
ebo pacientov na dialýze sú k dispozícii len obmedzené klinické údaje. Pri liečbe pacientov s ťažkým alebo terminálnym poškodením obličiek je
potrebné dôkladné bezpečnostné monitorovanie (pozri časti 4.4 a 5.2).
Pediatrická populácia
U pacientov do prvého roku života je k dispozícii len obmedzené množstvo údajov. Pri liečbe týchto pacientov je potrebné postupovať opatrne (pozri časť 5.2).
Spôsob podávania
Len na subkutánne použitie.
Tento liek sa dodáva ako roztok pripravený na použitie v jednorazovej injekčnej liekovke.
• Požadovaný objem Oxluma sa má vypočítať na základe odporúčanej dávky stanovenej podľa telesnej hmotnosti, ako je uvedené v tabuľke 1.
• Ak je dávka väčšia ako 0,5 ml (94,5 mg), bude potrebná viac ako jedna injekčná liekovka.
• Maximálny prijateľný objem jednorazovej injekcie je 1,5 ml. Ak sú potrebné dávky väčšie ako
1,5 ml, majú sa podávať vo forme viacerých injekcií (celková dávka rovnomerne rozdelená na jednotlivé striekačky, pričom každá injekcia obsahuje približne rovnaký objem), aby sa minimalizoval nepríjemný pocit v mieste podania vyvolaný objemom podanej injekcie.
• Zabráňte tomu, aby sa liek dostal do špičky ihly skôr, ako bude ihla v subkutánnom priestore.
• Tento liek má byť podaný subkutánne injekciou do brucha, ramien alebo stehien.
• V prípade nasledujúcich injekcií alebo dávok sa odporúča striedanie miest podania.
• Tento liek sa nemá podávať do zjazveného tkaniva ani do oblastí, ktoré sú začervenané, zapálené alebo opuchnuté.
Oxlumo má podávať lekár. Pokyny týkajúce sa lieku pred jeho podaním, pozri časť 6.6.
4.3 Kontraindikácie
Závažná precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Ťažké alebo terminálnepoškodenieobličiek
Liečba lumasiranom zvyšuje hladiny glykolátu v plazme, čo môže zvýšiť riziko metabolickej acidózy
alebo spôsobiť zhoršenie už existujúcej metabolickej acidózy u pacientov s ťažkým alebo terminálnym poškodením obličiek. Z tohto dôvodu majú byť títo pacienti sledovaní, či sa u nich nevyskytujú
prejavy alebo príznaky metabolickej acidózy.
Stredná alebo závažnáporuchafunkciepečene
U pacientov so strednou alebo závažnou poruchou funkcie pečenie môže dôjsť ku zníženiu účinnosti.
Preto sa má u týchto pacientov sledovať účinnosť (pozri časť 5.2),
Pomocná látka (obsah sodíka)
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednom ml, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne klinické liekové interakčné štúdie (pozri časť 5.2). Súbežnépoužitiespyridoxínom
Súbežné použitie pyridoxínu významne neovplyvnilo farmakodynamiku ani farmakokinetiku
lumasiranu.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii žiadne údaje o použití lumasiranu u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). O
užívaní tohto lieku počas gravidity sa má uvažovať s prihliadnutím na očakávaný zdravotný prínos pre ženu a prípadné riziká pre plod.
Dojčenie
Nie je známe, či sa lumasiran vylučuje do ľudského mlieka. Riziko u novorodencov alebo dojčiat
nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Oxlumom, sa musí urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne údaje o vplyve lumasiranu na fertilitu u ľudí. V štúdiách na zvieratách sa
nezistil žiadny vplyv na fertilitu samcov ani samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Oxlumo nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluNajčastejšie hlásenou nežiaducou reakciou bola reakcia v mieste podania injekcie (32 %).
Tabuľkový prehľadnežiaducichreakciíNežiaduce reakcie súvisiace s lumasiranom získané z klinických štúdií sú uvedené nižšie v tabuľke.
Nežiaduce reakcie sú klasifikované podľa preferovaných termínov (PTs, preferred terms) tried orgánových systémov (SOC, system organ class) databázy MedDRA a uvedené podľa frekvencie ich
výskytu. Frekvencia nežiaducich reakcií je vyjadrená podľa nasledovných kategórií: veľmi časté
(≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); veľmi zriedkavé (< 1/10 000).
