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok a rozpúšťadlo na injekčný roztok. Biely až sivobiely prášok.
Rozpúšťadlo je číra, bezfarebná tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Ruconest je indikovaný dospelým a dospievajúcim s dedičným angioedémom (hereditary angioedema,
HAE) v dôsledku deficiencie inhibítora C1 esterázy na liečbu akútnych záchvatov angioedému.
4.2 Dávkovanie a spôsob podávania
Liečba liekom Ruconest sa má začať pod vedením a dohľadom lekára so skúsenosťami v oblasti diagnostiky a liečby dedičného angioedému.
Dávkovanie
- Telesná hmotnosť do 84 kg
Jedna intravenózna injekcia 50 jednotiek/kg telesnej hmotnosti.
- Telesná hmotnosť 84 kg a viac
Jedna intravenózna injekcia 4 200 jednotiek (dve liekovky).
Vo väčšine prípadov stačí na vyliečenie akútneho záchvatu angioedému jedna dávka lieku Ruconest. V prípade nedostatočnej klinickej reakcie sa môže podať ďalšia dávka (50 jednotiek/kg telesnej hmotnosti až do 4 200 jednotiek) (pozri časť 5.1).
V priebehu 24 hodín je možné podať maximálne dve dávky.
Výpočet dávky
Určite telesnú hmotnosť pacienta.
- Telesná hmotnosť do 84 kg
V prípade pacientov do 84 kg vypočítajte požadovaný objem dávky, ktorá sa má podať, podľa
nasledujúceho vzorca:
Objem, ktorý sa má
podať (ml) =
telesná hmotnosť(kg)krát50(jednotiek/kg)
150 (jednotiek/ml) =
telesnáhmotnosť(kg)
3
- Telesná hmotnosť 84 kg a viac
V prípade pacientov s hmotnosťou 84 kg a viac je požadovaný objem, ktorý sa má podať, 28 ml, čo zodpovedá množstvu 4 200 jednotiek (2 liekovky).
Pediatrická populácia
Bezpečnosť a účinnosť Ruconestu u detí (vo veku 0 až 12 rokov) nebola doteraz stanovená.
St arš í paci ent i (≥ 65 rokov)
Údaje o pacientoch starších ako 65 rokov sú obmedzené.
Neexistuje žiadne odôvodnenie odlišných reakcií na liek Ruconest v prípade pacientov starších ako 65
rokov.
Porucha f unkc i e obl i či ek
V prípade pacientov s poruchou funkcie obličiek nie je potrebné upravovať dávku, keďže konestat
alfa nepodlieha renálnemu klírensu.
Porucha f unkc i e pe če ne
Neexistujú klinické skúsenosti s liekom Ruconest u pacientov s poruchou funkcie pečene. Porucha funkcie pečene môže predĺžiť plazmový polčas konestatu alfa, nie je však dôvod na klinické obavy. Nie je možné uviesť odporúčania na úpravu dávkovania.
Spôsob podávania
Na intravenózne použitie.
Ruconest musí byť podávaný zdravotníckym pracovníkom, kým pacient (alebo opatrovateľ) nie je
spôsobilý podávať liek po riadnom zaškolení a so súhlasom zdravotníckeho pracovníka.
Pokyny na rekonštitúciu Ruconestu pred podaním, pozri časť 6.6.
Požadovaný objem rekonštituovaného roztoku sa má podávať ako pomalá intravenózna injekcia
v priebehu približne 5 minút.
4.3 Kontraindikácie
· Známa alebo predpokladaná alergia na králiky (pozri časť 4.4).
· Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Konestat alfa je odvodený z mlieka transgenických králikov a obsahuje stopy králičieho proteínu.
Pred začatím liečby Ruconestom je potrebné opýtať sa pacientov na predchádzajúci kontakt s králikmi a na prejavy a príznaky, ktoré by naznačovali alergickú reakciu.
Hypersenzitívne reakcie sa nedajú vylúčiť.
Pacienti musia byť počas celej doby podávania lieku dôkladne monitorovaní a pozorovaní na prítomnosť akýchkoľvek príznakov hypersenzitivity. Pacienti musia byť informovaní o včasných príznakoch hypersenzitívnych reakcií vrátane žihľavky, generalizovanej urtikárie, zvierania v hrudi, sipotu, hypotenzie a anafylaxie. Ak sa tieto príznaky vyskytnú po podaní, pacienti musia upozorniť svojho lekára.
