
aktívnymi alebo suspektnými očnými alebo periokulárnymi infekciami. Pacienti s aktívnym ťažkým vnútroočným zápalom.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Reakcie súvisiace s intravitreálnou injekciou
Podanie intravitreálnych injekcií vrátane injekcií ranibizumabu sa spájalo s endoftalmitídou,
vnútroočným zápalom, regmatogénnym odlúčením sietnice, trhlinou v sietnici a iatrogénnou traumatickou kataraktou (pozri časť 4.8). Pri podaní ranibizumabu sa musia vždy dodržať náležité aseptické injekčné postupy. Okrem toho je potrebné pacientov sledovať počas týždňa po podaní injekcie, čo umožní včasnú liečbu v prípade infekcie. Pacienti majú byť poučení, aby bezodkladne oznámili akýkoľvek príznak, ktorý poukazuje na endoftalmitídu alebo na niektorú z vyššie uvedených
udalostí.
Zvýšenia vnútroočného tlaku
U dospelých sa v priebehu 60 minút po podaní injekcie ranibizumabu pozorovalo prechodné zvýšenie
vnútroočného tlaku (IOP). Zistilo sa aj trvalé zvýšenie IOP (pozri časť 4.8). Vnútroočný tlak aj perfúzia hlavy zrakového nervu sa musia monitorovať a náležite liečiť.
Pacientov je potrebné informovať o prejavoch týchto potenciálnych nežiaducich reakcií a poučiť ich, aby informovali svojho lekára, ak u nich vzniknú príznaky ako bolesť oka alebo zvýšenie nepríjemných pocitov, zhoršujúce sa sčervenanie oka, neostré alebo zhoršené videnie, zvýšený počet malých čiastočiek v zornom poli, alebo zvýšená citlivosť na svetlo (pozri časť 4.8).
Bilaterálna liečba
Obmedzené údaje o bilaterálnom použití ranibizumabu (vrátane podania v ten istý deň) nenaznačujú
zvýšené riziko systémových nežiaducich udalostí v porovnaní s unilaterálnou liečbou.
I
m
unog
enita
Ranibizumab môže byť imunogénny. Keďže je možnosť zvýšenej systémovej expozície u osôb s
DME, nemožno vylúčiť zvýšené riziko vzniku precitlivenosti u tejto populácie pacientov. Pacienti majú byť tiež poučení, aby hlásili akékoľvek zvýšenie závažnosti vnútroočného zápalu, ktoré môže byť klinickým príznakom zodpovedajúcim tvorbe protilátok vo vnútri oka.
Súbežné použitie s inými anti-VEGF (vaskulárny endoteliálny rastový faktor)
Ranibizumab sa nemá podávať súbežne s inými anti-VEGF liekmi (systémovými alebo okulárnymi).
Prerušenie liečby ranibizumabom u dospelých
Dávka sa nemá podať a v liečbe sa nemá pokračovať skôr ako počas najbližšej plánovanej návštevy
v prípade:
• poklesu najlepšie korigovanej zrakovej ostrosti (BCVA) o ≥ 30 písmen v porovnaní s posledným stanovením zrakovej ostrosti;
• intraokulárneho tlaku ≥ 30 mmHg;
• trhliny v sietnici;
• subretinálneho krvácania postihujúceho stred fovey, alebo ak rozsah krvácania je ≥ 50 %
celkovej plochy lézie;
• uskutočneného alebo plánovaného intraokulárneho chirurgického zákroku počas uplynulých
alebo nasledujúcich 28 dní.
Trhlina v pigmentovom epiteli sietnice
Rizikové faktory, ktoré sa spájajú so vznikom trhliny v pigmentovom epiteli sietnice po anti-VEGF
liečbe pri vlhkej VPDM a prípadne aj pri iných formách CNV, zahŕňajú veľké a/alebo vysoko uložené odlúčenie pigmentového epitelu sietnice. Pri začatí liečby ranibizumabom je potrebná opatrnosť
u pacientov s týmito rizikovými faktormi pre trhliny v pigmentovom epiteli sietnice.
Regmatogénne odlúčenie sietnice alebo makulárne diery u dospelých
Liečba sa má ukončiť u osôb s regmatogénnym odlúčením sietnice alebo makulárnymi dierami 3.
alebo 4. stupňa.
Skupiny pacientov, u ktorých sú skúsenosti obmedzené
S liečbou osôb s DME spôsobeným diabetom typu I sú len obmedzené skúsenosti. Ranibizumab sa
neskúmal u pacientov, ktorí v minulosti dostali intravitreálne injekcie, u pacientov s aktívnymi systémovými infekciami alebo u pacientov so sprievodnými očnými ochoreniami, napr. s odlúčením sietnice alebo makulárnou dierou. Skúsenosti s liečbou ranibizumabom u pacientov s diabetom s HbA1c vyšším ako 108 mmol/mol (12 %) sú obmedzené a u pacientov s nekontrolovanou hypertenziou nie sú žiadne skúsenosti. Pri liečbe takýchto pacientov má lekár vziať do úvahy tento nedostatok informácií.
Nie je dostatok údajov, z ktorých by bolo možné vyvodiť závery o účinku ranibizumabu u pacientov s
RVO, u ktorých dôjde k ireverzibilnej strate zrakovej funkcie pre ischémiu.
Údaje o účinku ranibizumabu sú obmedzené u pacientov s PM, ktorí v minulosti podstúpili neúspešnú fotodynamickú liečbu verteporfínom (vPDT). Zatiaľ čo sa pozoroval zhodný účinok u osôb so subfoveálnymi a juxtafoveálnymi léziami, nie je tiež dostatok údajov, z ktorých by bolo možné usudzovať na účinok ranibizumabu u osôb s PM, ktoré majú extrafoveálne lézie.
Systémové účinky po intravitreálnom použití
Po intravitreálnej injekcii inhibítorov VEGF sa zaznamenali systémové nežiaduce udalosti vrátane
krvácaní mimo oka a artériových tromboembolických príhod.
Údaje o bezpečnosti liečby u pacientov s DME, makulárnym edémom po RVO a s CNV v dôsledku PM, ktorí majú v anamnéze cievnu mozgovú príhodu alebo tranzitórne ischemické ataky, sú obmedzené. Pri liečbe takýchto pacientov je potrebné postupovať opatrne (pozri časť 4.8).
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Adjuvantné použitie fotodynamickej liečby (PDT) verteporfínom a ranibizumabu pri vlhkej VPDM a
PM, pozri časť 5.1.
Adjuvantné použitie laserovej fotokoagulácie a ranibizumabu pri DME a BRVO, pozri časti 4.2 a 5.1.
V klinických skúšaniach liečby poškodenia zraku v dôsledku DME súbežná liečba tiazolidíndiónmi neovplyvnila výsledky týkajúce sa zrakovej ostrosti alebo hrúbky čiastkového poľa centrálnej sietnice (CSTF) u pacientov liečených ranibizumabom.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/antikoncepcia u žien
Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby.
Gravidita
Nie sú dostupné klinické údaje o použití ranibizumabu v gravidite. Štúdie na makakoch krabožravých
nepreukázali priame alebo nepriame škodlivé účinky z hľadiska gravidity alebo vývinu embrya/plodu (pozri časť 5.3). Systémová expozícia ranibizumabu po podaní do oka je nízka, ale vzhľadom na jeho mechanizmus účinku sa ranibizumab musí považovať za potenciálne teratogénny a embryo-
/fetotoxický. Preto sa ranibizumab nemá používať v gravidite, pokiaľ očakávaný prínos nie je väčší ako možné riziko pre plod. Ženám, ktoré chcú otehotnieť a boli liečené ranibizumabom, sa odporúča počkať s počatím dieťaťa aspoň 3 mesiace od poslednej dávky ranibizumabu.
Dojčenie
Na základe veľmi obmedzených údajov sa ranibizumab môže v nízkych hladinách vylučovať do
materského mlieka. Účinok ranibizumabu na dojčeného novorodenca/dojča nie je známy. Ako
preventívne opatrenie sa počas používania ranibizumabu neodporúča dojčiť.