Tabuľka 2: Nežiaduce reakcieTrieda orgánových systémov
| Nežiaduca reakcia
| Frekvencia
|
Poruchy gastrointestinálneho traktu
| Bolesť bruchaa
| Veľmi časté
|
Celkové poruchy a reakcie v mieste podania
| Reakcia v mieste podania injekcieb
| Veľmi časté
|
a Zahŕňa bolesť brucha, bolesť v hornej časti brucha, bolesť v dolnej časti brucha, nepríjemný pocit v bruchu a citlivosť brucha.
b Zahŕňa reakciu v mieste podania injekcie, erytém v mieste podania injekcie, bolesť v mieste
podania injekcie, svrbenie v mieste podania injekcie, opuch v mieste podania injekcie, nepríjemný pocit v mieste podania injekcie, sfarbenie v mieste podania injekcie, hrču v mieste podania injekcie,
stvrdnutie v mieste podania injekcie, vyrážku v mieste podania injekcie, podliatinu v mieste
podania injekcie, hematóm v mieste podania injekcie a exfoliáciu kože v mieste podania injekcie.
Popis vybraných nežiaducich reakciíReakcie v mieste podania injekcieV placebom kontrolovaných a otvorených klinických štúdiách boli reakcie v mieste podania injekcie hlásené u 26 z 81 pacientov (32,1 %), pričom sa vyskytli u 10 % injekcií. Najčastejšie hlásené príznaky boli erytém, bolesť, pruritus a opuch. Väčšina reakcií v mieste podania injekcie začala v deň podania, 7 pacienti mali reakcie v mieste podania injekcie, ktoré začali 5 alebo viac dní po podaní (vyskytli sa u 1,6 % injekcií). Reakcie v mieste podania injekcie boli všeobecne mierne, vymizli do dvoch dní a nespôsobili prerušenie ani ukončenie liečby.
Bolesť bruchaV placebom kontrolovanej štúdii bola bolesť brucha hlásená u 1 z 13 (7,7 %) pacientov liečených placebom a 4 z 26 (15,4 %) pacientov liečených lumasiranom. V placebom kontrolovaných
a otvorených klinických štúdiách hlásilo 17 z 81 pacientov (21,0 %) bolesť brucha vrátane bolesti v hornej alebo dolnej časti brucha, nepríjemného pocitu v bruchu alebo citlivosti brucha. Väčšina
udalostí boli mierne, prechodné a vymizli bez liečby. Žiadne neviedli k prerušeniu liečby.
ImunogenitaU pacientov s PH1 a zdravých dobrovoľníkov, ktorým sa podávalo Oxlumo, bolo 6 zo 100 (6,0 %) jedincov pozitívne testovaných na protilátky proti liekom (ADA, anti-drug-antibodies). Titre ADA boli nízke a všeobecne prechodné bez vplyvu na účinnosť, bezpečnosť, farmakokinetický alebo farmakodynamický profil lieku.
Pediatrická populáciaBezpečnostný profil lumasiranu bol podobný u pediatrických pacientov (vo veku od 4 mesiacov do
17 rokov) a dospelých pacientov s PH1.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania sa odporúča sledovanie pacienta podľa lekárskej indikácie z hľadiska akýchkoľvek prejavov a príznakov nežiaducich reakcií a začatie vhodnej symptomatickej liečby.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: zatiaľ nepridelená, ATC kód: zatiaľ nepridelený.
Mechanizmus účinkuLumasiran je dvojvláknová malá interferujúca ribonukleová kyselina (siRNA, small interfering
ribonucleic acid), ktorá znižuje hladiny enzýmu glykolát oxidázy (GO) zacielením na mediátorovú ribonukleovú kyselinu (mRNA, gene messenger ribonucleic acid) génu oxidázy hydroxykyseliny
1 (
HAO1) v hepatocytoch prostredníctvom RNA interferencie. Znížené hladiny enzýmu GO redukujú
množstvo dostupného glyoxylátu, substrátu na produkciu oxalátu. To vedie k zníženiu hladín oxalátu v moči a plazme, čo je hlavnou príčinou prejavov choroby u pacientov s PH1. Pretože je sekvencia
enzýmu GO v protismere expresie génu deficientného enzýmu alanín: glyoxylátaminotransferáza
(AGT), ktorý spôsobuje PH1, mechanizmus účinku lumasiranu je nezávislý od príčinnej mutácie génu
AGXT.
Klinická účinnosťÚčinnosť lumasiranu sa skúmala v randomizovanej dvojito zaslepenej, placebom kontrolovanej
klinickej štúdii u pacientov vo veku 6 rokov a starších s PH1 (ILLUMINATE-A) a v jednoramennej klinickej štúdii u pacientov mladších ako 6 rokov s PH1 (ILLUMINATE-B).
