
ťažkou poruchou funkcie pečene (pozri časť 5.2).
Spôsob podávania
Liek POTELIGEO sa podáva intravenózne. Má sa podávať iba intravenóznou infúziou aspoň 60
minút. V prípade reakcie súvisiacej s infúziou pozri vyššie uvedené odporúčania.
Pokyny na riedenie lieku pred podaním pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Kožné reakcie
Pacienti, ktorí dostávali mogamulizumab, mali liekovú vyrážku (liekovú erupciu), z ktorých niektoré
boli ťažké a/alebo závažné.
Pri podávaní mogamulizumabu pacientom s T-bunkovými lymfómami inými ako MF a SS boli hlásené závažné kožné reakcie, vrátane Stevensovho-Johnsonovho syndrómu (SJS) a toxickej epidermálnej nekrolýzy (TEN), u menej ako 1 % pacientov počas klinických skúšaní, ale boli hlásené aj v postmarketingovom období, a niektoré z týchto prípadov mali fatálne následky. U pacientov sa majú podrobne sledovať príznaky a prejavy, ktoré poukazujú na SJS alebo TEN. Ak sa vyskytnú, používanie lieku POTELIGEO sa má prerušiť a liečba sa nemá znovu začať, pokým sa nevylúči SJS alebo TEN a kožná reakcia neustúpi na 1. stupeň alebo nižšie. Ak sa vyskytne SJS/TEN, má sa podať vhodná medikamentózna liečba. Informácie o úprave dávky pozri v časti 4.2.
R
eakcie súvisiace s infúziou
Akútne reakcie súvisiace s infúziou (IRR) sa zistili u pacientov liečených mogamulizumabom. IRR
boli väčšinou mierne alebo stredne závažné, hoci bolo hlásených aj niekoľko závažných reakcií
(3. stupeň). Väčšina IRR sa vyskytla počas alebo krátko po podaní prvej infúzie (všetky do 24 hodín po podaní) a výskyt sa znižoval počas ďalšej liečby.
Pacienti sa majú pozorne sledovať počas a po podaní infúzie. Ak sa objaví anafylaktická reakcia, podávanie mogamulizumabu sa má okamžite a natrvalo prerušiť a má sa podať vhodná medikamentózna liečba.
Ak sa vyskytne IRR, infúzia sa má prerušiť a začať vhodnú liečbu.
Po zmiznutí príznakov sa infúzia môže znovu spustiť, ale pomalšie. Pozri časť 4.2 s informáciami o
premedikácii a úprave dávkovania.
Infekcie
U osôb s MF alebo SS, ktorí sú liečení mogamulizumabom, je zvýšené riziko vzniku závažných infekcií a/alebo vírusovej reaktivácie. Kombinácia mogamulizumabu so systémovými liekmi modulujúcimi imunitu alebo inými licencovanými liečbami MF a SS sa neskúmala, a preto sa neodporúča najmä vzhľadom na riziko vzniku závažných infekcií u pacientov liečených mogamulizumabom. Topické steroidy alebo nízke dávky systémových kortikosteroidov sa smú užívať počas liečby mogamulizumabom, avšak riziko vzniku závažných infekcií a/alebo vírusovej reaktivácie môže byť vyššie v prípade súbežného podávania systémových imunosupresívnych látok. U pacientov sa majú sledovať prejavy a príznaky infekcií a majú sa okamžite liečiť.
Pred začatím liečby mogamulizumabom treba pacientom urobiť testy na hepatitídu B. Pacientom, ktorí budú mať pozitívny test na aktuálnu/predchádzajúcu infekciu vírusom hepatitídy B, sa odporúčajú konzultácia so skúseným lekárom na liečbu hepatitídy B, aby sa poradili o vhodných preventívnych opatreniach proti reaktivácii hepatitídy B.
Komplikáciealogénnejtransplantáciehematopoetickýchkmeňovýchbuniek(HSCT)popodanímogamulizumabu
Komplikácie vrátane ťažkej choroby graft versus host (GVHD) sa zistili u pacientov s T-bunkovými
lymfómami inými ako MF a SS, ktorí dostali po podaní mogamulizumabu alogénnu HSCT.
Vyššie riziko komplikácie pri traFsplantácii sa zistilo, keď sa mogamulizumab podal v krátkom časovom rámci (približne 50 dní) pred HSCT. Dôkladne sledujte u pacientov prvé dôkazy komplikácií súvisiacich s transplantáciou.
Bezpečnosť liečby mogamulizumabom po autológnej alebo alogénnej HSCT sa neskúmala. Syndróm rozpadu nádoru
U pacientov, ktorí dostávali mogamulizumab, sa zistil syndróm rozpadu nádoru (TLS). TLS bol
zistený najčastejšie počas prvého mesiaca liečby. Riziko vzniku TLS majú pacienti s rýchlou proliferáciou nádoru a vysokým nádorovým zaťažením. Pacienti sa majú dôkladne sledovať prostredníctvom príslušných laboratórnych a klinických testov stavu elektrolytov, hydratácie a funkcie obličiek najmä v prvom mesiaci liečby a liečiť podľa najlepšej lekárskej praxe. Manažment TLS môže zahŕňať agresívnu hydratáciu, úpravu abnormalít elektrolytov, antihyperurikemickú liečbu a podpornú starostlivosť.
