
r môže ovplyvniť expozíciu piperacilínu/tazobaktámu, amfotericínu B a mikafungínu. Možná interakcia medzi letermovirom a týmito liekmi sa neskúmala. Existuje teoretické riziko zníženej expozície kvôli indukcii, ale veľkosť účinku a tým ani klinický význam nie sú v súčasnosti známe.
Lieky metabolizované CYP3A
Letermovir je stredne silný inhibítor CYP3A in vivo. Súbežné podávanie PREVYMISU s perorálnym midazolamom (substrát CYP3A) vedie k 2- až 3-násobne zvýšeným plazmatickým koncentráciám
midazolamu. Súbežné podávanie PREVYMISU môže viesť ku klinicky významným zvýšeniam plazmatických koncentrácií súbežne podávaných substrátov CYP3A (pozri časti 4.3, 4.4 a 5.2).
- Príkladmi takýchto liekov sú niektoré imunosupresíva (napr. cyklosporín, takrolimus, sirolimus), inhibítory HMG-CoA reduktázy a amiodarón (pozri tabuľku 1). Pimozid a námeľové alkaloidy sú kontraindikované (pozri časť 4.3).
Veľkosť inhibičného účinku na CYP3A je závislá od cesty podávania letermoviru a od toho, či sa súbežne používa aj cyklosporín.
V dôsledku časovo závislej inhibície a simultánnej indukcie nie je možné dosiahnuť čistý inhibičný účinok na enzýmy skôr, ako po 10-14 dňoch. Čas potrebný na dosiahnutie rovnovážneho stavu určitého ovplyvneného lieku bude taktiež ovplyvňovať čas potrebný na dosiahnutie plného účinku na
plazmatické koncentrácie. Po ukončení liečby trvá 10-14 dní kým inhibičný účinok vymizne. Ak sa použije sledovanie hladín, tak sa to odporúča počas prvých 2 týždňov po začatí a ukončení liečby letermovirom (pozri časť 4.4) a takisto aj po zmene cesty podávania letermoviru.
Lieky transportované OATP1B1/3
Letermovir je inhibítor transportérov OATP1B1/3. Podávanie PREVYMISU môže viesť ku klinicky významnému zvýšeniu plazmatických koncentrácií súbežne podávaných liekov, ktoré sú substrátmi OATP1B1/3.
- Príkladmi takýchto liekov sú inhibítory HMG-CoA reduktázy, fexofenadín, repaglinid a glyburid
(pozri tabuľku 1). Pri porovnávaní režimu letermoviru podávaného bez cyklosporínu je účinok výraznejší po i.v. ako po perorálnom letermovire.
Rozsah inhibície OATP1B1/3 na súbežne podávané lieky je pravdepodobne väčší, ak sa PREVYMIS
podáva súbežne s cyklosporínom (silný inhibítor OATP1B1/3). To sa má vziať do úvahy, ak sa režim letermoviru zmení počas liečby substrátom OATP1B1/3.
Lieky metabolizované CYP2C9 a/alebo CYP2C19
Súbežné podávanie PREVYMISU s vorikonazolom (substrát CYP2C19) vedie k významne zníženým plazmatickým koncentráciám vorikonazolu, čo naznačuje, že letermovir je induktor CYP2C19.
CYP2C9 je pravdepodobne tiež indukovaný. Letermovir potenciálne znižuje expozíciu substrátom
CYP2C9 a/alebo CYP2C19, čo môže viesť k subterapeutickým hladinám.
- Príkladmi takýchto liekov sú warfarín, fenytoín, vorikonazol, diazepam, lanzoprazol, omeprazol, esomeprazol, pantoprazol, tilidín, tolbutamid (pozri tabuľku 1).
Očakáva sa, že účinok bude menej výrazný pri perorálnom letermovire bez cyklosporínu ako pri i.v.
letermovire s cyklosporínom alebo bez neho alebo pri perorálnom letermovire s cyklosporínom. To sa má vziať do úvahy, ak sa režim letermoviru mení počas liečby substrátom CYP2C9 alebo CYP2C19. Ohľadom časového priebehu interakcie pozri tiež všeobecné informácie o indukcii vyššie.
Lieky metabolizované CYP2C8
Letermovir inhibuje CYP2C8 in vitro, ale na základe svojho indukčného potenciálu môže CYP2C8 aj indukovať. Čistý účinok in vivo nie je známy.
-Príkladom lieku, ktorý je eliminovaný hlavne prostredníctvom CYP2C8, je repaglinid (pozri tabuľku
1). Súbežné používanie repaglinidu a letermoviru s cyklosporínom alebo bez neho sa neodporúča.
Lieky transportované P-gp v črevách
Letermovir je induktor intestinálneho P-gp. Podávanie PREVYMISU môže viesť ku klinicky významnému zníženiu plazmatických koncentrácií súbežne podávaných liekov, ktoré sú vo významnej
miere transportované P-gp v črevách, ako sú dabigatran a sofosbuvir.
Lieky metabolizované CYP2B6, UGT1A1 alebo transportované BCRP alebo OATP2B1
Letermovir je všeobecný induktor in vivo, ale taktiež sa pozorovalo, že inhibuje CYP2B6, UGT1A1, BCRP a OATP2B1 in vitro. Čistý účinok in vivo nie je známy. Plazmatické koncentrácie liekov, ktoré
sú substrátmi týchto enzýmov alebo transportérov, sa preto môžu zvýšiť alebo znížiť, ak sa podávajú
súbežne s letermovirom. Odporúča sa ďalšie sledovanie, pozri súhrn charakteristických vlastností takýchto liekov.
- Príkladmi liekov metabolizovaných CYP2B6 sú bupropión a efavirenz.
- Príkladmi liekov metabolizovaných UGT1A1 sú raltegravir a dolutegravir.
- Príkladmi liekov transportovaných BCRP sú rosuvastatín a sulfasalazín.
- Príkladom lieku transportovaného OATP2B1 je celiprolol.
Lieky transportované renálnym transportérom OAT3
Údaje in vitro naznačujú, že letermovir je inhibítor OAT3; letermovir môže preto inhibovať OAT3 in vivo. Plazmatické koncentrácie liekov transportovaných OAT3 sa môžu zvýšiť.
- Príkladmi liekov transportovaných OAT3 sú ciprofloxacín, tenofovir, imipeném a cilastatín.
Všeobecné
informácie
Ak sa úpravy dávky súbežne podávaných liekov vykonajú z dôvodu liečby PREVYMISOM, dávky sa majú opätovne upraviť po dokončení liečby PREVYMISOM. Úprava dávky môže byť potrebná aj pri zmene cesty podávania alebo pri zmene imunosupresíva.
V tabuľke 1 je uvedený zoznam potvrdených alebo potenciálne klinicky významných liekových interakcií. Uvedené liekové interakcie sú založené na štúdiách vykonaných s PREVYMISOM alebo sú predpokladanými liekovými interakciami, ktoré sa môžu objaviť pri PREVYMISE (pozri časti 4.3,
4.4, 5.1 a 5.2).
Tabuľka 1: Interakcie a odporúčania na dávky pri podávaní s inými liekmi. Všimnite si, že tabuľka nie je rozsiahla, ale uvádza príklady klinicky významných interakcií. Taktiež pozrite všeobecný text vyššie ohľadom liekových interakcií.
Pokiaľ to nie je inak uvedené, interakčné štúdie boli uskutočnené s perorálnym letermovirom bez cyklosporínu. Prosím, všimnite si, že interakčný potenciál a klinické následky sa môžu líšiť v závislosti od toho, či sa letermovir podáva perorálne alebo i.v. a či sa súbežne používa cyklosporín. Pri zmene cesty podávania alebo ak sa mení imunosupresívum, sa má opakovane prezrieť odporúčanie ohľadom súbežného podávania.
Súbežne podávaný liek
Antimykotiká
Vplyv na koncentráciu†
Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
flukonazol Interakcia sa neskúmala.
Očakáva sa:
↔ flukonazolu
↔ letermoviru
Nevyžaduje sa žiadna úprava dávky.
posakonazol‡
(300 mg jednorazová dávka)/ letermovir (480 mg denne) vorikonazol‡
(200 mg dvakrát denne)/ letermovir (480 mg denne)
Antivirotiká
aciklovir‡
(400 mg jednorazová dávka)/ letermovir
(480 mg denne)
↔ posakonazolu
AUC 0,98 (0,82; 1,17) Cmax 1,11 (0,95; 1,29)
↓ vorikonazolu
AUC 0,56 (0,51; 0,62) Cmax 0,61 (0,53; 0,71)
(indukcia CYP2C9/19)
↔ acikloviru
AUC 1,02 (0,87; 1,2) Cmax 0,82 (0,71; 0,93)
Nevyžaduje sa žiadna úprava dávky.
Ak je súbežné podávanie nevyhnutné, počas prvých 2 týždňov po začatí alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru alebo imunosupresíva sa odporúča TDM pre vorikonazol.
Nevyžaduje sa žiadna úprava dávky.

valaciklovir Interakcia sa neskúmala.
Očakáva sa:
↔ valacikloviru
Nevyžaduje sa žiadna úprava dávky.
Súbežne podávaný liek
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
Inhibítory HMG-CoA reduktázy
atorvastatín‡
(20 mg jednorazová dávka)/ letermovir
(480 mg denne)
simvastatín, pitavastatín, rosuvastatín
fluvastatín, pravastatín
Imunosupresívacyklosporín
(50 mg jednorazová dávka)/ letermovir
(240 mg denne)
cyklosporín
(200 mg jednorazová dávka)/ letermovir (240 mg denne)
↑ atorvastatínu
AUC 3,29 (2,84; 3,82) Cmax 2,17 (1,76; 2,67)
(inhibícia CYP3A, OATP1B1/3)
Interakcia sa neskúmala. Očakáva sa:
↑ koncentrácií inhibítorov
HMG-CoA reduktázy
(inhibícia CYP3A, OATP1B1/3)
Interakcia sa neskúmala. Očakáva sa:
↑ koncentrácií inhibítorov
HMG-CoA reduktázy
(inhibícia OATP1B1/3
a/alebo BCRP)
↑ cyklosporínu
AUC 1,66 (1,51; 1,82) Cmax 1,08 (0,97; 1,19) (inhibícia CYP3A)
↑ letermoviru
AUC 2,11 (1,97; 2,26) Cmax 1,48 (1,33; 1,65) (inhibícia OATP1B1/3)
Nežiaduce udalosti súvisiace so statínmi ako napr. myopatia sa majú pozorne sledovať. Dávka atorvastatínu nemá presiahnuť 20 mg denne, ak sa podáva súbežne
s PREVYMISOM#.
Hoci sa to neskúmalo, očakáva sa, že ak sa
PREVYMIS podáva súbežne
s
cyklosporínom, rozsah zvýšenia plazmatických koncentrácií atorvastatínu bude
väčší ako so samotným PREVYMISOM. Ak
sa PREVYMIS podáva súbežne s cyklosporínom, atorvastatín je
kontraindikovaný.
Letermovir môže podstatne zvýšiť plazmatické koncentrácie týchto statínov. Súbežné používanie so samotným PREVYMISOM sa neodporúča.
Ak sa PREVYMIS podáva súbežne
s cyklosporínom, používanie týchto statínov je kontraindikované.
Letermovir môže zvýšiť plazmatické koncentrácie statínov.
Ak sa PREVYMIS podáva súbežne s týmito statínmi, môže byť potrebné zníženie dávky statínu#. Nežiaduce udalosti súvisiace so statínmi ako napr. myopatia sa majú pozorne sledovať.
Ak sa PREVYMIS podáva súbežne
s cyklosporínom, pravastatín sa neodporúča, kým u fluvastatínu môže byť potrebné zníženie dávky#. Nežiaduce udalosti súvisiace so statínmi ako napr. myopatia sa majú pozorne sledovať.
Ak sa PREVYMIS podáva súbežne
s cyklosporínom, dávkovanie PREVYMISU
sa má znížiť na 240 mg jedenkrát denne (pozri časti 4.2 a 5.1).
Počas liečby PREVYMISOM, pri zmene cesty podávania PREVYMISU a pri jeho vysadení je potrebné vykonávať časté sledovanie koncentrácií cyklosporínu v celej krvi a dávka cyklosporínu sa má náležite upraviť#.
Súbežne podávaný liek
mofetilmykofenolát (1 g jednorazová dávka)/ letermovir (480 mg denne)
sirolimus‡
(2 mg jednorazová dávka)/ letermovir (480 mg denne)
takrolimus
(5 mg jednorazová dávka)/ letermovir (480 mg denne) takrolimus
(5 mg jednorazová
dávka)/ letermovir (80 mg dvakrát denne)
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
↔ kyseliny mykofenolovej
AUC 1,08 (0,97; 1,20) Cmax 0,96 (0,82; 1,12)
↔ letermoviru
AUC 1,18 (1,04; 1,32) Cmax 1,11 (0,93; 1,34)
↑ sirolimu
AUC 3,40 (3,01; 3,85) Cmax 2,76 (2,48; 3,06)
(inhibícia CYP3A) Interakcia sa neskúmala.
Očakáva sa:
↔ letermoviru
↑ takrolimu
AUC 2,42 (2,04; 2,88) Cmax 1,57 (1,32; 1,86) (inhibícia CYP3A)
↔ letermoviru
AUC 1,02 (0,97; 1,07) Cmax 0,92 (0,84; 1,00)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
Nevyžaduje sa žiadna úprava dávky.
Počas liečby PREVYMISOM, pri zmene cesty podávania PREVYMISU a pri jeho vysadení je potrebné vykonávať časté sledovanie koncentrácií sirolimu v celej krvi a dávka sirolimu sa má náležite upraviť#.
Pri začatí a vysadení súbežného podávania cyklosporínu s PREVYMISOM sa odporúča časté sledovanie koncentrácií sirolimu.
Ak sa PREVYMIS podáva súbežne
s cyklosporínom, pre špecifické odporúčania na dávkovanie pri používaní sirolimu
s cyklosporínom pozri tiež súhrn
charakteristických vlastností sirolimu.
Ak sa PREVYMIS podáva súbežne s cyklosporínom, rozsah zvýšenia koncentrácií sirolimu môže byť väčší ako so samotným PREVYMISOM.
Počas liečby PREVYMISOM, pri zmene cesty podávania PREVYMISU a pri jeho vysadení je potrebné vykonávať časté sledovanie koncentrácií takrolimu v celej krvi a dávka takrolimu sa má náležite upraviť#.
Perorálne kontraceptíva