V prípade anafylaktických reakcií alebo šoku je potrebné podať pohotovostnú liečbu.
Hoci sa skrížená reaktivita medzi kravským a králičím mliekom považuje za nepravdepodobnú, možnosť takejto skríženej reaktivity u pacienta s preukázanou klinickou alergiou na kravské mlieko sa nedá vylúčiť a pacient sa má kvôli prejavom a príznakom hypersenzitivity po podaní Ruconestu sledovať. Pacienti s alergiou na kravské mlieko majú byť informovaní o tom, že môžu mať reakciu na Ruconest. Na vylúčenie skríženej reaktivity medzi kravským a králičím mliekom sa môže vykonať kožný vpichový (prick) test.
Sodík
Každá injekčná liekovka obsahuje približne 19,5 mg sodíka. Má sa to vziať do úvahy u pacientov na diéte s kontrolovaným obsahom sodíka.
Domácaliečba a samopodanie
K dispozícii sú obmedzené údaje v súvislosti s používaním tohto lieku v domácom prostredí alebo
s jeho samopodaním (vlastným podaním pacientom). Potenciálne riziká spojené s domácou liečbou súvisia so samotným podávaním lieku, ako aj zvládaním nežiaducich účinkov, najmä precitlivenosti. O domácej liečbe jednotlivých pacientov musí rozhodnúť ošetrujúci lekár, ktorý musí zabezpečiť príslušné zaškolenie pacienta a kontrolu používania v určitých intervaloch.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Vedecká literatúra uvádza interakciu tkanivového aktivátora plazminogénu(tPA) a C1INH
obsahujúceho lieky. Liek Ruconest sa nesmie podávať zároveň s tPA.
4.6 Fertilita, gravidita a laktácia
Gravidita a dojčenie
S použitím lieku Ruconest v prípade gravidných a dojčiacich žien nie sú žiadne skúsenosti.
V jednej štúdii na zvieratách sa preukázala reprodukčná toxicita (pozri časť 5.3). Liek Ruconest sa neodporúča používať počas gravidity alebo laktácie s výnimkou prípadu, keď ošetrujúci lekár vyhodnotí, že prínosy prevažujú nad možnými rizikami.
Fertilita
Nie sú k dispozícii žiadne údaje o účinkoch lieku Ruconest na mužskú ani ženskú fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Na základe známej farmakológie a profilu vedľajších účinkov lieku Ruconest sa neočakávajú žiadne nežiaduce účinky na schopnosť viesť vozidlá a obsluhovať stroje. Počas užívania lieku Ruconest však boli nahlásené bolesti hlavy alebo vertigo, tie sa však môžu vyskytnúť aj v dôsledku záchvatu HAE. Pacientom sa musí odporučiť, aby neriadili vozidlá a neobsluhovali stroje, ak sa u nich vyskytne bolesť hlavy alebo vertigo.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
V klinických štúdiách s Ruconestom sa pozoroval jeden prípad hypersenzitivity. Najčastejším nežiaducim účinkom pozorovaným po podaní Ruconestu je bolesť hlavy.
Tabuľkovýsúhrnnežiaducichreakcií
Klinické skúsenosti podporujúce bezpečnosť lieku Ruconest obsahujú 300 podaní (83 podaní
zdravým osobám alebo pacientom asymptomatickým na HAE a 217 podaní 119 pacientom s HAE). V nasledujúcej tabuľke sa uvádza zoznam všetkých nežiaducich účinkov, ktoré sa vyskytli počas 7 dní po liečbe liekom Ruconest, ako boli hlásené v šiestich liečebných štúdiách.
Nežiaduce účinky boli zvyčajne miernej až strednej závažnosti. Výskyt nežiaducich účinkov bol podobný pre všetky skupiny s dávkou a po opakovaných podaniach sa nezvýšil.
Frekvencie nežiaducich účinkov uvedených nižšie sú definované nasledujúcim spôsobom:
veľmi časté (≥ 1/10),
časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Nežiaduce účinky
Časté Menej časté
Poruchy nervového systému bolesť hlavy vertigo parestézia
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Poruchy kože a podkožného
tkaniva
Celkové poruchy a reakcie v mieste podania
podráždenie hrdla
hnačka
nauzea
žalúdočná nevoľnosť orálna parestézia urtikária
opuch
H
l
ásenie
podozrení
na
nežiaduce
reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.'