Fertilita
Nie sú dostupné žiadne údaje o vplyve na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liečba môže vyvolať dočasné poruchy videnia, čo môže ovplyvniť schopnosť viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8). Pacienti, u ktorých sa vyskytnú tieto príznaky, nesmú viesť vozidlá alebo obsluhovať stroje, kým tieto dočasné poruchy zraku neustúpia.
4
.8 Nežiaduce účinky
Zhrnutie profilu bezpečnostiVäčšina nežiaducich reakcií hlásených po podaní ranibizumabu súvisí s postupom intravitreálnej
injekcie.
Najčastejšie hlásené nežiaduce reakcie týkajúce sa očí po podaní injekcie ranibizumabu sú: bolesť oka, hyperémia oka, zvýšený vnútroočný tlak, vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, opacity v sklovci, krvácanie do spojovky, podráždenie oka, pocit cudzieho telieska v očiach, zvýšené slzenie, blefaritída, suché oko a svrbenie oka.
Najčastejšie hlásené nežiaduce reakcie netýkajúce sa očí sú bolesť hlavy, nazofaryngitída a bolesť kĺbov.
Medzi menej často hlásené, ale závažnejšie nežiaduce reakcie patria endoftalmitída, slepota, odlúčenie
sietnice, trhlina v sietnici a iatrogénna traumatická katarakta (pozri časť 4.4).
Nežiaduce reakcie, ktoré sa vyskytli po podaní ranibizumabu v klinických skúšaniach, sú zhrnuté v tabuľke nižšie.
Tabuľkový zoznam nežiaducich reakcií#Nežiaduce reakcie sú zatriedené podľa orgánových systémov a frekvencie podľa nasledujúcich
konvencií: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Infekcie a nákazy
|
Veľmi časté
| Nazofaryngitída
|
Časté
| Infekcia močových ciest*
|
 Poruchy krvi a lymfatického systémuČasté
Poruchy krvi a lymfatického systémuČasté Anémia
 Poruchy imunitného systémuČasté
Poruchy imunitného systémuČasté Precitlivenosť
 Psychické poruchyČasté
Psychické poruchyČasté Úzkosť
Poruchy nervového systémuVeľmi časté Bolesť hlavy
Poruchy oka
|
Veľmi časté
| Vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, bolesť oka, opacity v sklovci, krvácanie do spojovky,
podráždenie oka, pocit cudzieho telieska v očiach, zvýšené
slzenie, blefaritída, suché oko, hyperémia oka, svrbenie oka.
|
Časté
| Degenerácia sietnice, porucha sietnice, odlúčenie sietnice, trhlina v sietnici, odlúčenie pigmentového epitelu sietnice, trhlina v pigmentovom epiteli sietnice, znížená zraková ostrosť, krvácanie
do sklovca, porucha sklovca, uveitída, iritída, iridocyklitída,
katarakta, subkapsulárna katarakta, opacifikácie zadného puzdra šošovky, bodkovitá keratitída, abrázia rohovky, zápal prednej
|
|
očnej komory, neostré videnie, krvácanie v mieste podania injekcie, krvácanie do oka, konjunktivitída, alergická konjunktivitída, výtok z oka, fotopsia, fotofóbia, nepríjemné pocity v oku, edém mihalnice, bolesť mihalnice, hyperémia
spojoviek.
|
M
enej časté
|
Slepota, endoftalmitída, hypopyon, hyféma, keratopatia, adhézia dúhovky, depozity v rohovke, edém rohovky, strie rohovky, bolesť v mieste podania injekcie, podráždenie v mieste podania injekcie, abnormálne pocity v oku, podráždenie mihalnice.
|
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté Kašeľ
Poruchy gastrointestinálneho traktuČasté Nauzea
Poruchy kože a podkožného tkanivaČasté Alergické reakcie (exantém, urtikária, pruritus, erytém)
 Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté Artralgia
 Laboratórne a funkčné vyšetreniaVeľmi časté
Laboratórne a funkčné vyšetreniaVeľmi časté Zvýšenie vnútroočného tlaku
# Nežiaduce reakcie boli definované ako nežiaduce udalosti (u najmenej 0,5 percentuálneho bodu pacientov), ktoré sa vyskytli častejšie (najmenej 2 percentuálne body) u pacientov liečených ranibizumabom 0,5 mg, ako u pacientov, ktorí dostávali kontrolnú liečbu (simulované podanie alebo PDT verteporfínom).
* pozorované iba u populácie s DME
Nežiaduce reakcie súvisiace so skupinou liekovV klinických skúšaniach fázy III pri vlhkej VPDM bola celková frekvencia krvácaní mimo oka, čo je
nežiaduca udalosť potenciálne súvisiaca so systémovou inhibíciou VEGF (vaskulárneho endoteliálneho rastového faktora), mierne zvýšená u pacientov liečených ranibizumabom. Charakteristika rôznych krvácaní však nebola zhodná. Po intravitreálnom použití inhibítorov VEGF existuje teoretické riziko arteriálnych tromboembolických príhod vrátane cievnej mozgovej príhody a infarktu myokardu. V klinických skúšaniach ranibizumabu sa pozorovala nízka incidencia arteriálnych tromboembolických príhod u pacientov s VPDM, DME, PDR, RVO a CNV a neboli významné rozdiely medzi skupinami liečenými ranibizumabom v porovnaní s kontrolnou liečbou.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieBoli hlásené prípady náhodného predávkovania v klinických skúšaniach pri vlhkej VPDM a z údajov po uvedení lieku na trh. Nežiaduce reakcie súvisiace s týmito hlásenými prípadmi boli zvýšenie vnútroočného tlaku, prechodná slepota, znížená zraková ostrosť, edém rohovky, bolesť rohovky
a bolesť oka. Ak dôjde k predávkovaniu, je potrebné sledovať a liečiť vnútroočný tlak, ak to ošetrujúci lekár považuje za potrebné.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5
.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: oftalmologiká, látky s antineovaskularizačným účinkom, ATC kód:
S01LA04
Ranivisio je podobný biologický liek. Podrobné informácie sú dostupné na internetovej stránke
Európskej agentúry pre lieky
http://www.ema.europa.eu.MechanizmusúčinkuRanibizumab je fragment humanizovanej rekombinantnej monoklonálnej protilátky, ktorého cieľom je
ľudský vaskulárny endoteliálny rastový faktor A (VEGF-A). Viaže sa s vysokou afinitou na izoformy VEGF-A (napr. VEGF110, VEGF121 a VEGF165), a tým zabraňuje vzniku väzby VEGF-A na jeho receptory VEGFR-1 a VEGFR-2. Naviazanie VEGF-A na jeho receptory má za následok proliferáciu endotelových buniek a neovaskularizáciu, ako aj únik tekutín z ciev, čo všetko sa považuje za faktory prispievajúce k progresii neovaskulárnej formy vekom podmienenej degenerácie makuly, patologickej
myopie a CNV alebo poškodeniu zraku spôsobeného buď diabetickým makulárnym edémom, alebo
makulárnym edémom po RVO u dospelých.
Klinická účinnosť a bezpečnosťLiečbavlhkejVPDMPri vlhkej VPDM sa klinická bezpečnosť a účinnosť ranibizumabu hodnotila v troch randomizovaných, dvojito maskovaných, simulovane alebo aktívne kontrolovaných klinických skúšaniach trvajúcich 24 mesiacov u pacientov s neovaskulárnou VPDM. Do týchto klinických skúšaní bolo zaradených celkovo 1 323 pacientov (879 v aktívnej a 444 v kontrolnej skupine).
V klinickom skúšaní FVF2598g (MARINA) 716 pacientov s minimálne klasickou alebo okultnou léziou bez klasických lézií bolo randomizovaných v pomere 1:1:1, aby dostávali každý mesiac injekcie ranibizumabu 0,3 mg, ranibizumabu 0,5 mg, alebo simulované injekcie.
