
bol zaradený obmedzený počet pacientov vo veku
65 rokov a starších na stanovenie toho, či reagujú inak ako mladší jedinci (pozri časť 5.1). U pacientov vo veku 65 rokov a starších nie je potrebná úprava dávky.
Pediatrická populácia
U dospievajúcich (12-17 rokov) je dávkovanie rovnaké ako u dospelých.
Bezpečnosť a účinnosť krému ruxolitinibu u detí vo veku do 12 rokov neboli stanovené. K dispozícii
nie sú žiadne údaje.
Spôsob podávania
Krém je určený len na dermálne použitie.
Najmenej 2 hodiny po aplikácii krému ruxolitinibu sa treba vyhnúť umývaniu liečenej kože. Krém sa nemá aplikovať na pery, aby sa zabránilo jeho požitiu.
Pacientov treba poučiť, aby si po aplikácii krému umyli ruky, ak ruky nie sú liečenou oblasťou. Ak
pacientovi aplikuje krém niekto iný, má si po aplikácii umyť ruky.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Gravidita a dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Krém nie je určený na očné, perorálne ani intravaginálne použitie (pozri časť 4.2). V prípade náhodnej
expozície očí alebo slizníc sa má krém dôkladne utrieť a/alebo opláchnuť vodou.
Dlhodobá bezpečnosť
Bezpečnostný profil dlhodobého používania krému s obsahom ruxolitinibu na liečbu vitiliga nie je
známy. Krém s obsahom ruxolitinibu sa má používať na čo najmenšej potrebnej oblasti kože a nemajú
sa prekročiť odporúčania pre dávkovanie (časť 4.2).
N
emelanómová rakovina kože
U pacientov liečených lokálne aplikovaným ruxolitinibom sa hlásili prípady nemelanómovej rakoviny
kože (Non-Melanoma Skin Cancers, NMSCs), predovšetkým karcinómy bazálnych buniek. Väčšina z týchto pacientov mala rizikové faktory ako je predchádzajúca fototerapia alebo NMSC. Príčinný vzťah s lokálne aplikovaným ruxolitinibom sa nestanovil. Odporúčajú pravidelné kožné vyšetrenia
u všetkých pacientov, predovšetkým u pacientov s rizikovými faktormi.
Pomocné látky soznámymúčinkom
Propylénglykol
Tento liek obsahuje 150 mg propylénglykolu (E1520) v jednom grame krému, ktorý môže vyvolať podráždenie kože.
Cetylalkohol a stearylalkohol
Tento liek obsahuje cetylalkohol a stearylalkohol, ktoré môžu vyvolať miestne kožné reakcie (napr.
kontaktnú dermatitídu).
Parahydroxybenzoáty
Tento liek obsahuje metyl-parahydroxybenzoát (E218) a propyl-parahydroxybenzoát, ktoré môžu vyvolať alergické reakcie (možno oneskorené).
Butylhydroxytoluén
Tento liek obsahuje butylhydroxytoluén (E321), ktorý môže vyvolať mieste kožné reakcie (napr.
kontaktnú dermatitídu) alebo podráždenie očí a slizníc.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s lokálne aplikovaným ruxolitinibom.
Potenciál interakcií s ruxolitinibom sa z dôvodu obmedzenej systémovej expozície po lokálnej
aplikácii považuje za nízky.
Na základe údajov in vitro sa ruxolitinib vylučuje predovšetkým prostredníctvom metabolizmu cytochrómu P450 3A4 (CYP3A4). V špeciálnych klinických farmakologických štúdiách, ktoré zahŕňali súbežné podávanie silných alebo stredne silných inhibítorov alebo induktorov CYP3A4, sa hodnotil interakčný potenciál s perorálne podávaným ruxolitinibom. Plazmatická hodnota AUC je približne dvojnásobná pri súbežnom podávaní silného inhibítora CYP3A4, zatiaľ čo pri súbežnom podávaní stredne silného inhibítora CYP3A4 sa pozorovalo len mierne zvýšenie.
Používanie krému s obsahom ruxolitinibu v kombinácii s inými lokálne aplikovanými liekmi používanými na liečbu vitiliga sa nehodnotilo a súbežná aplikácia na rovnakú oblasť kože sa neodporúča.