Poruchy srdca
V klinickom skúšaní sa zistil jeden prípad akútneho infarktu myokardu u pacienta s MF/SS, ktorý dostával mogamulizumab. V klinických skúšaniach sa u pacientov s inými T-bunkovými lymfómami
zistila stresová kardiomyopatia (jeden prípad) a akútny infarkt myokardu (jeden prípad). Účastníci
mali anamnézu s rôznymi rizikovými faktormi. Pacienti, ktorí majú rizikové faktory súvisiace s chorobami srdca, sa majú sledovať a treba prijať vhodné preventívne opatrenia.
V
eľkobunková
tr
ansformácia
(
L
C
T
)
Existuje iba obmedzené množstvo údajov o pacientoch s LCT.
Ďalšie
Mogamulizumab sa nepodáva subkutánne alebo intramuskulárne, rýchlym intravenóznym podaním ani ako intravenózny bolus.
Tento liek obsahuje menej ako 1 mmol sodíka v jednej dávke, t. j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia u mužov a u žien
Ženy vo fertilnom veku a muži v reproduktívnom veku musia používať účinnú antikoncepciu počas liečby liekom POTELIGEO až po dobu 6 mesiacov po liečbe.
Gravidita
Nie sú k dispozícii žiadne údaje o použití mogamulizumabu u gravidných žien. Hoci mogamulizumab prechádza cez placentárnu bariéru u makaka dlhochvostého, okrem farmakologických účinkov na
plody, štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej
toxicity (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu mogamulizumabu počas gravidity.
Dojčenie
Nie je známe, či sa mogamulizumab vylučuje do ľudského mlieka. Je známe, že ľudské imunoglobulíny IgG sa vylučujú do materského mlieka počas prvých dní po pôrode, koncentrácie
ktorých sa čoskoro znižujú, takže sa nedá vylúčiť riziko u dieťaťa počas tohto krátkeho obdobia.
Neskôr sa liek POTELIGEO môže používať počas dojčenia, ak je to klinicky nutné.
Fertilita
Nie sú dostupné žiadne klinické údaje o vplyve mogamulizumabu na ľudskú plodnosť. Neboli
vykonané žiadne špeciálne štúdie na zvieratách, aby sa posúdil vplyv mogamulizumabu na plodnosť. Neboli zistené žiadne nežiaduce účinky na mužské a ženské pohlavné orgány v skúšaniach toxicity po opakovanej dávke u makakov dlhochvostých (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Mogamulizumab má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní mogamulizumabu sa môže vyskytnúť únava (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrnprofilubezpečnosti
Najčastejšie hlásené závažné nežiaduce reakcie boli zápal pľúc, horúčka, reakcie súvisiace s infúziou a celulitída.
Najčastejšie hlásené nežiaduce reakcie boli reakcie súvisiace s infúziou a vyrážky (lieková erupcia);
väčšina týchto reakcií nebola závažná a bola 1. alebo 2. stupňa.
Ťažké nežiaduce reakcie 4. stupňa zahŕňali zlyhanie dýchania (1,1 %) a reakcie 5. stupňa boli
polyomyozitída a otrava krvi (každá 0,5 %).
Z
o
z
nam
nežiaducich
r
eakcií
z
oradených
do
tabuľky
Nežiaduce reakcie sú zoradené podľa triedy orgánových systémov a kategórií frekvencie, ktoré sú
definované nasledovne: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až
<1/100), zriedkavé (≥1/10 000 až <1/1 000) a veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov). V každej skupine frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
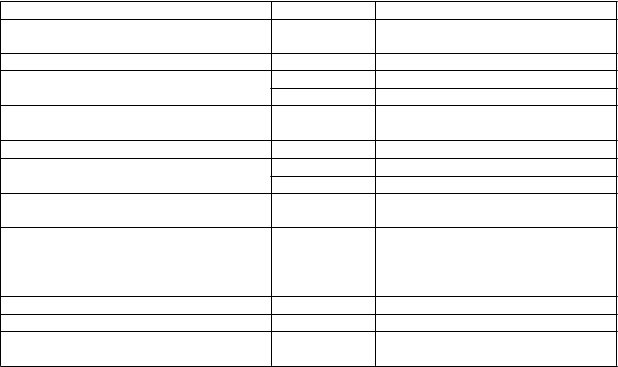 Tabuľka 1: Nežiaduce reakcie u pacientov, ktorí dostávali liek POTELIGEO (N = 184) Trieda orgánových systémov (SOC) Frekvencia Nežiaduci účinok
Tabuľka 1: Nežiaduce reakcie u pacientov, ktorí dostávali liek POTELIGEO (N = 184) Trieda orgánových systémov (SOC) Frekvencia Nežiaduci účinokPoruchy krvi a lymfatického systému Časté Anémia, neutropénia, leukopénia, trombocytopénia
Poruchy endokrinného systému Časté Hypotyroidizmus
Poruchy gastrointestinálneho traktu Veľmi časté Zápcha, hnačka, nevoľnosť, stomatitída
Časté Zvracanie
Celkové poruchy a reakcie v mieste podania Veľmi časté Únava, periférny edém, horúčka
Poruchy pečene a žlčových ciest Menej časté Akútna hepatitída, hepatitída
a
