
>Vakcinácia
Nie sú dostupné žiadne údaje týkajúce sa odpovede na očkovanie živými vakcínami u pacientov
užívajúcich upadacitinib. Použitie živých atenuovaných vakcín počas liečby upadacitinibom alebo krátko pred ňou sa neodporúča. Pred začatím liečby upadacitinibom sa odporúča, aby pacienti absolvovali všetky očkovania vrátane profylaktických vakcinácií proti vírusu herpes zoster v súlade so súčasnými smernicami pre imunizáciu (pozri časť 5.1 pre údaje o inaktivovanej pneumokokovej polysacharidovej konjugovanej vakcíne (13-valentnej, adsorbovanej) a súbežnom použití
s upadacitinibom).
Malignita
U pacientov s reumatoidnou artritídou je zvýšené riziko výskytu malignít vrátane lymfómu.
Imunomodulačné lieky môžu zvýšiť riziko výskytu malignít vrátane lymfómu. Klinické údaje sú v súčasnosti obmedzené a prebiehajú dlhodobé štúdie.
V klinických štúdiách s upadacitinibom sa pozorovali malignity. Pred začatím liečby u pacientov
so známou malignitou inou ako úspešne liečená nemelanómová rakovina kože (non-melanoma skin cancer, NMSC) alebo pri zvažovaní pokračovania liečby upadacitinibom u pacientov, u ktorých došlo k rozvoju malignity, sa majú zvážiť riziká a prínosy liečby upadacitinibom.
Nemelanómová rakovina kože
U pacientov liečených upadacitinibom sa hlásili prípady NMSC. U pacientov so zvýšeným rizikom rakoviny kože sa odporúča pravidelné vyšetrenie kože.
Hematologické abnormality
V klinických skúšaniach sa u ≤ 1 % pacientov hlásil celkový počet neutrofilov (ANC) < 1 x 109
buniek/l, celkový počet lymfocytov (ALC) < 0,5 x 109 buniek/l a hladina hemoglobínu < 8 g/dl (pozri časť 4.8). S liečbou sa nemá začínať alebo liečba sa má dočasne prerušiť u pacientov s ANC < 1 x 109
buniek/l, ALC < 0,5 x 109 buniek/l alebo hladinou hemoglobínu < 8 g/dl, pozorovaných počas
rutinných vyšetrení pacientov (pozri časť 4.2).
Kardiovaskulárne riziko
Pacienti s reumatoidnou artritídou majú zvýšené riziko kardiovaskulárnych porúch. Pacienti liečení
upadacitinibom majú mať kontrolované rizikové faktory (napr. hypertenziu, hyperlipidémiu) ako súčasť bežnej štandardnej starostlivosti.
Divertikulitída
Prípady divertikulitídy boli hlásené v klinických skúšaniach a zo zdrojov po uvedení lieku na trh.
Divertikulitída môže spôsobiť gastrointestinálnu perforáciu. Upadacitinib sa má používať
s opatrnosťou u pacientov s divertikulárnym ochorením a obzvlášť u pacientov chronicky liečených súbežnými liekmi spojenými so zvýšeným rizikom divertikulitídy: nesteroidné protizápalové lieky, kortikosteroidy a opioidy. Pacienti s novým nástupom abdominálnych prejavov a symptómov majú byť ihneď vyšetrení, aby sa včas rozpoznala divertikulitída a zabránilo sa gastrointestinálnej perforácii.
Lipidy
Liečba upadacitinibom bola spojená so zvýšeniami lipidových parametrov v závislosti od dávky,
vrátane celkového cholesterolu, cholesterolu s nízkou hustotou lipoproteínov (LDL) a cholesterolu
s vysokou hustotou lipoproteínov (HDL) (pozri časť 4.8). Zvýšené hladiny LDL cholesterolu poklesli na hladiny pred liečbou v reakcii na liečbu statínmi, aj keď sú dôkazy obmedzené. Vplyv týchto zvýšení lipidových parametrov na kardiovaskulárnu morbiditu a mortalitu nebol stanovený (pokyny na monitorovanie pozri časť 4.2).
Zvýšenia pečeňovýchtransamináz
Liečba upadacitinibom bola v porovnaní s placebom spojená so zvýšeným výskytom zvýšení
pečeňových enzýmov.
Pečeňové transaminázy sa majú vyhodnotiť na začiatku liečby a následne v súlade so štandardnou starostlivosťou o pacienta. Na identifikáciu možných prípadov poškodenia pečene, vyvolaného liekom, sa odporúča bezodkladné vyšetrenie príčiny zvýšenia pečeňových enzýmov.
Ak sa počas rutinného vyšetrenia pacienta pozorujú zvýšenia ALT alebo AST a existuje podozrenie na poškodenie pečene vyvolané liekom, liečba upadacitinibom sa má prerušiť dovtedy, kým sa táto diagnóza nevylúči.
Venózna tromboembólia
U pacientov používajúcich inhibítory JAK, vrátane upadacitinibu, boli hlásené prípady hlbokej žilovej
trombózy (deep venous thrombosis, DVT) a pľúcnej embólie (PE). Medzi rizikové faktory, ktoré sa majú brať do úvahy pri určovaní rizika DVT/PE u pacientov, patria vek, obezita, DVT/PE
v anamnéze, pacienti podstupujúci veľký chirurgický zákrok a dlhodobá imobilizácia. U pacientov s vysokým rizikom DVT/PE sa má upadacitinib používať s opatrnosťou. Ak sa objavia klinické prejavy DVT/PE, liečba upadacitinibom sa má prerušiť a pacienti majú byť bezodkladne vyšetrení
a následne náležite liečení.
Staršie osoby
Pri podávaní dávky upadacitinibu 30 mg jedenkrát denne pacientom vo veku 65 rokov a starším existuje zvýšené riziko nežiaducich reakcií. Odporúčaná dávka pri dlhodobom používaní je u tejto populácie pacientov 15 mg jedenkrát denne (pozri časti 4.2 a 4.8).
Reakcie zprecitlivenosti
U pacientov dostávajúcich upadacitinib boli hlásené závažné reakcie z precitlivenosti, ako je
anafylaxia a angioedém. V prípade výskytu klinicky významnej reakcie z precitlivenosti prerušte podávanie upadacitinibu a začnite vhodnú liečbu (pozri časti 4.3 a 4.8).
4.5 Liekové a iné interakcie
Potenciál iných liekov ovplyvniť farmakokinetickévlastnosti upadacitinibu
Upadacitinib je metabolizovaný najmä prostredníctvom CYP3A4. Z tohto dôvodu môžu byť
plazmatické expozície lieku ovplyvnené liekmi, ktoré silne inhibujú alebo indukujú CYP3A4.
Súbežné podávanie s inhibítormi CYP3A4
Expozícia upadacitinibu je zvýšená, ak sa súbežne podáva so silnými inhibítormi CYP3A4 (ako napr. ketokonazol, itrakonazol, pozakonazol, vorikonazol, klaritromycín a grapefruit). V klinickej štúdii viedlo súbežné podávanie upadacitinibu s ketokonazolom k 70 % zvýšeniu Cmax a 75 % zvýšeniu AUC upadacitinibu. U pacientov dlhodobo liečených silnými inhibítormi CYP3A4 sa má upadacitinib
15 mg jedenkrát denne používať s opatrnosťou. U pacientov s atopickou dermatitídou dlhodobo
liečených silnými inhibítormi CYP3A4 sa upadacitinib v dávke 30 mg jedenkrát denne neodporúča. U pacientov s ulceróznou kolitídou, ktorí užívajú silné inhibítory CYP3A4, je odporúčaná úvodná dávka 30 mg jedenkrát denne (maximálne 16 týždňov) a odporúčaná udržiavacia dávka 15 mg jedenkrát denne (pozri časť 4.2). Pri dlhodobom používaní silných inhibítorov CYP3A4 sa má zvážiť použitie alternatívnych liečiv. Počas liečby upadacitinibom je potrebné vyhnúť sa konzumácii potravín alebo nápojov obsahujúcich grapefruit.
Súbežné podávanie s induktormi CYP3A4
Expozícia upadacitinibu je znížená, ak sa súbežne podáva so silnými induktormi CYP3A4 (ako napr. rifampicín a fenytoín), čo môže viesť k zníženému terapeutickému účinku upadacitinibu. V klinickej štúdii viedlo súbežné podanie upadacitinibu a opakovaných dávok rifampicínu (silný induktor CYP3A4) k približne 50 % zníženiu Cmax a 60 % zníženiu AUC upadacitinibu. Ak sa upadacitinib podáva súbežne so silnými induktormi CYP3A4, je potrebné u pacientov sledovať zmeny v aktivite ochorenia.
Metotrexát a lieky upravujúce pH (napr. antacidá alebo inhibítory protónovej pumpy) nemajú žiadny účinok na plazmatické expozície upadacitinibu.
Potenciál upadacitinibu ovplyvniť farmakokinetickévlastnosti iných liekov
Opakované podávanie dávok upadacitinibu 30 mg alebo 45 mg jedenkrát denne zdravým jedincom
malo obmedzený účinok na plazmatické expozície midazolamu (citlivý substrát pre CYP3A4)
(24 - 26 % zníženie AUC a Cmax midazolamu), čo naznačuje, že upadacitinib 30 mg alebo 45 mg jedenkrát denne môže mať slabý indukčný účinok na CYP3A. V klinickej štúdii sa po opakovanom podávaní dávky upadacitinibu 30 mg jedenkrát denne zdravým jedincom znížila AUC rosuvastatínu
o 33 %, AUC atorvastatínu o 23 % a Cmax rosuvastatínu o 23 %. Upadacitinib nemal žiadny významný účinok na Cmax atorvastatínu ani na plazmatické expozície orto-hydroxyatorvastatínu (hlavný aktívny
metabolit atorvastatínu). Podanie opakovaných dávok upadacitinibu 45 mg jedenkrát denne zdravým
jedincom viedlo k obmedzenému zvýšeniu AUC a Cmax dextrometorfánu (citlivý substrát CYP2D6)
o 30 % resp. 35 %, čo naznačuje, že dávka upadacitinibu 45 mg jedenkrát denne má slabý inhibičný účinok na CYP2D6. Pri súbežnom podávaní upadacitinibu a substrátov CYP3A, substrátov CYP2D6, rosuvastatínu alebo atorvastatínu sa neodporúča žiadna úprava dávky.
Upadacitinib nemá žiadne významné účinky na plazmatické expozície etinylestradiolu, levonorgestrelu, metotrexátu ani liečiv, ktoré sú substrátmi pre metabolizmus prostredníctvom CYP1A2, CYP2B6, CYP2C9 alebo CYP2C19.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženy vo fertilnom veku je potrebné poučiť, aby počas liečby a počas 4 týždňov od poslednej dávky
upadacitinibu používali účinnú antikoncepciu. Pediatrické pacientky a/alebo ich rodičov/opatrovateľov
treba informovať, aby kontaktovali ošetrujúceho lekára, ak pacientka dostane počas užívania upadacitinibu prvú menštruáciu.
Gravidita
K dispozícii nie sú žiadne alebo len obmedzené údaje týkajúce sa používania upadacitinibu
u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Upadacitinib bol teratogénny u potkanov a králikov s účinkami na kosti u plodov potkana a s účinkami na srdce
u plodov králika pri expozícii in utero.
Upadacitinib je v gravidite kontraindikovaný (pozri časť 4.3).
Ak pacientka počas užívania upadacitinibu otehotnie, rodičia majú byť informovaní o možnom riziku pre plod.
Dojčenie
Nie je známe, či sa upadacitinib/metabolity vylučujú do materského mlieka. Dostupné
farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie upadacitinibu do mlieka
(pozri časť 5.3).
Nie je možné vylúčiť riziko pre novorodencov/dojčatá.
Upadacitinib sa nemá používať počas dojčenia. Rozhodnutie o tom, či sa má prerušiť dojčenie alebo ukončiť liečba upadacitinibom, sa má vykonať na základe posúdenia prínosu dojčenia pre dieťa
a prínosu liečby pre ženu.
Fertilita
Účinok upadacitinibu na fertilitu u ľudí nebol hodnotený. Štúdie na zvieratách nepreukázali účinky
s ohľadom na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Upadacitinib nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
V placebom kontrolovaných klinických skúšaniach s reumatoidnou artritídou, psoriatickou artritídou
a axiálnou spondyloartritídou boli najčastejšie hlásenými nežiaducimi reakciami (≥ 2 %pacientov v prípade najmenej jednej indikácie s najvyššou mierou výskytu spomedzi uvedených indikácií)
u upadacitinibu 15 mg infekcie horných dýchacích ciest (19,5 %), zvýšená kreatínfosfokináza (CPK)
(8,6 %), zvýšená alaníntransamináza (4,3 %), bronchitída (3,9 %), nevoľnosť (3,5 %), neutropénia
(2,8 %), kašeľ (2,2 %), zvýšená aspartát transamináza (2,2 %) a hypercholesterolémia (2,2 %).
V placebom kontrolovaných klinických skúšaniach s atopickou dermatitídou boli najčastejšie hlásenými nežiaducimi reakciami (≥ 2 % pacientov) u upadacitinibu 15 mg alebo 30 mg infekcie horných dýchacích ciest (25,4 %), akné (15,1 %), herpes simplex (8,4 %), bolesť hlavy (6,3 %), zvýšená CPK (5,5 %), kašeľ (3,2 %), folikulitída (3,2 %), bolesť brucha (2,9 %), nevoľnosť (2,7 %), neutropénia (2,3 %), pyrexia (2,1 %) a chrípka (2,1 %).
Najčastejšie závažné nežiaduce reakcie boli závažné infekcie (pozri časť 4.4).
Bezpečnostný profil upadacitinibu pri dlhodobej liečbe bol vo všetkých indikáciách všeobecne podobný bezpečnostnému profilu počas placebom kontrolovaného obdobia.
V placebom kontrolovaných klinických štúdiách úvodnej a udržiavacej dávky pri ulceróznej kolitíde boli najčastejšie hlásenými nežiaducimi reakciami (≥ 3 % pacientov) u upadacitinibu 45 mg, 30 mg alebo 15 mg infekcia horných dýchacích ciest (19,9 %), zvýšená hladina CPK v krvi (7,6 %), akné (6,3 %), neutropénia (6,0 %), vyrážka (5,2 %), herpes zoster (4,4 %), hypercholesterolémia (4,0 %), folikulitída (3,6 %), herpes simplex (3,2 %) a chrípka (3,2 %).
Tabuľkový zoznamnežiaducichreakciíNasledujúci zoznam nežiaducich reakcií je založený na skúsenostiach z klinických štúdií. Frekvencia
nežiaducich reakcií uvedených nižšie je definovaná pomocou nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100). Frekvencie v tabuľke 3 sú založené na vyššej miere výskytu nežiaducich reakcií hlásených v klinických skúšaniach s liekom RINVOQ pri liečbe reumatického ochorenia (15 mg), atopickej dermatitídy (15 mg a 30 mg) alebo ulceróznej kolitídy (15 mg, 30 mg a 45 mg). Ak sa medzi indikáciami pozorovali výrazné rozdiely vo frekvencii, sú uvedené v poznámkach pod tabuľkou.
Tabuľka 3 Nežiaduce reakcieTrieda
orgánových systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Infekcie a nákazy
| infekcie horných dýchacích ciest (upper respiratory tract infections, URTI)a
| bronchitídaa,b
herpes zoster herpes simplexa folikulitída chrípka
infekcia močových ciest
| pneumónia
orálna kandidóza
divertikulitída
|
Poruchy krvi a lymfatického systému
|
| anémia neutropénia lymfopénia
|
|
Poruchy imunitného systému
|
| žihľavkac
| závažné reakcie
z precitlivenostia,e
|
Poruchy metabolizmu a výživy
|
| hypercholesterolémiab
hyperlipidémiaa,b
| hypertriglyceridémia
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
| kašeľ
|
|
Poruchy
gastrointestinálneho traktu
|
| bolesť bruchaa,d
nevoľnosť
|
|
Poruchy kože a podkožného tkaniva
| aknéc,d
| vyrážkaa
|
|
Celkové poruchy a reakcie v mieste podania
|
| únava pyrexia
|
|
Laboratórne a funkčné vyšetrenia
|
| zvýšená hladina CPK
v krvi
zvýšená hladina ALTb
zvýšená hladina ASTb zvýšenie telesnej hmotnosti
|
|
Poruchy nervového
systému
|
| bolesť hlavy
|
|
a Prezentované ako zoskupený názov
b V skúšaniach s atopickou dermatitídou bola frekvencia bronchitídy, hypercholesterolémie, hyperlipidémie, zvýšenej ALT a zvýšenej AST menej častá.
c V skúšaniach s reumatickým ochorením bola frekvencia častá pre akné a menej častá pre urtikáriu.
d V skúšaniach s ulceróznou kolitídou bola frekvencia častá pre akné; frekvencia výskytu
|

bolesti brucha bola menej častá pri upadacitinibe ako pri placebe.
e Závažné reakcie z precitlivenosti vrátane anafylaktickej reakcie a angioedému
OpisvybranýchnežiaducichreakciíReumatoidnáartiritídaInfekcieV placebom kontrolovaných klinických štúdiách so sprievodnou liečbou DMARD bola frekvencia výskytu infekcie počas 12/14 týždňov v skupine s upadacitinibom 15 mg 27,4 % v porovnaní s 20,9 % v skupine s placebom. V metotrexátom (MTX) kontrolovaných štúdiách bola frekvencia výskytu infekcie počas 12/14 týždňov v skupine s upadacitinibom 15 mg v monoterapii 19,5 % v porovnaní
s 24,0 % v skupine s placebom. Celková dlhodobá miera výskytu infekcií v skupine s upadacitinibom
15 mg vo všetkých piatich klinických štúdiách fázy 3 (2630 pacientov) bola 93,7 udalostí na
100 pacientorokov.