Iné lokálne používané lieky používané na liečbu iných ochorení na rovnakej oblasti kože sa majú aplikovať minimálne 2 hodiny po aplikácii krému s obsahom ruxolitinibu. To platí aj pre používanie opaľovacích krémov a emoliencií.
4.6 Fertilita, gravidita a laktácia
Antikoncepcia u žien vofertilnomveku
Ženy vo fertilnom veku musia počas liečby a 4 týždne po ukončení liečby používať účinnú
antikoncepciu.
G
r
avidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití ruxolitinibu u gravidných žien.
Údaje týkajúce sa systémovej absorpcie lokálne aplikovaného ruxolitinibu počas gravidity nie sú k dispozícii. Môžu sa vyskytovať aj individuálne faktory (napr. poškodená kožná bariéra, nadmerné používanie), ktoré prispievajú k zvýšenej systémovej expozícii.
Štúdie na zvieratách preukázali, že ruxolitinib je po perorálnom podaní embryotoxický a fetotoxický. U potkanov ani králikov sa nepozorovala teratogenita (pozri časť 5.3). Opzelura je kontraindikovaná
počas gravidity (pozri časť 4.3).
DojčenieK dispozícii nie sú žiadne údaje o prítomnosti ruxolitinibu v ľudskom mlieku, účinkoch na dojčené
dieťa ani účinkoch na tvorbu mlieka po lokálnej aplikácii Opzelury. Po perorálnom podaní ruxolitinibu laktujúcim potkanom boli ruxolitinib a/alebo jeho metabolity v mlieku prítomné
v 13-násobne vyššej koncentrácii, ako je plazmatická koncentrácia u matky. V štúdiách na mladých potkanoch viedlo perorálne podávanie ruxolitinibu k dopadom na rast a rozmery kostí (pozri časť 5.3). Opzelura je kontraindikovaná počas dojčenia (pozri časť 4.3) a liečba sa musí ukončiť približne
4 týždne pred začatím dojčenia.
FertilitaNie sú k dispozícii žiadne údaje o účinku ruxolitinibu na fertilitu u ľudí. V štúdiách na zvieratách na
nepozoroval žiadny účinok perorálne podávaného ruxolitinibu na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeKrém s obsahom ruxolitinibu nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá
a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluNajčastejšou nežiaducou reakciou je akné na mieste aplikácie (5,8 %).
Tabuľkový zoznamnežiaducichreakciíNežiaduce reakcie sú zoradené pod nadpismi frekvencií s ako prvou najvyššou frekvenciou podľa
nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Tabuľka 1: Nežiaduce reakcieTrieda orgánových systémov
| Frekvencia
| Nežiaduca reakcia
|
Celkové poruchy a reakcie v mieste podania
| časté
| akné na mieste aplikácie
|
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Predávkovanie po dermálnom použití je nepravdepodobné. Ak sa aplikovalo príliš veľa krému, nadbytok sa dá utrieť.
V prípadoch náhodnej expozície očí, ústnej sliznice či intravaginálnej expozície sa má krém dôkladne utrieť a/alebo opláchnuť vodou (pozri časti 4.2 a 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné dermatologiká, liečivá na dermatitídy s výnimkou kortikosteroidov, ATC kód: D11AH09
Mechanizmus účinku
Ruxolitinib je inhibítor Janusovej kinázy (JAK) so selektivitou izoforiem JAK1 a JAK2. Intracelulárna
signalizácia JAK zahŕňa aktiváciu STAT (signálne transduktory a aktivátory transkripcie) na cytokínových receptoroch a nadväzujúcu moduláciu génovej exprimácie. Predpokladá sa, že
autoimunitné IFNγ produkujúce cytotoxické T-lymfocyty priamo zodpovedajú za deštrukciu
melanocytov pri vitiligu u ľudí. Aktivácia cytotoxických lymfocytov na koži s léziami je
sprostredkovaná cytokínmi závislými od IFNγ, ako je CXCL10. „Downstream“ signalizácia IFNγ je závislá od JAK1/2 a liečba ruxolitinibom znižuje hladiny CXCL10 u pacientov s vitiligom.
Klinická účinnosť abezpečnosť
Do dvoch dvojito zaslepených, randomizovaných, nosičom kontrolovaných štúdií s identickým
dizajnom (TRuE-V1 a TRuE-V2) bolo zaradených celkom 674 pacientov s vitiligom na tvári
a celkovou telesnou oblasťou postihnutou vitiligom (na tvári a ostatných častiach tela) nepresahujúcou
10 % BSA, s rozšírením ochorenia na začiatku štúdie v rozsahu od 3,2 % do 10,1 % BSA, vo veku
12 rokov a starších (10,7 % pacientov bolo vo veku 12 až 17 rokov a 6,7 % bolo vo veku 65 rokov
a starších). Ženy tvorili 53,1 % pacientov, 81,9 % pacientov boli belosi, 4,7 % černosi a 4,2 % Ázijci.
Väčšina pacientov mala typ pleti podľa Fitzpatrickovej škály III, IV, V alebo VI (67,5 %).
V oboch štúdiách pacientov randomizovali v pomere 2:1 na liečbu krémom ruxolitinibu alebo aplikáciu nosiča dvakrát denne počas 24 týždňov s postihnutou BSA neprekračujúcou 10 %, po nich nasledovalo ďalších 28 týždňov liečby krémom ruxolitinibu dvakrát denne. Primárnym koncovým ukazovateľom účinnosti bol podiel pacientov, ktorí dosiahli 75 % repigmentácie v tvárovom indexe skórovania oblasti postihnutej vitiligom (Facial Vitiligo Area Scoring Index, F-VASI75) v 24. týždni. Kľúčové sekundárne koncové ukazovatele zahŕňali podiel pacientov, ktorí dosiahli 90 % repigmentácie v F-VASI (F-VASI90), 50 % zlepšenie celkového telového indexu skórovania oblasti
postihnutej vitiligom (Total body Vitiligo Area Scoring Index, T-VASI50) a skóre stupnice nápadnosti
vitiliga (Vitiligo Noticeability Scale, VNS) 4 z 5 (vitiligo „oveľa menej nápadné“ alebo „už nenápadné“).
V oboch štúdiách sa pozorovala repigmentácia liečených lézií vitiliga a superiorita krému s obsahom ruxolitinibu nad nosičovým krémom, ako preukázali štatisticky významné rozdiely v mierach odpovede na F-VASI75/90, T-VASI50 a skóre VNS 4 z 5 v 24. týždni (tabuľka 2).
Rozdiel v liečebnom účinku oproti nosiču sa numericky objavil už v 12. týždni. Pokračujúca repigmentácia hodnotená podľa stupnice VASI a VNS sa pozorovala do 52. týždňa u pacientov, ktorí od začiatku štúdie nepretržite pokračovali v aplikovaní krému s obsahom ruxolitinibu dvakrát denne. Podiely pacientov, ktorí dosiahli F-VASI75 počas 52-týždňového liečebného obdobia v združených údajoch zo štúdií TRuE-V1 a TRuE-V2, sú zobrazené na obrázku 1.
Podobné odpovede na liečbu v 52. týždni sa pozorovali u pacientov, ktorí prešli z nosiča na ruxolitinib
(obrázok 1).
Tabuľka 2: Percento pacientov s vitiligom, ktorí dosiahli primárny a kľúčové sekundárnekoncové ukazovatele v 24. týždni (Intent-
To-Treat)a
| TRuE-V1
| TRuE-V2
|
Opzelura
| Nosič
| Opzelura
| Nosič
|
(N = 221)
| (N = 109)
| (N = 222)
| (N = 109)
|
F-VASI75 (%)
| 29,8
| 7,4
| 30,9
| 11,4
|
Rozdiel v mierach odpovede (95 % IS)
| 22,3b
(14,214; 30,471)
| -
| 19,5c
(10,537; 28,420)
| -
|
F-VASI90 (%)
| 15,3
| 2,2
| 16,3
| 1,3
|
Rozdiel v mierach odpovede (95 % IS)
| 13,2d
(7,497; 18,839)
| -
| 15,0e
(9,250; 20,702)
| -
|
T-VASI50 (%)
| 20,6
| 5,1
| 23,9
| 6,8
|
Rozdiel v mierach odpovede (95 % IS)
| 15,5d
(8,339; 22,592)
| -
| 17,1c
(9,538; 24,721)
| -
|
VNS 4 alebo 5 (%)
| 24,5
| 3,3
| 20,5
| 4,9
|
Rozdiel v mierach odpovede (95 % IS)
| 21,2c
(14,271; 28,143)
| -
| 15,5d
(8,515; 22,561)
| -
|
a Primárne a kľúčové sekundárne výsledky sa upravili metódou viacnásobnej imputácie.
b Hodnota p < 0,0001
c Hodnota p < 0,001 d Hodnota p < 0,005 e Hodnota p < 0,01
O
brázok 1: Podiel pacientov, ktorí dosiahli F-VASI75 počas 52-týždňového liečebného obdobia (Intent-
To-Treat) – združené údaje zo štúdií TRuE-
V1 a TRuE-
V2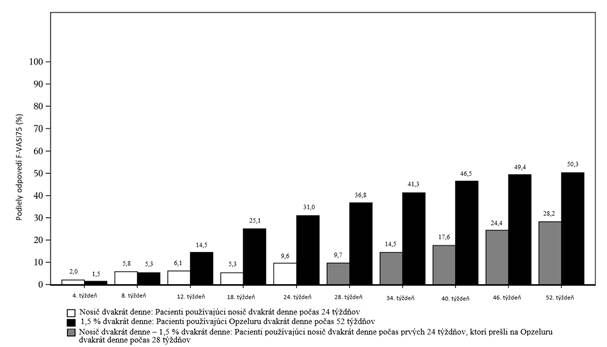
V 52. týždni sa v združenej populácii ITT pozorovala miera odpovede na F-VASI90, T-VASI50
a VNS 30,3 %, 51,1 % a 36,3 % v uvedenom poradí.
Pediatrická populáciaDo pivotných štúdií sa zaradilo celkom 72 dospievajúcich (12-18 rokov, n = 55 krém s ruxolitinibom,
n = 17 nosič). Dospievajúci liečení ruxolitinibom preukázali rovnaké miery odpovede v primárnych a kľúčových sekundárnych koncových ukazovateľoch v 24. týždni ako dospelí vo veku 18-65 rokov. Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Opzelurou v jednej alebo vo viacerých podskupinách pediatrickej populácie na liečbu vitiliga (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaFarmakokinetické vlastnosti krému s ruxolitinibom sa skúmali u 429 jedincov s vitiligom vo veku
12 rokov a starších (12,6 % bolo vo veku 12-17 rokov) s priemerným ± STD postihnutím BSA
7,31 ± 2,02 % (rozsah 3,2 % až 10,0 %). Jedinci si aplikovali približne 1,58 mg/cm2 krému
ruxolitinibu (rozsah dávky bol približne 0,18 gramov až 8,4 gramov krému ruxolitinibu na jednu
aplikáciu) na rovnaké oblasti kože dvakrát denne počas 24 týždňov.
Priemerná ± STD minimálna plazmatická koncentrácia v rovnovážnom stave bola 56,9 ± 62,6 nM s premietnutou hodnotou AUC0-12 h 683 ± 751 h*nM, čo je približne 25 % pozorovanej priemernej hodnoty AUC0-12 h v rovnovážnom stave (2 716 h*nM) po podávaní 15 mg dvakrát denne zdravým účastníkom. Priemerná (geometrický priemer) lokálna biologická dostupnosť krému ruxolitinibu
u účastníkov s vitiligom v združených údajoch dvoch štúdií fázy 3 bola 9,27 % (5,78 %).'
DistribúciaNa základe štúdie
in vitro sa ruxolitinib viaže z 97 % na ľudské plazmatické proteíny, najviac na
albumín.
B
i
otransformácia
Ruxolitinib sa metabolizuje prostredníctvom CYP3A4 a v menšej miere prostredníctvom CYP2C9.
Eliminácia
Priemerný polčas eliminácie perorálne podaného ruxolitinibu je približne 3 hodiny. Priemerný zjavný
koncový polčas ruxolitinibu po lokálnej aplikácii Opzelury sa odhadoval u 9 dospelých
a dospievajúcich pacientov s ≥ 25 % postihnutím BSA atopickou dermatitídou a je to približne
116 hodín, čo odzrkadľuje skôr pomalú absorpciu lieku ako rýchlosť eliminácie lieku.
Osobitné populácie
Porucha funkcie obličiek
Odhadovaná hodnota AUC upravená na farmakologickú aktivitu ruxolitinibu a metabolitov sa
v prípade ochorenia obličiek v konečnom štádiu (End Stage Renal Disease, ESRD) zvyšuje približne dvojnásobne. Kvôli chýbajúcim údajom týkajúcim sa bezpečnosti sa ako preventívne opatrenie Opzelura nemá používať u pacientov s ESRD.
Porucha funkcie pečene
Aj keď bola hodnota AUC po perorálnom podaní ruxolitinibu pacientom s poruchou funkcie pečene zvýšená, neexistoval jasný vzťah medzi závažnosťou poruchy funkcie pečene a zvýšením hodnoty AUC. Odporúčanie pri dávkovaní u pacientov s poruchou funkcie pečene nie je potrebné.
5.3 Predklinické údaje o bezpečnosti
Ruxolitinib sa hodnotil vo farmakologických štúdiách bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a reprodukčnej toxicity a karcinogenity po perorálnom podaní. Dodatočné štúdie sa vykonali po dermálnej aplikácii u miniprasiat a myší. Cieľové orgány spojené s farmakologickým účinkom ruxolitinibu v štúdiách s opakovaným perorálnym podávaním zahŕňajú kostnú dreň, periférne krvné a lymfoidné tkanivá. U psov sa zaznamenali infekcie všeobecne súvisiace s imunosupresiou. Hraničné hodnoty expozície (na základe hodnoty AUC neviazaného lieku) na úrovni hladín bez pozorovaného nepriaznivého účinku v štúdiách chronickej toxicity boli približne 6- a 200-násobne vyššie u samcov a samíc potkanov a 10-násobne vyššie u psov, ako je systémová expozícia
pozorovaná u pacientov s vitiligom, ktorí si aplikovali 1,5 % krém ruxolitinibu dvakrát denne.
V telemetrickej štúdii sa u psov zaznamenal nežiaduci pokles krvného tlaku spolu so zrýchlením tepu a v respiračnej štúdii na potkanoch sa zaznamenal nežiaduci pokles v minútovom objeme. Hraničné hodnoty (na základe hodnoty Cmax neviazaného lieku) na úrovni hladín bez pozorovaného nepriaznivého účinku v štúdiách na psoch a potkanoch boli približne 300-násobne a 100-násobne vyššie, v uvedenom poradí, ako je systémová expozícia pozorovaná u pacientov s vitiligom, ktorí si aplikovali 1,5 % krém ruxolitinibu dvakrát denne. V hodnotení neurofarmakologických účinkov ruxolitinibu u potkanov sa nezaznamenali žiadne nežiaduce účinky.
3-mesačná štúdia opakovaného dermálneho podávania odhalila znížený počet lymfocytov u myší. Hraničné hodnoty (na základe hodnoty AUC neviazaného lieku) na úrovni hladín bez pozorovaného nepriaznivého účinku boli približne 10-násobne vyššie u samcov a 24-násobne vyššie u samíc myší, ako je systémová expozícia pozorovaná u pacientov s vitiligom, ktorí si aplikovali 1,5 % krém s ruxolitinibom dvakrát denne. V 9-mesačnej štúdii toxicity po dermálnom použití sa u miniprasiat tiež zaznamenalo zníženie počtu periférnych lymfocytov bez pozorovaného nepriaznivého účinku. Hraničné hodnoty (na základe hodnoty AUC neviazaného lieku) na úrovni hladín bez pozorovaného nepriaznivého účinku boli približne 3-násobne vyššie ako je systémová expozícia pozorovaná
u pacientov s vitiligom, ktorí si aplikovali 1,5 % krém ruxolitinibu dvakrát denne. Tento účinok sa
nepozoroval v 3-mesačnej štúdii toxicity po dermálnom použití u miniprasiat. Po lokálnom podaní liekovej formy 1,5 % krému ruxolitinibu dvakrát denne po dobu až 9 mesiacov sa nepozoroval žiadny dôkaz systémovej toxicity u miniprasiat rasy Gottingen.
V štúdiách na mladých potkanoch viedlo perorálne podávanie ruxolitinibu k vplyvu na rast a rozmery kostí. Znížený rast kostí sa pozoroval pri dávkach ≥ 5 mg/kg/deň, ak sa liek začal podávať 7. deň po narodení (porovnateľné s novorodencom človeka) a ≥ 15 mg/kg/deň, ak sa podávanie začalo 14. alebo
21. deň po narodení (porovnateľné s dieťaťom človeka vo veku 1-3 roky). Pri dávkach ≥ 30 mg/kg/deň
sa pozorovali fraktúry a predčasné usmrtenie u potkanov, ak sa liečba začala v 7. deň po narodení. Na základe hodnoty AUC neviazaného lieku bola expozícia na úrovni hladiny bez pozorovaného nepriaznivého účinku (No Observed Adverse Effect Level, NOAEL) u mladých potkanov, ktorým sa liek podával už 7. deň po narodení, približne 20-násobne vyššia ako u dospelých pacientov s vitiligom, zatiaľ čo znížený rast kostí a fraktúry sa vyskytli pri expozíciách 22- až 150-násobne vyšších ako
u dospelých pacientov s vitiligom, v uvedenom poradí. Účinky boli vo všeobecnosti závažnejšie
u samcov a ak sa podávanie začalo v skoršom období po narodení. Okrem dopadu na vývoj kostí boli účinky ruxolitinibu u mladých potkanov podobné ako u dospelých potkanov. Mladé potkany sú na
toxicitu spôsobenú ruxolitinibom citlivejšie ako dospelé potkany.
V štúdiách embryofetálneho vývoja viedlo perorálne podávanie ruxolitinibu potkanom a králikom počas gravidity k zníženej hmotnosti plodov a zvýšenej poimplantačnej strate pri dávkach spojených s toxicitou u matiek. U potkanov a králikov neexistovali dôkazy teratogénnych účinkov. Hraničné hodnoty (na základe hodnoty AUC neviazaného lieku) na úrovni hladín bez pozorovaného
nepriaznivého účinku v štúdiách vývojovej toxicty na potkanoch boli približne 25-násobne vyššie ako
je systémová expozícia pozorovaná u pacientov s vitiligo, ktorí si aplikovali 1,5 % krém s ruxolitinibom dvakrát denne. Nezaznamenali sa žiadne účinky perorálne podávaného ruxolitinibu na fertilitu u samcov ani samíc potkanov. V štúdiách pre- a postnatálneho vývoja sa pozorovalo mierne predĺžené obdobie gravidity, znížený počet implantačných miest a znížený počet vrhnutých mláďat.
U mláďat sa pozorovala znížená začiatočná telesná hmotnosť a krátke obdobie zníženého priemerného
nárastu telesnej hmotnosti. U laktujúcich potkanov sa ruxolitinib a/alebo jeho metabolity vylučovali do mlieka v koncentrácii 13-násobne vyššej ako je plazmatická koncentrácia u matky. Ruxolitinib nebol mutagénny ani klastogénny. Ruxolinitib nepreukazoval karcinogénny potenciál po lokálnom podávaní u myší ani po perorálnom podávaní u Sprague-Dawley potkanov a Tg.rasH2 myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
butylhydroxytoluén (ako antioxidant v parafíne, biely, mäkký) (E321)
cetylalkohol dimetikón (E900)
edetát disodný (E385)
samoemulgujúci glycerol-stearát makrogol
triacylglyceroly, so stredne dlhým reťazcom
metyl-parahydroxybenzoát (E218) parafín (E905), tekutý, ľahký parafín (E905), biely, mäkký fenoxyetanol
polysorbát 20 (E432)
propylénglykol (E1520) propyl-parahydroxybenzoát čistená voda
stearylalkohol
xantánová guma (E415)
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
21 mesiacov
Po prvom otvorení: 6 mesiacov.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote neprevyšujúcej 30 ºC.
6.5 Druh obalu a obsah baleniaHliníková tuba s vnútorným lakovým poťahom a s polypropylénovým prepichovacím viečkom.
Tuba s obsahom 100 g. Jedna tuba v každej škatuli.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIIncyte Biosciences Distribution B.V. Paasheuvelweg 25
1105 BP Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLOEU/1/23/1726/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.