4.9 PredávkovanieK dispozícii nie sú žiadne klinické informácie o predávkovaní.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné hematologické liečivá, liečivá používané pri dedičnom angioedéme, ATC kód: B06AC04.
Plazmový proteín C1INH je hlavným regulátorom aktivácie kontaktných a komplementárnych systémov
in vivo. Pacienti s HAE majú heterozygótnu deficienciu plazmového proteínu C1INH. Preto môžu trpieť nekontrolovanou aktiváciou kontaktných a komplementárnych systémov s tvorbou zápalových mediátorov, ktoré sa klinicky prejavujú ako výskyt akútnych záchvatov angioedému.
Konestat alfa, rekombinantný ľudský komplementárny komponent 1 (C1) inhibítora esterázy (rhC1INH), je analógom ľudského C1INH a získava sa z mlieka králikov, vyjadruje kódovanie génov pre ľudské C1INH. Sekvencia aminokyselín konestatu alfa je rovnaká ako sekvencia endogénneho C1INH.
C1INH využíva inhibičný účinok na niekoľkých proteázach (cieľových proteázach) kontaktných a komplementárnych systémov. Účinok konestatu alfa na nasledujúcich cieľových proteázach sa hodnotil in vitro: aktivované C1s, kalikreín, faktor XIIa a faktor XIa. Inhibičná kinetika je porovnateľná s kinetikou pozorovanou pri ľudskom C1INH odvodenom z plazmy.
Komplementárny komponent (proteín) C4 je substrátom pre aktivované C1. Pacienti s HAE majú nízke hladiny C4 v obehu. Čo sa týka C1INH odvodeného z plazmy, farmakodynamické účinky konestatu alfa na C4 vykazujú zotavenie komplementárnej homeostázy závislé od dávky v prípade pacientov s HAE na úrovni aktivity plazmového C1INH väčšie než 0,7 jednotiek/ml, čo je najnižší limit normálneho rozsahu. V prípade pacientov s HAE zvyšuje Ruconest v dávke 50 jednotiek/kg úroveň aktivity plazmového C1INH na úroveň vyššiu ako 0,7 jednotiek/ml na približne 2 hodiny (pozri časť 5.2).
Účinnosť a bezpečnosť lieku Ruconest pri liečbe akútnych záchvatov angioedému v prípade pacientov s HAE bola vyhodnotená v dvoch dvojito zaslepených randomizovaných placebom kontrolovaných
a štyroch otvorených klinických štúdiách. Dávky vyhodnotené v klinických štúdiách boli v rozsahu jednej liekovky 2 100 jednotiek (zodpovedajúcej 18 - 40 jednotiek/kg) až 50 a 100 jednotiek/kg. Účinnosť lieku Ruconest pri liečbe akútnych záchvatov angioedému bola preukázaná výrazne kratším časom do začiatku úľavy od príznakov a času nastúpenia minimálnych príznakov, ako aj malým počtom zlyhaní liečby. V nasledujúcej tabuľke sa uvádzajú výsledky (primárne a sekundárne parametre) dvoch randomizovaných kontrolovaných skúšok:
Štúdia Liečba
C1-1205 RCT 100 jednotiek/kg n = 13
50 jednotiek/kg n = 12
Čas (v min.) do
začiatku úľavy,
priemerná hodnota (95 % IS)
68 (62, 132)
p = 0,001
122 (72, 136)
p < 0,001
Čas (v min.) do
minimálnych príznakov, priemerná hodnota (95 % IS)
245 (125, 270)
p = 0,04
247 (243, 484)
Fyziologický roztok
n = 13
258 (240, 720) 1101 (970, 1494)
C1-1304 RCT 100 jednotiek/kg n = 16
62 (40, 75)
p = 0,003
480 (243, 723)
p = 0,005
Fyziologický roztok
n = 16
508 (70, 720) 1440 (720, 2885)
Výsledky otvorených štúdií boli konzistentné s uvedenými zisteniami a podporujú opakované
užívanie lieku Ruconest pri liečbe následných záchvatov angioedému.
