
imunosupresívne dávky kortikosteroidov alebo inú imunosupresívnu liečbu. Antibiotiká sa majú profylakticky použiť na zabránenie vzniku oportúnnych infekcií u pacientov, ktorí dostávajú imunosupresívnu liečbu.
Pri opätovnom výskyte akejkoľvek závažnej imunitne podmienenej nežiaducej reakcie a pri výskyte akejkoľvek život ohrozujúcej imunitne podmienenej nežiaducej reakcie sa musí liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom natrvalo ukončiť.
Imunitnepodmienenápneumonitída
Pri monoterapii nivolumabom alebo pri nivolumabe v kombinácii s ipilimumabom sa pozorovala závažná pneumonitída alebo intersticiálne ochorenie pľúc vrátane smrteľných prípadov (pozri časť 4.8). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov pneumonitídy, ako sú
rádiografické zmeny (napr. fokálne „groud glass“ opacity, škvrnité infiltráty), dyspnoe a hypoxia.
Infekcie a etiológie súvisiacich ochorení sa majú vylúčiť.
Pri pneumonitíde 3. alebo 4. stupňa sa musí liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom natrvalo ukončiť a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 2 až 4 mg/kg/deň.
Pri (symptomatickej) pneumonitíde 2. stupňa sa má podávanie nivolumabu alebo nivolumabu v kombinácii s ipilimumabom prerušiť a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 1 mg/kg/deň. Po zlepšení sa môže liečba nivolumabom alebo nivolumabom v
kombinácii s ipilimumabom obnoviť po postupnom znížení dávky kortikosteroidu. Ak aj napriek začatiu liečby kortikosteroidmi dôjde k zhoršeniu alebo sa stav nezlepší, dávka kortikosteroidu sa má zvýšiť na dávku zodpovedajúcu metylprednizolónu na 2 až 4 mg/kg/deň a liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť.
Imunitnepodmienenákolitída
Pri monoterapii nivolumabom alebo pri nivolumabe v kombinácii s ipilimumabom sa pozorovala závažná hnačka alebo kolitída (pozri časť 4.8). Pacienti majú byť sledovaní z dôvodu hnačky a ďalších príznakov kolitídy, ako je bolesť brucha a hlien alebo krv v stolici. Majú sa vylúčiť infekcie a etiológie súvisiacich ochorení.
Z dôvodu hnačky alebo kolitídy 4. stupňa sa musí liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom natrvalo ukončiť a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 1 až 2 mg/kg/deň.
Z dôvodu hnačky alebo kolitídy 3. stupňa sa má prerušiť monoterapia nivolumabom a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 1 až 2 mg/kg/deň. Po zlepšení sa môže monoterapia nivolumabom obnoviť po postupnom znížení dávky kortikosteroidu. Ak aj napriek začatiu liečby kortikosteroidmi dôjde k zhoršeniu alebo sa stav nezlepší, monoterapia nivolumabom sa musí natrvalo ukončiť. Hnačka alebo kolitída 3. stupňa pozorované pri nivolumabe v kombinácii s ipilimumabom si vyžaduje trvalé ukončenie liečby a začatie liečby kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 1 až 2 mg/kg/deň.
Z dôvodu hnačky alebo kolitídy 2. stupňa sa má prerušiť podávanie nivolumabu alebo nivolumabu v kombinácii s ipilimumabom. Ak hnačka alebo kolitída pretrvávajú majú sa liečiť kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 0,5 až 1 mg/kg/deň. Ak je to potrebné, po zlepšení sa môže liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom obnoviť po postupnom znížení dávky kortikosteroidu. Ak aj napriek začatiu liečby kortikosteroidmi dôjde k zhoršeniu stavu alebo sa stav nezlepší, dávka kortikosteroidu sa má zvýšiť na dávku zodpovedajúcu metylprednizolónu až 1 až
2 mg/kg/deň a liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť.
Imunitnepodmienenáhepatitída
Pri monoterapii nivolumabom alebo pri nivolumabe v kombinácii s ipilimumabom sa pozorovala závažná hepatitída (pozri časť 4.8). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov hepatitídy, ako sú vzostupy transaminázy a celkového bilirubínu. Majú sa vylúčiť infekcie a etiológie súvisiacich ochorení.
Pri vzostupe transamináz alebo celkového bilirubínu 3. alebo 4. stupňa sa musí liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom natrvalo ukončiť a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 1 až 2 mg/kg/deň.
Pri vzostupe transamináz alebo celkového bilirubínu 2. stupňa sa má podávanie nivolumabu alebo nivolumabu v kombinácii s ipilimumabom prerušiť. Pretrvávajúce zvýšenia v týchto laboratórnych hodnotách sa majú liečiť kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 0,5 až
1 mg/kg/deň. Ak je to potrebné, po zlepšení sa môže liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom obnoviť po postupnom znížení dávky kortikosteroidu. Ak aj napriek začatiu liečby kortikosteroidmi dôjde k zhoršeniu alebo sa stav nezlepší, má sa dávka kortikosteroidu zvýšiť na dávku zodpovedajúcu metylprednizolónu až 1 až 2 mg/kg/deň a liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť.
Imunitnepodmienenánefritídaadysfunkciaobličiek
Pri liečbe monoterapiou alebo pri nivolumabe v kombinácii s ipilimumabom sa pozorovali závažná nefritída a dysfunkcia obličiek (pozri časť 4.8). Pacienti majú byť sledovaní z dôvodu prejavov a príznakov nefridíty alebo dysfunkcie obličiek. U väčšiny pacientov sú prítomné asymptomatické zvýšenia kreatinínu v sére. Majú sa vylúčiť etiológie súvisiacich ochorení.
Z dôvodu vzostupu kreatinínu v sére 4. stupňa sa musí liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom natrvalo ukončiť a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 1 až 2 mg/kg/deň.
Z dôvodu vzostupu kreatinínu v sére 2. alebo 3. stupňa sa má podávanie nivolumabu alebo nivolumabu v kombinácii s ipilimumabom prerušiť a má sa začať liečba kortikosteroidmi v dávke zodpovedajúcej metylprednizolónu 0,5 až 1 mg/kg/deň. Po zlepšení sa môže liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom obnoviť po postupnom znížení dávky kortikosteroidu. Ak aj napriek začatiu liečby kortikosteroidmi dôjde k zhoršeniu alebo sa stav nezlepší, má sa dávka kortikosteroidu zvýšiť na dávku zodpovedajúcu metylprednizolónu až 1 až
2 mg/kg/deň a liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť.
Imunitnepodmienenéendokrinopatie
Pri monoterapii nivolumabom alebo pri nivolumabe v kombinácii s ipilimumabom sa pozorovali závažné endokrinopatie zahŕňajúce hypotyreoidizmus, hypertyreoidizmus, nedostatočnosť nadobličiek (vrátane sekundárnej adrenokortikálnej insuficiencie), hypofyzitídu (vrátane hypopituitarizmu), diabetes mellitus a diabetickú ketoacidózu (pozri časť 4.8).
Pacienti majú byť monitorovaní z dôvodu klinických prejavov a príznakov endokrinopatií a z
dôvodu hyperglykémie a zmien vo funkcii štítnej žľazy (na začiatku liečby, periodicky počas liečby a podľa toho, ako je indikované na základe klinického hodnotenia). U pacientov sa môže vyskytnúť únava, bolesť hlavy, zmeny duševného stavu, bolesť brucha, nezvyčajné prejavy čriev a hypotenzia alebo nešpecifické príznaky, ktoré môžu pripomínať iné príčiny, ako sú metastázy v mozgu alebo základné ochorenie. Pokiaľ sa nezistila iná etiológia, príznaky alebo prejavy endokrinopatií sa majú považovať za imunitne podmienené.
Z dôvodu symptomatického hypotyreoidizmu sa má liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom prerušiť a ak je to potrebné má sa začať substitučné podávanie
tyreoidného hormónu. Z dôvodu symptomatického hypertyreoidizmu sa má liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom prerušiť a ak je to potrebné má sa začať tyreostatická liečba. Pri podozrení akútneho zápalu štítnej žľazy sa má zvážiť aj podanie kortikosteroidov v dávke zodpovedajúcej metylprednizolónu 1 až 2 mg/kg/deň. Ak je to potrebné, po zlepšení sa môže liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom obnoviť po postupnom znížení
dávky kortikosteroidu. V sledovaní funkcie štítnej žľazy sa má pokračovať, aby sa zaručilo, že sa používa vhodné substitučné podávanie hormónu. Liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť pri život ohrozujúcom hypertyreoidizme alebo hypotyreoidizme.
Z dôvodu symptomatickej nedostatočnosti nadobličiek 2. stupňa sa má liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom prerušiť a ak je to potrebné má sa začať substitučné podávanie fyziologického kortikosteroidu. Liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť pri závažnej (3. stupeň) alebo život ohrozujúcej (4. stupeň) nedostatočnosti nadobličiek. V sledovaní funkcie nadobličiek a hladín hormónov sa má pokračovať, aby sa zaručilo, že sa používa vhodné substitučné podávanie kortikosteroidu.
Z dôvodu symptomatickej hypofyzitídy 2. alebo 3. stupňa sa má liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom prerušiť a ak je to potrebné má sa začať substitučné podávanie hormónu. Pri podozrení akútneho zápalu hypofýzy sa má zvážiť aj podanie kortikosteroidov v dávke zodpovedajúcej metylprednizolónu 1 až 2 mg/kg/deň. Ak je to potrebné, po zlepšení sa môže liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom obnoviť po postupnom znížení dávky kortikosteroidu. Liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť pri život ohrozujúcej (4. stupeň) hypofyzitíde. V sledovaní funkcie hypofýzy a hladín hormónov sa má pokračovať, aby sa zaručilo, že sa používa vhodné substitučné podávanie hormónu.
Z dôvodu symptomatického diabetu sa má liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom prerušiť a ak je to potrebné má sa začať substitučné podávanie inzulínu. V sledovaní cukru v krvi sa má pokračovať, aby sa zaručilo, že sa používa vhodné substitučné podávanie inzulínu. Liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa musí natrvalo ukončiť pri život ohrozujúcej cukrovke.
Imunitnepodmienenékožnénežiaducereakcie
Pri liečbe nivolumabom v kombinácii s ipilimumabom a menej často pri nivolumabe monoterapii sa pozorovala závažná vyrážka (pozri časť 4.8). Pri vyrážke 3. stupňa sa má liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom prerušiť a pri vyrážke 4. stupňa sa má ukončiť. Závažná vyrážka sa má liečiť vysokými dávkami kortikosteroidu v dávke zodpovedajúcej metylprednizolónu
1 až 2 mg/kg/deň.
Pozorovali sa zriedkavé prípady SJS a TEN, niektoré z nich so smrteľným následkom. Ak sa objavia príznaky alebo prejavy SJS alebo TEN, liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom sa má ukončiť a pacient sa má poslať na špecializovanú jednotku na posúdenie a liečbu. Ak sa u pacienta pri používaní nivolumabu alebo nivolumabu v kombinácii s ipilimumabom vyvinul SJS alebo TEN, odporúča sa trvalé ukončenie liečby (pozri časť 4.2).
Ak sa zvažuje použitie nivolumabu u pacientov, ktorí mali v minulosti pri predchádzajúcej liečbe inými imunostimulačnými protinádorovými látkami závažnú alebo život ohrozujúcu kožnú nežiaducu reakciu, je potrebná opatrnosť.
Ďalšieimunitnepodmienenénežiaducereakcie
V klinických skúšaniach pri rôznych dávkach a typoch nádorov sa u menej ako 1 % pacientov liečených nivolumabom v monoterapii alebo nivolumabom v kombinácii s ipilimumabom hlásili nasledovné imunitne podmienené nežiaduce reakcie: pankreatitída, uveitída, demyelinizácia, autoimunitná neuropatia (zahŕňajúca parézu tvárového a odťahujúceho nervu), Guillainov-Barrého syndróm, myastenický syndróm, encefalitída, gastritída, sarkoidóza, duodenitída, myozitída, myokarditída a rabdomyolýza. Po uvedení lieku na trh sa hlásili prípady Vogtovho-Koyanagiho- Haradovho syndrómu (pozri časť 4.8).
Z dôvodu podozrenia imunitne podmienených nežiaducich reakcií sa má vykonať zodpovedajúce vyhodnotenie, aby sa potvrdila etiológia alebo aby sa vylúčili iné príčiny. Na základe závažnosti nežiaducej reakcie sa má liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom prerušiť a majú sa podať kortikosteroidy. Po zlepšení sa môže liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom obnoviť po postupnom znížení dávky kortikosteroidu. Z
dôvodu akejkoľvek závažnej imunitne podmienenej nežiaducej reakcie, ktorá sa opakovane vyskytne a z dôvodu akejkoľvek život ohrozujúcej imunitne podmienenej nežiaducej reakcie sa liečba nivolumabom alebo nivolumabom v kombinácii s ipilimumabom musí natrvalo ukončiť.
Po nivolumabe alebo nivolumabe v kombinácii s ipilimumabom sa hlásili zriedkavé prípady myotoxicity (myozitída, myokarditída a rabdomyolýza) niektoré so smrteľným následkom. Ak sa u pacienta vyvinú prejavy a príznaky myotoxicity, má sa vykonať dôkladné sledovanie a pacient sa má bez omeškania odoslať špecialistovi na posúdenie a liečbu. Na základe závažnosti myotoxicity sa má podávanie nivolumabu alebo nivolumabu v kombinácii s ipilimumabom prerušiť alebo ukončiť (pozri časť 4.2) a má sa začať vhodná liečba.
U pacientov liečených inhibítormi PD-1 bola po uvedení lieku na trh hlásená rejekcia transplantovaného solídneho orgánu. Liečba nivolumabom môže u príjemcov transplantovaného solídneho orgánu zvýšiť riziko rejekcie. U týchto pacientov treba zvážiť prínos liečby nivolumabom v porovnaní s rizikom možnej rejekcie orgánu.
Infúznereakcie
V klinických skúšaniach s nivolumabom alebo s nivolumabom v kombinácii s ipilimumabom sa hlásili závažné infúzne reakcie (pozri časť 4.8). V prípade závažnej alebo život ohrozujúcej infúznej reakcie sa musí infúzia nivolumabu alebo nivolumabu v kombinácii s ipilimumabom ukončiť a musí sa podať
zodpovedajúca medicínska liečba. Pacientom s miernou alebo stredne závažnou infúznou reakciou sa nivolumab alebo nivolumab v kombinácii s ipilimumabom môže podávať pri dôkladnom sledovaní a po použití premedikácie v súlade s lokálnymi liečebnými postupmi profylaxie infúznych reakcií.
Opatrenia špecifické pre ochorenie
Pokročilý melanóm
Pacienti s východiskovým skóre výkonnostného stavu ≥ 2, aktívnymi metastázami v mozgu alebo autoimunitným ochorením a pacienti, ktorí dostali systémové imunosupresíva pred zaradením do
štúdie boli z klinických skúšaní s nivolumabom alebo s nivolumabom v kombinácii s ipilimumabom
vylúčení (pozri časti 4.5 a 5.1). Pacienti s okulárnym/uveálnym melanómom boli z klinických skúšaní s melanómom vylúčení. Navyše zo štúdie CA209037 boli vyradení pacienti, ktorí mali nežiaducu reakciu 4. stupňa, ktorá súvisela s liečbou proti CTLA-4 (pozri časť 5.1). V prípade chýbajúcich údajov sa má u týchto populácií nivolumab použiť s opatrnosťou po starostlivom posúdení pomeru potenciálneho prínosu oproti riziku na individuálnom základe.
Čo sa týka nivolumabu monoterapie, predĺžené PFS pre kombináciu nivolumabu s ipilimumabom je potvrdené iba u pacientov s malou expresiou PD-L1 v nádore. U pacientov s vysokou expresiou PD- L1 (PD-L1 ≥ 1 %) v nádore bolo zlepšenie OS podobné medzi nivolumabom v kombinácii s ipilimumabom a nivolumabom v monoterapii. Pred začatím liečby kombináciou, sa lekárom odporúča starostlivo vyhodnotiť každého pacienta a charakteristiky nádoru so zohľadnením pozorovaných prínosov a toxicity kombinácie v porovnaní s nivolumabom monoterapiou (pozri časti 4.8 a 5.1).
Použitie nivolumabu u pacientov s melanómom s rýchlo progredujúcim ochorením
Pred začatím liečby u pacientov s rýchlo progredujúcim ochorením majú lekári zvážiť oneskorený nástup účinku nivolumabu (pozri časť 5.1).
Adjuvantná liečba melanómu
K dispozícii nie sú údaje o adjuvantnej liečbe pacientov s melanómom s nasledovnými rizikovými faktormi (pozri časti 4.5 a 5.1)
• pacienti s predchádzajúcim autoimunitným ochorením a akýmkoľvek stavom vyžadujúcim si systémovú liečbu buď kortikosteroidmi (≥ 10 mg prednizónu alebo jeho ekvivalentu denne) alebo inými imunosupresívnymi liekmi,
• pacienti s predchádzajúcou liečbou melanómu (okrem pacientov s chirurgickým výkonom, s
adjuvantnou rádioterapiou po neurochirurgickej resekcii z dôvodu lézií v centrálnom nervovom systéme a s predchádzajúcou adjuvantnou liečbou interferónom ukončenou ≥ 6 mesiacov pred randomizáciou),
• pacienti liečení predchádzajúcou liečbou protilátkou anti-PD-1, anti-PD-L1, anti-PD-L2, anti- CD137, alebo anti CTLA-4 (vrátane ipilimumabu alebo akejkoľvek inej protilátky alebo liečiva špecificky zameranému na súbežnú stimuláciu T buniek alebo kontrolný bod dráh),
• jedinci vo veku do 18 rokov.
Pri absencii údajov sa má v týchto populáciách nivolumab používať s opatrnosťou po starostlivom zvážení možného prínosu oproti riziku na individuálnom základe.
Nemalobunkový karcinóm pľúc
Pacienti s východiskovým skóre výkonnostného stavu ≥ 2, aktívnymi metastázami v mozgu alebo autoimunitným ochorením, symptomatickým intersticiálnym ochorením pľúc a pacienti, ktorí dostali systémové imunosupresíva pred zaradením do štúdie boli z klinických skúšaní s NSCLC vylúčení (pozri časti 4.5 a 5.1). V prípade chýbajúcich údajov sa má u týchto populácií nivolumab použiť
s opatrnosťou po starostlivom posúdení pomeru potenciálneho prínosu oproti riziku na individuálnom
základe.
Pred začatím liečby u pacientov s horšími prognostickými znakmi a/alebo agresívnym ochorením majú lekári zvážiť oneskorený nástup účinku nivolumabu. Pri neskvamóznom NSCLC sa pozoroval vyšší počet úmrtí v priebehu 3 mesiacov pri nivolumabe v porovnaní s docetaxelom. Faktory spojené so skorými úmrtiami boli horšie prognostické faktory a/alebo agresívnejšie ochorenie v kombinácii
s nízkou expresiou PD-L1 v nádore alebo bez jeho expresie (pozri časť 5.1).
Karcinóm z renálnych buniek
Pacienti so súbežnými metastázami v mozgu, aktívnym autoimunitným ochorením či zdravotnými stavmi vyžadujúcimi si systémovú imunosupresiu alebo s ich akoukoľvek anamnézou boli z pivotného
skúšania s RCC vylúčení (pozri časti 4.5 a 5.1). Pri nedostupnosti údajov sa má v týchto populáciách nivolumab používať s opatrnosťou po starostlivom zvážení pomeru potenciálneho prínosu oproti riziku na individuálnom základe.
Klasický Hodgkinov lymfóm
Pacienti s aktívnym autoimunitným ochorením a symptomatickým intersticiálnym ochorením pľúc boli z klinických skúšaní s cHL vylúčení (pozri časť 5.1). Pri absencii údajov sa má nivolumab používať v týchto populáciách s opatrnosťou po dôkladnom zvážení pomeru potenciálneho prínosu oproti riziku na individuálnej báze.
Komplikáciealogénnejtransplantáciehematopoietickýchkmeňovýchbuniek(Haematopoietic Stem
Cell Transplant, HSCT) pri klasickom Hodgkinovomlymfóme
Predbežné výsledky z následných návštev pacientov s cHL podstupujúcich alogénnu HSCT po predchádzajúcej expozícii nivolumabu preukázali vyšší než očakávaný počet prípadov akútnej reakcie štepu proti hostiteľovi (acute graft-versus-host-disease, aGVHD) a mortality súvisiacej s
transplantáciou (transplant related mortality, TRM). Pokiaľ nebudú dostupné ďalšie údaje, od prípadu
k prípadu sa má vykonať dôkladné zváženie možných prínosov HSCT a možného zvýšenia rizika komplikácií súvisiacich s transplantáciou (pozri časť 4.8).
Po uvedení lieku na trh sa u pacientov liečených nivolumabom po alogénnej HSCT hlásil náhly nástup a závažná GVHD, niektoré so smrteľným následkom. Liečba nivolumabom môže zvýšiť riziko závažnej GVHD a úmrtia u pacientov, ktorí predtým podstúpili alogénnu HSCT, najmä u tých, ktorí majú prechádzajúcu GVHD v anamnéze. U týchto pacientov sa má zvážiť prínos liečby nivolumabom oproti možnému riziku (pozri časť 4.8).
Karcinóm hlavy a krku
Pacienti s východiskovým stavom výkonnostného skóre ≥ 2, aktívnymi metastázami v mozgu alebo leptomeningeálnymi metastázami, aktívnym autoimunitným ochorením, medicínskymi stavmi vyžadujúcimi si systémovú imunosupresiu, alebo s karcinóm nosohltana alebo slinnej žľazy ako primárnych nádorových miest boli z klinického skúšania s SCCHN vylúčení (pozri časti 4.5 a 5.1). V týchto populáciách sa má pri absencii údajov nivolumab používať s opatrnosťou po starostlivom zvážení pomeru potenciálneho prínosu oproti riziku na individuálnom základe.
Pred začatím liečby u pacientov s horšími prognostickými znakmi a/alebo agresívnym ochorením majú lekári zvážiť oneskorený nástup účinku nivolumabu. Pri karcinóme hlavy a krku sa pozoroval vyšší počet úmrtí v priebehu 3 mesiacov pri nivolumabe v porovnaní s docetaxelom. Faktory spojené so skorými úmrtiami boli výkonostný stav podľa ECOG, rýchlo progredujúce ochorenie
po predchádzajúcej liečbe platinou a vysoká nádorová záťaž.
Uroteliálny karcinóm
Pacienti s východiskovým stavom výkonnostného skóre ≥ 2, aktívnymi metastázami v mozgu alebo leptomeningeálnymi metastázami, aktívnym autoimunitným ochorením alebo medicínskymi stavmi vyžadujúcimi si systémovú imunosupresiu, boli z klinických skúšaní s uroteliálnym karcinómom
vylúčení (pozri časti 4.5 a 5.1). V týchto populáciách sa má pri absencii údajov nivolumab používať s
opatrnosťou po starostlivom zvážení pomeru potenciálneho prínosu oproti riziku na individuálnom základe.
