
otehotnieť alebo otehotnie, semaglutid sa má vysadiť. Semaglutid sa má vysadiť aspoň
2 mesiace pred plánovanou graviditou kvôli dlhému polčasu premeny (pozri časť 5.2).
Dojčenie
U potkanov v období laktácie sa semaglutid vylučoval do mlieka. Keďže riziko pre dojčené dieťa sa
nemôže vylúčiť, semaglutid sa nemá používať počas dojčenia.
Fertilita
Účinok semaglutidu na fertilitu u ľudí nie je známy. Semaglutid neovplyvnil samčiu fertilitu
u potkanov. U samíc potkanov sa pozorovalo predĺženie estrálneho cyklu a mierne zníženie počtu ovulácií pri dávkach spojených so znížením telesnej hmotnosti matky (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Semaglutid nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Keď sa používa v kombinácii so sulfonylureou alebo s inzulínom, pacientov je potrebné upozorniť,
aby prijali opatrenia na zabránenie vzniku hypoglykémie počas vedenia vozidiel a obsluhy strojov
(pozri časť 4.4).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
V 8 skúšaniach fázy 3a bolo semaglutidu v dávke do 1 mg vystavených 4 792 pacientov. Najčastejšie
hlásené nežiaduce reakcie v klinických skúšaniach boli gastrointestinálne poruchy vrátane nauzey (veľmi časté), hnačky (veľmi časté) a vracania (časté). Vo všeobecnosti boli tieto reakcie mierne alebo stredne závažné a trvali krátko.
Tabuľkový zoznam nežiaducich reakcií
Tabuľka 1 uvádza zoznam nežiaducich reakcií zistených vo všetkých klinických skúšaniach fázy 3
(vrátane skúšania zameraného na dlhodobé kardiovaskulárne výsledky) a z postmarketingových hlásení u pacientov s diabetom mellitus 2. typu (ďalej opísané v časti 5.1). Frekvencie nežiaducich reakcií (okrem komplikácií pri diabetickej retinopatii, pozri poznámku pod tabuľkou 1) sú určené na súbore klinických skúšaní fázy 3a s vylúčením skúšania sledujúceho kardiovaskulárne ukazovatele (ďalšie podrobnosti pozri v texte uvedenom pod tabuľkou).
Reakcie sú uvedené nižšie a sú rozdelené podľa triedy orgánových systémov a absolútnej frekvencie. Frekvencie sú definované nasledovne: veľmi časté: (≥1/10); časté: (≥1/100 až <1/10); menej časté: (≥1/1 000 až <1/100); zriedkavé: (≥1/10 000 až <1/1 000); veľmi zriedkavé: (<1/10 000) a neznáme:
nedá sa odhadnúť z dostupných údajov. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Trieda orgánových systémov podľa databázy MedDRA
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Neznáme
| Poruchy imunitného systému
|
|
| Precitlivenosťc
| Anafylak- tická reakcia
|
| Poruchy
metabolizmu
a výživy
| Hypoglykémiaa pri použití s inzulínom
alebo
so sulfonylureou
| Hypoglykémiaa pri použití s inými
perorálnymi antidiabetikami
(PAD)
Znížená chuť do
jedla
|
|
|
| Poruchy nervového systému
|
| Závrat
| Porucha chuti
|
|
| Poruchy oka
|
| Komplikácie spojené
s diabetickou retinopatioub
|
|
|
| Poruchy srdca a srdcovej činnosti
|
|
| Zvýšená srdcová frekvencia
|
|
| Poruchy gastrointesti-
nálneho traktu
| Nauzea
Hnačka
| Vracanie
Bolesť brucha Abdominálna distenzia
Zápcha
Dyspepsia Gastritída Gastroezofageálny reflux
Grganie
Nadúvanie
| Akútna pankreatitída
Oneskorené vyprázdňovanie
žalúdka
|
|
| Poruchy pečene
a žlčových ciest
|
| Cholelitiáza
|
|
|
| Poruchy kože a podkožného tkaniva
|
|
|
|
| Angioedémd
| Celkové poruchy
a reakcie v mieste
podania
|
| Únava
| Reakcie v mieste
podania
injekcie
|
|
| Laboratórne a funkčné
vyšetrenia
|
| Zvýšená hladina lipázy
Zvýšená hladina
amylázy Znížená telesná hmotnosť
|
|
|
|
|
|
Tabuľka 1 Frekvencia výskytu nežiaducich reakcií pri semaglutide a) Hypoglykémia definovaná ako závažná (vyžadujúca asistenciu inej osoby) alebo symptomatická v kombinácii s glykémiou <3,1 mmol/l.
b) Komplikácie diabetickej retinopatie zahŕňajú: retinálnu fotokoaguláciu, liečbu intravitreálnymi implantátmi, vitreálnu hemorágiu, slepotu súvisiacu s diabetom (menej časté). Frekvencia je určená na základe skúšania
sledujúceho kardiovaskulárne ukazovatele.
c) Združený termín zahŕňajúci aj nežiaduce udalosti súvisiace s precitlivenosťou, ako sú vyrážka a žihľavka.
d) Z postmarketingových hlásení.
2-ročnékardiovaskulárneukazovateleaskúšaniebezpečnosti
V populácii s vysokým kardiovaskulárnym rizikom bol profil nežiaducich reakcií podobný ako u ostatných skúšaní fázy 3a (opísané v časti 5.1).
Opis vybraných nežiaducich reakcií
Hypoglykémia
Pri používaní semaglutidu ako monoterapie neboli pozorované žiadne prípady závažnej hypoglykémie. Závažná hypoglykémia sa primárne vyskytovala pri kombinácii semaglutidu so sulfonylureou (1,2 % účastníkov, 0,03 prípad/pacientorok) alebo s inzulínom (1,5 % účastníkov, 0,02 prípad/pacientorok). Málo prípadov (0,1 % účastníkov, 0,001 prípad/pacientorok) bolo pozorovaných pri semaglutide
v kombinácii s perorálnymi antidiabetikami inými ako sulfonylurea.
Hypoglykémia klasifikovaná podľa Americkej asociácie pre diabetes (ADA, American Diabetes Association) sa vyskytla u 11,3 % (0,3 udalosti/pacientorok) pacientov, keď sa 1 mg semaglutidu pridal k inhibítoru SGLT2 v SUSTAIN 9 v porovnaní s 2,0 % (0,04 udalosti/pacientorok) pacientov liečených placebom. Závažná hypoglykémia bola hlásená u 0,7 % (0,01 udalosti/pacientorok) a 0 % pacientov, v uvedenom poradí.
V 40-týždňovom skúšaní fázy 3b u pacientov užívajúcich semaglutid v dávke 1 mg a 2 mg sa väčšina epizód hypoglykémie (45 zo 49 epizód) vyskytla pri užívaní semaglutidu v kombinácii so sulfonylureou alebo s inzulínom. Celkovo sa nezvýšilo riziko vzniku hypoglykémie pri semaglutide v dávke 2 mg.
Gastrointestinálnenežiaducereakcie
Nauzea sa vyskytla u 17 % pacientov liečených semaglutidom 0,5 mg a u 19,9 % pacientov liečených semaglutidom 1,0 mg, hnačka u 12,2 % a 13,3 %, a vracanie u 6,4 % a 8,4 %. Väčšina prípadov bola mierne až stredne závažná a trvala krátko. Tieto prípady viedli k ukončeniu liečby u 3,9 % a u 5 % pacientov. Prípady boli najčastejšie hlásené počas prvých mesiacov liečby.
U pacientov s nízkou telesnou hmotnosťou sa môže prejaviť pri liečbe semaglutidom viac gastrointestinálnych nežiaducich reakcií.
V 40-týždňovom skúšaní fázy 3b u pacientov užívajúcich semaglutid v dávke 1 mg a 2 mg sa vyskytla nauzea v podobných podieloch pacientov pri liečbe semaglutidom v dávke 1 mg a 2 mg. Hnačka
a vracanie sa vyskytli u vyššieho podielu pacientov pri liečbe semaglutidom v dávke 2 mg než
v prípade semaglutidu v dávke 1 mg. Gastrointestinálne nežiaduce reakcie viedli k prerušeniu liečby v podobných podieloch v liečebných skupinách užívajúcich semaglutid v dávke 1 mg a v dávke 2 mg.
Pri súbežnom použití s inhibítorom SGLT2 v SUSTAIN 9 sa zápcha a gastroezofágový reflux vyskytli u 6,7%, respektíve 4% pacientov liečených semaglutidom 1 mg, v porovnaní so žiadnymi udalosťami
u pacientov liečených placebom. Prevalencia týchto udalostí sa časom neznižovala.
Akútnapankreatitída
Z potvrdených hlásení v klinických skúšaniach fázy 3a bola frekvencia akútnej pankreatitídy 0,3 % v prípade semaglutidu a 0,2 % v prípade komparátora. V dvojročnom klinickom skúšaní sledujúcom kardiovaskulárne ukazovatele bola frekvencia posudkom potvrdenej akútnej pankreatitídy 0,5 % v prípade semaglutidu a 0,6 % v prípade placeba (pozri časť 4.4).