V placebom kontrolovaných klinických štúdiách so sprievodnou liečbou DMARD bola frekvencia výskytu závažnej infekcie počas 12/14 týždňov v skupine s upadacitinibom 15 mg 1,2 % v porovnaní s 0,6 % v skupine s placebom. V metotrexátom (MTX) kontrolovaných štúdiách bola frekvencia výskytu závažnej infekcie počas 12/14 týždňov v skupine s upadacitinibom 15 mg v monoterapii
0,6 % v porovnaní s 0,4 % v skupine s MTX. Celková dlhodobá miera výskytu závažných infekcií v skupine s upadacitinibom 15 mg vo všetkých piatich klinických štúdiách fázy 3 (2630 pacientov) bola 3,8 udalostí na 100 pacientorokov. Najčastejšou závažnou infekciou bola pneumónia. Miera výskytu závažných infekcií pri dlhodobej expozícii zostala ustálená.
Oportúnne infekcie (okrem tuberkulózy)V placebom kontrolovaných klinických štúdiách so sprievodnou liečbou DMARD bola frekvencia výskytu oportúnnych infekcií počas 12/14 týždňov v skupine s upadacitinibom 15 mg 0,5 %
v porovnaní s 0,3 % v skupine s placebom. V MTX kontrolovaných štúdiách sa neobjavili žiadne
prípady oportúnnych infekcií počas 12/14 týždňov v skupine s upadacitinibom 15 mg a objavili sa u 0,2 % v skupine s MTX. Celková dlhodobá miera výskytu oportúnnych infekcií v skupine
s upadacitinibom 15 mg vo všetkých piatich klinických štúdiách fázy 3 bola 0,6 udalostí na 100
pacientorokov.
Dlhodobá miera výskytu herpes zoster v skupine s upadacitinibom 15 mg vo všetkých piatich klinických štúdiách fázy 3 bola 3,7 udalostí na 100 pacientorokov. Väčšina prípadov herpes zoster zahŕňala jeden dermatóm a nebola závažná.
Zvýšenia pečeňových transaminázV placebom kontrolovaných štúdiách so sprievodnou liečbou DMARD až do 12/14 týždňov boli u pacientov liečených upadacitinibom 15 mg pozorované zvýšenia alaníntransaminázy (ALT)
o ≥ 3-násobok hornej hranice normálu (upper limit of normal, ULN) pri minimálne jednom stanovení u 2,1 % pacientov a aspartáttransaminázy u 1,5 % pacientov v porovnaní s 1,5 % pre alaníntransaminázu a 0,7 % pre aspartát transaminázu u pacientov, ktorí dostávali placebo. Väčšina
prípadov zvýšení pečeňových transamináz bola asymptomatická a prechodná.
V MTX kontrolovaných štúdiách až do 12/14 týždňov boli pozorované zvýšenia o ≥ 3-násobok ULN pri minimálne jednom stanovení hladiny ALT u 0,8 % pacientov a hladiny AST u 0,4 % pacientov liečených upadacitinibom 15 mg v porovnaní s 1,9 % pre ALT a 0,9 % pre AST u pacientov, ktorí dostávali placebo.
Vzorec a výskyt zvýšenia hladiny ALT/AST zostali v priebehu času ustálené vrátane dlhodobých predĺžených štúdií.
Zvýšenia hladín lipidov
Liečba upadacitinibom 15 mg bola spojená so zvýšeniami lipidových parametrov, vrátane celkového cholesterolu, triglyceridov, LDL cholesterolu a HDL cholesterolu. Neobjavila sa žiadna zmena v pomere LDL/HDL. Zvýšenia boli pozorované v 2. až 4. týždni liečby a zostali ustálené pri dlhodobej liečbe. Medzi pacientmi v kontrolovaných štúdiách s východiskovými hodnotami pod špecifikovanými limitmi sa pozorovali nasledovné frekvencie zmeny nad špecifikované limity
v minimálne jednom prípade počas 12/14 týždňov (vrátane pacientov, u ktorých sa objavili izolované prípady zvýšenej hodnoty):
· Celkový cholesterol ≥ 5,17 mmol/l (200 mg/dl): 62 % v skupine s upadacitinibom 15 mg vs.
31 % v skupine s placebom
· LDL cholesterol ≥ 3,36 mmol/l (130 mg/dl): 42 % v skupine s upadacitinibom 15 mg vs. 19 %
v skupine s placebom
· HDL cholesterol ≥ 1,03 mmol/l (40 mg/dl): 89 % v skupine s upadacitinibom 15 mg vs. 61 %
v skupine s placebom
· Triglyceridy ≥ 2,26 mmol/l (200 mg/dl): 25 % v skupine s upadacitinibom 15 mg vs. 15 %
v skupine s placebom
Kreatínfosfokináza
V placebom kontrolovaných štúdiách DMARD počas najmenej 12/14 týždňov sa pozorovali zvýšenia hodnôt CPK. Zvýšenia hodnôt CPK o > 5-násobok hornej hranice normálu (ULN) boli do
12./14. týždňa hlásené u 1 % pacientov v skupine s upadacitinibom 15 mg a u 0,3 % pacientov
v skupine s placebom. Väčšina zvýšení hodnôt o > 5-násobok ULN bola prechodná a nevyžadovala vysadenie liečby. Priemerné hodnoty CPK sa do 4. týždňa zvýšili s priemerným zvýšením o 60 U/l v 12. týždni a následne zostali stabilné na zvýšenej hodnote vrátane predĺženej liečby.
Neutropénia
V placebom kontrolovaných štúdiách so sprievodnou liečbou DMARD počas najmenej 12/14 týždňov došlo k poklesom počtu neutrofilov pod 1 x 109 buniek/l pri minimálne jednom stanovení u 1,1 % pacientov v skupine s upadacitinibom 15 mg a < 0,1 % pacientov v skupine s placebom. V klinických štúdiách sa pri ANC < 1 x 109 buniek/l liečba prerušila (pozri časť 4.2). Priemerný počet neutrofilov sa počas 4 až 8 týždňov znížil. Postupom času vrátane predĺženej liečby zostali poklesy počtu neutrofilov ustálené na hodnote nižšej, ako boli na začiatku.
Psoriatická artritída
Celkový bezpečnostný profil pozorovaný u pacientov s aktívnou psoriatickou artritídou, ktorí boli
liečení upadacitinibom 15 mg, bol konzistentný s bezpečnostným profilom pozorovaným u pacientov
s reumatoidnou artritídou. U pacientov liečených upadacitinibom v kombinácii s liečbou MTX bol v porovnaní s pacientmi liečenými v monoterapii pozorovaný vyšší výskyt závažných infekcií
(2,6 prípadu na 100 pacientorokov resp. 1,3 prípadu na 100 pacientorokov) a zvýšenia pečeňových
transamináz (zvýšenia ALT 3. stupňa a vyšší výskyt 1,4 % resp. 0,4 %).
Axiálna spondyloartritída
Celkový bezpečnostný profil pozorovaný u pacientov s aktívnou axiálnou spondyloartritídou, ktorí
boli liečení upadacitinibom 15 mg, bol konzistentný s bezpečnostným profilom pozorovaným
u pacientov s reumatoidnou artritídou. Neboli identifikované žiadne nové bezpečnostné zistenia.
Atopická
dermatitída
Infekcie
V placebom kontrolovanom období klinických štúdií bola počas 16 týždňov v skupinách
s upadacitinibom 15 mg a 30 mg frekvencia výskytu infekcie 39 %, resp. 43 % v porovnaní s 30 %
v skupine s placebom. V skupinách s upadacitinibom 15 mg a 30 mg bola dlhodobá miera výskytu infekcií 98,5; resp. 109,6 prípadu na 100 pacientorokov.
V placebom kontrolovaných klinických štúdiách bola počas 16 týždňov v skupinách s upadacitinibom
15 mg a 30 mg frekvencia výskytu závažnej infekcie 0,8 %; resp. 0,4 % v porovnaní s 0,6 % v skupine s placebom. Dlhodobá miera výskytu závažných infekcií bola v skupinách s upadacitinibom 15 mg
a 30 mg 2,3; resp. 2,8 prípadu na 100 pacientorokov.
Oportúnne infekcie (okrem tuberkulózy)
V placebom kontrolovanom období klinických štúdií boli všetky hlásené oportúnne infekcie (okrem TBC a herpes zoster) herpetický ekzém. Počas 16 týždňov bola v skupinách s upadacitinibom 15 mg a 30 mg frekvencia herpetického ekzému 0,7 %; resp. 0,8 % v porovnaní s 0,4 % v skupine
s placebom. V skupinách s upadacitinibom 15 mg a 30 mg bola dlhodobá miera výskytu herpetického
ekzému 1,6; resp. 1,8 prípadu na 100 pacientorokov. Pri liečbe upadacitinibom 30 mg bol hlásený jeden prípad kandidózy pažeráka.
Dlhodobá miera výskytu herpes zoster v skupinách s upadacitinibom 15 mg a 30 mg bola 3,5 prípadu;
resp. 5,2 prípadu na 100 pacientorokov. Väčšina prípadov herpes zoster zahŕňala jeden dermatóm a nebola závažná.
Laboratórne abnormality
Od dávky závislé zmeny zvýšení ALT a/alebo zvýšení AST (≥ 3-násobok ULN), lipidových parametrov, hodnôt CPK (> 5-násobok ULN) a neutropénie (ANC < 1 x 109 buniek/l) spojené s liečbou upadacitinibom boli podobné tým, ktoré boli pozorované v klinických štúdiách
s reumatickými chorobami.
Malé zvýšenia LDL cholesterolu boli pozorované po 16. týždni v štúdiách s atopickou dermatitídou.
Ulcerózna kolitída
Celkový bezpečnostný profil pozorovaný u pacientov s ulceróznou kolitídou sa vo všeobecnosti
zhodoval s bezpečnostným profilom pozorovaným u pacientov s reumatoidnou artritídou.
Pri úvodnej liečbe v trvaní 16 týždňov v porovnaní s 8 týždňami bol pozorovaný vyšší výskyt herpes zoster.
Infekcie
V placebom kontrolovaných štúdiách úvodnej dávky bola frekvencia výskytu infekcie počas
8-týždňového obdobia v skupine užívajúcej upadacitinib 45 mg 20,7 % v porovnaní so skupinou
užívajúcou placebo, kde bol výskyt infekcie 17,5 %. V placebom kontrolovanej štúdii udržiavacej dávky bola frekvencia výskytu infekcie počas obdobia 52 týždňov v skupine užívajúcej upadacitinib
15 mg a 30 mg 38,4 % resp. 40,6 % v porovnaní so skupinou užívajúcou placebo, kde bol výskyt
infekcie 37,6 %. Dlhodobá miera výskytu infekcií pri podávaní dávky upadacitinibu 15 mg a 30 mg bola 73,8 resp. 82,6 udalostí na 100 pacientorokov.
V placebom kontrolovaných štúdiách úvodnej dávky bola frekvencia výskytu závažnej infekcie počas
8-týždňového obdobia v skupine užívajúcej upadacitinib 45 mg aj v skupine užívajúcej placebo 1,3 %. Počas 8-týždňového predĺženého obdobia liečby upadacitinibom 45 mg neboli pozorované žiadne ďalšie závažné infekcie. V placebom kontrolovanej štúdii udržiavacej dávky bola frekvencia výskytu
závažnej infekcie počas obdobia 52 týždňov v skupine užívajúcej upadacitinib 15 mg a 30 mg 3,2 % resp. 2,4 % v porovnaní so skupinou užívajúcou placebo, kde bol výskyt infekcie 3,3 %. Dlhodobá miera výskytu závažných infekcií pri podávaní dávky upadacitinibu 15 mg a 30 mg bola 4,1 resp.
3,9 udalostí na 100 pacientorokov. Najčastejšie hlásenou závažnou infekciou v úvodnej a udržiavacej fáze bol zápal pľúc spôsobený infekciou COVID-19.
Oportúnne infekcie (okrem tuberkulózy)
V placebom kontrolovaných štúdiách úvodnej dávky v trvaní 8 týždňov bola frekvencia výskytu oportúnnej infekcie (okrem tuberkulózy a herpes zoster) v skupine užívajúcej upadacitinib 45 mg
0,4 % v porovnaní so skupinou užívajúcou placebo, kde bola frekvencia výskytu 0,3 %. Počas
8-týždňového predĺženého obdobia liečby upadacitinibom 45 mg neboli pozorované žiadne ďalšie oportúnne infekcie (okrem tuberkulózy a herpes zoster). V placebom kontrolovanej štúdii udržiavacej dávky počas obdobia 52 týždňov bola frekvencia výskytu oportúnnej infekcie (okrem tuberkulózy
a herpes zoster) v skupine užívajúcej upadacitinib 15 mg a 30 mg 0,8 % resp. 0,4 %, v porovnaní so skupinou užívajúcou placebo, kde bol výskyt 0,8 %. Dlhodobá miera výskytu oportúnnych infekcií (okrem tuberkulózy a herpes zoster) pri dávke upadacitinibu 15 mg a 30 mg bola 0,6 resp. 0,3 udalostí na 100 pacientorokov.
V placebom kontrolovaných štúdiách úvodnej dávky v trvaní 8 týždňov bola frekvencia výskytu herpes zoster v skupine užívajúcej upadacitinib 45 mg 0,6 % v porovnaní so skupinou užívajúcou placebo, kde bol výskyt infekcie 0 %. Frekvencia výskytu herpes zoster počas 16-týždňovej liečby upadacitinibom 45 mg bola 3,9 %. V placebom kontrolovanej štúdii udržiavacej dávky bola frekvencia výskytu herpes zoster počas obdobia 52 týždňov v skupine užívajúcej upadacitinib 15 mg a 30 mg
4,4 % resp. 4,0 %, v porovnaní so skupinou užívajúcou placebo, kde bol výskyt 0 %. Dlhodobá miera výskytu herpes zoster pri dávke upadacitinibu 15 mg a 30 mg bola 5,7 resp. 6,3 udalostí na
100 pacientorokov.
Laboratórne abnormality
V klinických skúšaniach úvodnej a udržiavacej dávky boli laboratórne zmeny zvýšení ALT a/alebo zvýšení AST (≥ 3 x ULN), hodnôt CPK (> 5 x ULN) a neutropénie (ANC < 1 x 109 buniek/l) súvisiace s liečbou upadacitinibom vo všeobecnosti podobné zmenám pozorovaným v klinických štúdiách reumatolického ochorenia a atopickej dermatitídy. Boli pozorované od dávky závislé zmeny týchto laboratórnych parametrov súvisiace s liečbou upadacitinibom 15 mg a 30 mg.
V placebom kontrolovaných štúdiách úvodnej dávky v trvaní do 8 týždňov sa zníženia počtu lymfocytov pod hodnotu 0,5 x 109 buniek/l pri minimálne jednom meraní vyskytli u 2,0 % a 0,8 % pacientov v skupine užívajúcej upadacitinib 45 mg resp. placebo. V placebom kontrolovaných štúdiách udržiavacej dávky v trvaní do 52 týždňov sa zníženia počtu lymfocytov pod hodnotu
0,5 x 109 buniek/l pri minimálne jednom meraní vyskytli u 1,6 %, 0,8 % a 0,8 % pacientov v skupine užívajúcej upadacitinib 15 mg, 30 mg resp. placebo. V klinických štúdiách sa liečba prerušila ako
reakcia na ALC < 0,5 x 109 buniek/l (pozri časť 4.2). Počas liečby upadacitinibom neboli pozorované žiadne významné zmeny priemeru počtu lymfocytov.
Počas 8-týždňovej liečby upadacitinibom 45 mg boli pozorované zvýšenia lipidových parametrov, ktoré boli pri dlhodobejšej liečbe upadacitinibom 15 mg a 30 mg vo všeobecnosti stabilné. Medzi pacientmi v placebom kontrolovaných štúdiách úvodnej dávky s východiskovými hodnotami nižšími ako stanovené limity boli pozorované nasledujúce frekvencie pacientov s posunom nad stanovené limity v minimálne jednom prípade počas 8 týždňov (vrátane pacientov, u ktorých sa objavili izolované prípady zvýšenej hodnoty):
· Celkový cholesterol ≥ 5,17 mmol/l (200 mg/dl): 49 % vs. 11 % v skupine s upadacitinibom 45 mg resp. s placebom
· LDL cholesterol ≥ 3,36 mmol/l (130 mg/dl): 27 % vs. 9 % v skupine s upadacitinibom 45 mg resp. s placebom
· HDL cholesterol ≥ 1,03 mmol/l (40 mg/dl): 79 % vs. 36 %v skupine s upadacitinibom 45 mg resp. s placebom
· Triglyceridy ≥ 2,26 mmol/l (200 mg/dl): 6 % vs. 4 % v skupine s upadacitinibom 45 mg resp. s placebom
Staršie osobyNa základe obmedzených údajov u pacientov s atopickou dermatitídou vo veku 65 rokov a starších
bola pri dávke upadacitinibu 30 mg vyššia miera výskytu celkových nežiaducich reakcií v porovnaní s dávkou 15 mg.