Pacientinadiéteskontrolovanýmpríjmomsodíka
Každý ml tohto lieku obsahuje 0,1 mmol (alebo 2,5 mg) sodíka. Musí sa to zohľadniť pri liečbe pacientov na diéte s kontrolovaným príjmom sodíka.
Bezpečnostnákartapacienta
Všetci lekári predpisujúci OPDIVO musia byť oboznámení s Informáciami pre lekára a Pokynmi liečby. Predpisujúci lekár musí s pacientom prediskutovať riziká liečby OPDIVOM. Pri každom predpísaní lieku dostane pacient Bezpečnostnú kartu pacienta.
4.5 Liekové a iné interakcie
Nivolumab je ľudská monoklonálna protilátka, preto sa takéto farmakokinetické štúdie interakcií nevykonali. Keďže monoklonálne protilátky nie sú metabolizované prostredníctvom enzýmov cytochrómu P450 (CYP) ani inými enzýmami metabolizujúcimi liečivá, nepredpokladá sa, že inhibícia alebo indukcia týchto enzýmov pri súbežnom podávaní liekov ovplyvní farmakokinetiku nivolumabu.
Iné formy interakcie
Systémováimunosupresia
Je potrebné sa vyhnúť používaniu systémových kortikosteroidov a iných imunosupresív na začiatku pred začatím liečby nivolumabom, pretože majú potenciál zasahovať do farmakodynamickej aktivity. Systémové kortikosteroidy a iné imunosupresíva sa však môžu použiť po začatí liečby nivolumabom na liečbu imunitne podmienených nežiaducich reakcií. Podľa predbežných výsledkov sa zdá, že systémová imunosupresia po začatí liečby nivolumabom nezabraňuje reakcii na nivolumab.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití nivolumabu u gravidných žien. Štúdie na zvieratách preukázali embryofetálnu toxicitu (pozri časť 5.3). Je známe, že ľudský IgG4 prechádza placentárnou bariérou a nivolumab je IgG4; preto prichádza do úvahy, že nivolumab sa bude prenášať z matky na vyvíjajúci sa plod. Nivolumab sa neodporúča počas gravidity a u žien vo fertilnom veku nepoužívajúcich účinnú antikoncepciu, pokiaľ klinický prínos neprevažuje nad možným rizikom.
Po poslednej dávke nivolumabu sa má počas minimálne 5 mesiacov používať účinná antikoncepcia.
Dojčenie
Nie je známe, či sa nivolumab vylučuje do materského mlieka u ľudí. Keďže sa veľa liekov vrátane protilátok môže vylučovať do materského mlieka u ľudí, riziko u novorodencov/dojčiat nemožno vylúčiť. Rozhodnutie, či ukončiť dojčenie alebo či prerušiť liečbu nivolumabom sa musí urobiť po zohľadnení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Štúdie na hodnotenie účinku nivolumabu na fertilitu neboli vykonané. Preto účinok nivolumabu na fertilitu u mužov a žien nie je známy.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Na základe jeho farmakodynamických vlastností, nie je pravdepodobné, že nivolumab ovplyvňuje schopnosť viesť vozidlá a obsluhovať stroje. Vzhľadom na možné nežiaduce reakcie, ako je únava (pozri časť 4.8), majú byť pacienti upozornení na to, aby boli obozretní pri vedení vozidiel alebo obsluhe strojov, pokiaľ si nie sú istí, že na nich nivolumab nepôsobí nepriaznivo.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Nivolumab používaný ako monoterapia alebo v kombinácii s ipilimumabom
V zhromaždenom súbore údajov nivolumabu s dávkou 3 mg/kg ako monoterapie pokrývajúcej rôzne typy nádorov (n = 2 578) s minimálnym následným sledovaním v rozmedzí od 2,3 do 28°mesiacov boli najčastejšie nežiaduce reakcie (≥ 10 %) únava (30 %), vyrážka (17 %), svrbenie (13 %), hnačka (13 %) a nauzea (12 %). Väčšina nežiaducich reakcií bolo miernych až stredne závažných (1. alebo
2. stupňa). Pri minimálne 24 mesačnom následnom sledovaní sa pri NSCLC nezistili žiadne nové bezpečnostné signály.
V zhromaždenom súbore údajov nivolumabu s dávkou 1 mg/kg v kombinácii s ipilimumabom s dávkou 3 mg/kg pri melanóme (n = 448) s minimálnym následným sledovaním v rozmedzí od 6 do
28°mesiacov boli najčastejšie (≥ 10 %) nežiaduce reakcie vyrážka (52 %), únava (46 %),
hnačka (43 %), svrbenie (36 %), nevoľnosť (26 %), pyrexia (19 %), znížená chuť do jedla (16 %), hypotyreoidizmus (16 %), kolitída (15 %), vracanie (14 %), artralgia (13 %), bolesť brucha (13 %), bolesť hlavy (11 %) a dyspnoe (10 %). Väčšina nežiaducich reakcií bola mierna až stredne závažná (1. alebo 2. stupeň).
U pacientov liečených nivolumabom v dávke 1 mg/kg v kombinácii s ipilimumabom v dávke 3 mg/kg v štúdii CA209067 malo 154/313 (49 %) prvý nástup nežiaducich reakcií 3. alebo 4. stupňa počas úvodnej fázy kombinácie. U 147 pacientov v tejto skupine, ktorí pokračovali v liečbe vo fáze s jedným liečivom sa u 47 (32 %) vyskytla minimálne jedna nežiaduca reakcia 3. alebo 4. stupňa počas fázy s jedným liečivom.
Nivolumab používaný ako monoterapia na adjuvantnú liečbu melanómu
V súbore údajov o nivolumabe v dávke 3 mg/kg ako monoterapia na adjuvantnú liečbu melanómu
(n = 452) boli najčastejšie nežiaduce reakcie (≥ 10 %) únava (46 %), vyrážka (29 %), hnačka (24 %), svrbenie (23 %), nauzea (15 %), artralgia (13 %), muskuloskeletálna bolesť (11 %) a hypotyreoidizmus (11 %). Väčšina nežiaducich reakcií bola mierna až stredne závažná
(1. alebo 2. stupňa).
Zoznamnežiaducichreakciívtabuľke
Nežiaduce reakcie hlásené v zhromaždenom súbore údajov u pacientov liečených nivolumabom monoterapiou (n = 2 578) a u pacientov liečených nivolumabom v kombinácii s ipilimumabom
(n = 448) sú uvedené v tabuľke 4. Tieto reakcie sú uvedené podľa tried orgánového systému a podľa frekvencie. Frekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(nemožno odhadnúť z dostupných údajov po uvedení lieku na trh). V rámci jednotlivých skupín
frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 4: Nežiaduce reakcie
Nivolumab monoterapia Nivolumab v kombinácii s ipilimumabom
Infekcie a nákazy
Časté infekcia horných dýchacích ciest pneumónia, infekcia horných dýchacích ciest
Menej časté pneumóniaa, bronchitída bronchitída
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Zriedkavé histiocytová nekrotizujúca lymfadenitída
(Kikuchiho lymfadenitída)
Poruchy krvi a lymfatického systému
Veľmi časté neutropéniaa,b
Časté eozinofília
Menej časté eozinofília
Poruchy imunitného systému
Časté reakcie spojené s infúziouc, precitlivenosťc
reakcie spojené s infúziou, precitlivenosť
Menej časté sarkoidóza
Zriedkavé anafylaktická reakciac
Neznáma rejekcia transplantovaného solídneho
orgánuh
Poruchy endokrinného systému
rejekcia transplantovaného solídneho orgánuh
Veľmi časté hypotyreoidizmus
Časté hypotyreoidizmus, hypertyreoidizmus nedostatočnosť nadobličiek, hypopituitarizmus, hypofyzitída, hypertyreoidizmus, tyroiditída
Menej časté nedostatočnosť nadobličiek, hypopituitarizmus, hypofyzitída, tyroiditída, diabetes mellitus
Zriedkavé diabetická ketoacidóza
Poruchy metabolizmu a výživy
diabetická ketoacidózac, diabetes mellitusc
Veľmi časté znížená chuť do jedla
Časté znížená chuť do jedla dehydratácia
Menej časté dehydratácia, metabolická acidóza
Neznáme syndróm spôsobený rozpadom nádorui syndróm spôsobený rozpadom nádorui
Poruchy pečene a žlčových ciest
Časté hepatitídac
Menej časté hepatitídac Zriedkavé cholestáza Poruchy nervového systému
Veľmi časté bolesť hlavy
Časté periférna neuropatia, bolesť hlavy, závrat
Menej časté polyneuropatia, autoimunitná neuropatia (zahŕňajúca parézu tvárového a odťahujúceho nervu)
Zriedkavé Guillainov-Barrého syndróm, demyelinizácia, myastenický syndróm, encefalitídaa,c
periférna neuropatia, závrat
Guillainov-Barrého syndróm, polyneuropatia, neuritída, ochrnutie lýtkového nervu, autoimunitná neuropatia (zahŕňajúca parézu tvárového a odťahujúceho nervu), encefalitídac
 Poruchy oka
Poruchy oka
Časté uveitída, rozmazané videnie
Menej časté uveitída, rozmazané videnie, suché oko
Neznáme Vogtov-Koyanagiho-Haradov syndrómh Vogtov-Koyanagiho-Haradov syndrómh
Poruchy srdca a srdcovej činnosti
Časté tachykardia
Menej časté tachykardia, perikardiálne ochoreniaj arytmia (vrátane ventrikulárnej arytmie)d, atriálna fibrilácia, myokarditídaa,f
Zriedkavé arytmia (vrátane ventrikulárnej arytmie)d, atriálna fibrilácia, myokarditídaa,f
Neznáme perikardiálne ochoreniaj
Poruchy ciev
Časté hypertenzia hypertenzia
Zriedkavé vaskulitída
Poruchy dýchacej sústavy, hrudníka a mediastína
Veľmi časté dyspnoe
Časté pneumonitídaa,c, dyspnoea, kašeľ pneumonitídaa,c, pľúcna embóliaa, kašeľ
Menej časté pleurálna efúzia pleurálna efúzia
Zriedkavé infiltrácia pľúc
Poruchy gastrointestinálneho traktu
Veľmi časté hnačka, nevoľnosť kolitídaa, hnačka, vracanie, nevoľnosť, bolesť brucha
Časté kolitídaa, stomatitída, vracanie, bolesť brucha, zápcha, sucho v ústach
stomatitída, pankreatitída, zápcha, sucho v ústach
Menej časté pankreatitída, gastritída perforácia črevaa, gastritída, duodenitída
Zriedkavé dvanástnikový vred
Poruchy kože a podkožného tkaniva
Veľmi časté vyrážkae, svrbenie vyrážkae, svrbenie
Časté vitiligo, suchá koža, erytém, alopécia vitiligo, suchá koža, erytém, alopécia, urtikária
Menej časté multiformný erytém, psoriáza, rosacea, urtikária
Zriedkavé toxická epidermálna nekrolýzaa,f, Stevensov-Johnsonov syndróma,f
psoriáza
toxická epidermálna nekrolýzaa,f, Stevensov-Johnsonov syndrómf
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Veľmi časté artralgia
Časté muskuloskeletálna bolesťg, artralgia muskuloskeletálna bolesťg
Menej časté reumatická polymyalgia, artritída spondylartropatia, Sjogrenov syndróm, artritída, myopatia, myozitída (vrátane polymyozitídy)a,f, rabdomyolýzaa,f
Zriedkavé Sjogrenov syndróm, myopatia, myozitída (vrátane polymyozitídy)a,f, rabdomyolýzaa,f
Poruchy obličiek a močových ciest
Časté zlyhanie obličiek (vrátane akútneho poškodenia obličiek)a,c
Menej časté tubulointersticiálna nefritída, zlyhanie obličiek (vrátane akútneho poškodenia obličiek)a,c
Celkové poruchy a reakcie v mieste podania
tubulointersticiálna nefritída
Veľmi časté únava únava, pyrexia
Časté pyrexia, edém (vrátane periférneho edému)
edém (vrátane periférneho edému), bolesť
Menej časté bolesť, bolesť na hrudi bolesť na hrudi
Laboratórne a funkčné vyšetreniab
Veľmi časté zvýšená AST, zvýšená ALT, zvýšená alkalická fosfatáza, zvýšená lipáza, zvýšená amyláza, hypokalciémia, zvýšený kreatinín, hyperglykémiac, lymfopénia, leukopénia, trombocytopénia, anémia, hyperkalciémia, hyperkaliémia, hypokaliémia, hypomagneziémia, hyponatriémia
Časté zvýšený celkový bilirubín, hypoglykémia, hypermagneziémia, hypernatriémia, úbytok telesnej hmotnosti
zvýšená AST, zvýšená ALT, zvýšený celkový bilirubín, zvýšená alkalická fosfatáza, zvýšená lipáza, zvýšená amyláza, zvýšený kreatinín, hyperglykémiac, hypoglykémia, lymfopénia, leukopénia, neutropénia, trombocytopénia, anémia, hypokalciémia, hyperkaliémia, hypokaliémia, hypomagneziémia, hyponatriémia
hyperkalciémia, hypermagneziémia, hypernatriémia, úbytok telesnej hmotnosti
a V ukončených alebo prebiehajúcich klinických štúdiách sa hlásili smrteľné prípady
b Frekvencie laboratórnych termínov vyjadrujú podiel pacientov, u ktorých sa vyskytlo zhoršenie v laboratórnych meraniach oproti východiskovým hodnotám. Pozri nižšie „Opis vybraných nežiaducich
reakcií; Laboratórne abnormality“.
c V ukončených alebo prebiehajúcich klinických štúdiách sa hlásili život ohrozujúce prípady.
d V populácii s metastatickým melanómom po liečbe inhibítorom CTLA4/BRAF bola frekvencia nežiaducich udalostí triedy orgánového systému poruchy srdca a srdcovej činnosti bez ohľadu na príčinnú
súvislosť vyššia v skupine s nivolumabom než v skupine s chemoterapiou. Miera výskytu na
100 pacientorokov expozície boli 9,3 oproti 0; závažné srdcové udalosti sa hlásili u 4,9 % pacientov v skupine s nivolumabom oproti 0 v skupine podľa voľby skúšajúceho. V populácii s metastatickým melanómom bez predchádzajúcej liečby bola frekvencia kardiálnych nežiaducich udalostí nižšia v skupine s nivolumabom než v skupine s dakarbazínom. Okrem arytmií (atriálna fibrilácia, tachykardia a ventrikulárna arytmia) boli všetky nežiaduce udalosti posúdené skúšajúcimi ako nesúvisiace
s nivolumabom.
e Vyrážka je kompozitný názov, ktorý zahŕňa makulopapulárnu vyrážku, erytematóznu vyrážku, svrbivú vyrážku, folikulárnu vyrážku, makulárnu vyrážku, morbiliformnú vyrážku, papulárnu vyrážku, pustulárnu vyrážku, papuloskvamóznu vyrážku, vezikulárnu vyrážku, generalizovanú vyrážku, dermatitídu,
akneiformnú dermatitídu, alergickú dermatitídu, atopickú dermatitídu, bulóznu dermatitídu, exfoliatívnu
dermatitídu, psoriaziformnú dermatitídu, liekovú vyrážku a pemfigoid.
f Hlásené tiež v štúdiách mimo súhrnného súboru údajov. Frekvencia sa zakladá na programe veľkej expozície.
g Muskuloskeletálna bolesť je kompozitný názov, ktorý zahŕňa bolesť chrbta, bolesť kosti, muskuloskeletálnu bolesť na hrudi, muskuloskeletalnu ťažkosť, myalgiu, bolesť šije, bolesť v končatine a
bolesť chrbtice.
h Udalosť po uvedení lieku na trh (pozri tiež časť 4.4)
i Hlásený v klinických štúdiách a po uvedení lieku na trh.
j Perikardiálne ochorenia je kompozitný názov, ktorý zahŕňa perikarditídu, perikardiálnu efúziu, kardiálnu tamponádu a Dresslerov syndróm.
Celkový bezpečnostný profil nivolumabu v dávke 3 mg/kg pri adjuvantnej liečbe melanómu (n = 452)
bol zhodný s tým, ktorý sa stanovil u jednotlivých typov nádorov pri monoterapii nivolumabom.
OpisvybranýchnežiaducichreakciíNivolumab alebo nivolumab v kombinácii s ipilimumabom sa spája s imunitne podmienenými nežiaducimi reakciami. Vo väčšine prípadov imunitne podmienené nežiaduce reakcie odzneli po vhodnej medicínskej liečbe. Pri imunitne podmienenej kolitíde bolo potrebné trvalé ukončenie liečby u väčšej časti pacientov, ktorí dostávali nivolumab v kombinácii s ipilimumabom ako u tých, ktorí dostávali nivolumab v monoterapii (16 % a 0,8 %, v uvedenom poradí), pri imunitne podmienenej
hepatitíde (9 % a 1 %) a pri imunitne podmienených endokrinopatiách (2,7 % a 0,1 %). U pacientov, u ktorých sa vyskytla udalosť, boli potrebné vysoké dávky kortikosteroidov (zodpovedajúce minimálne
40 mg prednizónu) na liečbu imunitne podmienenej kolitídy u väčšej časti pacientov, ktorí dostávali
režim kombinácie ako u pacientov, ktorí dostávali nivolumab v monoterapii (46 % a 15 %, v uvedenom poradí), na liečbu imunitne podmienenej hepatitídy (46 % a 21 %), imunitne podmienených endokrinopatií (27 % a 7 %, v uvedenom poradí) a imunitne podmienenej kožnej nežiaducej reakcie (7 % a 4 %, v uvedenom poradí). Postupy liečby týchto nežiaducich reakcií sú uvedené v časti 4.4.
Imunitne podmienená pneumonitída
U pacientov liečených nivolumabom v monoterapii bola incidencia pneumonitídy vrátane intersticiálneho ochorenia pľúc a infiltrácie pľúc 3,4 % (87/2 578). Väčšina hlásených prípadov bola
1. stupňa závažnosti u 0,8 % (21/2 578) alebo 2. stupňa u 1,7 % (44/2 578) pacientov. Prípady
3. stupňa sa hlásili u 0,7 % (19/2 578) a 4. stupňa u < 0,1 % (1/2 578) pacientov. V týchto štúdiách sa hlásili prípady 5. stupňa u < 0,1 % (2/2 578) pacientov. Medián času do nástupu bol 3,6 mesiaca (rozsah: 0,2 – 19,6). K vyriešeniu došlo u 63 pacientov (72,4 %) s mediánom času do vyriešenia
6,1 týždňa (rozsah: 0,1+ – 96,7+); + označuje cenzurované sledovanie.
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bola incidencia pneumonitídy vrátane intersticiálneho ochorenia pľúc 7,8 % (35/448). Prípady 2. stupňa sa hlásili u 4,7 % (21/448),
3. stupňa u 1,1 % (5/448) a 4. stupňa u 0,2 % (1/448) pacientov. Jedna pneumonitída z prípadov
3. stupňa sa zhoršila v priebehu 11 dní so smrteľným koncom. Medián času do nástupu
bol 2,6 mesiaca (rozsah: 0,7 – 12,6). K vyriešeniu došlo u 33 pacientov (94,3 %) s mediánom času do vyriešenia 6,1 týždňa (rozsah: 0,3 – 35,1+).
Imunitne podmienená kolitída
U pacientov liečených nivolumabom v monoterapii bola incidencia hnačky, kolitídy alebo častých pohybov čriev 13,1 % (339/2 578). Väčšina hlásených prípadov bola 1. stupňa závažnosti u 8,5 %
(220/2 578) alebo 2. stupňa u 3,0 % (78/2 578) pacientov. Prípady 3. stupňa sa hlásili u 1,6 %
(41/2 578) pacientov. V týchto štúdiách sa nehlásili žiadne prípady 4. alebo 5. stupňa. Medián času
do nástupu bol 1, 8mesiaca (rozsah: 0,0 – 26,6). K vyriešeniu došlo u 296 pacientov (88,1 %)
s mediánom času do vyriešenia 2,1 týždňa (rozsah: 0,1 – 124,4+).
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bola incidencia hnačky alebo kolitídy 46,7 % (209/448). Prípady 2. stupňa sa hlásili u 13,6 % (61/448), 3. stupňa u 15,8 % (71/448) a 4. stupňa u 0,4 % (2/448) pacientov. Nehlásili sa žiadne prípady 5. stupňa. Medián času do nástupu bol 1,2 mesiaca (rozsah: 0,0-22,6). K vyriešeniu došlo u 186 pacientov (89,4 %) s mediánom času do vyriešenia 3,0 týždne (rozsah: 0,1-159,4+).
Imunitne podmienená hepatitída
U pacientov liečených nivolumabom v monoterapii bola incidencia abnormalít testov funkcie pečene
6,7 % (173/2 578). Väčšina hlásených prípadov bola 1. stupňa závažnosti u 3,5 % (91/2 578) alebo
2. stupňa u 1,2 % (32/2 578) pacientov. Prípady 3. stupňa sa hlásili u 1,6 % (41/2 578) a 4. stupňa u 0,3 % (9/2 578) pacientov. V týchto štúdiách sa nehlásili žiadne prípady 5. stupňa. Medián času do nástupu bol 2,1 mesiaca (rozsah: 0,0 – 27,6). K vyriešeniu došlo u 132 pacientov (76,7 %)
s mediánom času do vyriešenia 5,9 týždňa (rozsah: 0,1 – 82,6+).
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bola incidencia abnormalít testov funkcie pečene 29,5 % (132/448). Prípady 2. stupňa sa hlásili u 6,7 % (30/448), 3. stupňa u 15,4 % (69/448) a 4. stupňa u 1,8 % (8/448) pacientov. Nehlásili sa žiadne prípady 5. stupňa. Medián času do nástupu bol 1,5 mesiaca (rozsah: 0,0-30,1). K vyriešeniu došlo u 124 pacientov (93,9 %) s mediánom času do vyriešenia 5,1 týždňa (rozsah: 0,1-106,9).
Imunitne podmienená nefritída a dysfunkcia obličiek
U pacientov liečených nivolumabom v monoterapii bola incidencia nefritídy alebo dysfunkcie obličiek
2,8 % (71/2 578). Väčšina hlásených prípadov bola 1. stupňa závažnosti u 1,6 % (41/2 578) alebo
2. stupňa u 0,7 % (18/2 578) pacientov. Prípady 3. stupňa sa hlásili u 0,4 % (11/2 578) a 4. stupňa u < 0,1 % (1/ 2 578) pacientov. V týchto štúdiách sa nehlásili žiadne prípady nefritídy alebo dysfunkcie obličiek 5. stupňa. Medián času do nástupu bol 2,3 mesiaca (rozsah: 0,0 – 18,2). K
vyriešeniu došlo u 42 pacientov (61,8 %) s mediánom času do vyriešenia 12,1 týždňa (rozsah: 0,3+ –
79,1+).