V klinickom skúšaní FVF2587g (ANCHOR) 423 pacientov s prevažne klasickými CNV léziami bolo randomizovaných v pomere 1:1:1, aby dostávali každý mesiac ranibizumab 0,3 mg, každý mesiac ranibizumab 0,5 mg, alebo PDT verteporfínom (pri zaradení do skúšania a potom každé 3 mesiace, ak fluoresceínová angiografia ukázala pretrvávanie alebo recidívu úniku tekutín z ciev).
Kľúčové merané parametre sú zhrnuté v Tabuľke 1 a na Obrázku 1.
Tabuľka 1 Výsledky po 12 mesiacoch a 24 mesiacoch v klinickom skúšaní FVF2598g(MARINA) a FVF2587g (ANCHOR)
|
| FVF2598g (MARINA)
| FVF2587g (ANCHOR)
|
Meraný parameter
| Mesiac
| Simulované podanie (n=238)
| Ranibizumab
0,5 mg
(n=240)
| PDT verteporfínom (n=143)
| Ranibizuma b 0,5 mg (n=140)
|
Strata < 15 písmen zrakovej ostrosti (%)a (zachovanie vízu, primárny
koncový ukazovateľ)
| 12. mesiac
| 62 %
| 95 %
| 64 %
| 96 %
|
24. mesiac
| 53 %
| 90 %
| 66 %
| 90 %
|
Zisk ≥ 15 písmen zrakovej ostrosti (%)a
| 12. mesiac
| 5 %
| 34 %
| 6 %
| 40 %
|
24. mesiac
| 4 %
| 33 %
| 6 %
| 41 %
|
Priemerná zmena zrakovej ostrosti
| 12. mesiac
| -10,5 (16,6)
| +7,2 (14,4)
| -9,5 (16,4)
| +11,3 (14,6)
|
24. mesiac
| -14,9 (18,7)
| +6,6 (16,5)
| -9,8 (17,6)
| +10,7 (16,5)
|

(písmená) (SD)a a p< 0,01
Obrázok 1 Priemerná zmena zrakovej ostrosti od východiskovej hodnoty do 24. mesiaca v klinickom skúšaní FVF2598g (MARINA) a v klinickom skúšaní FVF2587g (ANCHOR)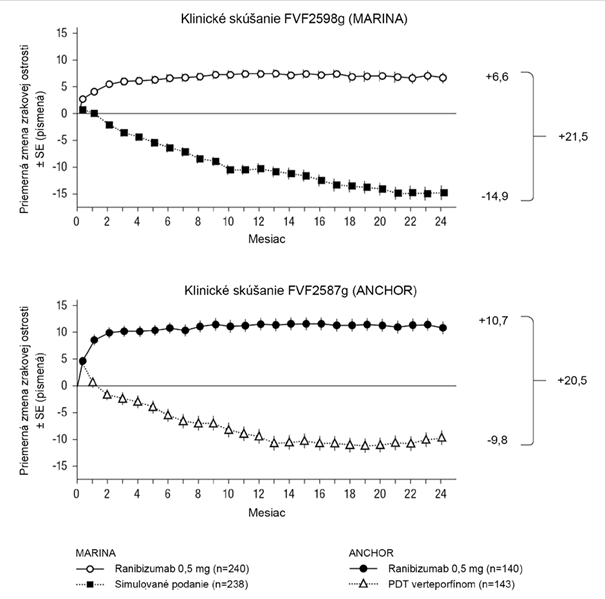
Výsledky oboch klinických skúšaní ukázali, že pokračujúca liečba ranibizumabom môže predstavovať prínos aj pre pacientov, ktorí stratili ≥ 15 písmen najlepšie korigovanej zrakovej ostrosti (BCVA)
v prvom roku liečby.
V skúšaniach MARINA aj ANCHOR sa pozoroval štatisticky významný prínos pre funkcie súvisiace so zrakom udávaný pacientmi pri liečbe ranibizumabom v porovnaní s kontrolnou skupinou, čo sa stanovilo prostredníctvom NEI VFQ-25.
V klinickom skúšaní FVF3192g (PIER) 184 pacientov so všetkými formami neovaskulárnej VPDM bolo randomizovaných v pomere 1:1:1, aby dostávali ranibizumab 0,3 mg, ranibizumab 0,5 mg, alebo simulované injekcie raz za mesiac v 3 po sebe idúcich dávkach, po ktorých nasledovalo podávanie dávky každé 3 mesiace. Od 14. mesiaca klinického skúšania sa u pacientov so simulovaným podaním povolilo podávanie ranibizumabu a od 19. mesiaca boli možné častejšie podania. Pacienti liečení ranibizumabom v PIER dostali priemerne 10 kompletných liečebných cyklov.
Po počiatočnom zvýšení zrakovej ostrosti (po podávaní raz za mesiac) sa zraková ostrosť pacientov v priemere zhoršovala pri podávaní raz za štvrťrok a vrátila sa na východiskovú hodnotu v 12. mesiaci, pričom tento účinok sa zachoval u väčšiny pacientov liečených ranibizumabom (82 %) do 24. mesiaca. Obmedzené údaje u osôb, ktorým sa podávala simulovaná liečba a ktoré neskôr dostávali ranibizumab, naznačili, že včasný začiatok liečby sa môže spájať s lepším zachovaním zrakovej ostrosti.
Údaje z dvoch štúdií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309), ktoré sa vykonali po registrácii lieku, potvrdili účinnosť ranibizumabu, ale nepreukázali prídavný účinok pri kombinovanom podaní verteporfínu (Visudyne PDT) a ranibizumabu oproti monoterapii ranibizumabom.
LiečbapoškodeniazrakuvdôsledkusekundárnejCNVpriPMKlinická bezpečnosť a účinnosť ranibizumabu u pacientov s poškodením zraku v dôsledku CNV pri PM sa stanovili na základe údajov z 12 mesiacov dvojito maskovaného, kontrolovaného pivotného klinického skúšania F2301 (RADIANCE). V tomto skúšaní bolo 277 pacientov randomizovaných v pomere 2:2:1 do nasledujúcich skupín:
• Skupina I (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „stability“, definovanými ako žiadna zmena BCVA v porovnaní s dvomi predchádzajúcimi mesačnými hodnoteniami).
• Skupina II (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „aktivity choroby“, definovanými ako zhoršenie zraku, ktoré možno pripísať intra- alebo subretinálnej tekutine alebo aktívnemu presakovaniu v dôsledku CNV lézie, stanovené prostredníctvom optickej koherentnej tomografie a/alebo fluorescenčnej angiografie).
• Skupina III (vPDT – pacienti mali od 3. mesiaca povolené dostať liečbu ranibizumabom).
V skupine II, ktorá zodpovedá odporúčanému dávkovaniu (pozri časť 4.2), 50,9 % pacientov potrebovalo 1 alebo 2 injekcie, 34,5 % potrebovalo 3 až 5 injekcií a 14,7 % potrebovalo 6 až
12 injekcií počas 12 mesiacov trvania skúšania. V skupine II nepotrebovalo injekcie 62,9 % pacientov v druhom 6-mesačnom období skúšania.
Najdôležitejšie výsledky RADIANCE sú zhrnuté v Tabuľke 2 a na Obrázku 2.
 Tabuľka 2 Výsledky po 3. a 12. mesiaci (RADIANCE)
Tabuľka 2 Výsledky po 3. a 12. mesiaci (RADIANCE)
Skupina I
ranibizumab
0
,5 mg „stabilita zraku“
Skupina II
ranibizumab
0
,5 mg „aktivita choroby“
Skupina III
v
PDT
b
(n=105) (n=116) (n=55)
3
. mesiac
Priemerná zmena BCVA od 1. do
3. mesiaca v porovnaní s východiskovou hodnotoua (písmená)
Podiel pacientov, ktorí získali:
≥ 15 písmen, alebo dosiahli ≥ 84 písmen
BCVA
12. mesiac
Počet injekcií do 12. mesiaca:
+10,5 +10,6 +2,2
38,1 % 43,1 % 14,5 %
Priemer 4,6 3,5 N/A Stred 4,0 2,5 N/A
Priemerná zmena BCVA od 1. do
12. mesiaca v porovnaní s východiskovou hodnotou (písmená)
Podiel pacientov, ktorí získali:
+12,8 +12,5 N/A
≥ 15 písmen, alebo dosiahli ≥ 84 písmen
53,3 % 51,7 % N/A
BCVA
a p< 0,00001 v porovnaní s kontrolou vPDT
b Porovnávacia kontrola do 3. mesiaca. Pacienti randomizovaní do skupiny vPDT mali od 3. mesiaca
povolené dostať liečbu ranibizumabom (v skupine III dostalo ranibizumab od 3. mesiaca 38 pacientov)
Obrázok 2 Priemerná zmena od východiskovej hodnoty BCVA do 12. mesiaca (RADIANCE)
Zlepšenie zraku sprevádzal pokles hrúbky stredu sietnice.
Prínos liečby hlásený pacientmi sa pozoroval vo väčšej miere v skupinách ranibizumabu v porovnaní s
vPDT (hodnota p< 0,05) z hľadiska zlepšenia kombinovaného skóre a niekoľkých podškál NEI VFQ-
25 (celkové videnie, aktivity vyžadujúce videnie do blízka, duševné zdravie a odkázanosť na iných).
LiečbapoškodeniazrakuvdôsledkuCNV(inejakosekundárnejpriPMavlhkejVPDM)Klinická bezpečnosť a účinnosť ranibizumabu u pacientov s poškodením zraku v dôsledku CNV sa stanovili na základe údajov z 12 mesiacov dvojito maskovaného, simulovaným podaním kontrolovaného pivotného klinického skúšania G2301 (MINERVA). V tomto skúšaní bolo
178 dospelých pacientov randomizovaných v pomere 2:1 na podávanie:
• ranibizumab 0,5 mg na začiatku skúšania, po ktorom nasledoval individuálne prispôsobený režim dávkovania určovaný aktivitou choroby, stanovenej prostredníctvom zrakovej ostrosti a/alebo anatomických parametrov (napr. zhoršenie zrakovej ostrosti, intra/subretinálna tekutina, krvácanie alebo presakovanie);
• simulované podanie injekcie na začiatku skúšania, po ktorom nasledoval individuálne prispôsobený režim liečby určovaný aktivitou choroby.
Po 2 mesiacoch všetci pacienti dostávali otvorenú liečbu ranibizumabom podľa potreby.
Najdôležitejšie výsledky MINERVA sú zhrnuté v Tabuľke 3 a na Obrázku 3. Pozorovalo sa zlepšenie
zraku, ktoré sprevádzal pokles hrúbky centrálnej časti sietnice počas obdobia 12 mesiacov.
Priemerný počet injekcií podaných počas 12 mesiacov bol 5,8 v skupine ranibizumabu a 5,4 u tých pacientov v skupine simulovaného podania, ktorí spĺňali podmienky pre podávanie ranibizumabu od
2. mesiaca. V skupine simulovaného podania nedostalo 7 z 59 pacientov v období 12 mesiacov liečbu
ranibizumabom do skúšaného oka.
Tabuľka 3 Výsledky po 2. mesiaci (MINERVA)
| Ranibizumab
0,5 mg (n=119)
| Simulované podanie (n=59)
|
Priemerná zmena BCVA po 2. mesiaci v porovnaní s východiskovou hodnotoua
| 9,5 písmen
| -0,4 písmena
|
Pacienti, ktorí po 2. mesiaci získali ≥ 15 písmen alebo dosiahli 84 písmen v porovnaní s východiskovou hodnotou
| 31,4 %
| 12,3 %
|
Pacienti, ktorí po 2. mesiaci nestratili > 15 písmen v porovnaní s východiskovou hodnotou
| 99,2 %
| 94,7 %
|
Pokles CSFTb po 2. mesiaci v porovnaní s východiskovou hodnotou a
| 77 µm
| -9,8 µm
|
a Jednostranné p < 0,001 pre porovnanie s kontrolným simulovaným podaním
b CSFT – hrúbka centrálnej časti sietnice
Obrázok 3 Priemerná zmena od východiskovej hodnoty BCVA do 12. mesiaca (MINERVA)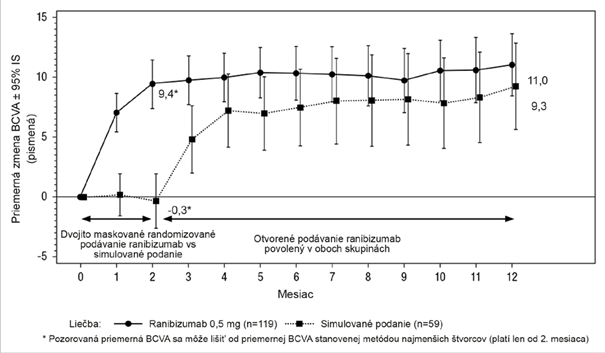
Pri porovnaní ranibizumabu a kontrolného simulovaného podania po 2. mesiaci sa pozoroval zhodný
účinok liečby celkove, ako aj naprieč podskupinami podľa východiskovej etiológie.
Tabuľka 4 Účinok liečby celkovo a naprieč podskupinami podľa východiskovej etiológieCelkovo a podľa východiskovej etiológie
| Účinok liečby oproti
simulovanému podaniu
[písmená]
| Počet pacientov [n] (liečba + simulované podanie)
|
Celkovo
| 9,9
| 178
|
Angioidné pruhy
| 14,6
| 27
|
Pozápalová retinochoroidopatia
| 6,5
| 28
|
Centrálna serózna chorioretinopatia
|
5,0
|
23
|
Idiopatická chorioretinopatia
|
11,4
|
63
|
Rôzne etiológiea
|
10,6
|
37
|
a zahŕňa rôzne etiológie s nízkou frekvenciou výskytu, ktoré nie sú obsiahnuté v iných podskupinách
V pivotnom klinickom skúšaní G2301 (MINERVA) dostalo na začiatku skúšania päť dospievajúcich pacientov vo veku 12 až 17 rokov s poškodením zraku v dôsledku CNV otvorenú liečbu ranibizumabom 0,5 mg, po ktorej nasledoval individuálne prispôsobený režim liečby ako u dospelej populácie. BCVA sa zlepšovala od východiskovej hodnoty do 12. mesiaca u všetkých piatich pacientov a bola v rozmedzí od 5 do 38 písmen (priemer 16,6 písmen). Zlepšenie zraku sprevádzali stabilizácia alebo pokles hrúbky centrálnej časti sietnice počas obdobia 12 mesiacov. Priemerný počet injekcií ranibizumabu podaný počas 12 mesiacov do skúšaného oka bol 3 (rozmedzie od 2 do 5). Celkovo sa liečba ranibizumabom dobre znášala.
LiečbapoškodeniazrakuvdôsledkuDMEBezpečnosť a účinnosť ranibizumabu sa vyhodnotili v troch randomizovaných, kontrolovaných štúdiách trvajúcich najmenej 12 mesiacov. Do týchto štúdií bolo zaradených celkovo 868 pacientov (708 s aktívnou liečbou a 160 ako kontrola).
V štúdii fázy II, D2201 (RESOLVE), dostávalo 151 pacientov ranibizumab (6 mg/ml, n=51,
10 mg/ml, n=51) alebo simulovanú liečbu (n=49) intravitreálnymi injekciami raz za mesiac. Priemerná zmena BCVA od 1. mesiaca do 12. mesiaca v porovnaní s východiskovou hodnotou bola +7,8 (±7,72) písmen u všetkých pacientov liečených ranibizumabom (n=102) v porovnaní s -0,1 (±9,77) písmen
u pacientov pri simulovanej liečbe; priemerná zmena BCVA po 12. mesiaci oproti východiskovej hodnote bola 10,3 (±9,1) písmen v porovnaní s -1,4 (±14,2) písmenami (p< 0,0001 pre rozdiel
v liečbe).