Infekcie a nákazy Veľmi časté Infekcie
Časté Infekcie horných dýchacích ciest
Úrazy, otravy a komplikácie liečebného
postupu Veľmi časté Reakcie súvisiace s infúziou
Alanínaminotransferáza zvýšená,
Laboratórne a funkčné vyšetrenia Časté
aspartátaminotransferáza zvýšená, alkalínfosfatáza v krvi zvýšená, počet lymfocytov znížený
Poruchy metabolizmu a výživy Menej časté Syndróm rozpadu nádoru
Poruchy nervového systému Veľmi časté Bolesť hlavy
Poruchy kože a podkožného tkaniva Veľmi časté Lieková erupcia (vrátane kožnej vyrážky)
a Folikulitída, celulitída, kandidóza, zápal pľúc, otrava krvi, infekcia kože, zápal vonkajšieho ucha, pásový opar,
stafylokoková infekcia kože, infekcie močových ciest, herpes a cytomegalovírus
Opis vybraných nežiaducich reakcií
Kožné reakcie
Pacienti, ktorí dostávali liek POTELIGEO, mali liekovú vyrážku (liekovú erupciu), z ktorých niektoré
boli ťažké a/alebo závažné. Väčšina kožných reakcií súvisiacich s liečbou bola 1. alebo 2. stupňa a so stupňom liekovej vyrážky ≥3, ktoré sa vyskytli u 4,3 % pacientov. Nebol určený žiadny trend pre oneskorený začiatok udalosti liekovej erupcie a vyrážok, vyskytli sa udalosti so skorým aj oneskoreným začiatkom.
Reakcie súvisiace s infúziou
Reakcie súvisiace s infúziou sa zistili u 33 % pacientov liečených liek POTELIGEO. Väčšina reakcií na liečbu súvisiacich s infúziou bola 1. alebo 2. stupňa a vyskytla sa počas alebo krátko po podaní
prvej infúzie. Závažné reakcie (3. stupeň) mali 4 % pacientov.
Výskyt reakcií súvisiacich s infúziou bol najvyšší po podaní prvej infúzie (28,8 % osôb) a znížil sa na
≤3,8 % osôb po dvoch a viac ako dvoch infúziách.
Prerušenie infúzií sa vyskytlo približne u 6 % pacientov, z ktorých sa väčšina (približne 90 %)
vyskytla počas prvého cyklu liečby mogamulizumabom.
Menej ako 1 % pacientov, ktorí boli liečení v Štúdii 0761-010, prerušilo liečbu v dôsledku reakcií
súvisiacich s infúziou.
Závažné infekcie
Pacienti s MF alebo SS majú zvýšené riziko vzniku závažných infekcií v dôsledku narušenia integrity kože spôsobenej kožným ochorením ako aj imunosupresívnych účinkov extrakutánnych ochorení, pričom liečba mogamulizumabom môže toto riziko zvýšiť. Závažné infekcie vrátane otravy krvi, zápalu pľúc a kožných infekcií malo 14,3 % osôb, ktorí dostávali mogamulizumab. Oneskorený začiatok udalosti po prvej dávke bol výrazne odlišný. Väčšina pacientov sa z infekcie zotavila.
V klinickom skúšaní (0761-010) boli hlásené 2 zlyhania dýchania s fatálnymi následkami u pacientov s ťažkým zápalom pľúc, ktorý sa vyskytol po viac ako 9 mesiacoch po začatí liečby mogamulizumabom.
ImunogenicitaTak ako pri všetkých terapeutických proteínoch, aj tu existuje možnosť imunogenicity. Malé percento
pacientov, ktorí dostávali liek POTELIGEO, malo pozitívne výsledky na liečbou spustené (liečbou vyvolané alebo liečbou povzbudené ) protilátky proti mogamulizumabu. Nezistili sa žiadne odpovede pozitívnych neutralizovaných protilátok.
Bezpečnosť po poslednej dávkeZ 320 osôb, ktorí boli v Štúdii 0761-010 vystavení mogamulizumabu, malo 21 (6,6 %) aspoň jednu závažnú nežiaducu liekovú reakciu (SADR), ktorá sa vyskytla počas 90 dní po poslednom podaní skúšaného lieku.
Z toho boli SADR, hlásené od viac ako jedného pacienta, označené ako infekcie a nákazy SOC
(7 [2,2 %] pacientov), Celkové poruchy a reakcie v mieste podania (5 [1,6 %] pacientov), Poruchy dýchacej sústavy, hrudníka a mediastína (4 [1,3 %] pacienti), Poruchy kostrovej a svalovej sústavy a spojivového tkaniva (3 [0,9 %] pacienti), Poruchy pečene a žlčových ciest (2 [0,6 %] pacienti) a Úrazy, otravy a komplikácie liečebného postupu (2 [0,6 %] pacienti). Pre všetky ostatné SOC bol SADR nahlásený u jedného pacienta (0,3 %).
Bezpečnostný profil zistený počas 90 dní po poslednom podaní mogamulizumabu zodpovedá bezpečnostnému profilu zistenému počas obdobia skúšania lieku.