ILLUMINATE-ACelkom 39 pacientov s PH1 bolo randomizovaných v pomere 2:1 a dostali subkutánne dávky lumasiranu alebo placeba počas 6-mesačného dvojito zaslepeného placebom kontrolovaného obdobia. Do štúdie boli zaradení pacienti vo veku 6 rokov a starší s odhadovanou rýchlosťou glomerulárnej filtrácie (eGFR) ≥ 30 ml/min/1,73 m² a dostali 3 úvodné dávky 3 mg/kg lumasiranu alebo placeba podávané jedenkrát mesačne, po ktorých nasledovali štvrťročné udržiavacie dávky 3 mg/kg lumasiranu alebo placeba (pozri časť 4.2). Po 6-mesačnom dvojito zaslepenom období liečby pacienti vrátane tých, ktorí boli pôvodne zaradení do skupiny s placebom, vstúpili do predĺženého obdobia s podávaním lumasiranu.
Počas 6-mesačného dvojito zaslepeného placebom kontrolovaného obdobia 26 pacientov dostalo lumasiran a 13 pacientov dostalo placebo. Stredný vek pacientov pri prvej dávke bol 14,9 roka
(v rozsahu od 6,1 do 61,0 rokov), 66,7 % boli muži a 76,9 % boli belosi. Medián 24-hodinového
vylučovania oxalátu močom korigovaný na plochu povrchu tela (BSA, body surface area) na začiatku bol 1,72 mmol/24 h/1,73 m², medián pomeru oxalátu a kreatinínu v spotovom moči na začiatku bol
0,21 mmol/mmol a stredná hladina oxalátu v plazme na začiatku bola 13,1 µmol/l. Celkovo 33,3 % pacientov malo normálnu funkciu obličiek (eGFR ≥ 90 ml/min/1,73 m²), 48,7 % malo miernu poruchu funkcie obličiek (eGFR 60 až < 90 ml/min/1,73 m²) a 18 % malo stredne ťažkú poruchu funkcie obličiek (eGFR 30 až < 60 ml/min/1,73 m²). U pacientov zaradených do štúdie bola u 84,6 % zaznamenaná anamnéza symptomatických udalosti obličkových kameňov a u 53,8 % bola zaznamenaná anamnéza nefrokalcinózy na začiatku. Liečebné ramená boli na začiatku vyvážené
s ohľadom na vek, hladinu oxalátu v moči a eGFR.
Primárnym koncovým ukazovateľom bolo percentuálne zníženie 24-hodinového vylučovania oxalátu v moči korigovaného na plochu povrchu tela v priemere od 3 do 6 mesiacov v porovnaní
s východiskovými hodnotami. Lumasiran bol spájaný so štatisticky významným znížením o 65,4 %
v 24-hodinovom oxaláte v moči korigovanom na plochu povrchu tela v porovnaní s 11,8 % v skupine s placebom, čo predstavuje rozdiel 53,5 % (95 % CI: 44,8, 62,3; p < 0,0001). V súlade s primárnym koncovým ukazovateľom bolo v 6. mesiaci pozorované zníženie pomeru oxalátu a kreatinínu
v spotovom moči v ramene s lumasiranom o 60,5 %, v porovnaní s 8,5 % zvýšením v ramene
s placebom. Ďalej, pacienti liečení lumasiranom mali rýchly a trvalý pokles 24-hodinového oxalátu v moči korigovaného na plochu povrchu tela, ako je znázornené na obrázku 1.
Obrázok 1: ILLUMINATE-A: Percentuálna zmena oproti východiskovej hodnotev 24-hodinovom oxaláte v moči korigovanom na plochu povrchu tela podľa mesiaca

BL M1 M2 M3 M4 M5 M6
Počet pacientov:
Kontrolné vyšetrenie v rámci štúdie

Liečebná skupina —■— Lumasiran —r— Placebo
■
|
N =
|
26
|
24
|
26
|
24
|
23
|
25
|
25
|
r
|
N =
|
13
|
13
|
12
|
13
|
13
|
13
|
13
|
Skratky: BL (baseline) = východisková hodnota; BSA = plocha povrchu tela; M = mesiac;
SEM (standard error of mean) = štandardná chyba priemeru.
Výsledky sú vykreslené ako priemer (±SEM) percentuálnej zmeny oproti východiskovej hodnote.
V 6. mesiaci dosiahol vyšší podiel pacientov liečených lumasiranom normálne alebo takmer normálne hladiny 24-hodinového oxalátu v moči korigovaného na BSA (≤ 1,5 × ULN) v porovnaní s pacientmi liečenými placebom, ako je uvedené v tabuľke 3.
Tabuľka 3: ILLUMINATE-A: Výsledky sekundárneho koncového ukazovateľa počas6-mesačného dvojito zaslepeného placebom kontrolovaného obdobiaKoncové ukazovatele
| Lumasiran
(N = 26)
| Placebo
(N = 13)
| Liečebný rozdiel
(95 % Cl)
| p-hodnota
|
Podiel pacientov
s 24-hodinovými hladinami oxalátu v moči na úrovni alebo pod ULN‡
| 0,5 (0,3; 0,7)§
| 0 (0; 0,2)§
| 0,5 (0,2; 0,7)¶
| 0,001#
|
K
oncové ukazovatele
|
L
umasiran
(
N = 26)
|
Placebo
(
N = 13)
|
L
i
ečebný rozdiel
(
95 % Cl)
|
p-hodnota
|
Podiel pacientov
s 24-hodinovými hladinami oxalátu v moči na úrovni alebo pod 1,5 × ULN‡
|
0,8 (0,6; 1,0)§
|
0 (0; 0,2)§
|
0,8 (0,5; 0,9)¶
|
< 0,0001#
|
Percentuálne zníženie oxalátu v plazme oproti východiskovej hodnote*Þ
|
39,8 (2,9)†
|
0,3 (4,3)†
|
39,5 (28,9; 50,1)
|
< 0,0001
|
Skratky: ULN (upper limit of normal) = horná hranica normálnych hodnôt; SEM = štandardná chyba priemeru
Výsledky sú založené na teste tandemovej hmotnostnej spektrometrie s kvapalinovou chromatografiou
(LC-MS/MS, liquid chromatography tandem mass spectrometry).
* Odhad na základe priemeru strednej hodnoty percentuálneho zníženia v 3., 4., 5. a 6. mesiaci vypočítaného
metódou najmenších štvorcov (LS, least square) za použitia zmiešaného modelu pre opakované merania.
† Stredná hodnota LS (SEM).
‡ ULN = 0,514 mmol/24 h/1,73 m² pre 24-hodinový oxalát v moči korigovaný na BSA.
§ 95 % interval spoľahlivosti na základe Clopper Pearsonovho presného intervalu spoľahlivosti.
¶ Vypočítané pomocou Newcombovej metódy na základe Wilsonovho skóre.
# p-hodnota je založená na Cochran-Mantel-Haenszelovom teste stratifikovanom podľa východiskovej hodnoty
24-hodinového oxalátu v moči korigovaného na BSA (≤ 1,70 oproti > 1,70 mmol/24 h/1,73 m²).
Þ Analyzované u 23 pacientov s lumasiranom a 10 pacientov s placebom, ktorí mali východiskové hladiny, ktoré umožňovali zníženie.
Zníženie 24-hodinového oxalátu v moči korigovaného na BSA oproti východiskovej hodnote
u pacientov s PH1 užívajúcich lumasiran v porovnaní s placebom bolo podobné vo všetkých vopred určených podskupinách vrátane veku, pohlavia, rasy, poruchy funkcie obličiek, východiskového
použitia pyridoxínu (vitamín B6) a anamnézy symptomatických udalostí obličkových kameňov
(obrázok 2).
Obrázok 2: ILLUMINATE-A: Percentuálna zmena oproti východiskovej hodnotev 24-hodinovom oxaláte v moči korigovanom na BSA, analýza podskupín
Podskupina
|
Lumasiran – Placebo
|
Lumasiran
|
Placebo
|
|
| (N)
| (N)
|
Celkovo
|
| 26
| 13
|
Vek pri skríningu
|
|
|
|
6 – < 12 rokov
|
| 9
| 7
|
12 – < 18 rokov
|
| 5
| 1
|
≥ 18 rokov
|
| 12
| 5
|
Pohlavie
|
|
|
|
Muž
|
| 18
| 8
|
Žena
|
| 8
| 5
|
Rasa
|
|
|
|
Biela
|
| 21
| 9
|
Iná ako biela
|
| 5
| 4
|
Východiskové použitie pyridoxínu
|
|
|
|
Áno
|
| 13
| 9
|
Nie
|
| 13
| 4
|
Východisková hodnota 24-hodinového oxalátu v moči korigovaného na BSA
|
|
|
|
≤ 1,70 mmol/24 h/1,73 m²
|
| 11
| 7
|
> 1,70 mmol/24 h/1,73 m²
|
| 15
| 6
|
Východisková hodnota eGFR
|
|
|
|
< 60 ml/min/1,73 m²
|
| 4
| 3
|
≥ 60 ml/min/1,73 m²
|
| 22
| 10
|
Anamnéza symptomatických udalostí obličkových kameňov počas celého života
|
|
|
|
Áno
|
| 23
| 10
|
Nie
|
| 3
| 3
|

V prospech lumasiranu V prospech placeba
Znížené hladiny oxalátu pozorované v dvojito zaslepenom období sa udržiavali 12 mesiacov počas
predĺženého obdobia štúdie.
Hodnota eGFR a udalosti súvisiace s obličkovými kameňmi (hlásené podľa udalostí na 100 osobodní) sa hodnotili prostredníctvom dvojito zaslepených a predĺžených období celkovo 12 mesiacov. Hodnota eGFR zostala stabilná u pacientov, ktorým bol podávaný lumasiran. V ramene lumasiranu bola miera udalostí súvisiacich s obličkovými kameňmi hlásených 12 mesiacov pred súhlasom
0,87 (95 % CI: 0,70; 1,08). Pozorované udalosti počas dvojito zaslepeného obdobia a prvých
6 mesiacov predĺženého obdobia boli v miere 0,30 (95 % CI: 0,17; 0,51) a 0,23 (95 % CI: 0,13; 0,43)
v uvedenom poradí. V ramene placeba miera udalostí súvisiacich s obličkovými kameňmi hlásených
12 mesiacov pred súhlasom bola 0,15 (95 % CI: 0,07; 0,31) a miera pozorovaných udalostí počas dvojito zaslepeného obdobia bola 0,18 (95 % CI: 0,07; 0,48). Počas prvých 6 mesiacoch liečby
lumasiranom v predĺženom období bola miera 0,05 (95 % CI: 0,01; 0,32) udalostí pozorovaných
u pacientov, ktorým bolo predtým podávané placebo. Pokiaľ ide o nefrokalcinózu, k dispozícii sú údaje za 6-mesačné dvojito zaslepené obdobie. Z 34 pacientov s východiskovou hodnotou
a obličkovými ultrazvukmi v 6. mesiaci sa u 3 z 22 v skupine s lumasiranom preukázalo zlepšenie
nefrokalcinózy a u 1 z 12 v skupine s placebom sa preukázalo zhoršenie nefrokalcinózy. Žiadny
z ďalších pacientov liečených lumasiranom (n = 19) alebo placebom (n = 11) nevykazoval zmenu nefrokalcinózy.
ILLUMINATE-BCelkovo 18 pacientov bolo zaradených a liečených lumasiranom v rámci prebiehajúcej multicentrickej jednoramennej štúdie pacientov s PH1 (ILLUMINATE-B). Do štúdie boli zaradení pacienti mladší ako
6 rokov s hodnotou eGFR > 45 ml/min/1,73 m² u pacientov vo veku 12 mesiacov a starších
a s normálnym sérovým kreatinínom u pacientov mladších ako 12 mesiacov. Pri podaní prvej dávky v primárnej 6-mesačnej analýze boli 3 pacienti s hmotnosťou menšou ako 10 kg, 12 pacientov
s hmotnosťou 10 kg až menej ako 20 kg a 3 pacienti s hmotnosťou 20 kg a viac. Stredný vek pacientov
pri prvej dávke bol 51,4 mesiaca (v rozsahu od 4,0 do 74,0 mesiacov), 55,6% bolo žien a 88,9% bolo
bielej rasy. Stredná hodnota bodového pomeru oxalátu a kreatinínu v moči na začiatku bola
0,47 mmol/mmol.
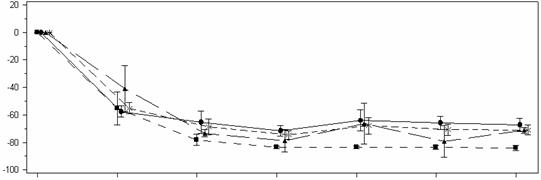
V 6. mesiaci dosiahli pacienti liečení lumasiranom zníženie o 72,0% (95 % CI: 66,4; 77,5) v bodovom pomere oxalátu a kreatinínu v moči oproti východiskovej hodnote (priemer za 3. až 6. mesiac), čo je primárny koncový ukazovateľ. Lumasiran bol spojený s rýchlym a trvalým znížením bodového pomeru oxalátu a kreatinínu v moči (obrázok 3), čo bolo podobné vo všetkých hmotnostných skupinách. Percentuálne zníženie vylučovania oxalátu v moči bolo v súlade s údajmi
z ILLUMINATE-A.
Obrázok 3: ILLUMINATE-B: Percentuálna zmena bodového pomeru oxalátu a kreatinínu v moči oproti východiskovej hodnote podľa mesiaca
'
BL M1 M2 M3 M4 M5 M6

Návšteva
Počet pacientov:
Skupina v ILLUMINATE-B s počiatočnou
hmotnosťou - – -■- – – < 10 kg —●— 10 až < 20 kg – –▲– – ≥ 20 kg
-----Û----- Všetci liečení
lumasiranom
■
|
N =
|
3
|
3
|
3
|
3
|
3
|
3
|
3
|
●
|
N =
|
12
|
12
|
12
|
12
|
12
|
12
|
12
|
▲
|
N =
|
3
|
3
|
3
|
3
|
3
|
3
|
3
|
Û
|
N =
|
18
|
18
|
18
|
18
|
18
|
18
|
18
|
Deväť pacientov dosiahlo takmer normalizáciu (≤ 1,5 × ULN) vrátane 1 pacienta, ktorý dosiahol
normalizáciu (≤ ULN), v 6. mesiaci v bodovom pomere oxalátu a kreatinínu v moči.
Okrem toho od začiatku do 6. mesiaca (priemer 3. až 6. mesiaca) bolo pozorované priemerné zníženie oxalátu v plazme o 31,7 % (95 % CI: 23,9; 39,5). Počas 6-mesačného obdobia hodnota eGFR zostala stabilná a boli hlásené 2 neskoršie udalosti obličkových kameňov u 2 pacientov v porovnaní so
4 udalosťami obličkových kameňov u 3 pacientov v 12-mesačnom období pred súhlasom. Štrnásť
z 18 pacientov malo na začiatku nefrokalcinózu. Údaje renálneho ultrazvuku v 6. mesiaci naznačovali zlepšenie u 8 pacientov vrátane 3 s dvojstranným zlepšením. U žiadneho z 18 pacientov sa nevyskytol nový nástup ani zhoršenie nefrokalcinózy.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s lOxlumom
v jednej alebo viacerých podskupinách pediatrickej populácie pri liečbe hyperoxalúrie (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po subkutánnom podaní sa lumasiran rýchlo absorbuje, pričom stredný čas (rozsah) na dosiahnutie
maximálnej plazmatickej koncentrácie (tmax) je 4,0 (0,5 až 12,0) hodiny. U detí a dospelých s PH1
≥ 20 kg maximálna plazmatická koncentrácia lumasiranu (Cmax) a plocha pod krivkou koncentrácie od času nula po poslednú merateľnú koncentráciu po podaní dávky (AUC0–last) po odporúčanej dávke
lumasiranu 3 mg/kg boli 529 (205 až 1130) ng/ml a 7400 (2890 až 10700) ng h/ml v uvedenom poradí.
U detí s hmotnosťou menej ako 20 kg hodnoty Cmax a AUC0–last lumasiranu po odporúčanej dávke lumasiranu 6 mg/kg boli 912 (523 až 1760) a 7960 (5920 až 13300). Koncentrácie lumasiranu boli
merateľné do 24 až 48 hodín po podaní dávky.
Distribúcia
Vo vzorkách plazmy zdravých dospelých je väzba lumasiranu na bielkoviny mierna až vysoká (77 až
85 %) pri klinicky relevantných koncentráciách. U dospelého pacienta s PH1 je populačný odhad pre zdanlivý centrálny distribučný objem (Vd/F) lumasiranu 4,9 l. Lumasiran sa po subkutánnom podaní primárne distribuuje do pečene.
Biotransformácia
Lumasiran sa metabolizuje endo- a exonukleázami na oligonukleotidy kratších dĺžok. Štúdie in vitro
naznačujú, že lumasiran nie je metabolizovaný enzýmami CYP450.
Eliminácia
Podľa súhrnných údajov od zdravých dospelých jedincov a pacientov s PH1 vo veku > 6 rokov sa
lumasiran primárne eliminuje z plazmy absorpciou pečene, pričom iba 7 až 26 % podanej dávky sa vylučuje v moči ako lumasiran. Priemerný (% CV) terminálny plazmatický polčas lumasiranu je 5,2 (47,0 %) hodiny. Populačný odhad pre zdanlivý plazmatický klírens bol 26,5 l/h pre typického dospelého s hmotnosťou 70 kg. Priemerný renálny klírens lumasiranu bol malý a pohyboval sa
v rozmedzí od 2,0 do 3,4 l/h u pediatrických a dospelých pacientov s PH1.
Linearita/nelinearita
Lumasiran vykazoval lineárnu až mierne nelineárnu časovo nezávislú farmakokinetiku v plazme po
jednorazových subkutánnych dávkach v rozmedzí od 0,3 do 6 mg/kg a viacnásobných dávkach
1 a 3 mg/kg raz mesačne alebo 3 mg/kg štvrťročne. Po opakovanom podávaní raz mesačne alebo štvrťročne nedošlo k hromadeniu lumasiranu v plazme.
Farmakokinetický/farmakodynamický vzťah
Plazmatické koncentrácie lumasiranu neodrážajú rozsah či trvanie farmakodynamickej aktivity
lumasiranu. Rýchla a cielená absorpcia lumasiranu pečeňou vedie k rýchlemu poklesu plazmatických koncentrácií. V pečeni vykazuje lumasiran dlhý polčas, čo vedie k udržiavaniu farmakodynamického
účinku počas mesačného alebo štvrťročného dávkovacieho intervalu.
Interakcie
Štúdie in vitro naznačujú, že lumasiran nie je substrátom ani inhibítorom enzýmov cytochrómu
P450 (CYP). Neočakáva sa, že by lumasiran inhiboval alebo indukoval enzýmy CYP, alebo moduloval aktivitu transportérov liekov.
Osobitné skupiny pacientov
Starší pacienti
Neuskutočnili sa žiadne štúdie u pacientov vo veku ≥ 65 rokov. Vek nebol významným kovariantom vo farmakokinetike lumasiranu.
Pohlavie a rasa
V klinických štúdiách nebol žiadny rozdiel v plazmatickej expozícii ani farmakodynamike lumasiranu na základe pohlavia alebo rasy.
Porucha funkcie pečene
Neuskutočnili sa žiadne štúdie u pacientov s poruchou funkcie pečene (pozri časť 4.2). Obmedzené farmakokinetické údaje u pacientov s miernym a prechodným zvýšením celkového bilirubínu (celkový
bilirubín > 1,0 až 1,5 × ULN) preukázali porovnateľnú plazmatickú expozíciu lumasiranu a podobnú farmakodynamiku ako u pacientov s normálnou funkciou pečene. V dosiaľ publikovanej literatúre sa
uvádza znížená expresia asialoglykoproteínových receptorov v pečeni, t. j. receptorov zodpovedných za príjem lumasiranu, u pacientov s poškodením pečene. Preedklinické údaje naznačujú, že táto skutočnosť pravdepodobne neovplyvňuje jeho vychytávanie pečeňou alebo jeho farmakodynamiku pri
terapeutických dávkach. Klinický význam týchto údajov nie je známy.
Porucha funkcie obličiek
Pacienti s miernou poruchou funkcie obličiek (eGFR 60 až < 90 ml/min/1,73 m²) mali porovnateľnú plazmatickú expozíciu lumasiranu a podobnú farmakodynamiku ako pacienti s normálnou funkciou
obličiek (eGFR ≥ 90 ml/min/1,73 m²). U pacientov so strednou poruchou funkcie obličiek
(eGFR 30 až < 60 ml/min/1,73 m²) bola hodnota Cmax podobná ako u pacientov s normálnou funkciou obličiek; podľa obmedzených údajov bola hodnota AUC bola o 25 % vyššia. V prípade pacientov so závažnou poruchou funkcie obličiek (eGFR 15 až < 30 ml/min/1,73 m²), ochorením obličiek
v terminálnom štádiu (eGFR < 15 ml/min/1,73 m²) alebo pacientov na dialýze sú k dispozícii len obmedzené klinické údaje (pozri časť 4.2). U pacientov s ochorením obličiek v terminálnom štádiu na
dialýze, ktorí spadajú do tej istej váhovej kategórie, sa spozorovalo prechodné 3- až 7-násobné
zvýšenie hodnoty Cmax a 2- až 3,5-násobné zvýšenie AUC0–last (pozri časť 5.2
Farmakokinetický/farmakodynamický vzťah). Koncentrácie v plazme však klesnú pod úroveň, ktorú je
možné detegovať do 24 až 48 hodín, podobne ako u pacientov bez poruchy funkcie obličiek.