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bola incidencia nefritídy alebo dysfunkcie obličiek 5,1 % (23/448). Prípady 2. stupňa sa hlásili u 1,6 % (7/448), 3. stupňa u 0,9 % (4/448) a 4. stupňa u 0,7 % (3/448) pacientov. Nehlásili sa žiadne prípady 5. stupňa. Medián času do nástupu bol 2,6 mesiaca (rozsah: 0,5 – 21,8). K vyriešeniu došlo u 21 pacientov (91,3 %) s mediánom času do vyriešenia 2,1 týždňa (rozsah: 0,1 – 125,1+).
Imunitnepodmienenéendokrinopatie
U pacientov liečených nivolumabom v monoterapii bola incidencia ochorení štítnej žľazy vrátane hypotyreoidizmu alebo hypertyreoidizmu 9,6 % (248/2 578). Väčšina hlásených prípadov bola
1. stupňa závažnosti u 4,2 % (107/2 578) alebo 2. stupňa u 5,4 % (139/2 578) pacientov. Ochorenia štítnej žľazy 3. stupňa sa hlásili u < 0,1 % (2/2 578) pacientov. Hlásila sa hypofyzitída (1 1. stupňa;
2 2. stupňa, 5 3. stupňa a 1 4. stupňa), hypopituitarizmus (4 2.stupňa a 1 3.stupňa), nedostatočnosť
nadobličiek (vrátane sekundárnej adrenokortikálnej nedostatočnosti) (1 1. stupňa; 9 2. stupňa a
5 3. stupňa), diabetes mellitus (vrátane diabetes mellitus 1. typu) (3 2. stupňa a 1 3. stupňa) a diabetická ketoacidóza (2 3. stupňa). V týchto štúdiách sa nehlásili žiadne prípady 5. stupňa. Medián času do nástupu týchto endokrinopatií bol 2,8 mesiaca (rozsah: 0,3 – 29,1). K vyriešeniu došlo u
117 pacientov (42,9 %). Čas do vyriešenia bol v rozmedzí od 0,4 do 144,1+ týždňa.
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bola incidencia ochorení štítnej
žľazy 25,2 % (113/448). Ochorenia štítnej žľazy 2. stupňa sa hlásili u 14,5 % (65/448) a 3. stupňa u
1,3 % (6/448) pacientov. Hypofyzitída (vrátane lymfocytárnej hypofyzitídy) 2. stupňa sa vyskytla u
5,8 % (26/448) a 3. stupňa u 2,0 % (9/448) pacientov. Hypopituitarizmus 2. stupňa a 3. stupňa sa vyskytol u 0,4 % (2/448) a u 0,7 % (3/448) pacientov, v uvedenom poradí. Nedostatočnosť
nadobličiek (vrátane sekundárnej adrenokortikálnej nedostatočnosti) 2. stupňa, 3. stupňa a 4. stupňa sa vyskytla u 1,6 % (7/448), 1,3 % (6/448) a 0,2 % (1/448) pacientov, v uvedenom poradí. Diabetes mellitus 1. stupňa, 2. stupňa, 3. stupňa a 4. stupňa, ako aj diabetická ketoacidóza 4. stupňa sa každé hlásilo u 0,2 % (1/448) pacientov. Nehlásili sa žiadne prípady 5. stupňa endokrinopatie. Medián času
do nástupu týchto endokrinopatií bol 1,9 mesiaca (rozsah: 0,0 – 28,1). K vyriešeniu došlo u
64 pacientov (45,4 %). Čas do vyriešenia bol v rozmedzí od 0,4 do 155,4+ týždňa.
Imunitne podmienenékožnénežiaducereakcie
U pacientov liečených nivolumabom v monoterapii bola incidencia vyrážky 26,4 % (680/2 578). Väčšina hlásených prípadov bola 1. stupňa závažnosti u 20,1 % (518/2 578) pacientov. Prípady
2. stupňa sa hlásili u 5,1 % (131/2 578) a 3. stupňa u 1,2 % (31/2 578) pacientov. V týchto štúdiách sa
nehlásili žiadne prípady 4. alebo 5. stupňa. Medián času do nástupu bol 1,4 mesiaca (rozsah: 0,0 –
27,9). K vyriešeniu došlo u 428 pacientov (63,8 %) s mediánom času do vyriešenia 17,1 týždňa (0,1-
150,0+).
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bola incidencia vyrážky
65,0 % (291/448). Prípady 2. stupňa sa hlásili u 20,3 % (91/448) a 3. stupňa u 7,6 % (34/448) pacientov. Nehlásili sa žiadne prípady 4. alebo 5. stupňa. Medián času do nástupu bol 0,5 mesiaca (rozsah: 0,0 – 19,4). K vyriešeniu došlo u 191 pacientov (65,9 %) s mediánom času do vyriešenia
11,4 týždňa (rozsah: 0,1 – 150,1+).
Pozorovali sa zriedkavé prípady SJS a TEN, niektoré z nich so smrteľným následkom (pozri časti 4.2 a 4.4).
Infúznereakcie
U pacientov liečených nivolumabom v monoterapii bola incidencia precitlivenosti/infúznych reakcií
4,7 % (121/2 578) vrátane 6 prípadov 3. stupňa a 2 4. stupňa.
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bola incidencia precitlivenosti/infúznych reakcií 3,8 % (17/448); všetky boli 1. alebo 2. stupňa závažnosti. Prípady
2. stupňa sa hlásili u 2,2 % (10/448) pacientov. Nehlásili sa žiadne prípady 3.-5. stupňa.
Komplikácie
a
logénnej
HSCT
p
ri
klasickom
Hodgkinovom
lymfóme
Po použití nivolumabu pred alogénnou HSCT a po nej sa hlásil náhly nástup GVHD (pozri časť 4.4).
U 49 hodnotených pacientov z dvoch štúdií s cHL, ktorí podstúpili alogénnu HSCT po ukončení monoterapie nivolumabom sa hlásili akútne GVHD 3. alebo 4. stupňa u 13/49 pacientov (26,5 %). Hyperakútne GVHD, definované ako akútne GVHD vyskytujúce sa v priebehu 14 dní po infúzii kmeňových buniek, sa hlásili u troch pacientov (6 %). Febrilný syndróm vyžadujúci si liečbu steroidom bez identifikovaného dôvodu infekcie sa hlásil u šiestich pacientov (12 %) počas prvých
6 týždňov po transplantácii, traja pacienti reagovali na liečbu steroidmi. Hepatovenózne okluzívne ochorenie sa vyskytlo u jedného pacienta, ktorý zomrel na GVHD a multiorgánové zlyhanie. Deväť zo
49 pacientov (18,4 %) zomrelo na komplikácie alogénnej HSCT po podaní nivolumabu. 49 pacientov malo medián následných návštev z dodatočných alogénnych HSCT 5,6 mesiaca (rozsah: 0 –
19 mesiacov).
Laboratórneabnormality
U pacientov liečených nivolumabom v monoterapii bol podiel pacientov, u ktorých došlo k posunu laboratórnych abnormalít do 3. alebo 4. stupňa od východiskového stavu, nasledovný: 5,2 % z dôvodu anémie (všetky 3. stupňa), 1,0 % z dôvodu trombocytopénie, 1,0 % z dôvodu leukopénie, 10,0 % z dôvodu lymfopénie, 1,1 % z dôvodu neutropénie, 2,1 % z dôvodu zvýšenej alkalickej fosfatázy, 2,7 %
z dôvodu zvýšenej AST, 2,2 % z dôvodu zvýšenej ALT, 1,2 % z dôvodu zvýšeného celkového
bilirubínu, 0,9 % z dôvodu zvýšeného kreatinínu, 3,8 % z dôvodu hyperglykémie, 1,0 % z dôvodu hypoglykémie, 3,5 % z dôvodu zvýšenej amylázy, 7,9 % z dôvodu zvýšenej lipázy, 6,4 % z dôvodu hyponatriémie, 1,8 % z dôvodu hyperkaliémie, 1,5 % z dôvodu hypokaliémie, 1,2 % z dôvodu hyperkalciémie, 0,7 % z dôvodu hypermagneziémie, 0,5 % z dôvodu hypomagneziémie, 0,7 % z dôvodu hypokalciémie a 0,1 % z dôvodu hypernatriémie.
U pacientov liečených nivolumabom v kombinácii s ipilimumabom bol podiel pacientov, u ktorých došlo k zhoršeniu laboratórnych abnormalít do 3. alebo 4. stupňa od východiskového stavu nasledovný: 2,8 % z dôvodu anémie (všetky 3. stupňa); 1,2 % z dôvodu trombocytopénie; 0,5 % z dôvodu leukopénie; 6,7 % z dôvodu lymfopénie; 0,7 % z dôvodu neutropénie; 4,3 % z dôvodu zvýšenej alkalickej fosfatázy; 12,4 % z dôvodu zvýšenej AST; 15,3 % z dôvodu zvýšenej ALT;
1,2 % z dôvodu zvýšeného celkového bilirubínu; 2,4 % z dôvodu zvýšeného kreatinínu; 5,3 % z dôvodu hyperglykémie, 8,7 % z dôvodu zvýšenej amylázy; 19,5 % z dôvodu zvýšenej lipázy; 1,2 % z dôvodu hypokalciémie, z dôvodu hypernatriémie a hyperkalciémie každý 0,2 %;, 0,5 % z
dôvodu hyperkaliémie, 0,3 % z dôvodu hypermagneziémie, 4,8 % z dôvodu hypokaliémie a 9,5 % z dôvodu hyponatriémie.
Imunogenita
Z 2 022 pacientov, ktorí sa liečili nivolumabom v monoterapii 3 mg/kg každé 2 týždne a boli hodnotení na prítomnosť protilátok proti lieku bolo 231 pacientov (11,4 %) pozitívne testovaných na vznik protilátok proti lieku pri liečbe a pätnásť pacientov (0,7 %) bolo pozitívne testovaných na
neutralizujúce protilátky.
Z 394 pacientov, ktorí sa liečili nivolumabom v kombinácii s ipilimumabom a boli hodnotení na prítomnosť protilátok proti nivolumabu bolo 149 pacientov (37,8 %) pozitívne testovaných na vznik protilátok proti nivolumabu pri liečbe a 18 pacientov (4,6 %) bolo pozitívne testovaných na neutralizujúce protilátky.
Hoci sa v prítomnosti protilátok proti nivolumabu zvýšil klírens nivolumabu o 24 %, na základe farmakokinetiky a analýzy odpovede pri expozícii ako pri monoterapii, tak aj pri kombinácii sa v prítomnosti protilátok proti nivolumabu nezistil žiaden dôkaz straty účinnosti alebo zmeneného profilu toxicity.
Staršípacienti
Medzi staršími (≥ 65 rokov) a mladšími pacientmi (< 65 rokov) sa nehlásili žiadne celkové rozdiely v bezpečnosti. Údaje od pacientov s NSCLC, SCCHN a adjuvantným melanómom vo veku 75 rokov alebo starších sú príliš obmedzené na vyvodenie záverov v tejto populácii (pozri časť 5.1). Údaje od
pacientov s cHL vo veku 65 rokov alebo starších sú príliš obmedzené na vyvodenie záverov o tejto populácii (pozri časť 5.1).
Poruchafunkciepečenealebo obličiekV štúdii s neskvamóznym NSCLC (CA209057) bol bezpečnostný profil u pacientov s východiskovou poruchou funkcie obličiek alebo pečene porovnateľný s tým, ktorý bol v celkovej populácii. Z dôvodu malej veľkosti vzorky v podskupinách sa majú tieto výsledky interpretovať s opatrnosťou.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických skúšaniach sa nehlásili žiadne prípady predávkovania. V prípade predávkovania majú byť pacienti dôkladne sledovaní z dôvodu prejavov alebo príznakov nežiaducich reakcií a okamžite sa má začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, monoklonálne protilátky. ATC kód: L01XC17
MechanizmusúčinkuNivolumab je ľudská monoklonálna protilátka (human monoclonal antibody, HuMAb) immunoglobulín G4 (IgG4), ktorá sa viaže na receptor programovanej smrti-1 (programmed death-1, PD-1) a blokuje jeho interakciu s PD-L1 a PD-L2. Receptor PD-1 je negatívny regulátor aktivity
T-buniek, pre ktorý je preukázané, že sa podieľa na kontrole imunitných odpovedí T-buniek. Spojenie PD-1 s ligandmi PD-L1 a PD-L2, sú exprimované v bunkách, obsahujúcich antigén, a ktoré môžu byť exprimované nádorovými alebo inými bunkami v mikroprostredí nádoru, má za následok inhibíciu proliferácie T-buniek a sekréciu cytokínov. Nivolumab zosilňuje odpovede T-buniek vrátane protinádorových odpovedí, prostredníctvom blokády PD-1 väzbou na ligandy PD-L1 a PD-L2. Na syngénnych modeloch myší viedla blokáda aktivity PD-1 k zníženiu rastu nádoru.
Kombinovaná inhibícia sprostredkovaná nivolumabom (anti-PD-1) a ipilimumabom (anti-CTLA-4) mala za následok pri metastatickom melanóme zlepšené odpovede proti nádoru. Na syngénnych modeloch nádorov myší viedla dvojitá blokáda PD-1 a CTLA-4 k synergickej protinádorovej aktivite.
Klinická účinnosť a bezpečnosťNa základe modelovania dávky/expozície vo vzťahoch k účinnosti a bezpečnosti neexistujú žiadne
klinicky významné rozdiely v účinnosti a bezpečnosti medzi dávkou nivolumabu 240 mg každé
2 týždne alebo 3 mg/kg každé 2 týždne. Navyše, pri pokročilom melanóme a RCC sa na základe týchto vzťahov nezistili žiadne klinicky významné rozdiely medzi dávkou nivolumabu 480 mg každé
4 týždne alebo 3 mg/kg každé 2 týždne.
MelanómLiečba pokročilého melanómuRandomizovanáštúdia3.fázyoproti dakarbazínu(CA209066)Bezpečnosť a účinnosť nivolumabu 3 mg/kg na liečbu pokročilého (neresekovateľného alebo metastatického) melanómu sa hodnotili v randomizovanej, dvojito zaslepenej štúdii 3. fázy (CA209066). Do štúdie boli zaradení dospelí pacienti (18 roční alebo starší) s potvrdeným predtým
neliečeným melanónom v štádiu III alebo IV s divokým typom BRAF a so skóre výkonnostného stavu
0 alebo 1 podľa ECOG. Pacienti s aktívnym autoimunitným ochorením, okulárnym melanómom alebo s aktívnymi metastázami v mozgu alebo s leptomeningeálnymi metastázami boli zo štúdie vyradení.
Celkovo bolo randomizovaných 418 pacientov buď na intravenózne podávanie nivolumabu
(n = 210) v dávke 3 mg/kg počas 60 minút každé 2 týždne alebo na dakarbazín (n = 208) v dávke
1000 mg/m2 každé 3 týždne. Randomizácia bola rozdelená podľa stavu PD-L1 v nádore a stupňa M (M0/M1a/M1b oproti M1c). Liečba pokračovala dovtedy, kým sa pozoroval klinický prínos alebo kým bola liečba tolerovaná. Liečba po progresii ochorenia bola povolená u pacientov, ktorí mali klinický
prínos a nemali podľa posúdenia skúšajúceho významné nežiaduce účinky pri študovanom liečive.
Hodnotenia nádoru pomocou kritérií hodnotenia odpovede solídnych tumorov (Response Evaluation Criteria in Solid Tumours, RECIST), verzia 1.1, sa vykonali 9 týždňov po randomizácii a pokračovali každých 6 týždňov počas prvého roka a potom neskôr každých 12 týždňov. Primárnym koncovým ukazovateľom účinnosti bolo OS. Kľúčovými sekundárnymi koncovými ukazovateľmi účinnosti boli PFS hodnotené skúšajúcim a miera objektívnej odpovede (objective response rate, ORR).
Základnou charakteristikou bola rovnováha medzi uvedenými dvoma skupinami. Medián veku bol 65 rokov (rozsah: 18-87), 59 % bolo mužov a 99,5 % bolo belochov. Väčšina pacientov mala skóre výkonnostného stavu ECOG 0 (64 %) alebo 1 (34 %). Pri vstupe do štúdie malo šesťdesiatjeden percent pacientov stupeň ochorenia M1c. Sedemdesiatštyri percent pacientov malo kožný melanóm a
11 % malo mukózny melanóm; 35 % pacientov malo melanóm pozitívny na PD-L1 ((
≥ 5 % expresia na membráne nádorovej bunky). Šestnásť percent pacientov predtým dostalo adjuvantnú liečbu; najčastejšou adjuvantnou liečbou bol interferón (9 %). Pri vstupe do štúdie mali štyri percentá pacientov metastázy v mozgu v anamnéze a 37 % pacientov malo východiskovú hladinu LDH vyššiu než ULN.
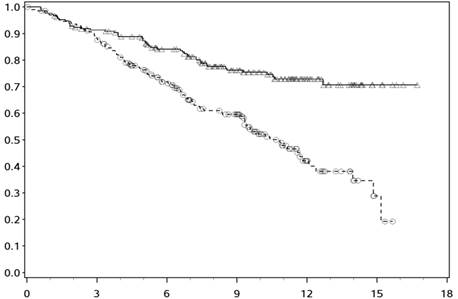
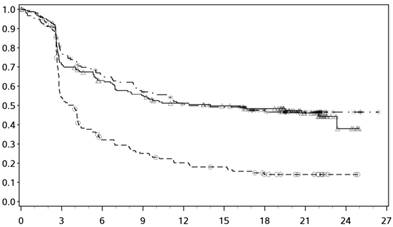
Kaplanove-Meierove krivky OS sú uvedené na obrázku 1.
Obrázok 1:
Kaplanove-Meierove krivky OS (CA209066)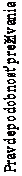
Počet jedincov s rizikom
Nivolumab
Celkové prežívanie (mesiace)
210 185 150 105 45 8 0
Dakarbazín
208 177 123 82 22 3 0
──∆─── Nivolumab (udalosti: 50/210), medián a 95 % CI: N.A.
- - -¦- - - Dakarbazín (udalosti: 96/208), medián a 95 % CI: 10,84 (9,33; 12,09)
Pozorovaný prínos OS bol zhodne preukázaný v podskupinách pacientov vrátane východiskového výkonnostného stavu ECOG, štádia M, metastáz v mozgu v anamnéze a východiskovej hladiny LDH. Prínos prežívania sa pozoroval bez ohľadu na to, či mali pacienti nádory, ktoré boli označené ako
PD – L1 negatívne alebo PD – L1 pozitívne (expresia na membráne nádorovej bunky znížená o 5 %
alebo 10 %).
Dostupné údaje naznačujú, že nástup účinku nivolumabu je oneskorený, takže tento prínos nivolumabu oproti chemoterapii môže trvať 2-3 mesiace.
Výsledky účinnosti sú uvedené v tabuľke 5.
Tabuľka 5: Výsledky účinnosti (CA209066)
nivolumab
(n = 210)
dakarbazín
(n = 208)


 C
elkové prežívanie
C
elkové prežívanie
Udalosti 50 (23,8 %) 96 (46,2 %)
Pomer rizika 0,42
99,79 % CI (0,25, 0,73)
95 % CI (0,30; 0,60)
p-hodnota < 0,0001
Medián (95 % CI) Nedosiahnuté 10,8 (9,33; 12,09)
Výskyt (95 % CI)
Pri 6 mesiacoch 84,1 (78,3; 88,5) 71,8 (64,9; 77,6)
Pri 12 mesiacoch 72,9 (65,5; 78,9) 42,1 (33,0; 50,9)
Prežívanie bez progresieUdalosti 108 (51,4 %) 163 (78,4 %)
Pomer rizika 0,43
95 % CI (0,34; 0,56)
p-hodnota < 0,0001
Medián (95 % CI) 5,1 (3,48; 10,81) 2,2 (2,10; 2,40) Výskyt (95 % CI)
Pri 6 mesiacoch 48,0 (40,8; 54,9) 18,5 (13,1; 24,6)
Pri 12 mesiacoch 41,8 (34,0; 49,3) NA
Objektívna odpoveď 84 (40,0 %) 29 (13,9 %)
(95 % CI) (33,3; 47,0) (9,5; 19,4) Pomer šancí (95 % CI) 4,06 (2,52; 6,54)
p-hodnota < 0,0001
Kompletná odpoveď (CR) 16 (7,6 %) 2 (1,0 %) Parciálna odpoveď (PR) 68 (32,4 %) 27 (13,0 %) Stabilizácia ochorenia (SD) 35 (16,7 %) 46 (22,1 %)
Medián trvania odpovedeMesiace (rozsah) Nedosiahnuté (0+– 12,5+) 6,0 (1,1 – 10,0+)
Medián času do odpovedeMesiace (rozsah) 2,1 (1,2 – 7,6) 2,1 (1,8 – 3,6)
„+“ označuje cenzurované sledovanie.
Randomizovanáštúdia3.fázyoproti chemoterapii(CA209037)Bezpečnosť a účinnosť nivolumabu 3 mg/kg na liečbu pokročilého (neresekovateľného alebo metastatického) melanómu sa hodnotili v 3. fáze, randomizovanej, otvorenej štúdie (CA209037). Štúdia zahŕňala dospelých pacientov, ktorí mali progresívne ochorenie alebo boli po liečbe
ipilimumabom a ak mali pozitívnu mutáciu BRAF V600 taktiež s progresívnych ochorením alebo po
liečbe inhibítorom BRAF kinázy. Pacienti s aktívnym autoimunitným ochorením, okulárnym melanómom, aktívnymi metastázami v mozgu alebo leptomeningeálnymi metastázami alebo so známymi predchádzajúcimi nežiaducimi reakciami vysokého stupňa v súvislosti s ipilimumabom v
anamnéze (4. stupeň podľa CTCAE v4.0) s výnimkou vyriešenej nauzey, únavy, infúznych reakcií alebo endokrinopatií boli zo štúdie vyradení.
Celkovo bolo randomizovaných 405 pacientov, aby dostávali buď nivolumab (n = 272) podávaný intravenózne počas 60 minút v dávke 3 mg/kg každé 2 týždne alebo chemoterapiu (n = 133), ktorou bola podľa voľby skúšajúceho buď dakarbazín (1000 mg/m2 každé 3 týždne) alebo karboplatina
(AUC 6 každé 3 týždne) a paklitaxel (175 mg/m2 každé 3 týždne). Randomizácia bola rozdelená podľa
stavu BRAF a PD-L1 v nádore a najlepšej odpovede na predchádzajúcu liečbu ipilimumabom.
Súbežnými primárnymi koncovými ukazovateľmi účinnosti boli potvrdená ORR u prvých
120 pacientov liečených nivolumabom, meraná nezávislou rádiologickou hodnotiacou komisiou (independent radiology review committee, IRRC) pomocou RECIST, verzia 1.1 a porovnanie OS pri nivolumabe a chemoterapii. Ďalšie koncové ukazovatele zahŕňajú trvanie a čas odpovede.
