
r />s neliečenou latentnou TBC v minulosti sa má zvážiť protituberkulózna liečba.
H
ematologické abnormality
U menej ako 1 % pacientov v klinických skúšaniach bol hlásený absolútny počet neutrofilov (ANC)
< 1 x 109 buniek/l, absolútny počet lymfocytov (ALC) < 0,5 x 109 buniek/l a hladina hemoglobínu
< 8 g/dl. U pacientov, u ktorých boli počas štandardnej liečby pozorované hodnoty
ANC < 1 x 109 buniek/l, ALC < 0,5 x 109 buniek/l alebo hladina hemoglobínu < 8 g/dl, sa nemá začať
s liečbou, alebo sa má liečba dočasne prerušiť (pozri časť 4.2).
U starších pacientov s reumatoidnou artritídou riziko lymfocytózy stúpa. Hlásené boli zriedkavé prípady lymfoproliferatívnych porúch.
Reaktivácia vírusu
V klinických skúšaniach bola hlásená reaktivácia vírusu, vrátane prípadov reaktivácie herpetického
vírusu (napr. herpes zoster, herpes simplex) (pozri časť 4.8). Herpes zoster bol hlásený častejšie
u pacientov ≥ 65 rokov, ktorí už boli liečení oboma biologickými aj konvenčnými DMARD. Ak sa
u pacienta objaví herpes zoster, liečba Olumiantom sa má dočasne prerušiť až do vyliečenia epizódy.
Skríning na vírusovú hepatitídu sa má uskutočniť v súlade s klinickými smernicami pred začiatkom liečby Olumiantom. Pacienti s preukázanou infekciou aktívnej hepatitídy B alebo C boli z klinických skúšaní vylúčení. Pacienti s pozitívnym výsledkom na protilátky hepatitídy C, ale negatívnym výsledkom na vírus RNA hepatitídy C, mohli byť zaradení do klinického skúšania. Do klinického skúšania mohli byť zaradení aj pacienti s povrchovými protilátkami hepatitídy B a jadrovými protilátkami hepatitídy B, bez povrchového antigénu hepatitídy B; títo pacienti majú byť sledovaní na expresiu DNA vírusu hepatitídy B (HBV). Ak sa zistí HBV DNA, je potrebné poradiť sa
s hepatológom, aby sa zistilo, či je prerušenie liečby odôvodnené.
Očkovanie
U pacientov užívajúcich baricitinib nie sú k dispozícii žiadne údaje o odpovedi na očkovanie živými
alebo inaktivovanými vakcínami. Neodporúča sa očkovanie živými, oslabenými vakcínami v priebehu alebo bezprostredne pred liečbou Olumiantom. Ak sa pred liečbou Olumiantom zvažuje očkovanie proti varicella zoster, majú sa dodržiavať Medzinárodné liečebné pokyny na očkovanie pacientov
s reumatoidnou artritídou.
Lipidy
U pacientov liečených baricitinibom bolo v porovnaní s pacientmi s placebom hlásené od dávky
závislé zvýšenie tukových parametrov v krvi (pozri časť 4.8). Zvýšený LDL cholesterol klesol po
liečbe statínmi na úroveň pred liečbou. Lipidové parametre sa majú hodnotiť približne 12 týždňov po
začiatku liečby Olumiantom, a následne sa pacienti majú liečiť v súlade s medzinárodnými klinickými smernicami pre hyperlipidémiu. Nebol zistený vplyv zvýšenia lipidových parametrov na kardiovaskulárnu morbiditu a mortalitu.
Zvýšenie hodnoty pečeňových transamináz
U menej ako 1 % pacientov v klinických skúšaniach bolo hlásené zvýšenie hodnôt
alanínaminotransferázy (ALT) a aspartátaminotransferázy (AST) na ≥ 5 a ≥ 10-násobok hornej hranice normy (ULN). U dovtedy neliečených pacientov mala kombinácia s metotrexátom v porovnaní
s liečbou baricitinibom v monoterapii za následok zvýšenie frekvencie výskytu zvýšených hodnôt
pečeňových transamináz (pozri časť 4.8). Ak sa počas štandardnej liečby pacienta pozoruje zvýšenie ALT alebo AST a existuje podozrenie na liekom spôsobené poškodenie pečene, liečba Olumiantom sa má dočasne prerušiť, kým sa táto diagnóza nevylúči.
Z
hubné nádory
U pacientov s reumatoidnou atritídou sa zvyšuje riziko výskytu zhubných nádorov, vrátane lymfómov.
Imunomodulačné lieky môžu zvyšovať riziko vzniku zhubných nádorov, vrátane lymfómov. Na vyhodnotenie možného výskytu zhubných nádorov po expozícii baricitinibu nemáme dostatok klinických údajov. Vyhodnocovanie dlhodobej bezpečnosti ešte prebieha.
Laboratórne monitorovanie
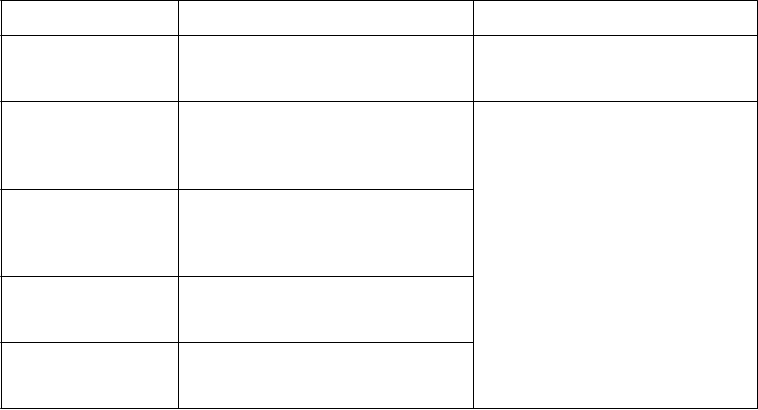
Tabuľka č. 1. Pokyny na laboratórne meranie a monitorovanie
Laboratórne meranie Postup Pokyny na monitorovanie
Lipidové parametre
Absolútny počet
neutrofilov (ANC)
Absolútny počet
lymfocytov (ALC) Hemoglobín (Hb)
Pečeňové transaminázy
Pacienti sa majú liečiť v súlade s medzinárodnými klinickými smernicami pre hyperlipidémiu.
Ak je ANC < 1 x 109 buniek/l, liečba sa
má prerušiť a môže sa začať znova vtedy, ak sa ANC vráti na hodnotu vyššiu ako je uvedené.
Ak je ALC < 0,5 x 109 buniek/l, liečba sa
má prerušiť a môže sa začať znova
vtedy, ak sa ALC vráti na hodnotu vyššiu ako je uvedené.
Ak je Hb < 8 g/dl, liečba sa má prerušiť a môže sa začať znova vtedy, ak sa Hb
vráti na hodnotu vyššiu ako je uvedené. Ak je podozrenie na poškodenie pečene vyvolané liekmi, liečba sa má dočasne prerušiť.
12 týždňov od začiatku liečby a potom v súlade s medzinárodnými klinickými smernicami pre hyperlipidémiu.
Pred začiatkom liečby a potom
v súlade so štandardnou liečbou
pacienta.
 I
m
unosupresívne lieky
I
m
unosupresívne lieky
Kombinácia s biologickými DMARD ani s inými inhibítormi Janus kinázy (JAK) sa neodporúča,
pretože nemožno vylúčiť riziko ďalšej imunosupresie. Údaje týkajúce sa užívania baricitinibu so silnými imunosupresívami (napr. azatioprinom, takrolimom, cyklosporínom) sú obmedzené a pri užívaní týchto kombinácií sa vyžaduje opatrnosť (pozri časť 4.5).
4.5 Liekové a iné interakcieFarmakodynamické interakcieImunosupresívne lieky:Kombinácia s biologickými DMARD ani inými JAK inhibítormi nebola skúmaná. Užívanie baricitinibu so silnými imunosupresívnymi liekmi ako napríklad azatioprinom, takrolimom alebo
cyklosporínom bolo v klinických skúšaniach s baricitinibom obmedzené a nemožno vylúčiť riziko
ďalšej imunosupresie (pozri časť 4.4).
Možnosti vplyvu iných liekov na farmakokinetiku baricitinibuTransportéryIn vitro je baricitinib substrátom pre organický aniónový transportér (OAT)3, P-glykoproteín (Pgp),
proteín rezistencie voči rakovine prsníka (breast cancer resistance protein - BCRP) a proteín viacliekovej a toxickej extrúzie (multidrug and toxic extrusion protein - (MATE)2-K).
Vo farmakologickom klinickom skúšaní malo dávkovanie probenecidu (inhibítor OAT3 so silným inhibičným potenciálom) za následok približne 2-násobný nárast AUC(0-∞) bezo zmeny tmax alebo Cmax baricitinibu. Dávka 2 mg raz denne sa preto odporúča pacientom užívajúcim inhibítory OAT3 so
silným inhibičným potenciálom, ako je napr. probenecid, 2 mg raz denne (pozri časť 4.2). Neuskutočnilo sa žiadne farmakologické klinické skúšanie s inhibítormi OAT3 so slabším inhibičným potenciálom. Prekurzor leflunomid sa rýchlo mení na teriflunomid, ktorý je slabým inhibítorom OAT3, a preto môže viesť k zvýšeniu expozície baricitinibu. Keďže sa neuskutočnili cielené interakčné štúdie, je pri súbežnom podávaní leflunomidu a teriflunomidu s baricitinibom potrebná opatrnosť. Súbežné užívanie OAT3 inhibítorov ibuprofenu a diklofenaku môže viesť ku zvýšenej expozícii baricitinibu, hoci inhibičný potenciál OAT3 je nižší v porovnaní s probenecidom, a preto sa neočakáva klinicky významná interakcia. Súčasné podávanie baricitinibu s cyklosporínom (inhibítorom Pgp/BCRP) alebo metotrexátom (substrátom niekoľkých transportérov vrátane OATP1B1, OAT1, OAT3, BCRP, MRP2, MRP3 a MRP4) nemalo za následok žiadny klinicky významný účinok na expozíciu baricitinibu.
Enzýmy cytochrómu P450
In vitro je baricitinib substrát enzýmu cytochrómu P450 (CYP)3A4, hoci menej ako 10 % dávky sa metabolizuje oxidáciou. Vo farmakologických klinických skúšaniach nemalo súbežné užívanie baricitinibu s ketokonazolom (silným inhibítorom CYP3A) za následok žiadny klinicky významný účinok na farmakokinetiku baricitinibu. Súbežné užívanie baricitinibu s flukonazolom (stredne silným inhibítorom CYP3A/CYP2C19/CYP2C9) alebo s rifampicínom (silným induktorom CYP3A) nemalo za následok žiadne klinicky významné zmeny na expozíciu baricitinibu.
Lieky upravujúce žalúdočné pH
Zvýšenie žalúdočného pH pri užívaní omeprazolu nemalo žiadny klinický vplyv na expozíciu baricitinibu.
Možnosti vplyvu baricitinibu na farmakokinetiku iných liekov
Transportéry
In vitro baricitinib inhiboval OAT1, OAT3, organický katiónový transportér (OCT) 1, OCT2, OATP1B3, BCRP a MATE1 a MATE2-K. Klinicky významné zmeny vo farmakokinetike liekov, ktoré sú substrátmi týchto transportérov, okrem substrátov OCT1, nie sú pravdepodobné. Nemožno vylúčiť, že inhibícia OCT1 baricitinibom je klinicky relevantná, ale v súčasnosti nie sú známe žiadne substráty selektívne pre OCT1, pre ktoré by sa dali predpovedať klinicky významné interakcie.
Vo farmakologických klinických skúšaniach sa nevyskytli žiadne klinicky významné účinky na vystavenie pôsobeniu týmto liekom pri súčasnom podávaní baricitinibu s digoxínom (substrátom Pgp) ani s metotrexátom (substrátom niekoľkých transportérov).
Enzýmy cytochrómu P450
Vo farmakologických klinických skúšaniach nemalo súčasné podávanie baricitinibu so substrátmi CYP3A simvastatínom, etinylestradiolom alebo levonorgestrelom za následok žiadne klinicky významné zmeny vo farmakokinetike týchto liečiv.
4.6 Fertilita, gravidita a laktácia
Gravidita
Bolo dokázané, že dráha JAK/STAT sa podieľa na bunkovej adhézii a bunkovej polarite, ktorá môže
mať vplyv na skorý embryonálny vývin. Nie sú k dispozícii dostatočné údaje o užívaní baricitinibu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Baricitinib
bol teratogénny u potkanov a králikov. Štúdie na zvieratách naznačujú, že baricitinib môže mať vo
vyšších dávkach nežiaduci vplyv na vývin kostí in utero.
Olumiant je počas gravidity kontraindikovaný (pozri časť 4.3). Ženy vo fertilnom veku musia počas liečby a najmenej 1 týždeň po liečbe používať účinnú antikoncepciu. Ak pacientka otehotnie
v priebehu užívania Olumiantu, rodičia majú byť informovaní o možnom riziku pre plod.
D
ojčenie
Nie je známe, či sa baricitinib/ jeho metabolity vylučujú do ľudského materského mlieka. Dostupné
farmakodynamické/toxikologické údaje preukázali vylučovanie baricitinibu do mlieka u zvierat (pozri
časť 5.3).
Nedá sa vylúčiť riziko pre novorodencov/dojčatá a Olumiant sa počas dojčenia nemá užívať. Je potrebné sa rozhodnúť, či ukončiť dojčenie alebo ukončiť liečbu Olumiantom, pričom je potrebné vziať do úvahy prínos dojčenia pre dieťa a prínos liečby pre ženu.
Fertilita
Štúdie na zvieratách naznačujú, že liečba baricitinibom môže počas liečby potenciálne znižovať
ženskú fertilitu, ale nemá žiadny účinok na mužskú spermatogenézu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Olumiant nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšími nežiaducimi liekovými reakciami (ADR) vyskytujúcimi sa u ≥ 2 % pacientov liečených
Olumiantom v monoterapii alebo v kombinácii s bežnými syntetickými DMARD bol zvýšený LDL
cholesterol (33,6 %), infekcie horných dýchacích ciest (14,7 %) a nevoľnosť (2,8 %). Infekcie hlásené
pri liečbe Olumiantom zahŕňali herpes zoster.
Prehľad nežiaducichreakciívtabuľke
V klinických skúšaniach zameraných na reumatoidnú artritídu bolo Olumiantom liečených celkovo
3 464 pacientov, čo predstavuje 4 214 pacientorokov expozície. Z nich bolo 2 166 pacientov s reumatoidnou artritídou najmenej rok vystavených účinkom Olumiantu. Na vyhodnotenie
bezpečnosti Olumiantu v porovnaní s placebom počas 16 týždňov od začiatku liečby bolo
integrovaných šesť placebom kontrolovaných klinických skúšaní (997 pacientov s dávkou 4 mg raz denne a 1 070 pacientov s placebom).
Tabuľka č. 2. Nežiaduce reakcie
Frekvencia výskytu: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až 1/100).
Trieda orgánových
systémov Veľmi časté Časté Menej časté
Infekcie a nákazy infekcie horných
dýchacích ciesta
Poruchy krvi
a lymfatického systému
herpes zoster,
herpes simplexb
gastroenteritída
infekcie močových ciest
trombocytóza
>600 x 109 buniek/lc
neutropénia
<1 x 109 buniek/lc

Poruchy metabolizmu a výživy
Poruchy gastrointestinálneho traktu
hypercholesterolémiac hypertriglyceridémiac
nevoľnosť
T
rieda orgánových
systémov
V
eľmi časté Časté Menej časté
Poruchy pečene
a žlčových ciest
Poruchy kože a podkožného tkaniva Laboratórne
a funkčné vyšetrenia
zvýšené ALT
≥ 3 x ULNc
zvýšené AST
≥ 3 x ULNc
akné
zvýšenie hmotnosti zvýšená kreatínfosfokináza
>5 x ULNc

a Pojem, ktorý zahŕňa (akútnu sinusitídu, epiglotitídu, laryngitídu, nazofaryngitídu, orofaryngeálnu
bolesť, faryngitídu, faryngotonzilitídu, rinitídu, sinusitídu, tonzilitídu, tracheitídu, infekciu horných dýchacích ciest).
b Pojem, ktorý zahŕňa (herpetický ekzém, herpes simplex, očný herpes simplex, orálny herpes).
c Zahŕňa zmeny zistené počas laboratórneho monitorovania (pozri text nižšie).
Opis vybraných nežiaducich reakciíNevoľnosťU dovtedy neliečených pacientov bola frekvencia nevoľnosti v priebehu 52 týždňov vyššia
pri kombinovanej liečbe metotrexátom a Olumiantom (9,3 %) oproti liečbe metotrexátom
v monoterapii (6,2 %) alebo Olumiantom v monoterapii (4,4 %). Nevoľnosť bola najčastejšia
v priebehu prvých 2 týždňov liečby.
InfekcieV kontrolovaných klinických skúšaniach v trvaní do 16 týždňov bola miera výskytu všetkých infekcií
(miera výskytu pacientov s ≥ 1 príhodou na 100 pacientorokov expozície) 101 s Olumiantom
v porovnaní s 83 v skupine s placebom. Väčšina infekcií bola mierna až stredne závažná. V klinických
skúšaniach, ktoré zahŕňali obe dávky, boli infekcie hlásené u 31,9 % pacientov v skupine so 4 mg,
28,8 % v skupine s 2 mg a 24,1 % pacientov v skupine s placebom v období do 16 týždňov. Hlásená miera výskytu ADR súvisiacich s infekciou u Olumiantu v porovnaní s placebom bola: infekcie horných dýchacích ciest (14,7 % oproti 11,7 %), infekcie močových ciest (3,4 % oproti 2,7 %), gastroenteritída (1,6 % oproti 0,8 %), herpes simplex (1,8 % oproti 0,7 %) a herpes zoster (1,4 % oproti 0,4 %). U dovtedy neliečených pacientov v období do 52 týždňov bola frekvencia výskytu infekcií horných dýchacích ciest vyššia pri kombinovanej liečbe metotrexátom a Olumiantom
(26,0 %) v porovnaní s metotrexátom v monoterapii (22,9 %) alebo s Olumiantom v monoterapii
(22,0 %). Miera výskytu závažných infekcií bola s Olumiantom (1,1 %) podobná ako pri placebe (1,2 %). Najčastejšie závažné infekcie pri liečbe Olumiantom boli herpes zoster a celulitída. Miera výskytu závažných infekcií ostala počas dlhodobej expozície stabilná. Celková miera výskytu závažných infekcií v programe klinického skúšania bola 3,2 na 100 pacientorokov.
Zvýšené pečeňové transaminázyV kontrolovaných klinických skúšaniach v trvaní do 16 týždňov sa pozorovalo zvýšenie alanínaminotransferázy (ALT) a aspartátaminotransferázy (AST) ≥ 3x horný limit normy (ULN)
u 1,4 % a 0,8 % pacientov liečených Olumiantom, v porovnaní s 1,0 % a 0,8 % pacientov liečených
placebom. Väčšina prípadov zvýšenia hodnôt transamináz bolo asymptomatických a prechodných.
U dovtedy neliečených pacientov mala kombinácia Olumiantu s potenciálne hepatotoxickými liekmi, akým je napríklad metotrexát, za následok nárast frekvencie výskytu tohto zvýšenia. V období do
52 týždňov bola frekvencia výskytu zvýšených hodnôt ALT a AST ≥ 3 x ULN vyššia
pri kombinovanej liečbe metotrexátom a Olumiantom (7,5 % a 3,8 %) v porovnaní s metotrexátom v monoterapii (2,9 % a 0,5 %) alebo s Olumiantom v monoterapii (1,9 % a 1,3 %).
Charakter a výskyt zvýšených hodnôt ALT/AST ostali v priebehu času stabilné aj v dlhodobom
predĺžení klinického skúšania.
Zvýšenie hladiny lipidov
Liečba baricitinibom súvisela so zvýšením lipidových parametrov závislých od dávky vrátane celkového cholesterolu, triglyceridov, LDL cholesterolu a HDL cholesterolu. Nevyskytla sa žiadna
zmena v pomere LDL/HDL. V 12. týždni sa pozorovalo zvýšenie hodnôt a potom sa ustálili na vyššej
úrovni ako pri vstupnom vyšetrení, vrátane dlhodobého predĺženia klinického skúšania.
V kontrolovaných klinických skúšaniach trvajúcich najviac 16 týždňov bola pozorovaná nasledujúca miera výskytu pri Olumiante v porovnaní s placebom:
· zvýšený celkový cholesterol ≥ 5,17 mmol/l: 49,1 % s Olumiantom oproti 15,8 % pri placebe v uvedenom poradí
· zvýšený LDL cholesterol ≥ 3,36 mmol/l: 33,6 % s Olumiantom oproti 10,3 % pri placebe v uvedenom poradí
· zvýšený HDL cholesterol ≥ 1,55 mmol/l: 42,7 % s Olumiantom oproti 13,8 % pri placebe v uvedenom poradí
· zvýšená hladina triglyceridov ≥ 5,65 mmol/l: 0,4 % s Olumiantom oproti 0,5 % pri placebe v uvedenom poradí
V klinických skúšaniach, ktoré zahŕňali obe dávky, bola pri zvýšenom celkovom cholesterole
≥ 5,17 mmol/l hlásenom u 48,8 %, 34,7 % a 17,8 % pacientov pozorovaná súvislosť s dávkou lieku v trvaní 16 týždňov v skupinách liečby s dávkami 4 mg, 2 mg a s placebom, v uvedenom poradí.
Zvýšená hladina LDL cholesterolu klesla na hladinu pred liečbou ako odpoveď na liečbu statínmi.
Kreatínfosfokináza (CPK)V kontrolovaných klinických skúšaniach v trvaní najviac 16 týždňov sa často vyskytovali zvýšené
hodnoty CPK. Významné zvýšenie (> 5 x ULN) sa vyskytlo u 0,8 % pacientov liečených Olumiantom aj u 0,3 % pacientov liečených placebom. Súvislosť s dávkou lieku bola pozorovaná pri zvýšenej CPK
≥ 5 x ULN normálu hlásenej u 1,5 %, 0,8 % a 0,6 % pacientov v 16. týždni v skupinách s dávkami
4 mg, 2 mg a v skupine s placebom, v uvedenom poradí. Väčšina prípadov bola prechodná
a nevyžadovala ukončenie liečby. V klinických skúšaniach sa nevyskytli žiadne potvrdené prípady rabdomyolýzy. Zvýšená CPK bola pozorovaná vo 4. týždni a potom ostala stabilná na vyšších
hodnotách ako pri vstupom vyšetrení, vrátane dlhodobého predĺženia klinického skúšania.
NeutropéniaV kontrolovaných klinických skúšaniach v trvaní najviac 16 týždňov sa u 0,3 % pacientov liečených Olumiantom objavilo zníženie počtu neutrofilov pod 1 x 109 buniek/l v porovnaní s 0 % pacientov liečených placebom. Medzi zníženým počtom neutrofilov a výskytom závažných infekcií nebola jasná súvislosť. V klinických skúšaniach však bola liečba prerušená ako odpoveď na ANC
< 1 x 109 buniek/l. Charakter a výskyt zníženého počtu neutrofilov ostal v priebehu času stabilný na hodnote nižšej ako pri vstupnom vyšetrení, vrátane dlhodobého predĺženia klinického skúšania.
TrombocytózaV kontrolovaných klinických skúšaniach v trvaní 16 týždňov sa vyskytol zvýšený počet krvných doštičiek nad 600 x 109 buniek/l u 2,0 % pacientov liečených Olumiantom v dávke 4 mg a u 1,1 % pacientov liečených placebom. Medzi zvýšeným počtom krvných doštičiek a nežiaducimi účinkami z trombotickej príčiny nebola pozorovaná žiadna súvislosť. Charakter a výskyt zvýšeného počtu krvných doštičiek ostal v priebehu času stabilný na hodnote vyššej ako pri vstupnom vyšetrení, vrátane dlhodobého predĺženia klinického skúšania.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
V klinických skúšaniach sa počas 10 dní podávali jednotlivé dávky do 40 mg a opakované dávky do
20 mg denne bez prejavov toxicity, ktorá by obmedzila dávkovanie. Nežiaduce účinky boli porovnateľné s nežiaducimi účinkami pozorovanými pri nižších dávkach a neboli zistené žiadne konkrétne toxické prejavy. Farmakokinetické údaje o jednotlivej dávke 40 mg u zdravých dobrovoľníkov naznačujú, že sa do 24 hodín očakáva vyše 90 % eliminácia podanej dávky. V prípade predávkovania sa odporúča, aby bol pacient sledovaný na prejavy a príznaky nežiaducich reakcií. Pacienti, u ktorých sa objavia nežiaduce reakcie, majú dostať vhodnú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: selektívne imunosupresíva, ATC kód: L04AA37
Mechanizmus účinku
Baricitinib je selektívny a reverzibilný inhibítor Janus kinázy (JAK)1 a JAK2. V testoch na
izolovaných enzýmoch baricitinib inhiboval aktivity JAK1, JAK2, tyrozínkinázy 2 a JAK3
s hodnotami IC50 5,9; 5,7, 53 a > 400 nM, v uvedenom poradí.
Janus kinázy (JAK) sú enzýmy, ktoré prenášajú signály vo vnútri bunky z receptorov pre množstvo cytokínov a rastových faktorov na bunkovom povrchu, sú zapojené do hematopoézy, zápalu a funkcie imunitného systému. V rámci vnútrobunkovej signálnej dráhy JAK fosforylujú a tým aktivujú signálne transduktory a aktivátory transkripcie (STAT), čím dochádza k aktivácii génovej expresie vo vnútri bunky. Baricitinib moduluje tieto signálne dráhy čiastočnou inhibíciou enzymatickej aktivity JAK1
a JAK2, čím sa zníži fosforylácia a aktivácia STAT.
Farmakodynamické účinky
Inhibícia fosforylácie STAT3 indukovanej interleukínom IL-6
Podávanie baricitinibu malo za následok od dávky lieku závislú inhibíciu fosforylácie STAT3 indukovanej IL-6 v krvi zdravých osôb s maximálnou inhibíciou pozorovanou 2 hodiny po podaní dávky, ktorá sa do 24 hodín vrátila na hodnotu blízku základnej hodnote.
Imunoglobulíny
Priemerné hodnoty sérových IgG, IgM a IgA klesli do 12 týždňov od začiatku liečby Olumiantom
a ostali stabilné na hodnote nižšej ako boli základné hodnoty počas najmenej 104 týždňov. U väčšiny
pacientov sa zmeny v imunoglobulínoch vyskytovali v normálnom referenčnom rozsahu.
Lymfocyty
Priemerný absolútny počet lymfocytov sa zvýšil do 1 týždňa od začiatku liečby Olumiantom, vrátil sa na základnú hodnotu do 24. týždňa a potom ostal stabilný počas najmenej 104 týždňov. U väčšiny pacientov sa zmeny počtu lymfocytov vyskytovali v normálnom referenčnom rozsahu.
C-reaktívny proteín
U pacientov s reumatoidnou artritídou bol pozorovaný pokles C-reaktívneho proteínu v sére (CRP) už v 1. týždni po začiatku liečby Olumiantom a počas celého podávania lieku sa táto hodnota udržala.
Kreatinín
Baricitinib indukoval vyššie priemerné zvýšenie hladiny sérového kreatinínu s hodnotou 3,8 µmol/l po
dvoch týždňoch liečby v porovnaní s placebom, hodnoty ktorého potom ostali stabilné až do
104. týždňa liečby. Môže to byť v dôsledku baricitinibovej inhibície sekrécie kreatinínu v renálnych tubuloch. Preto odhad glomerulárnej filtrácie na základe sérového kreatinínu môže byť mierne
znížený, bez skutočného zníženia funkcie obličiek alebo výskytu renálnych nežiaducich účinkov.
K
l
i
n
i
cká účinnosť abezpečnosť
Účinnosť a bezpečnosť Olumiantu podávaného raz denne bola hodnotená v 4 randomizovaných,
dvojito zaslepených, multicentrických klinických skúšaniach fázy III u pacientov so stredne závažnou až závažnou aktívnou reumatoidnou artritídou diagnostikovanou v súlade s kritériami ACR/EULAR
2010 (pozri tabuľku č. 3). Zaradení boli pacienti starší ako 18 rokov. Pri vstupnom vyšetrení sa vyžadovala prítomnosť najmenej 6 bolestivých a 6 opuchnutých kĺbov. Všetci pacienti, ktorí absolvovali tieto klinické skúšania, boli vhodní na zaradenie do dlhodobého predĺženia klinického
skúšania s nepretržitou liečbou v trvaní najviac 4 roky.
Klinické skúšanie RA-BEGIN u pacientov predtým neliečených MTX pomáha cieľovej populácii pacientov s nedostatočnou odpoveďou alebo neznášanlivosťou iných DMARD (časť 4.1). Tabuľka č. 3. Zhrnutie klinického skúšania (KS)
Názov KS
(trvanie)
Populácia
(počet)
Liečebné ramená Zhrnutie výsledkov kľúčových meraní
RA-BEGIN (52 týždňov)
RA-BEAM (52 týždňov)
RA-BUILD (24 týždňov)
RA- BEACON (24 týždňov)
neliečení MTX1
(584)
MTX-IR2
(1305)
cDMARD-IR3
(684)
TNF-IR4
(527)
· Olumiant 4 mg QD
· Olumiant 4 mg QD + MTX
· MTX
· Olumiant 4 mg QD
· Adalimumab 40 mg SC Q2W
· placebo
Všetci pacienti užívajú aj MTX
· Olumiant 4 mg QD
· Olumiant 2 mg QD
· placebo
Ak sú na začiatku KS stabilní na
cDMARD, užívajú aj cDMARD5
· Olumiant 4 mg QD
· Olumiant 2 mg QD
· placebo
Užívajú aj cDMARD5
· Primárny koncový ukazovateľ: ACR20
v 24. týždni
· Fyzické funkcie (HAQ-DI)
· RTG progresia (mTSS)
· Nízka aktivita ochorenia a remisia (SDAI)
· Primárny koncový ukazovateľ:ACR20
v 12. týždni
· Fyzické funkcie (HAQ-DI)
· RTG progresia (mTSS)
· Nízka aktivita ochorenia a remisia (SDAI)
· Ranná stuhnutosť kĺbov
· Primárny koncový ukazovateľ: ACR20
v 12. týždni
· Fyzické funkcie (HAQ-DI)
· Nízka aktivita ochorenia a remisia (SDAI)
· RTG progresia (mTSS)
· Ranná stuhnutosť kĺbov
· Primárny koncový ukazovateľ: ACR20
v týždni č. 12
· Fyzické funkcie (HAQ-DI)
· Nízka aktivita ochorenia a remisia (SDAI)

Skratky: QD = raz denne; Q2W = raz za 2 týždne; SC = subkutánne; ACR = American College of Rheumatology; SDAI = zjednodušený index aktivity ochorenia; HAQ-DI = Dotazník hodnotenia zdravia - index postihnutia; mTSS = modifikované celkové Sharpovo skóre
1 Pacienti, ktorým boli podané menej ako 3 dávky metotrexátu (MTX); dovtedy neliečení konvenčnými ani biologickými DMARD.
2 Pacienti, ktorí mali nedostatočnú odpoveď na MTX (+/- iné cDMARD); neliečení biologickými
liekmi.
3 Pacienti, ktorí mali nedostatočnú odpoveď alebo s neznášanlivosťou na ≥ 1 cDMARD; neliečení
biologickými liekmi.
4 Pacienti, ktorí mali nedostatočnú odpoveď alebo s neznášanlivosťou na ≥ 1 bDMARD; vrátane najmenej jedného inhibítora TNF .
5 Najčastejšie súbežne užívané cDMARD zahŕňali MTX, hydroxychlorochín, leflunomid
a sulfasalazín.
Klinická odpoveď:Vo všetkých klinických skúšaniach mali pacienti liečení Olumiantom 4 mg raz denne štatisticky
významne vyššiu odpoveď ACR20, ACR50 a ACR70 v 12. týždni v porovnaní s placebom, MTX
alebo adalimumabom (pozri tabuľku č. 4). Doba nástupu účinku bola rýchla pri meraniach s významne vyššími odpoveďami pozorovanými už v 1. týždni. Pozorovaná bola nepretržitá, nemenná miera odpovede, pričom odpovede ACR20/50/70 sa udržali ešte najmenej 2 roky vrátane dlhodobého predĺženia klinického skúšania.
Liečba Olumiantom 4 mg v monoterapii alebo v kombinácii s cDMARD mala za následok významnejšie zlepšenie všetkých jednotlivých zložiek ACR vrátane počtu bolestivých a opuchnutých kĺbov, celkového hodnotenia pacienta a lekára, HAQ-DI, hodnotenia bolesti a CRP, v porovnaní s placebom alebo MTX v monoterapii. V klinickom skúšaní RA-BEAM mala liečba Olumiantom za následok významnejšie zlepšenie celkového hodnotenia pacienta a lekára, HAQ-DI, hodnotenia bolesti a CRP v 12., 24. a 52. týždni ako pri adalimumabe.
V placebom kontrolovaných klinických skúšaniach, v ktorých nebol požadovaný MTX bolo 501 pacientov randomizovaných na 2 mg alebo 4 mg baricitinibu, užívali MTX ako doplnkovú liečbu a 303 pacientov užívalo iné konvenčné DMARD ako MTX (približne polovica s MTX a polovica bez). Najčastejším súbežne podávaným DMARD u týchto subjektov boli MTX (79 % pacientov), hydroxychlorochín (19 %), leflunomid (11 %) a sulfasalazín (9 %). V žiadnej podskupine sa
nepozorovali významné rozdiely v účinnosti či bezpečnosti definované typom súbežne podávaného
DMARD v kombinácii s baricitinibom.
Remisia a nízka aktivita ochorenia
Štatisticky významne vyššie percento pacientov liečených Olumiantom 4 mg v porovnaní s placebom
alebo MTX dosiahlo remisiu, ako je to definované podľa SDAI Ł 3,3 a CDAI Ł 2,8, v 12. a 24. týždni
(tabuľka č. 4).
Vo všetkých 4 klinických skúšaniach dosiahlo nižšiu aktivitu ochorenia alebo remisiu významne vyššie percento pacientov liečených Olumiantom 4 mg v porovnaní s placebom alebo MTX (DAS28-ESR alebo DAS28-hsCRP Ł 3,2 a DAS28-ESR alebo DAS28-hsCRP < 2,6) v 12. a 24. týždni.
Vyššia miera výskytu remisie v porovnaní s placebom bola pozorovaná už vo 4. týždni. Ak berieme do úvahy aj údaje z dlhodobého klinického skúšania, miera výskytu remisie a nízkej aktivity ochorenia sa udržala najmenej 2 roky.
Tabuľka č. 4: Odpoveď, remisia a fyzická aktivita
Klinické skúšanie
RA-BEGIN Pacienti neliečení MTX
RA-BEAM
Pacienti s MTX-IR
RA-BUILD
Pacienti s cDMARD-IR
RA-BEACON
Pacienti s TNF-IR
Skupina
liečby
MTX OLU
4 mg
OLU
4 mg
+ MTX
PBO OLU
4 mg
ADA
40 mg
Q2W
PBO OLU
2 mg
OLU
4 mg
PBO OLU
2 mg
OLU
4 mg
N 210 159 215 488 487 330 228 229 227 176 174 177
ACR20:12. týždeň 59 % 79 %*** 77 %*** 40 % 70 %***† 61 %*** 39 % 66 %*** 62 %*** 27 % 49 %*** 55 %***
24. týždeň 62 % 77 %** 78 %*** 37 % 74 %***† 66 %*** 42 % 61 %*** 65 %*** 27 % 45 %*** 46 %***
52. týždeň 56 % 73 %*** 73 %*** 71 %†† 62 %
ACR50:12. týždeň 33 % 55 %*** 60 %*** 17 % 45 %***†† 35 %*** 13 % 33 %*** 34 %*** 8 % 20 %** 28 %***
24. týždeň 43 % 60 %** 63 %*** 19 % 51 %*** 45 %*** 21 % 41 %*** 44 %*** 13 % 23 %* 29 %***
52. týždeň 38 % 57 %*** 62 %*** 56 %† 47 %
ACR70:12. týždeň 16 % 31 %*** 34 %*** 5 % 19 %***† 13 %*** 3 % 18 %*** 18 %*** 2 % 13 %*** 11 %**
24. týždeň 21 % 42 %*** 40 %*** 8 % 30 %***† 22 %*** 8 % 25 %*** 24 %*** 3 % 13 %*** 17 %***'
52. týždeň 25 % 42 %*** 46 %*** 37 % 31 %
DAS28-hsCRP Ł
3,2:
12. týždeň 30 % 47 %*** 56 %*** 14 % 44 %***†† 35 %*** 17 % 36 %*** 39 %*** 9 % 24 %*** 32 %***
24. týždeň 38 % 57 %*** 60 %*** 19 % 52 %*** 48 %*** 24 % 46 %*** 52 %*** 11 % 20 %* 33 %***
52. týždeň 38 % 57 %*** 63 %*** 56 %† 48 %
DAS28-ESR Ł
3,2:12. týždeň 15 % 21 % 34 %*** 7 % 24 %*** 21 %*** 7 % 21 %*** 22 %*** 4 % 13 %** 12 %**
24. týždeň 23 % 36 %** 39 %*** 10 % 32 %*** 34 %*** 10 % 29 %*** 32 %*** 7 % 11 % 17 %**
52. týždeň 27 % 36 % 45 %*** 39 % 36 %
SDAI Ł
3,3:12. týždeň 6 % 14 %* 20 %*** 2 % 8 %*** 7 %*** 1 % 9 %*** 9 %*** 2 % 2 % 5 %
24. týždeň 10 % 22 %** 23 %*** 3 % 16 %*** 14 %*** 4 % 17 %*** 15 %*** 2 % 5 % 9 %**
52. týždeň 13 % 25 %** 30 %*** 23 % 18 %
CDAI Ł
2,8:12. týždeň 7 % 14 %* 19 %*** 2 % 8 %*** 7 %** 2 % 10 %*** 9 %*** 2 % 3 % 6 %
24. týždeň 11 % 21 %** 22 %** 4 % 16 %*** 12 %*** 4 % 15 %*** 15 %*** 3 % 5 % 9 %*
52. týždeň 16 % 25 %* 28 %** 22 % 18 %
HAQ-DI Minimálny klinicky významný rozdiel (zníženie HAQ-DI skóre o ≥ 0,30):12. týždeň 60 % 81 %*** 77 %*** 46 % 68 %*** 64 %*** 44 % 60 %*** 56 %** 35 % 48 %* 54 %***
24. týždeň 66 % 77 %* 74 % 37 % 67 %***† 60 %*** 37 % 58 %*** 55 %*** 24 % 41 %*** 44 %***
52. týždeň 53 % 65 %* 67 %** 61 % 55 %
Poznámka: Percento respondérov v každom časovom intervale randomizovaných na začiatku liečby (N). Pacienti, ktorí ukončili liečbu alebo dostali záchrannú terapiu, boli potom považovaní za non-respondérov. Skratky: ADA = adalimumab; MTX = metotrexát; OLU = Olumiant; PBO = placebo
* p ≤ 0,5; ** p ≤ 0,01; *** p ≤ 0,001 oproti placebu (v klin. skúšaní RA-BEGIN oproti MTX)
† p ≤ 0,05; †† p ≤ 0,01; ††† p ≤ 0,001 oproti adalimumabu
R
TG odpoveď
Účinok Olumiantu na progresiu štrukturálneho poškodenia kĺbov bol hodnotený pomocou RTG
v klinických skúšaniach RA-BEGIN, RA-BEAM a RA-BUILD a bol vyhodnotený podľa modifikovaného celkového Sharpovho skóre (mTSS) a jeho zložiek, skóre erózie a skóre zúženia kĺbovej štrbiny.
Liečba Olumiantom 4 mg mala za následok štatisticky významnú inhibíciu progresie poškodenia
kĺbovej štrbiny (tabuľka č. 5). Analýzy skóre erózie a skóre zúženia kĺbovej štrbiny sa zhodovali
s celkovými skóre. Percento pacientov bez RTG progresie (zmena mTSS ≤ 0) bolo významne vyššie s Olumiantom 4 mg ako s placebom v 24. a 52. týždni.
Tabuľka č. 5. RTG zmeny
Klinické
skúšanie
RA-BEGIN
Pacienti neliečení MTX
RA-BEAM
Pacienti s MTX-IR
RA-BUILD
Pacienti s cDMARD-IR
Skupina
liečby
MTX OLU
4 mg
OLU
4 mg
+ MTX
PBOa OLU
4 mg
ADA
40 mg
Q2W
PBO OLU
2 mg
OLU
4 mg
 M
o
difikované celkové Sharpovo skóre, priemerná zmena od vstupných údajov:
M
o
difikované celkové Sharpovo skóre, priemerná zmena od vstupných údajov:
24. týždeň 0,61 0,39 0,29* 0,90 0,41*** 0,33*** 0,70 0,33* 0,15**
52. týždeň 1,02 0,80 0,40** 1,80 0,71*** 0,60***
Skóre erózie, priemerná zmena od vstupných údajov:24. týždeň 0,47 0,33 0,26* 0,61 0,29*** 0,24*** 0,47 0,30 0,11**
52. týždeň 0,81 0,55 0,34** 1,23 0,51*** 0,42***
Skóre zúženia kĺbovej štrbiny, priemerná zmena od vstupných údajov:24. týždeň 0,14 0,06 0,03 0,29 0,12** 0,10** 0,23 0,03* 0,04*
52. týždeň 0,21 0,25 0,06 0,58 0,21*** 0,19**
Percento pacientov bez RTG progresieb:24. týždeň 68 % 76 % 81 %** 70 % 81 %*** 83 %*** 74 % 72 % 80 %
52. týždeň 66 % 69 % 80 %** 70 % 79 %** 81 %**
Skratky: ADA = adalimumab; MTX = metotrexát; OLU = Olumiant; PBO = placebo
a Údaje o placebe v 52. týždni odvodené pomocou lineárnej extrapolácie
b Žiadna progresia definovaná ako zmena mTSS ≤ 0.
* p ≤ 0,05; ** p ≤ 0,01; *** p ≤ 0,001 oproti placebu (v klin. skúšaní RA-BEGIN oproti MTX)
Odpoveď fyzických funkcií a výsledky súvisiace so zdravímLiečba Olumiantom 4 mg v monoterapii alebo v kombinácii s cDMARD mala za následok významnejšie zlepšenie fyzických funkcií ako pri všetkých komparátoroch (placebe, MTX, adalimumabe), ako bolo zistené pomocou HAQ-DI v 12., 24. a 52. týždni. Percento pacientov, ktorí dosiahli klinicky významné zlepšenie (HAQ-DI ≥ 0,30) bolo vyššie pri liečbe Olumiantom oproti placebu alebo MTX v 12. týždni (tabuľka č. 4). Zlepšenie bolo pozorované už v 1. týždni
a v klinických skúšaniach RA-BEGIN a RA-BEAM sa udržalo až 52 týždňov.
Liečba Olumiantom 4 mg v monoterapii alebo v kombinácii s cDMARD mala za následok významnejšie zlepšenie bolesti oproti všetkým komparátorom (placebo, MTX, adalimumab), ako to bolo namerané na vizuálnej analógovej stupnici 0 - 100 v 12. týždni. Štatisticky významná redukcia bolesti bola pozorovaná už v 1. týždni v klinických skúšaniach RA-BEGIN a RA-BEAM a udržala sa počas 52 týždňov.
V klinických skúšaniach RA-BEAM a RA-BUILD mala liečba Olumiantom 4 mg za následok významné zlepšenie priemerného trvania a závažnosti rannej stuhnutosti kĺbov oproti placebu alebo adalimumabu podľa hodnotenia denných elektronických diárov pacienta v trvaní 12 týždňov.
Vo všetkých klinických skúšaniach hlásili pacienti liečení Olumiantom zlepšenie kvality života hlásenej pacientom, na základe skóre fyzickej zložky dotazníka na hodnotenie kvality života Short Form (36) Health Survey (SF-36) a únavy, ako bolo zistené podľa únavového skóre Funkčného hodnotenia liečby chronických ochorení (FACIT-F).
Olumiant 4 mg v porovnaní s 2 mg
Rozdiely v účinnosti pri podávaní 4 mg a 2 mg dávok boli najvýznamnejšie v populácii bDMARD-IR (RA-BEACON), v ktorej bolo pozorované štatisticky významné zlepšenie zložiek ACR v počte
opuchnutých kĺbov, v počte bolestivých kĺbov a v ESR pri 4 mg Olumiantu v porovnaní s placebom v 24 týždni, ale nie pri 2 mg Olumiantu v porovnaní s placebom. Okrem toho nástup účinku v oboch
klinických skúšaniach, RA-BEACON aj RA-BUILD, bol rýchlejší a miera účinku bola obvykle vyššia v skupinách s dávkou 4 mg oproti skupinám s dávkou 2 mg.
V dlhodobom predĺžení klinického skúšania boli pacienti z klinických skúšaní RA-BEAM,
RA-BUILD a RA-BEACON, ktorí dosiahli trvalo nízku aktivitu ochorenia alebo remisiu (CDAI ≤ 10)
po najmenej 15 mesiacoch liečby Olumiantom 4 mg raz denne, opakovane randomizovaní v pomere
1:1 dvojito zaslepeným spôsobom na pokračovanie v liečbe 4 mg lieku raz denne alebo na zníženie dávky na 2 mg raz denne. U väčšiny pacientov sa udržala nízka aktivita ochorenia alebo remisia na
základe skóre CDAI:
· v 12. týždni: 234/251 (93 %) pokračovanie so 4 mg oproti 207/251 (82 %) zníženiu na 2 mg
(p ≤ 0,001)
· v 24. týždni: 163/191 (85 %) pokračovanie so 4 mg oproti 144/189 (76 %) zníženiu na 2 mg
(p ≤ 0,05)
· v 48. týždni: 57/73 (78 %) pokračovanie so 4 mg oproti 51/86 (59 %) zníženiu na 2 mg (p ≤ 0,05)
Väčšina pacientov, ktorí po znížení dávky lieku stratili status nízkej aktivity ochorenia alebo remisie, mohli znovu získať kontrolu nad ochorením potom, ako sa dávka vrátila na hodnotu 4 mg.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Olumiantom
v jednej alebo vo viacerých podskupinách pediatrickej populácie s chronickou idiopatickou artritídou
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po perorálnom podávaní baricitinibu bolo v rozsahu terapeutickej dávky pozorované zvýšenie systémovej expozície úmerné dávke lieku. FK baricitinibu sa lineárne mení s časom.
Absorpcia
Po perorálnom podávaní sa baricitinib rýchlo absorboval s mediánom tmax približne 1 hodiny (rozpätie
0,5 – 3,0 hod) a s absolútnou biologickou dostupnosťou približne 79 % (CV = 3,94 %). Príjem potravy
viedol k zníženej expozícii o najviac 14 %, zníženiu Cmax o najviac 18 % a omeškaniu tmax
o 0,5 hodiny. Podávanie s jedlom nesúviselo s klinicky významným ovplyvnením expozície.
Distribúcia
Priemerný distribučný objem po podaní intravenóznej infúzie bol 76 l, čo naznačuje distribúciu
baricitinibu do tkanív. Približne 50 % baricitinibu sa viaže na plazmatické proteíny.
Biotransformácia
Metabolizmus baricitinibu je sprostredkovaný CYP3A4, pričom sa zistilo, že menej ako 10 % dávky
sa metabolizuje. V plazme sa nezistili žiadne metabolity. Vo farmakologickom klinickom skúšaní sa baricitinib vylučoval predovšetkým ako nezmenené liečivo močom (69 %) a stolicou (15 %) a boli
identifikované iba 4 menšie oxidačné metabolity (3 v moči; 1 v stolici), ktoré tvorili približne 5 % a
1 % dávky v uvedenom poradí. In vitro je baricitinib substrátom CYP3A4, OAT3, Pgp, BCRP a MATE2-K aj inhibítorom transportérov OAT1, OAT3, OCT1, OCT2, OATP1B3, BCRP, MATE1 a MATE2-K, ale klinicky významné interakcie s liekmi, ktoré sú substrátmi týchto transportérov okrem substrátov OCT1, nie sú pravdepodobné (pozri časť 4.5).
Eliminácia
Renálna eliminácia je hlavným mechanizmom vylučovania baricitinibu prostredníctvom glomerulárnej
filtrácie a aktívnej sekrécie cez OAT3, Pgp, BCRP a MATE2-K. Vo farmakologickom klinickom skúšaní sa približne 75 % podávanej dávky lieku vylúčilo močom, zatiaľ čo asi 20 % dávky sa vylúčilo stolicou. Priemerný zdanlivý klírens (CL/F) u pacientov s reumatoidnou artritídou bol
9,42 l/hod (CV = 34,3 %) a eliminačný polčas bol 12,5 hod (CV = 27,4 %). Cmax v rovnovážnom stave je 1,4-násobne vyššia a AUC je v rovnovážnom stave 2,0-násobne vyššia u pacientov s reumatoidnou artritídou v porovnaní so zdravými osobami.
Porucha funkcie obličiek
Zistilo sa, že funkcia obličiek má významný vplyv na expozíciu baricitinibu. Priemerný pomer AUC
u pacientov so stredne závažnou až závažnou poruchou funkcie obličiek je 1,41 (90 % CI: 1,15-1,74) oproti pacientom s normálnou funkciou obličiek, ktorý je 2,22 (90 % CI: 1,81-2,73). Priemerný pomer Cmax u pacientov so stredne závažnou až závažnou poruchou funkcie obličiek je 1,16 (90 %
CI: 0,92-1,45) oproti pacientom s normálnou funkciou obličiek, ktorých Cmax je 1,46 (90 %
CI: 1,17-1,83). Pre odporúčané dávkovania, pozri časť 4.2.
Porucha funkcie pečene
U pacientov so stredne závažnou až závažnou poruchou funkcie pečene sa nezistil žiadny klinicky
významný vplyv na FK baricitinibu. U pacientov so závažnou poruchou funkcie pečene sa užívanie baricitinibu neskúmalo.
Starší
Vek ≥ 65 rokov alebo ≥ 75 rokov nemá vplyv na expozíciu baricitinibu (Cmax a AUC).
Pediatrická populácia
Bezpečnosť, účinnosť ani farmakokinetika baricitinibu v pediatrickej populácii ešte neboli stanovené
(pozri časť 4.2).
Iné vnútorné faktory
Telesná hmotnosť, pohlavie, rasa ani národnosť nemali klinicky významný vplyv na FK baricitinibu.
Priemerný vplyv vnútorných faktorov na parametre FK (AUC a Cmax) sa obvykle nachádzal v rámci interindividuálnej variability FK baricitinibu. Preto na základe týchto pacientskych faktorov nie je potrebná úprava dávky lieku.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, genotoxicity
a karcinogénneho potenciálu, neodhalili žiadne osobitné riziko pre ľudí.
U myší, potkanov a psov bol pozorovaný pokles počtu lymfocytov, eozinofilov a bazofilov ako aj lymfoidná deplécia v orgánoch/tkanivách imunitného systému. U psov pri expozíciách približne 7- násobne vyšších ako pri expozícii u ľudí boli pozorované oportúnne infekcie súvisiace s demodikózou (svrab). U myší, potkanov a psov pri expozíciách približne 6- až 36-násobne vyšších ako expozícia
u ľudí bol pozorovaný pokles počtu červených krviniek. U niektorých psov aj u kontrolných zvierat
bol pozorovaný nízky výskyt degenerácie sternálnej rastovej doštičky, pričom vzhľadom na závažnosť
degenerácie existuje vzťah medzi dávkou a účinkom. V súčasnosti nie je známe, či je to klinicky významné.
V štúdiách reprodukčnej toxikológie na potkanoch a králikoch baricitinib preukázal redukciu rastu/hmotnosti plodu a vznik kostrových malformácií (pri približne 10- a 39-krát vyššej expozícii ako u ľudí, v uvedenom poradí). Pri expozícii 2-krát vyššej ako u ľudí neboli na základe AUC pozorované žiadne nežiaduce účinky na plod.
V kombinovaných štúdiách fertility na potkaních samcoch/samiciach baricitinib znižoval celkovú reprodukčnú výkonnosť (indexy poklesu fertility a počatia). U potkaních samíc sa vyskytol pokles počtu žltých teliesok a implantačných miest, nárast predimplantačných strát a/alebo nežiaducich účinkov na vnútromaternicové prežitie embryí. Keďže sa u potkaních samcov nevyskytli žiadne účinky na spermatogenézu (podľa histopatologického hodnotenia) ani na cieľové ukazovatele semena
/spermií, pokles celkovej reprodukčnej výkonnosti bol pravdepodobne výsledkom pôsobenia na samice.
Baricitinib bol objavený v mlieku laktujúcich potkanov. V štúdii zameranej na prenatálny
a postnatálny vývin bol pozorovaný pokles hmotnosti mláďat a pokles postnatálneho prežitia pri expozíciách 4- a 21-krát vyšších ako sú expozície u ľudí, v uvedenom poradí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrá tabliet
· mikrokryštalická celulóza
· sodná soľ kroskarmelózy
· stearan horečnatý
· manitol
Obalová vrstva
· červený oxid železitý (E172)
· lecitín (sójový) (E322)
· makrogol
· polyvinylalkohol
· mastenec
· oxid titaničitý (E171)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Polyvinylchlorid/polyetylén/polychlorotrifluoroetylén - hliníkové blistre v škatuliach so 14, 28, 35, 56,
84 alebo 98 filmom obalenými tabletami.
Polyvinylchlorid/hliník/orientovaný polyamid - hliníkové perforované blistre s jednorazovou dávkou v škatuliach po 28 x 1 alebo 84 x 1 filmom obalená tableta.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky na likvidáciu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIEli Lilly Nederland B.V., Papendorpseweg 83, 3528BJ Utrecht, Holandsko.
8. REGISTRAČNÉ ČÍSLAOlumiant 2 mg filmom obalené tabletyEU/1/16/1170/001
EU/1/16/1170/002
EU/1/16/1170/003
EU/1/16/1170/004
EU/1/16/1170/005
EU/1/16/1170/006
EU/1/16/1170/007
EU/1/16/1170/008
Olumiant 4 mg filmom obalené tabletyEU/1/16/1170/009
EU/1/16/1170/010
EU/1/16/1170/011
EU/1/16/1170/012
EU/1/16/1170/013
EU/1/16/1170/014
EU/1/16/1170/015
EU/1/16/1170/016
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
cholesterolu.