Komplikácie
spojené
s
d
iabetickou
retinopatiou
2-ročné klinické skúšanie skúmalo 3 297 pacientov s diabetom 2. typu s vysokým kardiovaskulárnym rizikom, dlhým trvaním diabetu a slabo kompenzovanou glykémiou. V tomto skúšaní sa posudzované prípady komplikácií spojených s diabetickou retinopatiou vyskytli u väčšieho počtu pacientov liečených semaglutidom (3 %) v porovnaní s placebom (1,8 %). Pozorovalo sa to u pacientov liečených inzulínom so známou diabetickou retinopatiou. Rozdiel v liečbe sa prejavil na začiatku
a pretrvával počas skúšania. Systematické hodnotenie komplikácií diabetickej retinopatie sa vykonalo len počas skúšania zameraného na kardiovaskulárne ukazovatele. V klinických skúšaniach do 1 roka zahŕňajúcich 4 807 pacientov s diabetom 2. typu boli nežiaduce účinky spojené s diabetickou retinopatiou hlásené v podobných počtoch u účastníkov liečených semaglutidom (1,7 %)
a komparátormi (2,0 %).
PrerušenievdôsledkunežiaducehoúčinkuIncidencia prerušenia liečby v dôsledku nežiaducich účinkov bola 6,1 % u pacientov liečených semaglutidom 0,5 mg a 8,7 % u pacientov liečených semaglutidom 1 mg, oproti 1,5 % u placeba. Najčastejšie nežiaduce účinky vedúce k prerušeniu boli gastrointestinálne.
ReakcievmiestepodaniainjekcieReakcie v mieste podania injekcie (napr. vyrážka v mieste podania injekcie, erytém) boli hlásené
u 0,6 % pacientov dostávajúcich semaglutid 0,5 mg a u 0,5 % pacientov dostávajúcich semaglutid
1 mg. Tieto reakcie boli väčšinou mierne.
ImunogenicitaV súlade s potenciálne imunogénnymi vlastnosťami liekov obsahujúcimi proteíny alebo peptidy sa
u pacientov po liečbe semaglutidom môžu vytvoriť protilátky. Podiel pacientov s pozitívnym testom na protilátky proti semaglutidu v akomkoľvek čase po začiatku bol nízky (1–3 %) a na konci skúšania žiadni pacienti nemali neutralizačné protilátky proti semaglutidu ani protilátky proti semaglutidu
s neutralizačným účinkom na endogénny GLP-1.
ZvýšenásrdcováfrekvenciaZvýšenie srdcovej frekvencie bolo pozorované pri agonistoch receptora GLP-1. Vo fáze 3a klinického skúšania bolo pozorované u jedincov liečených liekom Ozempic priemerné zvýšenie o 1 až 6 úderov za minútu (bpm) z východiskovej hodnoty 72 až 76 úderov za minútu. V dlhodobom klinickom skúšaní u jedincov s kardiovaskulárnymi rizikovými faktormi malo 16 % jedincov liečených liekom Ozempic nárast srdcovej frekvencie o >10 úderov za minútu v porovnaní s 11 % jedincov na placebe po 2 rokoch liečby.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePočas klinických skúšaní boli hlásené predávkovania do 4 mg v jednorazovej dávke a do 4 mg za týždeň. Najčastejšie hlásenou nežiaducou reakciou bola nauzea. Všetci pacienti sa zotavili bez komplikácií.
Na predávkovanie semaglutidom neexistuje špecifické antidotum. V prípade predávkovania sa má začať vhodná podporná liečba podľa klinických prejavov a príznakov pacienta. Môže byť potrebné predĺženie obdobia sledovania a liečby týchto príznakov berúc do úvahy dlhý polčas premeny semaglutidu trvajúci približne 1 týždeň (pozri časť 5.2).
5
. FARMAKOLOGICKÉ VLASTNOSTI
5
.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antidiabetiká, analógy glukagónu podobného peptidu-1 (GLP-1), ATC
kód: A10BJ06
Mechanizmus účinku
Semaglutid je analóg GLP-1 s 94 % sekvenčnou homológiou s ľudským GLP-1. Semaglutid pôsobí
ako agonista GLP-1 receptora, selektívne sa viaže a aktivuje GLP-1 receptor, cieľ natívneho GLP-1.
GLP-1 je fyziologický hormón, ktorý zohráva viacero úloh pri regulácii glukózy a chuti do jedla
a v kardiovaskulárnom systéme. Účinky na glukózu a na chuť do jedla sú špecificky sprostredkované receptormi GLP-1 v pankrease a mozgu.
Semaglutid znižuje glykémiu spôsobom závislým od glukózy stimuláciou sekrécie inzulínu a znížením sekrécie glukagónu, keď je glykémia vysoká. Mechanizmus znižovania glykémie zahŕňa aj mierne spomalenie vyprázdňovania žalúdka v skorej postprandiálnej fáze. Počas hypoglykémie semaglutid znižuje sekréciu inzulínu a nenarúša sekréciu glukagónu.
Semaglutid znižuje telesnú hmotnosť a množstvo telesného tuku prostredníctvom zníženia energetického príjmu vrátane celkového zníženia chuti do jedla. Okrem toho, semaglutid znižuje preferenciu jedla s vysokým obsahom tukov.
GLP-1 receptory sú tiež lokalizované v srdci, cievach, imunitnom systéme a obličkách.
Semaglutid mal v klinických skúšaniach prínosný účinok na plazmatické lipidy, znižoval systolický krvný tlak a znižoval zápal. V štúdiách na zvieratách semaglutid spomaľoval rozvoj aterosklerózy zabraňujúc progresii aortálneho plaku a redukoval zápal v plaku.
Farmakodynamické účinky
Všetky farmakodynamické hodnotenia boli vykonané po 12 týždňoch liečby (vrátane eskalácie dávky)
v ustálenom stave pri podávaní semaglutidu 1 mg jedenkrát týždenne.
Glukózanalačnoa postprandiálnaglukóza
Semaglutid znižuje koncentrácie glukózy nalačno a po jedle. U pacientov s diabetom 2. typu viedla liečba semaglutidom 1 mg k zníženiu hladín glukózy v zmysle absolútnej zmeny od začiatku (mmol/l) a relatívneho zníženia v porovnaní s placebom (%) u glukózy nalačno (1,6 mmol/l; zníženie o 22 %), glukózy 2 hodiny po jedle (4,1 mmol/l; zníženie o 37 %), priemernej koncentrácie glukózy za
24 hodín (1,7 mmol/l; zníženie o 22 %) a výkyvov postprandiálnej glukózy po 3 jedlách (0,6–
1,1 mmol/l) v porovnaní s placebom. Semaglutid znížil glukózu nalačno po prvej dávke.
Funkciabetabuniekasekréciainzulínu
Semaglutid zlepšuje funkciu beta buniek. V porovnaní s placebom zlepšil semaglutid inzulínovú odpoveď prvej a druhej fázy 3-, a 2-násobne, v uvedenom poradí, a zvýšil maximálnu sekrečnú kapacitu beta buniek u pacientov s diabetom 2. typu. Okrem toho liečba semaglutidom zvýšila
v porovnaní s placebom koncentrácie inzulínu nalačno.
Sekréciaglukagónu
Semaglutid znižuje koncentrácie glukagónu nalačno a po jedle. U pacientov s diabetom 2. typu viedol semaglutid k nasledujúcim relatívnym zníženiam glukagónu v porovnaní s placebom: glukagón nalačno (8–21 %), postprandiálna glukagónová odpoveď (14–15 %) a priemerná koncentrácia glukagónu za 24 hodín (12 %).
S
ekrécia
inzulínu
a
g
lukagónu
závislá
o
d
g
lukózy
Semaglutid znížil vysokú glykémiu stimuláciou sekrécie inzulínu a znížením sekrécie glukagónu spôsobom závislým od glukózy. Pri semaglutide je rýchlosť sekrécie inzulínu u pacientov s diabetom
2. typu porovnateľná so sekréciou u zdravých jedincov.
Počas indukovanej hypoglykémie semaglutid v porovnaní s placebom nezmenil opačné regulačné odpovede zvýšenia glukagónu a neovplyvnil zníženie C-peptidu u pacientov s diabetom 2. typu.
Vyprázdňovaniežalúdka
Semaglutid spôsobil malé oneskorenie začiatku vyprázdňovania žalúdka po jedle, čím znížil rýchlosť, ktorou sa glukóza po jedle objavuje v krvnom obehu.
Chuťdojedla,energetickýpríjemavýberjedla
Semaglutid v porovnaní s placebom znížil energetický príjem 3 po sebe nasledujúcich ad libitum jedál o 18–35 %. Bolo to podporené semaglutidom indukovanou supresiou chuti do jedla v stave nalačno, ako aj po jedle, zlepšenou kontrolou jedenia, menšou chuťou do jedla a relatívne nižšou preferenciou jedla s vysokým obsahom tuku.
Lipidynalačnoapostprandiálnelipidy
Semaglutid v porovnaní s placebom znižoval koncentrácie triglyceridov a VLDL (lipoproteíny s veľmi nízkou hustotou) cholesterolu nalačno o 12 %, resp. 21 %, v uvedenom poradí. Postprandiálna
odpoveď triglyceridov a VLDL cholesterolu na jedlo s vysokým obsahom tuku sa znížila o >40 %.
Srdcováelektrofyziológia(QTc)
Účinok semaglutidu na srdcovú repolarizáciu bol testovaný v skúšaní QTc. Semaglutid nepredlžoval intervaly QTc pri dávkach až do 1,5 mg v ustálenom stave.
Klinická účinnosť a bezpečnosť
Zlepšenie kontroly glykémie, ako aj zníženie kardiovaskulárnej morbidity a mortality sú integrovanou
súčasťou liečby diabetu 2. typu.
Účinnosť a bezpečnosť semaglutidu 0,5 mg a 1 mg jedenkrát týždenne boli hodnotené v šiestich randomizovaných kontrolovaných skúšaniach fázy 3a, ktoré zahŕňali 7 215 pacientov s diabetes mellitus 2. typu (4 107 liečených semaglutidom). Päť skúšaní (SUSTAIN 1–5) malo ako primárny cieľ hodnotenie glykemickej účinnosti, zatiaľ čo jedno skúšanie (SUSTAIN 6) malo ako primárny cieľ kardiovaskulárny ukazovateľ.
Účinnosť a bezpečnosť semaglutidu 2 mg jedenkrát týždenne boli hodnotené v skúšaní fázy 3b
(SUSTAIN FORTE), do ktorého bolo zaradených 961 pacientov.
Navyše bolo vykonané skúšanie fázy 3b (SUSTAIN 7) zahŕňajúce 1 201 pacientov na porovnanie účinnosti a bezpečnosti semaglutidu 0,5 mg a 1 mg jedenkrát týždenne s dulaglutidom 0,75 mg a
1,5 mg jedenkrát týždenne, v uvedenom poradí. Skúšanie fázy 3b (SUSTAIN 9) bolo vykonané na zistenie účinnosti a bezpečnosti semaglutidu ako doplnku k liečbe inhibítorom SGLT2.
Liečba semaglutidom preukázala ustálené, štatisticky superiórne a klinicky významné zníženie HbA1c a telesnej hmotnosti za 2 roky v porovnaní s placebom a aktívnou kontrolnou liečbou (sitagliptín, inzulín glargín, exenatid ER a dulaglutid).
Účinnosť semaglutidu nebola ovplyvnená vekom, pohlavím, rasou, etnickou príslušnosťou, začiatočným BMI, začiatočnou telesnou hmotnosťou (kg), trvaním diabetu ani úrovňou poruchy funkcie obličiek.
Výsledky sa zameriavajú na obdobie počas liečby u všetkých randomizovaných účastníkov (analýzy na základe opakovaných meraní zmiešaných modelov alebo viacnásobného pripočítania).
Navyše bolo vykonané skúšanie fázy 3b (SUSTAIN 11) na zistenie účinnosti semaglutidu v porovnaní s inzulínom aspartátom, oba ako doplnok k liečbe metformínom a optimalizovaným inzulínom glargínom (U100).
Podrobné informácie sú uvedené nižšie.
SUSTAIN1 – MonoterapiaV 30-týždňovom dvojito zaslepenom placebom kontrolovanom skúšaní 388 pacientov, neadekvátne kompenzovaných diétou a cvičením, bolo randomizovaných do skupín semaglutidu 0,5 mg alebo semaglutidu 1 mg jedenkrát týždenne alebo placeba.
Tabuľka 2 SUSTAIN 1: Výsledky v 30. týždni
| Semaglutid
0,5 mg
| Semaglutid
1 mg
| Placebo
|
Populácia so zámerom liečby (Intent-to-
Treat, ITT) (N)
| 128
| 130
| 129
|
HbA1c (%)
|
|
|
|
Východisková hodnota (priemer)
| 8,1
| 8,1
| 8,0
|
Zmena oproti východiskovej hodnote v 30. týždni
| -1,5
| -1,6
| 0
|
Rozdiel oproti placebu [95 % CI]
| -1,4 [-1,7; -1,1]a
| -1,5 [-1,8; -1,2]a
| –
|
Pacienti (%) dosahujúci HbA1c <7 %
| 74
| 72
| 25
|
Glykémia nalačno (mmol/l)
|
|
|
|
Východisková hodnota (priemer)
| 9,7
| 9,9
| 9,7
|
Zmena oproti východiskovej hodnote v 30. týždni
| -2,5
| -2,3
| -0,6
|
Telesná hmotnosť (kg)
|
|
|
|
Východisková hodnota (priemer)
| 89,8
| 96,9
| 89,1
|
Zmena oproti východiskovej hodnote v 30. týždni
| -3,7
| -4,5
| -1,0
|
Rozdiel oproti placebu [95 % CI]
| -2,7 [-3,9; -1,6]a
| -3,6 [-4,7; -2,4]a
| -
|
ap <0,0001 (2-stranný) pre superioritu
SUSTAIN2 – Semaglutidvs.sitagliptínobidvavkombináciis1–2perorálnymiantidiabetikami(metformína/alebotiazolidíndióny)V 56-týždňovom dvojito zaslepenom aktívne kontrolovanom skúšaní bolo 1 231 pacientov randomizovaných do skupín semaglutidu 0,5 mg jedenkrát týždenne, semaglutidu 1 mg jedenkrát týždenne alebo sitagliptínu 100 mg jedenkrát denne, všetci s kombináciou s metformínom (94 %) a/alebo tiazolidíndiónmi (6 %).
| Semaglutid
0,5 mg
| Semaglutid
1 mg
| Sitagliptín
100 mg
| Populácia so zámerom liečby (Intent-to-
Treat, ITT) (N)
| 409
| 409
| 407
| HbA1c (%)
|
|
|
| Východisková hodnota (priemer)
| 8,0
| 8,0
| 8,2
| Zmena oproti východiskovej hodnote v 56. týždni
| -1,3
| -1,6
| -0,5
| Rozdiel oproti sitagliptínu [95 %
CI]
| -0,8 [-0,9; -0,6]a
| -1,1 [-1,2; -0,9]a
| –
| Pacienti (%) dosahujúci HbA1c <7 %
| 69
| 78
| 36
| Glykémia nalačno (mmol/l)
|
|
|
| Východisková hodnota (priemer)
| 9,3
| 9,3
| 9,6
| Zmena oproti východiskovej hodnote v 56. týždni
| -2,1
| -2,6
| -1,1
|
|
|
Tabuľka 3 SUSTAIN 2: Výsledky v 56. týždni
|
Semaglutid
0
,5 mg
|
Semaglutid
1 mg
|
Sitagliptín
10
0 mg
|
Telesná hmotnosť (kg)
|
|
|
|
Východisková hodnota (priemer)
|
89,9
|
89,2
|
89,3
|
Zmena oproti východiskovej hodnote v 56. týždni
|
-4,3
|
-6,1
|
-1,9
|
Rozdiel oproti sitagliptínu [95 %
CI]
|
-2,3 [-3,1; -1,6]a
|
-4,2 [-4,9; -3,5]a
|
–
|
ap <0,0001 (2-stranný) pre superioritu
,
, ,
, ,
, , ,

, ,
. ,
, ,
, , ,
, , ,
, ,
, ,
, ,
,
,
,
,
, ,
,
,
,
,
, ,
,
,

,
, ,

,
Čas od randomizácie (týždeň)
Čas od randomizácie (týždeň)
Semaglutid 0,5 mg
Semaglutid 1 mg
Semaglutid 0,5 mg
Sitagliptín
Semaglutid 1 mg
Sitagliptín
Obrázok 1 Priemerná zmena HbA
1
c
(%) a telesnej hmotnosti (kg) oproti východiskovej hodnote
do 56. týždňa
S
USTAIN
7 – Semaglutid
v
s.
du
laglutid
ob
idva
v
kombinácii s metformínom
V 40-týždňovom otvorenom skúšaní 1 201 pacientov na metformíne bolo randomizovaných 1:1:1:1
jedenkrát týždenne na semaglutid 0,5 mg, dulaglutid 0,75 mg, semaglutid 1 mg alebo dulaglutid
1,5 mg v uvedenom poradí. Toto skúšanie porovnávalo 0,5 mg semaglutidu s 0,75 mg dulaglutidu a
1 mg semaglutidu s 1,5 mg dulaglutidu.
Gastrointestinálne poruchy boli najčastejšími nežiaducimi udalosťami, a prejavili sa v podobnom pomere pacientov používajúcich semaglutid 0,5 mg (129 pacientov [43 %]), semaglutid 1 mg (133
[44 %]), a dulaglutid 1,5 mg (143 [48 %]); menej pacientov malo gastrointestinálne poruchy s dulaglutidom 0,75 mg (100 [33 %]).
V 40. týždni zvýšenie pulzovej frekvencie pri semaglutid (0,5 mg a 1 mg) a pri dulaglutide (0,75 mg a
1,5 mg) bolo 2,4, 4,0 a 1,6, 2,1 úderov/min, v uvedenom poradí.
| Semaglutid
0,5 mg
| Semaglutid
1 mg
| Dulaglutid
0,75 mg
| Dulaglutid
1,5 mg
| Populácia so zámerom liečby
(Intent-to-Treat, ITT) (N)
| 301
| 300
| 299
| 299
| HbA1c (%)
|
|
|
|
| Východisková hodnota (priemer)
| 8,3
| 8,2
| 8,2
| 8,2
| Zmena oproti východiskovej hodnote v 40. týždni
| -1,5
| -1,8
| -1,1
| -1,4
| Rozdiel oproti dulaglutidu
[95% CI]
| -0,4b
[-0,6, -0,2]a
| -0,4c
[-0,6, -0,3]a
| -
| -
| Pacienti (%) dosahujúci HbA1c
<7%
| 68
| 79
| 52
| 67
| Glykémia nalačno (mmol/l)
|
|
|
|
| Východisková hodnota (priemer)
| 9,8
| 9,8
| 9,7
| 9,6
| Zmena oproti východiskovej hodnote v 40. týždni
| -2,2
| -2,8
| -1,9
| -2,2
|
|
|
Tabuľka 4 SUSTAIN 7: Výsledky v 40. týždni
|
Semaglutid
0
,5 mg
|
Semaglutid
1 mg
|
Dulaglutid
0
,75 mg
|
Dulaglutid
1
,5 mg
|
Telesná hmotnosť (kg)
|
|
|
|
|
Východisková hodnota (priemer)
|
96,4
|
95,5
|
95,6
|
93,4
|
Zmena oproti východiskovej hodnote v 40. týždni
|
-4,6
|
-6,5
|
-2,3
|
-3,0
|
Rozdiel oproti dulaglutidu
[95% CI]
|
-2,3b
[-3,0, -1,5]a
|
-3,6c
[-4,3, -2,8]a
|
-
|
-
|
ap <0,0001 (2-stranný) pre superioritu
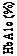
b semaglutid 0,5 mg vs dulaglutid 0,75 mg
c semaglutid 1 mg vs dulaglutid 1,5 mg
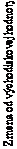
,
. ,
, ,
, ,
, ,
, ,
| .
|
| .
|
|
.
|
|
.
|
Čas od randomizácie (týždeň)
|
|
|
|
, ,
,
|
|
| ,
|
| ,
| ,
|
| ,
| ,
|
| ,
| ,
|
| ,
| ,
|
| ,
| ,
| .
| ,
| ,
| .
| ,
| ,
| .
| ,
|
|
|
, . , , ,
, ,
, , ,
Čas od randomizácie (týždeň)
Semaglutid 0,5 mg
Semaglutid 1 mg
Semaglutid 0,5 mg
Semaglutid 1 mg
Dulaglutid 0,75 mg Dulaglutid 1,5 mg
Dulaglutid 0,75 mg Dulaglutid 1,5 mg
Obrázok 2 Priemerná zmena HbA
1
c
(%) a telesnej hmotnosti (kg) oproti východiskovej hodnote
do 40. týždňa
S
USTAIN
3
– Semaglutid
v
s. exenatid ER obidva v kombinácii s metformínom
a
l
ebo
metformínom
so
sulfonylureou
V 56-týždňovom otvorenom skúšaní 813 pacientov liečených len metformínom (49 %), metformínom so sulfonylureou (45 %) alebo iným (6 %) bolo randomizovaných do skupiny semaglutidu 1 mg alebo exenatidu ER 2 mg jedenkrát týždenne.
Tabuľka 5 SUSTAIN 3: Výsledky v 56. týždni
| Semaglutid
1 mg
| Exenatid ER
2 mg
|
Populácia so zámerom liečby (Intent-to-
Treat, ITT) (N)
| 404
| 405
|
HbA1c (%)
|
|
|
Východisková hodnota (priemer)
| 8,4
| 8,3
|
Zmena oproti východiskovej hodnote v 56. týždni
| -1,5
| -0,9
|
Rozdiel oproti exenatidu [95 % CI]
| -0,6 [-0,8; -0,4]a
| -
|
Pacienti (%) dosahujúci HbA1c <7 %
| 67
| 40
|
Glykémia nalačno (mmol/l)
|
|
|
Východisková hodnota (priemer)
| 10,6
| 10,4
|
Zmena oproti východiskovej hodnote v 56. týždni
| -2,8
| -2,0
|
Telesná hmotnosť (kg)
|
|
|
Východisková hodnota (priemer)
| 96,2
| 95,4
|
Zmena oproti východiskovej hodnote v 56. týždni
| -5,6
| -1,9
|
Rozdiel oproti exenatidu [95 % CI]
| -3,8 [-4,6; -3,0]a
| -
|
ap < 0,0001 (2-stranný) pre superioritu
S
USTAIN
4 – Semaglutid
v
s.
inzulín
g
l
a
rgín
o
b
idva
v
kombinácii
s
1–
2
p
erorálnymi
an
tidiabetikami
(metformín
a
lebo
metformín
a
sulfonylurea)
V 30-týždňovom otvorenom porovnávacom skúšaní 1 089 pacientov bolo randomizovaných do skupín semaglutidu 0,5 mg jedenkrát týždenne, semaglutidu 1 mg jedenkrát týždenne alebo inzulínu glargínu jedenkrát denne na pozadí metformínu (48 %) alebo metformínu a sulfonylurey (51 %).
Tabuľka 6 SUSTAIN 4: Výsledky v 30. týždni
| Semaglutid
0,5 mg
| Semaglutid
1 mg
| Inzulín glargín
|
Populácia so zámerom liečby (Intent-to-Treat, ITT) (N)
| 362
| 360
| 360
|
HbA1c (%)
|
|
|
|
Východisková hodnota (priemer)
| 8,1
| 8,2
| 8,1
|
Zmena oproti východiskovej hodnote v 30. týždni
| -1,2
| -1,6
| -0,8
|
Rozdiel oproti inzulínu glargín [95 %
CI]
| -0,4 [-0,5; -0,2]a
| -0,8 [-1,0; -0,7]a
| –
|
Pacienti (%) dosahujúci HbA1c <7 %
| 57
| 73
| 38
|
Glykémia nalačno (mmol/l)
|
|
|
|
Východisková hodnota (priemer)
| 9,6
| 9,9
| 9,7
|
Zmena oproti východiskovej hodnote v 30. týždni
| -2,0
| -2,7
| -2,1
|
Telesná hmotnosť (kg)
|
|
|
|
Východisková hodnota (priemer)
| 93,7
| 94,0
| 92,6
|
Zmena oproti východiskovej hodnote v 30. týždni
| -3,5
| -5,2
| +1,2
|
Rozdiel oproti inzulínu glargín [95 %
CI]
| -4,6 [-5,3; -4,0]a
| -6,34 [-7,0; -5,7]a
| –
|
ap < 0,0001 (2-stranný) pre superioritu
SUSTAIN5 – Semaglutidvs.placeboobidvavkombináciisbazálnyminzulínomV 30-týždňovom dvojito zaslepenom placebom kontrolovanom skúšaní 397 pacientov nedostatočne kontrolovaných bazálnym inzulínom s metformínom alebo bez neho bolo randomizovaných do skupín semaglutidu 0,5 mg jedenkrát týždenne, semaglutidu 1 mg jedenkrát týždenne alebo placeba.
Tabuľka 7 SUSTAIN 5: Výsledky v 30. týždni
| Semaglutid
0,5 mg
| Semaglutid
1 mg
| Placebo
|
Populácia so zámerom liečby (Intent-to-
Treat, ITT) (N)
| 132
| 131
| 133
|
HbA1c (%)
|
|
|
|
Východisková hodnota (priemer)
| 8,4
| 8,3
| 8,4
|
Zmena oproti východiskovej hodnote v 30. týždni
| -1,4
| -1,8
| -0,1
|
Rozdiel oproti placebu [95 % CI]
| -1,4 [-1,6; -1,1]a
| -1,8 [-2,0; -1,5]a
| –
|
Pacienti (%) dosahujúci HbA1c <7 %
| 61
| 79
| 11
|
Glykémia nalačno (mmol/l)
|
|
|
|
Východisková hodnota (priemer)
| 8,9
| 8,5
| 8,6
|
Zmena oproti východiskovej hodnote v 30. týždni
| -1,6
| -2,4
| -0,5
|
Telesná hmotnosť (kg)
|
|
|
|
Východisková hodnota (priemer)
| 92,7
| 92,5
| 89,9
|
Zmena oproti východiskovej hodnote v 30. týždni
| -3,7
| -6,4
| -1,4
|
Rozdiel oproti placebu [95 % CI]
| -2,3 [-3,3; -1,3]a
| -5,1 [-6,1; -4,0]a
| –
|
ap < 0,0001 (2-stranný) pre superioritu
S
USTAIN
FORTE
– Semaglutid
2
mg
vs.
semaglutid
1
mg
V 40-týždňovom dvojito zaslepenom skúšaní 961 pacientov nedostatočne kontrolovaných metformínom so sulfonylureou alebo bez nej bolo randomizovaných na užívanie semaglutidu 2 mg jedenkrát týždenne alebo semaglutidu 1 mg jedenkrát týždenne.
Liečba semaglutidom 2 mg viedla k štatistický nadradenej redukcii hodnoty HbA1c po 40 týždňoch liečby v porovnaní so semaglutidom 1 mg.
Tabuľka 8 SUSTAIN FORTE: Výsledky v 40. týždni
| Semaglutid
1 mg
| Semaglutid
2 mg
|
Populácia so zámerom liečby (Intent-to-
Treat, ITT) (N)
| 481
| 480
|
HbA1c (%)
|
|
|
Východisková hodnota (priemer)
| 8,8
| 8,9
|
Zmena oproti východiskovej hodnote v 40. týždni
| -1,9
| -2,2
|
Rozdiel oproti semaglutidu 1 mg
[95 % CI]
| -
| -0,2 [-0,4; -0,1]a
|
Pacienti (%) dosahujúci HbA1c <7 %
| 58
| 68
|
Glykémia nalačno (mmol/l)
|
|
|
Východisková hodnota (priemer)
| 10,9
| 10,7
|
Zmena oproti východiskovej hodnote v 40. týždni
| -3,1
| -3,4
|
Telesná hmotnosť (kg)
|
|
|
Východisková hodnota (priemer)
| 98,6
| 100,1
|
Zmena oproti východiskovej hodnote v 40. týždni
| -6,0
| -6,9
|
Rozdiel oproti semaglutidu 1 mg
[95 % CI]
|
| -0,9 [-1,7; -0,2]b
|
ap < 0,001 (2-stranný) pre superioritu
bp < 0,05 (2-stranný) pre superioritu
SUSTAIN9 – Semaglutidvs.placeboakodoplnokkinhibítoruSGLT2±metformínaleboSUV 30-týždňovom dvojito zaslepenom placebom kontrolovanom skúšaní bolo 302 pacientov nedostatočne kontrolovaných inhibítorom SGLT2 s metformínom alebo bez neho alebo sulfonylureou randomizovaných do skupín 1 mg semaglutidu raz týždenne alebo placeba.
| Semaglutid
1 mg
| Placebo
| Populácia so zámerom liečby (Intent-to-
Treat, ITT) (N)
| 151
| 151
| HbA1c (%)
|
|
| Východisková hodnota (priemer)
| 8,0
| 8,1
| Zmena oproti východiskovej hodnote v 30. týždni
| -1,5
| -0,1
| Rozdiel oproti placebu [95 % CI]
| -1,4 [-1,6; -1,2]a
| -
| Pacienti (%) dosahujúci HbA1c <7 %
| 78,7
| 18,7
| Glykémia nalačno (mmol/l)
|
|
| Východisková hodnota (priemer)
| 9,1
| 8,9
| Zmena oproti východiskovej hodnote v 30. týždni
| -2,2
| 0,0
|
|
|
Tabuľka 9 SUSTAIN 9: Výsledky v 30. týždni
|
Semaglutid
1 mg
|
Placebo
|
Telesná hmotnosť (kg)
|
|
|
Východisková hodnota (priemer)
|
89,6
|
93,8
|
Zmena oproti východiskovej hodnote v 30. týždni
|
-4,7
|
-0,9
|
Rozdiel oproti placebu [95 % CI]
|
-3,8 [-4,7; -2,9]a
|
-
|
ap < 0,0001 (2-stranný) pre superioritu, upravené vzhľadom na multiplicitu na základe hierarchického testovania hodnoty HbA1c a telesnej hmotnosti
SUSTAIN11- Semaglutidvs.inzulínaspartátakodoplnokkinzulínuglargínu+metformínV 52-týždňovom otvorenom skúšaní bolo 1 748 jedincov s nedostatočne kontrolovaným T2D po 12- týždňovom úvodnom období na inzulíne glargíne a metformíne randomizovaných 1:1, aby dostávali buď semaglutid jedenkrát týždenne (0,5 mg alebo 1,0 mg) alebo inzulín aspartát trikrát denne. Zahrnutá populácia mala priemerné trvanie diabetu 13,4 roka a priemerný HbA1c 8,6 %, s cieľovým HbA1c 6,5 – 7,5 %.
Liečba semaglutidom viedla k zníženiu HbA1c v 52. týždni (-1,5 % pre semaglutid oproti -1,2 % pre inzulín aspartát).
Počet ťažkých hypoglykemických epizód v oboch liečebných ramenách bol nízky (4 epizódy so semaglutidom vs. 7 epizód s inzulínom aspartátom).
Priemerná východisková telesná hmotnosť sa znížila so semaglutidom (-4,1 kg) a zvýšila sa s inzulínom aspartátom (+2,8 kg) a odhadovaný rozdiel v liečbe bol -6,99 kg (95 % CI -7,41 až -6,57) v
52. týždni.
KombináciasosulfonylureouakomonoterapiouV skúšaní SUSTAIN 6 (pozri časť „Kardiovaskulárne ochorenie“) bolo na začiatku 123 pacientov na monoterapii so sulfonylureou. Začiatočné HbA1c bolo 8,2 %, 8,4 % a 8,4 % so semaglutidom 0,5 mg, semaglutidom 1 mg, resp. s placebom, v uvedenom poradí. V 30. týždni bola zmena HbA1c -1,6 %, -
1,5 %, resp. 0,1 % so semaglutidom 0,5 mg, semaglutidom 1 mg, resp. s placebom, v uvedenom poradí.
Kombinácia s premixovanýminzulínom±1–2 PADV skúšaní SUSTAIN 6 (pozri časť „Kardiovaskulárne ochorenie“) bolo na začiatku 867 pacientov na liečbe premixovaným inzulínom (s PAD alebo bez neho/bez nich). Začiatočné HbA1c bolo 8,8 %,
8,9 % a 8,9 % so semaglutidom 0,5 mg, semaglutidom 1 mg, resp. s placebom, v uvedenom poradí.
V 30. týždni bola zmena HbA1c -1,3 %, -1,8 %, resp. -0,4 % u semaglutidu 0,5 mg, semaglutidu 1 mg,
resp. placeba, v uvedenom poradí.
Kardiovaskulárne ochorenieV 104-týždňovom dvojito zaslepenom skúšaní (SUSTAIN 6) 3 297 pacientov s diabetom 2. typu s vysokým kardiovaskulárnym rizikom randomizovaných buď do skupiny semaglutidu 0,5 mg jedenkrát týždenne, semaglutidu 1 mg jedenkrát týždenne alebo odpovedajúcich na placebo ako prídavku k štandardnej starostlivosti pozorovaných počas 2 rokov. Celkovo skúšanie dokončilo 98 % pacientov a zdravotný stav bol na konci skúšania známy u 99,6 % pacientov.
Skúmaná populácia bola rozdelená podľa veku nasledovne: 1 598 pacientov (48,5 %) ≥65 rokov,
321 (9,7 %) ≥75 rokov a 20 (0,6 %) ≥85 rokov. 2 358 pacientov malo normálnu funkciu alebo miernu poruchu funkcie obličiek, 832 stredne závažnú poruchu funkcie obličiek a 107 závažnú poruchu funkcie obličiek alebo finálne štádium ochorenia obličiek. V skúšaní bolo 61 % mužov, priemerný vek bol 65 rokov a priemerný BMI bol 33 kg/m2. Priemerná doba trvania diabetu bola 13,9 roka.
Primárny cieľový ukazovateľ bol čas od randomizácie do prvého výskytu významných nežiaducich kardiovaskulárnych udalostí (MACE): kardiovaskulárne úmrtie, nefatálny infarkt myokardu alebo nefatálna cievna mozgová príhoda
Celkový počet primárnych komponentných cieľových ukazovateľov MACE bol 254 vrátane
108 (6,6 %) pri semaglutide a 146 (8,9 %) pri placebe. Výsledky primárnych a sekundárnych kardiovaskulárnych cieľových ukazovateľov nájdete na obrázku 4. Liečba semaglutidom viedla k zníženiu rizika o 26 % v primárnom kompozitnom cieľovom ukazovateli–úmrtie
kvôli kardiovaskulárnej príčine, nefatálnemu infarktu myokardu alebo nefatálnej cievnej mozgovej príhode. Celkový počet úmrtí kvôli kardiovaskulárnej príčine bol 90, kvôli nefatálnemu infarktu myokardu bol 111a kvôli nefatálnej cievnej mozgovej príhode bol 71 z čoho bolo 44 (2,7 %), 47
(2,9 %) a 27 (1,6 %) v uvedenom poradí, pri semaglutide (obrázok 4). Zníženie rizika v primárnom
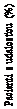
kompozitnom ukazovateli bolo spôsobené hlavne poklesom výskytu nefatálnej cievnej mozgovej príhody (39 %) a nefatálneho infarktu myokardu (26 %) (obrázok 3).
HR: 0,74
95% CI 0,58: 0,95
S
e
maglutid
Placebo
Počet rizikových účastníkov
Čas od randomizácie (týždeň)
Obrázok 3 Kaplan-Meierov graf času do prvého výskytu kompozitného ukazovateľa:
kardiovaskulárne úmrtie, nefatálny infarkt myokardu alebo nefatálna cievna mozgová príhoda
(SUSTAIN 6)
FAS
Primárny cieľový ukazovateľ – MACE
K
omp
o
n
e
n
ty MACE Kardiovaskulárne úmrtie Nefatálna mozgová príhoda Nefatálny infarkt myokardu
Ďalšie sekundárne cieľové ukazovatele
Hazard Ratio
(95% CI)
0,74 (0,58- 0,95)
0,98 (0,65-1,48)
0,61 (0,38-0,99)
0,74 (0,51-1,08)
Semaglutid
N (%)
1648 (100)
108 (6,6)
44 (2,7)
27 (1,6)
47 (2,9)
Placebo
N (%)
1649 (100)
146 (8,9)
46 (2,8)
44 (2,7)
64
(3,9)
Všetky príčiny úmrtia 1,05 (0,74-1,50)
62 (3,8)
60 (3,6)
 0.2 1 5
V prospech semaglutidu V prospech placeba
Obrázok 4: Pruhový graf: analýza času do prvého prejavu kompozitného ukazovateľa, jeho
komponenty a všetky príčiny úmrtia (SUSTAIN 6)
0.2 1 5
V prospech semaglutidu V prospech placeba
Obrázok 4: Pruhový graf: analýza času do prvého prejavu kompozitného ukazovateľa, jeho
komponenty a všetky príčiny úmrtia (SUSTAIN 6)
Vyskytlo sa 158 prípadov novej alebo zhoršujúcej sa nefropatie. Pomer rizík [95 % CI] pre čas do nefropatie (nový nástup perzistentnej makroalbuminúrie, perzistentné zdvojnásobenie kreatinínu
v sére, potreba kontinuálnej renálnej substitučnej terapie a úmrtie kvôli ochoreniu obličiek) bol
0,64 [0,46; 0,88] v dôsledku nového nástupu perzistentnej makroalbuminúrie.
TelesnáhmotnosťPo roku liečby dosiahol úbytok telesnej hmotnosti ≥5 % a ≥10 % väčší počet účastníkov so semaglutidom 0,5 mg (46 % a 13 %) a 1 mg (52–62 % a 21–24 %) v porovnaní s aktívnymi komparátormi sitagliptínom (18 % a 3 %) a exenatidom ER (17 % a 4 %).