V klinickom skúšaní fázy III, D2301 (RESTORE), bolo randomizovaných 345 pacientov v pomere
1:1:1, aby dostávali 0,5 mg ranibizumabu ako monoterapiu a simulovanú laserovú fotokoaguláciu, kombináciu 0,5 mg ranibizumabu a laserovej fotokoagulácie, alebo simulovanú injekciu a laserovú fotokoaguláciu. 240 pacientov, ktorí predtým ukončili 12-mesačné klinické skúšanie RESTORE, bolo zaradených do otvorenej, multicentrickej extenzie skúšania trvajúcej 24 mesiacov (RESTORE Extension). Pacienti dostávali 0,5 mg ranibizumabu
pro re nata (PRN) do toho istého oka ako
v základnom klinickom skúšaní (D2301, RESTORE).
Kľúčové merané parametre sú zhrnuté v Tabuľke 5 (RESTORE a Extension) a na Obrázku 4 (RESTORE).
Obrázok 4 Priemerná zmena zrakovej ostrosti v čase oproti východiskovej hodnote v štúdii
D2301 (RESTORE)
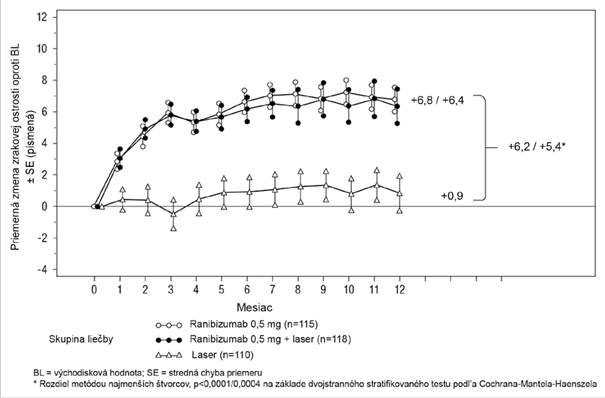
Účinok po 12 mesiacoch sa zhodoval u väčšiny podskupín. Avšak pre osoby s východiskovou hodnotou BCVA > 73 písmen a makulárnym edémom s hrúbkou centrálnej retiny < 300 µm sa liečba ranibizumabom v porovnaní laserovou fotokoaguláciou nezdala prínosom.
Tabuľka 5 Výsledky po 12 mesiacoch v klinickom skúšaní D2301 (RESTORE) a po36 mesiacoch v skúšaní D2301-E1 (RESTORE Extension) Meraný parameter po 12 mesiacoch v porovnaní s východiskovou hodnotou v skúšaní D2301 (RESTORE)
| Ranibizumab
0,5 mg n=115
| Ranibizumab
0,5 mg + laser n=118
| Laser
n=110
|
Priemerná zmena BCVA od 1. do
12. mesiacaa (±SD)
| 6,1 (6,4)a
| 5,9 (7,9)a
| 0,8 (8,6)
|
Priemerná zmena BCVA po
12. mesiacoch (±SD)
| 6,8 (8,3)a
| 6,4 (11,8)a
| 0,9 (11,4)
|
Zisk ≥ 15 písmen alebo BCVA
≥ 84 písmen po 12 mesiacoch (%)
| 22,6
| 22,9
| 8,2
|
Priemerný počet injekcií (0. – 11. mesiac)
| 7,0
| 6,8
| 7,3 (simulované podanie)
|
|
Meraný parameter po 36 mesiacoch v skúšaní D2301-E1 (RESTORE Extension) v porovnaní s východiskovou hodnotou v skúšaní D2301 (RESTORE)
|
Predtým ranibizumab
0,5 mg n=83
|
Predtým ranibizumab
0,5 mg + laser n=83
|
Predtým laser
n=74
|
Priemerná zmena BCVA po 24 mesiacoch
(SD)
|
7,9 (9,0)
|
6,7 (7,9)
|
5,4 (9,0)
|
Priemerná zmena BCVA po 36 mesiacoch
(SD)
|
8,0 (10,1)
|
6,7 (9,6)
|
6,0 (9,4)
|
Zisk ≥ 15 písmen alebo BCVA
≥ 84 písmen po 36 mesiacoch (%)
|
27,7
|
30,1
|
21,6
|
Priemerný počet injekcií (12. –
35. mesiac)*
|
6,8
|
6,0
|
6,5
|
ap< 0,0001 pre porovnania skupín ranibizumabu oproti skupine laseru.
n v D2301-E1 (RESTORE Extension) je počet pacientov s východiskovou hodnotou (0. mesiac)
v D2301 (RESTORE) a tiež s hodnotou z návštevy po 36. mesiaci.
* Podiel pacientov, ktorí nepotrebovali liečbu ranibizumabom počas fázy extenzie, bol 19 % v skupine predchádzajúceho podávania ranibizumabu, 25 % v skupine predchádzajúceho podávania ranibizumabu + liečby laserom a 20 % v skupine predchádzajúcej liečby laserom.
Štatisticky významný pacientmi udávaný prínos vzhľadom na väčšinu funkcií súvisiacich so zrakom sa pozoroval pri liečbe ranibizumabom (s laserom, alebo bez neho) v porovnaní s kontrolnou skupinou, čo sa stanovilo prostredníctvom NEI VFQ-25. Rozdiely medzi druhmi liečby sa nezistili podľa iných podškál tohto dotazníka.
Profil dlhodobej bezpečnosti ranibizumabu, ktorý sa pozoroval v 24-mesačnej extenzii klinického skúšania, sa zhoduje so známym profilom bezpečnosti ranibizumabu.
V klinickom skúšaní fázy IIIb, D2304 (RETAIN), 372 pacientov bolo randomizovaných v pomere
1:1:1, aby dostávali:
• 0,5 mg ranibizumabu súbežne s laserovou fotokoaguláciou v režime podávania a predlžovania intervalov medzi podaniami (treat-and-extend, TE),
• 0,5 mg ranibizumabu v monoterapii v režime TE,
• 0,5 mg ranibizumabu v monoterapii v režime PRN.
Vo všetkých skupinách sa ranibizumab podával raz za mesiac až do dosiahnutia stabilnej BCVA pri
najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Pri TE sa ranibizumab podával
s intervalmi v liečbe 2 – 3 mesiace. Vo všetkých skupinách sa obnovilo podávanie každý mesiac pri
poklese BCVA v dôsledku progresie DME a pokračovalo sa v ňom až do opätovného dosiahnutia
stabilnej BCVA.
Počet plánovaných návštev s podaním liečby po začiatočných 3 injekciách bol 13 pri režime TE a 20 pri režime PRN. Pri oboch režimoch TE si viac ako 70 % pacientov udržalo ich BCVA pri priemernej frekvencii návštev ≥ 2 mesiace.
Kľúčové merané parametre sú zhrnuté v Tabuľke 6.
Tabuľka 6 Výsledky v klinickom skúšaní D2304 (RETAIN)
Meraný parameter v porovnaní s
východiskovou
hodnotou
|
Ranibizumab
0,5 mg + laser pri TE
n=117
|
Samotný ranibizumab
0,5 mg pri TE
n=125
|
Ranibizumab
0,5 mg pri PRN
n=117
|
Priemerná zmena
BCVA od 1. do
12. mesiaca (SD)
|
5,9 (5,5) a
|
6,1 (5,7) a
|
6,2 (6,0)
|
Priemerná zmena
BCVA od 1. do
24. mesiaca (SD)
|
6,8 (6,0)
|
6,6 (7,1)
|
7,0 (6,4)
|
Priemerná zmena
BCVA po
24 mesiacoch (SD)
|
8,3 (8,1)
|
6,5 (10,9)
|
8,1 (8,5)
|
Zisk ≥ 15 písmen alebo
BCVA ≥ 84 písmen po
24 mesiacoch (%)
|
25,6
|
28,0
|
30,8
|
Priemerný počet
injekcií (0.-23. mesiac)
|
12,4
|
12,8
|
10,7
|
ap< 0,0001 pre stanovenie neinferiority oproti PRN
V klinických skúšaniach pri DME sprevádzalo zlepšenie BCVA postupom času zníženie priemernej
CSFT vo všetkých skupinách liečby.