Medián veku bol 60 rokov (rozsah: 23-88). Šesťdesiatštyri percent pacientov bolo mužov a 98 % bolo belochov. Skóre výkonnostného stavu podľa ECOG 0 malo 61 % pacientov a 1 39 % pacientov. Väčšina (75 %) pacientov mala pri vstupe do štúdie stupeň ochorenia M1c. Sedemdesiattri percent pacientov malo kožný melanóm a 10 % malo mukózny melanóm. Počet predtým liečených systémovou liečbou bol 1 z 27 % pacientov, 2 z 51 % pacientov a > 2 z 21 % pacientov. Dvadsaťdva percent pacientov malo nádory, ktoré boli testované na pozitívnu mutáciu BRAF a 50 % pacientov malo nádory, ktoré boli považované za PD-L1 pozitívne. Šesťdesiatštyri percent pacientov nemalo predchádzajúci klinický prínos (CR/PR alebo SD) po liečbe ipilimumabom. Základnou charakteristikou bola rovnováha medzi skupinami s výnimkou počtov pacientov, ktorí mali metastázy v mozgu v anamnéze (19 % v skupine s nivolumabom a 13 % v skupine s chemoterapiou) a pacienti s LDH vyššou než ULN pri východiskovom stave (51 % a 35 %, v uvedenom poradí).
V čase tejto finálnej analýzy ORR sa analyzovali výsledky od 120 pacientov liečených nivolumabom a
47 pacientov liečených chemoterapiou, ktorí mali minimálne 6 mesačné následné sledovanie. Výsledky účinnosti sú uvedené v tabuľke 6.
Tabuľka 6: Najlepšia celková odpoveď, čas a trvanie odpovede (CA209037)
nivolumab
(n = 120)
chemoterapia
(n = 47)
 Potvrdená objektívna odpoveď (IRRC)
Potvrdená objektívna odpoveď (IRRC) 38 (31,7 %) 5 (10,6 %)
(95 % CI) (23,5; 40,8) (3,5; 23,1)
Kompletná odpoveď (CR) 4 (3,3 %) 0
Parciálna odpoveď (PR) 34 (28,3 %) 5 (10,6 %)
Stabilizácia ochorenia (SD) 28 (23,3 %) 16 (34,0 %)
Medián trvania odpovedeMesiace (rozsah) Nedosiahnutý 3,6 (Nedostupný)
Medián času do odpovedeMesiace (rozsah) 2,1 (1,6-7,4) 3,5 (2,1-6,1)
Dostupné údaje naznačujú, že nástup účinku nivolumabu je oneskorený, takže tento prínos
nivolumabu oproti chemoterapii môže trvať 2-3 mesiace.
Aktualizovanáanalýza(24-mesačnénáslednésledovanie)U všetkých randomizovaných pacientov bolo ORR v skupine s nivolumabom 27,2 % (95 % CI: 22,0;
32,9) a v skupine s chemoterapiou 9,8 % (95 % CI: 5,3; 16,1). Mediány trvaní odpovedí boli
31,9 mesiaca (rozsah: 1,4+-31,9) a 12,8 mesiaca (rozsah: 1,3+-13,6+), v uvedenom poradí. PFS HR pre nivolumab oproti chemoterapii bol 1,03 (95 % CI: 0,78; 1,36). ORR a PFS boli hodnotené IRRC podľa RECIST verzia 1.1.V záverečnej analýze OS nebol štatisticky významný rozdiel medzi
nivolumabom a chemoterapiou. Primárna analýza OS nebola upravená tak, aby zahŕňala následné
terapie, 54 (40,6 %) pacientov v skupine s chemoterapiou následne dostávalo anti-PD1 liečbu. OS
mohlo byť ovplyvnené z dôvodu úbytku pacientov zo štúdie, nevyváženosti následných terapií a
rozdielov vo východiskových faktoroch. V skupine s nivolumabom malo viac pacientov horšie prognostické faktory (zvýšené LDH a metastázy v mozgu) ako v skupine s chemoterapiou.
Účinnosť pomocou stavu BRAF: Objektívne odpovede na nivolumab (podľa definície súbežného primárneho koncového ukazovateľa) sa pozorovali u pacientov s melanómom s pozitívnou mutáciou BRAF alebo bez nej. ORR v podskupine s pozitívnou mutáciou BRAF bolo 17 % (95 % CI: 8,4; 29,0) po nivolumabe a 11 % (95 % CI: 2,4; 29,2) po chemoterapii a v podskupine s divokým typom BRAF bolo 30 % (95 % CI: 24,0; 36,7) a 9 % (95 % CI: 4,6; 16,7), v uvedenom poradí..
PFS HR po nivolumabe oproti chemoterapii bolo 1,58 (95 % CI: 0,87; 2,87) u pacientov s pozitívnou mutáciou BRAF a 0,82 (95 % CI: 0,60; 1,12) u pacientov s divokým typom BRAF. OS HR po nivolumabe oproti chemoterapii bolo 1,32 (95 % CI: 0,75; 2,32) u pacientov s pozitívnou mutáciou BRAF a 0,83 (95 % CI: 0,62; 1,11) u pacientov s divokým typom BRAF.
Účinnosť pomocou expresie PD-L1 v nádore: Objektívne odpovede na nivolumab sa pozorovali bez ohľadu na expresiu PD-L1 v nádore. Úloha tohto biomarkera (expresia PD-L1 v nádore) však nie je úplne objasnená.
U pacientov s expresiou PD-L1 v nádore ≥ 1 % bolo ORR 33,5 % po nivolumabe
(n = 179; 95 % CI: 26,7; 40,9) a 13,5 % po chemoterapii (n = 74; 95 % CI: 6,7; 23,5). U pacientov s expresiou PD-L1 v nádore < 1 % bolo ORR podľa IRRC 13,0 % (n = 69; 95 % CI: 6,1; 23,3) a
12,0 % (n = 25; 95 % CI: 2,5; 31,2), v uvedenom poradí.
PFS HR po nivolumabe oproti chemoterapii bolo 0,76 (95 % CI: 0,54; 1,07) u pacientov s expresiou
PD-L1 v nádore ≥ 1 % a 1,92 (95 % CI: 1,05; 3,5) u pacientov s expresiou PD-L1 v nádore < 1 %.
OS HR po nivolumabe oproti chemoterapii bolo 0,69 (95 % CI: 0,49; 0,96) u pacientov s expresiou
PD-L1 v nádore ≥ 1 % a 1,52 (95 % CI: 0,89; 2,57) u pacientov s expresiou PD-L1 v nádore < 1 %.
Tieto analýzy podskupín sa majú interpretovať s opatrnosťou so zohľadnením malej veľkosti podskupín a absencie štatisticky významného rozdielu OS v celej randomizovanej populácii.
Otvorenáštúdia1.fázysozvyšovanímdávky(MDX1106-03)
Bezpečnosť a znášanlivosť nivolumabu sa skúmali v 1. fáze, otvorenej štúdie so zvyšovaním dávky na rôznych typoch nádorov vrátane malígneho melanómu. Z 306 predtým liečených pacientov
zaradených do štúdie malo 107 melanóm a dostalo nivolumab v dávke 0,1 mg/kg, 0,3 mg/kg, 1 mg/kg,
3 mg/kg alebo 10 mg/kg maximálne 2 roky. V tejto populácii pacientov sa hlásila objektívna odpoveď u 33 pacientov (31 %) s mediánom trvania odpovede 22,9 mesiaca (95 % CI: 17,0; NR). Medián PFS bol 3,7 mesiaca (95 % CI: 1,9; 9,3). Medián OS bol 17,3 mesiaca (95 % CI: 12,5; 37,8) a odhadované pomery OS boli 42 % (95 % CI: 32; 51) pri 3 rokoch, 35 % (95 % CI: 26, 44) pri 4 rokoch a 34 %
(95 % CI: 25, 43) pri 5 rokoch (minimálne následné sledovanie 45 mesiacov).
Randomizovanáštúdia3.fázysnivolumabomvkombináciisipilimumabomalebonivolumabakomonoterapia oproti ipilimumabu ako monoterapii (CA209067)
Bezpečnosť a účinnosť nivolumabu v kombinácii s ipilimumabom alebo monoterapia nivolumabu oproti ipilimumabu na liečbu pokročilého (neresekovateľného alebo metastatického) melanómu sa hodnotili v 3. fáze, randomizovanej, dvojito zaslepenej štúdie (CA209067). Rozdiely medzi dvoma
skupinami obsahujúcimi nivolumab sa hodnotili popisne. Do štúdie boli zaradení dospelí pacienti s
potvrdeným neresekovateľným III. štádiom alebo IV. štádiom melanómu. Pacienti mali mať skóre výkonnostného stavu ECOG 0 alebo 1. Boli zaradení pacienti, ktorí nedostali predchádzajúcu systémovú protinádorovú liečbu na neresekovateľný alebo metastatický melanóm. Predchádzajúca adjuvantná alebo neoadjuvantná liečba bolo povolená, ak sa ukončila minimálne 6 týždňov pred randomizáciou. Pacienti s aktívnym autoimunitným ochorením, okulárnym/uveálnym melanómom alebo s aktívnymi metastázami v mozgu alebo aktívnymi leptomeningeálnymi metastázami boli zo štúdie vyradení.
Celkovo bolo randomizovaných 945 pacientov, aby dostávali nivolumab v kombinácii s ipilimumabom (n = 314), nivolumab v monoterapii (n = 316) alebo ipilimumab v monoterapii
(n = 315). Pacienti v skupine s kombináciou dostávali nivolumab v dávke 1 mg/kg počas 60 minút a ipilimumab 3 mg/kg počas 90 minút, ktoré sa podávali intravenózne každé 3 týždne počas prvých
4 dávok, potom nasledoval nivolumab v dávke 3 mg/kg ako monoterapia každé 2 týždne. Pacienti v skupine s nivolumabom v monoterapii dostávali nivolumab v dávke 3 mg/kg každé 2 týždne. Pacienti v skupine s komparátorom dostávali ipilimumab v dávke 3 mg/kg a placebo v súlade s dávkovaním nivolumabu intravenózne každé 3 týždne 4 dávky, potom nasledovalo placebo každé 2 týždne. Randomizácia bola stratifikovaná podľa expresie PD-L1 (≥ 5 % oproti < 5 % expresia na membráne
nádorových buniek), stavu BRAF a štádia M podľa systému štádií Amerického spoločného výboru pre
rakovinu (American Joint Committee on Cancer, AJCC). Liečba pokračovala dovtedy, kým sa
pozoroval klinický prínos alebo pokým bola liečba tolerovaná. Hodnotenia nádoru sa vykonali
12 týždňov po randomizácii a potom každých 6 týždňov počas prvého roka a potom každých
12 týždňov. Merané súbežné primárne koncové ukazovatele boli prežívanie bez progresie a OS.
Hodnotili sa aj ORR a trvanie odpovede.
Východiskové charakteristiky boli medzi troma liečenými skupinami vyvážené. Medián veku
bol 61 rokov (rozsah: 18 až 90 rokov), 65 % pacientov bolo mužov a 97 % bolo belochov. Skóre výkonnostného stavu ECOG bolo 0 (73 %) alebo 1 (27 %). Pri vstupe do štúdie mala väčšina pacientov podľa AJCC IV. štádium ochorenia (93 %); 58 % malo ochorenie v štádiu M1c. Dvadsaťdva percent pacientov dostalo predchádzajúcu adjuvantnú liečbu. Tridsaťdva percent pacientov malo pri melanóme pozitívnu mutáciu BRAF; 26,5 % pacientov malo expresiu PD-L1 na membráne nádorových buniek ≥ 5 %. Pri vstupe do štúdie mali štyri percentá pacientov metastázy v mozgu v anamnéze a 36 % pacientov malo východiskovú hladinu LDH vyššiu ako ULN. Pacienti s kvantifikovateľnou expresiou PD-L1 v nádore boli rovnomerne distribuovaní v troch liečených skupinách. Expresia PD-L1 v nádore bola stanovená pomocou analýzy PD-L1 IHC 28-8 pharmDx.
Výsledky PFS (s minimálnym následným sledovaním 18 mesiacov) sú uvedené na obrázku 2 (všetky randomizované populácie), obrázku 3 (hladina PD-L1 v nádore pod 5 %) a obrázku 4 (hladina PD-L1 v nádore pod 1 %).
 Obrázok 2: Prežívanie bez progresie (CA209067)
Obrázok 2: Prežívanie bez progresie (CA209067)
Prežívanie bez progresie podľa skúšajúceho (mesiace)
Počet jedincov s rizikom
Nivolumab + Ipilimumab
314 219 174 156 133 126 103 48 8 0
Nivolumab
316 177 148 127 114 104 94 46 8 0
Ipilimumab
315 137 78 58 46 40 25 15 3 0
- - -*- - - - Nivolumab+ipilimumab (udalosti: 161/314), medián a 95 % CI: 11,50 (8,90; 22,18).
Miera PFS po 12 mesiacoch a 95 % CI: 49 % (44; 55)
──∆─── Nivolumab (udalosti: 183/316), medián a 95 % CI: 6,87 (4,34; 9,46).
Miera PFS po 12 mesiacoch a 95% CI: 42 % (36; 47)
- - -¦- - - Ipilimumab (udalosti: 245/315), medián a 95 % CI: 2,89 (2,79; 3,42).
Miera PFS po 12 mesiacoch a 95 % CI: 18 % (14; 23)
Nivolumab+ipilimumab oproti ipilimumabu (primárna analýza) - HR (99,5 % CI): 0,42 (0,32; 0,56); p-hodnota:
< 0,0001
Nivolumab oproti ipilimumabu (primárna analýza) - HR (99,5 % CI): 0,55 (0,42; 0,73); p-hodnota: < 0,0001
Nivolumab+ipilimumab oproti nivolumabu (deskriptívna analýza) - HR (95 % CI): 0,76 (0,62; 0,95)
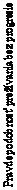 Obrázok 3: Prežívanie bez progresie pomocou expresie PD-L1: hladina pod 5 % (CA209067)
expresia PD-L1 < 5 %
Obrázok 3: Prežívanie bez progresie pomocou expresie PD-L1: hladina pod 5 % (CA209067)
expresia PD-L1 < 5 %
Prežívanie bez progresie (mesiace)
Počet jedincov s rizikom
Nivolumab + Ipilimumab
210 142 113 101 86 81 69 31 5 0
Nivolumab
208 108 89 75 69 62 55 29 7 0
Ipilimumab
202 82 45 34 26 22 12 7 0 0
- - -*- - - - Nivolumab+Ipilimumab (udalosti: 111/210), medián a 95 % CI: 11,10 (7,98; 22,18)
──∆─── Nivolumab (udalosti: 125/208), medián a 95 % CI: 5,32 (2,83; 7,06)
- - -¦- - - Ipilimumab (udalosti: 159/202), medián a 95 % CI: 2,83 (2,76; 3,09)
Nivolumab+Ipilimumab oproti Ipilimumabu- pomer rizika: 0,42 (0,33; 0,54) Nivolumab oproti Ipilimumabu - pomer rizika: 0,57 (0,45; 0,72) Nivolumab+Ipilimumab oproti Nivolumabu - pomer rizika: 0,74 (0,58; 0,96)
expresia PD-L1 ≥ 5 %Prežívanie bez progresie (mesiace)
Počet jedincov s rizikom
Nivolumab + Ipilimumab
68 53 44 39 33 31 22 13 3 0
Nivolumab
80 57 51 45 39 37 36 16 1 0
Ipilimumab
75 40 21 17 14 12 8 6 2 0
- - -*- - - - Nivolumab+Ipilimumab (udalosti: 29/68), medián a 95 % CI: N.A. (9,72; N.A.)
──∆─── Nivolumab (udalosti: 38/80), medián a 95 % CI: 21,95 (8,90; N.A.)
- - -¦- - - Ipilimumab (udalosti: 57/75), medián a 95 % CI: 3,94 (2,79; 4,21)
Nivolumab+Ipilimumab oproti Ipilimumabu - pomer rizika: 0,35 (0,22; 0,55) Nivolumab oproti Ipilimumabu - pomer rizika: 0,41 (0,27; 0,62) Nivolumab+Ipilimumab oproti Nivolumabu - pomer rizika: 0,87 (0,54; 1,41)
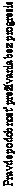 Obrázok 4: Prežívanie bez progresie pomocou expresie PD-L1: hladina pod 1 % (CA209067)
expresia PD-L1 < 1 %
Obrázok 4: Prežívanie bez progresie pomocou expresie PD-L1: hladina pod 1 % (CA209067)
expresia PD-L1 < 1 %
Prežívanie bez progresie (mesiace)
Počet jedincov s rizikom
Nivolumab + Ipilimumab
123 82 65 59 50 46 41 18 4 0
Nivolumab
117 50 43 35 33 29 27 11 3 0
Ipilimumab
113 39 20 15 12 10 4 3 0 0
- - -*- - - - Nivolumab+Ipilimumab (udalosti: 63/123), medián a 95 % CI: 11,24 (6,93; 23,03)
──∆─── Nivolumab (udalosti: 77/117), medián a 95 % CI: 2,83 (2,76; 5,13)
- - -¦- - - Ipilimumab (udalosti: 87/113), medián a 95 % CI: 2,79 (2,66; 2,96)
Nivolumab+Ipilimumab oproti Ipilimumabu - pomer rizika: 0,39 (0,28; 0,54) Nivolumab oproti Ipilimumabu - pomer rizika: 0,65 (0,48; 0,88) Nivolumab+Ipilimumab oproti Nivolumabu - pomer rizika: 0,60 (0,43; 0,84)
expresia PD-L1 ≥ 1 %
e i
es r g o r p
ez b a
ni a v
í
ž
e
pr ť s o
bn do
o
p
de v a r P
|
|

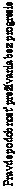
Počet jedincov s rizikom
Nivolumab + Ipilimumab
Prežívanie bez progresie (mesiace)
155 113 92 81 69 66 50 26 4 0
Nivolumab
171 115 97 85 75 70 64 34 5 0
Ipilimumab
164 83 46 36 28 24 16 10 2 0
- - -*- - - - Nivolumab+Ipilimumab (udalosti: 77/155), medián a 95 % CI: 12,35 (8,74; N.A.)
──∆─── Nivolumab (udalosti: 86/171), medián a 95 % CI: 14,00 (7,03; N.A.)
- - -¦- - - Ipilimumab (udalosti: 129/164), medián a 95 % CI: 3,91 (2,83; 4,17)
Nivolumab+Ipilimumab oproti Ipilimumabu - pomer rizika: 0,42 (0,31; 0,55) Nivolumab oproti Ipilimumabu - pomer rizika: 0,44 (0,34; 0,58) Nivolumab+Ipilimumab oproti Nivolumabu - pomer rizika: 0,94 (0,69; 1,28)
Finálna analýza OS sa vykonala, keď mali všetci pacienti minimálne následné sledovanie
28 mesiacov. Výsledky OS pri dodatočnej analýze vykonané pri minimálnom následnom sledovaní
36 mesiacov ukázali zhodné výsledky s pôvodnou analýzou. Výsledky OS z tejto analýzy následného sledovania sú uvedené na obrázku 5 (všetci randomizovaní), na obrázku 6 (hladina PD-L1 v nádore pod 1 %) a v tabuľke 7 (hladina PD-L1 v nádore pod 5 %).
Analýza OS nebola upravená tak, aby zohľadnila následne podané terapie. Následné systémové terapie boli podané 31,8 % pacientov s kombinovanou liečbou, 44,3 % s nivolumabom v monoterapii a 62,2 % v skupinách s ipilimumabom. Následné imunoterapie (vrátane anti-PD1 liečby, protilátkou anti-CTLA-4 alebo iné imunoterapie) boli podané 14,6 % pacientov s kombinovanou liečbou, 29,1 % s nivolumabom v monoterapii a 44,1 % v skupinách s ipilimumabom.
Obrázok 5 Celkové prežívanie (CA209067) - Minimálne následné sledovanie 36 mesiacov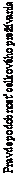
Počet jedincov s rizikom
Nivolumab + Ipilimumab
Celkové prežívanie (mesiace)
314 292 265 247 226 221 209 200 198 192 186 180 177 131 27 3 0
Nivolumab
316 292 265 244 230 213 201 191 181 175 171 163 156 120 28 0 0
Ipilimumab
315 285 253 227 203 181 163 148 135 128 113 107 100 68 20 2 0
- - -*- - - - Nivolumab+ipilimumab (udalosti: 139/314), medián a 95 % CI: N.A. (38,18; N.A.)
Miera OS a 95 % CI pri 12 mesiacoch: 73 % (68; 78), 24 mesiacoch: 64 % (59; 69) a 36 mesiacoch: 58 % (52; 63)
──∆─── Nivolumab (udalosti: 158/316), medián a 95 % CI: 37,59 mesiacov (29,08; N.A.)
Miera OS a 95 % CI pri 12 mesiacoch: 74 % (69; 79), 24 mesiacoch: 59 % (53; 64) a 36 mesiacoch: 52 % (46; 57)
- - -¦- - - Ipilimumab (udalosti: 206/315), medián a 95 % CI: 19,94 mesiacov (16,85; 24,61)
Miera OS a 95 % CI pri 12 mesiacoch: 67 % (61; 72), 24 mesiacoch: 45 % (39; 50) a 36 mesiacoch: 34 % (29; 39)
Nivolumab+ipilimumab oproti ipilimumabu (primárna analýza) - HR (95 % CI): 0,55 (0,45; 0,69); p-
hodnota: < 0,0001
Nivolumab oproti ipilimumabu (primárna analýza) - HR (95 % CI): 0,65 (0,53; 0,80); p-hodnota: < 0,0001
Nivolumab+ipilimumab oproti nivolumabu (deskriptívna analýza) - HR (95 % CI): 0,85 (0,68; 1,07)
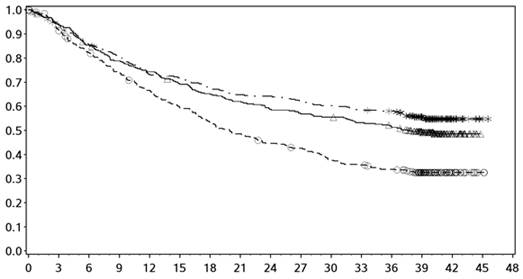
Obrázok 6: Celkové prežívanie pomocou expresie PD-L1: hladina pod 1 % (CA209067) - Minimálne následné sledovanie 36 mesiacov
expresia PD-L1 < 1 %
Počet jedincov s rizikom
Nivolumab + Ipilimumab
Celkové prežívanie (mesiace)
123 113 102 91 82 82 79 74 74 72 70 67 65 50 11 2 0
Nivolumab
117 103 86 76 73 65 62 59 57 55 53 51 49 37 7 0 0
Ipilimumab
113 96 87 79 71 61 57 50 44 43 36 34 33 24 8 1 0
- - -*- - - - Nivolumab+Ipilimumab (udalosti: 59/123), medián a 95 % CI: N.A. (26,45; N.A.)