etinylestradiol (EE) (0,03 mg)/ levonorgestrel (LNG)‡
(0,15 mg) jednorazová dávka/ letermovir (480 mg denne)
↔ EE
AUC 1,42 (1,32; 1,52) Cmax 0,89 (0,83; 0,96)
↔ LNG
AUC 1,36 (1,30; 1,43) Cmax 0,95 (0,86; 1,04)
Nevyžaduje sa žiadna úprava dávky.
Súbežne podávaný liek
Iné perorálne kontraceptívne steroidy so systémovým účinkom
Antidiabetiká
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku) Riziko ↓ koncentrácií kontraceptívnych steroidov
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
Letermovir môže znížiť plazmatické koncentrácie iných perorálnych kontraceptívnych steroidov a ovplyvniť tým ich účinnosť.
Na zabezpečenie adekvátneho kontraceptívneho účinku perorálnym
kontraceptívom sa majú zvoliť lieky s obsahom EE a LNG.
repaglinid Interakcia sa neskúmala.
Očakáva sa:
↑ alebo ↓ koncentrácií repaglinidu
(indukcia CYP2C8, inhibícia CYP2C8
a OATP1B)
glyburid Interakcia sa neskúmala.
Očakáva sa:
↑ koncentrácií glyburidu
(inhibícia OATP1B1/3, inhibícia CYP3A, indukcia CYP2C9)
Antiepileptiká (pozri tiež všeobecný text)fenytoín Interakcia sa neskúmala.
Očakáva sa:
↓ koncentrácií fenytoínu
(indukcia CYP2C9/19)
↓ koncentrácií letermoviru
Letermovir môže zvýšiť alebo znížiť plazmatické koncentrácie repaglinidu. (Čistý účinok nie je známy).
Súbežné používanie sa neodporúča. Očakáva sa, že ak sa PREVYMIS podáva
súbežne s
cyklosporínom, plazmatické
koncentrácie repaglinidu sa zvýšia v dôsledku ďalšej inhibície OATP1B cyklosporínom. Súbežné používanie sa neodporúča#. Letermovir môže zvýšiť plazmatické koncentrácie glyburidu.
Počas prvých 2 týždňov po začatí alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru sa odporúča časté sledovanie koncentrácií glukózy.
Ak sa PREVYMIS podáva súbežne
s
cyklosporínom, pre špecifické odporúčania na dávkovanie pozri tiež súhrn
charakteristických vlastností glyburidu.
Letermovir môže znížiť plazmatické koncentrácie fenytoínu.
Ak sa fenytoín podáva súbežne s liečbou PREVYMISOM, je potrebné vykonávať časté sledovanie koncentrácií fenytoínu. Počas prvých 2 týždňov po začatí alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru alebo imunosupresíva sa odporúča TDM.
Zníženie koncentrácií letermoviru môže viesť k strate účinnosti.
Súbežne podávaný liek
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
Perorálne antikoagulanciá
warfarín Interakcia sa neskúmala.
Očakáva sa:
↓ koncentrácií warfarínu
(indukcia CYP2C9)
dabigatran Interakcia sa neskúmala.
Očakáva sa:
↓ koncentrácií dabigatranu
(indukcia intestinálneho P- gp)
Sedatíva
Letermovir môže znížiť plazmatické koncentrácie warfarínu.
Ak sa warfarín podáva súbežne s liečbou PREVYMISOM, je potrebné vykonávať časté sledovanie medzinárodného normalizovaného pomeru (International Normalised Ratio, INR)#. Počas prvých 2 týždňov po začatí
alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru alebo imunosupresíva sa odporúča sledovanie. Letermovir môže znížiť plazmatické koncentrácie dabigatranu a môže znížiť účinnosť dabigatranu. Súbežnému používaniu dabigatranu je potrebné sa vyhnúť kvôli riziku zníženej účinnosti dabigatranu.
Ak sa PREVYMIS podáva súbežne s cyklosporínom, dabigatran je kontraindikovaný.
midazolam
(1 mg jednorazová dávka i.v.)/
letermovir (240 mg
jedenkrát denne p.o.)
midazolam
(2 mg jednorazová dávka p.o.)/ letermovir (240 mg jedenkrát denne p.o.)
↑ midazolamu i.v.:
AUC 1,47 (1,37; 1,58) Cmax 1,05 (0,94; 1,17)
p.o.:
AUC 2,25 (2,04; 2,49) Cmax 1,72 (1,54; 1,92)
(inhibícia CYP3A)
Počas súbežného podávania PREVYMISU s midazolamom je potrebné vykonávať pozorné klinické sledovanie pre útlm dýchania a/alebo predĺženú sedáciu. Má sa zvážiť úprava dávky midazolamu#. Zvýšenie plazmatickej koncentrácie midazolamu môže
byť väčšie, ak sa perorálny midazolam podáva s letermovirom v klinickej dávke oproti skúmanej dávke.
Agonisty opioidných receptorov
Príklady: alfentanil, fentanyl
Interakcia sa neskúmala. Očakáva sa:
↑ koncentrácií opiátov metabolizovaných CYP3A
(inhibícia CYP3A)
Počas súbežného podávania sa odporúča časté sledovanie pre nežiaduce reakcie súvisiace
s týmito liekmi. Môže byť potrebná úprava dávky opiátov metabolizovaných CYP3A# (pozri časť 4.4). Sledovanie sa odporúča aj pri zmene cesty podávania. Ak sa PREVYMIS podáva súbežne s
cyklosporínom, rozsah zvýšenia plazmatických koncentrácií opiátov metabolizovaných CYP3A môže byť väčší. Počas súbežného podávania PREVYMISU
v kombinácii s
cyklosporínom a alfentanilom alebo fentanylom je potrebné vykonávať
pozorné klinické sledovanie pre útlm
dýchania a/alebo predĺženú sedáciu. Pozri príslušný súhrn charakteristických vlastností
(pozri časť 4.4).
Súbežne podávaný liek
Antiarytmiká
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
amiodarón Interakcia sa neskúmala.
Očakáva sa:
↑ koncentrácií amiodarónu
(primárne inhibícia CYP3A a inhibícia alebo indukcia CYP2C8)
chinidín Interakcia sa neskúmala.
Očakáva sa:
↑ koncentrácií chinidínu
(inhibícia CYP3A)
Lieky na kardiovaskulárny systém
Letermovir môže zvýšiť plazmatické koncentrácie amiodarónu.
Počas súbežného podávania sa odporúča časté sledovanie pre nežiaduce reakcie súvisiace
s amiodarónom. Ak sa amiodarón podáva súbežne s PREVYMISOM, e potrebné
vykonávať pravidelné sledovanie koncentrácií amiodarónu#.
Letermovir môže zvýšiť plazmatické koncentrácie chinidínu.
Počas podávania PREVYMISU s chinidínom je potrebné vykonávať pozorné klinické sledovanie. Pozri príslušný súhrn charakteristických vlastností#.
digoxín‡
(0,5 mg jednorazová dávka)/ letermovir (240 mg dvakrát denne)
↔ digoxínu
AUC 0,88 (0,80; 0,96) Cmax 0,75 (0,63; 0,89)
(indukcia P-gp)
Nevyžaduje sa žiadna úprava dávky.
Inhibítory protónovej pumpy
omeprazol Interakcia sa neskúmala.
Očakáva sa:
↓ omeprazolu
(indukcia CYP2C19) Interakcia sa neskúmala.
Očakáva sa:
↔ letermoviru
pantoprazol Interakcia sa neskúmala.
Očakáva sa:
↓ pantoprazolu
(pravdepodobne v dôsledku indukcie CYP2C19)
Letermovir môže znížiť plazmatické koncentrácie substrátov CYP2C19.
Môže byť potrebné klinické sledovanie a úprava dávky.
Letermovir môže znížiť plazmatické koncentrácie substrátov CYP2C19.
Môže byť potrebné klinické sledovanie a úprava dávky.
Interakcia sa neskúmala.
Očakáva sa:
↔ letermoviru
*Táto tabuľka neobsahuje všetky informácie.
† ↓ = pokles, ↑ = zvýšenie
↔ =žiadna klinicky významná zmena
‡ Štúdia jednocestnej interakcie hodnotiaca vplyv letermoviru na súbežne podávaný liek.
# Pozri príslušný súhrn charakteristických vlastností.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii nie sú žiadne údaje týkajúce sa použitia letermoviru u gravidných žien. Štúdie na
zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
PREVYMIS sa neodporúča počas gravidity a u žien, ktoré môžu otehotnieť a nepoužívajú antikoncepciu.
Dojčenie
Nie je známe, či sa letermovir vylučuje do ľudského mlieka.
Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie letermoviru do mlieka (pozri časť 5.3).
Riziko u novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu PREVYMISOM sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
U potkanov sa neobjavili žiadne účinky na fertilitu samíc. Ireverzibilná testikulárna toxicita a porucha
fertility sa pozorovala u samcov potkana, ale nepozorovala sa u samcov myší ani samcov opíc.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
PREVYMIS môže mať malý vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje. U niektorých pacientov bola počas liečby PREVYMISOM hlásená únava a vertigo, ktoré môžu ovplyvniť schopnosť pacienta viesť vozidlá a obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Hodnotenie bezpečnosti PREVYMISU bolo založené na klinickom skúšaní fázy 3 (P001) u príjemcov
HSCT, ktorí dostávali PREVYMIS alebo placebo počas 14 týždňov po transplantácii a ktorí sa z dôvodu bezpečnosti sledovali až do konca 24. týždňa po transplantácii (pozri časť 5.1).
Najčastejšie hlásenými nežiaducimi reakciami objavujúcimi sa u minimálne 1 % osôb v skupine
s PREVYMISOM a vo vyššej frekvencii ako pri placebe boli nevoľnosť (7,2 %), hnačka (2,4 %)
a vracanie (1,9 %).
Najčastejšie hlásenými nežiaducimi reakciami, ktoré viedli k vysadeniu PREVYMISU, boli nevoľnosť
(1,6 %), vracanie (0,8 %) a bolesť brucha (0,5 %).
Tabuľkový súhrnnežiaducichreakcií
Nasledujúce nežiaduce reakcie sa zistili u pacientov užívajúcich PREVYMIS v klinických skúšaniach.
Nežiaduce reakcie sú uvedené nižšie podľa triedy orgánových systémov a frekvencie. Frekvencie sú

definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) alebo veľmi zriedkavé (< 1/10 000).
Tabuľka 2: Nežiaduce reakcie zistené pri PREVYMISEFrekvencia Nežiaduce reakciePoruchy imunitného systémumenej časté hypersenzitivita
Poruchy metabolizmu a výživymenej časté znížená chuť do jedla
Poruchy nervového systémumenej časté dysgeúzia, bolesť hlavy
Poruchy ucha a labyrintumenej časté vertigo
Poruchy gastrointestinálneho traktučasté nevoľnosť, hnačka, vracanie menej časté bolesť brucha
Poruchy pečene a žlčových ciestmenej časté zvýšená alanínaminotransferáza, zvýšená aspartátaminotransferáza
Poruchy kostrovej a svalovej sústavy a spojivového tkanivamenej časté svalové spazmy
Poruchy obličiek a močových ciestmenej časté zvýšený kreatinín v krvi
Celkové poruchy a reakcie v mieste podaniamenej časté únava, periférny edém
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeexistuje žiadna skúsenosť s predávkovaním PREVYMISOM u ľudí. Počas klinických skúšaní fázy
1 dostávalo 86 zdravých osôb dávky pohybujúce sa v rozmedzí od 720 mg/deň do 1 440 mg/deň
PREVYMISU počas až 14 dní. Profil nežiaducich reakcií bol podobný ako pri klinickej dávke
480 mg/deň. Pri predávkovaní PREVYMISOM neexistuje žiadne špecifické antidotum. V prípade predávkovania sa odporúča sledovanie pacienta pre nežiaduce reakcie a začatie vhodnej
symptomatickej liečby.
Nie je známe, či dialýza bude viesť k významnému odstráneniu PREVYMISU zo systémovej cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antivirotiká na systémové použitie, priamo pôsobiace antivirotiká, ATC
kód: J05AX18
Mechanizmus účinku
Letermovir inhibuje komplex DNA terminázy CMV, ktorý je potrebný na štiepenie a zbalenie
vírusovej progénovej DNA. Letermovir ovplyvňuje tvorbu genómov so správnou dĺžkou jednotiek a narúša dozrievanie viriónov.
Antivírusováaktivita
Stredná hodnota EC50 letermoviru proti vzorke klinických izolátov CMV v bunkovej kultúre modelu
infekcie bola 2,1 nmol/l (rozmedzie = 0,7 nmol/l až 6,1 nmol/l, n=74).
Vírusová rezistencia
V bunkovej kultúre
Gény UL56 a UL89 CMV kódujú podjednotky DNA terminázy CMV. V bunkovej kultúre boli potvrdené zmutované vírusy CMV so zníženou citlivosťou na letermovir. Mutácie sú lokalizované
v UL56 a vyskytujú sa na aminokyselinových zvyškoch 231 až 369 (V231A, V231L, V236L, V236M,
E237D, L241P, T244K, T244R, L257I, F261C, F261L, F261S, Y321C, C325F, C325R, C325W, C325Y, M329T, R369G, R369M, R369S). Hodnoty EC50 pre tieto mutácie sú 13- až 8 262-násobne vyššie ako hodnoty pre referenčný divoký typ vírusu. Žiadne známe mutácie spôsobujúce rezistenciu voči letermoviru nie sú lokalizované v UL89.
V klinických štúdiách
V skúšaní fázy 2b hodnotiacom letermovir v dávkach 60, 120 alebo 240 mg/deň alebo placebo až do
84 dní u 131 príjemcov HSCT sa na vzorkách získaných od 12 osôb liečených letermovirom,
u ktorých došlo k zlyhaniu profylaxie a u ktorých boli vzorky dostupné na analýzu, vykonala analýza
DNA sekvencie zvolenej oblasti UL56 (aminokyseliny 231 až 369). U jednej osoby (ktorá dostávala
60 mg/deň) sa objavil genotypový variant (GV) rezistentný voči letermoviru (V236M).
V skúšaní fázy 3 (P001) sa na vzorkách získaných od 22 osôb liečených letermovirom v populácii FAS, u ktorých došlo k zlyhaniu profylaxie a u ktorých boli vzorky dostupné na analýzu, vykonala analýza DNA sekvencie celých kódujúcich oblastí UL56 a UL89. U jednej osoby sa objavil GV rezistentný voči letermoviru (V236M).
Skrížená rezistencia
Skrížená rezistencia je nepravdepodobná s liekmi s rozdielnym mechanizmom účinku. Letermovir je
plne účinný proti populáciám vírusov so substitúciami spôsobujúcimi rezistenciu voči inhibítorom DNA polymerázy CMV (ganciklovir, cidofovir a foskarnet). Tieto inhibítory DNA polymerázy sú plne účinné proti populáciám vírusov so substitúciami spôsobujúcimi rezistenciu voči letermoviru.
Elektrofyziológia srdca
Vplyv letermoviru v dávkach až do 960 mg podávaných i.v. na QTc interval sa hodnotil
v randomizovanom, placebom a aktívne kontrolovanom (moxifloxacín 400 mg perorálne) dôkladnom skúšaní QT s jednorazovou dávkou a 4 obdobiami prekríženia u 33 zdravých osôb. Letermovir po i.v.
dávke 960 mg nepredlžuje QTc interval v klinicky významnom rozsahu s plazmatickými
koncentráciami približne 2-násobne vyššími ako pri i.v. dávke 480 mg.
Klinická účinnosťabezpečnosť
Dospelí CMV-séropozitívni príjemcovia [R+] alogénneho štepu krvotvorných kmeňových buniek
Na vyhodnotenie profylaxie letermovirom ako preventívnej metódy pri infekcii alebo ochorení vyvolanom CMV sa účinnosť letermoviru hodnotila v multicentrickom, dvojito zaslepenom, placebom kontrolovanom skúšaní fázy 3 (P001) u dospelých CMV-séropozitívnych príjemcov [R+] alogénneho HSCT. Osoby boli randomizované (2:1) na užívanie buď letermoviru v dávke 480 mg jedenkrát denne upravenej na 240 mg, ak sa podávala súbežne s cyklosporínom, alebo na placebo. Randomizácia bola
stratifikovaná skúšajúcim pracoviskom a rizikom (vysoké oproti nízkemu) reaktivácie CMV v čase vstupu do štúdie. Letermovir sa začal podávať po HSCT (0. – 28. deň po transplantácii) a v podávaní sa pokračovalo do konca 14. týždňa po transplantácii. Letermovir sa podával buď perorálne alebo i.v.; dávka letermoviru bola rovnaká bez ohľadu na cestu podávania. Osoby boli sledované do konca 24. týždňa po transplantácii pre primárny cieľový ukazovateľ účinnosti s pokračovaním sledovania do konca 48. týždňa po transplantácii.
Osoby boli sledované pre DNA CMV týždenne až do konca 14. týždňa a následne každé dva týždne až do konca 24. týždňa po transplantácii so začatím štandardnej preemptívnej terapie CMV, ak sa hladina DNA CMV v krvi považovala za klinicky významnú. V sledovaní osôb sa pokračovalo do konca 48. týždňa po transplantácii.
Spomedzi 565 liečených osôb 373 osôb dostávalo letermovir (vrátane 99 osôb, ktoré dostali minimálne jednu i.v. dávku) a 192 dostávalo placebo (vrátane 48 osôb, ktorí dostali minimálne jednu i.v. dávku). Medián času do začatia podávania letermoviru bol 9 dní po transplantácii. Tridsaťsedem percent (37 %) osôb malo prijatý štep na začiatku. Medián veku bol 54 rokov (rozmedzie: 18 až 78 rokov); 56 (15 %) osôb bolo vo veku 65 rokov a starších; 58 % bolo mužov; 82 % bolo bielej; 10 % bolo ázijskej; 2 % boli čiernej alebo africkej a 7 % bolo hispánskej alebo latinskoamerickej rasy. Na začiatku 50 % osôb dostávalo myeloablatívny režim, 52 % dostávalo cyklosporín a 42 % dostávalo takrolimus. Najčastejšími primárnymi dôvodmi pre transplantáciu boli akútna myeloidná leukémia (38 %), myeloblastický syndróm (15 %) a lymfóm (13 %). Dvanásť percent (12 %) osôb bolo na začiatku pozitívnych na DNA CMV.
Na začiatku malo 31 % osôb vysoké riziko reaktivácie na základe definície jedným alebo viacerými z nasledujúcich kritérií: darca s príbuzným ľudským leukocytovým antigénom (HLA) (súrodenec)
s minimálne jednou nezhodou na jednom z nasledujúcich troch lókusov na géne HLA: HLA-A, -B
alebo –DR, haploidentický darca; nepríbuzný darca s minimálne jednou nezhodou na jednom
z nasledujúcich štyroch lókusov na géne HLA: HLA-A, -B, -C a -DRB1; použitie pupočníkovej krvi ako zdroja kmeňových buniek; použitie štepov zbavených T-buniek ex vivo; reakcia štepu proti príjemcovi (Graft-Versus-Host Disease, GVHD) 2. alebo vyššieho stupňa vyžadujúca systémové kortikosteroidy.
Primárny cieľový ukazovateľ účinnosti
Primárny cieľový ukazovateľ účinnosti klinicky významnej infekcie CMV v štúdii P001 sa definoval ako výskyt DNA CMV v krvi vyžadujúci preemptívnu terapiu proti CMV (pre-emptive therapy, PET)
alebo výskyt ochorenia CMV v koncovom orgáne. Použila sa metóda pacient, ktorý nedokončil štúdiu
= zlyhanie (Non-Completer=Failure, NC=F), kde osoby, ktoré prerušili štúdiu pred 24. týždňom po transplantácii alebo u nich nebol dostupný výsledok v 24. týždni po transplantácii, sa započítali ako
zlyhania.
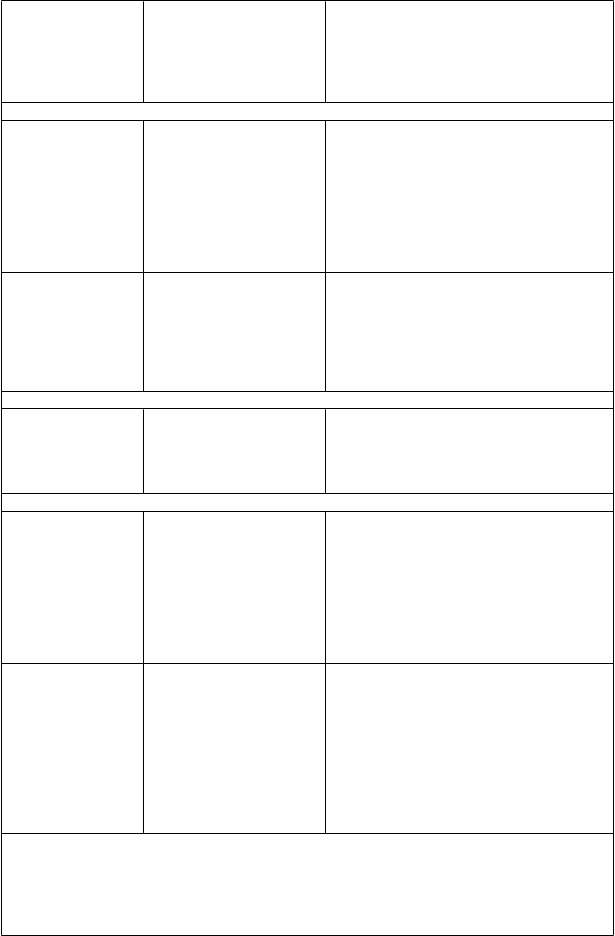
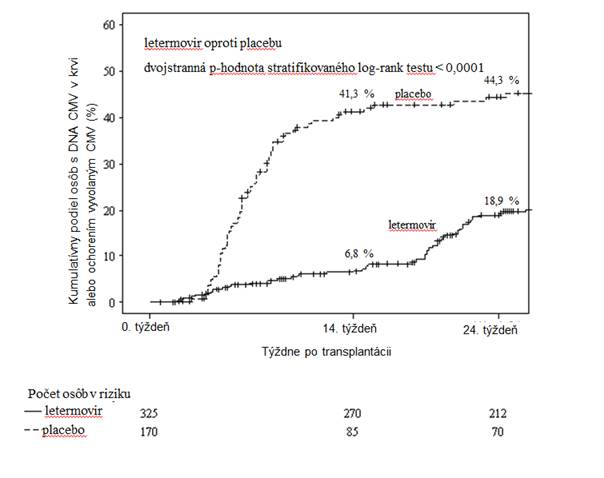
Pri letermovire sa preukázala vyššia účinnosť oproti placebu v analýze primárneho cieľového ukazovateľa, ako je uvedené v tabuľke 3. Odhadovaný rozdiel liečby -23,5 % bol štatisticky významný (jednostranná p-hodnota < 0,0001).
Tabuľka 3: P001: Výsledky účinnosti u príjemcov HSCT (metóda NC = F, populácia FAS).
letermovir placebo
(N = 325) (N = 170) Parameter n (%) n (%)
Primárny cieľový ukazovateľ účinnosti
(podiel pacientov so zlyhaním profylaxie počas 24
týždňov)
Dôvody zlyhania†
122 (37,5) 103 (60,6)

Klinicky významná infekcia CMV 57 (17,5) 71 (41,8) DNA CMV v krvi vyžadujúca PET proti CMV 52 (16,0) 68 (40,0) Ochorenie CMV v koncovom orgáne 5 (1,5) 3 (1,8)
Prerušenie štúdie 56 (17,2) 27 (15,9) Chýbajúci výsledok 9 (2,8) 5 (2,9)
Rozdiel liečby upravený na skupinu (letermovir- placebo)§
Rozdiel (95% IS) -23,5 (-32,5; -14,6)
p-hodnota < 0,0001
† Kategórie zlyhania sa vzájomne vylučujú a sú založené na hierarchii kategórií v uvedenom poradí.
§ 95% IS a p-hodnota pre rozdiely liečby v percentuálnom vyjadrení odpovede sa vypočítali pomocou Mantelovej-Haenszelovej metódy upravenej na skupiny s rozdielom porovnávaným pomocou harmonického priemeru veľkosti vzorky v ramene pre každú skupinu (vysoké alebo nízke riziko). Na
vyjadrenie štatistickej významnosti sa použila jednostranná p-hodnota ≤ 0,0249.
FAS = celý analyzovaný súbor (full analysis set); FAS zahŕňa randomizované osoby, ktoré dostali minimálne jednu dávku skúšaného lieku a vylučuje osoby s detegovateľnou DNA CMV na začiatku.
Metóda na spracovanie chýbajúcich hodnôt: metóda NC = F (pacient, ktorý nedokončil štúdiu =
zlyhanie). Pri metóde NC=F sa zlyhanie definovalo ako všetky osoby s klinicky významnou infekciou
CMV alebo ktoré predčasne vystúpili zo štúdie alebo u nich nebol dostupný výsledok v návštevnom okne v 24. týždni po transplantácii.
N = počet osôb v každej liečebnej skupine.
n (%) = počet (percento) osôb v každej podkategórii.
Poznámka: Podiel osôb s detegovateľnou vírusovou DNA CMV v deň 1, u ktorých sa rozvinula klinicky významná infekcia CMV počas 24 týždňov po transplantácii bol v skupine s letermovirom
64,6 % (31/48) v porovnaní s 90,9 % (20/22) v skupine s placebom. Odhadovaný rozdiel (95% IS pre
rozdiel) bol -26,1 % (-45,9 %, -6,3 %) s nominálnou jednostrannou p-hodnotou < 0,0048.
Faktory súvisiace s výskytom DNA CMV v krvi po 14. týždni po transplantácii medzi osobami
liečenými letermovirom zahŕňali vysoké riziko pre reaktiváciu CMV na začiatku, GVHD, používanie kortikosteroidov a sérum donora negatívne na prítomnosť CMV.
Obrázok 1: P001: Kaplanova-Meierova krivka času do začatia PET proti CMV alebo do vzniku ochorenia CMV v koncovom orgáne počas 24 týždňov po transplantácii u príjemcov HSCT (populácia FAS)
Neobjavili sa žiadne rozdiely vo výskyte prijatia štepu alebo v čase do jeho prijatia medzi skupinou s PREVYMISOM a skupinou s placebom.
Účinnosť bola naprieč podskupinami konzistentne v prospech letermoviru vrátane nízkeho a vysokého rizika pre reaktiváciu CMV, prípravných režimov a súbežných režimov obsahujúcich imunosupresíva (pozri obrázok 2).
Obrázok 2: P001: Forestova krivka podielu osôb, u ktorých sa začala PET proti CMV alebo s ochorením CMV v koncovom orgáne počas 24 týždňov po transplantácii podľa vybraných podskupín (metóda NC=F, populácia FAS)

Celkovo (N=325, 170)
Skupiny podľa rizika


Vysoké riziko (n=102, 45) Nízke riziko (n=223, 125)
Pôvod kmeňových buniek

Periférna krv (n=241, 117)

Kostná dreň (n=72, 43)
Nezhoda s darcom
Zhodný príbuzný (n=108, 58)


Nezhodný príbuzný (n=52, 18) Zhodný nepríbuzný (n=122, 70)
Nezhodný nepríbuzný (n=43, 24)
Haploidentický darca

Áno (n=49, 17) Nie (n=276, 153)
Prípravný režim

Myeloablatívny (n=154, 85)



Príprava so zníženou intenzitou (n=86, 48) Nemyeloablatívny (n=85, 37)

Immunosupresívny režim cyklosporín A (n=162, 90) takrolimus (n=145, 69)
-70 -60 -50 -40 -30 -20 -10 0 10 20
 V prospech letermoviru
V prospech placeba
V prospech letermoviru
V prospech placeba
Rozdiel medzi letermovirom - placebom (%) a 95% IS
NC=F, pacient, ktorý nedokončil štúdiu = zlyhanie (Non-Completer=Failure). Pri metóde NC=F sa osoby, ktoré prerušili
štúdiu pred 24. týždňom po transplantácii alebo u ktorých nebol dostupný výsledok v 24. týždni po transplantácii, započítali ako zlyhania.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s PREVYMISOM
v jednej alebo vo viacerých podskupinách pediatrickej populácie pre profylaxiu infekcie cytomegalovírusom (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika letermoviru bola opísaná po perorálnom a i.v. podaní u zdravých osôb a príjemcov HSCT. Expozícia letermoviru sa zvyšovala viac ako proporcionálne dávke pri perorálnom aj i.v. podaní. Pravdepodobný mechanizmus je saturácia/autoinhibícia OATP1B1/3.
U zdravých osôb bol geometrický priemer AUC v rovnovážnom stave 71 500 ng•hod/ml
a geometrický priemer Cmax v rovnovážnom stave bol 13 000 ng/ml pri letermovire podávanom perorálne v dávke 480 mg jedenkrát denne.
Letermovir dosiahol rovnovážny stav za 9 až 10 dní s akumulačným pomerom 1,2 pre AUC a 1,0 pre
Cmax.

U príjemcov HSCT bola AUC letermoviru odhadnutá pomocou analýz populačnej farmakokinetiky
s použitím údajov z fázy 3 (pozri tabuľku 4). Rozdiely v expozícii medzi liečebnými režimami nie sú klinicky významné; účinnosť bola v rámci rozsahu expozícií pozorovaných v štúdii P001 konzistentná.
Tabuľka 4: Hodnoty AUC letermoviru (ng•hod/ml) u príjemcov HSCT Liečebný režim Medián (90% predikčný interval)*480 mg perorálne, bez cyklosporínu 34 400 (16 900, 73 700)
480 mg i.v., bez cyklosporínu 100 000 (65 300, 148 000)
240 mg perorálne, s cyklosporínom 60 800 (28 700, 122 000)
240 mg i.v., s cyklosporínom 70 300 (46 200, 106 000)
* Post-hoc predpovede populácie z analýzy populačnej FK s použitím údajov z fázy 3
AbsorpciaLetermovir sa rýchlo vstrebával s mediánom času do maximálnej plazmatickej koncentrácie (Tmax) 45
minút až 2,25 hodín a koncentrácie klesali bifázicky. U príjemcov HSCT bola biologická dostupnosť
letermoviru odhadnutá na približne 35 % pri perorálnom letermovire v dávke 480 mg jedenkrát denne podávanej bez cyklosporínu. Interindividuálna variabilita pre biologickú dostupnosť bola odhadnutá na približne 37 %.
Vplyv cyklosporínuU príjemcov HSCT spôsobilo súbežné podávanie cyklosporínu zvýšenie plazmatických koncentrácií letermoviru v dôsledku inhibície OATP1B. Biologická dostupnosť letermoviru bola odhadnutá na približne 85 % pri letermovire v dávke 240 mg jedenkrát denne perorálne podávanom pacientom súbežne s cyklosporínom.
Ak sa letermovir podáva súbežne s cyklosporínom, odporúčaná dávka letermoviru je 240 mg jedenkrát denne (pozri časť 4.2).
Vplyv potravyU zdravých osôb perorálne podanie 480 mg jednorazovej dávky letermoviru so štandardným jedlom s vysokým obsahom tukov a kalórií nemalo žiadny vplyv na celkovú expozíciu (AUC) a viedlo
k približne 30 % zvýšeniu maximálnych hladín (Cmax) letermoviru. Letermovir sa môže podávať perorálne s jedlom alebo bez jedla tak, ako sa podával aj v klinických štúdiách (pozri časť 4.2).
DistribúciaNa základe analýz populačnej farmakokinetiky sa priemerný distribučný objem v rovnovážnom stave
odhadol na 45,5 l po intravenóznom podaní u príjemcov HSCT.
Letermovir sa v rozsiahlej miere (98,2 %) viaže na ľudské plazmatické bielkoviny nezávisle od rozmedzia koncentrácií (3 až 100 mg/l) hodnoteného
in vitro. Určitá saturácia sa pozorovala pri nižších koncentráciách. Pomer rozdelenia letermoviru medzi krv a plazmu je 0,56 a je nezávislý od rozmedzia koncentrácií (0,1 až 10 mg/l) hodnoteného
in vitro.
V predklinických štúdiách distribúcie sa letermovir distribuoval do orgánov a tkanív s najvyššími koncentráciami pozorovanými v gastrointestinálnom trakte, žlčovode a pečeni a s nízkymi koncentráciami v mozgu.
Biotransformácia
Väčšia časť zložiek súvisiacich s letermovirom v plazme je vo forme nezmenenej materskej látky
(96,6 %). V plazme sa nezistili žiadne významné metabolity. Letermovir sa čiastočne eliminuje prostredníctvom glukuronidácie sprostredkovanej UGT1A1/1A3.
Eliminácia
Priemerný zdanlivý terminálny polčas letermoviru u zdravých osôb je približne 12 hodín pri i.v.
letermovire v dávke 480 mg. Hlavné cesty eliminácie letermoviru sú biliárna exkrécia a takisto aj priama glukuronidácia. Tento proces zahŕňa transportéry pečeňového vychytávania OATP1B1 a 3
nasledované glukuronidáciou katalyzovanou UGT1A1/3.
Na základe analýz populačnej farmakokinetiky sa zdanlivý klírens letermoviru v rovnovážnom stave odhaduje na 4,84 l/hod po intravenóznom podaní 480 mg u príjemcov HSCT. Interindividuálna variabilita pre klírens sa odhaduje na 24,6 %.
Vylučovanie
Po perorálnom podaní rádioaktívne značeného letermoviru sa 93,3 % izotopom značeného liečiva
vylúčilo stolicou. Väčšia časť letermoviru sa vylúčila žlčou vo forme nezmenenej materskej látky
s malým podielom (6 % dávky) vo forme acylglukuronidového derivátu v stolici. Acylglukuronid je v stolici nestabilný. Vylučovanie letermoviru močom bolo zanedbateľné (< 2 % dávky).
Farmakokinetika vosobitnýchskupináchpacientov
Porucha funkcie pečene
AUC neviazaného letermoviru bola približne o 81 % vyššia u osôb so stredne ťažkou (Childova- Pughova trieda B [CP-B], skóre 7 – 9) a 4-násobne vyššia u osôb s ťažkou (Childova-Pughova trieda C [CP-C], skóre 10 – 15) poruchou funkcie pečene v porovnaní so zdravými osobami. Zmeny expozície letermoviru u osôb so stredne ťažkou poruchou funkcie pečene nie sú klinicky významné.
Výrazné zvýšenia expozície neviazanému letermoviru sa očakávajú u pacientov so stredne ťažkou poruchou funkcie pečene v kombinácii so stredne ťažkou alebo ťažkou poruchou funkcie obličiek (pozri časť 4.2).
Porucha funkcie obličiek
AUC neviazaného letermoviru bola približne o 115 % vyššia u osôb so stredne ťažkou (eGFR 31,0 až
56,8 ml/min/1,73 m2) a o 81 % vyššia u osôb s ťažkou (eGFR 11,9 až 28,1 ml/min/1,73 m2) poruchou funkcie obličiek v porovnaní so zdravými osobami. Zmeny expozície letermoviru z dôvodu stredne
ťažkej alebo ťažkej poruchy funkcie obličiek sa nepovažujú za klinicky významné. Osoby s ESRD sa neskúmali.
Telesná hmotnosť
Na základe analýz populačnej farmakokinetiky sa odhaduje, že AUC letermoviru je o 18,7 % nižšia
u osôb s telesnou hmotnosťou 80 – 100 kg v porovnaní s osobami s telesnou hmotnosťou 67 kg. Tento rozdiel nie je klinicky významný.
Rasa
Na základe analýz populačnej farmakokinetiky sa odhaduje, že AUC letermoviru je o 33,2 % vyššia u príslušníkov ázijskej rasy v porovnaní s príslušníkmi bielej rasy. Táto zmena nie je klinicky
významná.
Pohlavie
Na základe analýz populačnej farmakokinetiky neexistuje žiadny rozdiel vo farmakokinetike letermoviru u žien v porovnaní s mužmi.
Staršie osoby
Na základe analýz populačnej farmakokinetiky neexistuje žiadny vplyv veku na farmakokinetiku letermoviru. Na základe veku sa nevyžaduje žiadna úprava dávky.
5.3 Predklinické údaje o bezpečnosti
Všeobecná toxicita
Ireverzibilná testikulárna toxicita sa zaznamenala len u potkanov pri systémových expozíciách (AUC)
≥ 3-násobku expozícií u ľudí pri odporúčanej dávke pre ľudí (recommended human dose, RHD). Táto toxicita bola charakterizovaná degeneráciou tubulov v semenníkoch a oligospermiou a zvyškami
buniek v nadsemenníkoch s poklesom hmotnosti semenníkov a nadsemenníkov. Pri expozíciách (AUC) podobných expozíciám u ľudí pri RHD sa u potkanov nepozorovala testikulárna toxicita. Testikulárna toxicita sa nepozorovala pri najvyšších dávkach skúmaných pri expozíciách u myší až do
4-násobku a u opíc až do 2-násobku expozícií u ľudí pri RHD. Význam pre ľudí nie je známy.
Je známe, že hydroxypropylbetadex môže vyvolať vakuoláciu obličiek u potkanov, ak sa podáva intravenózne v dávkach väčších ako 50 mg/kg/deň. Vakuolácia sa zaznamenala v obličkách potkanov, ktorým sa letermovir podával intravenózne v dávke 1 500 mg/kg/deň vo forme s cyklodextrínovou pomocnou látkou hydroxypropylbetadexom.
Karcinogenita
Štúdie karcinogenity sa s letermovirom nevykonali.
Mutagenita
Letermovir nebol genotoxický v sérii testov in vitro alebo in vivo vrátane testov mikrobiálnej
mutagenézy, chromozomálnej aberácie na ovariálnych bunkách čínskeho škrečka a v štúdii myšacieho mikrojadierka in vivo.
Reprodukcia
Fertilita
V štúdiách fertility a skorého embryonálneho vývinu u potkanov sa neobjavili žiadne účinky letermoviru na fertilitu samíc. U samcov potkana sa pozorovala znížená koncentrácia spermií, znížená pohyblivosť spermií a pokles fertility pri systémových expozíciách ≥ 3-násobku AUC u ľudí pri RHD (pozri Všeobecná toxicita).
U opíc, ktorým sa podával letermovir, sa neobjavil žiadny dôkaz testikulárnej toxicity na základe histopatologického hodnotenia, stanovenia veľkosti semenníkov, rozboru hormónov v krvi (folikulostimulačný hormón, inhibín B a testosterón) a hodnotení spermií (počet spermií, pohyblivosť a morfológia) pri systémových expozíciách približne 2-násobku AUC u ľudí pri RHD.
Vývin
U potkanov sa zaznamenala maternotoxicita (vrátane zníženého prírastku telesnej hmotnosti) pri
dávke 250 mg/kg/deň (približne 11-násobok AUC pri RHD); u mláďat sa zaznamenal pokles hmotnosti plodu s oneskorenou osifikáciou, mierne opuchnutými plodmi a zvýšeným výskytom skrátených pupočníkových šnúr a zmeny a malformácie stavcov, rebier a panvy. Pri dávke
50 mg/kg/deň (približne 2,5-násobok AUC pri RHD) sa nezaznamenali žiadne účinky na matku a vývin.
U králikov sa zaznamenala maternotoxicita (vrátane mortality a potratov) pri dávke 225 mg/kg/deň (približne 2-násobok AUC pri RHD); u mláďat sa zaznamenal zvýšený výskyt malformácií a zmien stavcov a rebier.
V štúdii pre- a postnatálneho vývinu sa letermovir perorálne podával gravidným potkanom. Nepozorovala sa žiadna vývinová toxicita až do najvyššej skúmanej expozície (2-násobok AUC pri RHD).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
mikrokryštalická celulóza (E460)
sodná soľ kroskarmelózy (E468)
povidón (E1201)
koloidný oxid kremičitý bezvodý (E551)
stearan horečnatý (E470b)
Obal tablety
monohydrát laktózy
hypromelóza (E464) oxid titaničitý (E171) triacetín (E1518)
žltý oxid železitý (E172)
červený oxid železitý (len pre 480 mg tablety) (E172)
karnaubský vosk (E903)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
30 mesiacov
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne špeciálne teplotné podmienky na uchovávanie. Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
240mgfilmomobalenétablety
Polyamid/ hliník/PVC - hliníková blistrová karta. Každá škatuľka obsahuje štyri (4) papierové karty, obsahujúce jednu blistrovú kartu so 7 tabletami pre celkovo 28 tabliet.
480mgfilmomobalenétablety
Polyamid/ hliník/PVC - hliníková blistrová karta. Každá škatuľka obsahuje štyri (4) papierové karty obsahujúce jednu blistrovú kartu so 7 tabletami pre celkovo 28 tabliet.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU
Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/17/1245/001
EU/1/17/1245/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky agentúry
http://www.ema.europa.eu.

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUPREVYMIS 240 mg koncentrát na infúzny roztok
PREVYMIS 480 mg koncentrát na infúzny roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEPREVYMIS240mgkoncentrátnainfúznyroztokKaždá injekčná liekovka obsahuje 240 mg (12 ml v jednej injekčnej liekovke) letermoviru. Každý ml obsahuje 20 mg letermoviru.
PREVYMIS480mgkoncentrátnainfúznyroztokKaždá injekčná liekovka obsahuje 480 mg (24 ml v jednej injekčnej liekovke) letermoviru. Každý ml obsahuje 20 mg letermoviru.
PomocnálátkasoznámymúčinkomTento liek obsahuje 23 mg (1,0 mmol) sodíka v jednej 240 mg injekčnej liekovke, čo zodpovedá
1,15 % WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
Tento liek obsahuje 46 mg (2,0 mmol) sodíka v jednej 480 mg injekčnej liekovke, čo zodpovedá
2,30 % WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
Každá 240 mg dávka (12 ml injekčná liekovka) tohto lieku obsahuje 1 800 mg hydroxypropylbetadexu
(cyklodextrín).
Každá 480 mg dávka (24 ml injekčná liekovka) tohto lieku obsahuje 3 600 mg hydroxypropylbetadexu
(cyklodextrín).
Ďalšie informácie, pozri časť 4.2.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAKoncentrát na infúzny roztok (sterilný koncentrát) Číra, bezfarebná tekutina
pH 7 až 8
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikáciePREVYMIS je indikovaný na profylaxiu reaktivácie cytomegalovírusu (CMV) a ochorenia vyvolaného CMV dospelým CMV-séropozitívnym príjemcom [R+] alogénneho štepu krvotvorných kmeňových buniek (allogeneic haematopoietic stem cell transplant, HSCT).
Je potrebné vziať do úvahy oficiálne usmernenie týkajúce sa správneho používania antivirotík.
4.2 Dávkovanie a spôsob podávania
Liečbu PREVYMISOM má začať lekár so skúsenosťami s liečením pacientov, ktorí podstúpili transplantáciu alogénneho štepu krvotvorných kmeňových buniek.
Dávkovanie
PREVYMIS je tiež dostupný vo forme na perorálne použitie (240 mg a 480 mg filmom obalené
tablety).
PREVYMIS tablety a koncentrát na infúzny roztok sa môžu vzájomne zamieňať na základe rozhodnutia lekára a nie je potrebná žiadna úprava dávky.
Odporúčaná dávka PREVYMISU je 480 mg jedenkrát denne.
PREVYMIS sa má začať podávať po HSCT. PREVYMIS sa môže začať podávať v deň transplantácie a najneskôr 28 dní po transplantácii. PREVYMIS sa môže začať podávať pred prijatím štepu alebo po ňom. Profylaxia PREVYMISOM má pokračovať počas 100 dní po transplantácii.
Bezpečnosť a účinnosť používania letermoviru počas viac ako 100 dní sa v klinických skúšaniach neskúmali. Predĺžená profylaxia letermovirom nad 100 dní po transplantácii môže byť prínosná
u niektorých pacientov s vysokým rizikom neskorej reaktivácie CMV (pozri časť 5.1). Používanie profylaxie letermovirom počas viac ako 100 dní vyžaduje dôsledné vyhodnotenie pomeru prínosu
a rizika.
Úprava dávkovania
Ak sa PREVYMIS podáva súbežne s cyklosporínom, dávkovanie PREVYMISU sa má znížiť na
240 mg jedenkrát denne (pozri časti 4.5 a 5.2).
· Ak sa cyklosporín začne podávať po začatí podávania PREVYMISU, nasledujúca dávka
PREVYMISU sa má znížiť na 240 mg jedenkrát denne.
· Ak sa cyklosporín vysadí po začatí podávania PREVYMISU, nasledujúca dávka PREVYMISU sa má zvýšiť na 480 mg jedenkrát denne.
· Ak sa podávanie cyklosporínu dočasne preruší z dôvodu vysokých hladín cyklosporínu, nie je potrebná žiadna úprava dávky PREVYMISU.
Vynechaná dávka
Ak sa vynechá dávka, má sa podať pacientovi čím skôr. Ak už je čas na nasledujúcu dávku, preskočte vynechanú dávku a vráťte sa k pravidelnému režimu. Nepodávajte dvojnásobnú dávku alebo viac ako
je predpísaná dávka.
Osobitné skupinypacientov
Staršie osoby
Na základe veku sa nevyžaduje žiadna úprava dávky PREVYMISU (pozri časti 5.1 a 5.2).
Porucha funkcie pečene
Nevyžaduje sa žiadna úprava dávky PREVYMISU z dôvodu miernej (Childova-Pughova trieda A) až stredne ťažkej (Childova-Pughova trieda B) poruchy funkcie pečene. PREVYMIS sa neodporúča
u pacientov s ťažkou (Childova-Pughova trieda C) poruchou funkcie pečene (pozri časť 5.2).
Kombinovaná porucha funkcie pečene a obličiek
PREVYMIS sa neodporúča u pacientov so stredne ťažkou poruchou funkcie pečene v kombinácii so stredne ťažkou alebo ťažkou poruchou funkcie obličiek (pozri časť 5.2).
Porucha funkcie obličiek
U pacientov s miernou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek sa neodporúča žiadna úprava dávky PREVYMISU. U pacientov s koncovým štádiom ochorenia obličiek (End Stage Renal
Disease, ESRD) s dialýzou alebo bez nej nie je možné stanoviť odporúčanie na dávkovanie. Účinnosť a bezpečnosť u pacientov s ESRD neboli preukázané.
PREVYMIS koncentrát na infúzny roztok obsahuje hydroxypropylbetadex. Pre dávke 480 mg letermoviru sa predpokladá, že očakávaná klinická expozícia hydroxypropylbetadexu s intravenózne podaným letermovirom bude približne 3 600 mg/deň. V štúdiách u ľudí pri intravenóznom podávaní letermoviru s trvaním liečby až do 47 dní sa nevyskytli žiadne prípady poškodenia obličiek spôsobené hydroxypropylbetadexom. U pacientov so stredne ťažkou alebo ťažkou poruchou funkcie obličiek (klírens kreatinínu menej ako 50 ml/min) dostávajúcich PREVYMIS by mohlo dôjsť k akumulácii hydroxypropylbetadexu (pozri časť 5.3). U týchto pacientov sa majú starostlivo sledovať hladiny sérového kreatinínu.
Pediatrická populácia
Bezpečnosť a účinnosť PREVYMISU u pacientov mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje (pozri časť 5.1).
Spôsob podávania
Len na intravenózne použitie.
PREVYMIS koncentrát na infúzny roztok sa pred podaním musí zriediť (pozri časť 6.6).
PREVYMIS sa má podávať len formou intravenóznej infúzie (i.v.). PREVYMIS sa nemá podávať ako pretlaková infúzia alebo bolus.
Po zriedení sa má PREVYMIS podať ako intravenózna infúzia cez periférny alebo centrálny venózny katéter pri celkovom čase podania približne 60 minút. Má sa podať celý obsah intravenózneho vaku.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Súbežné podávanie s pimozidom (pozri časti 4.4 a 4.5).
Súbežné podávanie s námeľovými alkaloidmi (pozri časti 4.4 a 4.5).
Ak sa letermovir kombinuje s cyklosporínom:
Súbežné používanie dabigatranu, atorvastatínu, simvastatínu, rosuvastatínu alebo pitavastatínu je kontraindikované (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovanie DNACMV
Bezpečnosť a účinnosť letermoviru boli stanovené u pacientov s negatívnym výsledkom testu na DNA
CMV pred začatím profylaxie. DNA CMV sa sledovala týždenne až do konca 14. týždňa po transplantácii a následne každé dva týždne až do konca 24. týždňa. V prípadoch klinicky významnej hladiny DNA CMV v krvi alebo ochorenia vyvolaného CMV sa profylaxia letermovirom zastavila
a začalo sa so štandardnou preemptívnou terapiou (pre-emptive therapy, PET) alebo liečbou. U pacientov, u ktorých sa začala profylaxia letermovirom a následne bola zistená pozitivita
počiatočného testu na DNA CMV, sa mohlo pokračovať v profylaxii, ak neboli splnené kritériá pre
PET (pozri časť 5.1).
Riziko nežiaducichreakciíalebozníženéhoterapeutickéhoúčinkuzdôvoduliekovýchinterakcií
Súbežné používanie PREVYMISU a určitých liekov môže viesť k známym alebo potenciálne
významným liekovým interakciám, z ktorých niektoré môžu viesť:
· k možným klinicky významným nežiaducim reakciám spôsobeným vyššou expozíciou súbežne používaným liekom alebo letermoviru.
· k významnému poklesu plazmatických koncentrácií súbežne používaného lieku, čo môže viesť k zníženému terapeutickému účinku súbežne používaného lieku.
Opatrenia na predchádzanie alebo zvládnutie týchto známych alebo potenciálne významných liekových interakcií vrátane odporúčaní na dávkovanie (pozri časti 4.3 a 4.5), pozri v tabuľke 1.
Liekové interakcie
PREVYMIS sa má používať s opatrnosťou s liekmi, ktoré sú substrátmi CYP3A s úzkymi
terapeutickými rozsahmi (napr. alfentanil, fentanyl a chinidín), pretože súbežné podávanie môže viesť k zvýšeniam plazmatických koncentrácií substrátov CYP3A. Odporúča sa pozorné sledovanie a/alebo úprava dávky súbežne podávaných substrátov CYP3A (pozri časť 4.5).
Počas prvých 2 týždňov po začatí a ukončení liečby letermovirom (pozri časť 4.5) a takisto po zmene cesty podávania letermoviru sa všeobecne odporúča zvýšené sledovanie hladín cyklosporínu,
takrolimu, sirolimu.
Letermovir je stredne silný induktor enzýmov a transportérov. Indukcia môže viesť k zníženým plazmatickým koncentráciám niektorých metabolizovaných a transportovaných liekov (pozri časť 4.5). Odporúča sa preto terapeutické sledovanie hladín liekov (therapeutic drug monitoring, TDM) pre vorikonazol a fenytoín. Súbežnému používaniu dabigatranu je potrebné sa vyhnúť kvôli riziku
zníženia účinnosti dabigatranu.
Letermovir môže zvýšiť plazmatické koncentrácie liekov transportovaných OATP1B1/3 ako sú napríklad mnohé statíny (pozri časť 4.5 a tabuľku 1).
Pomocné látky
PREVYMIS 240 mg koncentrát na infúzny roztok obsahuje 23 mg (alebo 1,0 mmol) sodíka v dávke.
To sa má vziať do úvahy u pacientov na diéte s kontrolovaným obsahom sodíka.
PREVYMIS 480 mg koncentrát na infúzny roztok obsahuje 46 mg (alebo 2,0 mmol) sodíka v dávke. To sa má vziať do úvahy u pacientov na diéte s kontrolovaným obsahom sodíka.
4.5 Liekové a iné interakcie
Všeobecnéinformácieorozdielochvexpozíciimedzirôznymiliečebnýmirežimamiletermoviru
-Odhadované plazmatické expozície letermoviru sú odlišné v závislosti od použitého režimu dávkovania (pozri tabuľku v časti 5.2). Klinické následky liekových interakcií letermoviru budú preto závisieť od toho, aký režim letermoviru sa použije a či sa letermovir kombinuje s cyklosporínom alebo nie.
-Kombinácia cyklosporínu a letermoviru môže viesť k výraznejším alebo ďalším účinkom na súbežne podávané lieky v porovnaní s letermovirom samotným (pozri tabuľku 1).
Vplyv inýchliekovnaletermovir
Cesty eliminácie letermoviru in vivo sú biliárna exkrécia a glukuronidácia. Relatívny význam týchto
ciest nie je známy. Obidve cesty eliminácie zahŕňajú aktívne vychytávanie hepatocytmi prostredníctvom transportérov pečeňového vychytávania OATP1B1/3. Glukuronidácia letermoviru je po vychytávaní sprostredkovaná UGT1A1 a 3. Zdá sa, že letermovir taktiež podlieha efluxu sprostredkovanému P-gp a BCRP v pečeni a črevách (pozri časť 5.2).
Induktoryenzýmovmetabolizujúcichliekyalebotransportérov
Očakáva sa, že súbežné podávanie PREVYMISU s induktormi môže viesť k zníženým plazmatickým koncentráciám letermoviru.
Pre perorálny letermovir bez cyklosporínu:
Súbežná liečba stredne silnými a silnými induktormi môže viesť k subterapeutickej expozícii letermoviru.
Pre perorálny letermovir s cyklosporínom alebo intravenózny letermovir s cyklosporínom alebo bez neho:
Silné induktory môžu viesť k subterapeutickej expozícii letermoviru.
- Príkladmi silných induktorov sú rifampicín, fenytoín, karbamazepín, ľubovník bodkovaný
(Hypericum perforatum), rifabutín a fenobarbital.
- Príkladmi stredne silných induktorov sú tioridazín, modafinil, ritonavir, lopinavir, efavirenz a etravirín.
Ďalší vplyvinýchliekovnaletermovirrelevantnýprikombinovaníscyklosporínom
Inhibítory OATP1B1 alebo 3
Súbežné podávanie PREVYMISU s liekmi, ktoré inhibujú transportéry OATP1B1/3, môže viesť k zvýšeným plazmatickým koncentráciám letermoviru. Ak sa PREVYMIS podáva súbežne
s cyklosporínom (silný inhibítor OATP1B1/3), odporúčaná dávka PREVYMISU je 240 mg jedenkrát
denne (pozri tabuľku 1 a časti 4.2 a 5.2). Odporúča sa opatrnosť, ak sa ďalšie inhibítory OATP1B1/3
pridávajú k letermoviru kombinovanému s cyklosporínom.
- Príkladmi inhibítorov OATP1B1 sú rifampicín, gemfibrozil, erytromycín, klaritromycín a viaceré inhibítory proteázy (atazanavir, lopinavir, ritonavir, simeprevir).
Inhibítory P-gp/BCRP
Výsledky in vitro naznačujú, že letermovir je substrátom P-gp/BCRP. Neočakáva sa, že zmeny plazmatických koncentrácií letermoviru z dôvodu inhibície P-gp/BCRP budú klinicky významné.
Odporúča sa však opatrnosť, ak sa inhibítory P-gp/BCRP pridávajú k letermoviru kombinovanému
s cyklosporínom.
- Príkladmi inhibítorov P-gp/BCRP sú klaritromycín, erytromycín, azitromycín, itrakonazol, ketokonazol, verapamil, chinidín, fluvoxamín, ranolazín a niektoré inhibítory HIV-proteázy.
Vplyv letermovirunainélieky
Lieky eliminované hlavne prostredníctvom metabolizmu alebo ovplyvnené aktívnym transportom
Letermovir je všeobecný induktor enzýmov a transportérov in vivo. Pokiaľ určitý enzým alebo transportér nie je inhibovaný (pozri nižšie), možno očakávať indukciu. Letermovir môže preto viesť
k zníženej plazmatickej expozícii a môže znížiť účinnosť tých súbežne podávaných liekov, ktoré sú
eliminované hlavne prostredníctvom metabolizmu alebo aktívnym transportom.
Veľkosť indukčného účinku je závislá od cesty podávania letermoviru a od toho, či sa súbežne používa aj cyklosporín. Plný indukčný účinok sa očakáva 10-14 dní po začatí liečby letermovirom. Čas potrebný na dosiahnutie rovnovážneho stavu určitého ovplyvneného lieku bude taktiež ovplyvňovať čas potrebný na dosiahnutie plného účinku na plazmatické koncentrácie.
Letermovir je inhibítor CYP3A, CYP2C8, CYP2B6, BCRP, UGT1A1, OATP2B1 a OAT3 in vitro v koncentráciách významných in vivo. Štúdie in vivo sú dostupné a vyhodnocujú čistý účinok na CYP3A4, P-gp, OATP1B1/3 a ďalej na CYP2C19. Čistý účinok in vivo na ďalšie uvedené enzýmy a transportéry nie je známy. Podrobné informácie sú uvedené nižšie.
Nie je známe, či letermovir môže ovplyvniť expozíciu piperacilínu/tazobaktámu, amfotericínu B a mikafungínu. Možná interakcia medzi letermovirom a týmito liekmi sa neskúmala. Existuje teoretické riziko zníženej expozície kvôli indukcii, ale veľkosť účinku a tým ani klinický význam nie sú v súčasnosti známe.
Lieky metabolizované CYP3A
Letermovir je stredne silný inhibítor CYP3A in vivo. Súbežné podávanie PREVYMISU s perorálnym midazolamom (substrát CYP3A) vedie k 2- až 3-násobne zvýšeným plazmatickým koncentráciám midazolamu. Súbežné podávanie PREVYMISU môže viesť ku klinicky významným zvýšeniam plazmatických koncentrácií súbežne podávaných substrátov CYP3A (pozri časti 4.3, 4.4 a 5.2).
- Príkladmi takýchto liekov sú niektoré imunosupresíva (napr. cyklosporín, takrolimus, sirolimus), inhibítory HMG-CoA reduktázy a amiodarón (pozri tabuľku 1). Pimozid a námeľové alkaloidy sú kontraindikované (pozri časť 4.3).
Veľkosť inhibičného účinku na CYP3A je závislá od cesty podávania letermoviru a od toho, či sa súbežne používa aj cyklosporín.
V dôsledku časovo závislej inhibície a simultánnej indukcie nie je možné dosiahnuť čistý inhibičný účinok na enzýmy skôr, ako po 10-14 dňoch. Čas potrebný na dosiahnutie rovnovážneho stavu určitého ovplyvneného lieku bude taktiež ovplyvňovať čas potrebný na dosiahnutie plného účinku na plazmatické koncentrácie. Po ukončení liečby trvá 10-14 dní kým inhibičný účinok vymizne. Ak sa použije sledovanie hladín, tak sa to odporúča počas prvých 2 týždňov po začatí a ukončení liečby letermovirom (pozri časť 4.4) a takisto aj po zmene cesty podávania letermoviru.
Lieky transportované OATP1B1/3
Letermovir je inhibítor transportérov OATP1B1/3. Podávanie PREVYMISU môže viesť ku klinicky významnému zvýšeniu plazmatických koncentrácií súbežne podávaných liekov, ktoré sú substrátmi OATP1B1/3.
- Príkladmi takýchto liekov sú inhibítory HMG-CoA reduktázy, fexofenadín, repaglinid a glyburid
(pozri tabuľku 1). Pri porovnávaní režimu letermoviru podávaného bez cyklosporínu je účinok výraznejší po i.v. ako po perorálnom letermovire.
Rozsah inhibície OATP1B1/3 na súbežne podávané lieky je pravdepodobne väčší, ak sa PREVYMIS
podáva súbežne s cyklosporínom (silný inhibítor OATP1B1/3). To sa má vziať do úvahy, ak sa režim letermoviru zmení počas liečby substrátom OATP1B1/3.
Lieky metabolizované CYP2C9 a/alebo CYP2C19
Súbežné podávanie PREVYMISU s vorikonazolom (substrát CYP2C19) vedie k významne zníženým plazmatickým koncentráciám vorikonazolu, čo naznačuje, že letermovir je induktor CYP2C19.
CYP2C9 je pravdepodobne tiež indukovaný. Letermovir potenciálne znižuje expozíciu substrátom
CYP2C9 a/alebo CYP2C19, čo môže viesť k subterapeutickým hladinám.
- Príkladmi takýchto liekov sú warfarín, fenytoín, vorikonazol, diazepam, lanzoprazol, omeprazol, esomeprazol, pantoprazol, tilidín, tolbutamid (pozri tabuľku 1).
Očakáva sa, že účinok bude menej výrazný pri perorálnom letermovire bez cyklosporínu ako pri i.v. letermovire s cyklosporínom alebo bez neho alebo pri perorálnom letermovire s cyklosporínom. To sa má vziať do úvahy, ak sa režim letermoviru mení počas liečby substrátom CYP2C9 alebo CYP2C19.
Ohľadom časového priebehu interakcie pozri tiež všeobecné informácie o indukcii vyššie.
Lieky metabolizované CYP2C8
Letermovir inhibuje CYP2C8 in vitro, ale na základe svojho indukčného potenciálu môže CYP2C8 aj indukovať. Čistý účinok in vivo nie je známy.
- Príkladom lieku, ktorý je eliminovaný hlavne prostredníctvom CYP2C8, je repaglinid (pozri tabuľku
1). Súbežné používanie repaglinidu a letermoviru s cyklosporínom alebo bez neho sa neodporúča.
Lieky transportované P-gp v črevách
Letermovir je induktor intestinálneho P-gp. Podávanie PREVYMISU môže viesť ku klinicky významnému zníženiu plazmatických koncentrácií súbežne podávaných liekov, ktoré sú vo významnej
miere transportované P-gp v črevách, ako sú dabigatran a sofosbuvir.
Lieky metabolizované CYP2B6, UGT1A1 alebo transportované BCRP alebo OATP2B1
Letermovir je všeobecný induktor in vivo, ale taktiež sa pozorovalo, že inhibuje CYP2B6, UGT1A1, BCRP a OATP2B1 in vitro. Čistý účinok in vivo nie je známy. Plazmatické koncentrácie liekov, ktoré sú substrátmi týchto enzýmov alebo transportérov, sa preto môžu zvýšiť alebo znížiť, ak sa podávajú
súbežne s letermovirom. Odporúča sa ďalšie sledovanie, pozri súhrn charakteristických vlastností takýchto liekov.
- Príkladmi liekov metabolizovaných CYP2B6 sú bupropión a efavirenz.
- Príkladmi liekov metabolizovaných UGT1A1 sú raltegravir a dolutegravir.
- Príkladmi liekov transportovaných BCRP sú rosuvastatín a sulfasalazín.
- Príkladom lieku transportovaného OATP2B1 je celiprolol.
Lieky transportované renálnym transportérom OAT3
Údaje in vitro naznačujú, že letermovir je inhibítor OAT3; letermovir môže preto inhibovať OAT3 in vivo. Plazmatické koncentrácie liekov transportovaných OAT3 sa môžu zvýšiť.
- Príkladmi liekov transportovaných OAT3 sú ciprofloxacín, tenofovir, imipeném a cilastatín.
Všeobecnéinformácie
Ak sa úpravy dávky súbežne podávaných liekov vykonajú z dôvodu liečby PREVYMISOM, dávky sa majú opätovne upraviť po dokončení liečby PREVYMISOM. Úprava dávky môže byť potrebná aj pri zmene cesty podávania alebo pri zmene imunosupresíva.
V tabuľke 1 je uvedený zoznam potvrdených alebo potenciálne klinicky významných liekových interakcií. Uvedené liekové interakcie sú založené na štúdiách vykonaných s PREVYMISOM alebo sú predpokladanými liekovými interakciami, ktoré sa môžu objaviť pri PREVYMISE (pozri časti 4.3,
4.4, 5.1 a 5.2).
Tabuľka 1: Interakcie a odporúčania na dávky pri podávaní s inými liekmi. Všimnite si, že tabuľka nie je rozsiahla, ale uvádza príklady klinicky významných interakcií. Taktiež pozrite všeobecný text vyššie ohľadom liekových interakcií.
Pokiaľ to nie je inak uvedené, interakčné štúdie boli uskutočnené s perorálnym letermovirom bez cyklosporínu. Prosím, všimnite si, že interakčný potenciál a klinické následky sa môžu líšiť v závislosti od toho, či sa letermovir podáva perorálne alebo i.v. a či sa súbežne používa cyklosporín. Pri zmene cesty podávania alebo ak sa mení imunosupresívum, sa má opakovane prezrieť odporúčanie ohľadom súbežného podávania.
Súbežne podávaný liek
Antimykotiká
Vplyv na koncentráciu†
Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
flukonazol Interakcia sa neskúmala.
Očakáva sa:
↔ flukonazolu
↔ letermoviru
Nevyžaduje sa žiadna úprava dávky.


posakonazol‡
(300 mg jednorazová dávka)/ letermovir (480 mg denne) vorikonazol‡
(200 mg dvakrát denne)/ letermovir (480 mg denne)
Antivirotikáaciklovir‡
(400 mg jednorazová dávka)/ letermovir
(480 mg denne)
↔ posakonazolu
AUC 0,98 (0,82; 1,17) Cmax 1,11 (0,95; 1,29)
↓ vorikonazolu
AUC 0,56 (0,51; 0,62) Cmax 0,61 (0,53; 0,71)
(indukcia CYP2C9/19)
↔ acikloviru
AUC 1,02 (0,87; 1,2) Cmax 0,82 (0,71; 0,93)
Nevyžaduje sa žiadna úprava dávky.
Ak je súbežné podávanie nevyhnutné, počas prvých 2 týždňov po začatí alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru alebo imunosupresíva sa odporúča TDM pre vorikonazol.
Nevyžaduje sa žiadna úprava dávky.
Súbežne podávaný liek
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
valaciklovir Interakcia sa neskúmala.
Očakáva sa:
↔ valacikloviru
Inhibítory HMG-CoA reduktázy
Nevyžaduje sa žiadna úprava dávky.
atorvastatín‡
(20 mg jednorazová dávka)/ letermovir (480 mg denne)
simvastatín, pitavastatín, rosuvastatín
fluvastatín, pravastatín
↑ atorvastatínu
AUC 3,29 (2,84; 3,82) Cmax 2,17 (1,76; 2,67)
(inhibícia CYP3A, OATP1B1/3)
Interakcia sa neskúmala. Očakáva sa:
↑ koncentrácií inhibítorov
HMG-CoA reduktázy
(inhibícia CYP3A, OATP1B1/3)
Interakcia sa neskúmala. Očakáva sa:
↑ koncentrácií inhibítorov
HMG-CoA reduktázy
(inhibícia OATP1B1/3
a/alebo BCRP)
Nežiaduce udalosti súvisiace so statínmi ako napr. myopatia sa majú pozorne sledovať. Dávka atorvastatínu nemá presiahnuť 20 mg denne, ak sa podáva súbežne
s PREVYMISOM#.
Hoci sa to neskúmalo, očakáva sa, že ak sa
PREVYMIS podáva súbežne
s
cyklosporínom, rozsah zvýšenia plazmatických koncentrácií atorvastatínu bude väčší ako so samotným PREVYMISOM. Ak
sa PREVYMIS podáva súbežne s cyklosporínom, atorvastatín je
kontraindikovaný.
Letermovir môže podstatne zvýšiť plazmatické koncentrácie týchto statínov. Súbežné používanie so samotným PREVYMISOM sa neodporúča.
Ak sa PREVYMIS podáva súbežne
s cyklosporínom, používanie týchto statínov je kontraindikované.
Letermovir môže zvýšiť plazmatické koncentrácie statínov.
Ak sa PREVYMIS podáva súbežne s týmito statínmi, môže byť potrebné zníženie dávky statínu#. Nežiaduce udalosti súvisiace so statínmi ako napr. myopatia sa majú pozorne sledovať.
Ak sa PREVYMIS podáva súbežne
s cyklosporínom, pravastatín sa neodporúča, kým u fluvastatínu môže byť potrebné zníženie dávky#. Nežiaduce udalosti súvisiace so statínmi ako napr. myopatia sa majú pozorne sledovať.
Súbežne podávaný liek
Imunosupresíva
cyklosporín
(50 mg jednorazová dávka)/ letermovir
(240 mg denne)
cyklosporín
(200 mg jednorazová dávka)/ letermovir
(240 mg denne)
mofetilmykofenolát (1 g jednorazová dávka)/ letermovir (480 mg denne)
sirolimus‡
(2 mg jednorazová dávka)/ letermovir
(480 mg denne)
takrolimus
(5 mg jednorazová dávka)/ letermovir (480 mg denne) takrolimus
(5 mg jednorazová dávka)/ letermovir (80 mg dvakrát
denne)
Vplyv na koncentráciu†
Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)↑ cyklosporínu
AUC 1,66 (1,51; 1,82) Cmax 1,08 (0,97; 1,19) (inhibícia CYP3A)
↑ letermoviru
AUC 2,11 (1,97; 2,26) Cmax 1,48 (1,33; 1,65)
(inhibícia OATP1B1/3)
↔ kyseliny mykofenolovej
AUC 1,08 (0,97; 1,20) Cmax 0,96 (0,82; 1,12)
↔ letermoviru
AUC 1,18 (1,04; 1,32) Cmax 1,11 (0,93; 1,34)
↑ sirolimu
AUC 3,40 (3,01; 3,85) Cmax 2,76 (2,48; 3,06)
(inhibícia CYP3A) Interakcia sa neskúmala.
Očakáva sa:
↔ letermoviru
↑ takrolimu
AUC 2,42 (2,04; 2,88) Cmax 1,57 (1,32; 1,86) (inhibícia CYP3A)
↔ letermoviru
AUC 1,02 (0,97; 1,07) Cmax 0,92 (0,84; 1,00)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOMAk sa PREVYMIS podáva súbežne
s cyklosporínom, dávkovanie PREVYMISU
sa má znížiť na 240 mg jedenkrát denne (pozri časti 4.2 a 5.1).
Počas liečby PREVYMISOM, pri zmene cesty podávania PREVYMISU a pri jeho vysadení je potrebné vykonávať časté sledovanie koncentrácií cyklosporínu v celej krvi a dávka cyklosporínu sa má náležite upraviť#.
Nevyžaduje sa žiadna úprava dávky.
Počas liečby PREVYMISOM, pri zmene cesty podávania PREVYMISU a pri jeho vysadení je potrebné vykonávať časté sledovanie koncentrácií sirolimu v celej krvi a dávka sirolimu sa má náležite upraviť#.
Pri začatí a vysadení súbežného podávania cyklosporínu s PREVYMISOM sa odporúča časté sledovanie koncentrácií sirolimu.
Ak sa PREVYMIS podáva súbežne
s cyklosporínom, pre špecifické odporúčania na dávkovanie pri používaní sirolimu
s cyklosporínom pozri tiež súhrn
charakteristických vlastností sirolimu.
Ak sa PREVYMIS podáva súbežne s
cyklosporínom, rozsah zvýšenia koncentrácií sirolimu môže byť väčší ako so samotným PREVYMISOM.
Počas liečby PREVYMISOM, pri zmene cesty podávania PREVYMISU a pri jeho vysadení je potrebné vykonávať časté sledovanie koncentrácií takrolimu v celej krvi a dávka takrolimu sa má náležite upraviť#.
Súbežne podávaný liek
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
Perorálne kontraceptíva
etinylestradiol (EE) (0,03 mg)/ levonorgestrel (LNG)‡
(0,15 mg) jednorazová dávka/ letermovir (480 mg denne)
Iné perorálne kontraceptívne steroidy so systémovým účinkom
Antidiabetiká
↔ EE
AUC 1,42 (1,32; 1,52) Cmax 0,89 (0,83; 0,96)
↔ LNG
AUC 1,36 (1,30; 1,43) Cmax 0,95 (0,86; 1,04)
Riziko ↓ koncentrácií kontraceptívnych steroidov
Nevyžaduje sa žiadna úprava dávky.
Letermovir môže znížiť plazmatické koncentrácie iných perorálnych kontraceptívnych steroidov a ovplyvniť tým ich účinnosť.
Na zabezpečenie adekvátneho kontraceptívneho účinku perorálnym
kontraceptívom sa majú zvoliť lieky s obsahom EE a LNG.
repaglinid Interakcia sa neskúmala.
Očakáva sa:
↑ alebo ↓ koncentrácií repaglinidu
(indukcia CYP2C8, inhibícia CYP2C8
a OATP1B)
glyburid Interakcia sa neskúmala.
Očakáva sa:
↑ koncentrácií glyburidu
(inhibícia OATP1B1/3, inhibícia CYP3A, indukcia CYP2C9)
Letermovir môže zvýšiť alebo znížiť plazmatické koncentrácie repaglinidu. (Čistý účinok nie je známy).
Súbežné používanie sa neodporúča. Očakáva sa, že ak sa PREVYMIS podáva
súbežne s
cyklosporínom, plazmatické
koncentrácie repaglinidu sa zvýšia v dôsledku ďalšej inhibície OATP1B cyklosporínom. Súbežné používanie sa neodporúča#. Letermovir môže zvýšiť plazmatické koncentrácie glyburidu.
Počas prvých 2 týždňov po začatí alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru sa odporúča časté sledovanie koncentrácií glukózy.
Ak sa PREVYMIS podáva súbežne
s
cyklosporínom, pre špecifické odporúčania na dávkovanie pozri tiež súhrn
charakteristických vlastností glyburidu.
'
Súbežne podávaný liek
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
Antiepileptiká (pozri tiež všeobecný text)
fenytoín Interakcia sa neskúmala.
Očakáva sa:
↓ koncentrácií fenytoínu
(indukcia CYP2C9/19)
↓ koncentrácií letermoviru
Perorálne antikoagulanciá
warfarín Interakcia sa neskúmala.
Očakáva sa:
↓ koncentrácií warfarínu
(indukcia CYP2C9)
dabigatran Interakcia sa neskúmala.
Očakáva sa:
↓ koncentrácií dabigatranu
(indukcia intestinálneho P- gp)
Sedatíva
Letermovir môže znížiť plazmatické koncentrácie fenytoínu.
Ak sa fenytoín podáva súbežne s liečbou PREVYMISOM, je potrebné vykonávať časté sledovanie koncentrácií fenytoínu. Počas prvých 2 týždňov po začatí alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru alebo imunosupresíva sa odporúča TDM.
Zníženie koncentrácií letermoviru môže viesť k strate účinnosti.
Letermovir môže znížiť plazmatické koncentrácie warfarínu.
Ak sa warfarín podáva súbežne s liečbou PREVYMISOM, je potrebné vykonávať časté sledovanie medzinárodného normalizovaného pomeru (International Normalised Ratio, INR)#. Počas prvých 2 týždňov po začatí
alebo ukončení liečby letermovirom a takisto po zmene cesty podávania letermoviru alebo imunosupresíva sa odporúča sledovanie. Letermovir môže znížiť plazmatické koncentrácie dabigatranu a môže znížiť účinnosť dabigatranu. Súbežnému používaniu dabigatranu je potrebné sa vyhnúť kvôli riziku zníženej účinnosti dabigatranu.
Ak sa PREVYMIS podáva súbežne s cyklosporínom, dabigatran je kontraindikovaný.
midazolam
(1 mg jednorazová dávka i.v.)/
letermovir (240 mg
jedenkrát denne p.o.)
midazolam
(2 mg jednorazová dávka p.o.)/ letermovir (240 mg jedenkrát denne p.o.)
↑ midazolamu i.v.:
AUC 1,47 (1,37; 1,58) Cmax 1,05 (0,94; 1,17)
p.o.:
AUC 2,25 (2,04; 2,49) Cmax 1,72 (1,54; 1,92)
(inhibícia CYP3A)
Počas súbežného podávania PREVYMISU s midazolamom je potrebné vykonávať pozorné klinické sledovanie pre útlm dýchania a/alebo predĺženú sedáciu. Má sa zvážiť úprava dávky midazolamu#. Zvýšenie plazmatickej koncentrácie midazolamu môže
byť väčšie, ak sa perorálny midazolam podáva s letermovirom v klinickej dávke oproti skúmanej dávke.
Súbežne podávaný liek
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
Agonisty opioidných receptorov
Príklady: alfentanil, fentanyl
Antiarytmiká
Interakcia sa neskúmala. Očakáva sa:
↑ koncentrácií opiátov metabolizovaných CYP3A
(inhibícia CYP3A)
Počas súbežného podávania sa odporúča časté sledovanie pre nežiaduce reakcie súvisiace
s týmito liekmi. Môže byť potrebná úprava dávky opiátov metabolizovaných CYP3A# (pozri časť 4.4). Sledovanie sa odporúča aj pri zmene cesty podávania. Ak sa PREVYMIS podáva súbežne s cyklosporínom, rozsah zvýšenia plazmatických koncentrácií opiátov metabolizovaných CYP3A môže byť väčší. Počas súbežného podávania PREVYMISU
v kombinácii s cyklosporínom a alfentanilom alebo fentanylom je potrebné vykonávať
pozorné klinické sledovanie pre útlm dýchania a/alebo predĺženú sedáciu. Pozri
príslušný súhrn charakteristických vlastností
(pozri časť 4.4).
amiodarón Interakcia sa neskúmala.
Očakáva sa:
↑ koncentrácií amiodarónu
(primárne inhibícia CYP3A a inhibícia alebo indukcia CYP2C8)
chinidín Interakcia sa neskúmala.
Očakáva sa:
↑ koncentrácií chinidínu
(inhibícia CYP3A)
Lieky na kardiovaskulárny systém
Letermovir môže zvýšiť plazmatické koncentrácie amiodarónu.
Počas súbežného podávania sa odporúča časté sledovanie pre nežiaduce reakcie súvisiace
s amiodarónom. Ak sa amiodarón podáva
súbežne s PREVYMISOM, je potrebné vykonávať pravidelné sledovanie koncentrácií amiodarónu#.
Letermovir môže zvýšiť plazmatické koncentrácie chinidínu.
Počas podávania PREVYMISU s chinidínom je potrebné vykonávať pozorné klinické sledovanie. Pozri príslušný súhrn charakteristických vlastností#.
digoxín‡
(0,5 mg jednorazová dávka)/ letermovir
(240 mg dvakrát denne)
↔ digoxínu
AUC 0,88 (0,80; 0,96) Cmax 0,75 (0,63; 0,89)
(indukcia P-gp)
Nevyžaduje sa žiadna úprava dávky.
Inhibítory protónovej pumpy
omeprazol Interakcia sa neskúmala.
Očakáva sa:
↓ omeprazolu
(indukcia CYP2C19) Interakcia sa neskúmala.
Očakáva sa:
↔ letermoviru
Letermovir môže znížiť plazmatické koncentrácie substrátov CYP2C19.
Môže byť potrebné klinické sledovanie a úprava dávky.
Súbežne podávaný liek
Vplyv na koncentráciu† Priemerný pomer (90% interval spoľahlivosti) pre AUC, Cmax (pravdepodobný mechanizmus účinku)
Odporúčania týkajúce sa súbežného podávania s PREVYMISOM
pantoprazol Interakcia sa neskúmala.
Očakáva sa:
↓ pantoprazolu
(pravdepodobne v dôsledku indukcie CYP2C19)
Letermovir môže znížiť plazmatické koncentrácie substrátov CYP2C19.
Môže byť potrebné klinické sledovanie a úprava dávky.