Na základe obmedzených údajov u pacientov s ulceróznou kolitídou vo veku 65 rokov a starších bola pri dávke upadacitinibu 30 mg vyššia miera výskytu nežiaducich reakcií v porovnaní s dávkou 15 mg pri udržiavacej liečbe (pozri časť 4.4).
Pediatrická populáciaV štúdiách fázy 3 bolo liečených celkovo 343 dospievajúcich vo veku 12 až 17 rokov s atopickou
dermatitídou, z ktorých 167 bolo vystavených dávke 15 mg. Bezpečnostný profil upadacitinibu 15 mg u dospievajúcich bol podobný ako u dospelých. Bezpečnosť a účinnosť 30 mg dávky u dospievajúcich sa stále skúma.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieUpadacitinib bol podávaný v klinických štúdiách až do dávok, ktoré v dennej AUC zodpovedali dávke
60 mg s predĺženým uvoľňovaním jedenkrát denne. Nežiaduce reakcie boli porovnateľné
s nežiaducimi účinkami pozorovanými pri nižších dávkach a neboli zistené žiadne špecifické toxické účinky. Približne 90 % upadacitinibu v systémovom obehu sa vylúči do 24 hodín od podania dávky (v rámci rozmedzia dávok hodnoteného v klinickej štúdii). V prípade predávkovania sa odporúča
sledovať pacienta na prejavy a príznaky nežiaducich reakcií. Pacienti, u ktorých sa objavia nežiaduce reakcie, majú dostať náležitú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, selektívne imunosupresíva. ATC kód: L04AA44
Mechanizmus účinkuUpadacitinib je selektívny a reverzibilný inhibítor Janusovej kinázy (JAK). JAK sú intracelulárne
enzýmy, ktoré prenášajú signály cytokínov alebo rastových faktorov zapojených do širokej škály
bunkových procesov vrátane zápalových odpovedí, hematopoézy a imunitných reakcií. Do skupiny enzýmov JAK patria štyria zástupcovia JAK1, JAK2, JAK3 a TYK2, ktoré vo dvojiciach fosforylujú a aktivujú signálne transduktory a aktivátory transkripcie (signal transducers and activators of transcription, STAT). Táto fosforylácia následne moduluje génovú expresiu a bunkovú funkciu. JAK1 má dôležitú úlohu pri prenášaní signálov zápalových cytokínov, zatiaľ čo JAK2 je dôležitá pre dozrievanie erytrocytov a signály JAK3 hrajú dôležitú úlohu pri imunitnom mechanizme a funkcii lymfocytov.
V testoch na ľudských bunkách upadacitinib prednostne inhibuje signály JAK1 alebo JAK1/3
s funkčnou selektivitou oproti cytokínovým receptorom, ktoré prenášajú signál prostredníctvom párov JAK2. Atopická dermatitída je vyvolaná prozápalovými cytokínmi (vrátane IL-4, IL-13, IL-22, TSLP, IL-31 a IFN-y), ktoré prenášajú signály cestou JAK1. Inhibícia JAK1 upadacitinibom znižuje signalizáciu mnohých mediátorov, ktoré vyvolávajú prejavy a príznaky atopickej dermatitídy, ako sú ekzematózne kožné lézie a svrbenie. Prozápalové cytokíny (primárne IL-6, IL-7, IL-15 a IFNγ) prenášajú signály dráhou JAK1 a sú súčasťou patogenézy ulceróznej kolitídy. Inhibícia JAK1 upadacitinibom moduluje signalizáciu cytokínov závislých na JAK, čo je základ pre zápalovú záťaž
a prejavy a príznaky ulceróznej kolitídy.
Farmakodynamické účinky
Inhibícia fosforylácie STAT3 indukovanej IL-6 a fosforylácie STAT5 indukovanej IL-7
U zdravých dobrovoľníkov viedlo podanie upadacitinibu (lieková forma s okamžitým uvoľňovaním) k inhibícii fosforylácie STAT3 indukovanej IL-6 (JAK1/JAK2) a fosforylácie STAT5 indukovanej IL-7 (JAK1/JAK3) závislej od dávky a koncentrácie v krvi. Maximálna inhibícia bola pozorovaná
1 hodinu od podania dávky, do konca dávkovacieho intervalu sa upravila na takmer východiskovú úroveň.
Lymfocyty
U pacientov s reumatoidnou artritídou bola liečba upadacitinibom spojená s malým prechodným zvýšením priemerného ALC od východiskového stavu až do 36. týždňa, ktoré sa pri pokračovaní v liečbe upravilo na takmer východiskový stav.
hsCRP
U pacientov s reumatoidnou artritídou bola liečba upadacitinibom spojená s poklesmi priemerných hladín hsCRP od východiskového stavu už v 1. týždni, ktoré sa udržali pri pokračovaní v liečbe.
Vakcinačná štúdia
Vplyv upadacitinibu na humorálnu odpoveď po podaní inaktivovanej pneumokokovej polysacharidovej konjugovanej vakcíny (13-valentnej, adsorbovanej) sa hodnotil u 111 pacientov
s reumatoidnou artritídou pri stabilnej liečbe upadacitinibom 15 mg (n = 87) alebo 30 mg (n = 24).
97 % pacientov (n = 108) užívalo súbežne metotrexát. Primárny cieľový ukazovateľ bol podiel
pacientov s uspokojivou humorálnou odpoveďou definovanou ako ≥ 2-násobné zvýšenie koncentrácie protilátok od východiskovej hodnoty do 4. týždňa u najmenej 6 z 12 pneumokokových antigénov (1, 3,
4, 5, 6B, 7F, 9V, 14, 18C, 19A, 19F a 23F). Výsledky v 4. týždni preukázali uspokojivú humorálnu
odpoveď u 67,5 % (95 % CI: 57,4; 77,5) a 56,5 % (95 % CI: 36,3; 76,8) pacientov liečených upadacitinibom 15 mg resp. 30 mg.
Klinická účinnosťabezpečnosť
Reumatoidná artritída
Účinnosť a bezpečnosť upadacitinibu 15 mg jedenkrát denne boli hodnotené v piatich randomizovaných, dvojito zaslepených, multicentrických štúdiách fázy 3 u pacientov so stredne ťažkou až ťažkou reumatoidnou artritídou a ktorí spĺňali klasifikačné kritéria ACR/EULAR 2010 (pozri tabuľku 4). Zúčastniť sa mohli pacienti vo veku 18 rokov a starší. Na začiatku sa vyžadovala prítomnosť minimálne 6 bolestivých a 6 opuchnutých kĺbov a dôkaz systémového zápalu na základe zvýšenia hladiny hsCRP. Štyri štúdie zahŕňali dlhodobé predĺženia až do 5 rokov a jedna štúdia (SELECT-COMPARE) zahŕňala dlhodobé predĺženie až do 10 rokov.
Primárna analýza pri každej z týchto štúdií zahŕňala všetkých randomizovaných jedincov, ktorí dostali aspoň 1 dávku upadacitinibu alebo placeba, a pri kategorických cieľových ukazovateľoch boli pripočítaní aj pacienti bez odpovede.
V štúdiách fázy 3 bola účinnosť pozorovaná pri upadacitinibe 15 mg jedenkrát denne vo všeobecnosti podobná účinnosti, ktorá sa pozorovala pri upadacitinibe 30 mg jedenkrát denne.
Tabuľka 4 Súhrn klinických skúšaníNázov štúdie
| Populácia (n)
| Liečebné skupiny
| Kľúčové výsledné stanovenia
|
SELECT-EARLY
| bez predchádzajúcej liečby MTXa (947)
| · upadacitinib
15 mg
· upadacitinib
30 mg
· MTX Monoterapia
| · Primárny cieľový ukazovateľ:
klinická remisia (DAS28-CRP) v
24. týždni
· Nízka aktivita ochorenia (DAS28- CRP)
· ACR50
· Rádiografická progresia (mTSS)
· Fyzické funkcie (HAQ-DI)
· SF-36 PCS
|
SELECT-
MONOTHERAPY
| MTX-IRb
(648)
| · upadacitinib
15 mg
· upadacitinib
30 mg
· MTX Monoterapia
| · Primárny cieľový ukazovateľ:
nízka aktivita ochorenia (DAS28-
CRP) v 14. týždni
· Klinická remisia (DAS28-CRP)
· ACR20
· Fyzické funkcie (HAQ-DI)
· SF-36 PCS
· Ranná stuhnutosť
|
SELECT-NEXT
| csDMARD-IRc
(661)
| · upadacitinib
15 mg
· upadacitinib
30 mg
· placebo
Základná liečba csDMARD
| · Primárny cieľový ukazovateľ: nízka aktivita ochorenia (DAS28- CRP) v 12. týždni
· Klinická remisia (DAS28-CRP)
· ACR20
· Fyzické funkcie (HAQ-DI)
· SF-36 PCS
· Nízka aktivita ochorenia (CDAI)
· Ranná stuhnutosť
· FACIT-F
|
SELECT- COMPARE
| MTX-IRd
(1 629)
| · upadacitinib
15 mg
· placebo
· adalimumab
40 mg
Základná liečba
MTX
| · Primárny cieľový ukazovateľ:
klinická remisia (DAS28-CRP) v
12. týždni
· Nízka aktivita ochorenia (DAS28- CRP)
· ACR20
· Nízka aktivita ochorenia (DAS28- CRP) vs. adalimumab
· Rádiografická progresia (mTSS)
· Fyzické funkcie (HAQ-DI)
· SF-36 PCS
· Nízka aktivita ochorenia (CDAI)
· Ranná stuhnutosť
· FACIT-F
|
SELECT- BEYOND
| bDMARD-IRe
(499)
| · upadacitinib
15 mg
· upadacitinib
30 mg
| · Primárny cieľový ukazovateľ: nízka aktivita ochorenia (DAS28- CRP) v 12. týždni
· ACR20
|
Názo
v štúdie
|
Populáci
a (n)
|
Liečebn
é skupiny
|
Kľúčové výsledné stanovenia
|
|
|
· placebo
Základná liečba csDMARD
|
· Fyzické funkcie (HAQ-DI)
· SF-36 PCS
|
Skratky: ACR20 (alebo 50) = zlepšenie podľa Americkej reumatologickej asociácie o ≥ 20 % (alebo o ≥ 50 %); bDMARD = biologické chorobu modifikujúce antireumatikum, CRP = C-reaktívny proteín, DAS28 = Skóre aktivity ochorenia pre 28 kĺbov, mTSS = modifikované celkové Sharpovo skóre, csDMARD = konvenčné syntetické chorobu modifikujúce antireumatikum, HAQ-DI = index postihnutia na základe dotazníka hodnotiaceho zdravotný stav a index funkčnej neschopnosti, SF-36
PCS = Stručný formulár (36) prieskumu zdravia (SF-36) - súhrn telesných komponentov,
CDAI = index klinickej aktivity ochorenia, FACIT-F = Funkčné hodnotenie skóre liečby chronických
ochorení – skóre únavy, IR = pacient s nedostatočnou odpoveďou, MTX = metotrexát, n = počet randomizovaných jedincov
a. Pacienti predtým neužívali MTX alebo nedostali viac ako 3 týždňové dávky MTX
b Pacienti mali nedostatočnú odpoveď na MTX
c Pacienti, ktorí mali nedostatočnú odpoveď na csDMARD; pacienti s predchádzajúcou expozíciou najmenej jednému bDMARD boli vhodní (až do 20 % celkového počtu pacientov), ak mali buď
obmedzenú expozíciu (< 3 mesiace) alebo museli vysadiť bDMARD z dôvodu intolerancie
d Pacienti, ktorí mali nedostatočnú odpoveď na MTX; pacienti s predchádzajúcou expozíciou najviac jednému bDMARD (okrem adalimumabu) boli vhodní (až do 20 % celkového počtu pacientov), ak
mali buď obmedzenú expozíciu (< 3 mesiace) alebo museli bDMARD ukončiť z dôvodu intolerancie
e Pacienti, ktorí mali nedostatočnú odpoveď alebo intoleranciu na minimálne jedno bDMARD
|
Klinická
odpoveď
Remisia a nízka aktivita ochorenia
Nízku aktivitu ochorenia (DAS28-CRP ≤ 3,2) a klinickú remisiu (DAS28-CRP < 2,6) v štúdiách dosiahol významne vyšší podiel pacientov liečených upadacitinibom v porovnaní s placebom, MTX alebo adalimumabom (tabuľka 5). V štúdii SELECT-COMPARE sa v 12. týždni dosiahli významne vyššie podiely nízkej aktivity ochorenia v porovnaní s adalimumabom. Celkove bola nízka aktivita ochorenia a miera klinickej remisie konzistentné v skupinách pacientov s MTX alebo bez neho.
V 3. roku ostalo 297/651 (45,6 %) a 111/327 (33,9 %) pacientov na pôvodne randomizovanej liečbe upadacitinibom 15 mg resp. adalimumabom vštúdii SELECT COMPARE a 216/317 (68,1 %)
a 149/315 (47,3 %) pacientov ostalo na pôvodne randomizovanej liečbe upadacitinibom 15 mg resp.
monoterapiou MTX v štúdii SELECT-EARLY. Medzi pacientmi, ktorí ostali na svojej pôvodne pridelenej liečbe, sa nízka aktivita ochorenia a klinická remisia udržali počas 3 rokov.
Odpoveď ACRVo všetkých štúdiách sa odpoveď ACR20, ACR50 a ACR 70 v 12. týždni dosiahla u väčšieho počtu pacientov liečených upadacitinibom 15 mg v porovnaní s placebom, MTX alebo adalimumabom (tabuľka 5). Nástup účinku bol rýchly vo všetkých sledovaných parametroch s väčšou odpoveďou pozorovanou už v 1. týždni pre ACR20. Boli pozorované pretrvávajúce miery odpovede (s MTX alebo bez neho), pričom odpovede ACR20/50/70 boli zachované po dobu 3 rokov u pacientov, ktorí ostali
na pôvodne pridelenej liečbe.
Liečba upadacitinibom 15 mg samostatne alebo v kombinácii s csDMARD viedla k zlepšeniam
v jednotlivých zložkách ACR vrátane počtu bolestivých a opuchnutých kĺbov, celkového hodnotenia pacientom a lekárom, HAQ-DI, hodnotenia bolesti a hsCRP.
Tabuľk
a 5 Odpoveď na liečbu a remisia
Štúdia
|
SELECT EARLY bez
predchádz
a
j
ú
-
c
ej liečby MTX
|
SELECT MONO MTX-IR
|
SELECT NEXT csDMARD-IR
|
SELECT COMPARE MTX-IR
|
SELECT BEYOND bDMARD-IR
|
|
MTX
|
UPA
15 mg
|
MTX
|
UPA
15 mg
|
PBO
|
UPA
15 mg
|
PBO
|
UPA
15 mg
|
ADA
40 mg
|
PBO
|
UPA
15 mg
|
N
|
314
|
317
|
216
|
217
|
221
|
221
|
651
|
651
|
327
|
169
|
164
|
Týždeň
|
|
LDA DAS28-CRP ≤ 3,2 (% pacientov)
|
12a/14b
|
28
|
53g
|
19
|
45e
|
17
|
48e
|
14
|
45e,h
|
29
|
14
|
43e
|
24c/26d
|
32
|
60f
|
|
|
|
|
18
|
55g,h
|
39
|
|
|
48
|
39
|
59g
|
|
|
|
|
|
50h
|
35
|
|
|
CR DAS28-CRP < 2,6 (% pacientov)
|
12a/14b
|
14
|
36g
|
8
|
28e
|
10
|
31e
|
6
|
29e,h
|
18
|
9
|
29g
|
24c/26d
|
18
|
48e
|
|
|
|
|
9
|
41g,h
|
27
|
|
|
48
|
29
|
49g
|
|
|
|
|
|
38i
|
28
|
|
|
ACR2
0 (% pacientov)
|
12a/14b
|
54
|
76g
|
41
|
68e
|
36
|
64e
|
36
|
71e,j
|
63
|
28
|
65e
|
24c/26d
|
59
|
79g
|
|
|
|
|
36
|
67g,i
|
57
|
|
|
48
|
57
|
74g
|
|
|
|
|
|
65i
|
54
|
|
|
ACR5
0 (% pacientov)
|
12a/14b
|
28
|
52g
|
15
|
42g
|
15
|
38g
|
15
|
45g,h
|
29
|
12
|
34g
|
24c/26d
|
33
|
60e
|
|
|
|
|
21
|
54g,h
|
42
|
|
|
48
|
43
|
63g
|
|
|
|
|
|
49i
|
40
|
|
|
ACR7
0 (% pacientov)
|
12a/14b
|
14
|
32g
|
3
|
23g
|
6
|
21g
|
5
|
25g,h
|
13
|
7
|
12
|
24c/26d
|
18
|
44g
|
|
|
|
|
10
|
35g,h
|
23
|
|
|
48
|
29
|
51g
|
|
|
|
|
|
36h
|
23
|
|
|
CDA
I ≤ 10 (% pacientov)
|
12a/14b
|
30
|
46g
|
25
|
35 l
|
19
|
40e
|
16
|
40e,h
|
30
|
14
|
32g
|
24c/26d
|
38
|
56g
|
|
|
|
|
22
|
53g,h
|
38
|
|
|
48
|
43
|
60g
|
|
|
|
|
|
47h
|
34
|
|
|
Skratky: ACR20 (alebo 50 alebo 70) = zlepšenie podľa Americkej reumatologickej spoločnosti o ≥ 20 % (alebo o ≥ 50 % alebo o ≥ 70 %); ADA = adalimumab; CDAI = index klinickej aktivity ochorenia; CR = klinická remisia; CRP = C-reaktívny proteín, DAS28 = skóre aktivity ochorenia pre 28 kĺbov; IR = pacient s nedostatočnou odpoveďou, LDA = nízka aktivita ochorenia; MTX = metotrexát; PBO = placebo; UPA = upadacitinib
a SELECT-NEXT, SELECT-EARLY, SELECT-COMPARE, SELECT-BEYOND
b SELECT-MONOTHERAPY
c SELECT-EARLY
d SELECT-COMPARE
e mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. placebo alebo MTX
f mnohonásobné porovnávanie/multiplicita p ≤ 0,01 upadacitinib vs. placebo alebo MTX
g porovnanie nominálna p ≤ 0,001 upadacitinib vs. placebo alebo MTX
h porovnanie nominálna p ≤ 0,001 upadacitinib vs. adalimumab
i porovnanie nominálna p ≤ 0,01 upadacitinib vs. adalimumab
j porovnanie nominálna p < 0,05 upadacitinib vs adalimumab
k porovnanie nominálna p ≤ 0,01 upadacitinib vs. placebo alebo MTX
l porovnanie nominálna p ≤ 0,05 upadacitinib vs. MTX
Poznámka: Údaje v 48. týždni získané metódou Analýzy kompletného súboru (Full Analysis set, FAS)
u randomizovanej skupiny vrátane pacientov bez odpovede
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Rádiografická odpoveď
Inhibícia progresie štrukturálneho poškodenia kĺbov bola hodnotená pomocou modifikovaného celkového Sharpovho skóre (modified Total Sharp Score, mTSS) a jeho zložiek, skóre erózie a skóre zúženia kĺbnych štrbín v 24./26. týždni a 48. týždni v štúdii SELECT-EARLY a SELECT- COMPARE.