LiečbaPDRKlinická bezpečnosť a účinnosť ranibizumabu u pacientov s PDR bola posudzovaná v Protokole S, ktorý hodnotil liečbu intravitreálnymi injekciami ranibizumabu 0,5 mg v porovnaní s panretinálnou fotokoaguláciou (PRP). Primárny koncový ukazovateľ bola priemerná zmena zrakovej ostrosti v 2. roku. Navyše bola zmena závažnosti diabetickej retinopatie (DR) hodnotená na základe snímky očného pozadia pomocou skóre závažnosti DR (DRSS).
Protokol S bola multicentrická, randomizovaná, aktívne kontrolovaná, paralelne zadaná, neinferioritná štúdia fázy III, do ktorej bolo zaradených 305 pacientov (394 študovaných očí) s PDR, s DME alebo bez DME, na začiatku štúdie. Štúdia porovnávala intravitreálne injekcie ranibizumabu 0,5 mg so štandardnou liečbou PRP. Celkom 191 očí (48,5 %) bolo randomizovaných na ranibizumab 0,5 mg a
203 očí (51,5 %) očí bolo randomizovaných na PRP. Celkovo 88 očí (22,3 %) malo na začiatku liečby
DME: 42 (22,0 %) očí v skupine ranibizumab a 46 (22,7 %) očí v skupine PRP.
V tejto štúdii bola priemerná zmena zrakovej ostrosti v 2. roku +2,7 písmen v skupine ranibizumab, v porovnaní s -0,7 písmen v skupine PRP. Rozdiel metódou najmenších štvorcov bol 3,5 písmen
(95 % IS: [0,2 až 6,7]).
V 1. roku došlo u 41,8 % očí k ≥ 2-stupňovému zlepšeniu DRSS pri liečbe ranibizumabom (n=189),
v porovnaní s 14,6 % očí liečených PRP (n=199). Odhadovaný rozdiel medzi ranibizumabom
a laserom bol 27,4 % (95 % IS: [18,9; 35,9]).
Tabuľka 7 ≥ 2 alebo ≥ 3-stupňové zlepšenie alebo zhoršenie DRSS v 1. roku v Protokole S (LOCF Metóda)
Kategória zmeny od
z
a
čiatku liečby
|
Protokol S
|
Ranibizumab
0
,5 mg
'
(N=189)
|
PRP (N=199)
|
Rozdiel v podiele (%), IS
|
≥ 2-stupňové zlepšenie
|
n (%)
|
79 (41,8 %)
|
29 (14,6 %)
|
27,4 (18,9; 35,9)
|
≥ 3-stupňové zlepšenie
|
n (%)
|
54 (28,6 %)
|
6 (3,0 %)
|
25,7 (18,9; 32,6)
|
≥ 2-stupňové zhoršenie
|
n (%)
|
3 (1,6%)
|
23 (11,6 %)
|
-9,9
(-14,7; -5,2)
|
≥ 3-stupňové zhoršenie
|
n (%)
|
1 (0,5 %)
|
8 (4,0 %)
|
-3,4
(-6,3; -0,5)
|
DRSS = skóre závažnosti diabetickej retinopatie, n = počet pacientov spĺňajúcich podmienku pri kontrole, N = celkový počet očí v štúdii
|
V 1. roku v skupine liečenej ranibizumabom v Protokole S bolo ≥ 2-stupňové zlepšenie DRSS u očí
bez DME (39,9 %) a s DME (48,8 %) na začiatku liečby.
Analýza dvojročných údajov z Protokolu S preukázala, že 42,3 % (n=80) očí v skupine liečenej ranibizumabom malo ≥ 2-stupňové zlepšenie DRSS oproti východiskovým hodnotám v porovnaní s
23,1 % (n=46) očí v skupine PRP. V skupine liečenej ranibizumabom bolo ≥ 2-stupňové zlepšenie DRSS oproti východiskovým hodnotám pozorované u 58,5 % (n=24) očí s DME na začiatku liečby a u 37,8 % (n=56) očí bez DME na začiatku liečby.
Skóre závažnosti diabetickej retinopatie (DRSS) bolo tiež hodnotené v troch samostatných aktívne kontrolovaných DME štúdiách fázy III (ranibizumab 0,5 mg PRN vs laser), ktoré zahŕňali celkovo
875 pacientov, z ktorých približne 75 % bolo ázijského pôvodu. V metaanalýze týchto štúdií malo počas liečby ranibizumabom 48,4 % z 315 pacientov so stúpajúcim DRSS skóre, v podskupine pacientov s mierne závažnou neproliferatívnou DR (NPDR) alebo horšou na začiatku liečby,
≥ 2-stupňové zlepšenie DRSS v 12. mesiaci (n=192) vs 14,6 % pacientov liečených laserom (n=123). Odhadovaný rozdiel medzi liečbou ranibizumabom a laserom bol 29,9 % (95 % IS: [20,0; 39,7]).
U 405 pacientov so stúpajúcim DRSS, s miernou NPDR alebo lepšou, sa pozorovalo ≥ 2-stupňové
zlepšenie DRSS u 1,4 % v skupine ranibizumab a 0,9 % v skupine laser.
LiečbapoškodeniazrakuvdôsledkumakulárnehoedémupoRVOKlinická bezpečnosť a účinnosť ranibizumabu u pacientov s poškodením zraku v dôsledku makulárneho edému po RVO sa vyhodnotili v randomizovaných, dvojito maskovaných, kontrolovaných štúdiách BRAVO a CRUISE, do ktorých boli zaradené osoby s BRVO (n=397) a CRVO (n=392). V oboch štúdiách pacienti dostávali buď 0,3 mg, alebo 0,5 mg ranibizumabu, alebo simulované injekcie. Po 6 mesiacoch pacienti z kontrolných skupín simulovaného podania prešli na
0,5 mg ranibizumabu.
Kľúčové merané parametre z BRAVO a CRUISE sú zhrnuté v Tabuľke 8 a na Obrázkoch 5 a 6.
Tabuľka 8 Výsledky po 6 a 12 mesiacoch (BRAVO a CRUISE)
| BRAVO
| CRUISE
|
| Simulované podanie/ Ranibizumab
0,5 mg
(n=132)
| Ranibizumab
0,5 mg
(n=131)
| Simulované podanie/ Ranibizumab
0,5 mg
(n=130)
| Ranibizumab
0,5 mg
(n=130)
|
Priemerná zmena zrakovej ostrosti po 6 mesiacocha (písmená) (SD) (primárny koncový ukazovateľ)
| 7,3 (13,0)
| 18,3 (13,2)
| 0,8 (16,2)
| 14,9 (13,2)
|
Priemerná zmena BCVA po
12 mesiacoch (písmená) (SD)
| 12,1 (14,4)
| 18,3 (14,6)
| 7,3 (15,9)
| 13,9 (14,2)
|
Zisk ≥ 15 písmen zrakovej ostrosti po 6 mesiacocha (%)
| 28,8
| 61,1
| 16,9
| 47,7
|
Zisk ≥ 15 písmen zrakovej ostrosti po 12 mesiacoch (%)
| 43,9
| 60,3
| 33,1
| 50,8
|
Podiel (%), ktorý dostal záchrannú liečbu laserom počas 12 mesiacov
| 61,4
| 34,4
| NA
| NA
|
ap< 0,0001 pre obidve štúdie
Obrázok 5 Priemerná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca(BRAVO)
Obrázok 6 Priemerná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(CRUISE)
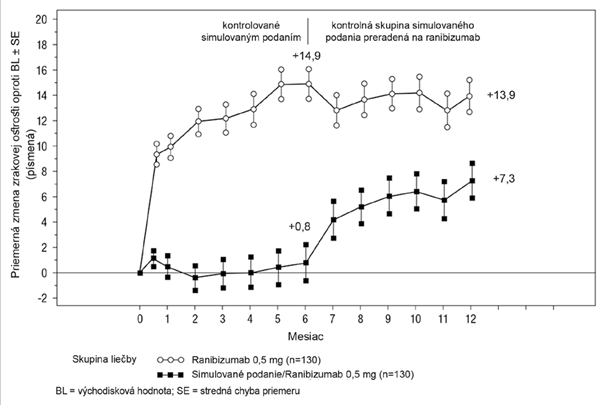
V oboch štúdiách zlepšenie zraku sprevádzalo kontinuálne a významné zmenšovanie makulárneho edému, merané ako hrúbka centrálnej retiny.
Pacienti s CRVO (CRUISE a extenzia štúdie HORIZON): Pacienti, ktorým sa počas prvých
6 mesiacov podávali simulované injekcie a potom dostávali ranibizumab, nedosiahli do 24. mesiaca porovnateľné zlepšenie zrakovej ostrosti (~6 písmen) ako pacienti, ktorí boli liečení ranibizumabom od začiatku štúdie (~12 písmen).
Štatisticky významný pacientmi udávaný prínos vzhľadom na aktivity vyžadujúce videnie do blízka a videnie do diaľky sa pozoroval pri liečbe ranibizumabom v porovnaní s kontrolnou skupinou, čo sa stanovilo prostredníctvom NEI VFQ-25.
Dlhodobá (24 mesiacov) klinická bezpečnosť a účinnosť ranibizumabu u pacientov so zhoršením zraku spôsobeným makulárnym edémom po RVO sa stanovili v štúdiách BRIGHTER (BRVO) a CRYSTAL (CRVO). Účastníci dostávali v oboch štúdiách 0,5 mg ranibizumabu v režime dávok PRN, určovanom individuálne prispôsobenými stabilizačnými kritériami. BRIGHTER bola randomizovaná, účinnou liečbou kontrolovaná štúdia s 3 skupinami, ktorá porovnávala 0,5 mg ranibizumabu podávaného ako monoterapia alebo v kombinácii s adjuvantnou laserovou fotokoaguláciou so samotnou laserovou fotokoaguláciou. Účastníci v skupine lasera mohli po 6 mesiacoch dostať 0,5 mg ranibizumabu. CRYSTAL bola štúdia s jednou skupinou monoterapie 0,5 mg ranibizumabu.
Kľúčové výsledné hodnoty z BRIGHTER a CRYSTAL sú uvedené v Tabuľke 9.
Tabuľka 9 Výsledky po 6 a 24 mesiacoch (BRIGHTER a CRYSTAL)
| BRIGHTER
| CRYSTAL
|
| Ranibizumab
0,5 mg
N=180
| Ranibizumab
0,5 mg + laser
N=178
| Laser* N=90
| Ranibizumab
0,5 mg
N=356
|
Priemerná zmena
BCVA po
6 mesiacocha
(písmená) (SD)
|
+14,8 (10,7)
|
+14,8 (11,13)
|
+6,0 (14,27)
|
+12,0 (13,95)
|
Priemerná zmena
BCVA po
24 mesiacochb
(písmená) (SD)
|
+15,5 (13,91)
|
+17,3 (12,61)
|
+11,6 (16,09)
|
+12,1 (18,60)
|
Zisk ≥ 15 písmen
BCVA po
24 mesiacoch (%)
|
52,8
|
59,6
|
43,3
|
49,2
|
Priemerný počet
injekcií (SD) (0. –
23. mesiac)
|
11,4 (5,81)
|
11,3 (6,02)
|
NA
|
13,1 (6,39)
|
a p< 0,0001 pre obe porovnania v BRIGHTER po 6 mesiacoch: Ranibizumab 0,5 mg oproti laseru a Ranibizumab 0,5 mg + laser oproti laseru.
b p< 0,0001 pre nulovú hypotézu v CRYSTAL, podľa ktorej priemerná zmena oproti
východiskovej hodnote po 24 mesiacoch je nula.
* Počínajúc 6. mesiacom bola povolená liečba 0,5 mg ranibizumabu (24 pacientov dostalo len
liečbu laserom).
|
V BRIGHTER preukázalo 0,5 mg ranibizumabu s adjuvantnou laserovou liečbou neinferioritu oproti
monoterapii ranibizumabom od východiskovej hodnoty do 24. mesiaca (95% IS -2,8; 1,4).
V oboch štúdiách sa po 1. mesiaci pozoroval rýchly a štatisticky významný pokles hrúbky centrálnej
časti sietnice. Tento účinok pretrval až do 24. mesiaca.
Účinok liečby ranibizumabom bol podobný bez ohľadu na prítomnosť ischémie sietnice. U pacientov s prítomnou ischémiou (N=46) alebo bez ischémie (N=133), ktorí v BRIGHTER dostali monoterapiu ranibizumabom, bola po 24. mesiacoch priemerná zmena oproti východiskovej hodnote +15,3 a +15,6 písmen, v uvedenom poradí. V CRYSTAL u pacientov s prítomnou ischémiou (N=53) alebo bez ischémie (N=300), ktorí dostali monoterapiu ranibizumabom, bola priemerná zmena oproti východiskovej hodnote +15,0 a +11,5 písmen, v uvedenom poradí.
Účinok vyjadrený ako zlepšenie zraku sa pozoroval bez ohľadu na trvanie choroby v BRIGHTER aj
CRYSTAL u všetkých pacientov, ktorí dostali monoterapiu 0,5 mg ranibizumabu. U pacientov
s trvaním choroby < 3 mesiace sa pozorovalo zvýšenie zrakovej ostrosti o 13,3 a 10,0 písmen po
1. mesiaci a o 17,7 a 13,2 písmen po 24. mesiaci v BRIGHTER a CRYSTAL, v uvedenom poradí. Zodpovedajúci zisk zrakovej ostrosti u pacientov s trvaním choroby ≥ 12 mesiacov v uvedených štúdiách bol 8,6 a 8,4 písmen. Má sa zvážiť začatie liečby v čase stanovenia diagnózy.
Profil dlhodobej bezpečnosti ranibizumabu pozorovaný v štúdiách trvajúcich 24 mesiacov sa zhoduje
so známym profilom bezpečnosti ranibizumabu.
Pediatrická populáciaBezpečnosť a účinnosť ranibizumabu sa zatiaľ nestanovili u detí a dospievajúcich.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s referenčným
liekom obsahujúcim ranibizumab vo všetkých podskupinách pediatrickej populácie pre neovaskulárnu
VPDM, poškodenie zraku v dôsledku DME, poškodenie zraku v dôsledku makulárneho edému po RVO, poškodenie zraku v dôsledku CNV a diabetickú retinopatiu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po intravitreálnom podaní ranibizumabu raz mesačne pacientom s neovaskulárnou VPDM boli sérové koncentrácie ranibizumabu vo všeobecnosti nízke, s maximálnymi hladinami (Cmax) spravidla pod koncentráciou ranibizumabu potrebnou na inhibovanie biologickej aktivity VEGF o 50 % (11 –
27 ng/ml, ako sa stanovilo in vitro v teste bunkovej proliferácie). Cmax bola úmerná dávke v rozmedzí dávok 0,05 až 1,0 mg/oko. Koncentrácie v sére obmedzeného počtu pacientov s DME naznačujú, že nemožno vylúčiť o niečo vyššiu systémovú expozíciu v porovnaní s expozíciou, aká sa pozorovala u pacientov s neovaskulárnou VPDM. Koncentrácie ranibizumabu v sére pacientov s RVO boli podobné alebo mierne vyššie v porovnaní s koncentráciami, ktoré sa pozorovali u pacientov s neovaskulárnou VPDM.
Na základe analýzy populačnej farmakokinetiky a vymiznutia ranibizumabu zo séra pacientov s neovaskulárnou VPDM liečených dávkou 0,5 mg, priemerný vitreálny eliminačný polčas ranibizumabu je približne 9 dní. Pri intravitreálnom podávaní ranibizumabu 0,5 mg/oko raz mesačne sa predpokladá, že Cmax ranibizumabu v sére, ktorá sa dosiahne približne 1 deň po podaní, bude vo všeobecnosti v rozmedzí medzi 0,79 a 2,90 ng/ml, a Cmin sa vo všeobecnosti predpokladá v rozmedzí medzi 0,07 a 0,49 ng/ml. Predpokladaná sérová koncentrácia ranibizumabu je približne 90 000- násobne nižšia ako vitreálna koncentrácia ranibizumabu.
Pacienti s poruchou funkcie obličiek: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky ranibizumabu u pacientov s poruchou funkcie obličiek. V populačnej farmakokinetickej analýze pacientov s neovaskulárnou VPDM malo 68 % (136 z 200) pacientov poruchu funkcie obličiek
(46,5 % ľahká [50 – 80 ml/min], 20 % stredne ťažká [30 – 50 ml/min] a 1,5 % ťažká [< 30 ml/min]). U
pacientov s RVO malo 48,2 % (253 z 525) poruchu funkcie obličiek (36,4 % ľahká, 9,5 % stredne
ťažká a 2,3 % ťažká). Systémový klírens bol trochu nižší, čo však nebolo klinicky významné.
Pacienti s poruchou funkcie pečene: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky
ranibizumabu u pacientov s poruchou funkcie pečene.
5.3 Predklinické údaje o bezpečnosti
Bilaterálne intravitreálne podávanie ranibizumabu opiciam rodu Cynomolgus v dávkach medzi
0,25 mg/oko a 2,0 mg/oko raz za 2 týždne až do 26 týždňov malo za následok účinky na oči závislé od
dávky.
Intraokulárne sa zaznamenalo od dávky závislé zosilnenie zápalu a zvýšenie počtu buniek v prednej očnej komore s maximom 2 dni po podaní injekcie. Závažnosť zápalovej odpovede sa spravidla znížila pri podaní ďalších injekcií alebo počas zotavenia. V zadnom segmente sa pozorovala vitreálna infiltrácia buniek a zákaly sklovca, ktoré tiež mali tendenciu závisieť od dávky a spravidla pretrvávali do konca liečebného obdobia. V štúdii trvajúcej 26 týždňov sa intenzita zápalu sklovca zvyšovala
s počtom injekcií. Po zotavení sa však pozorovali dôkazy reverzibility. Povaha a načasovanie zápalu zadného segmentu poukazuje na imunitne sprostredkovanú odpoveď protilátok, čo môže byť klinicky nevýznamné. Pri niektorých zvieratách sa pozoroval vznik katarakty po relatívne dlhom období intenzívneho zápalu, čo naznačuje, že zmeny na šošovke sú sekundárne po ťažkom zápale. Prechodné zvýšenie vnútroočného tlaku po podaní sa pozorovalo po intravitreálnych injekciách bez ohľadu na dávku.
Mikroskopické očné zmeny súviseli so zápalom a nepoukazovali na degeneratívne procesy.
V niektorých očiach sa zaznamenali granulomatózne zápalové zmeny na papile. Tieto zmeny
v zadnom segmente ustupovali a v niektorých prípadoch vymizli počas zotavovania.
Po intravitreálnom podaní sa nezistili žiadne známky systémovej toxicity. V podsúbore liečených
zvierat sa našli sérové a sklovcové protilátky voči ranibizumabu.
Nie sú dostupné údaje o karcinogenite alebo mutagenite.
U gravidných opíc nespôsobilo intravitreálne podávanie ranibizumabu, ktoré malo za následok maximálne systémové expozície 0,9- až 7-násobne vyššie ako najhorší prípad klinickej expozície, vývojovú toxicitu alebo teratogenitu a nemalo žiadny vplyv na hmotnosť alebo štruktúru placenty,
hoci ranibizumab sa vzhľadom na jeho mechanizmus účinku má považovať za potenciálne teratogénny
a embryo- a fetotoxický.
Neprítomnosť účinkov na vývoj embrya a plodu sprostredkovaných ranibizumabom pravdepodobne súvisí hlavne s neschopnosťou fragmentu Fab prestupovať cez placentu. Napriek tomu bol popísaný prípad vysokých hladín ranibizumabu v sére matky a prítomnosti ranibizumabu v sére plodu, čo naznačuje, že protilátka proti ranibizumabu fungovala ako transportná bielkovina (obsahujúca segment Fc) pre ranibizumab, čím sa znižoval klírens zo séra matky a umožňoval sa prestup cez placentu. Keďže sledovania vývoja embryí a plodov sa robili u zdravých gravidných zvierat a ochorenie
(napr. diabetes) môže meniť priepustnosť placenty pre fragment Fab, štúdia sa má interpretovať
s opatrnosťou.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Dihydrát α,α-trehalózy Monohydrát histidíniumchloridu Histidín
Polysorbát 20
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C).
Neuchovávajte v mrazničke.
Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Pred použitím sa môže neotvorená injekčná liekovka uchovávať pri izbovej teplote (25 °C) najviac
24 hodín.
6.5 Druh obalu a obsah balenia
Jedna injekčná liekovka (sklo typu I) so zátkou (chlórobutylová guma) obsahujúca 0,23 ml sterilného roztoku.
Veľkosť balenia jedna injekčná liekovka.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčná liekovka je určená len na jednorazové použitie. Po podaní injekcie sa všetok nespotrebovaný liek musí zlikvidovať. Akákoľvek injekčná liekovka, ktorá vykazuje známky poškodenia alebo
nesprávnej manipulácie, sa nesmie použiť. Sterilitu nemožno zaručiť, ak uzáver obalu nie je
neporušený.
Na prípravu a podanie intravitreálnej injekcie sú potrebné nasledujúce pomôcky na jednorazové použitie:
- ihla s 5 µm filtrom (18G)
- sterilná injekčná striekačka 1 ml (s ryskou pre objem 0,05 ml) a injekčná ihla (30G x ½″) pre
dospelých pacientov
Tieto pomôcky nie sú súčasťou tohto balenia.
Pri príprave Ranivisia na intravitreálne podanie dospelým dodržiavajte, prosím, nasledujúce pokyny:
1. Pred odobratím obsahu odstráňte viečko z injekčnej liekovky a očistite vonkajšiu časť zátky injekčnej liekovky (napr. tampónom napusteným 70 % alkoholom).
2. Asepticky nasaďte ihlu s 5 µm filtrom (18G x 1½″, 1,2 mm x 40 mm) na 1 ml injekčnú striekačku. Vtlačte hrubú ihlu s filtrom do stredu zátky liekovky, až sa ihla dotkne dna injekčnej liekovky.
3. Odoberte všetku tekutinu z injekčnej liekovky, pričom liekovka má byť vo zvislej polohe, mierne naklonená, aby sa uľahčilo úplné odobratie obsahu.
4. Dbajte na to, aby ste pri vyprázdnení injekčnej liekovky dostatočne potiahli piest, aby sa ihla s
filtrom úplne vyprázdnila.
5. Hrubú ihlu s filtrom nechajte v injekčnej liekovke a odpojte injekčnú striekačku od hrubej ihly s filtrom. Ihla s filtrom sa má po odobratí obsahu injekčnej liekovky zahodiť a nemá sa použiť na intravitreálnu injekciu.
6. Asepticky pevne nasaďte injekčnú ihlu (30G x ½″, 0,3 mm x 13 mm) na injekčnú striekačku.
7. Opatrne odstráňte kryt z injekčnej ihly bez toho, aby ste odpojili injekčnú ihlu od injekčnej striekačky.
Poznámka: Pri odstránení krytu pridržte násadec injekčnej ihly.
8. Opatrne vytlačte vzduch spolu s nadbytočným roztokom a upravte dávku na rysku označujúcu
0,05 ml na striekačke. Injekčná striekačka je pripravená na podanie injekcie.
Poznámka: Injekčnú ihlu neutierajte. Nepotiahnite piest injekčnej striekačky.
Po podaní injekcie nenasaďte kryt späť na injekčnú ihlu, ani ihlu neodpojte od injekčnej striekačky. Zahoďte použitú injekčnú striekačku spolu s ihlou do odpadovej nádoby na ostré predmety, alebo zlikvidujte v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Midas Pharma GmbH
Rheinstraße 49
D-55218 Ingelheim
Nemecko
8. REGISTRAČNÉ ČÍSLA
EU/1/22/1673/001
9
. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 25. augusta 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.