──∆─── Nivolumab (udalosti: 71/117), medián a 95 % CI: 23,46 mesiacov (13,01; 36,53)
- - -¦- - - Ipilimumab (udalosti: 77/113), medián a 95 % CI: 18,56 mesiacov (13,67; 23,20)
Nivolumab+Ipilimumab oproti Ipilimumabu - pomer rizika: 0,59 (0,42; 0,82) Nivolumab oproti Ipilimumab - pomer rizika: 0,84 (0,61; 1,16) Nivolumab+Ipilimumab oproti Nivolumabu - pomer rizika: 0,70 (0,49; 0,99)
expresia PD-L1 ≥ 1 % Celkové prežívanie (mesiace)
Počet jedincov s rizikom
Nivolumab + Ipilimumab
155 144 132 127 116 112 105 102 101 99 96 94 93 66 14 1 0
Nivolumab
171 165 158 148 139 131 122 117 112 109 108 102 99 76 18 0 0
Ipilimumab
164 155 137 125 113 101 88 82 76 73 67 64 58 38 10 0 0
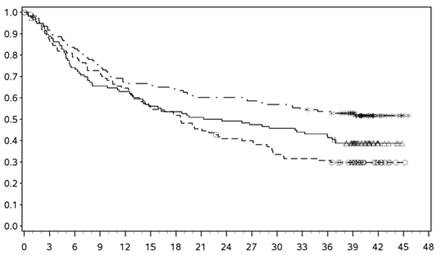
- - -*- - - - Nivolumab+Ipilimumab (udalosti: 63/155), medián a 95 % CI: N.A. (39,06; N.A.)
──∆─── Nivolumab (udalosti: 71/171), medián a 95 % CI: N.A. (40,21; N.A.)
- - -¦- - - Ipilimumab (udalosti: 103/164), medián a 95 % CI: 21,49 mesiacov (16,85; 29,08)
Nivolumab+Ipilimumab oproti Ipilimumabu - pomer rizika: 0,5 (0,40; 0,75) Nivolumab oproti Ipilimumabu - pomer rizika: 0,54 (0,40; 0,73) Nivolumab+Ipilimumab oproti Nivolumabu - pomer rizika: 1,02 (0,73; 1,43)
Tabuľka 7: Súhrn celkového prežívania pomocou expresie PD-L1: hladina pod 5 % - CA209067 - Minimálne následné sledovanie 36 mesiacov
Expresia
PD-L1 v nádore
n nivolumab + ipilimumab
Medián OS (95 % CI)
n ipilimumab
Medián OS (95 % CI)
Pomer rizika
(95 % CI)
< 5 % 210 NR (32,72; NR) 202 18,40 (13,70; 22,51) 0,56 (0,43; 0,72)
≥ 5 % 68 NR (39,06; NR) 75 28,88 (18,10; NR) 0,59 (0,36; 0,97)
nivolumab
Medián OS (95 % CI)
ipilimumab
Medián OS (95 % CI)
Pomer rizika
(95 % CI)
< 5 % 208 35,94 (23,06; NR) 202 18,40 (13,70; 22,51) 0,68 (0,53; 0,87)
≥ 5 % 80 NR (35,75; NR) 75 28,88 (18,10; NR) 0,60 (0,38; 0,95)
nivolumab + ipilimumab
Medián OS (95 % CI)
nivolumab
Medián OS (95 % CI)
Pomer rizika
(95 % CI)



< 5 % 210 NR (32,72; NR) 208 35,94 (23,06; NR) 0,82 (0,62; 1,08)
≥ 5 % 68 NR (39,06; NR) 80 NR (35,75; NR) 0,99 (0,59; 1,67)
NR = nedosiahnuté
Minimálne následné sledovanie pre analýzu ORR bolo 28 mesiacov. Výsledky sú zhrnuté v tabuľke 8.
Tabuľka 8: Objektívna odpoveď (CA209067)
nivolumab + ipilimumab (n = 314)
nivolumab
(n = 316)
ipilimumab
(n = 315)
Objektívna odpoveď 185 (59 %) 141 (45 %) 60 (19 %)
(95 % CI) (53,3; 64,4) (39,1; 50,3) (14,9; 23,8) Pomer šancí (oproti ipilimumabu) 6,50 3,54
(99,5 % CI) (3,81; 11,08) (2,10; 5,95)
Kompletná odpoveď (CR) 54 (17 %) 47 (15 %) 14 (4 %) Parciálna odpoveď (PR) 131 (42 %) 94 (30 %) 46 (15 %) Stabilizácia ochorenia (SD) 36 (12 %) 31 (10 %) 67 (21 %)
Trvanie odpovede
Medián (rozsah) mesiace
Nedosiahnutý +
(0+ – 33,3+) 31,1 (0
– 32,3+) 18,2 (0+
– 31,5+)
Rozsah trvania ≥ 12 mesiacov 64 % 70 % 53 %
Rozsah trvania ≥ 24 mesiacov 50 % 49 % 32 %
ORR (95 % CI) pomocou expresie PD-L1 v nádore
< 5 %
≥ 5 %
< 1 %
≥ 1 %
„+“ označuje cenzurované sledovanie.
56 % (49,2; 63,0)
n = 210
74 % (61,4; 83,5)
n = 68
55 % (45,2; 63,5)
n = 123
65 % (57,1; 72,6)
n = 155
42 % (35,5; 49,3)
n = 208
59 % (47,2; 69,6)
n = 80
35 % (26,5; 44,4)
n = 117
55 % (47,2; 62,6)
n = 171
18 % (12,8; 23,8)
n = 202
21 % (12,7; 32,3)
n = 75
19 % (11,9; 27,0)
n = 113
19 % (13,2; 25,7)
n = 164


Obe skupiny obsahujúce nivolumab preukázali signifikantný prínos PFS a OS a väčšiu ORR v
porovnaní so samotným ipilimumabom. Pozorované výsledky PFS po 18 mesiacoch následného sledovania a výsledky ORR a OS po 28 mesiacoch následného sledovania boli zhodne potvrdené v podskupinách pacientov vrátane východiskového výkonnostného stavu ECOG, stavu BRAF, štádia M, veku, metastáz v mozgu v anamnéze a východiskovej hladiny LDH. Toto pozorovanie sa zachovalo vo výsledkoch OS s minimálnym následným sledovaním 36 mesiacov.
U 128 pacientov, ktorí prerušili liečbu nivolumabom v kombinácii s ipilimumabom z dôvodu nežiaducej reakcie po 18 mesiacoch následného sledovania, bol medián PFS 16,7 mesiaca (95 % CI:
10,2; NA). U 131 pacientov, ktorí prerušili liečbu kombináciou z dôvodu nežiaducej reakcie po
28 mesiacoch následného sledovania bola ORR 71 % (93/131) s dosiahnutím kompletnej odpovede u
20 % (26/131) a medián OS sa nedosiahol.
Obe skupiny obsahujúce nivolumab preukázali vyššie miery objektívnej odpovede ako ipilimumab bez ohľadu na hladiny expresie PD-L1. Pri všetkých hladinách expresie PD-L1 v nádore boli ORR vyššie pri kombinácii nivolumabu a ipilimumabu v porovnaní s nivolumabom v monoterapii (Tabuľka 8) po
28 mesiacoch následného sledovania s najlepšou celkovou reakciou kompletnej odpovede korelujúcou so zlepšenou mierou prežívania.
Po 28 mesiacoch následného sledovania nebol dosiahnutý medián trvaní odpovede u pacientov s hladinou expresie PD-L1 v nádore ≥ 5 % v skupine s kombináciou (rozsah: 0+ – 31,6+), nebol dosiahnutý v skupine s nivolumabom v monoterapii (rozsah: 2,8 – 30,6+) a nebol dosiahnutý v skupine s ipilimumabom (rozsah: 1,4 – 30,6+). Pri expresii PD-L1 v nádore < 5 % nebol dosiahnutý medián trvaní odpovede v skupine s kombináciou (rozsah: 0+ – 33,3+), nebol dosiahnutý v skupine s nivolumabom v monoterapii (rozsah: 0+ – 32,3+) a v skupine s ipilimumabom v monoterapii bol
18,2 mesiaca (rozsah: 0,0+ – 31,5+).
Jasnú hladinu expresie PD-L1 nemožno spoľahlivo stanoviť, ak zohľadňujme príslušné koncové ukazovatele odpovede nádoru a PFS a OS. Výsledky z objasňujúcej multivariantnej analýzy identifikovali pacienta a charakteristiky nádoru (výkonnostný stav ECOG, štádium M, východisková hladina LDH, stav mutácie BRAF, stav PD-L1 a pohlavie), ktoré môžu prispievať ku výsledku prežívania.
Účinnosť pomocou stavu BRAF: Po 18 mesiacoch následného sledovania mali pacienti s pozitívnou mutáciou BRAF[V600] a divokým typom BRAF randomizovaní na nivolumab v kombinácii s ipilimumabom medián PFS 15,5 mesiaca (95 % CI: 8,0; NA) a 11,3 mesiaca (95 % CI: 8,3; 22,2), zatiaľ čo pacienti v skupine s nivolumabom v monoterapii mali medián PFS 5,6 mesiaca (95 % CI:
2,8; 9,3) a 7,1 mesiaca (95 % CI: 4,9; 14,3), v uvedenom poradí. Po 28 mesiacoch následného sledovania mali pacienti s pozitívnou mutáciou BRAF[V600] a divokým typom BRAF randomizovaní na nivolumab v kombinácii s ipilimumabom ORR 67,6 % (95 % CI: 57,7; 76,6; n = 102) a 54,7 %
(95 % CI: 47,8; 61,5; n = 212), zatiaľ čo pacienti v skupine s nivolumabom v monoterapii mali ORR
36,7 % (95 % CI: 27,2; 47,1; n = 98) a 48,2 % (95 % CI: 41,4; 55,0; n = 218), v uvedenom poradí. Po
28 mesiacoch následného sledovania sa medián OS nedosiahol v žiadnej skupine s obsahom nivolumabu bez ohľadu na stav BRAF. Pomery rizík OS pre nivolumab v kombinácii s ipilimumabom oproti nivolumabu v monoterapii boli 0,71 (95 % CI: 0,45; 1,13) u pacientov s pozitívnou mutáciou BRAF[V600] a 0,97 (95 % CI: 0,74; 1,28) u pacientov s divokým typom BRAF.
Randomizovaná2.fázaštúdiesnivolumabomvkombináciisipilimumabom aipilimumab(CA209069)Štúdia CA209069 bola randomizovaná, dvojito zaslepená štúdia 2. fázy porovnávajúca kombináciu nivolumabu a ipilimumabu so samotným ipilimumabom u 142 pacientov s pokročilým (neresekovateľným alebo metastatickým) melanómom s podobným kritériom na zaradenie ako do štúdie CA209067 a s primárnou analýzou u pacientov s melanómom s divokým typom BRAF
(77 % pacientov). Skúšajúcim hodnotené ORR bolo 61 % (95 % CI: 48,9; 72,4) v skupine s kombináciou (n = 72) oproti 11 % (95 % CI: 3,0; 25,4) v skupine s ipilimumabom (n = 37). Odhadované miery OS v 2. a 3. roku boli pre kombináciu (n = 73) 68 % (95 % CI: 56; 78) a 61 % (95 % CI: 49; 71), v uvedenom poradí a pre ipilimumab (n = 37) 53 % (95 % CI: 36; 68) a 44 % (95 % CI: 28; 60), v uvedenom poradí.
Adjuvantná liečba melanómu
Randomizovanáštúdia3.fázysnivolumabomoprotiipilimumabuvdávke10mg/kg(CA209238)
Bezpečnosť a účinnosť nivolumabu v dávke 3 mg/kg ako jednej látky na liečbu pacientov s úplne resekovaným melanómom sa hodnotili v randomizovanej, dvojito zaslepenej štúdii 3. fázy
(CA209238). Štúdia zahŕňala dospelých pacientov, ktorí mali skóre výkonnostného stavu podľa ECOG 0 alebo 1, so štádiom IIIB/C alebo so štádiom IV podľa Amerického spoločného výboru pre rakovinu (AJCC, American Joint Committee on Cancer), 7. vydanie, histologicky potvrdený melanóm, ktorý je úplne chirurgicky odstránený. Podľa AJCC 8. vydania to zodpovedalo pacientom s
postihnutím lymfatickej uzliny alebo metastázami. Pacienti boli zaradení bez ohľadu na ich stav PD-
L1 v nádore. Pacienti s predchádzajúcim autoimunitným ochorením a akýmkoľvek stavom vyžadujúcim si systémovú liečbu buď kortikosteroidmi (≥ 10 mg prednizónu alebo jeho ekvivalentu denne) alebo inými imunosupresívnymi liekmi, ako aj pacienti s predchádzajúcou liečbou melanómu (okrem pacientov s chirurgickým výkonom, adjuvantnou rádioterapiou po neurochirurgickej resekcii z dôvodu lézií v centrálnom nervovom systéme a s predchádzajúcou adjuvantnou liečbou interferónom ukončenou ≥ 6 mesiacov pred randomizáciou) s predchádzajúcou liečbou protilátkou anti-PD-1, anti- PD-L1, anti-PD-L2, anti-CD137 alebo anti CTLA-4 (vrátane ipilimumabu alebo akejkoľvek inej protilátky alebo liečivom špecificky zameraným na súbežnú stimuláciu T buniek alebo kontrolného bodu dráh) boli zo štúdie vyradení.
Celkovo bolo randomizovaných 906 pacientov na podávanie buď nivolumabu v dávke 3 mg/kg
(n = 453) podávaného každé 2 týždne alebo ipilimumabu v dávke 10 mg/kg (n = 453) podávaného každé 3 týždne počas 4 dávok, potom každých 12 týždňov so začiatkom na 24. týždeň až do jedného roka. Randomizácia bola stratifikovaná podľa expresie PD-L1 v nádore (≥ 5 % oproti
< 5 %/nestanovená) a stavu ochorenia podľa AJCC systému klasifikovania. Hodnotenia nádoru sa
vykonali každých 12 týždňov počas prvých 2 rokov, potom od tohto času každých 6 mesiacov. Primárny koncový ukazovateľ bol prežívanie bez rekurencie ochorenia (RFS, recurrence-free survival). RFS, hodnotené skúšajúcim, bolo definované ako čas medzi dátumom randomizácie a dátumom prvej rekurencie ochorenia (lokálna, regionálna alebo vzdialená metastáza), nového primárneho melanómu alebo úmrtia z dôvodu akejkoľvek príčiny, podľa toho čo sa vyskytlo prvé.
Východiskové charakteristiky boli celkovo vyvážené medzi dvoma skupinami. Medián veku bol
55 rokov (rozsah: 18-86), 58 % bolo mužov a 95 % bolo belochov. Východiskové skóre výkonnostného stavu podľa ECOG bolo 0 (90 %) alebo 1 (10 %). Väčšina pacientov mala podľa AJCC ochorenie v štádiu III (81 %) a 19 % malo ochorenie v štádiu IV. Štyridsaťosem percent pacientov malo makroskopické lymfatické uzliny a 32 % malo ulceráciu nádoru. Štyridsaťdva percent pacientov malo pozitívnu mutáciu BRAF V600, zatiaľ čo 45 % malo divý typ BRAF a 13 % malo neznámy stav BRAF. Čo sa týka expresie PD-L1 v nádore, podľa stanovenia pomocou metódy v klinickom skúšaní malo 34 % pacientov expresiu PD-L1 ≥ 5 % a 62 % malo < 5 %. U pacientov s merateľnou expresiou PD-L1 v nádore bolo rozdelenie pacientov do liečených skupín vyvážené. Expresia PD-L1 v nádore bola stanovená použitím metódy PD-L1 IHC 28-8 pharmDx.
Minimálne následné sledovanie bolo približne 24 mesiacov. V čase tejto analýzy nebolo OS dostupné. Výsledky RFS sú uvedené v tabuľke 9 a na obrázku 7 (všetky randomizované populácie).
Tabuľka 9: Výsledky účinnosti(CA209238)
 Prežívanie bez rekurencie
o
chorenia
nivolumab
(n = 453)
ipilimumab 10 mg/kg
(n = 453)
Prežívanie bez rekurencie
o
chorenia
nivolumab
(n = 453)
ipilimumab 10 mg/kg
(n = 453)
Udalosti 171 (37,7 %) 221 (48,8 %)
Pomer rizikaa 0,66
95 % CI (0,54; 0,81)
p-hodnota p < 0,0001
Medián (95 % CI) mesiace Nedostupnýb 24,08 (16,56; NR)
Miera (95 % CI) po
12 mesiacoch
Miera (95 % CI) po
18 mesiacoch
Miera (95 % CI) po
24 mesiacoch
70,4 (65,9; 74,4) 60,0 (55,2; 64,5)
65,8 (61,2; 70,0) 53,0 (48,1; 57,6)
62,6 (57,9; 67,0) 50,2 (45,3; 54,8)

a Získané z modelu stratifikovaných proporcionálnych rizík.
b Nedostupné, pretože medián bol nestabilný z dôvodu malého počtu pacientov a cenzurovania počas 24 mesiacov následného sledovania
Obrázok 7: Prežívanie bez rekurencie ochorenia (CA209238)

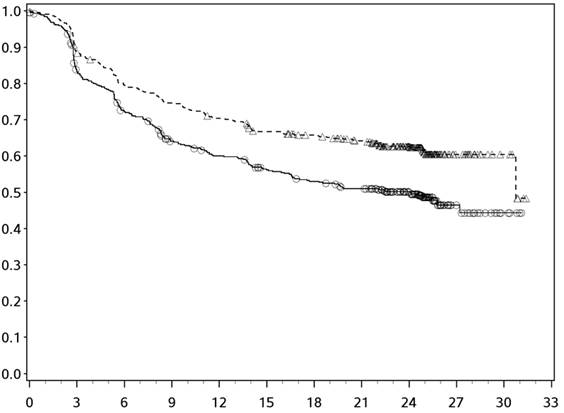
Počet jedincov s rizikom
Nivolumab
Prežívanie bez rekurencie ochorenia (mesiace)
453 394 353 331 311 291 280 264 205 28 7 0
Ipilimumab
453 363 314 270 251 230 216 204 149 23 5 0
- - - ∆- - - Nivolumab ──¦── Ipilimumab
Skúšanie preukázalo štatisticky významné zlepšenie RFS u pacientov randomizovaných do skupiny s nivolumabom v porovnaní so skupinou s ipilimumabom 10 mg/kg. Prínos RFS bol zhodne preukázaný v podskupinách vrátane expresie PD-L1 v nádore, stavu BRAF a štádia ochorenia.
Podľa hodnotenia pomocou validovaných a spoľahlivých stupníc merania, ako sú index užitočnosti EORTC QLQ-C30 a EQ-5D (EORTC, the European Organization for Research and Treatment of Cancer) a vizuálna analógová škála (VAS, visual analog scale) zostala kvalita života (QoL, quality of life) počas liečby nivolumabom stabilná a podobná východiskovým hodnotám.
Nemalobunkový karcinóm pľúc
Skvamózny NSCLC
Randomizovanáštúdia3.fázyoprotidocetaxelu(CA209017)
Bezpečnosť a účinnosť nivolumabu 3 mg/kg ako jedinej látky na liečbu pokročilého alebo metastatického skvamózneho NSCLC sa hodnotili v randomizovanej, otvorenej štúdii 3. fázy (CA209017). Do štúdie boli zaradení pacienti (18 roční alebo starší), ktorí mali progresiu ochorenia
počas alebo po jednom predchádzajúcom chemoterapeutickom režime založenom na platinovom
dublete a so skóre výkonnostného stavu 0 alebo 1 podľa ECOG. Pacienti boli zaradení bez ohľadu na ich stav PD-L1 v nádore. Pacienti s aktívnym autoimunitným ochorením, symptomatickým
intersticiálnym ochorením pľúc alebo s aktívnymi metastázami v mozgu boli zo štúdie vyradení. Pacienti s liečenými metastázami v mozgu boli vhodní, ak sa z neurologického hľadiska vrátili na východiskový stav minimálne 2 týždne pred zaradením a buď neužívali kortikosteroidy alebo boli na stabilnej dávke alebo sa im dávka znížila na dávku zodpovedajúcu prednizónu < 10 mg denne.
Celkovo bolo randomizovaných 272 pacientov, buď na intravenózne podávanie nivolumabu
v dávke 3 mg/kg počas 60 minút každé 2 týždne (n = 135) alebo na docetaxel (n = 137) 75 mg/m2 každé 3 týždne. Liečba pokračovala dovtedy, kým sa pozoroval klinický prínos alebo kým bola liečba tolerovaná. Hodnotenia nádoru pomocou RECIST, verzia 1.1, sa vykonali 9 týždňov po randomizácii
a potom pokračovali každých 6 týždňov.
Meraným primárnym koncovým ukazovateľom účinnosti bolo OS. Meranými kľúčovými sekundárnymi koncovými ukazovateľmi boli ORR a PFS hodnotené skúšajúcim. Ďalej sa hodnotili príznaky zlepšenia a celkového zdravotného stavu pomocou škály symptómov pri karcinóme pľúc (Lung Cancer Symptom Score, LCSS) indexom priemerných symptómov po záťaži a EQ-5D Vizuálnej analógovej škály (Visual Analogue Scale, EQ-VAS), v uvedenom poradí.
Východiskové charakteristiky boli celkovo vyvážené medzi dvoma skupinami. Medián veku
bol 63 rokov (rozsah: 39-85) s vekom 44 % ≥ 65 rokov a 11 % ≥ 75 rokov. Väčšina pacientov boli belosi (93 %) a muži (76 %). Tridsaťjeden percent malo progresívne ochorenie hlásené ako najlepšia odpoveď na ich posledný predchádzajúci režim a 45 % dostalo nivolumab počas 3 mesiacov po ukončení ich posledného predchádzajúceho režimu. Východiskové skóre výkonnostného stavu (ECOG) bolo 0 (24 %) alebo 1 (76 %).
Kaplanove-Meierove krivky OS sú uvedené na obrázku 8.
 Obrázok 8: Kaplanove-Meierove krivky OS (CA209017)
Obrázok 8: Kaplanove-Meierove krivky OS (CA209017) Celkové prežívanie (mesiace)
Počet jedincov s rizikom
Nivolumab 3 mg/kg
135 113 86 69 52 31 15 7 0
Docetaxel
137 103 68 45 30 14 7 2 0
──∆─── Nivolumab 3 mg/kg (udalosti: 86/135), medián a 95 % CI : 9,23 (7,33; 13,27)
- - -¦- - - Docetaxel (udalosti: 113/137), medián a 95 % CI : 6,01 (5,13; 7,33)
Pozorovaný prínos OS bol zhodne preukázaný v podskupinách pacientov. Prínos prežívania sa pozoroval bez ohľadu na to, či mali pacienti nádory, ktoré boli označené ako PD-L1 negatívne alebo PD-L1 pozitívne (expresia na membráne nádorovej bunky znížená o 1 %, 5 % alebo 10 %). Úloha tohto biomarkera (expresia PD-L1 v nádore), však nie je úplne objasnená. Pri minimálne
24,2 mesačnom následnom sledovaní zostal prínos OS trvalo potvrdený medzi podskupinami.
Štúdia CA209017 zahŕňala obmedzený počet pacientov ≥ 75 rokov (11 v skupine s nivolumabom a
18 v skupine s docetaxelom). Nivolumab preukázal numericky nižší účinok na OS (HR 1,85; 95 % CI:
0,76; 4,51), PFS (HR=1,76; 95 %-CI: 0,77; 4,05 ) a ORR (9,1 % oproti 16,7 %). Z dôvodu malého čísla vzorky, nemožno z týchto údajov vyvodiť definitívne závery.