Pri randomizovaných kontrolovaných skúškach sa pri 39/41 (95 %) pacientov liečených liekom Ruconest dosiahol čas do začiatku úľavy do 4 hodín. Pri otvorenej štúdii sa pri 114/119 (95 %) pacientov liečených jednou dávkou 50 jednotiek/kg dosiahol čas do začiatku úľavy do 4 hodín. Ďalšia dávka 50 jednotiek/kg sa podávala v prípade 13/133 (10 %) záchvatov.
Pediatrická populáciaDeväť dospievajúcich pacientov s HAE (vo veku od 13 do 17 rokov) sa liečilo dávkou 50 jednotiek/kg pri 26 akútnych záchvatoch angioedému a 7 pacientov (vo veku od 16 do 17 rokov) dávkou 2 100 jednotiek pri 24 akútnych záchvatoch angioedému. Účinnosť a bezpečnosť u dospievajúcich pacientov boli konzistentné s výsledkami u dospelých.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Ruconestom v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe akútnych záchvatov angioedému (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
D
i
stribúcia
Neuskutočnili sa žiadne štúdie o formálnej distribúcii. Objem distribúcie konestatu alfa bol približne
3 l, čo je porovnateľné s objemom plazmy.
Biotransformácia a eliminácia
Na základe údajov o zvieratách sa konestat alfa dostane z obehu pomocou pečene prostredníctvom receptorom sprostredkovanej endocytózy, po ktorej nasleduje úplná hydrolýza/degradácia.
Po podaní lieku Ruconest (50 jednotiek/kg) asymptomatickým pacientom s HAE sa pozorovalo Cmax =
1,36 jednotiek/ml. Eliminačný polčas konestatu alfa bol približne 2 hodiny.
Vylučovanie
Ku vylučovaniu nedochádza, keďže konestat alfa sa z obehu dostáva prostredníctvom receptorom sprostredkovanej endocytózy, po ktorej nasleduje úplná hydrolýza/degradácia v pečeni.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje nenaznačujú žiadne bezpečnostné riziko týkajúce sa používania konestatu alfa
u ľudí, a to na základe farmakologických štúdií bezpečnosti, toxicity po jednej dávke, dvojtýždňovej subchronickej toxicity a lokálnej tolerancie u rôznych druhov zvierat, vrátane potkanov, psov, králikov a makakov. Genotoxický a karcinogénny potenciál sa neočakáva.
Embryofetálne štúdie na potkanoch a králikoch; denné jednorazové dávky nosiča alebo
625 jednotiek/kg/podanie rhC1INH boli podávané intravenózne spáreným potkanom a králikom.
V štúdii na potkanoch neexistovali žiadne malformácie plodov, a to ani v skupine, ktorej sa podával konestat alfa, ani v kontrolnej skupine. V štúdii embryotoxicity králikov sa pozorovalo zvýšenie výskytu defektov srdcových ciev plodu (1,12 % v liečebnej skupine oproti 0,03 % v kontrolnej skupine pozorovanej v minulosti) u zvierat, ktorým sa podal konestat alfa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Injekčnáliekovka s práškom:
Sacharóza
Citrónan sodný (E331) Kyselina citrónová
Injekčnáliekovka s rozpúšťadlom:
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
4 roky.
Rekonštituovanýroztok
Chemická a fyzikálna stabilita pri používaní bola preukázaná počas 48 hodín pri teplote 5 ˚C a 25 ˚C. Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa nepodáva hneď, potom čas uchovávania pri používaní a podmienky pred používaním sú zodpovednosťou používateľa a normálne by nemal prekročiť 24 hodín pri teplote 2 °C až 8 °C, ak sa rekonštitúcia lieku nevykonala
v kontrolovaných a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Injekčnáliekovka s práškom:
Uchovávajte pri teplote neprevyšujúcej 25 °C. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Injekčnáliekovka s rozpúšťadlom: Uchovávajte pri teplote neprevyšujúcej 25 °C.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka s práškom: 2 100 jednotiek konestatu alfa, prášku, v injekčnej liekovke (sklo typu
1) so zátkou (silikonizovaná chlorobutylová guma) a odnímateľným viečkom (hliník a farebný plast).
Injekčná liekovka s rozpúšťadlom: 20 ml vody na injekciu v injekčnej liekovke (sklo typu 1) so zátkou
(chlórbutylová guma potiahnutá silikónom) a odnímateľným viečkom (hliník a farebný plast).
Súprava na podanie lieku:
· 1 injekčná liekovka s práškom
· 1 injekčná liekovka s rozpúšťadlom
· 2 adaptéry na injekčné liekovky
· 1 injekčná striekačka
· 1 infúzna súprava s 35 cm hadičkou a ihlou veľkosti 25 G
· 2 alkoholové tampóny
· 1 sterilný netkaný tampón
· 1 samolepiaca náplasť
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Príprava a zaobchádzanie s liekom
Každá injekčná liekovka Ruconestu je určená len na jedno použitie.
Ruconest je určený na intravenózne podanie po rekonštitúcii s vodou na injekciu. Rekonštitúciu,
kombinovanie a zmiešavanie roztokov je potrebné vykonávať za aseptických podmienok.
Rekonštitúcia
1. Každá injekčná liekovka Ruconestu (2100 U) sa má rekonštituovať so 14 ml rozpúšťadla.
2. Gumové zátky liekoviek s práškom a s rozpúšťadlom vydezinfikujte a na každú z týchto
liekoviek nasaďte adaptér, aby zapadol na hrdlo liekovky.
3. K adaptéru na liekovke s rozpúšťadlom pripojte injekčnú striekačku a otočte ju v smere hodinových ručičiek, kým nedôjde k jej zafixovaniu. Natiahnite 14 ml rozpúšťadla. Odpojte striekačku od adaptéra jej otočením proti smeru hodinových ručičiek a liekovku s adaptérom zlikvidujte.
4. Pripojte injekčnú striekačku s rozpúšťadlom k adaptéru na liekovke s práškom a otočte ju
v smere hodinových ručičiek, kým nedôjde k jej zafixovaniu. Rozpúšťadlo sa musí pridávať pomaly, aby nedošlo ku jeho prudkému nárazu na prášok a musí sa jemne premiešavať, aby sa minimalizovalo spenenie roztoku. Nechajte striekačku na adaptéri. Ak potrebujete pripraviť druhý roztok, zopakujte kroky 3 a 4 (použite druhú súpravu).
5. Rekonštituovaný roztok obsahuje 150 U/ml konestatu alfa a javí sa ako číry, bezfarebný roztok.
Rekonštituovaný roztok v každej liekovke sa má vizuálne skontrolovať na prítomnosť častíc a zmenu farby. Roztok obsahujúci častice a so zmeneným sfarbením sa nesmie použiť. Malé množstvá peny sú prípustné. Liek sa musí použiť okamžite (pozri časť 6.3).
Podanie1. Do injekčnej striekačky natiahnite požadované množstvo pripraveného roztoku. Nikdy nepresiahnite objem 14 ml na jednu injekčnú striekačku. Odpojte striekačku (striekačky) jej (ich) otočením proti smeru hodinových ručičiek a liekovku s adaptérom zlikvidujte.
2. Pripojte infúznu súpravu k injekčnej striekačke a otočte ju v smere hodinových ručičiek, kým nedôjde k jej zafixovaniu. Striekačku pridržte v polohe s hrotom smerujúcim nahor a jemne stlačte piest, čím naplníte infúznu súpravu roztokom.
3. Miesto vpichu dezinfikujte alkoholovým tampónom. Z ihly infúznej súpravy odstráňte kryt a opatrne zasuňte ihlu do žily.
4. Uistite sa, že je turniket uvoľnený. Jemne vstreknite roztok do žily – vstrekujte približne 5
minút.
5. Ak boli pripravené dve injekčné striekačky: zohnite hadičku, aby sa zabránilo spätnému toku, odskrutkujte prázdnu striekačku z infúznej súpravy (proti smeru hodinových ručičiek) a ihneď ju nahraďte druhou striekačkou. Pozvoľne vstreknite roztok druhej striekačky.
LikvidáciaBezpečne zlikvidujte použitú infúznu súpravu, všetok nepoužitý roztok, injekčnú striekačku a prázdnu injekčnú liekovku vo vhodnej nádobe na zdravotnícky odpad. V prípade nesprávnej likvidácie môžu tieto materiály spôsobiť iným ľuďom ujmu na zdraví. Pomôcky nepoužívajte opakovane.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPharming Group N.V. Darwinweg 24
NL-2333 CR LEIDEN Holandsko
8. REGISTRAČNÉ ČÍSLOEU/1/10/641/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.