Staršia populáciaBezpečnostný profil u starších pacientov (≥65 rokov) sa vo všeobecnosti zhodoval s dospelými pacientmi, okrem dermatologických reakcií a reakcií spojených s infúziou, ktoré boli častejšie spozorované u starších osôb.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
prílohe V.4.9 PredávkovanieNeexistuje žiadna informácia o predávkovaní mogamulizumabom. V prípade predávkovania sa má pacient dôkladne sledovať, vrátane vitálnych funkcií (aspoň 1 hodinu), a ak treba, má sa poskytnúť podporná starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastické a imunomodulačné látky, monoklonálne protilátky
ATC kód: L01XC25
MechanizmusúčinkuMogamulizumab je defukozylovaný humanizovaný imunoglobulín IgG kappa, ktorý výberovo viaže
CCR4, CC chemokínový receptor spriahnutý s G-proteínmi, potrebný na prenos lymfocytov do rôznych orgánov vrátane kože a vedie ku zníženiu počtu cieľových buniek. Receptor CCR4 sa nachádza na povrchu niektorých rakovinových buniek vrátane malignancií T-buniek ako MF a SS, v ktorých je inherentná expresia receptora CCR4.
Klinickáúčinnosťabezpečnosť
Účinnosť mogamulizumabu pri liečbe pacientov s mycosis fungoides (MF) alebo Sézaryho syndrómom (SS) bola stanovená v 3. fáze multicentrického otvoreného skúšania (0761-010) s 372
dospelými pacientmi randomizovanými 1:1, ktorí boli liečení buď mogamulizumabom, alebo
vorinostatom. V každej skupine bolo zapísaných 186 pacientov. Infúzia s mogamulizumabom bola
podaná v dávke 1 mg/kg raz za týždeň počas prvého 28-dňového cyklu (1., 8., 15. a 22. deň) a v 1. a
15. deň nasledujúcich 28-dňových cykloch. Vorinostat bol podaný v počiatočnej dávke 400 mg
perorálne raz za deň, v 1. deň 28-dňového cyklu. Pacienti s progresívnym ochorením alebo neprijateľnými toxicitami, ktorí užívali vorinostat, mohli prejsť na liečbu mogamulizumabom.
Prechádzajúci pacienti dostávali mogamulizumab až 46 mesiacov do zastavenia zberu údajov v
decembri 2016. Liečba mogamulizumabom pokračovala až do progresie ochorenia alebo neprijateľnej toxicity. Zo skúšania boli vylúčení pacienti s aktívnymi autoimunitnými chorobami, metastázami v centrálnom nervovom systéme a zdravotnými stavmi, ktoré vyžadujú užívanie systémových kortikosteroidov a iných imunosupresívnych liekov, alebo s infekciami, ktoré vyžadujú liečbu vrátane HIV alebo hepatitídy B a C. Pacienti so stavom ECOG ≥2 boli taktiež vylúčení. Na začiatku skúšania malo 38 % pacientov štádium ochorenia IB-II, 10 % štádium III, 52 % štádium IV. Toto skúšanie zahŕňa pacientov bez ohľadu na vstupnú hladinu expresie receptora CCR4 pri biopsii kože.
Primárnym ukazovateľom účinnosti bolo prežitie bez progresie ochorenia (PFS) založené na hodnotení skúšajúceho lekára podľa kritérií celkovej kompozitnej odpovede, ktorá brala do úvahy všetky možné kompartmenty postihnuté ochorením (koža, krv, lymfatické uzliny a vnútorné orgány). Odpoveď v koži a krvi sa hodnotila každé 4 týždne. Odpoveď v lymfatických uzlinách a vnútorných orgánoch sa hodnotila každé 4 týždne, potom 8 týždňov počas prvého roka a nakoniec každých 16 týždňov.
Všetci pacienti mali histologicky potvrdenú diagnózu mycosis fungoides (MF), 56,5 %, 53,2 %, alebo Sézaryho syndróm (SS), 43,5 %, 46,8 % v skupinách s mogamulizumabom a vorinostatom, v uvedenom poradí, a dostali predtým aspoň jednu systémovú liečbu. Najčastejšie užívanými
predošlými systémovými liečbami u pacientov v Európe sú bexarotén (70 %), interferón (59 %), metotrexát (49 %), extrakorporálna fotoferéza (ECP) (31 %) a gemcitabín/gemcitabínový režim
(28 %).
Medián dĺžky vystavenia mogamulizumabu bol 5,6 mesiacov (v rozsahu: <1 do 45,3 mesiacov), 56 % pacientov dostávalo mogamulizumab aspoň 6 cyklov a 25 % pacientov dostávalo mogamulizumab aspoň 12 cyklov.
Medián veku pacientov bol v čase skríningu 64 rokov (v rozsahu od 25 do 101 rokov), 49,5 % malo 65
rokov a viac, a 58,1 % boli muži.
Expresia receptora CCR4 sa hodnotila retrospektívne pri predliečebnej biopsii kože (formalínom fixovanej do parafínu vloženej) pomocou imunohistochémie. V skupine s mogamulizumabom boli vstupné hladiny expresie receptora CCR4 k dispozícii u 75 % pacientov (N=140), pričom receptor CCR4 sa zistil v ≥1 % lymfocytov u 100 % pacientov a u 134/140 (96 %) pacientov sa receptor CCR4 zistil v ≥10 % lymfocytov kože.