Pediatrická populácia
Pre deti mladšie ako 1 rok je k dispozícii len obmedzené množstvo údajov. U detí s hmotnosťou
< 20 kg bola hodnota Cmax lumasiranu 2-násobne vyššia v dôsledku nominálne vyššej dávky 6 mg/kg
a rýchlejšej absorpcie. Farmakodynamika lumasiranu bola porovnateľná u pediatrických pacientov (vo veku od 4 mesiacov do 17 rokov) a u dospelých, a to napriek prechodne vyšším plazmatickým koncentráciám u detí s hmotnosťou < 20 kg v dôsledku rýchlej a prevažujúcej distribúcie lumasiranu
v pečeni.
Telesná hmotnosť
Odporúčaný režim dávkovania spôsobil až 2-dvojnásobne vyššie hodnoty Cmax u detí s hmotnosťou
< 20 kg, ale hodnoty AUC boli podobné pre všetky študované hmotnosti (6,2 až 110 kg).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
U potkanov, ale nie u opíc, sa pozorovali mikroskopické zmeny v pečeni (napríklad hepatocelulárna vakuolácia, mitóza a zväčšenie bunkového jadra), ktoré boli sprevádzané znížením úrovní plazmového fibrinogénu a inými laboratórnymi zmenami. Príčina zjavnej špecifickosti pre hlodavce nie je známa
a jej význam pre ľudí je nejasný.
Lumasiran nepreukázal u potkanov žiadne nežiaduce účinky na plodnosť samcov a samíc ani na prenatálny a postnatálny vývoj. V štúdiách vývoja embryí a plodov u potkanov a králikov boli pozorované kostrové abnormality, avšak pri vystavení vysokým dávkam, ktoré bol násobkom
v porovnaní s liečebnou expozíciou u ľudí. Najvyššie hladiny, pri ktorých nebol spozorovaný nežiaduci účinok (NOAEL, No Observed Adverse Effect Level) boli približne 20- až 70-krát vyššie
(založené na každomesačnej expozícii).
Štúdia toxicity zameraná na zistenie rozsahu dávok u novorodencov potkanov nepreukázala zvýšenú citlivosť vyvíjajúcich sa potkanov na toxikológiu ani farmakológiu lumasiranu pri dvojnásobnej expozícii v porovnaní s liečebnou expozíciou u ľudí (založené na každomesačnej expozícii).
Nevykonali sa štúdie na zvieratách s cieľom hodnotenia karcinogénneho potenciálu lumasiranu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
hydroxid sodný (na úpravu pH) kyselina fosforečná (na úpravu pH) voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
Po prvom otvorení injekčnej liekovky sa má liek ihneď použiť.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Injekčnú liekovku uchovávajte vo vonkajšej škatuli na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Sklenená injekčná liekovka s gumenou zátkou potiahnutou fluoropolymérom a hliníkovým prekrytím s odklopným krytom. Každá injekčná liekovka obsahuje 0,5 ml injekčného roztoku.
Balenie obsahuje jednu liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom Tento liek je pripravený na použitie a určený len na jednorazové použitie. Len na subkutánne použitie
• Pred podaním lieku si pripravte materiál, ktorý nie je súčasťou balenia, ale je potrebný na podanie, pričom bude zahŕňať sterilnú striekačku (0,3 ml, 1 ml alebo 3 ml), ihlu veľkosti 18 G a ihlu veľkosti 25 G až 31 G.
• Požadovaný objem Oxluma sa má vypočítať na základe odporúčanej dávky stanovenej podľa telesnej hmotnosti (pozri časť 4.2).
• Na vytiahnutie Oxluma z injekčnej liekovky použite ihlu s kalibrom 18 G. Injekčná liekovka sa
má držať v zvislej polohe alebo naklonená v miernom uhle a plochý okraj ihly má smerovať nadol.
• Pri objemoch menších ako 0,3 ml sa odporúča sterilná striekačka 0,3 ml.
• Liek sa má podávať subkutánnou injekciou pomocou sterilnej ihly veľkosti 25 až 31 G s dĺžkou ihly 13 mm alebo 16 mm.
• Poznámka: Tento liek sa nemá naťahovať ihlou veľkosti 25 G až 31 G.
• Injekčné striekačky, ihly na natiahnutie roztoku a injekčné ihly sa majú použiť len raz.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAlnylam Netherlands B.V. Antonio Vivaldistraat 150
1083 HP Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/20/1496/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu