ONUREG 200 MG FILMOM OBALENÉ TABLETY tbl flm 7x200 mg (blis.OPA/PVC/Al)

>t v 2 po sebe nasledujúcich cykloch
Tabuľka 1: Úpravy dávky z dôvodu hematologických a nehematologických nežiaducich reakcií
K
r
it
é
r
i
u
m*
|
O
d
p
o
r
ú
č
a
n
ý postup
|
|
• Prerušte podávanie Onuregu. Liečebný cyklus obnovte v zníženej dávke 200 mg po úprave počtu trombocytov na 2. stupeň alebo nižší.
• Ak sa u pacienta po znížení dávky naďalej vyskytuje toxicita, skráťte dĺžku liečby o 7 dní.
• Ak po znížení dávky a skrátení režimu toxicita naďalej pretrváva
alebo sa opätovne objavuje, liečbu Onuregom ukončite.
|
N
e
vo
ľ
n
o
s
ť
, vracanie alebo hnačka 3. alebo vyššieho stupňa
|
• Prerušte podávanie Onuregu. Liečebný cyklus obnovte v rovnakej dávke hneď, ako sa toxicita upraví na 1. stupeň alebo nižší.
• Použite podpornú liečbu, ako je antiemetická liečba a liečte hnačku pri nástupe symptómov (pozri časť 4.4).
• V prípade opätovného výskytu udalosti prerušte podávanie dovtedy, kým nedôjde k jej úprave na 1. stupeň alebo nižší a dávku znížte na 200 mg.
• Ak sa u pacienta po znížení dávky naďalej vyskytuje toxicita, skráťte dĺžku liečby o 7 dní.
• Ak po znížení dávky a skrátení režimu toxicita naďalej pretrváva
alebo sa opätovne objavuje, liečbu Onuregom ukončite.
|
I
n
é nehematologické udalosti 3. alebo vyššieho stupňa
|
• Prerušte podávanie Onuregu a poskytnite lekársku starostlivosť podľa miestnych odporúčaní. Liečebný cyklus obnovte v rovnakej dávke hneď, ako sa toxicita upraví na 1. stupeň alebo nižší.
• V prípade opätovného výskytu toxicity prerušte podávanie Onuregu dovtedy, kým nedôjde k jej úprave na 1. stupeň alebo nižší a dávku znížte na 200 mg.
• Ak sa u pacienta po znížení dávky naďalej vyskytuje toxicita, skráťte dĺžku liečby o 7 dní.
• Ak po znížení dávky a skrátení režimu toxicita naďalej pretrváva
alebo sa opätovne objavuje, liečbu Onuregom ukončite.
|
* 1. stupeň je mierny, 2. stupeň je stredne závažný, 3. stupeň je závažný, 4. stupeň je život ohrozujúci.
Stupne toxicity sú v súlade so všeobecnými kritériami pre terminológiu nežiaducich udalostí, verzie 4.3, podľa Národného inštitútu pre rakovinu (
National Cancer Institute Common Terminology Criteria for Adverse Events, NCI-CTCAE v4.3).
Vynechané alebo oneskorené dávkyAk dôjde k vynechaniu dávky Onuregu alebo ak sa dávka neužije vo zvyčajnom čase, dávka sa má užiť čo najskôr v ten istý deň. Nasledujúca naplánovaná dávka sa má potom užiť vo zvyčajnom čase nasledujúci deň. Počas toho istého dňa sa nemajú užiť dve dávky.
Ak dôjde k vyvráteniu dávky, v ten istý deň sa nesmie užiť ďalšia dávka. Namiesto toho sa vráťte
k zvyčajnému času podania dávky nasledujúci deň.
Osobitné populácieStarší pacientiU pacientov vo veku nad 65 rokov sa neodporúčajú žiadne úpravy dávky (pozri časť 5.2).
Porucha funkcie obličiekOnureg sa môže podávať pacientom s miernou, stredne závažnou alebo závažnou poruchou funkcie
obličiek bez úpravy dávky na začiatku liečby (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s miernou poruchou funkcie pečene (celkový bilirubín (BIL) ≤ horný limit normálnej hodnoty (
upper limit of normal, ULN) a hladinou aspartátaminotransferázy (AST) > ULN alebo BIL 1
až 1,5 × ULN a akoukoľvek hladinou AST) sa neodporúča žiadna úprava dávky (pozri časť 5.2).
U pacientov so stredne závažnou (BIL > 1,5 až 3 × ULN) a závažnou poruchou funkcie pečene (BIL > 3 × ULN) sa majú častejšie sledovať nežiaduce reakcie a má sa vykonať príslušná úprava dávky (pozri tabuľku 1).
Pediatrická populácia
Bezpečnosť a účinnosť Onuregu u detí a dospievajúcich mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Onureg je na perorálne použitie.
Onureg sa môže užívať s jedlom alebo bez jedla. Tablety sa majú prehĺtať vcelku a zapíjať pohárom vody v približne rovnakom čase každý deň. Nemajú sa deliť, drviť, rozpúšťať ani žuvať (pozri
časť 6.6).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hematologická toxicita
Liečba Onuregom sa môže spájať s neutropéniou, trombocytopéniou a febrilnou neutropéniou (frekvencie pozri v časti 4.8). Na zvládnutie hematologických toxicít môže byť nevyhnutné prerušenie podávania, zníženie dávky alebo ukončenie liečby Onuregom. Pacientov je potrebné poučiť, aby
ihneď hlásili febrilné epizódy. Pacientov s nízkym počtom trombocytov je potrebné poučiť, aby hlásili včasné prejavy alebo symptómy krvácania. Na základe individuálnych charakteristík pacienta, odpovede na liečbu a v súlade s aktuálnymi klinickými usmerneniami sa má poskytnúť podporná liečba, ako sú antibiotiká a/alebo antipyretiká na liečbu infekcie/horúčky a pri neutropénii sa má podať GCSF (pozri časti 4.2 tabuľka 1).
Gastrointestinálna toxicita
Gastrointestinálne toxicity boli najčastejšími nežiaducimi reakciami u pacientov liečených Onuregom (pozri časť 4.8). Pacientom sa má podávať profylaktická antiemetická liečba počas prvých 2 cyklov liečby Onuregom (pozri časť 4.2). Hnačka sa má liečiť okamžite pri nástupe symptómov. Na zvládnutie gastrointestinálnych toxicít môže byť nevyhnutné prerušenie podávania, zníženie dávky alebo ukončenie liečby Onuregom (pozri časť 4.2).
Ženy vo fertilnom veku/Antikoncepciaumužovažien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a až do 6 mesiacov po
liečbe. Muži musia používať účinnú antikoncepciu počas liečby a až do 3 mesiacov po liečbe (pozri časť 4.6).
Intolerancia laktózy
Tablety Onuregu obsahujú laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej tablete, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
V prípade súbežného podania s inými antineoplastickými látkami sa odporúča opatrnosť a sledovanie, keďže nemožno vylúčiť antagonistický, aditívny ani synergický farmakodynamický účinok. Tieto účinky môžu závisieť od dávky, poradia alebo režimu dávkovania.
Pri súbežnom podávaní s inhibítorom protónovej pumpy (omeprazolom) bola expozícia Onuregu ovplyvnená minimálne. Pri súbežnom podávaní Onuregu s inhibítormi protónovej pumpy alebo
s inými liečivami upravujúcimi pH sa preto nevyžaduje úprava dávky.
Štúdia azacitidínu in vitro s ľudskými pečeňovými frakciami naznačila, že azacitidín nebol metabolizovaný izoformami cytochrómu P450 (CYP). Interakcie s induktormi alebo inhibítormi CYP sa preto považujú za nepravdepodobné (pozri časť 5.2).
Klinicky významné inhibičné alebo indukčné účinky azacitidínu na metabolizmus substrátov cytochrómu P450 nie sú pravdepodobné (pozri časť 5.2). Pri súbežnom podávaní Onuregu so substrátmi P-glykoproteínu (Pgp), proteínu rezistencie proti karcinómu prsníka (breast cancer resistance protein, BCRP), prenášačov organických aniónov (organic anion transporters, OAT) OAT1 a OAT3, polypeptidov prenášajúcich organické anióny (organic anion transporting polypeptides, OATP) OATP1B1 a OATP1B3 alebo prenášačov organických katiónov (organic cation transporter, OCT) OCT2 sa neočakávajú žiadne klinicky významné liekové interakcie.
Azacitidín nie je substrátom P-gp, preto sa neočakáva, že bude interagovať s induktormi alebo inhibítormi P-gp.
4.6 Fertilita, gravidita a laktácia
Ženyvo fertilnom veku/Antikoncepcia u mužovažien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a až do 6 mesiacov po
liečbe. Muži majú byť poučení, aby sa vyhli splodeniu dieťaťa počas liečby a že musia počas liečby
a ešte 3 mesiace po ukončení liečby používať účinnú antikoncepciu (pozri časti 4.4 a 5.3).
Gravidita
Nie sú k dispozícii dostatočné údaje o použití Onuregu u gravidných žien. Štúdie na myšiach
a potkanoch preukázali reprodukčnú a vývinovú toxicitu (pozri časť 5.3). Nie je známe potenciálne riziko pre ľudí. Na základe výsledkov zo štúdií na zvieratách a jeho mechanizmu účinku sa Onureg neodporúča počas gravidity (najmä počas prvého trimestra, ak to nie je jednoznačne nevyhnutné)
a u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu. Prínosy liečby sa majú v každom individuálnom prípade zvážiť vzhľadom na možné riziko pre plod. Ak pacientka alebo partnerka otehotnie počas užívania Onuregu, má byť informovaná o možnom riziku pre plod.
Dojčenie
Nie je známe, či sa azacitidín alebo jeho metabolity vylučujú do materského mlieka. Z dôvodu potenciálne závažných nežiaducich reakcií u dojčeného dieťaťa je dojčenie počas liečby Onuregom kontraindikované (pozri časť 4.3).
Fertilita
Nie sú k dispozícii žiadne údaje o vplyve azacitidínu na plodnosť ľudí. U zvierat boli dokumentované nežiaduce účinky azacitidínu na plodnosť samcov (pozri časť 5.3). Pacientom, ktorí si želajú splodiť dieťa, sa má odporučiť, aby pred začatím liečby Onuregom vyhľadali reprodukčné poradenstvo
a využili uchovanie vajíčok alebo spermií zmrazením.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Onureg má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri používaní Onuregu sa
hlásila únava. Preto sa pri vedení vozidiel alebo obsluhovaní strojov odporúča opatrnosť.
4
.
8 Nežiaduce účinky
Súh
r
n profilu bezpečnostiNajčastejšie nežiaduce reakcie sú nevoľnosť (64,8 %), vracanie (59,7 %), hnačka (50,4 %),
neutropénia (44,5 %), únava/asténia (44,1 %)5, zápcha (38,6 %), trombocytopénia (33,5 %), bolesť brucha (21,6 %)4, infekcia dýchacích ciest (17 %)2, artralgia (13,6 %), znížená chuť do jedla (12,7 %), febrilná neutropénia (11,9 %), bolesť chrbta (11,9 %), leukopénia (10,6 %), bolesť v končatine
(10,6 %) a pneumónia (10,2 %)1.
Závažné nežiaduce reakcie sa vyskytli u 16,1 % pacientov užívajúcich Onureg. Najčastejšie závažné nežiaduce reakcie sú febrilná neutropénia (6,8 %) a pneumónia (5,1 %)1.
K trvalému ukončeniu liečby Onuregom z dôvodu nežiaducej reakcie došlo u 6,8 % pacientov. Najčastejšími nežiaducimi reakciami vyžadujúcimi si trvalé ukončenie liečby sú nevoľnosť (2,1 %), hnačka (1,7 %) a vracanie (1,3 %).
K prerušeniam podávania z dôvodu nežiaducej reakcie došlo u 36,4 % pacientov užívajúcich Onureg. Nežiaduce reakcie vyžadujúce si prerušenie podávania zahŕňajú neutropéniu (19,9 %), trombocytopéniu (8,5 %), nevoľnosť (5,5 %), hnačku (4,2 %), vracanie (3,8 %), pneumóniu (3,4 %)1, leukopéniu (2,5 %), febrilnú neutropéniu (2,1 %) a bolesť brucha (2,1 %)4.
K zníženiu dávky z dôvodu obdobia výskytu nežiaducej reakcie došlo u 14 % pacientov užívajúcich
Onureg. Nežiaduce reakcie vyžadujúce si zníženie dávky zahŕňali neutropéniu (5,5 %), hnačku
(3,4 %), trombocytopéniu (1,7 %) a nevoľnosť (1,7 %).
Zoznam nežiaducich reakcií v tabuľkeV tabuľke 2 sú uvedené kategórie frekvencií nežiaducich reakcií hlásených v pivotnej štúdii
s Onuregom fázy 3. Onureg užívalo celkovo 236 pacientov. V skupine s Onuregom bol medián dĺžky
liečby 11,6 mesiacov (rozmedzie: 0,5 až 74,3 mesiacov).
Frekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000
až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(z dostupných údajov). V rámci každej skupiny frekvencie sú nežiaduce účinky uvedené v poradí klesajúcej závažnosti. Nežiaduce reakcie sú uvedené v tabuľke podľa najvyššej pozorovanej frekvencie.
Trieda orgánových systémov
| Frekvencia všetkých stupňova
| Infekcie a nákazy
| Veľmičasté
pneumónia1, 6, infekcia dýchacích ciest2
Časté
chrípka, infekcia močových ciest3, bronchitída, rinitída
| Poruchy krvi a lymfatického systému
| Veľmičasté
neutropénia, trombocytopénia6, febrilná neutropénia6, leukopénia
| Poruchy metabolizmu a výživy
| Veľmičasté
znížená chuť do jedla
| Psychické poruchy
| Časté
Úzkosť
| Poruchy gastrointestinálneho traktu
| Veľmičasté
nevoľnosť, vracanie, hnačka, zápcha, bolesť brucha4
| Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Veľmičasté
artralgia, bolesť chrbta, bolesť v končatine
|
|
|
Tabuľka 2: Nežiaduce reakcie u pacientov s AML užívajúcich udržiavaciu liečbu Onuregom
T
r
i
e
d
a orgánových systémov
|
F
rek
v
e
n
cia všetkých stupňov
a
|
C
e
l
kov
é poruchy a reakcie v mieste podania
|
V
e
ľ
mi
časté
únava/asténia5
|
L
a
b
o
r
a
t
ó
r
n
e a funkčné vyšetrenia
|
Č
a
s
t
é
pokles telesnej hmotnosti
|
a Všetky nežiaduce udalosti s minimálne 5 % pacientov v skupine s Onuregom a s frekvenciou o minimálne 2 % vyššou ako
v skupine s placebom.
1 Skupinové pojmy zahŕňajú pneumóniu, bronchopulmonálnu aspergilózu, infekciu pľúc, pneumóniu spôsobenú hubou
Pneumocystis jirovecii, atypickú pneumóniu, bakteriálnu pneumóniu a mykotickú pneumóniu.
2 Skupinové pojmy zahŕňajú infekciu horných dýchacích ciest, infekciu dýchacích ciest a vírus ovú infekciu dýchacích ciest.
3 Skupinové pojmy zahŕňajú infekciu močových ciest, bakteriálnu infekciu močových ciest, infekciu močových ciest
spôsobenú baktériou
Escherichia a cystitídu.
4 Skupinové pojmy zahŕňajú bolesť brucha, bolesť v hornej časti brucha, nepríjemný pocit v bruchu a gastrointestinálnu
bolesť.
5 Skupinové pojmy zahŕňajú únavu a asténiu.
6 Nežiaduce reakcie, z ktorých sa minimálne jedna považovala za život ohrozujúcu (ak bola výsledkom reakcie smrť, je
zahrnutá do prípadov smrti).
Opis vybraných nežiaducich reakciíHematologická toxicitaU pacientov liečených Onuregom boli často hlásenými nežiaducimi reakciami nový výskyt neutropénie alebo jej zhoršenie na 3. alebo vyšší stupeň (41,1 %), nový výskyt trombocytopénie alebo jej zhoršenie na 3. alebo vyšší stupeň (22,5 %) alebo nový výskyt febrilnej neutropénie alebo jej zhoršenie na 3. alebo vyšší stupeň (11,4 %). U pacientov užívajúcich Onureg sa prvý výskyt neutropénie 3. alebo 4. stupňa počas prvých 2 cyklov objavil u 19,9 % pacientov, prvý výskyt trombocytopénie 3. alebo 4. stupňa počas prvých 2 cyklov u 10,6 % pacientov a prvý výskyt febrilnej neutropénie 3. alebo 4. stupňa počas prvých 2 cyklov u 1,7 % pacientov. Pozri časť 4.2 ohľadom usmernení týkajúcich sa sledovania a liečby.
Gastrointestinálna toxicitaGastrointestinálne toxicity boli najčastejšími nežiaducimi reakciami u pacientov liečených Onuregom.
U pacientov liečených Onuregom sa vyskytla nevoľnosť (64,8 %), vracanie (59,7 %) a hnačka (50,4 %). U pacientov liečených Onuregom sa hnačka 3. alebo vyššieho stupňa objavila u 5,1 % pacientov, vracanie 3. alebo vyššieho stupňa u 3,0 % pacientov a nevoľnosť 3. alebo vyššieho stupňa
u 2,5 % pacientov. U pacientov užívajúcich Onureg sa prvý výskyt nevoľnosti 3. alebo 4. stupňa počas
prvých 2 cyklov objavil u 1,7 % pacientov, prvý výskyt vracania 3. alebo 4. stupňa počas prvých
2 cyklov u 3,0 % pacientov a prvý výskyt hnačky 3. alebo 4. stupňa počas prvých 2 cyklov u 1,3 %
pacientov. Pozri časť 4.2 ohľadom usmernení týkajúcich sa sledovania a liečby.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, ab y hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania sa má u pacienta náležite sledovať krvný obraz a podľa potreby sa mu má poskytnúť podporná liečba v súlade s miestnymi odporúčaniami. Nie je známe žiadne špecifické antidotum na predávkovanie Onuregom.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastické látky, antimetabolity, analógy pyrimidínu; ATC kód: L01BC07
Mechanizmus
ú
č
i
nk
u
Azacitidín je inhibítor DNA metyltransferázy a epigenetický modifikátor. Po absorpcii bunkami
a enzymatickej biostransformácii na nukleotidové trifosfáty sa azacitidín inkorporuje do DNA a RNA. Inkorporáciou azacitidínu do DNA AML buniek došlo prostredníctvom inhibície DNA
metyltransferáz a znížením metylácie DNA k modifikácii epigenetických dráh. To viedlo k zmene
v expresii génov vrátane opätovnej expresie génov regulujúcich potláčanie nádoru, imunitné dráhy, bunkový cyklus a diferenciáciu buniek. Inkorporáciou azacitidínu do RNA AML buniek dochádza
k inhibícii RNA metyltransferázy, zníženiu metylácie RNA, zníženiu stability RNA a k zníženiu
syntézy proteínov.
Klinickáúčinnosťabezpečnosť
Účinnosť a bezpečnosť Onuregu boli skúmané v multicentrickej, placebom kontrolovanej štúdii
3. fázy QUAZAR AML-001 (CC-486-AML-001) s dvojito zaslepeným, randomizovaným dizajnom s
paralelnými skupinami, ktorá hodnotila Onureg ako udržiavaciu liečbu u pacientov s AML
v porovnaní s placebom. Zaradení boli pacienti s AML de novo, s AML vyskytujúcou sa sekundárne po predchádzajúcej diagnóze myelodysplastických syndrómov (MDS) alebo chronickej myelomonocytickej leukémie (CMML); pacienti boli vo veku ≥ 55 rokov a dosiahli prvú kompletnú
remisiu (CR) alebo kompletnú remisiu s neúplnou úpravou krvného obrazu (incomplete remission,
CRi) počas 4 mesiacov (+/ 7 dní) po intenzívnej indukčnej chemoterapii s konsolidačnou liečbou alebo bez nej. Pacienti neboli v čase randomizácie vhodní na HSCT, čo zahŕňalo pacientov, ktorí nemali darcu transplantátu alebo ktorí si vybrali nepokračovať s HSCT.
V oboch liečených skupinách dostávali pacienti najlepšiu podpornú starostlivosť podľa uváženia jej nevyhnutnosti skúšajúcim. Najlepšia podporná starostlivosť okrem iného zahŕňala liečbu transfúziami erytrocytov (red blood cell, RBC), transfúziami trombocytov, použitie látky stimulujúcej erytropoézu, antibiotickú, protivírusovú a/alebo antimykotickú liečbu, GCSF, antiemetickú liečbu a podporu výživy.
Pacientom, ktorí po intenzívnej indukčnej liečbe s konsolidačnou liečbou alebo bez nej dosiahli CR/CRi, bol podaný Onureg v dávke 300 mg (N = 236) alebo placebo (N = 233) jedenkrát denne počas 1. až 14. dňa každého 28-dňového cyklu. V prípade relapsu ochorenia (5 % až 15 % blastov
v periférnej krvi alebo kostnej dreni), došlo na základe rozhodnutia lekára k predĺženiu dávkovacieho režimu na 21 dní v rámci opakujúcich sa 28-dňových liečebných cyklov. Liečba pokračovala do
progresie ochorenia (viac ako 15 % blastov pozorovaných v periférnej krvi alebo kostnej dreni) alebo do neprijateľnej toxicity.
Do skupiny liečenej Onuregom a placebom bolo v pomere 1:1 randomizovaných celkovo
472 pacientov. Základné demografické charakteristiky a charakteristiky ochorenia v populácii pacientov s AML boli medzi liečenými skupinami vyrovnané, ako je uvedené v tabuľke 3. Medián dĺžky liečby bol 11,6 mesiacov (rozmedzie: 0,5 až 74,3 mesiacov) v skupine s Onuregom oproti
5,7 mesiacom (rozmedzie: 0,7 až 68,5 mesiacov) v skupine s placebom. U celkovo 51 pacientov (21 %) užívajúcich Onureg a 40 pacientov (17 %) užívajúcich placebo došlo z dôvodu relapsu ochorenia AML k predĺženiu ich dávkovacieho režimu na 300 mg denne počas 21 dní.
Zo 469 liečených pacientov v štúdie fázy 3 bolo 61 % (285/469) vo veku 65 rokov alebo starších
a 11 % (51/469) bolo vo veku 75 rokov alebo starších. Medzi týmito pacientmi a mladšími pacientmi
sa celkovo nepozorovali žiadne rozdiely v bezpečnosti alebo účinnosti Onuregu.
P
a
r
a
meter
|
O
nu
re
g
(
N = 238)
|
P
l
a
ce
b
o
(
N = 234)
|
V
e
k (roky)
|
|
|
Medián (min., max.)
|
68,0 (55; 86)
|
68,0 (55; 82)
|
Veková kategória, n (%)
|
|
|
< 65 rokov
|
66 (27,7)
|
68 (29,1)
|
≥ 65 rokov až < 75 rokov
|
144 (60,5)
|
142 (60,7)
|
≥ 75 rokov
|
28 (11,8)
|
24 (10,3)
|
P
oh
l
av
i
e
, n (%)
|
|
|
Muži
|
118 (49,6)
|
127 (54,3)
|
Ženy
|
120 (50,4)
|
107 (45,7)
|
R
a
s
a
, n (%)
|
|
|
Belosi
|
216 (90,8)
|
197 (84,2)
|
Černosi alebo Afroameričania
|
2 (0,8)
|
6 (2,6)
|
Aziati
|
6 (2,5)
|
20 (8,5)
|
Iná
|
12 (5,0)
|
11 (4,7)
|
Nezískané alebo nenahlásené
|
2 (0,8)
|
0 (0)
|
S
t
a
v výkonnosti podľa ECOG,
n (%)
|
|
|
0
|
116 (48,7)
|
111 (47,4)
|
1
|
101 (42,4)
|
106 (45,3)
|
2
|
21 (8,8)
|
15 (6,4)
|
3
|
0 (0)
|
2 (0,9)
|
S
t
a
v cytogenetického rizika pri diagnóze, n (%)
|
|
|
Stredné riziko1
|
203 (85,3)
|
203 (86,6)
|
Nízke riziko2
|
35 (14,7)
|
31 (13,2)
|
Ú
v
o
d
n
á klasifikácia AML, n (%)
|
|
|
AML s opakujúcimi sa genetickými
poruchami
|
39 (16,4)
|
46 (19,7)
|
AML so zmenami súvisiacimi
s myelodyspláziou
|
49 (20,6)
|
42 (17,9)
|
Myeloidné novotvary súvisiace s liečbou
|
2 (0,8)
|
0 (0)
|
AML bez inej špecifikácie
|
148 (62,2)
|
145 (62,0)
|
Chýbajúci údaj
|
0 (0)
|
1 (0,4)
|
T
y
p AML, n (%)
|
|
|
Primárna (de novo)
|
213 (89,5)
|
216 (92,3)
|
Sekundárna
|
25 (10,5)
|
18 (7,7)
|
S
t
a
v MRD pri randomizácii
3
,
n (%)
|
|
|
Negatívny
|
133 (55,9)
|
111 (47,4)
|
Pozitívny
|
103 (43,3)
|
116 (49,6)
|
Chýbajúci údaj
|
2 (0,8)
|
7 (3,0)
|
|
|
T
a
b
u
ľ
k
a 3: Východiskové demografické charakteristiky a charakteristiky týkajúce sa ochorenia v štúdii CC-486-AML-001
AML = akútna myelogénna leukémia, MDS = myelodysplastický syndróm, CMML = chronická myelomonocytická
leukémia, ECOG = Východná spolupracujúca onkologická skupina (
Eastern cooperative oncology group), CR =
morfologická CR, CRi = morfologická kompletná remisia s neúplnou úpravou krvného obrazu
1 Stredné riziko bolo definované ako normálna cytogenetika +8, t(9; 11), inak nedefinované.
2 Nízke riziko bolo definované ako komplex (≥ 3 porúch): -5; 5q-; -7; 7q-; 11q23 - non t(9; 11); inv(3); t(3; 3); t(6;
9); alebo t(9; 22). Zdroj pre stredné a nízke riziko: Usmernenia Národnej komplexnej onkologickej siete pre klinickú prax v onkológii pri AML.
3Stav MRD v kostnej dreni bol meraný počas skríningového obdobia pomocou prietokovej cytometrie s citlivosťou
0,1 %.
Väčšina pacientov v skupine liečenej Onuregom (78 %) a aj v skupine liečenej placebom (82 %) po indukčnej liečbe užívala konsolidačnú liečbu; viac ako 90 % z týchto pacientov v každej liečenej skupine užilo po indukčnej liečbe 1 alebo 2 cykly konsolidačnej liečby (tabuľka 4).
Tabuľka 4: Konsolidačná liečba v štúdii CC-486-AML-001Parameter
| Onureg
(N = 238)
| Placebo
(N = 234)
|
Užívanie konsolidačnej liečby po indukčnej liečbe
|
|
|
Áno, n (%)
| 186 (78,2)
| 192 (82,1)
|
1 cyklus, n (%)
| 110 (46,2)
| 102 (43,6)
|
2 cykly, n (%)
| 70 (29,4)
| 77 (32,9)
|
3 cykly, n (%)
| 6 (2,5)
| 13 (5,6)
|
Nie, n (%)
| 52 (21,8)'
| 42 (17,9)
|
Stav CR / CRi pri randomizácii
|
|
|
CR, n (%)
| 183 (76,9)
| 177 (75,6)
|
CRi, n (%)
| 50 (21,0)
| 44 (18,8)
|
Bez CR/CRi a, n (%)
| 5 (2,1)
| 11 (4,7)
|
Chýbajúci údaj, n (%)
| 0 (0)
| 2 (0,9)
|
CR = kompletná remisia; CRi = morfologická CR s neúplnou úpravou krvného obrazu.
a Títo pacienti mali na začiatku menej ako 5 % blastov v kostnej dreni a počet ANC < 1 x 109 ako aj počet
trombocytov < 100 x 109.
Účinnosť Onuregu u dospelých pacientov s AML bola stanovená na základe celkového prežívania
(
overall survival, OS) a prežívania bez relapsu (
relapse-free survival, RFS).
Výsledky účinnosti sú zhrnuté v tabuľke 5.
Tabuľka 5: Výsledky účinnosti v štúdii CC-486-AML-001 (populácia ITT)Koncové ukazovatele
| Onureg
(N = 238)
| Placebo
(N = 234)
|
Celkové prežívanie
|
|
|
Udalosti OS, n (%)
| 158 (66,4)
| 171 (73,1)
|
Medián OS, v mesiacoch (95 % IS)
| 24,7 (18,7; 30,5)
| 14,8 (11,7; 17,6)
|
Pomer rizika (95 % IS) Hodnota p
| 0,69 (0,55; 0,86)
0,0009
|
Prežívania bez relapsu
|
|
|
Udalosti, n (%)
| 164 (68,9)
| 181 (77,4)
|
Medián RFS, v mesiacoch (95 % IS)
| 10,2 (7,9; 12,9)
| 4,8 (4,6; 6,4)
|
Pomer rizika (95 % IS)
hodnota p
| 0,65 (0,52; 0,81)
0,0001
|
Čas do relapsu
|
|
|
V relapse, n (%)
| 154 (64,7)
| 179 (76,5)
|
Medián času do relapsu
v mesiacoch, (95 % IS)
| 10,2 (8,3; 13,4)
| 4,9 (4,6; 6,4)
|
Čas do ukončenia liečby
|
|
|
Ukončenie liečby, n (%)
| 193 (81,1)
| 208 (88,9)
|
Medián času do ukončenia liečby,
v mesiacoch
(95 % IS)
| 11,4 (9,8; 13,6)
| 6,1 (5,1; 7,4)
|
Ukončenie liečby – relaps ochorenia, n (%)
| 143 (60,1)
| 180 (76,9)
|
IS = interval spoľahlivosti
V analýzach OS a RFS vykonaných vo vopred špecifikovanej podskupine sa preukázal konzistentný účinok liečby pri Onuregu naprieč demografickými podskupinami a podskupinami súvisiacimi
s ochorením vrátane východiskového cytogenetického rizika, počtu absolvovaných predchádzajúcich
cyklov konsolidačnej liečby a stavu CR/CRi.
Výsledky OS (pozri obrázok 1) a RFS (pozri obrázok 2) znázorňujú Kaplanove-Meierove krivky.
Obrázok 1: Kaplanova-Meierova krivka celkového prežívania: Onureg oproti placebu
(populácia ITT)
1,0
0,9
0,8
0,7
0,6
0,5
0,4
0,3
0,2
0,1
0,0
Medián OS: 14,8
Medián OS: 24,7
Stratifikovaný HR: 0,69 (95 % CI: 0,55-0,86)
Stratifikovaná log-rank hodnota p: 0,0009
Č
a
s (mesiace) od randomizácie
Placebo
Počet rizikových pacientov
Placebo
Obrázok 2: Kaplanova-Meierova krivka prežívania bez relapsu: Onureg oproti placebu
(populácia ITT)
1,0
0,9
0,8

0,7
0,6
0,5
0,4
0,3
0,2
0,1
0,0
Medián RFS: 4,8
Medián RFS: 10,2
Stratifikovaný HR: 0,65 (95 % CI: 0,52-0,81) Stratifikovaná log-rank hodnota p: 0,0001
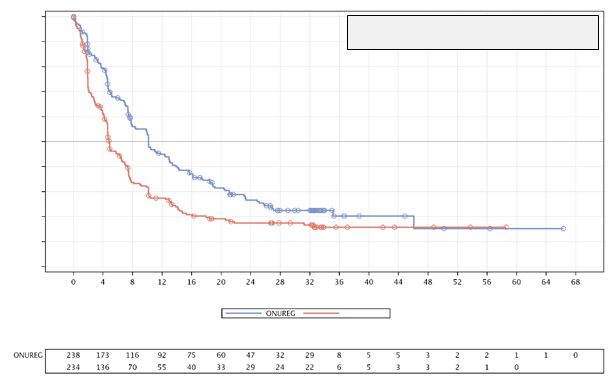
 Č
a
s (mesiace) od randomizácie
Č
a
s (mesiace) od randomizácie
Placebo
Počet rizikových pacientovPlacebo
U pacientov s dávkovacím režimom predĺženým na 300 mg počas 21 dní z dôvodu relapsu ochorenia bol medián OS (22,8 mesiacov pri Onuregu a 14,6 mesiacov pri placebe) a medián RFS (7,4 mesiacov pri Onuregu a 4,6 mesiacov pri placebe) porovnateľný s celkovými výsledkami štúdie.
Onureg preukázal priaznivý účinok liečby na OS v porovnaní s placebom u pacientov, ktorí dosiahli minimálne reziduálne ochorenie (minimal residual disease, MRD) a aj u pacientov, ktorí nedosiahli MRD. Účinok liečby na OS bol výraznejší u pacientov, ktorí dosiahli MRD(HR = 0,69; 95 % IS: 0,51;
0,93) ako u pacientov, ktorí nedosiahli MRD (HR = 0,81; 95 % IS: 0,59; 1,12).
Kvalita života súvisiaca so zdravím (Health related quality of life, HRQoL)
HRQoL bola hodnotená pomocou škály únavy na funkčné zhodnotenie liečby chronického ochorenia (Functional assessment of chronic illness therapy-fatigue scale, škála únavy-FACIT) a indexu utility zdravia (Five Dimensions Three Levels, EQ-5D-3L) a vizuálnej analógovej stupnice (visual analogue scale, VAS). Na začiatku mali pacienti nízku úroveň únavy a dobrú úroveň HRQoL, ktoré boli vo všeobecnosti porovnateľné s úrovňami v celkovej populácii podobného veku. V porovnaní
s východiskovým stavom ako aj s placebom sa táto úroveň HRQoL v priebehu času pri Onuregu udržiavala. Zistilo sa, že čas do definitívneho zhoršenia a aj podiel pacientov pociťujúcich klinicky významné zhoršenie je medzi pacientmi užívajúcimi Onureg a placebo podobný. Celkovo zistenia preukazujú, že HRQoL bola medzi skupinou liečenou Onuregom a skupinou liečenou placebom podobná, bez klinicky významného zhoršenia v priebehu času.
5.2 Farmakokinetické vlastnosti
Absorpcia
Expozícia bola vo všeobecnosti lineárna so zvýšeniami systémovej expozície priamo úmernými dávke;
pozorovala sa vysoká interindividuálna variabilita. Geometrický priemer (koeficient zmeny [%CV])
hodnoty Cmax po perorálnom podaní 300 mg jednorazovej dávky bol 145,1 ng/ml (63,7) a hodnota
AUC 241,6 ng h/ml (64,5). Opakované podávanie dávky v odporúčanom dávkovacom režime neviedlo k akumulácii liečiva. Absorpcia azacitidínu bola rýchla s mediánom Tmax 1 hodina po podaní dávky.
Priemerná biologická dostupnosť po perorálnom užití bola v porovnaní so subkutánnym (s.c.)
podaním približne 11 %.
Vplyv potravy
Vplyv potravy na expozíciu Onuregu bol minimálny. Onureg sa preto môže podávať s jedlom alebo bez jedla.
Distribúcia
Po perorálnom podaní osobe s telesnou hmotnosťou 70 kg bol geometrický priemer zdanlivého
distribučného objemu 12,6 l/kg. Väzba azacitidínu na plazmatické bielkoviny bola 6 až 12 %.
Biotransformácia
Na základe údajov in vitro sa nezdá, že je metabolizmus azacitidínu sprostredkovaný izoenzýmami cytochrómu P450 (CYP). Azacitidín podlieha spontánnej hydrolýze a deaminácii prostredníctvom cytidíndeaminázy.
Eliminácia
Geometrický priemer zdanlivého klírensu bol 1 242 l/h a geometrický priemer biologického polčasu bol približne 0,5 hodín. Po intravenóznom podaní azacitidínu značeného 14C 5 pacientom s nádorovým ochorením sa kumulatívne močom vylúčilo 85 % rádioaktívne značenej dávky. Stolicou sa vylúčilo
< 1 % podanej rádioaktívne značenej dávky počas 3 dní. V priemere 50 % rádioaktívne značenej dávky sa po subkutánnom podaní 14C-azacitidínu vylúčilo močom. Množstvo nezmeneného azacitidínu vylúčeného močom v porovnaní s dávkou bolo < 2 % buď po subkutánnom (s.c.) alebo perorálnom podaní. Vylučovanie stolicou po perorálnom podaní sa nestanovovalo.
F
a
r
m
a
kod
y
n
a
m
i
c
k
é
ú
č
i
n
k
y
Epigenetický regulačný účinok azacitidínu na zníženie celkovej metylácie DNA v krvi sa udržiaval pri predĺženej expozícii 300 mg denne podávanej počas 14 alebo 21 dní v rámci 28-dňového cyklu pri myeloidných nádorových ochoreniach vrátane pacientov s AML zo štúdie 1./2. fázy. Medzi plazmatickou expozíciou azacitidínu a farmakodynamickým účinkom zníženia celkovej metylácie DNA v krvi sa pozorovala pozitívna korelácia.
Osobitné populácie
Starší pacienti
V analýze populačnej farmakokinetiky (PK) 286 pacientov s AML nemal vek (46 až 93 rokov) klinicky významné účinky na PK Onuregu. Bez ohľadu na vek sa preto úprava dávky Onuregu nevyžaduje.
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene sa neuskutočnili žiadne formálne štúdie. Nie je
pravdepodobné, že porucha funkcie pečene ovplyvňuje PK v klinicky významnom rozsahu, keďže
azacitidín podlieha spontánnej hydrolýze a deaminácii prostredníctvom cytidíndeaminázy. V analýze populačnej PK sa stanovilo, že AST (8 až 155 U/l), ALT (5 až 185 U/l) a mierna porucha funkcie pečene (BIL ≤ ULN a AST > ULN alebo BIL 1 až 1,5 × ULN a akákoľvek hladina AST) nemá žiadne klinicky významné účinky na PK azacitidínu. Účinky stredne závažnej až ťažkej poruchy funkcie pečene (BIL > 1,5 × ULN a akákoľvek hladina AST) na PK azacitidínu nie sú známe.
Porucha funkcie obličiek
U pacientov s nádorovým ochorením sa porovnávala PK azacitidínu po subkutánnom podávaní dávky
75 mg/m2/deň (1. až 5. deň) u 6 pacientov s normálnou funkciou obličiek (CLcr > 80 ml/min) a u 6
pacientov so závažnou poruchou obličiek (CLcr < 30 ml/min). Závažná porucha funkcie obličiek zvýšila expozíciu azacitidínu o približne 70 % po jednorazovom podaní a o 41 % po opakovaných
subkutánnych podaniach. Toto zvýšenie expozície nekorelovalo so zvýšením miery výskytu
nežiaducich udalostí.
V analýze populačnej PK po podaní Onuregu v dávke 300 mg sa stanovilo, že pacienti s miernou
poruchou funkcie obličiek (CLcr: ≥ 60 až < 90 ml/min) mali plazmatickú AUC azacitidínu zvýšenú
o 19 %, pacienti so stredne závažnou poruchou funkcie obličiek (CLcr: ≥ 30 až < 60 ml/min) o 25 %
a pacienti so závažnou poruchou funkcie obličiek (CLcr: < 30 ml/min) o 38 %. Vplyv ťažkej poruchy
funkcie obličiek na Onureg bol podobný ako vo vyššie uvedenej klinickej štúdii s poruchou funkcie obličiek s injekčne podávaným azacitidínom (~40 % zvýšenie AUC). Expozícia azacitidínu (AUC) je približne o 75 % nižšia po perorálnom podaní v porovnaní s expozíciou dosiahnutou po s.c. podaní; zvýšenie expozície o približne 40 % po perorálnom podaní sa preto stále považuje za bezpečné
a znesiteľné. Preto sa u pacientov s miernou, stredne závažnou alebo závažnou poruchou funkcie
obličiek neodporúča žiadna úprava dávky Onuregu.
Rasa/etnický pôvod
Vplyvy rasy/etnického pôvodu na PK Onuregu nie sú známe.
5.3 Predklinické údaje o bezpečnosti
V 14-dňovej štúdii perorálnej toxicity na psoch sa mortalita objavila pri dávkach 8 a 16 mg/m2/deň. Maximálna tolerovaná dávka (maximum tolerated dose, MTD) bola 4 mg/m2/deň. Pri 1 alebo všetkých dávkach sa pozorovala pancytopénia, ktorá korelovala s hypopláziou kostnej drene, lymfoidná deplécia, dilatácia žľazy/lúmenu a jednobunková nekróza v kryptách sliznice tenkého a hrubého čreva a/alebo centrilobulárna hepatocelulárna vakuolizácia. Pri MTD sa tieto nálezy čiastočne alebo úplne
upravili po 3 týždňoch. Po parenterálnych podaniach azacitidínu v porovnateľných rozmedziach dávky sa mortalita a toxicita v podobných cieľových orgánoch pozorovala u hlodavcov, psov a opíc. Predklinické údaje zo štúdií toxicity opakových dávok s azacitidínom neodhalili žiadne osobitné riziko pre ľudí.
Azacitidín indukuje génové mutácie aj chromozómové aberácie v bunkových systémoch baktérií a cicavcov in vitro. Potenciálna karcinogenita azacitidínu sa hodnotila na myšiach a potkanoch. Azacitidín vyvolával vznik nádorov hematopoetického systému u samíc myší pri intraperitoneálnom podávaní 3-krát týždenne počas 52 týždňov. Zvýšený výskyt nádorov v lymforetikulárnom systéme, pľúcach, mliečnych žľazách a koži sa pozoroval u myší liečených azacitidínom podávaným intraperitoneálne počas 50 týždňov. Štúdia tumorogenicity na potkanoch odhalila zvýšený výskyt testikulárnych nádorov.
Štúdie zamerané na skorú embryotoxicitu u myší odhalili 44 % výskyt vnútromaternicových embryonálnych úmrtí (zvýšená resorpcia) po podaní jednej intraperitoneálnej injekcie azacitidínu počas organogenézy. U myší, ktorým sa podával azacitidín v čase uzatvorenia tvrdého podnebia alebo pred ním, sa zistili vývojové poruchy mozgu. U potkanov azacitidín nespôsobil žiadne nežiaduce reakcie pri podávaní pred uhniezdením oplodneného vajíčka, bol však jednoznačne embryotoxický pri podávaní počas organogenézy. Poškodenia plodu počas organogenézy u potkanov zahŕňali: anomálie centrálneho nervového systému (CNS) (exencefália/encefalokéla), anomálie končatín (mikromélia, vbočená noha, syndaktýlia, oligodaktýlia) a iné (mikroftalmia, mikrognatia, gastroschíza, edém a poruchy rebier).
Podávanie azacitidínu samcom myší pred párením s neliečenými samicami myší viedlo k zníženiu plodnosti a strate potomstva počas následného embryonálneho a postnatálneho vývinu. Liečba samcov potkanov viedla k zníženiu hmotnosti semenníkov a nadsemenníkov, zníženiu počtu spermií, zníženiu frekvencie gravidity, zvýšeniu počtu poškodených embryí a k zvýšeným stratám embryí
u oplodnených samíc (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Sodná soľ kroskarmelózy (E468)
Stearát horečnatý (E572) Manitol (E421)
Silicifikovaná mikrokryštalická celulóza (E460, E551)
Obal Onuregu 200 mg tablety Ružová Opadry II obsahuje: Hypromelózu (E464)
Oxid titaničitý (E171) Laktóza, monohydrát Polyetylénglykol/makrogoly (E1521)
Triacetín (E1518)
Červený oxid železitý (E172)
Obal Onuregu 300 mg tablety Hnedá Opadry II obsahuje: Hypromelózu (E464)
Oxid tataničitý (E171) Laktóza, monohydrát Polyetylénglykol/makrogoly (E1521)
Triacetín (E1518)
Červený oxid železitý (E172) Žltý oxid železitý (E172)
Čierny oxid železitý (E172)
6
.
2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Filmom obalené tablety sú balené do nylonových (OPA)/polyvinylchloridových (PVC) hliníkových blistrov s pretláčacou hliníkovou fóliou.
Veľkosť balenia so 7 alebo 14 filmom obalenými tabletami.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Onureg je cytotoxický liek. Ak sa prášok z filmom obalených tabliet dostane do kontaktu s kožou, koža sa má ihneď dôkladne umyť mydlom a vodou. Ak sa prášok dostane do kontaktu so sliznicami, miesto sa má dôkladne opláchnuť vodou.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Bristol-Myers Squibb Pharma EEIG Plaza 254
Blanchardstown Corporate Park 2
Dublin 15, D15 T867
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
Onureg 200 mg filmom obalené tablety
EU/1/21/1556/001
EU/1/21/1556/002
Onureg 300 mg filmom obalené tablety
EU/1/21/1556/003
EU/1/21/1556/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
10
. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.