V 40-týždňovom skúšaní oproti dulaglutidu sa dosiahol úbytok telesnej hmotnosti ≥5 % a ≥10 %
u viac účastníkov, ktorí používali semaglutid 0,5 mg (44 % a 14 %) v porovnaní s dulaglutidom
0,75 mg (23 % a 3 %) a pri používaní semaglutidu 1 mg (až 63 % a 27 %) v porovnaní s dulaglutidom
1,5 mg (30 % a 8 %).
Významný a pretrvávajúci pokles telesnej hmotnosti od začiatku do 104. týždňa bol pozorovaný
pri semaglutide 0,5 mg a 1 mg oproti placebu 0,5 mg a 1 mg, po pridaní k štandardnej starostlivosti (-
3,6 kg a -4,9 kg oproti -0,7 kg a -0,5 kg, v uvedenom poradí) v SUSTAIN 6.
KrvnýtlakVýznamné zníženia priemerného systolického krvného tlaku sa pozorovali vtedy, keď sa semaglutid
0,5 mg (3,5–5,1 mmHg) a 1 mg (5,4–7,3 mmHg) používal v kombinácii s perorálnymi antidiabetikami alebo bazálnym inzulínom. Čo sa týka diastolického krvného tlaku neboli žiadne významné rozdiely medzi semaglutidom a komparátormi. Pozorované zníženia systolického krvného tlaku pri semaglutide v dávke 2 mg a v dávke 1 mg v 40. týždni boli 5,3 mmHg a 4,5 mmHg v uvedenom poradí.'
PediatrickápopuláciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre Ozempic v jednej alebo vo viacerých vekových podskupinách pediatrickej populácie s diabetom 2. typu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiV porovnaní s natívnym GLP-1 má semaglutid predĺžený polčas premeny približne 1 týždeň, čo ho robí vhodným na subkutánne podávanie jedenkrát týždenne. Hlavným mechanizmom predĺženia je
väzba na albumín, čo vedie k zníženiu renálneho klírensu a ochrane pred metabolickou degradáciou. Okrem toho je semaglutid stabilizovaný proti degradácii enzýmom DPP-4.
Absorpcia
Maximálna koncentrácia sa dosiahla 1 až 3 dni po dávke. Expozícia v ustálenom stave sa dosiahla po
4–5 týždňoch podávania jedenkrát týždenne. Pacienti s diabetom 2. typu mali priemerné koncentrácie v ustálenom stave po subkutánnom podaní 0,5 mg a 1 mg semaglutidu približne 16 nmol/l, resp.
30 nmol/l v uvedenom poradí. V skúšaní porovnávajúcom semaglutid 1 mg a 2 mg boli priemerné koncentrácie v ustálenom stave 27 nmol/l a 54 nmol/l v uvedenom poradí. Expozícia semaglutidu
u dávok 0,5 mg, 1 mg a 2 mg vzrastala spôsobom úmerným dávke. Podobná expozícia sa dosiahla pri subkutánnom podaní semaglutidu do brucha, stehna alebo nadlaktia. Absolútna biologická dostupnosť subkutánneho semaglutidu bola 89 %.
Distribúcia
Pacienti s diabetom 2. typu mali priemerný objem distribúcie semaglutidu po subkutánnom podaní
približne 12,5 l. Semaglutid sa vo veľkej miere viazal na albumín v plazme (>99 %).
Biotransformácia
Pred vylúčením sa semaglutid vo veľkej miere metabolizuje proteolytickým štiepením kostry peptidu
a sekvenčnou beta-oxidáciou mastnej kyseliny bočného reťazca. Predpokladá sa, že na metabolizme semaglutidu sa podieľa enzým neutrálna endopeptidáza (NEP).
Eliminácia
V skúšaní s jednou subkutánnou dávkou rádioaktívne označeného semaglutidu sa zistilo, že primárne
cesty vylučovania materiálu súvisiaceho so semaglutidom boli močom a stolicou. Približne 2/3 materiálu súvisiaceho so semaglutidom sa vylúčili močom a približne 1/3 stolicou. Približne 3 % dávky sa vylúčili močom ako intaktný semaglutid. Pacienti s diabetom 2. typu mali klírens semaglutidu približne 0,05 l/h. Pri polčase eliminácie približne 1 týždeň bude semaglutid prítomný v krvnom obehu približne 5 týždňov po poslednej dávke.
Špecifická populácia
Staršie osoby
Podľa údajov zo skúšaní fázy 3a zahŕňajúcich pacientov vo veku 20–86 rokov, vek nemá žiadny vplyv na farmakokinetiku semaglutidu.
Pohlavie,rasaaetnickápríslušnosť
Pohlavie, rasa (biela, čierna alebo afro-americká, aziatská) a etnická príslušnosť (hispánska alebo latinsko-americká, nehispánska alebo nelatinsko-americká) nemajú žiadny vplyv na farmakokinetiku semaglutidu.
Telesnáhmotnosť
Telesná hmotnosť má vplyv na expozíciu semaglutidu. Vyššia telesná hmotnosť vedie k nižšej expozícii; rozdiel 20 % medzi telesnými hmotnosťami jedincov bude viesť k približnému rozdielu
16 % v expozícii. Dávky semaglutidu 0,5 mg a 1 mg poskytujú adekvátnu systémovú expozíciu v rozsahu telesnej hmotnosti 40–198 kg.
Poruchafunkcieobličiek
Porucha funkcie obličiek neovplyvnila farmakokinetiku semaglutidu klinicky významným spôsobom. To sa preukázalo s jednou dávkou semaglutidu 0,5 mg u pacientov s rôznymi stupňami poruchy funkcie obličiek (mierna, stredná, závažná alebo pacienti na dialýze) v porovnaní s účastníkmi
s normálnou funkciou obličiek. Tiež sa to preukázalo u účastníkov s diabetom 2. typu a s poruchou
funkcie obličiek na základe údajov zo štúdií fázy 3a, aj keď skúsenosti s pacientmi v konečnej fáze ochorenia obličiek sú obmedzené.
Poruchafunkciepečene
Porucha funkcie pečene nemala žiadny vplyv na expozíciu semaglutidu. Farmakokinetika semaglutidu sa hodnotila u pacientov s rôznymi stupňami poruchy funkcie pečene (mierna, stredná, závažná)
v porovnaní s účastníkmi s normálnou funkciou pečene v skúšaní s jednou dávkou semaglutidu
0,5 mg.
Pediatrickápopulácia
Semaglutid sa neskúmal u pediatrických pacientov.
Imunogenicita
K tvorbe protilátok proti semaglutidu pri liečbe semaglutidom v dávke 1 mg a 2,4 mg dochádzalo zriedka (pozri časť 4.8) a odpoveď pravdepodobne nemala vplyv na farmakokinetiku semaglutidu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní alebo genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
U hlodavcov boli pozorované neletálne tumory C-buniek štítnej žľazy a sú účinkom triedy agonistov receptora GLP-1. V 2-ročných štúdiách karcinogenity u potkanov a myší spôsoboval semaglutid tumory C-buniek štítnej žľazy pri klinicky významných expozíciách. Žiadne ďalšie tumory súvisiace s liečbou neboli pozorované. Tumory C-buniek u hlodavcov sú spôsobené negenotoxickým mechanizmom sprostredkovaným špecifickým receptorom pre GLP-1, na ktorý sú hlodavce obzvlášť citlivé. Je pravdepodobné, že význam u ľudí bude nízky, ale nedá sa úplne vylúčiť.
V štúdiách fertility u potkanov semaglutid neovplyvnil párenie ani samčiu fertilitu. U samíc potkanov sa pozorovalo predĺženie estrálneho cyklu a mierne zníženie corpora lutea (ovulácií) pri dávkach spojených s úbytkom telesnej hmotnosti matky.
V štúdiách embryonálneho a fetálneho vývoja na potkanoch spôsoboval semaglutid embryotixicitu pod úrovňou klinicky významných expozícií. Semaglutid spôsobil značné úbytky telesnej hmotnosti matky a znížil prežívanie a rast embryí. U plodov boli pozorované veľké kostrové a viscerálne malformácie vrátane účinkov na dlhé kosti, rebrá, stavce, chvost, krvné cievy a mozgové komory. Mechanistické hodnotenia ukázali, že embryotoxicita zahŕňa poruchu zásobovania embrya živinami cez žĺtkový vak potkanov sprostredkovanú receptorom GLP-1. V dôsledku medzidruhových rozdielov v anatómii a funkcii žĺtkového vaku a v dôsledku nedostatočnej expresie receptora GLP-1 v žĺtkovom vaku nehumánnych primátov je nepravdepodobné, že by mal tento mechanizmus význam u ľudí. Avšak, priamy účinok semaglutidu na plod nemožno vylúčiť.
V štúdiách vývojovej toxicity na králikoch a makakoch jávskych sa pri klinicky významných expozíciách pozorovali zvýšené incidencie potratov a mierne zvýšené incidencie fetálnych abnormalít. Nálezy súviseli s pozorovaným úbytkom telesnej hmotnosti matky až do 16 %. Nie je známe, či sú tieto účinky spojené so zníženou konzumáciou jedla matkou ako priamy účinok GLP-1.
Postnatálny rast a vývoj sa hodnotil na makakoch jávskych. Dojčatá boli mierne menšie pri narodení, ale počas obdobia dojčenia sa zotavili.
U juvenilných potkanov spôsobil semaglutid oneskorené pohlavné dospievanie u samcov aj samíc. Toto oneskorenie nemalo vplyv na fertilitu ani reprodukčnú kapacitu ani jedného z pohlaví a ani na schopnosť samíc udržať tehotenstvo.
6
. FARMACEUTICKÉ INFORMÁCIE
6
.1 Zoznam pomocných látok
dihydrát hydrogenfosforečnanu sodného propylénglykol
fenol
kyselina chlorovodíková (na úpravu pH)
hydroxid sodný (na úpravu pH)
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Pred prvým použitím
Ozempic0,25mg,0,5mga1mg
3 roky.
Ozempic2mg
2 roky.
Po prvom otvorení
Čas použiteľnosti počas používania: 6 týždňov.
Uchovávajte pri teplote do 30 °C alebo v chladničke (2 °C až 8 °C). Ozempic neuchovávajte v mrazničke. Keď pero nepoužívate, ponechajte kryt na pere na ochranu pred svetlom.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C až 8 °C). Neuchovávajte v blízkosti chladiacej jednotky. Ozempic neuchovávajte v mrazničke.
Na pere ponechajte kryt na ochranu pred svetlom.
Podmienky na uchovávanie po prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
1,5 ml alebo 3 ml sklenená náplň (sklo typu I) zavretá na jednom konci gumovým piestom (chlórbutyl) a na druhom konci hliníkovým krytom so vsadeným laminátovým gumovým uzáverom (brómbutyl/polyizoprén). Náplň je v jednorazovom naplnenom pere, ktoré je vyrobené
z polypropylénu, polyoxymetylénu, polykarbonátu a akrylonitrilbutadiénstyrénu.
Veľkosti balenia:
Ozempic0,25mginjekčnýroztok
Každé naplnené pero obsahuje 1,5 ml roztoku, podáva 4 dávky po 0,25 mg.
1 naplnené pero a 4 jednorazové ihly NovoFine Plus
Ozempic0,5mginjekčnýroztok
Každé naplnené pero obsahuje 1,5 ml roztoku, podáva 4 dávky po 0,5 mg.
1 naplnené pero a 4 jednorazové ihly NovoFine Plus
3 naplnené perá a 12 jednorazových ihiel NovoFine Plus
Ozempic1mginjekčnýroztokKaždé naplnené pero obsahuje 3 ml roztoku, podáva 4 dávky po 1 mg.
1 naplnené pero a 4 jednorazové ihly NovoFine Plus
3 naplnené perá a 12 jednorazových ihiel NovoFine Plus
Ozempic2mginjekčnýroztokKaždé naplnené pero obsahuje 3 ml roztoku, podáva 4 dávky po 2 mg.
1 naplnené pero a 4 jednorazové ihly NovoFine Plus
3 naplnené perá a 12 jednorazových ihiel NovoFine Plus
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomPacient má byť upozornený, aby ihlu po každom podaní injekcie zlikvidoval a aby pero uchovával bez nasadenej ihly. To môže zabrániť upchatiu ihiel, kontaminácii, infekcii, vytekaniu roztoku
a nepresnému dávkovaniu.
Pero je určené na použitie len pre jednu osobu.
Ozempic sa nemá používať, ak nie je číry a bezfarebný alebo takmer bezfarebný. Ozempic sa nemá používať, ak bol zmrazený.
Ozempic je možné podať pomocou jednorazových ihiel 30G, 31G a 32G s maximálnou dĺžkou 8 mm. Všetok nepoužitý liek a iný odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovo Nordisk A/S Novo Allé
DK-2880 Bagsværd
Dánsko
8. REGISTRAČNÉ ČÍSLAEU/1/17/1251/002
EU/1/17/1251/003
EU/1/17/1251/004
EU/1/17/1251/005
EU/1/17/1251/006
EU/1/17/1251/010
EU/1/17/1251/011
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 8. februára 2018
Dátum posledného predĺženia registrácie: 21.september 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.

Návod na použitie Ozempic 2 mg injekčný roztok v naplnenom pere
|
Pred použitím naplneného pera Ozempic si starostlivo
prečítajte tento návod.
Poraďte sa so svojím lekárom, zdravotnou sestrou alebo lekárnikom o tom, ako si správne podať Ozempic. Začnite tým, že skontrolujte pero, aby ste sa uistili, že obsahuje Ozempic 2 mg, potom si pozrite obrázky uvedené nižšie a oboznámte sa s rôznymi časťami pera a ihly.
Ak ste nevidiaci alebo máte slabý zrak a nemôžete čítať na počítadle dávky na pere, nepoužívajte toto pero bez pomoci. Požiadajte o pomoc osobu s dobrým zrakom, ktorá vie ako používať naplnené pero Ozempic. Toto je naplnené dávkovacie pero. Obsahuje 8 mg semaglutidu a môžete zvoliť len dávky 2 mg. Jedno
nepoužité pero obsahuje štyri dávky po 2 mg.
Použite tabuľku vo viečku škatuľky na sledovanie koľko injekcií ste si podali a kedy ste si ich podali.
Pero je určené na použitie s jednorazovými ihlami 30G,
31G a 32G s maximálnou dĺžkou 8 mm. Ihly NovoFine Plus sú súčasťou balenia.
|
Ozempic naplnené pero a ihla (vzor)
Vonkajší
K
r
y
t pera
k
r
y
t ihly
Vnútorný
k
r
y
t ihly
Ihla
Papierový
O
k
ienko pera štítok
Š
títok pera
Počítadlo dávky
U
k
a
z
o
va
teľ dávky
Volič dávky
S
y
m
b
o
l
Dávkovacie
k
o
n
troly
tlačidlo
p
r
ietoku
|
 Dôležité informácie
Dôležité informácie
Venujte zvláštnu pozornosť týmto poznámkam, pretože sú dôležité pre bezpečné používanie pera.
|
1
. Príprava pera s novou ihlou
|
• Skontrolujte názov a farbu štítku pera, aby ste sa uistili, že obsahuje Ozempic 2 mg. To je mimoriadne dôležité, ak používate viac ako jeden typ injekčne podávaného lieku. Použitie
nesprávneho lieku môže byť škodlivé pre vaše
zdravie.
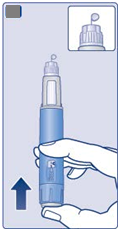
• Stiahnite kryt pera
|
A
|
• Skontrolujte, či je roztok v pere číry a bezfarebný. Pozrite sa do okienka pera. Ak roztok vyzerá zakalený alebo zafarbený, toto pero
nepoužívajte.
|
B
|


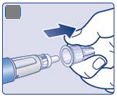

• Zoberte si novú ihlu.
• Skontrolujte, či nie je poškodený papierový štítok a vonkajší kryt ihly, čo by mohlo ovplyvniť sterilitu. Ak zistíte akékoľvek poškodenie, použite novú ihlu.
• Odtrhnite papierový štítok.
|
C
|
Uistite sa, že ste ihlu správne nasadili.
• Zatlačte ihlu rovno na pero.
• Otáčaním utiahnite.
|
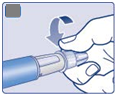
D
|
Ihla je prekrytá dvoma krytmi. Musíte odstrániť oba
kryty.
Ak zabudnete odstrániť oba kryty, nepodáte si žiaden liek.
• Stiahnite vonkajší kryt ihly a odložte si ho na neskôr. Budete ho potrebovať po podaní injekcie na bezpečné odstránenie ihly z pera.
|
E
|
• Odstráňte vnútorný kryt ihly a zahoďte ho. Ak sa ho budete snažiť znova nasadiť, môžete sa
ihlou neúmyselne pichnúť.
Na hrote ihly sa môže objaviť kvapka roztoku. To je normálne, ale aj tak musíte skontrolovať prietok, ak nové pero používate prvýkrát. Pozri krok 2 „Kontrola prietoku s každým novým perom”.
Nenasadzujte novú ihlu na pero, kým nie ste pripravený podať si injekciu.
|
F
|
Vždy použite novú ihlu na každé podanie injekcie.
To znižuje riziko upchatia ihiel, kontaminácie, infekcie a nepresného dávkovania.
|
Nikdy nepoužívajte ohnutú alebo poškodenú ihlu.
|
2
. Kontrola prietoku s každým novým perom
|
• Ak pero už používate, prejdite na krok 3
„Nastavenie dávky”. Pred podaním prvej injekcie každým novým perom vždy skontrolujte prietok.
• Otočte volič dávky na symbol kontroly prietoku ( ) tesne za hodnotou „0“. Uistite sa, že symbol kontroly prietoku je zarovnaný s ukazovateľom dávky. ) tesne za hodnotou „0“. Uistite sa, že symbol kontroly prietoku je zarovnaný s ukazovateľom dávky.
|
A
N
a
stavený symbol kontroly prietoku
|
• Držte pero ihlou smerom nahor.
Tlačte a držte dávkovacie tlačidlo stlačené, kým sa počítadlo dávky nevráti na 0. Hodnota 0 musí byť zarovno s ukazovateľom dávky.
Na hrote ihly sa má objaviť kvapka roztoku.
|
B
|

Na hrote ihly môže zostať malá kvapka, ktorá sa však injekciou nepodá.
Ak sa žiadna kvapka neobjaví, opakujte krok 2 „Kontrola prietoku s každým novým perom” najviac 6-krát. Ak sa ani potom neobjaví žiadna kvapka, vymeňte ihlu a opakujte krok 2 „Kontrola prietoku s každým novým perom” ešte raz.
Ak sa kvapka stále neobjavila, pero zlikvidujte a použite nové pero.
|
Predtým ako použijete nové pero prvýkrát, vždy sa uistite, že sa na hrote ihly objavila kvapka. Budete tak mať istotu, že je zabezpečený prietok roztoku.
Ak sa žiadna kvapka neobjaví, nepodáte si injekciou žiaden liek, hoci počítadlo dávky sa môže pohybovať. Môže to znamenať, že je ihla upchatá alebo poškodená.
Ak neskontrolujete prietok pred prvou injekciou každým novým perom, môže sa stať, že nedostanete predpísanú dávku a nedosiahnete cieľový účinok lieku Ozempic.
|
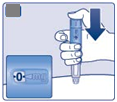
3
. Nastavenie dávky
|
• Otáčajte voličom dávky, aby ste si nastavili
2 mg.
Otáčajte až počítadlo dávky zastane a ukáže 1 mg.
|
A
n
a
stavené
2 mg
|
Len počítadlo dávky a ukazovateľ dávky ukazujú, že boli nastavené 2 mg.
Môžete zvoliť len 2 mg na dávku. Ak vaše pero obsahuje menej ako 2 mg, počítadlo dávky sa zastaví skôr, ako sa zobrazí 2.
Volič dávky rôzne kliká pri otáčaní dopredu, dozadu alebo pri zostávajúcich 2 mg. Nepočítajte kliknutia pera.
|
Pred injekčným podávaním tohto lieku vždy použite počítadlo dávky a ukazovateľ dávky, aby ste videli, že boli nastavené 2 mg.
Nepočítajte kliknutia pera.
2 mg musia byť presne zarovnané s ukazovateľom dávky, aby sa zaistilo podávanie správnej dávky.
|
|
Koľko roztoku zostáva
|
|
• Aby ste videli koľko roztoku zostáva, použite počítadlo dávky: Otáčajte voličom dávky až počítadlo dávky zastane. Ak ukáže 2, najmenej 2 mg zostáva v pere.
Ak počítadlo dávky zastane pred 2 mg, nezostáva dostatok roztoku na celú dávku 2 mg.
|
A
Počítadlo dávky zastalo: zostáva 2mg
|
 Ak v pere nezostáva dostatok roztoku na celú dávku, nepoužite ho. Použite nové pero Ozempic. Ak v pere nezostáva dostatok roztoku na celú dávku, nepoužite ho. Použite nové pero Ozempic.
|
4
. Injekčné podanie dávky
|
• Zaveďte ihlu do kože, ako vám ukázal lekár alebo zdravotná sestra.
• Presvedčte sa, že vidíte počítadlo dávky.
Neprekrývajte ho prstami. Mohlo by sa tým prerušiť podávanie injekcie.
|
A
|
• Tlačte a držte dávkovacie tlačidlo stlačené.
Sledujte, ako sa počítadlo dávky vracia na „0“.. Hodnota „0“ musí byť zarovno s ukazovateľom dávky. Potom môžete počuť alebo pocítiť kliknutie.
• Pokračujte v stláčaní dávkovacieho tlačidla, pričom držte ihlu v koži.
|
B
|
|
|
|
|
|
|

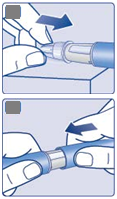
• Nechajte ihlu v koži a pomaly počítajte do 6.
• Ak ihlu vytiahnete skôr, môžete vidieť, ako z hrotu ihly vyteká prúd roztoku. V takom prípade sa nepodá celá dávka.
|
C
Počítajte pomaly:
1
-
2
-
3
-
4
-5-6
|
• Vytiahnite ihlu z kože. Potom môžete uvoľniť dávkovacie tlačidlo.
• Ak sa v mieste podania injekcie objaví krv, jemne naň pritlačte.
|
D
|
Po podávaní injekcie môžete na hrote ihly vidieť kvapku roztoku. Je to normálne a neovplyvní to dávku.
|
Vždy sledujte počítadlo dávky, aby ste vedeli, koľko mg si podávate. Držte dávkovacie tlačidlo úplne zatlačené, až kým sa počítadlo dávky nevráti na „0“.
Ako zistiť, že je ihla upchatá alebo poškodená
– Ak sa po neprerušenom stlačení dávkovacieho tlačidla nezobrazí na počítadle dávky 0, je možné, že ste použili upchatú alebo poškodenú ihlu.
– V takom prípade ste si nepodali žiadny liek – aj keď sa počítadlo dávky pohlo z pôvodnej dávky, ktorú ste nastavili.
Čo urobiť s upchatou ihlou
Vymeňte ihlu postupom uvedeným v kroku 5 „Po podaní injekcie” a zopakujte všetky kroky počnúc krokom 1 „Príprava pera s novou ihlou”. Presvedčte sa, že ste nastavili celú dávku, ktorú potrebujete.
Pri podávaní injekcie sa nikdy nedotýkajte počítadla dávky. Môže sa tým prerušiť podávanie injekcie.
|
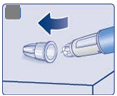
5
. Po podaní injekcie
|
• Po každej injekcii ihlu vždy zlikvidujte, aby ste zabezpečili pohodlné podanie injekcie a zabránili upchatiu ihiel. Ak je ihla upchatá, nepodáte si žiadny liek.
• Na rovnej ploche vsuňte hrot ihly do vonkajšieho krytu ihly bez toho, aby ste sa dotýkali ihly alebo vonkajšieho krytu ihly.
|
A
|
• Keď je ihla zakrytá, opatrne na ňu na doraz zatlačte vonkajší kryt ihly.
• Odskrutkujte ihlu a opatrne ju zlikvidujte podľa pokynov svojho lekára, zdravotnej sestry, lekárnika alebo národných predpisov.
|
B
|
• Po každom použití dajte kryt na pero, aby ste chránili roztok pred svetlom.
|
C
|
Keď je pero prázdne, zahoďte ho bez nasadenej ihly podľa pokynov lekára, zdravotnej sestry, lekárnika alebo národných predpisov.
|
Nikdy sa nepokúšajte dať vnútorný kryt ihly naspäť na ihlu. Mohli by ste sa ihlou pichnúť.
Po každom podaní injekcie vždy ihneď odstráňte ihlu z pera.
Môže to zabrániť upchaniu ihiel, kontaminácie, infekcie, vytekania roztoku a nepresného dávkovania.
|
Ďalšie dôležité informácie
|
• Pero a ihly vždy uchovávajte mimo dohľadu a dosahu iných osôb, najmä detí.
• Pero a ihly nikdy neposkytujte iným osobám.
• Opatrovatelia musia byť pri zaobchádzaní s použitými ihlami veľmi opatrní, aby zabránili poraneniu a prenosu infekcie.
|
Starostlivosť o pero
|
S perom zaobchádzajte opatrne. Hrubé zaobchádzanie alebo nesprávne používanie môže mať za následok nepresné dávkovanie. Ak sa to stane, môže dôjsť k tomu, že nedosiahnete cieľový účinok tohto lieku.
|
• Nenechávajte pero v aute alebo na inom mieste, kde môže byť príliš horúco alebo príliš chladno.
• Nepodávajte si injekčne Ozempic, ktorý bol zmrazený. Ak to spravíte, môže dôjsť k tomu, že nedosiahnete cieľový účinok tohto lieku.
• Nepodávajte si injekčne Ozempic, ktorý bol vystavený priamemu slnečnému žiareniu. Ak to spravíte, môže dôjsť k tomu, že nedosiahnete cieľový účinok tohto lieku.
• Pero nevystavujte prachu, nečistotám ani kvapalinám.
• Pero neumývajte, nenamáčajte ani nemažte. Môže sa čistiť jemným čistiacim prostriedkom pomocou vlhkej handričky.
• Nenechajte pero spadnúť ani naraziť na tvrdý povrch. Ak vám pero spadne alebo máte podozrenie na nejaký problém, nasaďte novú ihlu a pred podávaním injekcie skontrolujte prietok.
• Nepokúšajte sa pero znova napĺňať.
• Nepokúšajte sa pero opravovať ani rozoberať.
|
Príloha IV
Vedecké závery a dôvody zmeny podmienok rozhodnutia
(rozhodnutí) o registrácii
Vedecké závery
Vzhľadom na hodnotiacu správu Výboru pre hodnotenie rizík liekov (PRAC) o periodicky aktualizovaných správach o bezpečnosti lieku (PSUR) pre semaglutid dospel Výbor pre humánne lieky (CHMP) k týmto vedeckým záverom:
Vzhľadom na dostupné údaje z klinických skúšaní a spontánnych hlásení o dysgeuzii považuje výbor PRAC kauzálnu súvislosť medzi semaglutidom p.o. (Rybelsus) a dysgeuziou za prinajmenšom opodstatnenú možnosť. Výbor PRAC dospel k záveru, že informácie o liekoch obsahujúcich perorálny semaglutid (Rybelsus) majú byť zodpovedajúcim spôsobom zmenené.
Vzhľadom na dostupné údaje z klinických skúšaní o oneskorenom vyprázdňovaní žalúdka považuje výbor PRAC kauzálnu súvislosť medzi semaglutidom (Ozempic, Rybelsus a Wegovy) a oneskoreným vyprázdňovaním žalúdka za prinajmenšom opodstatnenú možnosť. Výbor PRAC dospel k záveru, že informácie o liekoch obsahujúcich semaglutid majú byť zodpovedajúcim spôsobom zmenené.
Výbor pre humánne lieky (CHMP) súhlasí s vedeckými závermi PRAC.
Dôvody zmeny podmienok rozhodnutia (rozhodnutí) o registráciiNa základe vedeckých záverov pre semaglutid je CHMP toho názoru, že pomer prínosu a rizika lieku (liekov) obsahujúceho (obsahujúcich) semaglutid je nezmenený za predpokladu, že budú prijaté navrhované zmeny v informáciách o lieku.
CHMP odporúča zmenu podmienok rozhodnutia o registrácii (rozhodnutí o registrácii).