Interakcia sa neskúmala.
Očakáva sa:
↔ letermoviru
*Táto tabuľka neobsahuje všetky informácie.
† ↓ = pokles, ↑ = zvýšenie
↔ =žiadna klinicky významná zmena
‡ Štúdia jednocestnej interakcie hodnotiaca vplyv letermoviru na súbežne podávaný liek.
# Pozri príslušný súhrn charakteristických vlastností.
PediatrickápopuláciaInterakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktáciaGraviditaK dispozícii nie sú žiadne údaje týkajúce sa použitia letermoviru u gravidných žien. Štúdie na
zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
PREVYMIS sa neodporúča počas gravidity a u žien, ktoré môžu otehotnieť a nepoužívajú antikoncepciu.
DojčenieNie je známe, či sa letermovir vylučuje do ľudského mlieka.
Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie letermoviru do mlieka (pozri časť 5.3).
Riziko u novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu PREVYMISOM sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
FertilitaU potkanov sa neobjavili žiadne účinky na fertilitu samíc. Ireverzibilná testikulárna toxicita a porucha
fertility sa pozorovala u samcov potkana, ale nepozorovala sa u samcov myší ani samcov opíc.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
PREVYMIS môže mať malý vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje. U niektorých pacientov bola počas liečby PREVYMISOM hlásená únava a vertigo, ktoré môžu ovplyvniť schopnosť pacienta viesť vozidlá a obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluHodnotenie bezpečnosti PREVYMISU bolo založené na klinickom skúšaní fázy 3 (P001) u príjemcov
HSCT, ktorí dostávali PREVYMIS alebo placebo počas 14 týždňov po transplantácii a ktorí sa z dôvodu bezpečnosti sledovali až do konca 24. týždňa po transplantácii (pozri časť 5.1).
Najčastejšie hlásenými nežiaducimi reakciami objavujúcimi sa u minimálne 1 % osôb v skupine
s PREVYMISOM a vo vyššej frekvencii ako pri placebe boli nevoľnosť (7,2 %), hnačka (2,4 %)
a vracanie (1,9 %).
Najčastejšie hlásenými nežiaducimi reakciami, ktoré viedli k vysadeniu PREVYMISU, boli nevoľnosť
(1,6 %), vracanie (0,8 %) a bolesť brucha (0,5 %).
Tabuľkový súhrnnežiaducichreakciíNasledujúce nežiaduce reakcie sa zistili u pacientov užívajúcich PREVYMIS v klinických skúšaniach.
Nežiaduce reakcie sú uvedené nižšie podľa triedy orgánových systémov a frekvencie. Frekvencie sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) alebo veľmi zriedkavé (< 1/10 000).
Tabuľka 2: Nežiaduce reakcie zistené pri PREVYMISEFrekvencia Nežiaduce reakciePoruchy imunitného systémumenej časté hypersenzitivita
Poruchy metabolizmu a výživymenej časté znížená chuť do jedla
Poruchy nervového systémumenej časté dysgeúzia, bolesť hlavy
Poruchy ucha a labyrintumenej časté vertigo
Poruchy gastrointestinálneho traktučasté nevoľnosť, hnačka, vracanie menej časté bolesť brucha
Poruchy pečene a žlčových ciestmenej časté zvýšená alanínaminotransferáza, zvýšená aspartátaminotransferáza
Poruchy kostrovej a svalovej sústavy a spojivového tkanivamenej časté svalové spazmy
Poruchy obličiek a močových ciestmenej časté zvýšený kreatinín v krvi
Celkové poruchy a reakcie v mieste podaniamenej časté únava, periférny edém
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Neexistuje žiadna skúsenosť s predávkovaním PREVYMISOM u ľudí. Počas klinických skúšaní fázy
1 dostávalo 86 zdravých osôb dávky pohybujúce sa v rozmedzí od 720 mg/deň do 1 440 mg/deň
PREVYMISU počas až 14 dní. Profil nežiaducich reakcií bol podobný ako pri klinickej dávke
480 mg/deň. Pri predávkovaní PREVYMISOM neexistuje žiadne špecifické antidotum. V prípade predávkovania sa odporúča sledovanie pacienta pre nežiaduce reakcie a začatie vhodnej
symptomatickej liečby.
Nie je známe, či dialýza bude viesť k významnému odstráneniu PREVYMISU zo systémovej cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antivirotiká na systémové použitie, priamo pôsobiace antivirotiká. ATC
kód: J05AX18
Mechanizmus účinku
Letermovir inhibuje komplex DNA terminázy CMV, ktorý je potrebný na štiepenie a zbalenie
vírusovej progénovej DNA. Letermovir ovplyvňuje tvorbu genómov so správnou dĺžkou jednotiek a narúša dozrievanie viriónov.
Antivírusová aktivita
Stredná hodnota EC50 letermoviru proti vzorke klinických izolátov CMV v bunkovej kultúre modelu
infekcie bola 2,1 nmol/l (rozmedzie = 0,7 nmol/l až 6,1 nmol/l, n=74).
Vírusová rezistencia
V bunkovej kultúre
Gény UL56 a UL89 CMV kódujú podjednotky DNA terminázy CMV. V bunkovej kultúre boli potvrdené zmutované vírusy CMV so zníženou citlivosťou na letermovir. Mutácie sú lokalizované
v UL56 a vyskytujú sa na aminokyselinových zvyškoch 231 až 369 (V231A, V231L, V236L, V236M, E237D, L241P, T244K, T244R, L257I, F261C, F261L, F261S, Y321C, C325F, C325R, C325W, C325Y, M329T, R369G, R369M, R369S). Hodnoty EC50 pre tieto mutácie sú 13- až 8 262-násobne vyššie ako hodnoty pre referenčný divoký typ vírusu. Žiadne známe mutácie spôsobujúce rezistenciu voči letermoviru nie sú lokalizované v UL89.
V klinických štúdiách
V skúšaní fázy 2b hodnotiacom letermovir v dávkach 60, 120 alebo 240 mg/deň alebo placebo až do
84 dní u 131 príjemcov HSCT sa na vzorkách získaných od 12 osôb liečených letermovirom,
u ktorých došlo k zlyhaniu profylaxie a u ktorých boli vzorky dostupné na analýzu, vykonala analýza
DNA sekvencie zvolenej oblasti UL56 (aminokyseliny 231 až 369). U jednej osoby (ktorá dostávala
60 mg/deň) sa objavil genotypový variant (GV) rezistentný voči letermoviru (V236M).
V skúšaní fázy 3 (P001) sa na vzorkách získaných od 22 osôb liečených letermovirom v populácii FAS, u ktorých došlo k zlyhaniu profylaxie a u ktorých boli vzorky dostupné na analýzu, vykonala analýza DNA sekvencie celých kódujúcich oblastí UL56 a UL89. U jednej osoby sa objavil GV rezistentný voči letermoviru (V236M).
Skrížená rezistencia
Skrížená rezistencia je nepravdepodobná s liekmi s rozdielnym mechanizmom účinku. Letermovir je
plne účinný proti populáciám vírusov so substitúciami spôsobujúcimi rezistenciu voči inhibítorom DNA polymerázy CMV (ganciklovir, cidofovir a foskarnet). Tieto inhibítory DNA polymerázy sú plne účinné proti populáciám vírusov so substitúciami spôsobujúcimi rezistenciu voči letermoviru.
Elektrofyziológia srdca
Vplyv letermoviru v dávkach až do 960 mg podávaných i.v. na QTc interval sa hodnotil
v randomizovanom, placebom a aktívne kontrolovanom (moxifloxacín 400 mg perorálne) dôkladnom skúšaní QT s jednorazovou dávkou a 4 obdobiami prekríženia u 33 zdravých osôb. Letermovir po i.v. dávke 960 mg nepredlžuje QTc interval v klinicky významnom rozsahu s plazmatickými koncentráciami približne 2-násobne vyššími ako pri i.v. dávke 480 mg.
Klinická účinnosťabezpečnosť
Dospelí CMV-séropozitívni príjemcovia [R+] alogénneho štepu krvotvorných kmeňových buniek
Na vyhodnotenie profylaxie letermovirom ako preventívnej metódy pri infekcii alebo ochorení vyvolanom CMV sa účinnosť letermoviru hodnotila v multicentrickom, dvojito zaslepenom, placebom kontrolovanom skúšaní fázy 3 (P001) u dospelých CMV-séropozitívnych príjemcov [R+] alogénneho HSCT. Osoby boli randomizované (2:1) na užívanie buď letermoviru v dávke 480 mg jedenkrát denne upravenej na 240 mg, ak sa podávala súbežne s cyklosporínom, alebo na placebo. Randomizácia bola stratifikovaná skúšajúcim pracoviskom a rizikom (vysoké oproti nízkemu) reaktivácie CMV v čase vstupu do štúdie. Letermovir sa začal podávať po HSCT (0. – 28. deň po transplantácii) a v podávaní
sa pokračovalo do konca 14. týždňa po transplantácii. Letermovir sa podával buď perorálne alebo i.v.; dávka letermoviru bola rovnaká bez ohľadu na cestu podávania. Osoby boli sledované do konca 24. týždňa po transplantácii pre primárny cieľový ukazovateľ účinnosti s pokračovaním sledovania do konca 48. týždňa po transplantácii.
Osoby boli sledované pre DNA CMV týždenne až do konca 14. týždňa a následne každé dva týždne až do konca 24. týždňa po transplantácii so začatím štandardnej preemptívnej terapie CMV, ak sa hladina DNA CMV v krvi považovala za klinicky významnú. V sledovaní osôb sa pokračovalo do konca 48. týždňa po transplantácii.
Spomedzi 565 liečených osôb 373 osôb dostávalo letermovir (vrátane 99 osôb, ktoré dostali minimálne jednu i.v. dávku) a 192 dostávalo placebo (vrátane 48 osôb, ktorí dostali minimálne jednu i.v. dávku). Medián času do začatia podávania letermoviru bol 9 dní po transplantácii. Tridsaťsedem percent (37 %) osôb malo prijatý štep na začiatku. Medián veku bol 54 rokov (rozmedzie: 18 až 78 rokov); 56 (15 %) osôb bolo vo veku 65 rokov a starších; 58 % bolo mužov; 82 % bolo bielej; 10 % bolo ázijskej; 2 % boli čiernej alebo africkej a 7 % bolo hispánskej alebo latinskoamerickej rasy. Na začiatku 50 % osôb dostávalo myeloablatívny režim, 52 % dostávalo cyklosporín a 42 % dostávalo takrolimus. Najčastejšími primárnymi dôvodmi pre transplantáciu boli akútna myeloidná leukémia (38 %), myeloblastický syndróm (15 %) a lymfóm (13 %). Dvanásť percent (12 %) osôb bolo na začiatku pozitívnych na DNA CMV.
Na začiatku malo 31 % osôb vysoké riziko reaktivácie na základe definície jedným alebo viacerými z nasledujúcich kritérií: darca s príbuzným ľudským leukocytovým antigénom (HLA) (súrodenec)
s minimálne jednou nezhodou na jednom z nasledujúcich troch lókusov na géne HLA: HLA-A, -B
alebo –DR, haploidentický darca; nepríbuzný darca s minimálne jednou nezhodou na jednom
z nasledujúcich štyroch lókusov na géne HLA: HLA-A, -B, -C a -DRB1; použitie pupočníkovej krvi ako zdroja kmeňových buniek; použitie štepov zbavených T-buniek ex vivo; reakcia štepu proti príjemcovi (Graft-Versus-Host Disease, GVHD) 2. alebo vyššieho stupňa vyžadujúca systémové kortikosteroidy.
Primárny cieľový ukazovateľ účinnosti
Primárny cieľový ukazovateľ účinnosti klinicky významnej infekcie CMV v štúdii P001 sa definoval ako výskyt DNA CMV v krvi vyžadujúci preemptívnu terapiu proti CMV (pre-emptive therapy, PET) alebo výskyt ochorenia CMV v koncovom orgáne. Použila sa metóda pacient, ktorý nedokončil štúdiu
= zlyhanie (Non-Completer=Failure, NC=F), kde osoby, ktoré prerušili štúdiu pred 24. týždňom po transplantácii alebo u nich nebol dostupný výsledok v 24. týždni po transplantácii, sa započítali ako
zlyhania.
Pri letermovire sa preukázala vyššia účinnosť oproti placebu v analýze primárneho cieľového ukazovateľa, ako je uvedené v tabuľke 3. Odhadovaný rozdiel liečby -23,5 % bol štatisticky významný (jednostranná p-hodnota < 0,0001).
Tabuľka 3: P001: Výsledky účinnosti u príjemcov HSCT (metóda NC = F, populácia FAS).
letermovir placebo
(N = 325) (N = 170) Parameter n (%) n (%)
Primárny cieľový ukazovateľ účinnosti
(podiel pacientov so zlyhaním profylaxie počas 24
týždňov)
Dôvody zlyhania†
122 (37,5) 103 (60,6)
Klinicky významná infekcia CMV 57 (17,5) 71 (41,8) DNA CMV v krvi vyžadujúca PET proti CMV 52 (16,0) 68 (40,0) Ochorenie CMV v koncovom orgáne 5 (1,5) 3 (1,8)
Prerušenie štúdie 56 (17,2) 27 (15,9) Chýbajúci výsledok 9 (2,8) 5 (2,9)
Rozdiel liečby upravený na skupinu (letermovir- placebo)§
Rozdiel (95% IS) -23,5 (-32,5; -14,6)

p-hodnota < 0,0001
† Kategórie zlyhania sa vzájomne vylučujú a sú založené na hierarchii kategórií v uvedenom poradí.
§ 95% IS a p-hodnota pre rozdiely liečby v percentuálnom vyjadrení odpovede sa vypočítali pomocou
Mantelovej-Haenszelovej metódy upravenej na skupiny s rozdielom porovnávaným pomocou harmonického priemeru veľkosti vzorky v ramene pre každú skupinu (vysoké alebo nízke riziko). Na vyjadrenie štatistickej významnosti sa použila jednostranná p-hodnota ≤ 0,0249.
FAS = celý analyzovaný súbor (full analysis set); FAS zahŕňa randomizované osoby, ktoré dostali minimálne jednu dávku skúšaného lieku a vylučuje osoby s detegovateľnou DNA CMV na začiatku.
Metóda na spracovanie chýbajúcich hodnôt: metóda NC = F (pacient, ktorý nedokončil štúdiu =
zlyhanie). Pri metóde NC=F sa zlyhanie definovalo ako všetky osoby s klinicky významnou infekciou
CMV alebo ktoré predčasne vystúpili zo štúdie alebo u nich nebol dostupný výsledok v návštevnom okne v 24. týždni po transplantácii.
N = počet osôb v každej liečebnej skupine.
n (%) = počet (percento) osôb v každej podkategórii.
Poznámka: Podiel osôb s detegovateľnou vírusovou DNA CMV v deň 1, u ktorých sa rozvinula klinicky významná infekcia CMV počas 24 týždňov po transplantácii bol v skupine s letermovirom
64,6 % (31/48) v porovnaní s 90,9 % (20/22) v skupine s placebom. Odhadovaný rozdiel (95% IS pre
rozdiel) bol -26,1 % (-45,9 %, -6,3 %) s nominálnou jednostrannou p-hodnotou < 0,0048.
Faktory súvisiace s výskytom DNA CMV v krvi po 14. týždni po transplantácii medzi osobami
liečenými letermovirom zahŕňali vysoké riziko pre reaktiváciu CMV na začiatku, GVHD, používanie kortikosteroidov a sérum donora negatívne na prítomnosť CMV.
Obrázok 1: P001: Kaplanova-Meierova krivka času do začatia PET proti CMV alebo do vzniku ochorenia CMV v koncovom orgáne počas 24 týždňov po transplantácii u príjemcov HSCT (populácia FAS)
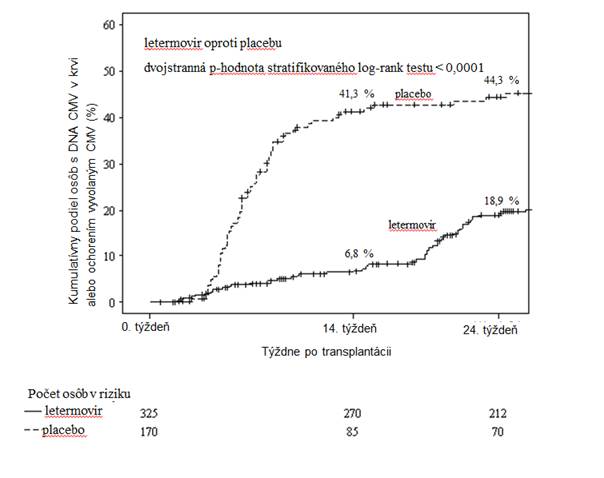
Neobjavili sa žiadne rozdiely vo výskyte prijatia štepu alebo v čase do jeho prijatia medzi skupinou s PREVYMISOM a skupinou s placebom.
Účinnosť bola naprieč podskupinami konzistentne v prospech letermoviru vrátane nízkeho a vysokého rizika pre reaktiváciu CMV, prípravných režimov a súbežných režimov obsahujúcich imunosupresíva (pozri obrázok 2).
Obrázok 2: P001: Forestova krivka podielu osôb, u ktorých sa začala PET proti CMV alebo s ochorením CMV v koncovom orgáne počas 24 týždňov po transplantácii podľa vybraných podskupín (metóda NC=F, populácia FAS)

Celkovo (N=325, 170)
Skupiny podľa rizika


Vysoké riziko (n=102, 45) Nízke riziko (n=223, 125)
Pôvod kmeňových buniek

Periférna krv (n=241, 117)

Kostná dreň (n=72, 43)
Nezhoda s darcom
Zhodný príbuzný (n=108, 58)


Nezhodný príbuzný (n=52, 18) Zhodný nepríbuzný (n=122, 70)
Nezhodný nepríbuzný (n=43, 24)
Haploidentický darca

Áno (n=49, 17) Nie (n=276, 153)
Prípravný režim

Myeloablatívny (n=154, 85)



Príprava so zníženou intenzitou (n=86, 48) Nemyeloablatívny (n=85, 37)

Immunosupresívny režim cyklosporín A (n=162, 90) takrolimus (n=145, 69)
-70 -60 -50 -40 -30 -20 -10 0 10 20
 V prospech letermoviru
V prospech placeba
V prospech letermoviru
V prospech placeba
Rozdiel medzi letermovirom - placebom (%) a 95% IS
NC=F, pacient, ktorý nedokončil štúdiu = zlyhanie (Non-Completer=Failure). Pri metóde NC=F sa osoby, ktoré prerušili
štúdiu pred 24. týždňom po transplantácii alebo u ktorých nebol dostupný výsledok v 24. týždni po transplantácii, započítali ako zlyhania.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s PREVYMISOM
v jednej alebo vo viacerých podskupinách pediatrickej populácie pre profylaxiu infekcie cytomegalovírusom (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika letermoviru bola opísaná po perorálnom a i.v. podaní u zdravých osôb a príjemcov HSCT. Expozícia letermoviru sa zvyšovala viac ako proporcionálne dávke pri perorálnom aj i.v. podaní. Pravdepodobný mechanizmus je saturácia/autoinhibícia OATP1B1/3.
U zdravých osôb bol geometrický priemer AUC v rovnovážnom stave 71 500 ng•hod/ml
a geometrický priemer Cmax v rovnovážnom stave bol 13 000 ng/ml pri letermovire podávanom perorálne v dávke 480 mg jedenkrát denne.
Letermovir dosiahol rovnovážny stav za 9 až 10 dní s akumulačným pomerom 1,2 pre AUC a 1,0 pre
Cmax.
U príjemcov HSCT bola AUC letermoviru odhadnutá pomocou analýz populačnej farmakokinetiky
s použitím údajov z fázy 3 (pozri tabuľku 4). Rozdiely v expozícii medzi liečebnými režimami nie sú klinicky významné; účinnosť bola v rámci rozsahu expozícií pozorovaných v štúdii P001 konzistentná.
Tabuľka 4: Hodnoty AUC letermoviru (ng•hod/ml) u príjemcov HSCT Liečebný režim Medián (90% predikčný interval)*480 mg perorálne, bez cyklosporínu 34 400 (16 900, 73 700)
480 mg i.v., bez cyklosporínu 100 000 (65 300, 148 000)
240 mg perorálne, s cyklosporínom 60 800 (28 700, 122 000)
240 mg i.v., s cyklosporínom 70 300 (46 200, 106 000)
* Post-hoc predpovede populácie z analýzy populačnej FK s použitím údajov z fázy 3
AbsorpciaLetermovir sa rýchlo vstrebával s mediánom času do maximálnej plazmatickej koncentrácie (Tmax) 45
minút až 2,25 hodín a koncentrácie klesali bifázicky. U príjemcov HSCT bola biologická dostupnosť
letermoviru odhadnutá na približne 35 % pri perorálnom letermovire v dávke 480 mg jedenkrát denne podávanej bez cyklosporínu. Interindividuálna variabilita pre biologickú dostupnosť bola odhadnutá na približne 37 %.
Vplyv cyklosporínuU príjemcov HSCT spôsobilo súbežné podávanie cyklosporínu zvýšenie plazmatických koncentrácií letermoviru v dôsledku inhibície OATP1B. Biologická dostupnosť letermoviru bola odhadnutá na približne 85 % pri letermovire v dávke 240 mg jedenkrát denne perorálne podávanom pacientom súbežne s cyklosporínom.
Ak sa letermovir podáva súbežne s cyklosporínom, odporúčaná dávka letermoviru je 240 mg jedenkrát denne (pozri časť 4.2).
Vplyv potravyU zdravých osôb perorálne podanie 480 mg jednorazovej dávky letermoviru so štandardným jedlom s vysokým obsahom tukov a kalórií nemalo žiadny vplyv na celkovú expozíciu (AUC) a viedlo
k približne 30 % zvýšeniu maximálnych hladín (Cmax) letermoviru. Letermovir sa môže podávať perorálne s jedlom alebo bez jedla tak, ako sa podával aj v klinických štúdiách (pozri časť 4.2).
DistribúciaNa základe analýz populačnej farmakokinetiky sa priemerný distribučný objem v rovnovážnom stave
odhadol na 45,5 l po intravenóznom podaní u príjemcov HSCT.
Letermovir sa v rozsiahlej miere (98,2 %) viaže na ľudské plazmatické bielkoviny nezávisle od rozmedzia koncentrácií (3 až 100 mg/l) hodnoteného
in vitro. Určitá saturácia sa pozorovala pri nižších koncentráciách. Pomer rozdelenia letermoviru medzi krv a plazmu je 0,56 a je nezávislý od rozmedzia koncentrácií (0,1 až 10 mg/l) hodnoteného
in vitro.
V predklinických štúdiách distribúcie sa letermovir distribuoval do orgánov a tkanív s najvyššími koncentráciami pozorovanými v gastrointestinálnom trakte, žlčovode a pečeni a s nízkymi koncentráciami v mozgu.
Biotransformácia
Väčšia časť zložiek súvisiacich s letermovirom v plazme je vo forme nezmenenej materskej látky
(96,6 %). V plazme sa nezistili žiadne významné metabolity. Letermovir sa čiastočne eliminuje prostredníctvom glukuronidácie sprostredkovanej UGT1A1/1A3.
Eliminácia
Priemerný zdanlivý terminálny polčas letermoviru u zdravých osôb je približne 12 hodín pri i.v.
letermovire v dávke 480 mg. Hlavné cesty eliminácie letermoviru sú biliárna exkrécia a takisto aj priama glukuronidácia. Tento proces zahŕňa transportéry pečeňového vychytávania OATP1B1 a 3
nasledované glukuronidáciou katalyzovanou UGT1A1/3.
Na základe analýz populačnej farmakokinetiky sa zdanlivý klírens letermoviru v rovnovážnom stave odhaduje na 4,84 l/hod po intravenóznom podaní 480 mg u príjemcov HSCT. Interindividuálna variabilita pre klírens sa odhaduje na 24,6 %.
Vylučovanie
Po perorálnom podaní rádioaktívne značeného letermoviru sa 93,3 % izotopom značeného liečiva
vylúčilo stolicou. Väčšia časť letermoviru sa vylúčila žlčou vo forme nezmenenej materskej látky
s malým podielom (6 % dávky) vo forme acylglukuronidového derivátu v stolici. Acylglukuronid je v stolici nestabilný. Vylučovanie letermoviru močom bolo zanedbateľné (< 2 % dávky).
Farmakokinetika vosobitnýchskupináchpacientov
Porucha funkcie pečene
AUC neviazaného letermoviru bola približne o 81 % vyššia u osôb so stredne ťažkou (Childova- Pughova trieda B [CP-B], skóre 7 – 9) a 4-násobne vyššia u osôb s ťažkou (Childova-Pughova trieda C [CP-C], skóre 10 – 15) poruchou funkcie pečene v porovnaní so zdravými osobami. Zmeny expozície letermoviru u osôb so stredne ťažkou poruchou funkcie pečene nie sú klinicky významné.
Výrazné zvýšenia expozície neviazanému letermoviru sa očakávajú u pacientov so stredne ťažkou poruchou funkcie pečene v kombinácii so stredne ťažkou alebo ťažkou poruchou funkcie obličiek (pozri časť 4.2).
Porucha funkcie obličiek
AUC neviazaného letermoviru bola približne o 115 % vyššia u osôb so stredne ťažkou (eGFR 31,0 až
56,8 ml/min/1,73 m2) a o 81 % vyššia u osôb s ťažkou (eGFR 11,9 až 28,1 ml/min/1,73 m2) poruchou funkcie obličiek v porovnaní so zdravými osobami. Zmeny expozície letermoviru z dôvodu stredne
ťažkej alebo ťažkej poruchy funkcie obličiek sa nepovažujú za klinicky významné. Osoby s ESRD sa neskúmali.
Telesná hmotnosť
Na základe analýz populačnej farmakokinetiky sa odhaduje, že AUC letermoviru je o 18,7 % nižšia
u osôb s telesnou hmotnosťou 80 – 100 kg v porovnaní s osobami s telesnou hmotnosťou 67 kg. Tento rozdiel nie je klinicky významný.
Rasa
Na základe analýz populačnej farmakokinetiky sa odhaduje, že AUC letermoviru je o 33,2 % vyššia u príslušníkov ázijskej rasy v porovnaní s príslušníkmi bielej rasy. Táto zmena nie je klinicky
významná.
Pohlavie
Na základe analýz populačnej farmakokinetiky neexistuje žiadny rozdiel vo farmakokinetike letermoviru u žien v porovnaní s mužmi.
Staršie osoby
Na základe analýz populačnej farmakokinetiky neexistuje žiadny vplyv veku na farmakokinetiku letermoviru. Na základe veku sa nevyžaduje žiadna úprava dávky.
5.3 Predklinické údaje o bezpečnosti
Všeobecná toxicita
Ireverzibilná testikulárna toxicita sa zaznamenala len u potkanov pri systémových expozíciách (AUC)
≥ 3-násobku expozícií u ľudí pri odporúčanej dávke pre ľudí (recommended human dose, RHD). Táto toxicita bola charakterizovaná degeneráciou tubulov v semenníkoch a oligospermiou a zvyškami
buniek v nadsemenníkoch s poklesom hmotnosti semenníkov a nadsemenníkov. Pri expozíciách (AUC) podobných expozíciám u ľudí pri RHD sa u potkanov nepozorovala testikulárna toxicita. Testikulárna toxicita sa nepozorovala pri najvyšších dávkach skúmaných pri expozíciách u myší až do
4-násobku a u opíc až do 2-násobku expozícií u ľudí pri RHD. Význam pre ľudí nie je známy.
Je známe, že hydroxypropylbetadex môže vyvolať vakuoláciu obličiek u potkanov, ak sa podáva intravenózne v dávkach väčších ako 50 mg/kg/deň. Vakuolácia sa zaznamenala v obličkách potkanov, ktorým sa letermovir podával intravenózne v dávke 1 500 mg/kg/deň vo forme s cyklodextrínovou pomocnou látkou hydroxypropylbetadexom.
Karcinogenita
Štúdie karcinogenity sa s letermovirom nevykonali.
Mutagenita
Letermovir nebol genotoxický v sérii testov in vitro alebo in vivo vrátane testov mikrobiálnej
mutagenézy, chromozomálnej aberácie na ovariálnych bunkách čínskeho škrečka a v štúdii myšacieho mikrojadierka in vivo.
Reprodukcia
Fertilita
V štúdiách fertility a skorého embryonálneho vývinu u potkanov sa neobjavili žiadne účinky letermoviru na fertilitu samíc. U samcov potkana sa pozorovala znížená koncentrácia spermií, znížená pohyblivosť spermií a pokles fertility pri systémových expozíciách ≥ 3-násobku AUC u ľudí pri RHD (pozri Všeobecná toxicita).
U opíc, ktorým sa podával letermovir, sa neobjavil žiadny dôkaz testikulárnej toxicity na základe histopatologického hodnotenia, stanovenia veľkosti semenníkov, rozboru hormónov v krvi (folikulostimulačný hormón, inhibín B a testosterón) a hodnotení spermií (počet spermií, pohyblivosť a morfológia) pri systémových expozíciách približne 2-násobku AUC u ľudí pri RHD.
Vývin
U potkanov sa zaznamenala maternotoxicita (vrátane zníženého prírastku telesnej hmotnosti) pri
dávke 250 mg/kg/deň (približne 11-násobok AUC pri RHD); u mláďat sa zaznamenal pokles hmotnosti plodu s oneskorenou osifikáciou, mierne opuchnutými plodmi a zvýšeným výskytom skrátených pupočníkových šnúr a zmeny a malformácie stavcov, rebier a panvy. Pri dávke
50 mg/kg/deň (približne 2,5-násobok AUC pri RHD) sa nezaznamenali žiadne účinky na matku a vývin.
U králikov sa zaznamenala maternotoxicita (vrátane mortality a potratov) pri dávke 225 mg/kg/deň (približne 2-násobok AUC pri RHD); u mláďat sa zaznamenal zvýšený výskyt malformácií a zmien stavcov a rebier.
V štúdii pre- a postnatálneho vývinu sa letermovir perorálne podával gravidným potkanom. Nepozorovala sa žiadna vývinová toxicita až do najvyššej skúmanej expozície (2-násobok AUC pri RHD).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
hydroxypropylbetadex (cyklodextrín)
chlorid sodný
hydroxid sodný (E524)
voda na injekcie
6.2 Inkompatibility
Inkompatibilné liečivá
PREVYMIS koncentrát na infúzny roztok je fyzikálne inkompatibilný s amiodaróniumchloridom,
amfotericínom B (lipozomálnym), aztreonamom, cefepímiumchloridom, ciprofloxacínom, cyklosporínom, diltiazemiumchloridom, filgrastimom, gentamicíniumsulfátom, levofloxacínom,
linezolidom, lorazepamom, midazolamiumchloridom, mofetilmykofenolát hydrochloridom,
ondansetrónom, palonosetrónom.
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6. Inkompatibilnémateriályintravenóznych vakovainfúznychsúprav
PREVYMIS koncentrát na infúzny roztok je inkompatibilný so súpravou hadičiek na i.v. podávanie
obsahujúcich polyuretán.
Tento liek sa nesmie používať s inými materiálmi intravenóznych vakov a infúznych súprav okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená liekovka: 30 mesiacov
Po otvorení: Použite okamžite
Uchovávanie zriedenéhoroztoku
Chemická a fyzikálna stabilita po zriedení pred použitím bola preukázaná počas 48 hodín pri 25 °C
a počas 48 hodín pri 2 °C až 8 °C.
Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa nepoužije okamžite, za čas
a podmienky uchovávania po zriedení pred použitím zodpovedá používateľ a zvyčajne by nemali presiahnuť 24 hodín pri 2 °C až 8 °C, pokiaľ riedenie neprebehlo v kontrolovaných a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie. Uchovávajte v pôvodnej škatuľke na ochranu pred svetlom.
Podmienky uchovávania po zriedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Liekovka z číreho skla typu I (30 ml) s 20 mm fluórom pokrytou chlorobutylovou zátkou s hliníkovým odklápacím viečkom obsahujúca 12 ml (slabozelené viečko) alebo 24 ml roztoku (tmavomodré
viečko).
Veľkosť balenia: 1 liekovka.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčné liekovky PREVYMIS sú určené len na jednorazové použitie. Príprava
Pokyny na prípravu a podávanie sú pre obidve dávky rovnaké.
PREVYMIS koncentrát na infúzny roztok sa musí pred intravenóznym použitím zriediť.
Pred zriedením skontrolujte obsah injekčnej liekovky kvôli zmene zafarbenia a prítomnosti pevných častíc. PREVYMIS koncentrát na infúzny roztok je číry bezfarebný roztok. Nepoužívajte injekčnú liekovku, ak je zafarbenie roztoku zmenené alebo obsahuje viditeľné častice.
Injekčnou liekovkou PREVYMISU netraste.
Obsah jednej injekčnej liekovky (buď 12 ml (240 mg dávka) alebo 24 ml (480 mg dávka)) PREVYMISU koncentrát na infúzny roztok na jednorazové použitie pridajte k 250 ml naplneného intravenózneho vaku obsahujúceho buď 0,9% chlorid sodný alebo 5% glukózu a zriedený roztok premiešajte jemným obrátením. Netraste ním.
Po zriedení je roztok PREVYMISU číry a je bezfarebný až žltý. Zmeny farby v rámci tohto rozsahu nemajú vplyv na kvalitu lieku. Vždy keď to roztok a obal umožňuje, zriedený roztok sa má pred podaním vizuálne skontrolovať pre prítomnosť pevných častíc a zmeny zafarbenia. Ak spozorujete zmenu zafarbenia alebo prítomnosť pevných častíc, roztok zlikvidujte. Ak sa obsah injekčnej liekovky pridá do 250 ml intravenózneho vaku, výsledná koncentrácia letermoviru bude v rozsahu 0,9 mg/ml (pre 240 mg dávku) a 1,8 mg/ml (pre 480 mg dávku).
Podávanie
Pozri časť 4.2.
Kompatibilnéintravenózneroztokyainélieky
PREVYMIS koncentrát na infúzny roztok je kompatibilný s 0,9% roztokom chloridu sodného a 5%
roztokom glukózy.
PREVYMIS sa nesmie podávať súbežne cez rovnakú intravenóznu hadičku (alebo kanylu) s inými liekmi a kombináciami rozpúšťadiel okrem tých, ktoré sú uvedené nižšie.
Zoznam kompatibilných liekov, ak sa PREVYMIS a lieky* pripravia v 0,9% roztoku chloridu sodného
• sodná soľ ampicilínu • flukonazol
• sodná soľ ampicilínu/sodná soľ sulbaktámu • ľudský inzulín
• antitymocytový globulín • síran horečnatý
• kaspofungín • metotrexát
• daptomycín • mikafungín
• fentanylcitrát
*Pozri súhrn charakteristických vlastností na potvrdenie kompatibility simultánneho súbežného podávania.
Z
o
znam kompatibilných liekov, ak sa PREVYMIS a lieky* pripravia v 5% roztoku glukózy
• amfotericín B (lipidový komplex)† • sodná soľ hydrokortizónsukcinátu
• anidulafungín • morfíniumsulfát
• sodná soľ cefazolínu • noradrenalíniumbitartrát
• ceftarolín • sodná soľ pantoprazolu
• sodná soľ ceftriaxónu • chlorid draselný
• doripeném • fosforečnan draselný
• famotidín • takrolimus
• kyselina listová • telavancín
• sodná soľ gancikloviru • tigecyklín
*Pozri súhrn charakteristických vlastností na potvrdenie kompatibility simultánneho súbežného podávania.
†Amfotericín B (lipidový komplex) je kompatibilný s PREVYMISOM. Amfotericín B (lipozomálny) je však inkompatibilný (pozri časť 6.2).
Kompatibilnémateriályintravenóznych vakovainfúznych súpravPREVYMIS je kompatibilný s nasledujúcimi materiálmi intravenóznych vakov a infúznych súprav. Všetky materiály intravenóznych vakov a infúznych súprav, ktoré nie sú uvedené nižšie, sa nemajú používať.
Materiály intravenóznych vakovPolyvinylchlorid (PVC), etylénvinylacetát (EVA) a polyolefín (polypropylén a polyetylén).
Materiály infúznych súpravPVC, polyetylén (PE), polybutadién (PBD), silikónová guma (SR), styrén-butadiénový kopolymér
(SDC), styrén-butadién-styrénový kopolymér (SBS), polystyrén (PS)
ZmäkčovadláDietylhexylftalát (DEHP), tris-(2-etylhexyl)-trimelitát (TOTM), butylbenzylftalát (BBP)
KatétreRöntgenkontrastný polyuretán
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN11 9BU
Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/17/1245/003
EU/1/17/1245/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky agentúry
http://www.ema.europa.eu.