Z pacientov randomizovaných pre vorinostat prešlo na mogamulizumab počas skúšania 136 pacientov (73,1 %). Dôvody prechodu na mogamulizumab boli progresia ochorenia (109 pacientov) a intolerancia liečby (27 pacientov). Počet infúzií s mogamulizumabom podaných prechádzajúcim pacientom bol v rozsahu od 1 do 94 (až 46 mesiacov liečby) do zastavenia zberu údajov v decembri
2016.
Po 6, 12, 18 a 24 mesiacoch po začatí randomizovaného skúšania bolo percento osôb prežívajúcich
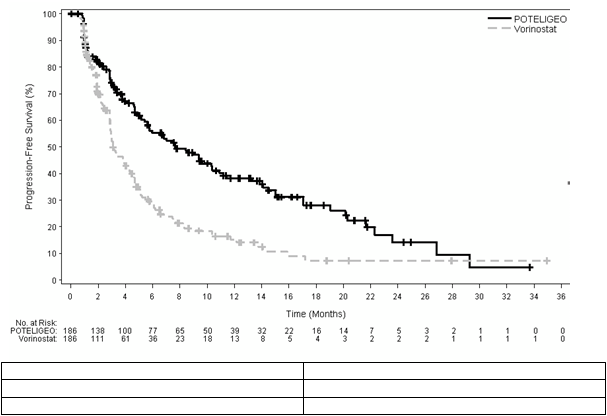
bez progresie ochorenia vyššie pri mogamulizumabe (v danom poradí 55,3 %, 38,3 %, 28,0 % a
14,1 % v uvedenom poradí) pri porovnaní s vorinostatom (v danom poradí 28,8 %, 15,3 %, 7,2 % a
7,2 % v uvedenom poradí). Medián PFS v skupine s mogamulizumabom bol 7,70 mesiacov (95 % CI:
5,67, 10,33) a 3,10 mesiacov (95 % CI: 2,87, 4,07) v skupine s vorinostatom s výslednou mierou rizika
0,53 (95 % CI: 0,41, 0,69), p<0,0001 (2-stranný stratifikovaný log rank test).
Kaplanovu-Meierovu krivku prežívania bez progresie ochorenia zobrazuje
Obrázok 1.
Obrázok 1: Zobrazenie Kaplanovej-Meierovej krivky prežívania bez progresie ochorenia, hodnotenie skúšajúcim lekárom, populácia (ITT)Progression-Free Survival Prežívanie bez progresie
No. at risk Počet ohrozených
Time (months) Čas (mesiace)
Kľúčovými sekundárnymi ukazovateľmi boli miera objektívnej odpovede (ORR), ORR po prechode,
trvanie odpovede (DOR) a zmeny od východiskovej hodnoty symptómov a funkčných škál Skindex-
29, funkčné zhodnotenie liečby rakoviny – všeobecné (FACT-G) fyzické a funkčné domény
celkového zdravia.
Celková objektívna odpoveď sa zaznamenala ako kompozitné skóre z hodnôt v každej časti, pričom na to, aby mohla byť uznaná, musela byť zistená v dvoch úspešných celkových hodnoteniach choroby (s rozostupom aspoň 8 týždňov v prvom roku a potom s rozostupom 16 týždňov). Pacienti boli zaradení do analýzy konkrétnych kompartmentov, ak mali pri vstupnom hodnotení ochorenie v danom kompartmente alebo ak mali akúkoľvek odpoveď v danom kompartmente po vstupnom hodnotení.
Tabuľka 2 sumarizuje ORR, DOR a odpoveď podľa kompartmentu. Skúšanie ukázalo štatisticky významné zlepšenia ORR a odpovede podľa kompartmentu v krvi, koži a lymfatických uzlinách pri porovnaní s vorinostatom. Odpoveď vo vnútorných orgánoch sa nedala vyhodnotiť vzhľadom na obmedzené údaje o účinnosti u osôb s ochorením vnútorných orgánov. Pomer prínosov a rizík mogamulizumabu u osôb s ochorením vnútorných orgánov nie je v súčasnosti stanovený z dôvodu nedostatku údajov.