Liečba upadacitinibom 15 mg viedla k významne väčšej inhibícii progresie štrukturálneho poškodenia kĺbov v porovnaní s placebom v kombinácii s MTX v štúdii SELECT-COMPARE a v monoterapii
v porovnaní s MTX v štúdii SELECT-EARLY (tabuľka 6). Analýzy skóre erózie a zúženia kĺbnych
štrbín boli totožné s celkovým skóre. Podiel pacientov bez rádiografickej progresie (zmena mTSS ≤ 0) bol významne vyšší pri upadacitinibe 15 mg v obidvoch štúdiách. Inhibícia progresie štrukturálneho poškodenia kĺbov bola zachovaná až do 96. týždňa v obidvoch štúdiách u pacientov, ktorí ostali na svojej pôvodne pridelenej liečbe upadacitinibom 15 mg (na základe dostupných výsledkov od 327 pacientov v štúdii SELECT-COMPARE a 238 pacientov v SELECT-EARLY).
Tabuľka 6 Rádiografické zmeny
Štúdia
| SELECT EARLY
bez predchádzajúcej liečby MTX
|
SELECT COMPARE MTX-IR
|
Liečebná skupina
|
MTX
| UPA
15 mg
|
PBOa
| UPA
15 mg
| ADA
40 mg
|
Modifikované celkové Sharpovo skóre, priemerná zmena od východiskového stavu
|
24.b/26.c týždeň
| 0,7
| 0,1f
| 0,9
| 0,2g
| 0,1
|
48. týždeň
| 1,0
| 0,03e
| 1,7
| 0,3e
| 0,4
|
Podiel pacientov bez rádiografickej progresied
|
24.b/26.c týždeň
| 77,7
| 87,5f
| 76,0
| 83,5f
| 86,8
|
48. týždeň
| 74,3
| 89,9e
| 74,1
| 86,4e
| 87,9
|
Skratky: ADA = adalimumab; IR = pacient s nedostatočnou odpoveďou; MTX = metotrexát; PBO =
placebo; UPA = upadacitinib
a všetky údaje týkajúce sa placeba v 48. týždni boli odvodené pomocou lineárnej extrapolácie
b SELECT-EARLY
c SELECT-COMPARE
d žiadna progresia definovaná ako zmena mTSS ≤ 0
e porovnanie nominálna p≤ 0,001 upadacitinib vs. placebo alebo MTX
f mnohonásobné porovnávanie/multiplicita p ≤ 0,01 upadacitinib vs. placebo alebo MTX
g mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. placebo alebo MTX
|
| | | | | | |
Odpoveď súvisiaca s fyzickými funkciami a výsledky vzťahujúce sa ku zdravotnému stavuLiečba upadacitinibom 15 mg samostatne alebo v kombinácii s csDMARD viedla k významne väčšiemu zlepšeniu fyzických funkcií v porovnaní so všetkými porovnávanými liekmi, merané pomocou HAQ-DI (pozri tabuľku 7). Zlepšenie HAQ-DI sa udržalo počas 3 rokov u pacientov, ktorí ostali na svojej pôvodne pridelenej liečbe upadacitinibom 15 mg na základe dostupných výsledkov zo štúdií SELECT COMPARE a SELECT EARLY.
Tabuľk
a 7 Priemerná zmena HAQ-DI oproti východiskovému stavu
a,b
-0,8
-0,9
-0,7
-0,6
-0,6
Štúdia
| SELECT
EARLY bez predchádzajúcej liečby MTX
|
SELECT MONO MTX-IR
|
SELECT NEXT csDMARD-IR
|
SELECT COMPARE MTX-IR
|
SELECT BEYOND BIO-IR
| Liečebná skupina
|
MTX
| UPA
15 mg
|
MTX
| UPA
15 mg
|
PBO
| UPA
15 mg
|
PBO
| UPA
15 mg
| ADA
40 mg
|
PBO
| UPA
15 mg
| N
| 313
| 317
| 216
| 216
| 220
| 216
| 648
| 644
| 324
| 165
| 163
| Skóre na začiatku, priemer
|
1,6
|
1,6
|
1,5
|
1,5
|
1,4
|
1,5
|
1,6
|
1,6
|
1,6
|
1,6
|
1,7
| 12.c/14.d
týždeň
|
-0,5
|
h
|
-0,3
|
g
|
-0,3
|
g
|
-0,3
|
g,i
|
-0,5
|
-0,2
|
g
| 24.e/26.f
týždeň
|
-0,6
|
g
|
|
|
|
|
-0,3
|
h,i
|
-0,6
|
|
| Skratky: ADA = adalimumab; HAQ-DI = index postihnutia na základe dotazníka hodnotiaceho zdravotný stav a index funkčnej neschopnosti; IR = pacient s nedostatočnou odpoveďou; MTX = metotrexát;
PBO = placebo; UPA = upadacitinib
a Uvedené údaje predstavujú priemerné hodnoty
b Index postihnutia na základe dotazníka hodnotiaceho zdravotný stav a index funkčnej neschopnosti:
0 = najlepší, 3 = najhorší; 20 otázok; 8 kategórií: obliekanie a starostlivosť o seba, vstávanie, stravovanie,
chôdza, hygiena, dosah, stisk a aktivity.
c SELECT-EARLY, SELECT-NEXT, SELECT-COMPARE, SELECT-BEYOND
d SELECT-MONOTHERAPY
e SELECT-EARLY
f SELECT-COMPARE
g mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. placebo alebo MTX
h porovnanie nominálna p ≤ 0,001 upadacitinib vs. placebo alebo MTX
i porovnanie nominálna p ≤ 0,01 upadacitinib vs. adalimumab
|
|
|
-0,7
-0,4
V štúdiách SELECT-MONOTHERAPY, SELECT-NEXT a SELECT-COMPARE viedla liečba
upadacitinibom 15 mg k významne väčšiemu zlepšeniu v priemernom trvaní rannej stuhnutosti kĺbov
v porovnaní s placebom alebo MTX.
V klinických štúdiách vykazovali pacienti liečení upadacitinibom významné zlepšenie v pacientmi hlásenej kvalite života, ktorá bola meraná pomocou Stručného formulára (36) prieskumu zdravia
(SF-36) v súhrne fyzických zložiek v porovnaní s placebom a MTX. Okrem toho vykazovali pacienti liečení upadacitinibom významné zlepšenie, ktoré bolo hodnotené Funkčným hodnotením skóre liečby chronických ochorení – skóre únavy (FACIT-F) v porovnaní s placebom.
Psoriatická artritída
Účinnosť a bezpečnosť upadacitinibu 15 mg jedenkrát denne boli hodnotené v dvoch randomizovaných, dvojito zaslepených, multicentrických, placebom kontrolovaných štúdiách fázy 3 u pacientov vo veku 18 rokov alebo starších so stredne ťažkou až ťažkou aktívnou psoriatickou artritídou. Všetci pacienti mali podľa klasifikačných kritérií pre psoriatickú artritídu (Classification Criteria for Psoriatic Arthritis, CASPAR) aktívnu psoriatickú artritídu minimálne 6 mesiacov, minimálne 3 bolestivé kĺby a minimálne 3 opuchnuté kĺby a aktívnu ložiskovú psoriázu alebo
ložiskovú psoriázu v anamnéze. Pri obidvoch štúdiách bol primárnym cieľovým ukazovateľom podiel pacientov, ktorí dosiahli odpoveď ACR20 v 12. týždni.
SELECT-PsA 1 bola 24-týždňová štúdia, ktorá skúmala 1705 pacientov s nedostatočnou odpoveďou alebo intoleranciou na aspoň jedno nebiologické DMARD. Na začiatku 1393 (82 %) pacientov používalo súbežne minimálne jedno nebiologické DMARD; 1084 (64 %) pacientov užívalo súbežne iba MTX a 311 (18 %) pacientov bolo liečených v monoterapii. Pacienti užívali upadacitinib 15 mg alebo 30 mg jedenkrát denne, adalimumab alebo placebo. V 24. týždni všetci pacienti randomizovaní do skupiny s placebom prešli zaslepenou metódou na upadacitinib 15 mg alebo 30 mg jedenkrát denne. SELECT-PsA 1 zahŕňala dlhodobé predĺženie až do 5 rokov.
SELECT-PsA 2 bola 24-týždňová štúdia, ktorá skúmala 642 pacientov s nedostatočnou odpoveďou alebo intoleranciou na aspoň jedno biologické DMARD. Na začiatku 296 (46 %) pacientov používalo súbežne minimálne jedno nebiologické DMARD; 222 (35 %) pacientov dostávalo súbežne iba MTX
a 345 (54 %) pacientov bolo liečených monoterapiou. Pacienti užívali upadacitinib 15 mg alebo 30 mg
jedenkrát denne alebo placebo. V 24. týždni všetci pacienti randomizovaní do skupiny s placebom prešli zaslepenou metódou na upadacitinib 15 mg alebo 30 mg jedenkrát denne. SELECT-PsA 2 zahŕňala dlhodobé predĺženie až do 3 rokov.
Klinická odpoveď
V oboch štúdiách dosiahol štatisticky významne väčší podiel pacientov liečených upadacitinibom
15 mg odpoveď ACR20 v porovnaní s placebom v 12. týždni (tabuľka 8). Nástup účinku bol rýchly vo všetkých sledovaných parametroch s väčšou odpoveďou pozorovanou už v 2. týždni pre ACR20.
Liečba upadacitinibom 15 mg viedla k zlepšeniam v jednotlivých zložkách ACR vrátane počtu citlivých/bolestivých a opuchnutých kĺbov, celkového hodnotenia pacientom a lekárom, HAQ-DI, hodnotenia bolesti a hsCRP v porovnaní s placebom.
V štúdii SELECT-PsA 1 dosiahol upadacitinib 15 mg neinferioritu v porovnaní s adalimumabom
v pomere pacientov, ktorí dosiahli odpoveď ACR20 v 12. týždni. Superioritu voči adalimumabu však nebolo možné preukázať.
V obidvoch štúdiách boli pozorované v primárnych a kľúčových sekundárnych cieľových ukazovateľoch konzistentné odpovede pri upadacitinibe samostatne alebo v kombinácii
s metotrexátom.
Účinnosť upadacitinibu 15 mg bola preukázaná bez ohľadu na hodnotené podskupiny vrátane východiskového BMI, východiskového hsCRP a počtu predchádzajúcich nebiologických DMARD (≤ 1 alebo > 1).
Tabuľk
a 8 Klinická odpoveď v štúdiách SELECT-PsA 1 a SELECT-PsA 2
Štúdia
|
SELECT-PsA 1
nebiologick
é DMARD-IR
|
SELECT-PsA 2
bDMAR
D
-
IR
|
Liečebná skupina
|
PBO
|
UPA
15 mg
|
ADA
40 mg
|
PBO
|
UPA
15 mg
|
N
|
423
|
429
|
429
|
212
|
211
|
ACR20, % pacientov (95 % CI)
|
12. týždeň
|
36 (32; 41)
|
71 (66; 75)f
|
65 (61; 70)
|
24 (18; 30)
|
57 (50; 64)
|
Rozdiel
v porovnaní
s placebom (95 % CI)
|
d,e
|
-
|
d,e
|
24. týždeň
|
45 (40; 50)
|
73 (69; 78)
|
67 (63; 72)
|
20 (15; 26)
|
59 (53; 66)
|
56. týždeň
|
|
74 (70; 79)
|
69 (64; 73)
|
|
60 (53; 66)
|
ACR50, % pacientov (95 % CI)
|
12. týždeň
|
13 (10; 17)
|
38 (33; 42)
|
38 (33; 42)
|
5 (2; 8)
|
32 (26; 38)
|
24. týždeň
|
19 (15; 23)
|
52 (48; 57)
|
44 (40; 49)
|
9 (6; 13)
|
38 (32; 45)
|
56. týždeň
|
|
60 (55; 64)
|
51 (47; 56)
|
|
41 (34; 47)
|
ACR70, % pacientov (95 % CI)
|
12. týždeň
|
2 (1; 4)
|
16 (12; 19)
|
14 (11; 17)
|
1 (0; 1)
|
9 (5; 12)
|
24. týždeň
|
5 (3; 7)
|
29 (24; 33)
|
23 (19; 27)
|
1 (0; 2)
|
19 (14; 25)
|
56. týždeň
|
|
41 (36; 45)
|
31 (27; 36)
|
|
24 (18; 30)
|
MDA, % pacientov (95 % CI)
|
12. týždeň
|
6 (4; 9)
|
25 (21; 29)
|
25 (21; 29)
|
4 (2; 7)
|
17 (12; 22)
|
24. týždeň
|
12 (9; 15)
|
37 (32; 41)e
|
33 (29; 38)
|
3 (1; 5)
|
25 (19; 31)e
|
56. týždeň
|
|
45 (40; 50)
|
40 (35; 44)
|
|
29 (23; 36)
|
Hodnotenie entezitídy (LEI = 0), % pacientov (95 % CI)
a
|
12. týždeň
|
33 (27; 39)
|
47 (42; 53)
|
47 (41; 53)
|
20 (14; 27)
|
39 (31; 47)
|
24. týždeň
|
32 (27; 39)
|
54 (48; 60)e
|
47 (42; 53)
|
15 (9; 21)
|
43 (34; 51)
|
56. týždeň
|
|
59 (53; 65)
|
54 (48; 60)
|
|
43 (34; 51)
|
Hodnotenie daktylitídy (LEI = 0), % pacientov (95 % CI)
a
|
12. týždeň
|
42 (33; 51)
|
74 (66; 81)
|
72 (64; 80)
|
36 (24; 48)
|
64 (51; 76)
|
24. týždeň
|
40 (31; 48)
|
77 (69; 84)
|
74 (66; 82)
|
28 (17; 39)
|
58 (45; 71)
|
56. týždeň
|
|
75 (68; 82)
|
74 (66; 82)
|
|
51 (38; 64)
|
PASI75, % pacientov (95 % CI)
c
|
16. týždeň
|
21 (16; 27)
|
63 (56; 69)e
|
53 (46; 60)
|
16 (10; 22)
|
52 (44; 61)e
|
24. týždeň
|
27 (21; 33)
|
64 (58; 70)
|
59 (52; 65)
|
19 (12; 26)
|
54 (45; 62)
|
56. týždeň
|
|
65 (59; 72)
|
61 (55; 68)
|
|
52 (44; 61)
|
PASI90, % pacientov (95 % CI)
c
|
16. týždeň
|
12 (8; 17)
|
38 (32; 45)
|
39 (32; 45)
|
8 (4; 13)
|
35 (26; 43)
|
24. týždeň
|
17 (12; 22)
|
42 (35; 48)
|
45 (38; 52)
|
7 (3; 11)
|
36 (28; 44)
|
56. týždeň
|
|
49 (42; 56)
|
47 (40; 54)
|
|
41 (32; 49)
|
Skratky: ACR20 (alebo 50 alebo 70) ≥ 20 % (alebo ≥ 50 % alebo ≥ 70 %) zlepšenie podľa
Americkej akadémie reumatológie, ADA = adalimumab; bDMARD = biologické chorobu modifikujúce antireumatikum; IR = pacient s nedostatočnou odpoveďou; MDA = minimálna aktivita ochorenia (minimal disease activity); PASI75 (alebo 90) = ≥ 75 % (alebo ≥ 90 %) zlepšenie oblasti psoriázy a indexu závažnosti; PBO = placebo; UPA = upadacitinib
Pacienti, ktorí prerušili randomizovanú liečbu alebo u ktorých chýbali údaje v týždni hodnotenia, boli v analýzach hodnotení ako neodpovedajúci na liečbu. Pokiaľ ide o MDA, hodnotenie entezitídy a daktylitídy v 24./56. týždni, jedinci, ktorým bola podaná záchranná liečba v 16. týždni boli v analýzach hodnotení ako neodpovedajúci na liečbu.
a U pacientov s východiskovou entezitídou (n = 241, 270 resp. 265 pre SELECT-PsA 1 a n = 144
resp. 133 pre SELECT-PsA 2)
b U pacientov s východiskovou daktylitídou (n = 126, 136 resp. 127 pre SELECT-PsA 1 a n = 64
|
|
|
35 (28; 41)
33 (24, 42)
resp. 55 pre SELECT-PsA 2)
c U pacientov s východiskovou hodnotou psoriázy BSA ≥ 3 % (n = 211, 214 resp. 211 pre
SELECT-PsA 1 a n = 131 resp. 130 pre SELECT-PsA 2)

d primárny cieľový ukazovateľ
e mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. placebo
f mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. adalimumab (test neinferiority)
Rádiografická odpoveďV štúdii SELECT-PsA 1 sa inhibícia progresie štrukturálneho poškodenia hodnotila rádiograficky
a vyjadrila sa ako zmena oproti východiskovej hodnote v modifikovanom celkovom Sharpovom skóre
(mTSS) a jeho zložkách, skóre erózie a skóre zúženia kĺbnych štrbín v 24. týždni.
Liečba upadacitinibom 15 mg viedla v 24. týždni k štatisticky významne väčšej inhibícii progresie štrukturálneho poškodenia kĺbov v porovnaní s placebom (tabuľka 9). Skóre erózie a skóre zúženia kĺbnych štrbín boli v súlade s celkovým skóre. Podiel pacientov bez rádiografickej progresie (zmena mTSS ≤ 0,5) bol v 24. týždni vyšší pri upadacitinibe 15 mg v porovnaní s placebom.
Tabuľka 9 Rádiografické zmeny v SELECT-PsA 1 Liečebná skupina
| PBO
| UPA
15 mg
| ADA
40 mg
|
Modifikované celkové Sharpovo skóre, priemerná zmena oproti východiskovému stavu
(95 % CI)
|
24. týždeň
| 0,25 (0,13; 0,36)
| -0,04 (-0,16; 0,07)c
| 0,01 (-0,11; 0,13)
|
56. týždeňa
| 0,44 (0,29; 0,59)
| -0,05 (-0,20; 0,09)
| -0,06 (-0,20; 0,09)
|
Podiel pacientov bez rádiografickej progresieb, % (95 % CI)
|
24. týždeň
| 92 (89; 95)
| 96 (94; 98)
| 95 (93; 97)
|
56. týždeňa
| 89 (86; 92)
| 97 (96; 99)
| 94 (92; 97)
|
Skratky: ADA = adalimumab; PBO = placebo; UPA = upadacitinib
a Všetky údaje týkajúce sa placeba v 56. týždni odvodené pomocou lineárnej extrapolácie
b Bez progresie definované ako zmena mTSS ≤ 0,5
c mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. placebo
|
Odpoveď súvisiaca s fyzickými funkciami a výsledky vzťahujúce sa ku zdravotnému stavuV štúdii SELECT-PsA 1 vykazovali pacienti liečení upadacitinibom 15 mg štatisticky významné zlepšenie fyzických funkcií oproti východiskovej hodnote na základe hodnotenia pomocou HAQ-DI v 12. týždni (-0,42 [95 % CI: -0,47, -0,37]) v porovnaní s placebom (-0,14 [95 % CI: -0,18, -0,09]); zlepšenie u pacientov liečených adalimumabom predstavovalo -0,34 (95 % CI: -0,38, -0,29). V štúdii SELECT-PsA 2 vykazovali pacienti liečení upadacitinibom 15 mg štatisticky významné zlepšenie oproti východiskovej hodnote v dotazníku HAQ-DI v 12. týždni (-0,30 [95 % CI: -0,37, -0,24]) v
porovnaní s placebom (-0,10 [95 % CI: -0,16, -0,03]). V oboch štúdiách sa zlepšenie fyzických funkcií udržalo do 56. týždňa.
Kvalita života súvisiaca so zdravím bola hodnotená pomocou SF-36v2. V obidvoch štúdiách došlo v 12. týždni u pacientov, ktorí užívali upadacitinib 15 mg k štatisticky významnému väčšiemu zlepšeniu skóre súhrnu fyzických zložiek oproti východiskovej hodnote v porovnaní s placebom.
V oboch štúdiách sa zlepšenia oproti východiskovej hodnote udržali do 56. týždňa.
U pacientov, ktorí užívali upadacitinib 15 mg, došlo v 12. týždni k štatisticky významnému zlepšeniu z hľadiska únavy oproti východiskovej hodnote, a to na základe merania skóre FACIT-F, v porovnaní s placebom v obidvoch štúdiách. V oboch štúdiách sa zlepšenia oproti východiskovej hodnote udržali do 56. týždňa.
Na začiatku bola psoriatická spondylitída hlásená u 31 % pacientov v štúdii SELECT-PsA 1 a u 34 % pacientov v štúdii SELECT-PsA 2. U pacientov so psoriatickou spondylitídou, ktorí sa liečili upadacitinibom v dávke 15 mg, došlo v 24. týždni k zlepšeniu skóre BASDAI (Bath Ankylosing Spondylitis Disease Activity Index, Bathov index aktivity ankylozujúcej spondylitídy) oproti východiskovej hodnote v porovnaní s placebom. V oboch štúdiách sa zlepšenia oproti východiskovej hodnote udržali do 56. týždňa.
Axiálna spondyloartritídaAxiálna spondyloartritída bez rádiografického dôkazuÚčinnosť a bezpečnosť upadacitinibu 15 mg jedenkrát denne sa hodnotili v randomizovanej, dvojito zaslepenej, multicentrickej, placebom kontrolovanej štúdii u pacientov vo veku 18 rokov alebo starších s aktívnou axiálnou spondyloartritídou bez rádiografického dôkazu. Štúdia SELECT-AXIS 2 (nr-axSpA) predstavovala 52-týždňové, placebom kontrolované skúšanie u 314 pacientov s aktívnou axiálnou spondyloartritídou bez rádiografického dôkazu s nedostatočnou odpoveďou na minimálne dva nesteroidné protizápalové lieky (NSAID) alebo s intoleranciou alebo kontraindikáciou NSAID.
Pacienti museli mať objektívne príznaky zápalu indikované zvýšeným C-reaktívnym proteínom (CRP)
(definované ako > horná hranica normálu) a/alebo sakroiliitídy na základe vyšetrenia magnetickou rezonanciou (MRI) a žiadny definitívny rádiografický dôkaz štrukturálneho poškodenia sakroiliakálnych kĺbov. Pacienti mali pri skríningu a návštevách na stanovenie východiskového stavu aktívne ochorenie, na základe skóre podľa Bathovho indexu aktivity ankylozujúcej spondylitídy (BASDAI) ≥ 4 a skóre v hodnotení celkovej bolesti chrbta pacientom ≥ 4 na číselnej hodnotiacej stupnici (NRS) 0 – 10. Pacienti mali vo východiskovom stave príznaky axiálnej spondyloartritídy bez rádiografického dôkazu v priemere 9,1 roka a 29,1 % pacientov súbežne používalo csDMARD.
32,9 % pacientov malo nedostatočnú odpoveď alebo intoleranciu na liečbu bDMARD. Pacienti užívali upadacitinib 15 mg jedenkrát denne alebo placebo. V 52. týždni všetci pacienti randomizovaní do skupiny s placebom prešli na upadacitinib 15 mg jedenkrát denne. Primárnym cieľovým
ukazovateľom bol podiel pacientov, ktorí v 14. týždni dosiahli odpoveď ASAS40 podľa hodnotenia Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis international Society, ASAS). Súčasťou štúdie bolo dlhodobé predĺženie až do 2 rokov. Doteraz sú k dispozícii
a uvádzajú sa iba údaje týkajúce sa účinnosti do 14. týždňa.
Klinická odpoveďV štúdii SELECT-AXIS 2 (nr-axSpA) dosiahol významne väčší podiel pacientov liečených
upadacitinibom 15 mg odpoveď ASAS40 v porovnaní s placebom v 14. týždni (tabuľka 10). Číselný rozdiel medzi liečebnými skupinami bol pozorovaný vo všetkých časových bodoch od 2. týždňa až do
14. týždňa.
Liečba upadacitinibom 15 mg viedla v porovnaní s placebom v 14. týždni k zlepšeniu jednotlivých zložiek ASAS (celkové hodnotenie aktivity ochorenia pacientom, hodnotenie celkovej bolesti chrbta, zápalu a funkcie) a ďalších parametrov aktivity ochorenia vrátane hsCRP.
Účinnosť upadacitinibu 15 mg bola preukázaná vo všetkých podskupinách vrátane pohlavia, východiskového BMI, trvania príznakov axiálnej spondyloartritídy bez rádiografického dôkazu, východiskovej hodnoty hsCRP, MRI sakroiliitídy a predchádzajúceho použitia bDMARD.
Tabuľka 10 Klinická odpoveď v SELECT-AXIS 2 (nr-axSpA) Liečebná skupina
| PBO
| UPA 15 mg
|
N
| 157
| 156
|
ASAS40, % pacientov (95 % CI)a
|
14. týždeň
| 22,5 (16,0; 29,1)
| 44,9 (37,1; 52,7)
|
Rozdiel v porovnaní
| 22,2 (12,1; 32,3)b
|
s placebom (95 % CI)
|
|
ASAS20, % pacientov (95 % CI)a
|
14. týždeň
|
43,8 (36,0; 51,5)
|
66,7 (59,3; 74,1)b
|
ASAS čiastočná remisia, % pacientov (95 % CI)
|
14. týždeň
|
7,6 (3,5; 11,8)
|
18,6 (12,5; 24,7)c
|
BASDA
I 50, % pacientov (95 % CI)
|
14. týždeň
|
22,1 (15,5; 28,6)
|
42,3 (34,6; 50,1)b
|
Zmena oproti východiskovej hodnote v ASDAS-CRP (95 % CI)
|
14. týždeň
|
-0,71 (-0,85; -0,56)
|
-1,36 (-1,50; -1,21)b
|
ASDAS neaktívne ochorenie, % pacientov (95 % CI)
|
14. týždeň
|
5,2 (1,7; 8,7)
|
14,1 (8,6; 19,6)c
|
ASDA
S nízka aktivita ochorenia, % pacientov (95 % CI)
|
14. týždeň
|
18,3 (12,2; 24,4)
|
42,3 (34,6; 50,1)b
|
Skratky: ASAS20 (alebo ASAS40) = ≥ 20 % (alebo ≥ 40 %) zlepšenie v hodnotení podľa Medzinárodnej spoločnosti pre spondyloartritídu; ASDAS-CRP = Skóre aktivity ankylozujúcej spondylitídy C-reaktívny proteín; BASDAI = Bathov index aktivity ankylozujúcej spondylitídy; PBO = placebo; UPA = upadacitinib
a Odpoveď ASAS20 (ASAS40) je definovaná ako ≥ 20 % (≥ 40 %) zlepšenie a absolútne
zlepšenie oproti východiskovej hodnote o ≥ 1 jednotku (≥ 2 jednotky) (rozmedzie 0 až 10)
v ≥ 3 zo 4 domén (celkové hodnotenie pacienta, celková bolesť chrbta, funkcia a zápal)
a žiadne zhoršenie potenciálnej zvyšnej domény (definované ako zhoršenie ≥ 20 % a ≥ 1 jednotka pre ASAS20 alebo definované ako zhoršenie > 0 jednotiek v prípade ASAS40). b mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. placebo
c mnohonásobné porovnávanie/multiplicita p ≤ 0,01 upadacitinib vs. placebo
Pri binárnych cieľových ukazovateľoch sú výsledky hodnotené metódou imputácie pacientov bez odpovede v spojitosti s viacnásobnou imputáciou. Pri kontinuálnych cieľových ukazovateľoch sú výsledky založené na priemernej zmene oproti východiskovému stavu, stanovené metódou najmenších štvorcov pomocou zmiešaných modelov pre opakované merania
|
Odpoveď súvisiaca s fyzickými funkciami a výsledky súvisiace so zdravím
Pacienti liečení upadacitinibom 15 mg preukázali významné zlepšenie fyzických funkcií oproti východiskovému stavu v porovnaní s placebom v rámci hodnotenia pomocou BASFI v 14. týždni.
Pacienti liečení upadacitinibom 15 mg preukázali významné zlepšenia celkovej bolesti chrbta a nočnej bolesti chrbta v porovnaní s placebom v 14. týždni.
Pacienti liečení upadacitinibom 15 mg preukázali významné zlepšenia kvality života súvisiacej so zdravím a celkového zdravia v porovnaní s placebom v rámci hodnotenia podľa ASQoL, resp. indexu zdravia ASAS v 14. týždni.
Objektívne meranie zápalovej aktivityPríznaky zápalu sa hodnotili pomocou MRI a vyjadrovali sa ako zmena oproti východiskovému stavu v skóre Kanadského konzorcia pre výskum spondyloartritídy (Spondyloarthritis Research Consortium of Canada, SPARCC) pre sakroiliakálne kĺby. V 14. týždni bolo pozorované významné zlepšenie zápalových príznakov na sakroiliakálnych kĺboch u pacientov liečených upadacitinibom 15 mg
v porovnaní s placebom.
Ankylozujúca spondylitída (AS, rádiografická axiálna spondyloartritída)Účinnosť a bezpečnosť upadacitinibu 15 mg jedenkrát denne sa hodnotili v dvoch randomizovaných, dvojito zaslepených, multicentrických, placebom kontrolovaných štúdiách u pacientov vo veku 18 rokov alebo starších s aktívnou ankylozujúcou spondylitídou na základe skóre podľa Bathovho indexu aktivity ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Disease Activity Index, BASDAI)
≥ 4 a skóre v hodnotení celkovej bolesti chrbta pacientom ≥ 4. Obe štúdie zahŕňali dlhodobé predĺženie až do 2 rokov.
Štúdia SELECT-AXIS 1 predstavovala 14-týždňové, placebom kontrolované skúšanie u 187 pacientov s ankylozujúcou spondylitídou s nedostatočnou odpoveďou na minimálne dvaNSAID alebo s intoleranciou alebo kontraindikáciou NSAID bez predchádzajúcej liečby biologickými DMARD. Pacienti mali vo východiskovom stave príznaky ankylozujúcej spondylitídy v priemere 14,4 rokov
a približne 16 % pacientov súbežne používalo csDMARD. Pacienti užívali upadacitinib 15 mg jedenkrát denne alebo placebo. V 14. týždni všetci pacienti randomizovaní do skupiny s placebom prešli na upadacitinib 15 mg jedenkrát denne. Primárnym cieľovým ukazovateľom bol podiel pacientov, ktorí v 14. týždni dosiahli odpoveď ASAS40 podľa hodnotenia Medzinárodnej spoločnosti pre spondyloartritídu (ASAS).
Štúdia SELECT-AXIS 2 (AS) predstavovala 14-týždňové, placebom kontrolované skúšanie
u 420 pacientov s ankylozujúcou spondylitídou s predchádzajúcou expozíciou bDMARD (77,4 % pacientov malo nedostatočnú účinnosť na blokátor faktora nekrotizujúceho nádory (TNF) alebo inhibítor interleukínu-17 (IL-17i), 30,2 % pacientov malo intoleranciu, 12,9 % pacientov malo predchádzajúcu expozíciu ale nie nedostatočnú účinnosť na dva bDMARD). Pacienti mali vo východiskovom stave príznaky ankylozujúcej spondylitídy v priemere 12,8 roka a približne 31 % pacientov bolo na súbežnej liečbe použitím csDMARD. Pacienti užívali upadacitinib 15 mg jedenkrát denne alebo placebo. V 14. týždni všetci pacienti randomizovaní do skupiny s placebom prešli na upadacitinib 15 mg jedenkrát denne. Primárnym cieľovým ukazovateľom bol podiel pacientov, ktorí
v 14. týždni dosiahli odpoveď ASAS40 podľa hodnotenia Medzinárodnej spoločnosti pre
spondyloartritídu (ASAS).
Klinická odpoveďV oboch štúdiách dosiahol významne väčší podiel pacientov liečených upadacitinibom 15 mg
odpoveď ASAS40 v porovnaní s placebom v 14. týždni (tabuľka 11). Číselný rozdiel medzi liečebnými skupinami bol pozorovaný od 2. týždňa v štúdii SELECT-AXIS a od 4. týždňa v štúdii SELECT-AXIS 2 (AS) podľa ASAS40.
Liečba upadacitinibom 15 mg viedla v porovnaní s placebom v 14. týždni k zlepšeniu jednotlivých zložiek ASAS (celkové hodnotenie aktivity ochorenia pacientom, hodnotenie celkovej bolesti chrbta, zápalu a funkcie) a ďalších parametrov aktivity ochorenia vrátane hsCRP.
Účinnosť upadacitinibu 15 mg bola preukázaná bez ohľadu na hodnotené podskupiny vrátane pohlavia, východiskového BMI, trvania príznakov AS, východiskovej hodnoty hsCRP
a predchádzajúceho použitia bDMARD.
Tabuľka 11 Klinická odpoveď
Štúdia
| SELECT-AXIS 1
bez predchádzajúcej liečby bDMARD
| SELECT-AXIS 2 (AS)
bDMARD-IR
|
Liečebná skupina
| PBO
| UPA 15 mg
| PBO
| UPA 15 mg
|
N
| 94
| 93
| 209
| 211
|
ASAS40, % pacientov (95 % CI)a,b
|
14. týždeň
| 25,5 (16,7; 34,3)
| 51,6 (41,5; 61,8)
| 18,2 (13,0; 23,4)
| 44,5 (37,8; 51,3)
|
Rozdiel v porovnaní
s placebom (95 % CI)
| 26,1 (12,6; 39,5)c
| 26,4 (17,9; 34,9)
|
ASAS20, % pacientov (95 % CI)a
|
14. týždeň
| 40,4 (30,5; 50,3)
| 64,5 (54,8; 74,2)e
| 38,3 (31,7; 44,9)
| 65,4 (59,0; 71,8)c
|
ASAS čiastočná remisia, % pacientov (95 % CI)
|
14. týždeň
| 1,1 (0,0; 3,1)
| 19,4 (11,3; 27,4)c
| 4,3 (1,6; 7,1)
| 17,5 (12,4; 22,7)c
|
BASDAI 50, % pacientov (95 % CI)
|
14. týždeň
| 23,4 (14,8; 32,0)
| 45,2 (35,0; 55,3)d
| 16,7 (11,7; 21,8)
| 43,1 (36,4; 49,8)c
|
Zmena oproti východiskovej hodnote v ASDAS-CRP (95% CI)
|
14. týždeň
|
-0,54 (-0,71; -0,37)
|
-1,45 (-1,62; -1,28)c
|
-0,49 (-0,62; -
0,37)
|
-1,52 (-1,64; -
1,39)c
|
ASDAS neaktívne ochorenie, % pacientov (95 % CI)
|
14. týždeň
|
0
|
16,1 (8,7; 23,6)e
|
1,9 (0,1; 3,8)
|
12,8 (8,3; 17,3)c
|
ASDAS nízka aktivita ochorenia, % pacientov (95 % CI)
|
14. týždeň
|
10,6 (4,4; 16,9)
|
49,5 (39,3; 59,6)f
|
10,1 (6,0; 14,2)
|
44,1 (37,4; 50,8)c
|
ASDA
S významné zlepšenie, % pacientov (95 % CI)
|
14. týždeň
|
5,3 (0,8; 9,9)
|
32,3 (22,8; 41,8)e
|
4,8 (1,9; 7,7)
|
30,3 (24,1; 36,5)e
|
a Odpoveď ASAS20 (ASAS40) je definovaná ako ≥ 20 % (≥ 40 %) zlepšenie a absolútne zlepšenie oproti východiskovej hodnote o ≥ 1 (≥ 2) jednotku (jednotky) (rozmedzie 0 až 10) v ≥ 3 zo 4 domén (celkové
hodnotenie pacienta, celková bolesť chrbta, funkcia a zápal) a žiadne zhoršenie potenciálnej zvyšnej domény (definované ako zhoršenie ≥ 20 % a ≥ 1 jednotka pre ASAS20 alebo definované ako zhoršenie > 0 jednotiek v prípade ASAS40).
b primárny cieľový ukazovateľ
c mnohonásobné porovnávanie/multiplicita p ≤ 0,001 upadacitinib vs. placebo
d mnohonásobné porovnávanie/multiplicita p ≤ 0,01 upadacitinib vs. placebo
e porovnanie bez kontroly multiplicity
f post-hoc analýza pre štúdiu SELECT-AXIS 1, bez kontroly multiplicity
Pri binárnych cieľových ukazovateľoch boli výsledky v 14. týždni hodnotené metódou imputácie pacientov bez odpovede (štúdia SELECT-AXIS 1) a metódou imputácie pacientov bez odpovede v spojení s metódou viacnásobnej imputácie (štúdia SELECT-AXIS 2 [AS]). Pri kontinuálnych cieľových ukazovateľoch sú výsledky v 14. týždni založené na priemernej zmene oproti východiskovému stavu, stanovené metódou najmenších štvorcov pomocou zmiešaných modelov pre opakované merania.
|
Na základe hodnotenia cieľových ukazovateľov uvedených v tabuľke 11 sa v štúdii SELECT-AXIS 1
udržala účinnosť počas obdobia 2 rokov.
Odpoveď súvisiaca s fyzickými funkciami a výsledky súvisiace so zdravímPacienti liečení upadacitinibom 15 mg preukázali v oboch štúdiách významné zlepšenie fyzických funkcií oproti východiskovému stavu v porovnaní s placebom v rámci hodnotenia pomocou zmeny Bathovho indexu aktivity ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Functional Index, BASFI) v 14. týždni. V štúdii SELECT-AXIS 1 bolo zlepšenie BASFI udržané počas obdobia
2 rokov.
V štúdii SELECT-AXIS 2 (AS) pacienti liečení upadacitinibom 15 mg preukázali významné zlepšenia celkovej bolesti chrbta a nočnej bolesti chrbta v porovnaní s placebom v 14. týždni.
V štúdii SELECT-AXIS 2 (AS) pacienti liečení upadacitinibom 15 mg preukázali významné zlepšenia kvality života súvisiacej so zdravím a celkového zdravia v porovnaní s placebom v rámci hodnotenia podľa ASQoL, resp. indexu zdravia ASAS v 14. týždni.
EntezitídaV štúdii SELECT-AXIS 2 (AS), pacienti s existujúcou entezitídou (n = 310) liečení upadacitinibom
15 mg preukázali významné zlepšenie entezitídy v porovnaní s placebom v rámci merania zmeny Maastrichtského skóre ankylozujúcej spondylitídy a entezitídy (Maastrich Ankylosing Spondylitis Enthsitis Score, MASES) v porovnaní s východiskovým stavom v 14. týždni.
Mobilita chrbticeV štúdii SELECT-AXIS 2 (AS), pacienti liečení upadacitinibom 15 mg preukázali významné zlepšenie mobility chrbtice v porovnaní s placebom v rámci merania zmeny Bathovho indexu aktivity ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Functional Index, BASFI) v porovnaní
s východiskovým stavom v 14. týždni.
Objektívne meranie zápalovej aktivity
Príznaky zápalu sa hodnotili pomocou MRI a vyjadrovali sa ako zmena oproti východiskovému stavu v skóre SPARCC pre chrbticu. V oboch štúdiách bolo v 14. týždni pozorované významné zlepšenie zápalových príznakov na chrbtici u pacientov liečených upadacitinibom 15 mg v porovnaní
s placebom. V štúdii SELECT-AXIS 1 sa zlepšenie zápalovej aktivity hodnotené MRI udržalo počas
2 rokov.
Atopická dermatitída
Účinnosť a bezpečnosť upadacitinibu 15 mg a 30 mg jedenkrát denne sa hodnotila v troch randomizovaných, dvojito zaslepených, multicentrických štúdiách fázy 3 (MEASURE UP 1, MEASURE UP 2 a AD UP) na celkovom počte 2 584 pacientov (vo veku 12 rokov a starších). Upadacitinib bol hodnotený u 344 dospievajúcich a 2 240 dospelých pacientov so stredne ťažkou až ťažkou atopickou dermatitídou (AD), ktorí nie sú adekvátne kontrolovaní lokálnymi liekmi. Na začiatku museli mať pacienti všetky z nasledovných parametrov: skóre v hodnotení Investigator's Global Assessment (vIGAAD) ≥ 3 v celkovom hodnotení AD (erytém, indurácia/papulácia
a sekrécia/tvorba krusty) na stupnici závažnosti od 0 do 4, skóre indexu oblasti a závažnosti ekzému
(EASI) ≥ 16 (zložené skóre hodnotiace rozsah a závažnosť erytému, edému/papulácie, škrabancov
a lichenifikácie na 4 rôznych miestach na tele), postihnutie minimálneho povrchu tela (BSA) ≥ 10 %
a týždenný priemer na číselnej stupnici najhoršieho svrbenia (NRS) ≥ 4.
Vo všetkých troch štúdiách dostávali pacienti upadacitinib v dávkach 15 mg, 30 mg jedenkrát denne alebo zodpovedajúce placebo počas 16 týždňov. V štúdii AD UP dostávali pacienti súbežne aj lokálne kortikosteroidy (TCS). Po ukončení dvojito zaslepeného obdobia mali pacienti pôvodne randomizovaní na liečbu upadacitinibom pokračovať v užívaní rovnakej dávky až do 260. týždňa. Pacienti v skupine s placebom boli znovu randomizovaní v pomere 1 : 1 na upadacitinib 15 mg alebo
30 mg do 260. týždňa.
Východiskové charakteristiky
V monoterapeutických štúdiách (MEASURE UP 1 a 2) malo 50,0 % pacientov východiskové skóre vIGA-AD 3 (stredné) a 50,0 % pacientov malo východiskové skóre vIGA-AD 4 (závažné). Priemerné východiskové skóre EASI bolo 29,3 a stredová východisková hodnota týždenného priemeru na číselnej stupnici najhoršieho svrbenia bola 7,3. V súbežnej štúdii TCS (AD UP) malo 47,1 % pacientov východiskové skóre vIGA-AD 3 (stredné) a 52,9 % pacientov malo východiskové skóre vIGA-AD 4 (závažné). Priemerné východiskové skóre EASI bolo 29,7 a stredová východisková hodnota týždenného priemeru na číselnej stupnici najhoršieho svrbenia bola 7,2.'
Klinická odpoveď
Štúdiemonoterapie(MEASUREUP1AMEASUREUP2)asúbežnáštúdiaTCS (ADUP)
Významne väčší podiel pacientov liečených upadacitinibom 15 mg alebo 30 mg dosiahol v 16. týždni
skóre vIGA-AD 0 alebo 1, EASI 75 alebo ≥ 4-bodové zlepšenie najhoršieho svrbenia NRS
v porovnaní s placebom. Dosiahlo sa aj rýchle zlepšenie hojenia kože a svrbenia (pozri tabuľku 12).
Obrázok 1 ukazuje podiel pacientov, ktorí dosiahli odpoveď EASI 75, a priemernú percentuálnu zmenu od východiskovej hodnoty pri najhoršom svrbení NRS do 16. týždňa v štúdiách MEASURE UP 1 a 2.
Tabuľk
a 12 Výsledky účinnosti upadacitinibu
Štúdia
|
MEASURE UP 1
|
MEASURE UP 2
|
AD UP
|
Liečebná skupina
|
PBO
|
UPA
15 mg
|
UPA
30 mg
|
PBO
|
UPA
15 mg
|
UPA
30 mg
|
PBO + TCS
|
UPA
15 mg
+ TCS
|
UPA
30 mg
+ TCS
|
Počet
randomizovaných jedincov
|
281
|
281
|
285
|
278
|
276
|
282
|
304
|
300
|
297
|
Cieľové ukazovatele v 16. týždni, % respondentov (95 % CI)
|
vIGA-AD 0/1a,b
(koprimárny)
|
8 (5,12)
|
48d
(42,54)
|
62d
(56,68)
|
5 (2,7)
|
39d
(33,45)
|
52d
(46,58)
|
11 (7,14)
|
40d
(34,45)
|
59d
(53,64)
|
EASI 75a
(koprimárny)
|
16
(12,21)
|
70d
(64,75)
|
80d
(75,84)
|
13
(9,17)
|
60d
(54,66)
|
73d
(68,78)
|
26
(21,31)
|
65d
(59,70)
|
77d
(72,82)
|
EASI 90a
|
8 (5,11)
|
53d
(47,59)
|
66d
(60,71)
|
5 (3,8)
|
42d
(37,48)
|
58d
(53,64)
|
13 (9,17)
|
43d
(37,48)
|
63d
(58,69)
|
EASI 100a
|
2 (0,3)
|
17d
(12,21)
|
27d
(22,32)
|
1 (0,2)
|
14d
(10,18)
|
19d
(14,23)
|
1 (0,3)
|
12e
(8,16)
|
23d
(18,27)
|
Najhoršie svrbenie NRSc (≥ 4-bodové zlepšenie)
|
12 (8,16)
|
52d
(46,58)
|
60d
(54,66)
|
9 (6,13)
|
42d
(36,48)
|
60d
(54,65)
|
15 (11,19)
|
52d
(46,58)
|
64d
(58,69)
|
Cieľov
é ukazovatele so skorým nástupom, % respondentov (95 % CI)
|
EASI 75a
(2. týždeň)
|
4 (1,6)
|
38d
(32,44)
|
47d
(42,53)
|
4 (1,6)
|
33d
(27,39)
|
44d
(38,50)
|
7 (4,10)
|
31d
(26,36)
|
44d
(38,50)
|
Najhoršie
svrbenie NRS (zlepšenie
o ≥ 4 body
v 1. týždni)c,f
|
0
(0,1)
|
15d
(11,19)
|
20d
(15,24)
|
1
(0,2)
|
7d
(4,11)
|
16d
(11,20)
|
3
(1,5)
|
12d
(8,16)
|
19d
(15,24)
|
Skratky: UPA = upadacitinib (RINVOQ); PBO = placebo
Jedinci, ktorým bola podaná záchranná liečba alebo u ktorých chýbali údaje, boli hodnotení ako neodpovedajúci na liečbu. Počet a percento pacientov, ktorí boli kvôli podaniu záchrannej liečby hodnotení ako neodpovedajúci na liečbu v ukazovateľoch EASI 75 a vIGA-AD 0/1 v16. týždni v skupinách
s placebom, upadacitibibom 15 mg resp. upadacitinibom 30 mg bolo 132 (47,0 %); 31 (11,0 %); 16 (5,6%)
v MEASURE UP 1; 119 (42,8 %); 24 (8,7 %); 16 (5,7 %) v MEASURE UP 2 a 78 (25,7 %); 15 (5,0 %); 14 (4,7 %) v AD UP.
a Na základe počtu randomizovaných jedincov
b Respondent bol definovaný ako pacient so skóre vIGA-AD 0 alebo 1 („čistý“ alebo „takmer čistý“) so znížením o ≥ 2 body na ordinálnej stupnici 0 až 4
c Výsledky uvedené v podskupine jedincov vhodných na posúdenie (pacienti s najhorším svrbením NRS ≥ 4
na začiatku)
d Štatisticky významné v porovnaní s placebom s p < 0,001
e p < 0,001 verzus placebo, bez kontroly multiplicity
f V rámci štúdií MEASURE UP 1 a 2 bolo pozorované štatisticky významné zlepšenie v porovnaní s placebom už 1. deň od začatia liečby upadacitinibom pri dávke 30 mg a 2 dni po začatí liečby
upadacitinibom pri dávke 15 mg
|
Obrázok 1 Podiel pacientov, ktorí dosiahli odpoveď EASI 75 a priemerná percentuálna zmena od východiskovej hodnoty pri najhoršom svrbení NRS v štúdiách MEASURE UP 1
a MEASURE UP 2
Podiel pacientov, ktorí dosiahli
odpove
ď
EAS
I 75
Pri
e
mern
á percentuálna zmena
 o
d východiskovej hodnoty pri najhoršom
svrben
í NRS
o
d východiskovej hodnoty pri najhoršom
svrben
í NRS


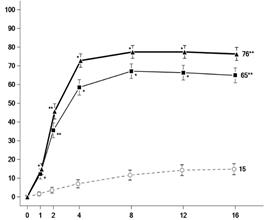 Týždn
e Týždne
Týždn
e Týždne
 Placeb
o
Placeb
o  Placebo
RINVOQ 15 mg QD RINVOQ 15 mg QD
Placebo
RINVOQ 15 mg QD RINVOQ 15 mg QD  RINVOQ 30 mg QD
RINVOQ 30 mg QD  RINVOQ 30 mg QD
RINVOQ 30 mg QD
*: p < 0,001 verzus placebo, bez kontroly multiplicity
**: štatisticky významné v porovnaní s placebom s p < 0,001
Účinky liečby v podskupinách (hmotnosť, vek, pohlavie, rasa a predchádzajúca systémová liečba imunosupresívami) boli v súlade s výsledkami v celkovej populácii štúdie.
Výsledky v 16. týždni sa zachovali až do 52. týždňa u pacientov liečených upadacitinibom 15 mg alebo 30 mg.
Kvalita života/výsledky hlásené pacientomTabuľka 13 Výsledky upadacitinibu hlásené pacientom v 16. týždniŠtúdia
| MEASURE UP 1
| MEASURE UP 2
|
Liečebná skupina
| PBO
| UPA
15 mg
| UPA
30 mg
| PBO
| UPA
15 mg
| UPA
30 mg
|
Počet randomizovaných pacientov
| 281
| 281
| 285
| 278
| 276
| 282
|
% respondentov (95 % CI)
|
ADerm-SS Skin Pain (bolesť kože)
(≥ 4-bodové zlepšenie)a
| 15 (10,20)
| 54e
(47,60)
| 63e
(57,69)
| 13 (9,18)
| 49e
(43,56)
| 65e
(59,71)
|
ADerm-IS Sleep (spánok)
(≥ 12-bodové zlepšenie) a,b
|
13 (9,18)
|
55e
(48,62)
|
66e
(60,72)
|
12 (8,17)
|
50e
(44,57)
|
62e
(56,69)
|
DLQI 0/1
|
4 (2,7)
|
30e
(25,36)
|
41e
(35,47)
|
5 (2,7)
|
24e
(19,29)
|
38e
(32,44)
|
Úzkosť podľa HADS < 8
a depresia podľa HADS < 8d
|
14 (8,20)
|
46e
(37,54)
|
49e
(41,57)
|
11 (6,17)
|
46e
(38,54)
|
56e
(48,64)
|
Skratky: UPA = upadacitinib (RINVOQ); PBO = placebo; DLQI = Dermatologický index kvality života; HADS = Stupnica hodnotenia úzkosti a depresie pri hospitalizácii
Jedinci, ktorým bola podaná záchranná liečba alebo u ktorých chýbali údaje, boli hodnotení
ako neodpovedajúci na liečbu.
Uvedené prahové hodnoty zodpovedajú minimálnemu klinicky dôležitému rozdielu (MCID)
a boli použité na stanovenie odpovede.
a Výsledky uvedené v podskupine pacientov vhodných na posúdenie (pacienti s hodnotiacim
skóre > MCID na začiatku).
b ADerm-IS Sleep hodnotí ťažkosti so zaspávaním, vplyv na spánok a nočné prebúdzanie spôsobené AD.
c Výsledky uvedené v podskupine pacientov vhodných na posúdenie (pacienti s DLQI > 1
na začiatku).
d Výsledky preukázané u podskupiny pacientov, ktorí sa mohli zúčastniť hodnotenia (pacienti s úzkosťou podľa HADS ≥ 8 alebo depresiou podľa HADS ≥ 8 na začiatku liečby)
e Štatisticky významné v porovnaní s placebom s p < 0,001
|
U
lcerózna kolitída
Účinnosť a bezpečnosť upadacitinibu boli hodnotené v troch multicentrických, dvojito zaslepených, placebom kontrolovaných klinických štúdiách fázy 3: dve replikované štúdie úvodnej dávky, UC-1
(U-ACHIEVE Induction) a UC-2 (U-ACCOMPLISH) a štúdia udržiavacej dávky UC-3 (U-ACHIEVE
Maintenance).
Aktivita ochorenia bola hodnotená na základe prispôsobeného Mayo skóre (adapted Mayo score, aMS, systém Mayo skóre s výnimkou Celkového posúdenia lekárom), ktoré je v rozsahu od 0 do 9 a má tri podskóre, kde je každému pridelené hodnotenie 0 (normálne) až 3 (najzávažnejšie): podskóre frekvencie stolice (stool frequency subscore, SFS), podskóre krvácania z konečníka (rectal bleeding subscore, RBS) a podskóre centrálne hodnotenej endoskopie (centrally-reviewed endoscopy subscore, ES).
 Štúdie úvodnej dávky (UC-1 a UC-2)
Štúdie úvodnej dávky (UC-1 a UC-2)V štúdiách UC-1 a UC-2 bolo randomizovaných 988 pacientov (473 resp. 515 pacientov) na upadacitinib 45 mg jedenkrát denne alebo placebo v trvaní 8 týždňov s pomerom pridelenia liečby
2 : 1 a zahrnutím do analýzy účinnosti. Všetci zaradení pacienti mali stredne ťažkú až ťažkú aktívnu
ulceróznu kolitídu definovanú ako hodnota aMS 5 až 9 a hodnota ES 2 alebo 3 a preukázanú neúspešnú liečbu vrátane nedostatočnej odpovede, straty odpovede, intolerancie predchádzajúcej konvenčnej a/alebo biologickej liečby. Neúspešná predchádzajúca liečba minimálne 1 biologickou liečbou (predchádzajúca neúspešná biologická liečba) bola pozorovaná u 52 % (246/473) resp. 51 % (262/515) pacientov. Neúspešná predchádzajúca liečba konvenčnou liečbou, nie však biologickou (bez predchádzajúcej neúspešnej biologickej liečby) bola pozorovaná u 48 % (227/473) resp. 49 % (253/515) pacientov.
Na začiatku liečby v štúdiách UC-1 a UC-2 39 % resp. 37 % pacientov užívalo kortikosteroidy, 1,1 % resp. 0,6 % pacientov dostávalo metotrexát a 68 % resp. 69 % pacientov dostávalo aminosalicyláty. Súbežné podávanie tiopurínu nebolo počas trvania štúdií povolené. Aktivita ochorenia pacientov bola stredne závažná (aMS ≥ 5, ≤ 7) u 61 % resp. 60 % pacientov a závažná (aMS > 7) u 39 %
resp. 40 % pacientov.
Primárnym cieľovým ukazovateľom bola klinická remisia podľa aMS v 8. týždni. V tabuľke 14 sú uvedené primárne a kľúčové sekundárne cieľové ukazovatele vrátane klinickej odpovede, hojenia sliznice, histologicko-endoskopického hojenia sliznice a hlbokého hojenia sliznice.
Tabuľka 14 Podiel pacientov, ktorí splnili primárne a kľúčové sekundárne cieľové ukazovatele účinnosti v 8. týždni v štúdiách úvodnej dávky UC-1 a UC-2
| UC-1
(U-ACHIEVE)
| UC-2
(U-ACCOMPLISH)
|
Cieľový ukazovateľ
|
PBO
N = 154
| UPA
45 mg
N = 319
| Rozdiel
liečby
(95 % CI)
|
PBO
N = 174
| UPA
45 mg
N = 341
| Rozdiel
liečby
(95 % CI)
|
Klinická remisiaa
| 4,8 %
| 26,1 %
| 21,6 %* (15,8; 27,4)
| 4,1 %
| 33,5 %
| 29,0 %* (23,2; 34,7)
|
Predchádzajúce zlyhanie biologickej liečby+
| 0,4 %
| 17,9 %
| 17,5 %
| 2,4 %
| 29,6 %
| 27,1 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
| 9,2 %
| 35,2 %
| 26,0 %
| 5,9 %
| 37,5 %
| 31,6 %
|
Klinická odpoveďb
| 27,3 %
| 72,6 %
| 46,3 %* (38,4; 54,2)
| 25,4 %
| 74,5 %
| 49,4 %* (41,7; 57,1)
|
Predchádzajúce zlyhanie biologickej liečby+
| 12,8 %
| 64,4 %
| 51,6 %
| 19,3 %
| 69,4 %
| 50,1 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
| 42,1 %
| 81,8 %
| 39,7 %
| 31,8 %
| 79,8 %
| 48,0 %
|
Hojenie sliznicec
| 7,4 %
| 36,3 %
| 29,3 %* (22,6; 35,9)
| 8,3 %
| 44,0 %
| 35,1 %* (28,6; 41,6)
|
Predchádzajúce zlyhanie biologickej liečby+
| 1,7 %
| 27,0 %
| 25,3 %
| 4,8 %
| 37,1 %
| 32,3 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
| 13,2 %
| 46,8 %
| 33,6 %
| 12,0 %
| 51,2 %
| 39,2 %
|
Histologicko- endoskopické hojenie slizniced
| 6,6 %
| 30,1 %
| 23,7 %* (17,5; 30,0)
| 5,9 %
| 36,7 %
| 30,1 %* (24,1; 36,2)
|
Predchádzajúce zlyhanie biologickej liečby+
| 1,4 %
| 22,7 %
| 21,3 %
| 4,6 %
| 30,7 %
| 26,1 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
| 11,8 %
| 38,2 %
| 26,4 %
| 7,2 %
| 42,9 %
| 35,7 %
|
Hlboké hojenie sliznicee
| 1,3 %
| 10,7 %
| 9,7 %* (5,7; 13,7)
| 1,7 %
| 13,5 %
| 11,3 %* (7,2; 15,3)
|
Predchádzajúce zlyhanie biologickej liečby+
| 0
| 6,5 %
| 6,5 %
| 1,1 %
| 9,2 %
| 8,1 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
| 2,6 %
| 15,4 %
| 12,8 %
| 2,4 %
| 17,9 %
| 15,5 %
|
|
UC
-
1
(
U
-
ACHIEVE)
|
UC
-
2
(
U
-
ACCOMPLISH)
|
Skratky: PBO = placebo; UPA = upadacitinib; aMS = prispôsobené Mayo skóre, založené na systéme
Mayo skóre (s výnimkou Celkového posúdenia lekárom), ktoré je v rozsahu od 0 do 9 a má tri
podskóre, kde je každému pridelené hodnotenie 0 (normálne) až 3 (najzávažnejšie): podskóre frekvencie stolice (stool frequency subscore, SFS), podskóre krvácania z konečníka (rectal bleeding subscore,
RBS) a podskóre centrálne hodnotenej endoskopie (centrally-reviewed endoscopy subscore, ES).
+Počet pacientov „Predchádzajúce zlyhanie biologickej liečby“ v štúdiách UC-1 a UC-2 je 78 resp. 89
v skupine s placebom a 168 resp. 173 v skupine s upadacitinibom 45 mg; počet pacientov „Bez predchádzajúceho zlyhania biologickej liečby“ v štúdiách UC-1 a UC-2 je 76 resp. 85 v skupine s placebom a 151 resp. 168 v skupine s upadacitinibom 45 mg.
*p < 0,001, upravený rozdiel liečby (95 % CI)
a Podľa aMS: SFS ≤ 1 a nie viac ako východisková hodnota, RBS = 0, ES ≤ 1 bez krehkosti (friability)
b Podľa aMS: zníženie o ≥ 2 body a ≥ 30 % v porovnaní s východiskovou hodnotou a zníženie RBS ≥ 1
v porovnaní s východiskovou hodnotou alebo absolútne RBS ≤ 1.
cES ≤ 1 bez krehkosti (friability)
d ES ≤ 1 bez krehkosti (friability) a Geboesov index ≤ 3,1 (znamenajúci infiltráciu neutrofilov v < 5 %
krýpt, žiadne poškodenie krýpt a žiadne erózie, ulcerácie alebo granulačné tkanivo.)
e ES = 0, Geboesov index < 2 (znamenajúci neprítomnosť neutrofilov v kryptách alebo lamina propria a žiadne zvýšenie počtu eozinofilov, žiadne poškodenie krýpt a žiadne erózie, ulcerácie alebo
granulačné tkanivo)
|
Aktivita ochorenia a príznaky
Čiastočne prispôsobené Mayo skóre (partial adapted Mayo score, paMS) sa skladá z SFS a RBS.
Symptomatická odpoveď podľa paMS je definovaná ako zníženie o ≥ 1 bod a ≥ 30 % v porovnaní s východiskovou hodnotou a zníženie RBS o ≥ 1 alebo absolútne RBS ≤ 1. Štatisticky významné zlepšenie pri porovnaní s placebom podľa paMS bolo pozorované už v 2. týždni (štúdia UC-1: 60,1 % v porovnaní s 27,3 % a štúdia UC-2: 63,3 % v porovnaní s 25,9 %).
Predĺžené obdobie úvodnej liečbyCelkovo 125 pacientov v štúdiách UC-1 a UC-2, ktorí nedosiahli klinickú odpoveď po 8. týždňoch
liečby upadacitinibom 45 mg jedenkrát denne, vstúpilo do 8-týždňového otvoreného predĺženého obdobia indukcie. Po liečbe ďalších 8 týždňov (celkom 16 týždňov) upadacitinibom 45 mg jedenkrát denne dosiahlo klinickú odpoveď podľa aMS 48,3 % pacientov. Z pacientov s odpoveďou na liečbu po
16-týždňovej liečbe upadacitinibom 45 mg jedenkrát denne si 35,7 % resp. 66,7 % pacientov udržalo klinickú odpoveď podľa aMS a 19,0 % resp. 33,3 % pacientov dosiahlo klinickú remisiu podľa aMS v 52. týždni udržiavacou liečbou s dávkou upadacitinibu 15 mg resp. 30 mg jedenkrát denne.
Štúdiaudržiavacejdávky(UC-3)Analýza účinnosti pre štúdiu UC-3 bola hodnotená u 451 pacientov, ktorí dosiahli klinickú odpoveď podľa aMS 8-týždňovou úvodnou liečbou s dávkou upadacitinibu 45 mg jedenkrát denne. Pacienti boli randomizovaní na užívanie dávky upadacitinibu 15 mg, 30 mg alebo placebo jedenkrát denne v trvaní až 52 týždňov.
Primárnym cieľovým ukazovateľom bola klinická remisia podľa aMS v 52. týždni. V tabuľke 15 sú uvedené sekundárne cieľové ukazovatele vrátane udržania klinickej odpovede, klinickej remisie bez užívania kortikosteroidov, hojenia sliznice, histologicko-endoskopického hojenia sliznice a hlbokého hojenia sliznice.
Tabuľk
a 15 Podiel pacientov, ktorí splnili primárne a kľúčové sekundárne
cieľové ukazovatele účinnosti v 52. týždni v štúdiách udržiavacej dávky
UC
-
3
|
PBO
N = 149
|
UPA
15 mg
N = 148
|
UPA
30 mg
N = 154
|
Rozdie
l liečby
15 mg
v porovnaní s
PBO (95 % CI)
|
Rozdie
l liečby
30 mg
v porovnaní s
PBO
(9
5 % CI)
|
Klinická remisia
a
|
12,1 %
|
42,3 %
|
51,7 %
|
30,7 %* (21,7; 39,8)
|
39,0 %* (29,7; 48,2)
|
Predchádzajúce zlyhanie biologickej liečby+
|
7,5 %
|
40,5 %
|
49,1 %
|
33,0 %
|
41,6 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
|
17,6 %
|
43,9 %
|
54,0 %
|
26,3 %
|
36,3 %
|
Udržanie klinickej remisie
b
|
N = 54
22,2 %
|
N = 47
59,2 %
|
N = 58
69,7 %
|
37,4 %* (20,3; 54,6)
|
47,0 %* (30,7; 63,3)
|
Predchádzajúce zlyhanie biologickej liečby
|
N = 22
13,6 %
|
N = 17
76,5 %
|
N = 20
73,0 %
|
62,8 %
|
59,4 %
|
Bez predchádzajúceho
zlyhania biologickej liečby
|
N = 32
28,1 %
|
N = 30
49,4 %
|
N = 38
68,0 %
|
21,3 %
|
39,9 %
|
Klinick
á remisia bez užívania kortikosteroidov
c
|
N = 54
22,2 %
|
N = 47
57,1 %
|
N = 58
68,0 %
|
35,4 %* (18,2; 52,7)
|
45,1 %* (28,7; 61,6)
|
Predchádzajúce zlyhanie
biologickej liečby
|
N = 22
13,6 %
|
N = 17
70,6 %
|
N = 20
73,0 %
|
57,0 %
|
59,4 %
|
Bez predchádzajúceho zlyhania biologickej liečby
|
N = 32
28,1 %
|
N = 30
49,4 %
|
N = 38
65,4 %
|
21,3 %
|
37,2 %
|
Hojenie sliznice
d
|
14,5 %
|
48,7 %
|
61,6 %
|
34,4 %*
(25,1; 43,7)
|
46,3 %*
(36,7; 55,8)
|
Predchádzajúce zlyhanie biologickej liečby+
|
7,8 %
|
43,3 %
|
56,1 %
|
35,5 %
|
48,3 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
|
22,5 %
|
53,6 %
|
66,6 %
|
31,1 %
|
44,1 %
|
Histologick
o
-
endoskopické hojenie sliznice
e
|
11,9 %
|
35,0 %
|
49,8 %
|
23,8 %* (14,8; 32,8)
|
37,3 %* (27,8; 46,8)
|
Predchádzajúce zlyhanie biologickej liečby+
|
5,2 %
|
32,9 %
|
47,6 %
|
27,7 %
|
42,4 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
|
20,0 %
|
36,9 %
|
51,8 %
|
16,9 %
|
31,8 %
|
Hlboké hojenie sliznice
f
|
4,7 %
|
17,6 %
|
19,0 %
|
13,0 %* (6,0; 20,0)
|
13,6 %* (6,6; 20,6)
|
Predchádzajúce zlyhanie biologickej liečby+
|
2,5 %
|
17,2 %
|
16,1 %
|
14,7 %
|
13,6 %
|
Bez predchádzajúceho zlyhania biologickej liečby+
|
7,5 %
|
18,0 %
|
21,6 %
|
10,6 %
|
14,2 %
|
Skratky: PBO = placebo; UPA = upadacitinib; aMS = prispôsobené Mayo skóresystém Mayo skóre, založené na systéme Mayo skóre (s výnimkou Celkového posúdenia lekárom), ktoré je v rozsahu od 0 do 9 a má tri podskóre, kde je každému pridelené hodnotenie 0 (normálne) až 3 (najzávažnejšie): podskóre frekvencie stolice (stool frequency subscore, SFS), podskóre krvácania z konečníka (rectal bleeding subscore, RBS) a podskóre centrálne hodnotenej endoskopie (centrally-reviewed endoscopy subscore, ES).
+Počet pacientov v skupine „Predchádzajúce zlyhanie biologickej liečby“ je 81, 71 a 73 (skupina placebo,
upadacitinib 15 mg a 30 mg). Počet pacientov v skupine „Bez predchádzajúceho zlyhania biologickej liečby“ je 68, 77 a 81 (skupina placebo, upadacitinib 15 mg a 30 mg).
* p < 0,001, upravený rozdiel liečby (95 % CI)
a Podľa aMS: SFS ≤ 1 a nie viac ako východisková hodnota, RBS = 0, ES ≤ 1 bez krehkosti
b Klinická remisia podľa aMS v 52. týždni medzi pacientmi, ktorí dosiahli klinickú remisiu na konci obdobia začatočnej liečby.
|
b Klinická remisia podľa aMS v 52. týždni a bez užívania kortikosteroidov ≥ 90 dní bezprostredne predchádzajúcich 52. týždňu medzi pacientmi, ktorí dosiahli klinickú remisiu na konci obdobia úvodnej liečby.
d ES ≤ 1 bez krehkosti
e ES ≤ 1 bez krehkosti a Geboesov index ≤ 3,1 (znamenajúci infiltráciu neutrofilov v < 5 % krýpt, žiadne poškodenie krýpt a žiadne erózie, ulcerácie alebo granulačné tkanivo.)
f ES = 0, Geboesov index < 2 (znamenajúci neprítomnosť neutrofilov v kryptách alebo lamina propria
a žiadne zvýšenie počtu eozinofilov, žiadne poškodenie krýpt a žiadne erózie, ulcerácie alebo granulačné tkanivo).
Príznaky ochoreniaSymtomatická remisia podľa paMS, definovaná ako SFS ˂ 1 a RBS = 0, sa dosiahla v priebehu času do 52 týždňa viac pacientov liečených obomi dávkami upadacitinibu 15 mg a 30 mg jedenkrát denne v porovnaní s pacientmi užívajúcimi placebo (obrázok 2).
Obrázok 2 Podiel pacientov so symptomatickou remisiou podľa čiastočného prispôsobenéhoMayo skóre v priebehu času v štúdiách udržiavacej dávky UC-364,5

57,4
17,5
Týždne

(N = 154154)
Placebo (N = 149) PA 15 mg jedenkrát denne (N = 148) UPA 30 mg jedenkrát denne
 Endoskopické vyšetrenie
Endoskopické vyšetrenie
Endoskopická remisia (normalizácia endoskopického vzhľadu sliznice) bola definovaná ako skóre ES na úrovni 0. V 8. týždni dosiahol endoskopickú remisiu významne vyšší podiel pacientov liečených upadacitinibom 45 mg jedenkrát denne v porovnaní s pacientmi užívajúcimi placebo (UC-1: 13,7 % vs. 1,3 %; UC-2: 18,2 % vs. 1,7 %). V štúdii UC-3 podstatne väčší podiel pacientov užívajúcich upadacitinib 15 mg a 30 mg jedenkrát denne v porovnaní s pacientmi užívajúcimi placebo dosiahol endoskopickú remisiu v 52. týždni (24,2 % a 25,9 % vs. 5,6 %). Udržanie hojenia sliznice v 52. týždni (ES ≤ 1 bez krehkosti) bolo pozorované u podstatne väčšieho podielu pacientov liečených upadacitinibom 15 mg a 30 mg jedenkrát denne v porovnaní s pacientmi užívajúcimi placebo (61,6 % a 69,5 % vs. 19,2 %) medzi pacientmi, ktorí dosiahli hojenie sliznice na konci úvodného obdobia.
Kvalita životaU pacientov liečených upadacitinibom bolo preukázané podstatne väčšie a klinicky významné zlepšenie kvality života súvisiacej so zdravím, merané celkovým skóre Dotazníka pre zápalové ochorenia čreva (Inflammatory Bowel Disease Questionnaire, IBDQ) v porovnaní s placebom. Zlepšenia boli pozorované v skóre všetkých 4 oblastí: systémové príznaky (vrátane únavy), sociálne fungovanie, emočné fungovanie a črevné príznaky (vrátanie bolesti brucha a nutkania na urgentnú stolicu). Zmeny celkového skóre IBDQ v 8. týždni v porovnaní s východiskovou hodnotou pre upadacitinib 45 mg jedenkrát denne v porovnaní s placebom boli 55,3 resp. 21,7 v štúdii UC-1 a 52,2
resp. 21,1 v štúdii UC-2. Zmeny celkového skóre IBDQ v 52. týždni v porovnaní s východiskovou hodnotou boli 49,2; 58,9 resp. 17,9 pre upadacitinib 15 mg, 30 mg jedenkrát denne, resp. placebo.
Pediatrická populáciaV troch štúdiách fázy 3 bolo randomizovaných celkom 344 dospievajúcich vo veku 12 až 17 rokov so
stredne ťažkou až ťažkou atopickou dermatitídou, ktorí dostávali buď 15 mg (N = 114), alebo 30 mg (N = 114) upadacitinibu, alebo zodpovedajúce placebo (N = 116 ) v monoterapii alebo v kombinácii s lokálnymi kortikosteroidmi. Účinnosť bola konzistentná medzi dospievajúcimi a dospelými. Bezpečnostný profil u dospievajúcich bol všeobecne podobný ako u dospelých, so zvýšeniami závislými od dávky, pokiaľ ide o mieru výskytu niektorých nežiaducich udalostí vrátane neutropénie a herpes zoster. V prípade oboch dávok bola miera výskytu neutropénie mierne vyššia
u dospievajúcich v porovnaní s dospelými. Miery výskytu herpes zoster u dospievajúcich bola pri
30 mg dávke porovnateľná s mierou výskytu u dospelých. Bezpečnosť a účinnosť 30 mg dávky u dospievajúcich sa stále skúmajú.
Tabuľka 16 Výsledky účinnosti upadacitinibu u dospievajúcich v 16. týždniŠtúdia
| MEASURE UP 1
| MEASURE UP 2
| AD UP
|
Liečebná skupina
| PBO
| UPA
15 mg
| PBO
| UPA
15 mg
| PBO +
TCS
| UPA
15 mg + TCS
|
Počet randomizovaných dospievajúcich jedincov
|
40
|
42
|
36
|
33
|
40
|
39
|
% respondentov (95 % CI)
|
vIGA-AD 0/1 a,b
| 8 (0,16)
| 38 (23,53)
| 3 (0,8)
| 42 (26,59)
| 8 (0,16)
| 31 (16,45)
|
EASI 75a
| 8 (0,17)
| 71 (58,85)
| 14 (3,25)
| 67 (51,83)
| 30 (16,44)
| 56 (41,72)
|
Najhoršie svrbenie
NRSc
(≥ 4-bodové zlepšenie)
| 15 (4,27)
| 45 (30,60)
| 3 (0,8)
| 33 (16,50)
| 13 (2,24)
| 42 (26,58)
|
Skratky: UPA = upadacitinib (RINVOQ); PBO = placebo
Jedinci, ktorým bola podaná záchranná liečba alebo u ktorých chýbali údaje, boli hodnotení
ako neodpovedajúci na liečbu.
a Na základe počtu randomizovaných jedincov
b Respondent bol definovaný ako pacient so skóre vIGA-AD 0 alebo 1 („čistý“ alebo
„takmer čistý“) so znížením o ≥ 2 body na ordinálnej stupnici 0 až 4.
c Výsledky uvedené v podskupine pacientov vhodných na posúdenie (pacienti s najhorším svrbením NRS ≥ 4 na začiatku).
|
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom RINVOQ
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri chronickej idiopatickej artritíde (zahŕňajúcej reumatoidnú artritídu, psoriatickú artritídu, spondyloartritídu a juvenilnú idiopatickú artritídu), atopickej dermatitíde a ulceróznej kolitíde (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiPlazmatické expozície upadacitinibu sú úmerné dávke v terapeutickom rozmedzí dávok. Rovnovážne plazmatické koncentrácie sa dosiahnu do 4 dní s minimálnou akumuláciou po opakovanom podávaní jedenkrát denne.
Absorpcia
Po perorálnom podaní upadacitinibu vo forme s predĺženým uvoľňovaním sa upadacitinib vstrebáva
s mediánom Tmax 2 až 4 hodiny. Súbežné podávanie upadacitinibu s jedlom s vysokým obsahom tuku nemalo klinicky významný účinok na expozície upadacitinibu (zvýšená AUC o 29 % a Cmax o 39 % až
60 %). V klinických skúšaniach bol upadacitinib podávaný bez ohľadu na jedlo (pozri časť 4.2).
Upadacitinib je substrátom pre efluxné transportéry P-gp a BCRP in vitro.
Distribúcia
52 % upadacitinibu sa viaže na plazmatické bielkoviny. Upadacitinib sa rozdeľuje v rovnakej miere
medzi plazmu a krvné bunkové zložky, ako naznačuje pomer krvi ku plazme 1.0.
Metabolizmus
Metabolizmus upadacitinibu je sprostredkovaný CYP3A4 s potenciálne malým príspevkom CYP2D6.
Farmakologická aktivita upadacitinibu sa pripisuje materskej molekule. V štúdii s rádioaktívne
značenou látkou u ľudí nezmenený upadacitinib zodpovedal za 79 % celkovej rádioaktivity v plazme pričom hlavný metabolit (produkt monooxidácie, po ktorej nasledovala glukuronidácia) zodpovedal za
13 % celkovej rádioaktivity v plazme. Pri upadacitinibe neboli zistené žiadne aktívne metabolity.
Eliminácia
Po podaní jednorazovej dávky roztoku obsahujúceho [14C]-upadacitinib s okamžitým uvoľňovaním sa
upadacitinib prednostne vylučoval vo forme nezmenenej materskej látky do moču (24 %) a stolice (38
%). Približne 34 % upadacitinibu bolo vylúčených vo forme metabolitov. Priemerný polčas eliminácie upadacitinibu sa pohyboval v rozmedzí od 9 do 14 hodín.
Špeciálne populácie
Porucha funkcie obličiek
AUC upadacitinibu bola o 18 % vyššia u jedincov s miernou poruchou funkcie obličiek (odhadovaná rýchlosť glomerulárnej filtrácie 60 až 89 ml/min/1,73 m2), o 33 % u jedincov so stredne ťažkou poruchou funkcie (odhadovaná rýchlosť glomerulárnej filtrácie 30 až 59 ml/min/1,73 m2) a o 44 %
u jedincov s ťažkou poruchou funkcie obličiek (odhadovaná rýchlosť glomerulárnej filtrácie 15 až
29 ml/min/1,73 m2). Cmax upadacitinibu bola podobná u jedincov s normálnou funkciou obličiek
a jedincov s poruchou funkcie obličiek. Mierne alebo stredne ťažká porucha funkcie obličiek nemá klinicky významný vplyv na expozíciu upadacitinibu (pozri časť 4.2).
Porucha funkcie pečene
Mierna (trieda A podľa Childa-Pugha) a stredne ťažká (trieda B podľa Childa-Pugha) porucha funkcie pečene nemá žiaden klinicky významný účinok na expozíciu upadacitinibu. AUC upadacitinibu bola vyššia o 28 % u jedincov s miernou poruchou funkcie pečene a o 24 % u jedincov so stredne ťažkou poruchou funkciou pečene v porovnaní s jedincami s normálnou funkciou pečene. Cmax upadacitinibu bola nezmenená u jedincov s miernou poruchou funkcie pečene a o 43 % vyššia u jedincov so stredne ťažkou poruchou funkcie pečene v porovnaní s jedincami s normálnou funkciou pečene. Upadacitinib nebol skúmaný u pacientov s ťažkou (trieda C podľa Childa-Pugha) poruchou funkcie pečene.
Pediatrická populácia
Farmakokinetika upadacitinibu nebola doteraz vyhodnocovaná u pediatrických pacientov
s reumatoidnou artritídou, psoriatickou artritídou, axiálnou spondyloartritídou a ulceróznou kolitídou
(pozri časť 4.2).
Farmakokinetika upadacitinibu a koncentrácie v rovnovážnom stave sú podobné u dospelých
a dospievajúcich vo veku 12 až 17 rokov s atopickou dermatitídou. Dávkovanie u dospievajúcich pacientov s hmotnosťou od 30 kg do < 40 kg sa stanovilo pomocou populačného farmakokinetického modelovania a simulácie.
Farmakokinetika upadacitinibu u pediatrických pacientov (vo veku < 12 rokov) s atopickou dermatitídou nebola stanovená.
Vnútorné faktory
Vek, pohlavie, telesná hmotnosť, rasa a etnicita nemali klinicky významný účinok na expozíciu upadacitinibu. Farmakokinetika upadacitinibu je u pacientov s reumatoidnou artritídou, psoriatickou artritídou, axiálnou spondyloartritídou, atopickou dermatitídou a ulceróznou kolitídou konzistentná.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje neodhalili žiadne špeciálne riziko pre ľudí na základe konvenčných farmakologických štúdií bezpečnosti.
V 2-ročnej štúdii karcinogenity u potkanov druhu Sprague-Dawley nebol upadacitinib pri expozíciách
(na základe AUC) približne 4- a 10-násobku klinickej dávky 15 mg, 2- a 5-násobku klinickej dávky
30 mg a 1,6- a 4-násobku klinickej dávky 45 mg u samcov a samíc potkana druhu Sprague-Dawley karcinogénny. Upadacitinib nebol karcinogénny v 26-týždňovej štúdii karcinogenity u transgénnych myší CByB6F1-Tg(HRAS)2Jic.
Upadacitinib nebol mutagénny ani genotoxický na základe výsledkov testov in vitro a in vivo pri génových mutáciách a chromozomálnych aberáciách.
Upadacitinib nemal v štúdii fertility a skorého embryonálneho vývoja žiadny vplyv na fertilitu samcov a samíc potkana pri expozíciách až do približne 16- a 31-násobku expozície pri maximálnej odporúčanej dávke u ľudí (MRHD) 45 mg u samcov a u samíc na základe hodnoty AUC. Zvýšenia fetálnej resorpcie súvisiace s dávkou. spojené s postimplantačnými stratami boli v tejto štúdii fertility
u potkanov pripisované vývojovým/teratogénnym účinkom upadacitinibu. Nepozorovali sa žiadne
nežiaduce účinky pri expozíciách nižších ako klinická expozícia (založené na AUC). Postimplantačné straty sa pozorovali pri expozíciách 8-násobne vyšších ako klinická expozícia pri MRHD 45 mg (založené na AUC).
V štúdiách embryo-fetálneho vývoja u zvierat bol upadacitinib teratogénny u potkanov aj králikov. Upadacitinib viedol k zvýšenému počtu skeletálnych malformácií u potkanov pri 1,6-; 0,8-
a 0,6-násobku klinickej expozície (založené na AUC) pri dávkach 15 mg, 30 mg resp. 45 mg
(MRHD). U králikov sa pozoroval vyšší výskyt kardiovaskulárnych malformácií pri 15-;
7,6- a 5,6-násobku klinickej expozície pri dávkach 15 mg, 30 mg resp. 45 mg(založené na AUC).
Po podaní upadacitinibu laktujúcim potkanom boli koncentrácie upadacitinibu v mlieku v priebehu času vo všeobecnosti podobné koncentráciám v plazme s približne 30-násobne vyššou expozíciou
v mlieku v porovnaní s plazmou matky. Približne 97 % látok súvisiacich s upadacitinibom v mlieku bola materská molekula upadacitinib.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah tablety
mikrokryštalická celulóza
hypromelóza
manitol kyselina vínna
koloidný oxid kremičitý bezvodý
stearát horečnatý
Filmový obal
polyvinylalkohol
makrogol mastenec
oxid titaničitý (E 171)
čierny oxid železitý (E 172) (len sila 15 mg)
červený oxid železitý (E 172)
žltý oxid železitý (E 172) (len sila 45 mg)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
RINVOQ 15 mg tablety s predĺženýmuvoľňovaním
Tablety s predĺženým uvoľňovaním v blistroch: 2 roky
Tablety s predĺženým uvoľňovaním vo fľaškách: 3 roky
RINVOQ 30 mg tablety s predĺženým uvoľňovaním
Tablety s predĺženým uvoľňovaním v blistroch: 2 roky
Tablety s predĺženým uvoľňovaním vo fľaškách: 3 roky
RINVOQ 45 mg tablety s predĺženým uvoľňovaním
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne špeciálne teplotné podmienky na uchovávanie.
Uchovávajte v pôvodnom blistri alebo fľaške na ochranu pred vlhkosťou. Fľašku uchovávajte dôkladne uzatvorenú.
6.5 Druh obalu a obsah balenia
RINVOQ 15 mg tablety s predĺženým uvoľňovaním
Polyvinylchloridové/polyetylénové/polychlorotrifluoroetylénové - hliníkové kalendárové blistre
v baleniach obsahujúcich 28 alebo 98 tabliet s predĺženým uvoľňovaním alebo multibalenia obsahujúce 84 (3 balenia po 28) tabliet s predĺženým uvoľňovaním.
HDPE fľašky s vysúšadlom a polypropylénovým uzáverom v škatuli obsahujúce 30 tabliet s predĺženým uvoľňovaním.
Veľkosť balenia: 1 fľaška (30 tabliet s predĺženým uvoľňovaním) alebo 3 fľašky (90 tabliet s predĺženým uvoľňovaním).
Na trh nemusia byť uvedené všetky veľkosti balenia.
RINVO
Q 30 mg tablety s predĺženým uvoľňovaním
Kalendárové blistre z polyvinylchloridu/polyetylénu/polychlórtrifluóretylénu a hliníka v baleniach
obsahujúcich 28 alebo 98 tabliet s predĺženým uvoľňovaním.
HDPE fľašky s vysúšadlom a polypropylénovým uzáverom v škatuli obsahujúce 30 tabliet s predĺženým uvoľňovaním.
Veľkosť balenia: 1 fľaška (30 tabliet s predĺženým uvoľňovaním) alebo 3 fľašky (90 tabliet s predĺženým uvoľňovaním).
Na trh nemusia byť uvedené všetky veľkosti balenia. RINVOQ45 mgtabletyspredĺženýmuvoľňovaním
Polyvinylchloridové/polyetylénové/polychlorotrifluoroetylénové - hliníkové kalendárové blistre
v baleniach obsahujúcich 28 tabliet s predĺženým uvoľňovaním.
HDPE fľašky s vysúšadlom a polypropylénovým uzáverom v škatuli obsahujúce 28 tabliet s predĺženým uvoľňovaním.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AbbVie Deutschland GmbH & Co. KG Knollstrasse
67061 Ludwigshafen
Nemecko
8. REGISTRAČNÉ ČÍSLO
EU/1/19/1404/001
EU/1/19/1404/002
EU/1/19/1404/003
EU/1/19/1404/004
EU/1/19/1404/005
EU/1/19/1404/006
EU/1/19/1404/007
EU/1/19/1404/008
EU/1/19/1404/009
EU/1/19/1404/010
EU/1/19/1404/011
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 16. december 2019
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.