Výsledky účinnosti sú uvedené v tabuľke 10.
Tabuľka 10: Výsledky účinnosti (CA209017) nivolumab (n = 135)
Primárna analýza
Minimálne následné sledovanie: 10,6 mesiaca
docetaxel
(n = 137)
C
elkové prežívanie
Udalosti 86 (63,7 %) 113 (82,5 %)
Pomer rizika 0,59
96,85 % CI (0,43; 0,81)
p-hodnota 0,0002
Medián (95 % CI) mesiace 9,23 (7,33; 13,27) 6,01 (5,13, 7,33) Výskyt (95 % CI) pri 12 mesiacoch 42,1 (33,7; 50,3) 23,7 (16,9, 31,1)
Potvrdená objektívna odpoveď 27 (20,0 %) 12 (8,8 %)
(95 % CI) (13,6; 27,7) (4,6; 14,8) Pomer šancí (95 % CI) 2,64 (1,27; 5,49)
p- hodnota 0,0083
Kompletná odpoveď (CR) 1 (0,7 %) 0
Parciálna odpoveď (PR) 26 (19,3 %) 12 (8,8 %)
Stabilizácia ochorenia (SD) 39 (28,9 %) 47 (34,3 %)
Medián trvania odpovede
Mesiace (rozsah) Nedosiahnuté (2,9 – 20,5+) 8,4 (1,4+ – 15,2+)
Medián času do odpovede
Mesiace (rozsah) 2,2 (1,6 – 11,8) 2,1 (1,8 – 9,5)
Prežívanie bez progresie
Udalosti 105 (77,8 %) 122 (89,1 %)
Pomer rizika 0,62
95 % CI (0,47; 0,81)
p- hodnota < 0,0004
Medián (95 % CI) (mesiace) 3,48 (2,14; 4,86) 2,83 (2,10; 3,52) Výskyt (95 % CI) pri 12 mesiacoch 20,8 (14,0; 28,4) 6,4 (2,9; 11,8)
Aktualizovaná analýza
Minimálne následné sledovanie: 24,2 mesiaca
Celkové prežívaniea
Udalosti 110 (81,4 %) 128 (93,4 %)
Pomer rizika 0,62
95 % CI (0,47; 0,80)
Výskyt (95 % CI)
pri 24 mesiacoch
22,9 (16,2; 30,3) 8 (4,3; 13,3)
Potvrdená objektívna odpoveď 20,0 % 8,8 %
(95 % CI) (13,6; 27,7) (4,6; 14,8)
Medián trvania odpovede
Mesiace (rozsah) 25,2 (2,9 – 30,4) 8,4 (1,4+ – 18,0+)
Prežívanie bez progresie
Výskyt (95 % CI)
pri 24 mesiacoch
15,6 (9,7; 22,7) Všetci pacienti mali buď progresiu, boli cenzurovaní, alebo sa stratili
pri následnom sledovaní
Výskyt zlepšenia príznakov súvisiacich s ochorením podľa meraní LCSS bol medzi skupinou s nivolumabom (18,5 %) a skupinou s docetaxelom (21,2 %) podobný. Priemerné EQ-VAS sa v priebehu času zvýšilo pre obe liečené skupiny, čo poukazuje na lepší celkový zdravotný stav pre pacientov, ktorí sa naďalej liečia.
Štúdia2.fázysjednouskupinou(CA209063)
Štúdia CA209063 bola otvorená štúdia s jednou skupinou, ktorá sa vykonala so 117 pacientmi s lokálne pokročilým alebo metastatickým skvamóznym NSCLC po dvoch alebo viacerých líniách liečby; inak boli použité rovnaké kritériá na zaradenie ako do štúdie CA209017. Nivolumab v dávke
3 mg/kg preukázal celkovú mieru odpovede u 14,5 % (95 % CI: 8,7; 22,2 %), medián OS
8,21 mesiaca (95 % CI: 6,05; 10,9) a medián PFS 1,87 mesiaca (95 % CI 1,77; 3,15). PFS sa hodnotilo pomocou RECIST, verzia 1.1. Odhadovaná 1-ročná miera prežívania bola 41 %.
Neskvamózny NSCLC
Randomizovanáštúdia3.fázyoprotidocetaxelu (CA209057)
Bezpečnosť a účinnosť nivolumabu 3 mg/kg ako jedinej látky na liečbu pokročilého alebo metastatického neskvamózneho NSCLC sa hodnotili v randomizovanej, otvorenej štúdii 3. fázy (CA209057). Do štúdie boli zaradení pacienti (18 roční alebo starší), ktorí mali progresiu ochorenia
počas jedného predchádzajúceho chemoterapeutického režimu založeného na platinovom dublete
alebo po ňom, ktorý mohol zahŕňať udržiavaciu liečbu a ktorí mali skóre výkonnostného stavu 0 alebo
1 podľa ECOG. U pacientov so známou EGFR mutáciou alebo translokáciou ALK bola povolená ďalšia línia liečby TKI (inhibítor tyrozínkinázy). Pacienti boli zaradení bez ohľadu na ich stav PD-L1
v nádore. Pacienti s aktívnym autoimunitným ochorením, symptomatickým intersticiálnym ochorením pľúc alebo s aktívnymi metastázami v mozgu boli zo štúdie vyradení. Pacienti s liečenými
metastázami v mozgu boli vhodní, ak sa z neurologického hľadiska vrátili na východiskový stav minimálne 2 týždne pred zaradením a buď neužívali kortikosteroidy alebo boli na stabilnej dávke alebo sa im dávka znížila na dávku zodpovedajúcu prednizónu < 10 mg denne.
Celkovo bolo randomizovaných 582 pacientov, buď na intravenózne podávanie nivolumabu
v dávke 3 mg/kg počas 60 minút každé 2 týždne (n = 292) alebo na docetaxel (n = 290) 75 mg/m2 každé 3 týždne. Liečba pokračovala dovtedy, kým sa pozoroval klinický prínos alebo kým bola liečba tolerovaná. Hodnotenia nádorov sa vykonali pomocou RECIST verzia 1.1. Meraným primárnym koncovým ukazovateľom účinnosti bolo OS. Meranými kľúčovými sekundárnymi koncovými
ukazovateľmi účinnosti boli ORR a PFS hodnotené skúšajúcim. Ďalej sa vykonali analýzy vopred špecifikovanej podskupiny na hodnotenie účinnosti expresie PD-L1 v nádore v preddefinovaných
hladinách 1 %, 5 % a 10 %. Hodnotenie podľa jednotlivých intervalov expresie PD-L1 nebolo
zahrnuté do vopred špecifikovanej analýzy z dôvodu malých veľkostí vzoriek v intervaloch.
Vzorky tkanív nádoru pred štúdiou boli pred randomizáciou systematicky zbierané na to, aby sa vykonali vopred naplánované analýzy účinnosti podľa expresie PD-L1 v nádore. Expresia PD-L1 v nádore sa stanovila pomocou analýzy PD-L1 IHC 28-8 pharmDx.
Medián veku bol 62 rokov (rozsah: 21 až 85) s vekom ≥ 65 rokov u 34 % a s vekom ≥ 75 rokov u 7 %. Väčšina pacientov boli belosi (92 %) a muži (55 %). Východiskové skóre výkonnostného stavu podľa ECOG bolo 0 (31 %) alebo 1 (69 %). Sedemdesiatdeväť percent pacientov boli bývalí/súčasní fajčiari.
Kaplanove-Meierove krivky OS sú uvedené na obrázku 9.
Obrázok 9: Kaplanove-Meierove krivky OS (CA209057)
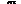
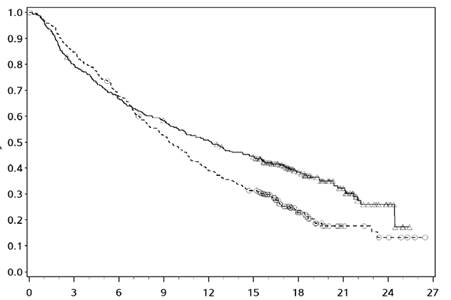
Počet jedincov s rizikom
Nivolumab 3 mg/kg
Celkové prežívanie (mesiace)
292 232 194 169 146 123 62 32 9 0
Docetaxel
290 244 194 150 111 88 34 10 5 0
──∆─── Nivolumab 3 mg/kg (udalosti: 190/292), medián a 95 % CI: 12,19 (9,66; 14,98)
- - -¦- - - Docetaxel (udalosti: 223/290), medián a 95 % CI: 9,36 (8,05; 10,68)
Skúšanie potvrdilo štatisticky významné zlepšenie OS u pacientov randomizovaných na nivolumab v porovnaní s docetaxelom pri vopred špecifikovanej predbežnej analýze, pri ktorej sa pozorovalo
413 udalostí (93 % plánovaného počtu udalostí pre finálnu analýzu). Výsledky účinnosti sú uvedené v tabuľke 11.
'
Tabuľka 11: Výsledky účinnosti (CA209057) nivolumab (n = 292)
Vopred špecifikovaná predbežná analýza
Minimálne následné sledovanie: 13,2 mesiaca
docetaxel
(n = 290)
C
elkové prežívanie
Udalosti 190 (65,1 %) 223 (76,9 %)
Pomer rizikaa 0,73 (95,92 % CI) (0,59; 0,89)
p-hodnotab 0,0015
Medián (95 % CI) mesiace 12,19 (9,66; 14,98) 9,36 (8,05; 10,68) Výskyt (95 % CI) pri 12 mesiacoch 50,5 (44,6; 56,1) 39,0 (33,3; 44,6)
Potvrdená objektívna odpoveď 56 (19,2 %) 36 (12,4%) (95 % CI) (14,8; 24,2) (8,8; 16,8) Pomer šancí (95% CI) 1,68 (1,07; 2,64)
p-hodnota 0,0246
Kompletná odpoveď (CR) 4 (1,4 %) 1 (0,3 %) Parciálna odpoveď (PR) 52 (17,8 %) 35 (12,1 %) Stabilizácia ochorenia (SD) 74 (25,3 %) 122 (42,1 %)
Medián trvania odpovedeMesiace (rozsah) 17,15 (1,8 – 22,6+) 5,55 (1,2+ – 15,2+)
Medián času do odpovedeMesiace (rozsah) 2,10 (1,2 – 8,6) 2,61 (1,4 – 6,3)
Prežívanie bez progresieUdalosti 234 (80,1 %) 245 (84,5 %)
Pomer rizika 0,92
95 % CI (0,77; 1,11)
p-hodnota 0,3932
Medián (95 % CI) (mesiace) 2,33 (2,17; 3,32) 4,21 (3,45; 4,86) Výskyt (95 % CI) pri 12 mesiacoch 18,5 (14,1; 23,4) 8,1 (5,1; 12,0)
Aktualizovaná analýzaMinimálne následné sledovanie: 24,2 mesiaca
Celkové prežívaniec
Udalosti 228 (78,1 %) 247 (85,1 %)
Pomer rizika 0,75
95 % CI (0,63; 0,91)
Výskyt (95 % CI) pri 24 mesiacoch 28,7 (23,6; 34,0) 15,8 (11,9; 20,3)
Potvrdená objektívna odpoveď 19,2 % 12,4 %
(95 % CI) (14,8; 24,2) (8,8; 16,8)
Medián trvania odpovedeMesiace (rozsah) 17,2 (1,8 – 33,7+) 5,6 (1,2+ – 16,8)
Prežívanie bez progresieVýskyt (95 % CI) pri 24 mesiacoch 11,9 (8,3; 16,2) 1,0 (0,2; 3,3)
a Získané z modelu stratifikovaných proporcionálnych rizík.
b P-hodnota je získaná z log-rank testu stratifikovaného pomocou prechádzajúcej udržiavacej liečby a línie liečby; zodpovedajúca účinnosť podľa O’Brienovej-Flemingovej hranice hladiny významnosti je 0,0408.
c Šestnásť pacientov (6 %) randomizovaných na docetaxel dostalo krížovo v ktoromkoľvek čase liečbu
nivolumabom.
“+” Označuje cenzurované sledovanie.
Kvantifikovateľná expresia PD-L1 v nádore sa merala u 79 % pacientov v skupine s nivolumabom a u 77 % pacientov v skupine s docetaxelom. Hladiny expresie PD-L1 v nádore boli medzi dvoma liečenými skupinami (nivolumab oproti docetaxelu) v rovnováhe pri všetkých preddefinovaných hladinách expresie PD-L1 v nádore ≥ 1 % (53 % oproti 55 %), ≥ 5 % (41 % oproti 38 %) alebo ≥ 10 % (37 % oproti 35 %).
Pacienti s expresiou PD-L1 v nádore pomocou všetkých preddefinovaných hladín expresie v skupine
s nivolumabom preukázali väčšiu pravdepodobnosť zlepšeného prežívania v porovnaní s docetaxelom, zatiaľ čo u pacientov s nízkou expresiou PD-L1 v nádore alebo bez jeho expresie bolo prežívanie podobné ako pri docetaxele. Z hľadiska ORR sa zvyšovanie expresie PD-L1 spájalo s väčšou ORR. V porovnaní s celkovou populáciou bol medián trvania odpovede predĺžený s nivolumabom oproti docetaxelu u pacientov bez expresie PD-L1 (18,3 mesiaca oproti 5,6 mesiaca) a u pacientov s
expresiou PD-L1 (16,0 mesiacov oproti 5,6 mesiaca).
Tabuľka 12 sumarizuje výsledky ORR a OS pomocou expresie PD-L1 v nádore.
 Tabuľka 12 ORR a OS pomocou expresie PD-L1 v nádore (CA209057)Expresia PD-L1 nivolumab docetaxelORR pomocou expresie PD-L1 v nádore
Tabuľka 12 ORR a OS pomocou expresie PD-L1 v nádore (CA209057)Expresia PD-L1 nivolumab docetaxelORR pomocou expresie PD-L1 v nádoreMinimálne následné sledovanie: 13,2 mesiaca
< 1 % 10/108 (9,3 %)
95 % CI: 4,5; 16,4
≥ 1 % 38/123 (30,9 %)
95 % CI: 22,9; 39,9
≥ 1 % až < 10 %a 6/37 (16,2 %)
95 % CI: 6,2; 32,0
≥ 10 % až < 50 %a 5/20 (25,0 %)
95 % CI: 8,7; 49,1
≥ 50 %a 27/66 (40,9 %)
95 % CI: 29,0; 53,7
15/101 (14,9 %)
95 % CI: 8,6; 23,3
15/123 (12,2 %)
95 % CI: 7,0; 19,3
5/ 44 (11,4 %)
95 % CI: 3,8; 24,6
7/33 (21,2 %)
95 % CI: 9,0; 38,9
3/46 (6,5 %)
95 % CI: 1,4; 17,9
Pomer šancí (95 % CI)0,59 (0,22; 1,48)
3,22 (1,60; 6,71)
1,51 (0,35; 6,85)
1,24 (0,26; 5,48)
9,92 (2,68; 54,09)
 OS pomocou expresie PD-L1 v nádore
OS pomocou expresie PD-L1 v nádore
Minimálne následné sledovanie: 13,2 mesiaca
Počet udalostí (počet pacientov) Nestratifikovaný pomer rizík (95 % CI)< 1 % 77 (108) 75 (101) 0,90 (0,66; 1,24)
≥ 1 % 68 (123) 93 (123) 0,59 (0,43; 0,82)
≥ 1 % až < 10 %a 27 (37) 30 (44) 1,33 (0,79; 2,24)
≥ 10 % až < 50 %a 11 (20) 26 (33) 0,61 (0,30; 1,23)
≥ 50 %a 30 (66) 37 (46) 0,32 (0,20; 0,53)
Aktualizovaná analýzaMinimálne následné sledovanie: 24,2 mesiaca
< 1 % 91 (108) 86 (101) 0,91 (0,67; 1,22)
≥ 1 % 87 (123) 103 (123) 0,62 (0,47; 0,83)
a Post-hoc analýza; výsledky sa majú interpretovať s opatrnosťou, pretože veľkosti vzoriek podskupín sú malé a v čase analýzy nebola metóda PD-L1 IHC 28-8 pharmDx analyticky validovaná pri hladinách expresie 10 % alebo 50 %.
Vyšší podiel pacientov zomrel v priebehu prvých 3 mesiacov v skupine s nivolumabom (59/292,
20,2 %) v porovnaní so skupinou s docetaxelom (44/290, 15,2 %). Výsledky post-hoc výskumnej multivariantnej analýzy poukazujú na to, že pacienti liečení nivolumabom s horšími prognostickými znakmi a/alebo agresívnym ochorením, ak sú v kombinácii s nižšou expresiou PD-L1 v nádore (napr.
< 50 %) alebo sú bez jeho expresie, môžu mať vyššie riziko úmrtia v priebehu prvých 3 mesiacov.
V analýze podskupiny sa nedokázal prínos prežívania v porovnaní s docetaxelom u pacientov, ktorí nikdy neboli fajčiarmi alebo ktorých nádory ukrývali EGFR aktivujúce mutácie; no z dôvodu malého počtu pacientov nemožno z týchto údajov vyvodiť žiadne definitívne závery.
Karcinóm z renálnych buniek
Bezpečnosť a účinnosť nivolumabu 3 mg/kg ako jedinej látky na liečbu pokročilého RCC so svetlou zložkou bunky sa hodnotili v randomizovanej, otvorenej štúdii 3. fázy (CA209025).
Do štúdie boli zaradení pacienti (18 roční alebo starší), ktorí mali progresiu ochorenia počas 1 alebo
2 predchádzajúcich antiangiogénnych liečebných režimoch alebo po nich a nemali viac ako 3 celkové predchádzajúce systémové liečebné režimy. Pacienti museli mať stav výkonnosti podľa Karnofského (Karnofsky Performance Score, KPS) ≥ 70 %. V tejto štúdii boli pacienti zaradení bez ohľadu na ich stav PD-L1 v nádore. Pacienti so súbežnými metastázami v mozgu, s predchádzajúcou liečbou inhibítorom cicavčej cieľovej kinázy rapamycínu (mammalian target of rapamycin inhibitor, mTOR), s aktívnym autoimunitným ochorením či zdravotnými stavmi vyžadujúcimi si systémovú imunosupresiu alebo s ich akoukoľvek anamnézou boli zo štúdie vyradení.
Celkovo bolo randomizovaných 821 pacientov, buď na intravenózne podávanie nivolumabu
v dávke 3 mg/kg počas 60 minút každé 2 týždne (n = 410) alebo na everolimus (n = 411) 10 mg denne podávaných perorálne. Liečba pokračovala dovtedy, kým sa pozoroval klinický prínos alebo kým bola liečba tolerovaná. Prvé hodnotenia nádorov sa vykonali 8 týždňov po randomizácii a potom pokračovali každých 8 týždňov počas prvého roka a potom každých 12 týždňov do progresie alebo do ukončenia liečby, podľa toho čo sa vyskytlo neskôr. V hodnoteniach nádoru sa pokračovalo po
ukončení liečby u pacientov, ktorí ukončili liečbu z iných dôvodov než progresia. Liečba po úvodnom
vyhodnotení skúšajúcim pomocou RECIST, verzia 1.1-definovanej progresie bola povolená, ak mal pacient klinický prínos a skúšané liečivo bolo tolerované podľa stanovenia skúšajúcim. Meraným primárnym koncovým ukazovateľom účinnosti bolo OS. Sekundárne hodnotenia účinnosti zahŕňali ORR a PFS hodnotené skúšajúcim.
Východiskové charakteristiky boli celkovo vyvážené medzi dvoma skupinami. Medián veku bol 62 rokov (rozsah: 18-88) s vekom ≥ 65 rokov u 40 % a s vekom ≥ 75 rokov u 9 %. Väčšina pacientov boli muži (75 %) a belosi (88 %), boli zastúpené všetky rizikové skupiny podľa MSKCC (Memorial Sloan Kettering Cancer Center, MSKCC) a 34 % pacientov malo východiskové KPS 70 až
80% a 66 % pacientov malo 90 až 100 %. Väčšina pacientov (72 %) sa liečilo s jednou
predchádzajúcou antiangiogénnou liečbou. Medián času trvania od stanovenia počiatočnej diagnózy
do randomizácie bol v oboch skupinách s nivolumabom aj everolimom 2,6 roka. Medán trvania liečby bol 5,5 mesiaca (rozsah: 0 – 29,6+ mesiaca) u pacientov liečených nivolumabom a 3,7 mesiaca
(rozsah: 6 dní – 25,7+ mesiaca) u pacienotv liečebých everolimom.
U 44 % pacientov sa po progresii pokračovalo v liečbe nivolumabom.
Kaplanove-Meierove krivky OS sú uvedené na obrázku 10.
Obrázok 10: Kaplanove-Meierove krivky OS (CA209025)
Počet jedincov s rizikom
Nivolumab
Celkové prežívanie (mesiace)
410 389 359 337 305 275 213 139 73 29 3 0
Everolimus
411 366 324 287 265 241 187 115 61 20 2 0

Nivolumab 3 mg/kg (príhody: 183/410), medián a 95 % CI: 25,00 (21,75; N.A.) Everolimus 10 mg (príhody: 215/411), medián a 95 % CI: 19,55 (17,64; 23,06)
Skúšanie potvrdilo štatisticky významné zlepšenie OS u pacientov randomizovaných na nivolumab v
porovnaní s everolimom pri vopred špecifikovanej predbežnej analýze, pri ktorej sa pozorovalo
398 udalostí (70 % plánovaného počtu udalostí pre finálnu analýzu) (Tabuľka 13 a Obrázok 10). Prínos OS sa pozoroval bez ohľadu na hladinu expresie PD-L1 v nádore.
Výsledky účinnosti sú uvedené v tabuľke 13.
Tabuľka 13:
Výsledky účinnosti (CA209025)nivolumabeverolimus
(n
= 410) (n
= 411) Celkové prežívanie
Udalosti 183 (45 %) 215 (52 %)
Pomer rizika 0,73
98,52 % CI (0,57; 0,93)
p-hodnota 0,0018
Medián (95 % CI) 25,0 (21,7; NE) 19,6 (17,6; 23,1)
Výskyt (95 % CI)
po 6 mesiacoch 89,2 (85,7; 91,8) 81,2 (77,0; 84,7)
po 12 mesiacoch 76,0 (71,5; 79,9) 66,7 (61,8; 71,0)
Objektívna odpoveď 103 (25,1 %) 22 (5,4 %)
(95 % CI) (21,0; 29,6) (3,4; 8,0)
Pomer šancí (95 % CI) 5,98 (3,68; 9,72)
p-hodnota < 0,0001
Kompletná odpoveď (CR) 4 (1,0 %) 2 (0,5 %) Parciálna odpoveď (PR) 99 (24,1 %) 20 (4,9 %) Stabilizácia ochorenia (SD) 141 (34,4 %) 227 (55,2 %)
Medián trvania odpovedeMesiace (rozsah) 11,99 (0,0 – 27,6+) 11,99 (0,0+ – 22,2+)
Medián času do odpovedeMesiace (rozsah) 3,5 (1,4-24,8) 3,7 (1,5-11,2)
Prežívanie bez progresieUdalosti 318 (77,6 %) 322 (78,3 %)
Pomer rizika 0,88
95 % CI (0,75; 1,03)
p-hodnota 0,1135
Medián (95 % CI) 4,6 (3,71; 5,39) 4,4 (3,71; 5,52)
“+” označuje cenzurované sledovanie. NE = neodhadnuteľné
Medián času do nástupu objektívnej odpovede bol 3,5 mesiaca (rozsah: 1,4 – 24,8 mesiaca) po začatí liečby nivolumabom. Štyridsaťdeväť (47,6 %) reagujúcich na liečbu malo pretvávajúce odpovede s trvaním v rozsahu od 0,0 – 27,6+ mesiaca.
Celkové prežívanie mohlo byť sprevádzané zlepšením symtómov spojených s ochorením v priebehu času a špecifickou QoL nesúvisiacou s ochorením podľa hodnotenia pomocou validovaných a spoľahlivých škál „Functional Assessment of Cancer Therapy-Kidney Symptom Index-Disease Related Symptoms“ (FKSI-DRS) a pomocou hodnotenia kvality života EuroQoL EQ-5D. Očividné významné zlepšenie symptómu (MID = o 2 body zmeny v skóre FKSI-DRS; p < 0,001) a čas do zlepšenia (HR = 1,66 (1,33; 2,08), p < 0,001) boli signifikatne lepšie u pacientov v skupine s nivolumabom. Keďže obe skupiny štúdie dostali aktívnu liečbu, údaje QoL sa majú interpretovať v kontexte dizajnu otvorenej štúdie a z tohto dôvodu je potrebná opatrnosť.
Klasický Hodgkinov lymfómBezpečnosť a účinnosť nivolumabu v dávke 3 mg/kg ako jednej látky na liečbu relabujúceho alebo refraktérneho (cHL) po ASCT sa hodnotila v dvoch multicentrických, otvorených štúdiách s jednou skupinou (CA209205 a CA209039).
CA209205 je prebiehajúca otvorená, multikohortová štúdia 2. fázy s jednou skupinou s nivolumabom na cHL. Zahŕňa 243 pacientov, ktorí mali ASCT; Kohorta A zahŕňala 63 (26 %) pacientov, ktorí sa
predtým neliečili brentuximab vedotinom; Kohorta B zahŕňala 80 (33 %) pacientov, ktorí dostali brentuximab vedotin po zlyhaní ASCT a Kohorta C zahŕňala 100 (41 %) pacientov, ktorí dostali brentuximab vedotin pred a/alebo po ASCT, z ktorých 33 (14 %) pacientov dostalo brentuximab vedotin iba pred ASCT. Všetci pacienti dostali monoterapiu nivolumabom v dávke 3 mg/kg intravenózne počas 60 minút každé 2 týždne. Prvé hodnotenia tumoru sa vykonali 9 týždňov po začatí liečby a potom pokračovali do progresie ochorenia alebo do ukončenia liečby. Primárnym meraným koncovým ukazovateľom účinnosti bola ORR podľa stanovenia IRRC. Dodatočné merania účinnosti zahŕňali trvanie odpovede, PFS a OS.
CA209039 je otvorená, multicentrická, so zvyšovaním dávky a multidávková štúdia 1b. fázy s nivolumabom na relabujúce/refraktérne hematologické malignity zahŕňajúca 23 pacientov s cHL liečených monoterapiou nivolumabom v dávke 3 mg/kg, z nich 15 pacientov dostalo predchádzajúcu liečbu brentuximab vedotinom ako záchrannú liečbu po ASCT, podobné s Kohortou B v štúdii CA209205. Prvé hodnotenia tumoru sa vykonali 4 týždne po začatí liečby a potom pokračovali do progresie ochorenia alebo do ukončenia liečby. Hodnotenia účinnosti zahŕňali hodnotenie ORR skúšajúcim, retrospektívne vyhodnotené IRRC, a trvanie odpovede.
Údaje od 80 pacientov zo štúdie CA209205 Kohorty B a od 15 pacientov zo štúdie CA209039, ktorí dostávali predchádzajúcu liečbu brentuximab vedotinom po ASCT boli zjednotené. Dodatočné údaje od 100 pacientov zo štúdie CA209205 Kohorty C, ktorá dostala brentuximab pred a/alebo po ASCT sú tiež poskytnuté. Východiskové charakteristiky boli medzi oboma štúdiami a kohortami podobné (pozri Tabuľku 14 nižšie).
Tabuľka 14: Východiskové charakteristiky pacientov v štúdii CA209205 Kohorta B,
Kohorta C a v štúdii CA209039
C
A2
0
92
0
5
Kohorta B a CA209039 (n = 95)
C
A2
0
92
0
5
Kohorta Ba
(n = 80)
CA209039
(n = 15)
CA209205
Cohort Cb
(n = 100)
Medián veku, roky (rozsah) 37,0 (18–72) 37,0 (18–72) 40,0 (24–54) 32,0 (19-69)
Pohlavie 61 (64 %)M / 34 (36 %)F
51 (64 %)M / 29
(36 %)F
10 (67 %)M / 5
(33 %)F
56 (56 %) M
44 (44 %) F
Stav ECOG
0 49 (52 %) 42 (52,5 %) 7 (47 %) 50 (50 %)
1 46 (48 %) 38 (47,5 %) 8 (53 %) 50 (50 %)
≥ 5 predchádzajúcich línií systémovej liečby
49 (52 %) 39 (49 %) 10 (67 %) 30 (30 %)
Predchádzajúca rádioterapia 72 (76 %) 59 (74 %) 13 (87 %) 69 (69 %)
Predchádzajúca ASCT
1 87 (92 %) 74 (92,5 %) 13 (87 %) 100 (100 %)
≥ 2 8 (8 %) 6 (7,5 %) 2 (13 %) 0 (0 %)
Roky od poslednej transplantácie do prvej dávky skúšanej liečby, medián (min- max)
3,5 (0,2 – 19,0) 3,4 (0,2 – 19,0) 5,6 (0,5 – 15,0) 1,7 (0,2-17,0)
a 18/80 (22,5 %) pacientov v štúdii CA209205 Kohorta B malo na začiatku prítomné B-symptómy.
b 25/100 (25 %) pacientov v štúdii CA209205 Kohorta C malo na začiatku prítomné B-symptómy.
Účinnosť z obidvoch štúdií hodnotila tá istá IRRC. Výsledky sú uvedené v tabuľke 15.
Tabuľka 15: Výsledky účinnosti u pacientov s relabujúcim/refraktérnym klasickým Hodgkinovým
lymfómom
Počet (n)/ minimálne následné sledovanie
(mesiace)
CA209205 Kohorta Ba
a CA209039 (n = 95/12,0)
CA209205 Kohorta Ba
(n = 80/12,0)
CA209039
(n = 15/12,0)
Objektívna odpoveď, n (%) ; (95 % CI) 63 (66 %); (56; 76) 54 (68 %); (56; 78) 9 (60 %); (32; 84) Úplná remisia (CR), n (%); (95 % CI) 6 (6 %); (2; 13) 6 (8 %); (3; 16) 0 (0 %); (0; 22) Čiastočná remisia (PR), n (%); (95 % CI) 57 (60 %); (49; 70) 48 (60 %); (48; 71) 9 (60 %); (32; 84)
Stabilné ochorenie, n (%) 22 (23) 17 (21) 5 (33)
Trvanie odpovede (mesiace)b
Medián (95 % CI)
Rozsah
13,1 (9,5; NE)
0,0+-23,1+
13,1 (8,7; NE)
0,0+-14,2+
12,0 (1,8; NE)
1,8-23,1+
Medián času do odpovede
Mesiace (rozsah)
Medián trvania následných návštev
Mesiace (rozsah)
Prežívanie bez progresie
2,0 (0,7-11,1) 2,1 (1,6-11,1) 0,8 (0,7-4,1)
15,8 (1,9-27,6) 15,4 (1,9-18,5) 21,9 (11,2-27,6)
Miera (95 % CI) po 12 mesiacoch 57 (45; 68)
55 (41; 66)
69 (37; 88)


“+” označuje cenzurované sledovanie.
a Následná návšteva prebiehala v čase podávania údajov.
b Údaje nestabilné z dôvodu obmedzeného trvania odpovede pre Kohortu B následkom cenzurovania. NE = neodhadnuteľné
Údaje z dlhšieho následného sledovania z Kohorty B (minimálne 20,5 mesiaca) a účinnosť Kohorty C
zo štúdie CA209205 sú uvedené nižšie v tabuľke 16.
Tabuľka 16: Aktualizované výsledky účinnosti u pacientov s relabujúcim/refraktérnym
klasickým Hodgkinovým lymfómom z dlhšieho následného sledovania štúdie CA209205
C
A2
0
92
0
5 Kohorta Ba CA209205 Kohorta Ca
Počet (n)/ minimálne následné sledovanie
(mesiace)
(n = 80/20,5) (n = 100/13,7)
b
Objektívna odpoveď, n (%) ; (95 % CI) 54 (68 %); (56; 78) 73 (73 %); (63; 81) Úplná remisia (CR), n (%); (95 % CI) 10 (13 %); (6; 22) 12 (12 %); (6; 20) Čiastočná remisia (PR), n (%); (95 % CI) 44 (55 %); (44; 66) 61 (61 %); (51; 71)
Stabilné ochorenie, n (%) 17 (21) 15 (15 %)
Trvanie odpovede u všetkých pacientov reagujúcich na liečbu (mesiace)c
Medián (95 % CI) 15,9 (7,8; 20,3) 14,5 (9,5; 16,6)
Rozsah 0,0+-21,0+ (0,0+, 16,8+)
Trvanie odpovede pri CR (mesiace)Medián (95 % CI) 20,3 (3,8; NE) 14,5 (8,2; NE)
Rozsah 1,6+-21,0+ (0,0+, 16,5+)
Trvanie odpovede pri PR (mesiace)Medián (95 % CI) 10,6 (6,8; 18,0) 13,2 (9,4; 16,6)
Rozsah 0,0+-20,7+ (0,0+, 16,8+)
Medián času do odpovedeMesiace (rozsah) 2,2 (1,6-9,1) 2,1 (0,8; 8,6)
Medián trvania následných návštevMesiace (rozsah) 22,7 (1,9-27,2) 16,2 (1,4, 20,4)
Prežívanie bez progresieMiera (95 % CI) po 12 mesiacoch 51 (38; 62) 49 (37; 60)
Miera (95 % CI) po 18 mesiacoch 47 (35; 59) –
Celkové prežívanieMedián Nedosiahnutý Nedosiahnutý
Miera (95 % CI) po 12 mesiacoch 95 (87; 98) 90 (82; 94) Miera (95 % CI) po 18 mesiacoch 91 (82; 96) –
“+” označuje cenzurované sledovanie.
a Následná návšteva prebiehala v čase podávania údajov.
b Pacienti v Kohorte C (n = 33), ktorí dostali brentuximab vedotin iba pred ASCT mali ORR 70 %
(95 % CI: 51; 84), CR 15 % (95 % CI: 5; 32), PR 55 % (95 % CI: 36; 72). Medián trvania odpovede bol
13,2 mesiaca (95 % CI: 8,2; NE)
c Stanovené pre jedincov s CR alebo PR
NE = neodhadnuteľné
Na začiatku štúdie boli B-symptómy prítomné u 22 % (53/243) pacientov v štúdii CA209205. Liečba nivolumabom mala za následok rýchle vyriešenie B-symptómov u 88,7 % (47/53) pacientov s mediánom času do vyriešenia 1,9 mesiaca.
V post-hoc analýze z 80 pacientov v štúdii CA209205 Kohorte B 37 nereagovalo na predchádzajúcu liečbu brentuximab vedotinom. U týchto 37 pacientov mala liečba nivolumabom za následok ORR
59,5 % (22/37). Medián trvania odpovede je 18,0 mesiacov (6,6; NE) u 22 pacientov reagujúcich na liečbu nivolumabom, u ktorých zlyhalo dosiahnutie odpovede na predchádzajúcu liečbu brentuximab vedotinom.
Skvamocelulárny karcinóm hlavy a krkuBezpečnosť a účinnosť nivolumabu 3 mg/kg ako jedinej látky na liečbu metastatického alebo recidivujúceho SCCHN sa hodnotili v randomizovanej, otvorenej štúdii 3. fázy (CA209141). Do štúdie boli zaradení pacienti (18 roční alebo starší) s histologicky potvrdenou rekurenciou
ochorenia alebo metastatickým SCCHN (ústna dutina, hltan, hrtan), so štádiom III/IV a s ochorením neprístupným pre lokálnu liečbu s kuratívnym zámerom (chirurgický výkon alebo rádioterapia s chemoterapiou alebo bez nej) a ktorí mali progresiu ochorenia počas alebo v priebehu 6 mesiacov podávania režimu liečby na báze platiny a mali skóre výkonnostného stavu podľa ECOG 0 alebo 1. Predchádzajúca liečba na báze platiny sa podávala buď ako adjuvantná, neoadjuvantná, primárna, liečba recidivujúceho alebo metastatického ochorenia. Pacienti boli zaradení bez ohľadu na ich stav PD-L1 v nádore alebo stav ľudského papilomavírusu (human papilloma virus, HPV). Pacienti s aktívnym autoimunitným ochorením, medicínskymi stavmi vyžadujúcimi si imunosupresiu, recidivujúcim alebo metastatickým karcinómom nosohltana, skvamocelulárnym karcinóm s neznámymi primárnymi histológiami, slinnej žľazy alebo s neskvamóznymi histológiami (napr. mukózny melanóm) alebo s aktívnymi metastázami v mozgu alebo leptomeningeálnymi metastázami
boli zo štúdie vyradení. Pacienti s liečenými metastázami v mozgu boli vhodní, ak sa z neurologického hľadiska vrátili na východiskový stav minimálne 2 týždne pred zaradením a buď neužívali kortikosteroidy alebo boli na stabilnej dávke alebo sa im dávka znížila na dávku zodpovedajúcu prednizónu < 10 mg denne.
Celkovo bolo randomizovaných 361 pacientov, buď na intravenózne podávanie nivolumabu
v dávke 3 mg/kg (n = 240) počas 60 minút každé 2 týždne alebo podľa voľby skúšajúceho buď na cetuximab (n = 15) v nasycovacej dávke 400 mg/m2, po ktorej nasledovala dávka 250 mg/m2 týždenne alebo na metotrexát (n = 52) 40 až 60 mg/m2 týždenne alebo na docetaxel (n = 54) 30 až
40 mg/m2 týždenne. Randomizácia bola stratifikovaná pomocou predchádzajúcej liečby cetuximabom. Liečba pokračovala dovtedy, kým sa pozoroval klinický prínos alebo kým bola liečba tolerovaná. Hodnotenia nádorov, podľa RECIST verzia 1.1, sa vykonali 9 týždňov po randomizácii a potom pokračovali každých 6 týždňov. Liečba po úvodnom vyhodnotení skúšajúcim pomocou RECIST, verzia 1.1-definovanej progresie bola povolená u pacientov, ktorí dostávali nivolumab, ak mal pacient
klinický prínos a skúšané liečivo bolo tolerované podľa stanovenia skúšajúcim. Meraným primárnym
koncovým ukazovateľom účinnosti bolo OS. Merané kľúčové sekundárne koncové ukazovatele účinnosti hodnotené skúšajúcim boli PFS a ORR. Ďalej sa vykonali analýzy vopred špecifikovanej podskupiny na hodnotenie účinnosti expresie PD-L1 v nádore v preddefinovaných hladinách 1 %, 5 % a 10 %.
Vzorky tkanív nádoru pred štúdiou boli pred randomizáciou systematicky zbierané na to, aby sa vykonali vopred naplánované analýzy účinnosti podľa expresie PD-L1 v nádore. Expresia PD-L1 v nádore sa stanovila pomocou medódy PD-L1 IHC 28-8 pharmDx.
Východiskové charakteristiky boli celkovo vyvážené medzi dvoma skupinami. Medián veku
bol 60 rokov (rozsah: 28 – 83) 31 % s vekom ≥ 65 rokov a 5 % s vekom ≥ 75 rokov, 83 % bolo mužov a 83 % bolo belochov. Východiskové skóre výkonnostného stavu podľa ECOG bolo 0 (20 %) alebo
1 (78 %), 77 % bolo bývalých/súčasných fajčiarov, 90 % malo IV. štádium ochorenia, 66 % malo dve alebo viac lézií, 45 %, 34 % a 20 % dostalo 1, 2, alebo 3 alebo viac predchádzajúcich línií systémovej liečby, v uvedenom poradí, a 25 % malo pozitívny stav HPV-16.
S minimálnym následným sledovaním 11,4 mesiaca skúšanie potvrdilo štatisticky významné zlepšenie OS u pacientov randomizovaných na nivolumab v porovnaní s voľbou skúšajúceho. Kaplanove- Meierove krivky OS sú uvedené na obrázku 11. Výsledky účinnosti sú uvedené v tabuľke 17.
 Obrázok 11: Kaplanove-Meierove krivky OS (CA209141)
Obrázok 11: Kaplanove-Meierove krivky OS (CA209141)
Počet jedincov s rizikom
Nivolumab
Celkové prežívanie (mesiace)
240 169 132 98 76 45 27 12 3
Voľba skúšajúceho
121 88 51 32 22 9 4 3 0
Nivolumab 3 mg/kg (príhody: 184/240), medián a 95 % CI: 7,72 (5,68; 8,77) Voľba skúšajúceho (príhody: 105/121), medián a 95 % CI: 5,06 (4,04; 6,24)
Tabuľka 17: Výsledky účinnosti (CA209141) nivolumab (n = 240)
voľba skúšajúceho
(n = 121)
C
elkové prežívanie
Udalosti 184 (76,7 %) 105 (86,8 %) Pomer rizikaa 0,71
(95 % CI) (0,55; 0,90)
p-hodnotab 0,0048
Medián (95 % CI) (mesiace) 7,72 (5,68; 8,77) 5,06 (4,04; 6,24) Výskyt (95 % CI) po 6 mesiacoch 56,5 (49,9; 62,5) 43,0 (34,0; 51,7) Výskyt (95 % CI) po 12 mesiacoch 34,0 (28,0; 40,1) 19,7 (13,0; 27,3) Výskyt (95 % CI) po 18 mesiacoch 21,5 (16,2; 27,4) 8,3 (3,6; 15,7)
Prežívanie bez progresieUdalosti 204 (85,0 %) 104 (86,0 %)
Pomer rizika 0,87
95 % CI (0,69; 1,11)
p-hodnota 0,2597
Medián (95 % CI) (mesiace) 2,04 (1,91; 2,14) 2,33 (1,97; 3,12) Výskyt (95 % CI) po 6 mesiacoch 21,0 (15,9; 26,6) 11,1 (5,9; 18,3) Výskyt (95 % CI) po 12 mesiacoch 9,5 (6,0; 13,9) 2,5 (0,5; 7,8)
Potvrdená objektívna odpoveďc 32 (13,3 %) 7 (5,8 %) (95 % CI) (9,3; 18,3) (2,4; 11,6) Pomer šancí (95 % CI) 2,49 (1,07; 5,82)
Kompletná odpoveď (CR) 6 (2,5 %) 1 (0,8 %) Parciálna odpoveď (PR) 26 (10,8 %) 6 (5,0 %) Stabilizácia ochorenia (SD) 55 (22,9 %) 43 (35,5 %)
Medián času do odpovedeMesiace (rozsah) 2,1 (1,8 – 7,4) 2,0 (1,9 – 4,6)
Medián trvania odpovedeMesiace (rozsah) 9,7 (2,8 – 20,3+) 4,0 (1,5+ – 8,5+)
a Získané z modelu stratifikovaných proporcionálnych rizík.
b P-hodnota je získaná z log-rank testu stratifikovaného pomocou predchádzajúcej liečby cetuximabom;
zodpovedajúca účinnosť podľa O’Brienovej-Flemingovej hranice hladiny významnosti je 0,0227.
c V skupine s nivolumabom boli dvaja pacienti s CR a sedem pacientov s PR, ktorí mali expresiu PD-L1
v nádore < 1 %.
Kvantifikovateľná expresia PD-L1 v nádore sa merala u 67 % pacientov v skupine s nivolumabom a u 82 % pacientov v skupine s voľbou skúšajúceho. Hladiny expresie PD-L1 v nádore boli medzi dvoma liečenými skupinami (nivolumab oproti voľbe skúšajúceho) vyrovnané pri
všetkých preddefinovaných hladinách expresie PD-L1 v nádore ≥ 1 % (55 % oproti 62 %), ≥ 5 %
(34 % oproti 43 %), alebo ≥ 10 % (27 % oproti 34 %).
Pacienti s expresiou PD-L1 v nádore pri všetkých preddefinovaných hladinách expresie v skupine s nivolumabom preukázali vyššiu pravdepodobnosť zlepšeného prežívania v porovnaní s voľbou skúšajúceho. Miera prínosu OS bola zhodná pri hladinách expresie PD-L1 v nádore ≥ 1 %, ≥ 5 % alebo ≥ 10 % (pozri tabuľka 18).
Tabuľka 18: OS pomocou expresie PD-L1 v nádore (CA209141)
Expresia PD-L1 nivolumab voľba skúšajúceho
OS pomocou expresie PD-L1 v nádore
Počet udalostí (počet pacientov) Nestratifikovaný pomer
rizika (95 % CI)
< 1 % 56 (73) 32 (38) 0,83 (0,54; 1,29)
≥ 1 % 66 (88) 55 (61) 0,53 (0,37; 0,77)
≥ 5 % 39 (54) 40 (43) 0,51 (0,32; 0,80)
≥ 10 % 30 (43) 31 (34) 0,57 (0,34; 0,95)
V prieskumnej post-hoc analýze s použitím nevalidovaného testu bola expresia PD-L1 v nádorových
bunkách aj expresia PD-L1 v imunitných bunkách súvisiacich s nádorom (tumour-associated immune cell, TAIC) analyzovaná vo vzťahu k miere účinku liečby nivolumabom v porovnaní s voľbou skúšajúceho. Táto analýza ukázala, že nie iba expresia PD-L1 v nádorových bunkách, ale aj expresia PD-L1 v TAIC sa javí súvisiaca s prínosom liečby nivolumabom v porovnaní s voľbou skúšajúceho (pozri tabuľka 19). Z dôvodu malého počtu pacientov v podkupinách a prieskumného charakteru analýzy, nemožno z týchto údajov vyvodiť definitívne závery.
Tabuľka 19: Účinnosť pomocou expresie PD-L1 v nádorových bunkách a v TAIC(CA209141)Medián OSa (mesiace) Medián PFSa (mesiace) ORR (%)HRb (95 % CI) HRb (95 % CI) (95 % CI)c
n
ivolumab voľba skúšajúceho
n
ivolumab voľba skúšajúceho
n
ivolumab voľba skúšajúceho
PD-L1 ≥ 1 %,
PD-L1+ TAIC výdatnád
(61 nivolumab,
47 voľba skúšajúceho)
PD-L1 ≥ 1 %,
PD-L1+ TAIC zriedkavád
(27 nivolumab,
14 voľba skúšajúceho)
PD-L1 < 1 %,
PD-L1+ TAIC výdatnád
(43 nivolumab,
25 voľba skúšajúceho)
PD-L1 < 1 %,
PD-L1+ TAIC zriedkavá
(27 nivolumab,
10 voľba skúšajúceho)
9,10 4,60 3,19 1,97 19,7 0
0,43 (0,28; 0,67) 0,48 (0,31; 0,75) (10,6; 31,8) (0; 7,5)
6,67 4,93 1,99 2,04 11,1 7,1
0,89 (0,44; 1,80) 0,93 (0,46; 1,88) (2,4; 29,2) (0,2; 33,9)
11,73 6,51 2,10 2,73 18,6 12,0
0,67 (0,38; 1,18) 0,96 (0,55; 1,67) (8,4; 33,4) (2,5; 31,2)
3,71 4,85 1,84 2,12 3,7 10,0
1,09 (0,50; 2,36) 1,91 (0,84; 4,36) (< 0,1; 19,0) (0,3; 44,5)

a OS a PFS boli odhadnuté použitím Kaplanovej-Meierovej metódy.
b Pomer rizika v každej podskupine odvodený z Coxovho modelu proporcionálnych rizík s liečbou iba ako kovarianciou.
c Interval spoľahlivosti pre ORR vypočítaný použitím Clopperovej-Pearsonovej metódy.
d PD-L1+ TAIC v mikroprostredí nádoru boli stanovené kvalitatívne a charakterizované ako “početná”, “stredne početná”, a “zriedkavá” na základe patologických vyhodnotení. Skupiny “početná” a “stredne
početná” boli skombinované na skupinu definovanú ako “výdatná”.
Pacienti s nádorom nosohltanu vyhodnoteným skúšajúcim ako primárnym miestom boli testovaní na HPV (stanovenie pomocou p16 imunohistochémie [IHC]). Prínos OS sa pozoroval bez ohľadu
na stav HPV (HPV-pozitívny: HR = 0,63; 95 % CI: 0,38; 1,04, HPV-negatívny: HR = 0,64; 95 % CI:
0,40; 1,03 a HPV-neznámy: HR = 0,78; 95 % CI: 0,55; 1,10).
Výsledky hlásené pacientom (patient-reported outcomes, PRO) sa hodnotili použitím EORTC QLQ- C30, EORTC QLQ-H&N35 a 3-úrovňovým EQ-5D. Po 15 týždňoch následného sledovania pacienti liečení nivolumabom vykazovali stabilné PRO, zatiaľ čo tí, ktorí sa liečili pridelenou voľbou skúšajúcim vykazovali signifikantný pokles vo fungovaní (napr. fyzickom, funkčnom, spoločenskom) a v zdravotnom stave ako aj zvýšenú symptomatológiu (napr. únavu, dýchavičnosť, stratu chuti
do jedla, bolesť, problémy so zmyslami, problémy so sociálnymi kontaktmi). Údaje PRO sa majú interpretovať v kontexte dizajnu otvorenej štúdie, a preto sa majú zohľadniť s opatrnosťou.
Uroteliálny karcinómOtvorená štúdia 2. fázy (CA209275)Bezpečnosť a účinnosť nivolumabu v dávke 3 mg/kg ako jedinej látky na liečbu pacientov s lokálne
pokročilým alebo metastatickým uroteliálnym karcinómom sa hodnotili v multicentrickej, otvorenej
štúdii 2. fázy s jednou skupinou (CA209275).
Do štúdie boli zaradení pacienti (18 roční alebo starší), ktorí mali progresiu ochorenia počas alebo po chemoterapii obsahujúcej platinu pri liečbe pokročilého alebo metastatického ochorenia alebo mali progresiu ochorenia v priebehu 12 mesiacov pri neoadjuvantnej alebo adjuvantnej liečbe chemoterapiou obsahujúcou platinu. Pacienti mali skóre výkonnostného stavu podľa ECOG 0 alebo 1 a boli zaradení bez ohľadu na ich stav PD-L1 v nádore. Pacienti s aktívnymi metastázami v mozgu alebo leptomeningeálnymi metastázami, s aktívnym autoimunitným ochorením alebo s medicínskymi stavmi vyžadujúcimi si systémovú imunosupresiu boli zo štúdie vyradení. Pacienti, ktorí dostali viac ako 2 predchádzajúce línie chemoterapie s metastázami v pečeni boli vylúčení.
Celkovo 270 pacientov, ktorí dostali intravenózne podávaný nivolumab v dávke 3 mg/kg počas
60 minút každé 2 týždne s minimálnym následným sledovaním 8,3 mesiaca, bolo hodnotených z hľadiska účinnosti. Liečba pokračovala dovtedy, kým sa pozoroval klinický prínos alebo kým bola liečba tolerovaná. Prvé hodnotenia nádorov sa vykonali 8 týždňov po začatí liečby a potom pokračovali každých 8 týždňov až do 48. týždňa, potom každých 12 týždňov do progresie ochorenia alebo do ukončenia liečby, podľa toho čo sa vyskylo neskôr. V hodnoteniach nádorov sa pokračovalo
po ukončení liečby u pacientov, ktorí ukončili liečbu z iných dôvodov ako progresia. Liečba po
úvodnom vyhodnotení skúšajúcim pomocou RECIST, verzia 1.1-definovanej progresie bola povolená, ak mal pacient klinický prínos, nemal rýchlu progresiu ochorenia a skúšané liečivo bolo tolerované podľa stanovenia skúšajúcim. Meraným primárnym koncovým ukazovateľom bolo ORR podľa stanovenia BICR (zaslepeným nezávislým centrálnym hodnotením, Blinded Independent Central Review). Dodatočné merania účinnosti zahŕňali trvanie odpovede, PFS a OS.
Medián veku bol 66 rokov (rozsah: 38 až 90), 55 % s vekom ≥ 65 rokov a 14 % s vekom ≥ 75 rokov. Väčšina pacientov boli belosi (86 %) a muži (78 %). Východiskový výkonnostný stav podľa ECOG bol 0 (54 %) alebo 1 (46 %).
 Tabuľka20:Výsledkyúčinnosti(CA209275)a nivolumab(n = 270)Potvrdená objektívna odpoveď
Tabuľka20:Výsledkyúčinnosti(CA209275)a nivolumab(n = 270)Potvrdená objektívna odpoveď 54 (20,0 %)
(95 % CI) (15,4; 25,3) Kompletná odpoveď (CR) 8 (3,0 %)
Parciálna odpoveď (PR) 46 (17,0 %)
Stabilizácia ochorenia (SD) 60 (22,2 %)
Medián trvania odpovedeb
Mesiace (rozsah) 10,4 (1,9+-12,0+)
Medián času do odpovedeMesiace (rozsah) 1,9 (1,6; 7,2)
Prežívanie bez progresieUdalosti (%) 216 (80 %) Medián (95 % CI) mesiace 2,0 (1,9; 2,6) Výskyt (95 % CI) po 6 mesiacoch 26,1 (20,9; 31,5)
Celkové prežívaniecUdalosti (%) 154 (57 %)
Medián (95 % CI) mesiace 8,6 (6,05; 11,27) Výskyt (95 % CI) po 12 mesiacoch 41,0 (34,8; 47,1)
Potvrdená objektívna odpoveď
(95 % CI)
Hladina expresie PD-L1 v nádore
< 1 % ≥ 1 %
Medián trvania odpovedeb
Mesiace (rozsah)
16 % (10,3; 22,7)
n = 146
25 % (17,7; 33,6)
n = 124
10,4 (3,7; 12,0+) Nedosiahnutý (1,9+; 12,0+)
Prežívanie bez progresie
Medián (95 % CI) mesiace 1,9 (1,8; 2,0) 3,6 (1,9; 3,7)
Výskyt (95 % CI) po
6 mesiacoch
22,0 (15,6; 29,2) 30,8 (22,7; 39,3)
C
elkové prežívanie
Medián (95 % CI) mesiace 5,9 (4,37; 8,08) 11,6 (9,10; NE)
Výskyt (95 % CI) po
12 mesiacoch
“+” označuje cenzurované sledovanie.
34,0 (26,1; 42,1) 49,2 (39,6; 58,1)

a medián následného sledovania 11,5 mesiaca.
b Údaje nestabilné z dôvodu obmedzeného trvania odpovede.
c zahŕňa 4 úmrtia súvisiace s liečivom: 1 pneumonitída, 1 akútne respiračné zlyhanie, 1 respiračné zlyhanie a
1 kardiovaskulárne zlyhanie. NE: neodhadnuteľné
Výsledky z post-hoc analýzy, prieskumnej analýzy naznačujú, že u pacientov s nízkou (napr. < 1 %) až žiadnou expresiou PD-L1 v nádore, inými charakteristikami pacienta (napr. metastázy v pečeni, viscerálne metastázy, východisková hladina hemoglobínu < 10 g/dl a výkonnostný stav podľa
ECOG = 1), môžu prispievať ku klinickému výsledku.
Otvorená štúdia 1./2. fázy (CA209032)CA209032 bola otvorená multikohortová štúdia 1./2. fázy, ktorá obsahovala kohortu 78 pacientov
(vrátane 18 jedincov, ktorí dostali plánovanú skríženú liečbu nivolumabom 3 mg/kg plus ipilimumabom v dávke 1 mg/kg v kombinácii) s podobnými kritériami na zaradenie ako štúdia CA209275, liečených nivolumabom monoterapiou v dávke 3 mg/kg na liečbu uroteliálneho karcinómu. Pri minimálnom následom sledovaní 9 mesiacov bolo potvrdené ORR vyhodnotené skúšajúcim 24,4 % (95 % CI: 15,3; 35,4). Medián trvania odpovede sa nedosiahol (rozsah: 4,4-16,6+
mesiaca). Medián OS bol 9,7 mesiaca (95 % CI:7,26; 16,16) a odhadované pomery OS boli 69,2 %
(CI: 57,7; 78,2) po 6 mesiacoch a 45,6 % (CI: 34,2; 56,3) po 12 mesiacoch.
Bezpečnosťaúčinnosťustaršíchpacientov
Medzi staršími (≥ 65 rokov) a mladšími pacientmi (< 65 rokov) sa nehlásili žiadne celkové rozdiely
v bezpečnosti alebo účinnosti. Údaje od pacientov s NSCLC, SCCHN a adjuvantným melanómom vo veku 75 rokov alebo starších sú príliš obmedzené na vyvodenie záverov v tejto populácii. Údaje od pacientov s cHL vo veku 65 rokov alebo starších sú príliš obmedzené na vyvodenie záverov o tejto populácii.
Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s nivolumabom vo viacerých podskupinách pediatrickej populácie na liečbu malígnych solídnych tumorov, malígnych neoplaziem lymfoidného tkaniva a malígnych neoplaziem centrálneho nervového systému (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika (PK) nivolumabu je lineárna v rozsahu dávok 0,1 až 10 mg/kg. Na základe analýzy populačnej PK bol geometrický priemerný klírens (CL) nivolumabu v rovnovážnom stave pri dávke
3 mg/kg každé 2 týždne 7,9 ml/h, terminálny polčas 25,0 dňa a priemerná expozícia 86,6 µg/ml.
CL nivolumabu u pacientov s cHL bol približne o 32 % nižší v porovnaní s pacientmi s NSCLC. Východiskový CL nivolumabu u pacientov s adjuvantným melanómom bol približne o 40 % nižší a rovnovážny stav CL bol približne o 20 % nižší v porovnaní s pacientmi s pokročilým melanómom. S dostupnými údajmi o bezpečnosti boli tieto zníženia CL klinicky nevýznamné.Metabolická dráha nivolumabu nie je popísaná. Očakáva sa, že nivolumab sa bude odbúravať na malé peptidy a aminokyseliny katabolickými dráhami rovnakým spôsobom ako endogénny IgG.
Nivolumab v kombinácii s ipilimumabom: Ak sa podával nivolumab v dávke 1 mg/kg v kombinácii s ipilimumabom v dávke 3 mg/kg, CL nivolumabu sa zvýšil o 29 %. Účinok nivolumabu na CL ipilimumabu sa nepozoroval.
Ak sa podávali v kombinácii, CL nivolumabu sa zvýšil o 24 % v prítomnosti protilátok proti nivolumabu. Protilátky proti ipilimumabu neovplyvnili CL ipilimumabu.
Osobitné populácie
Analýza populačnej PK nepoukázala na žiadny rozdiel v CL nivolumabu na základe veku, pohlavia,
rasy, typu solídneho nádoru, veľkosti nádoru a poruchy funkcie pečene. I napriek tomu, že stav ECOG, východisková hodnota pomeru glomerulárnej filtrácie (glomerular filtration rate, GFR), albumín, telesná hmotnosť a mierna porucha funkcie pečene mali vplyv na CL nivolumabu, účinok nebol klinicky významný. CL nivolumabu u pacientov s cHL bol približne o 32 % nižší v porovnaní s pacientmi s NSCLC. V dostupných údajoch o bezpečnosti nebolo toto zníženie CL klinicky významné.
Poruchafunkcieobličiek
V analýze populačnej PK sa hodnotil vplyv poruchy funkcie obličiek na CL nivolumabu u pacientov s miernou (GFR < 90 a ≥ 60 ml/min/1,73 m2; n = 379), stredne ťažkou (GFR < 60
a ≥ 30 ml/min/1,73 m2; n = 179) alebo ťažkou (GFR < 30 a ≥ 15 ml/min/1,73 m2; n = 2) poruchou funkcie obličiek v porovnaní s pacientmi s normálnou funkciou obličiek (GFR ≥ 90 ml/min/1,73 m2;
n = 342). Medzi pacientmi s miernou alebo stredne ťažkou poruchou funkcie obličiek a pacientmi s
normálnou funkciou obličiek sa nezistili žiadne klinicky významné rozdiely v CL nivolumabu. V tejto populácii sú údaje od pacientov s ťažkou poruchou funkcie obličiek na vyvodenie záverov príliš obmedzené (pozri časť 4.2).
Porucha
funkcie
p
ečene
V analýze populačnej PK sa hodnotil vplyv poruchy funkcie pečene na CL nivolumabu u pacientov s miernou poruchou funkcie pečene (celkový bilirubín 1,0 × až 1,5 × ULN alebo AST > ULN podľa
definovaných používaných kritérií dysfunkcie pečene Národným inštitútom pre výskum rakoviny;
n = 92) v porovnaní s pacientmi s normálnou funkciou pečene (celkový bilirubín a AST ≤ ULN; n = 804). Medzi pacientmi s mierou poruchou funkcie pečene a s normálnou funkciou pečene sa nezistili žiadne klinicky významné rozdiely v CL nivolumabu. Nivolumab sa neskúmal u pacientov so stredne ťažkou (celkový bilirubín > 1,5 × až 3 × ULN a akákoľvek AST) alebo ťažkou poruchou funkcie pečene (celkový bilirubín > 3 × ULN a akákoľvek AST) (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Blokáda PD-L1 signálov preukázala na modeloch gravidných myší narušenú znášanlivosť plodov a zvýšenú stratu plodov. Účinky nivolumabu na prenatálny a postnatálny vývoj sa hodnotili u opíc, ktoré dostali nivolumab dvakrát týždenne od nástupu organogenézy v prvom trimestri do pôrodu, pri hladinách expozície buď 8 alebo 35-násobne vyššej než sú hladiny pozorované pri klinickej dávke
3 mg/kg nivolumabu (na základe AUC). Na začiatku tretieho trimestra bol nárast straty plodov a zvýšená neonatálna mortalita závislé od dávky.
Zvyšná časť potomstva samíc liečených nivolumabom prežila do plánovaného ukončenia bez klinických prejavov spojených s liečbou, zmien v normálnom vývoji, účinkov na hmotnosť orgánov alebo makro a mikroskopických patologických zmien. Výsledky indexov rastu, rovnako ako aj teratogénne, neurobehaviorálne, imunologické a klinické patologické parametre počas 6-mesačného postnatálneho obdobia boli porovnateľné s kontrolnou skupinou. Na základe jeho mechanizmu účinku však expozícia nivolumabu v plode môže zvýšiť riziko vzniku imunitne podmienených ochorení alebo zmeniť normálnu imunitnú odpoveď a imunitne podmienené ochorenia sa hlásili u PD-1 knockoutovaných myší.
Štúdie fertility sa s nivolumabom nevykonali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dihydrát citrátu sodného chlorid sodný
manitol (E421)
kyselina pentetová (kyselina dietylentriaminpentaoctová)
polysorbát 80
hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
voda na injekciu
6.2 Inkompatibility
Chýbajú štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi. OPDIVO sa nesmie podávať súbežne infúziou v tej istej intravenóznej hadičke s inými liekmi.
6.3 Čas použiteľnosti
Neotvorenáinjekčnáliekovka
Injekčné liekovky so 40 mg/4 ml a 100 mg/10 ml: 3 roky
Injekčná liekovka s 240 mg/24 ml: 2 roky
Po
o
tvorení
Z mikrobiologického hľadiska sa má liek po otvorení ihneď podať infúziou alebo nariediť a podať infúziou.
Poprípraveinfúzie
Z mikrobiologického hľadiska sa má liek použiť okamžite.
Ak sa nepoužije okamžite, chemická a fyzikálna stabilita na používanie OPDIVA bola dokázaná na
24 hodín pri 2ºC až 8ºC pri ochrane pred svetlom a maximálne 8 hodín pri 20°C-25°C a izbovom svetle (toto 8-hodinové obdobie z celkových 24 hodín má zahŕňať obdobie podávania lieku).
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Zatvorená injekčná liekovka sa môže uchovávať pri kontrolovanej izbovej teplote do 25 °C pri dennom svetle až do 48 hodín.
Podmienky na uchovávanie po príprave infúzie, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
4 ml koncentrátu v 10 ml injekčnej liekovke (sklo typu I) so zátkou (potiahnutou butylkaučukom)
a tmavomodrým vyklápacím viečkom (hliník). Veľkosť balenia 1 injekčná liekovka.
10 ml koncentrátu v 10 ml injekčnej liekovke (sklo typu I) so zátkou (potiahnutou butylkaučukom)
a sivým vyklápacím viečkom (hliník). Veľkosť balenia 1 injekčná liekovka.
24 ml koncentrátu v 25 ml injekčnej liekovke (sklo typu I) so zátkou (potiahnutou butylkaučukom)
a červeným matným vyklápacím viečkom (hliník). Veľkosť balenia 1 injekčná liekovka. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Prípravu lieku má vykonať zaškolený personál v súlade s pravidlami správnej praxe, najmä s ohľadom na asepsu.
Príprava a podávanie
Výpočetdávky
Na podanie celkovej dávky pre pacienta môže byť potrebná viac než jedna injekčná liekovka
koncentrátu OPDIVA.
Monoterapia nivolumabom:
Predpísaná dávka pre pacienta je 240 mg alebo 480 mg, ktorá sa podáva bez ohľadu na telesnú hmotnosť v závislosti od indikácie (pozri časť 4.2).
Nivolumab v kombinácii s ipilimumabom:
Predpísaná dávka pre pacienta sa udáva v mg/kg. Na základe tejto predpísanej dávky vypočítajte celkovú dávku, ktorá sa má podať.
§ Celková dávka nivolumabu v mg = telesná hmotnosť pacienta v kg × predpísaná dávka v mg/kg.
§ Objem koncentrátu OPDIVA na prípravu dávky (ml) = celková dávka v mg vydelená 10 (sila koncentrátu OPDIVA je 10 mg/ml).
Príprava
infúzneho
roztoku
Pri príprave infúzneho roztoku dbajte na zabezpečenie aseptického zaobchádzania s liekom.
OPDIVO sa môže použiť na intravenózne podanie buď:
§ bez nariedenia, po prenesení do infúznej nádoby pomocou vhodnej sterilnej injekčnej striekačky;
alebo
§ po nariedení podľa nasledovných pokynov:
§ konečná koncentrácia infúzneho roztoku má byť v rozsahu medzi 1 a 10 mg/ml
§ celkový objem infúzie nesmie presiahnuť 160 ml. U pacientov vážiacich menej ako 40 kg
nesmie celkový objem infúzie presiahnuť 4 ml na kilogram telesnej hmotnosti pacienta.
Koncentrát OPDIVA sa môže nariediť buď s:
§ 0,9 % (9 mg/ml) injekčným roztokom chloridu sodného; alebo
§ 5 % (50 mg/ml) injekčným roztokom glukózy.
KROK 1
§ Skontrolujte koncentrát OPDIVA na prítomnosť cudzorodých častíc alebo zmenu farby. Injekčnú liekovku nepretrepávajte. Koncentrát OPDIVA je číry až opalescenčný, bezfarebný až svetložltý roztok. Injekčnú liekovku zlikvidujte, ak je roztok zakalený, je zafarbený alebo obsahuje pevné častice iné ako zopár priesvitných až-bielych častíc.
§ Odoberte potrebný objem koncentrátu OPDIVA pomocou vhodnej sterilnej injekčnej striekačky.
KROK 2
§ Koncentrát preneste do sterilnej, prázdnej sklenenej fľaše alebo intravenózneho vaku (s PVC alebo polyolefínu).
§ Ak je to aplikovateľné, narieďte potrebným objemom 0,9 % (9 mg/ml) injekčného roztoku chloridu sodného alebo 5 % (50 mg/ml) injekčného roztoku glukózy. Na jednoduchú prípravu možno koncentrát preniesť priamo aj do predplneného vaku s obsahom vhodného objemu injekčného roztoku 0,9 % (9 mg/ml) chloridu sodného alebo 5 % (50 mg/ml) injekčného roztoku glukózy. Infúzny roztok jemne premiešajte krúživým pohybom ruky. Nepretrepávajte.
Podávanie
Infúzny roztok OPDIVA sa nesmie podávať vo forme intravenóznej pretlakovej infúzie (tzv. i.v. push)
ani bolusovej injekcie.
Infúzny roztok OPDIVA podajte intravenózne počas 30 alebo 60 minút v závislosti od dávky.
Infúzny roztok OPDIVA sa nesmie podávať infúziou v tom istom čase v tej istej intravenóznej hadičke s ďalšími látkami. Na podanie infúzie použite samostatnú infúznu hadičku.
Použite infúznu súpravu a in-line, sterilný, nepyrogénny filter s nízkou afinitou k bielkovinám
(veľkosť pórov 0,2 μm až 1,2 μm).
Infúzny roztok OPDIVA je kompatibilný s PVC a polyolefínovými obalmi, sklenenými fľašami, PVC
infúznymi súpravami a s in-line filtrami s polyétersulfónovými membránami s veľkosťou pórov
0,2 μm až 1,2 μm.
Po podaní dávky nivolumabu prepláchnite infúznu hadičku 0,9 % (9 mg/ml) injekčným roztokom chloridu sodného alebo 5 % (50 mg/ml) injekčným roztokom glukózy.
Likvidácia
Nespotrebované množstvo infúzneho roztoku neuchovávajte na ďalšie použitie. Všetok nepoužitý liek
alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Bristol-Myers Squibb Pharma EEIG
Uxbridge Business Park
Sanderson Road Uxbridge UB8 1DH Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/15/1014/001
EU/1/15/1014/002
EU/1/15/1014/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. júna 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.