T
abuľka 2: Odpoveď počas obdobia randomizovanej liečby v štúdii 0761-010 (s úmyslom liečiť)
M
oga
m
u
lizumab
N=186
Vorinostat
N=186
M
iera celkovej objektívnej odpovede
(
potvrdené CR + PR, %)
28,0 4,8
95 % CI (21,6, 35,0) (2,2, 9,0) P-hodnotaa <0,0001
Trvanie odpovede (mesiace)
Medián (95 % CI) 14,1 (9,4, 19,2) 9,13 (4,7, -)
Odpoveď kompartmentu
Krv n=124 n=125
Miera odpovede (potvrdené CR + PR, %) 66,9 18,4
95 % CI (57,9, 75,1) (12,0, 26,3) P-hodnotaa <0,0001
Koža n=186 n=186
Miera celkovej objektívnej odpovede (potvrdené
CR + PR, %)
41,9 15,6
95 % CI (34,8, 49,4) (10,7, 21,6) P-hodnotaa <0,0001
Lymfatické uzliny n=136 n=133
Miera celkovej objektívnej odpovede (potvrdené
CR + PR, %)
15,4 3,8
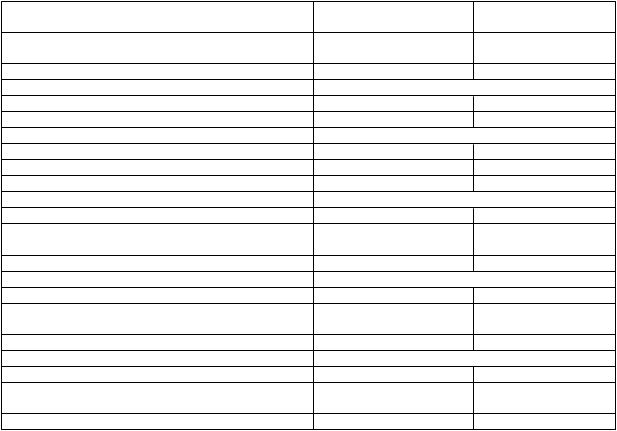
95 % CI (9,8, 22,6) (1,2, 8,6) P-hodnotaa 0,0008
Vnútorné orgány n=6 n=4
Miera celkovej objektívnej odpovede (potvrdené 0 0
CR + PR, %)
95 % CI (0,0, 45,9) (0,0, 60,2)
Poznámka: Miera objektívnej odpovede na základe Celkového skóre kompozitnej odpovede.
a: P-hodnota získaná z Cochran-Mantel-Haenszel testu prispôsobená na typ ochorenia, štádium a oblasť. CI = interval spoľahlivosti, CR = úplná odpoveď, PR = čiastočná odpoveď
Liečba mogamulizumabom priniesla 8 potvrdených úplných odpovedí (úplné očistenie všetkých postihnutých kompartmentov) v porovnaní s 0 pacientmi s vorinostatom: 4 z týchto 8 pacientov boli pôvodne randomizovaní na mogamulizumab a 4 počas skúšania na mogamulizumab prešli. Štyridsaťjeden zo 136 pacientov (30,1 %), ktorí prešli na mogamulizumab, malo buď čiastočnú, alebo úplnú odpoveď.
Existuje len obmedzené množstvo údajov o účinnosti u pacientov s nízkou expresiou (<10 %) receptora CCR4 v koži. V Štúdii 0761-010 bolo 10/290 vyhodnotiteľných pacientov s expresiou CCR4 (<10 %), z ktorých 6 bolo randomizovaných na mogamulizumab a 4 boli randomizovaní na vorinostat, ale neskôr prešli na mogamulizumab. U týchto 10 osôb s nízkou (<10 %) expresiou CCR4 sa nezistili žiadne potvrdené odpovede. Kompartmentové odpovede sa zistili u 3 z 10 vyhodnotiteľných osôb, ktorí boli v randomizovanej alebo prechodovej fáze liečení mogamulizumabom.
V porovnaní s 8,3 % pacientov liečených vorinostatom sa ORR potvrdila u 17,6 % pacientov so štádiom ochorenia IB/II ktorí boli liečení mogamulizumabom a Mieru odpovede hladiny kompartmentu (krv, koža, lymfatické uzliny) mali vyššiu ako pacienti liečení vorinostatom (Tabuľka 3). Celkovo medián obdobia prežitia bez progresie ochorenia v štádiu IB/II u osôb, ktorí'
boli liečení mogamulizumabom, bol 4,7 mesiacov v porovnaní s 3,9 mesiacmi u pacientov liečených vorinostatom (Tabuľka 4). Pre pacientov so štádiom choroby IB/II sa vzhľadom na dané obmedzené množstvo osôb s odpoveďou a nedostatočnosť údajov a príliš skoré údaje nedá urobiť žiadny záver
o trvaní odpovede.
Čas do odpovede hladiny kompartmentu u pacientov v štádiu IB/II bol približne 3 mesiace, čo zodpovedá času do odpovede u celkovej populácie ITT (približne 3 mesiace). Ak sa odpoveď hladiny kompartmentu alebo celková objektívna odpoveď nezistí do 3 mesiacov liečby, malo by sa zvážiť prerušenie liečby.
T
abuľka 3: Miera celkovej objektívnej odpovede a odpovede kompartmentu v počiatočných
št
ádiách ochorenia
M
oga
m
u
lizumab Vorinostat Rozdiel rizika (M vs. V) Štádium choroby IB/II N = 68 N = 72
M
iera celkovej objektívnej odpovede
(O
RR), n (%)
K
o
m
partment:
12 (17,6) 6 (8,3) 9,3
K
rv (n) 17 23
Miera odpovede (n, %) 8 (47,1) 4 (17,4) 29,7
95 % CIa (23,0, 72,2) (5,0, 38,8) (-2,2, 57,1)
Koža (n) 68 72
Miera odpovede (n, %) 19 (27,9) 14 (19,4) 8,5
95 % CIa (17,7, 40,1) (11,1, 38,8) (-8,3, 24,9)
Uzliny (n) 41 40
Miera odpovede (n, %) 4 (9,8) 1 (2,5) 7,3
95 % CIa (2,7, 23,1) (0,1, 13,2) (-14,3, 28,6)
M = mogamulizumab. V = vorinostat
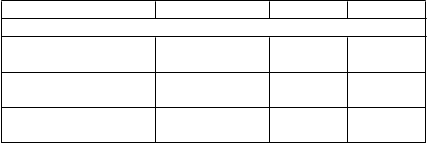
Tabuľka 4: Prežitie bez progresie ochorenia (PFS) podľa druhu liečby a štádia ochorenia
(obdobie randomizovanej liečby)
Mogamulizumab Vorinostat P hodnota
PF
S, mesiace
ITT Populácia 7,70 (5,67; 10,33) 3,10 (2,87;
4,07) IB/II 4,7 (2,9 – 7,47) 3,9 (2,87 –
4,73)
<0,0001
0,6790
III/IV 10,9 (7,03 –
15,03)
3,0 (2,83 –
3,87)
<0,0001
 ITT = s úmyslom liečiťPediatrická populácia
ITT = s úmyslom liečiťPediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom
POTELIGEO vo všetkých podskupinách pediatrickej populácie pre kutanózny T-bunkový lymfóm
(CTCL) (MF a SS sú podtypy CTCL). Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmakokinetické vlastnostiFarmakokinetika (FK) mogamulizumabu sa hodnotila u dospelých pacientov s T-bunkovou leukémiou/lymfómom (ATL) a CTCL v rozsahu dávky od 0,01 mg/kg do 1 mg/kg podanej ako viac dávok mogamulizumabu každý týždeň alebo každé 2 týždne a zahŕňal odporúčanú dávku 1,0 mg/kg a režim (1., 8., 15. a 22. deň počas prvého 28-dňového cyklu a 1. a 15. deň nasledujúcich 28-dňových cyklov). Populačná FK analýza zahŕňala 444 pacientov, ktorí dostávali mogamulizumab v šiestich klinických skúšaniach. Vystavenie sa mogamulizumabu sa úmerne zvyšovalo od dávky k dávke v rozsahu od 0,1 do 1,0 mg/kg.
AbsorpciaMogamulizumab sa podáva intravenóznou cestou, a preto je okamžite a úplne biologicky dostupný.
DistribúciaPodľa populačnej FK analýzy bol geometrický priemer [% variačného koeficientu (CV%)] hlavného distribučného objemu kompartmentu 3,57 l (20,1 %).
BiotransformáciaMetabolická dráha mogamulizumabu nebola definovaná. Predpokladá sa, že mogamulizumab sa rozkladá na malé peptidy a aminokyseliny v katabolických dráhach rovnakým spôsobom ako
endogénny imunoglobulín IgG.
Eliminácia
Podľa populačnej FK analýzy je geometrický priemer (% variačného koeficientu [CV%]) klírens (CL)
12,0 ml/h (83,7 %) a geometrický priemer polčasu eliminácie (t1/2) je 17 dní (65,5 %).
Linearita a akumulácia
Mogamulizumab preukazuje lineárny typ FK od dávky k dávke v rozsahu od 0,1 mg/kg do 1 mg/kg.
Podľa populačnej FK analýzy sa ustálený stav koncentrácií mogamulizumabu dosiahol po 12 týždňoch po opakovanom dávkovaní, ak sa dodržal odporúčaný režim, a systemická akumulácia bola 1,7- násobná. Pri analýze modelu sily nebola zjavná žiadna odchýlka od úmernosti dávky.
Poruchafunkcieobličiek
Vplyv poruchy funkcie obličiek na klírens mogamulizumabu sa hodnotil populačnou FK analýzou u pacientov s ľahkou (klírens kreatinínu [CrCL] medzi 60 a 89; n = 157), stredne ťažkou (CrCL medzi
59 a 30; n = 80) a ťažkou poruchou funkcie obličiek (CrCL menej ako 30 ml/min; n = 2). Medzi pacientmi s ľahkou až ťažkou poruchou funkcie obličiek a u pacientov s normálnou funkciou obličiek sa nenašli žiadne klinicky významné rozdiely v klírens mogamulizumabu.
Poruchafunkciepečene
Vplyv poruchy funkcie pečene na klírens mogamulizumabu sa hodnotil populačnou FK analýzou pacientov s ľahkou poruchou funkcie pečene (celkový bilirubín [TB] nižší alebo rovný ako horná hranica normálu [ULN] alebo AST vyššia ako ULN alebo 1 až 1,5-krát nižší TB ako ULN a akákoľvek AST; n = 80) alebo stredne ťažkou poruchou funkcie pečene (1,5 až 3-krát vyšší TB ako
ULN a akákoľvek AST; n=3). Medzi pacientmi s ľahkou až stredne ťažkou poruchou funkcie pečene a u pacientov s normálnou funkciou pečene sa nenašli žiadne klinicky významné rozdiely v klírens
mogamulizumabu. Mogamulizumab sa neskúšal u pacientov so závažnou poruchou funkcie pečene (3-
krát vyšší TB ako ULN alebo akákoľvek AST).
Ďalšieosobitné populácie
Vplyv rôznych premenných na FK mogamulizumabu sa hodnotil populačnou FK analýzou.
Nasledujúce faktory nemali žiadny klinický vplyv na CL mogamulizumabu: vek (v rozmedzí od 22 do
101 rokov), pohlavie, etnikum (iné ako japonské, k dispozícii sú len obmedzené údaje o inej etnickej populácii), porucha funkcie obličiek, ľahká alebo stredne závažná porucha funkcie pečene, podtyp ochorenia (mycosis fungoides (MF) alebo Sézaryho syndróm (SS)), stupeň expresie CCR4 alebo stav ECOG, avšak treba si uvedomiť, že pacienti s ECOG PS ≥2 boli z klinického skúšania vylúčení.
Farmakokinetický/farmakodynamickývzťah(y)
Účinnosť
Analýza na základe vystavenia a odpovede ukázala, že účinnosť nekorelovala s vystavením mogamulizumabu v hlavnom skúšaní. Účinnosť, ktorá sa merala zlepšením PFS založenom na
hodnotení skúšajúceho lekára, nesúvisela so zvyšujúcim sa vystavením mogamulizumabu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí. Neboli vykonané štúdie o karcinogenite ani genotoxicite mogamulizumabu. Neboli vykonané žiadne špeciálne štúdie na posúdenie možného vplyvu na plodnosť.
V toxikologických štúdiách opakovanej dávky sa nezistili sa žiadne toxické účinky spojené s mogamulizumabom na mužské alebo ženské pohlavné orgány u pohlavne zrelých opíc do 26 týždňov.
V štúdii o reproduktívnej a vývojovej toxicite u zvierat podávanie mogamulizumabu tehotným samiciam makaka dlhochvostého od začiatku organogenézy až po pôrod nepreukázalo možnú embryofetálnu letalitu, teratogenitu ani spomalenie rastu plodu. Vo všeobecnosti je známe, že
molekuly imunoglobulínu IgG prechádzajú placentárnou bariérou a boli zistené koncentrácie mogamulizumabu v plazme plodu. Farmakologická aktivita mogamulizumabu bola zaznamenaná u plodov, pretože bola zrejmá z poklesu expresie receptora CCR4 v lymfocytoch.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Monohydrát kyseliny citrónovej
Glycín
Polysorbát 80
Hydroxid sodný (na úpravu pH)
Kyselina chlorovodíková (na úpravu pH) Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi. Mogamulizumab
sa nemá podávať v jednej infúzii spolu s inými liekmi.
6.3 Čas použiteľnosti
Neotvorená injekčnáliekovka
3 roky
Po otvorení
Liek POTELIGEO neobsahuje konzervant. Po otvorení sa má okamžite rozriediť a naplniť (pozri časť
6.6).
Po príprave infúzie
Chemická a fyzikálna stabilita používania sa prejavila počas 24 hodín pri izbovej teplote (25°C) a
ambientnom osvetlení miestnosti.
Tieto časové hranice zahŕňajú uchovávanie infúzneho roztoku v infúznom vaku počas trvania infúzie.
Z mikrobiologického hľadiska sa musí pripravený roztok použiť okamžite.
Ak sa nepoužije okamžite, používateľ zodpovedá za čas stability používania a stav pred použitím, ktorý nesmie byť dlhší ako 4 hodiny pri teplote 2°C – 8°C, ak podanie roztoku prebehlo v kontrolovaných a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom. Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Objem 5 ml roztoku v 10 ml sklenenej injekčnej liekovke (1. typ skla) s gumenou zátkou, hliníkovým
tesnením a vyklápacím polypropylénovým viečkom.
Balenie s 1 injekčnou liekovkou.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Príprava
• Pred podaním skontrolujte infúzny roztok a presvedčte sa, či je naozaj čistý, bez viditeľných častíc a bezfarebný. Koncentrát POTELIGEO je číri až mierne opalescenčný bezfarebný roztok. Nepoužívajte koncentrát v injekčnej liekovke, ak je zakalený, bezfarebný a bez viditeľných častíc.
• Vypočítajte potrebné množstvo koncentrátu POTELIGEO a pripravte infúzny roztok s dávkou
1 mg/kg podľa telesnej hmotnosti pacienta (pozri časť 4.2). V aseptických podmienkach natiahnite potrebné množstvo infúzneho koncentrátu do striekačky a preneste ho do infúzneho vaku, ktorý obsahuje 9 mg/ml (0,9 %) roztoku chloridu sodného. Rozriedený roztok opatrne prevráťte a premiešajte. Netraste. Konečná koncentrácia v rozriedenom roztoku má byť medzi
0,1 mg/ml až 3,0 mg/ml.
• Každá injekčná liekovka je na jednorazové použitie. Zlikvidujte nepoužitý zvyšok v injekčnej
liekovke v súlade s miestnymi požiadavkami.
Podávanie• Rozriedený roztok je kompatibilný s polyvinylchloridovými (PVC) alebo polyolefinovými (PO)
infúznymi vakmi.
• Nemiešajte a nepodávajte koncentrát POTELIGEO v infúzii s inými koncentrátmi.
• Koncentrát POTELIGEO je určený iba na intravenózne použitie, nepodáva sa subkutáne, intramuskulárne, ako bolusová dávka alebo rýchlym intravenózným podaním.
• Infúzny koncentrát podávajte aspoň 60 minút formou intravenóznej infúzie, ktorá má sterilný
inline filter 0,22 mikrónu (alebo podobný) viažuci nízkomolekulové bielkoviny.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIKyowa Kirin Holdings B.V. Bloemlaan 2
2132NP Hoofddorp
Holandsko
8. REGISTRAČNÉ ČÍSLOEU/1/18/1335/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu