
· Ak sa hladiny CK a sérového kreatinínu upravia na východiskovú hodnotu, zvážiť
obnovenie liečby dávkou 200 mg každý
druhý deň a stanovovať hladiny CK počas
2 mesiacov raz týždenne a následne raz
mesačne; inak ukončiť liečbu natrvalo.

* Uvedené odporúčania pre úpravu dávky vychádzajú z Common Terminology Criteria for
Adverse Events (
CTCAE) v4.03, vypracované National Cancer Institute (USA). CTCAE je
štandardizovaná klasifikácia vedľajších účinkov, ktorá sa používa pri hodnotení liekov na liečbu
rakoviny.
Cr: kreatinín; ULN: horná hranica normálneho rozmedzia
Iné úpravy dávkyZvládnutie závažných alebo neprijateľných nežiaducich reakcií si môže vyžiadať prechodné prerušenie podávania (s následným znížením dávky, alebo bez neho) alebo ukončenie podávania.
Keď je potrebné prerušiť podávanie, zvážte opätovné začatie liečby Odomzom v rovnakej dávke po
ústupe nežiaducej reakcie na ≤1. stupeň.
Ak je potrebné znížiť dávku, potom sa dávka má znížiť na 200 mg každý druhý deň. Ak sa tá istá nežiaduca reakcia vyskytne po zmene na podávanie každý druhý deň a nezmierni sa, zvážte ukončenie liečby Odomzom.
Nástup plného účinku prerušenia podávania alebo úpravy dávky sonidegibu sa pri niekoľkých nežiaducich udalostiach pre dlhý polčas sonidegibu všeobecne očakáva po niekoľkých týždňoch (pozri časť 5.2).
Trvani e li ečby V klinických skúšaniach liečba Odomzom pokračovala do progresie choroby alebo neprijateľných príznakov toxicity. Prerušenia liečby až do 3 týždňov boli povolené na základe individuálnej znášanlivosti.
Prínos pokračujúcej liečby sa má pravidelne vyhodnocovať, pričom optimálne trvanie liečby sa líši
u každého individuálneho pacienta.
Osobitné populáciePacienti s poruchou funkcie obličiekSonidegib sa nesledoval v osobitnej farmakokinetickej štúdii u pacientov s poruchou funkcie obličiek.
Podľa dostupných údajov je vylučovanie sonidegibu obličkami zanedbateľné. Analýza farmakokinetiky populácie ukázala, že ľahká alebo stredne ťažká porucha funkcie obličiek nemala významný vplyv na zdanlivý klírens (CL/F) sonidegibu, čo naznačuje, že úprava dávky u pacientov
s poruchou funkcie obličiek nie je potrebná (pozri časť 5.2). Nie sú dostupné žiadne údaje o účinnosti a bezpečnosti u pacientov s ťažkou poruchou funkcie obličiek.
Pacienti s poruchou funkcie pečene
U pacientov s ľahkou (trieda A podľa Childa-Pugha) alebo stredne ťažkou (trieda B podľa
Childa-Pugha) poruchou funkcie pečene nie je potrebná úprava dávky. Sonidegib sa nesledoval u
pacientov s ťažkou poruchou funkcie pečene a má sa u týchto pacientov používať opatrne (pozri
časť 5.2).
Starší pacienti (≥65 rokov)
Údaje o bezpečnosti a účinnosti u pacientov vo veku 65 rokov a starších nenaznačujú, že by u týchto pacientov bola potrebná úprava dávky (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť Odomza u detí a dospievajúcich vo veku menej ako 18 rokov s bazocelulárnym karcinómom neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Odomzo sa užíva perorálne. Kapsuly sa musia prehltnúť celé. Nesmú sa hrýzť alebo drviť. Kapsuly sa
nesmú otvárať vzhľadom na riziko teratogenity (pozri časť 5.3).
Odomzo sa musí užívať najmenej dve hodiny po jedle a najmenej jednu hodinu pred nasledujúcim jedlom. Ak počas liečby dôjde k vracaniu, nie je povolené opakované podanie pacientovi pred nasledujúcou plánovanou dávkou.
Ak sa dávka vynechá, má sa užiť čo najskôr, ako sa to zistí, pokiaľ od plánovaného času užitia neuplynulo viac ako šesť hodín; v takom prípade má pacient počkať a užiť nasledujúcu plánovanú dávku.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Gravidita a dojčenie (pozri časti 4.4 a 4.6).
Ženy vo fertilnom veku, ktoré nekonajú v súlade s Programom prevencie gravidity pri Odomze (pozri
časti 4.4 a 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Nežiaduce udalosti súvisiace so svalmi
V pivotnej štúdii fázy II sa pozorovali svalové spazmy, myalgia, myopatia a prípady zvýšenia CK.
U väčšiny pacientov liečených Odomzom 200 mg denne, ktorí mali zvýšenie CK 2. alebo
vyššieho stupňa, sa svalové príznaky objavili ešte pred zvýšením CK. U väčšiny pacientov sa svalové
príznaky a zvýšenie CK po vhodných opatreniach upravili.
Všetci pacienti, ktorí začínajú liečbu Odomzom, musia byť informovaní o riziku nežiaducich udalostí súvisiacich so svalmi, vrátane možnej rabdomyolýzy. Musia byť poučení, aby okamžite hlásili akúkoľvek nevysvetliteľnú bolesť, citlivosť na dotyk alebo slabosť svalov, ktoré sa objavia počas liečby Odomzom, alebo ak príznaky pretrvávajú po prerušení liečby.
Hladiny CK sa majú skontrolovať pred začiatkom liečby a aj neskôr, pokiaľ je to klinicky indikované, napr. ak sú hlásené príznaky súvisiace so svalmi. Ak sa zistí klinicky významné zvýšenie CK, je potrebné vyšetriť funkciu obličiek (pozri časť 4.2).
Majú sa dodržiavať odporúčania pre úpravu dávky alebo prerušenie podávania (pozri časť 4.2). Pri zvýšení CK vysokého stupňa sa má v súlade s lokálnymi štandardnými liečebnými postupmi a odporúčaniami pre liečbu ako opatrenie zvážiť podporná liečba zahŕňajúca dostatočnú hydratáciu.
Ak sa Odomzo užíva v kombinácii s určitými liekmi, ktoré môžu zvýšiť potenciálne riziko vzniku toxických účinkov na svaly (napr. inhibítory CYP3A4, chlorochín, hydroxychlorochín, deriváty fibrátov, penicilamín, zidovudín, niacín a inhibítory HMG-CoA-reduktázy), pacienti majú byť starostlivo sledovaní pre príznaky súvisiace so svalmi (pozri časť 4.5).
Pacienti s neuromuskulárnymi poruchami (napr. zápalové myopatie, muskulárna dystrofia, amyotrofická laterálna skleróza, spinálna muskulárna atrofia) musia byť starostlivo sledovaní vzhľadom na zvýšené riziko toxických účinkov na svaly.
Embryofetálna smrť aleboťažkévrodenédefekty
Keď sa Odomzo podáva gravidným ženám, môže zapríčiniť smrť embrya/fétu alebo ťažké vrodené
defekty. V súlade s jeho mechanizmom účinku sa v štúdiách na zvieratách zistilo, že sonidegib je teratogénny a fetotoxický. Ženy užívajúce Odomzo nesmú byť gravidné alebo otehotnieť počas liečby a počas 20 mesiacov od skončenia liečby.
Kritériá definujúce ženu vofertilnomveku
Žena vo fertilnom veku je v Programe prevencie gravidity pri Odomze definovaná ako sexuálne zrelá
žena, ktorá
· mala menštruáciu kedykoľvek počas predchádzajúcich 12 po sebe nasledujúcich mesiacov,
· nepodstúpila hysterektómiu alebo obojstrannú ooforektómiu, alebo nemá lekárom potvrdené
trvalé predčasné zlyhanie ovárií,
· nemá genotyp XY, Turnerov syndróm alebo agenézu uteru,
· je amenoreická následkom liečby rakoviny vrátane liečby Odomzom.
Poradenstvo
Pre ženy v o f erti l nom ve ku
Odomzo je kontraindikované u žien vo fertilnom veku, ktoré nekonajú v súlade s Programom prevencie gravidity pri Odomze. Žena vo fertilnom veku musí chápať, že:
· Odomzo predstavuje riziko teratogenity pre nenarodené dieťa.
· Nesmie užívať Odomzo, ak je gravidná alebo plánuje otehotnieť.
· Musí mať negatívny výsledok testu na graviditu, vykonaného zdravotníckym pracovníkom
počas 7 dní pred začiatkom liečby Odomzom.
· Musí mať každý mesiac negatívny výsledok testu na graviditu počas liečby, aj keď je amenoreická.
· Nesmie otehotnieť počas užívania Odomza a počas 20 mesiacov od poslednej dávky.
· Musí byť schopná dodržiavať účinné antikoncepčné opatrenia.
· Musí počas užívania Odomza používať 2 odporúčané metódy antikoncepcie (pozri časť
„Antikoncepcia“ nižšie a časť 4.6), pokiaľ sa nezaviaže, že nebude mať pohlavný styk
(abstinencia).
· Musí informovať svojho lekára, ak sa počas liečby a počas 20 mesiacov od poslednej dávky
vyskytne niečo z uvedeného:
o otehotnie alebo si z akejkoľvek príčiny myslí, že môže byť tehotná,
o vynechala jej očakávaná menštruácia,
o prestala používať antikoncepciu, pokiaľ sa nezaviaže, že nebude mať pohlavný styk
(abstinencia),
o potrebuje zmeniť antikoncepciu.
· Nesmie dojčiť počas užívania Odomza a počas 20 mesiacov od poslednej dávky.
Pre mužov
Sonidegib môže prechádzať do semena. Aby sa zabránilo možnej expozícii u plodu počas gravidity, pacient musí chápať, že:
· Odomzo predstavuje riziko teratogenity pre nenarodené dieťa, ak má nechránený pohlavný styk s gravidnou ženou.
· Musí vždy používať odporúčanú antikoncepciu (pozri časť „Antikoncepcia“ nižšie a časť 4.6).
· Bude informovať svojho lekára, ak jeho partnerka otehotnie počas jeho liečby Odomzom alebo
počas 6 mesiacov od jeho poslednej dávky.
Pre zdravotníckych pracovníkov
Zdravotnícki pracovníci musia poučiť pacientov, aby pochopili a akceptovali všetky podmienky
Programu prevencie gravidity pri Odomze.
Antikoncepcia
Ženy v o fertilnom veku
Ženy vo fertilnom veku musia používať dve metódy odporúčanej antikoncepcie, vrátane jednej vysoko
účinnej metódy a jednej bariérovej metódy, počas užívania Odomza a počas 20 mesiacov od
ukončenia liečby (pozri časť 4.6).
M uži
Pacienti, vrátane tých, ktorí podstúpili vazektómiu, musia vždy používať pri pohlavnom styku s
partnerkou kondóm (so spermicídnou látkou, ak je dostupný) počas užívania Odomza a počas
6 mesiacov od ukončenia liečby (pozri časti 4.6 a 5.3).
Testovanie na graviditu
U žien vo fertilnom veku musí zdravotnícky pracovník počas 7 dní pred začiatkom liečby Odomzom a
raz mesačne počas liečby overiť ich stav testom na graviditu. Testy na graviditu majú mať podľa lokálnej dostupnosti minimálnu citlivosť 25 mIU/ml. V prípade gravidity sa liečba nesmie začať. Ak ku gravidite dôjde počas liečby, podávanie Odomza sa musí okamžite ukončiť (pozri časť 5.3). Pacientky, u ktorých sa počas liečby Odomzom vyskytne amenorea, majú počas trvania liečby pokračovať v každomesačnom testovaní na graviditu.
Obmedzenia predpisovania a výdaja ženám vo fertilnom veku
Prvé predpísanie a výdaj Odomza sa má uskutočniť počas 7 dní od testu na graviditu s negatívnym
výsledkom. Predpisovanie Odomza má byť obmedzené na 30 dní liečby, pričom na pokračovanie v liečbe je potrebný nový lekársky predpis.
Edukačný materiál
Aby sa zdravotníckym pracovníkom a pacientom pomohlo zabrániť expozícii embrya a fétu Odomzu,
držiteľ rozhodnutia o registrácii poskytne edukačné materiály (Program prevencie gravidity pri
Odomze), ktoré zdôraznia potenciálne riziká spojené s užívaním lieku.
Darcovstvo krvi
Pacienti majú byť poučení, aby počas užívania Odomza a počas najmenej 20 mesiacov od skončenia
liečby nedarovali krv.
Darovanie spermií
Pacienti nemajú darovať spermie počas užívania Odomza a počas najmenej 6 mesiacov od skončenia
liečby.
I
nterakcie
Je potrebné vyhnúť sa súbežnému podávaniu so silnými induktormi CYP (napr. rifampicínom,
karbamazepínom alebo fenytoínom), pretože nemožno vylúčiť riziko znížených koncentrácií v plazme a zníženej účinnosti sonidegibu (pozri aj časť 4.5).
Skvamocelulárny karcinóm kože (cuSCC)
U pacientov s pokročilým BCC je zvýšené riziko vzniku cuSCC. U pacientov s pokročilým BCC
liečených Odomzom boli hlásené prípady cuSCC. Nezistilo sa, či cuSCC súvisí s liečbou Odomzom.
Preto majú byť všetci pacienti počas užívania Odomza rutinne sledovaní a cuSCC sa má liečiť
v súlade so štandardmi starostlivosti.
Ďalšie bezpečnostnéopatrenia
Pacientov treba poučiť, aby tento liek nikdy nedali niekomu inému. Všetky kapsuly, ktoré na konci
liečby zostanú nepoužité, má pacient ihneď zlikvidovať v súlade s národnými požiadavkami (napr. vrátiť kapsuly lekárnikovi alebo lekárovi).
Pomocné látky
Kapsuly Odomzo obsahujú monohydrát laktózy. Pacienti so zriedkavými dedičnými problémami
galaktózovej intolerancie, laponského deficitu laktázy alebo glukózo-galaktózovej malabsorpcie
nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Sonidegib sa metabolizuje primárne prostredníctvom CYP3A4 a súbežné podávanie silných
inhibítorov alebo induktorov CYP3A4 môže významne zvýšiť alebo znížiť koncentrácie sonidegibu.
Látky, ktoré môžuzvýšiťkoncentráciu sonidegibu v plazme
U zdravých osôb súčasné podanie jednorazovej dávky 800 mg sonidegibu s ketokonazolom (200 mg
dvakrát denne počas 14 dní), silným inhibítorom CYP3A, zvýšilo AUC sonidegibu 2,25-násobne a Cmax 1,49- násobne v porovnaní s podaním samotného sonidegibu. Na základe simulácie, dlhšie súbežné podávanie silných inhibítorov CYP3A4 (napr. viac ako 14 dní) bude mať za následok viacnásobné zmeny expozície sonidegibu. Ak je potrebné súbežné použitie silného inhibítora CYP3A, dávka sonidegibu sa má znížiť na 200 mg každý druhý deň. Silné inhibítory CYP3A zahŕňajú okrem iného ritonavir, sachinavir, telitromycín, ketokonazol, itrakonazol, vorikonazol, posakonazol a nefazodón. Pacientov, ktorí užívajú niektorý z týchto liekov súbežne so sonidegibom, je potrebné dôsledne sledovať pre nežiaduce účinky.
Látky, ktoré môžuznížiť koncentráciu sonidegibu v plazme
U zdravých osôb súčasné podanie jednorazovej dávky 800 mg sonidegibu s rifampicínom (600 mg
denne počas 14 dní), silným induktorom CYP3A, znížilo AUC sonidegibu o 72 % a Cmax o 54 %
v porovnaní s podaním samotného sonidegibu. Súbežné podávanie sonidegibu so silnými induktormi
CYP3A znižuje koncentráciu sonidegibu v plazme. Je potrebné vyhnúť sa súbežnému podávaniu silných induktorov CYP3A; vrátane, ale nie výlučne karbamazepínu, fenobarbitalu, fenytoínu,
rifabutínu, rifampicínu a ľubovníka bodkovaného (Hypericum perforatum). Ak sa musí použiť silný
induktor CYP3A4 súbežne so sonidegibom, má sa zvážiť zvýšenie dennej dávky sonidegibu na
400-800 mg. Na základe farmakokinetických údajov sa predpokladá, že touto dávkou sonidegibu sa dosiahne AUC v rozmedzí, ktoré sa pozoruje bez induktorov, ak súbežné podávanie induktora netrvá
viac ako 14 dní. Dlhšia súbežná liečba induktorom sa neodporúča, pretože sa zníži expozícia
sonidegibu, čo môže znížiť jeho účinnosť. Dávka sonidegibu užívaná pred začatím liečby silným induktorom sa má použiť znovu, ak sa podávanie silného induktora skončí.
Lieky, ktoré zvyšujú pH v hornej časti gastrointestinálneho traktu, napr. inhibítory protónovej pumpy (PPI), môžu meniť rozpustnosť sonidegibu a potenciálne znížiť jeho biologickú dostupnosť. Nevykonala sa formálna klinická štúdia na vyhodnotenie vplyvu PPI na systémovú expozíciu sonidegibu. Avšak súbežné podávanie PPI alebo antagonistov H2-receptorov sa testovalo ako premenná v analýze farmakokinetiky populácie. Odhadlo sa, že PPI znižujú biologickú dostupnosť
na 0,69 (95 % IS: 0,60; 0,81), zatiaľ čo pri antagonistoch H2-receptorov sa odhaduje, že nemajú významný účinok na biologickú dostupnosť. Vplyv PPI alebo antagonistov H2-receptorov na účinnosť sonidegibu nie je známy.
Účinky sonidegibu nainélieky
Sonidegib je in vitro kompetitívny inhibítor CYP2B6 a CYP2C9, ktorý potenciálne zvyšuje
koncentrácie látok metabolizovaných týmito enzýmami. Sonidegib je tiež inhibítor proteínu rezistencie karcinómu prsníka (BCRP) (IC50 ~1,5µM). Pacientov, ktorí súbežne užívajú substráty
enzýmov CYP2B6 a CYP2C9 alebo transportéry BCRP, je potrebné starostlivo sledovať pre
nežiaduce reakcie. Je potrebné vyhnúť sa látkam, ktoré sú substrátmi enzýmov CYP2B6 a CYP2C9 s úzkym terapeutickým rozmedzím (napr. warfarín, acenokumarol, efavirenz, metadón) alebo substrátmi BCRP s úzkym terapeutickým rozmedzím (napr. metotrexát, mitoxantrón, irinotekan, topotekan).
Látky, ktoré môžuzhoršovaťudalosti súvisiace so svalmi
U pacientov užívajúcich Odomzo, ktorí užívajú aj lieky zvyšujúce riziko toxických účinkov
súvisiacich so svalmi, môže byť v dôsledku prekrývajúcich sa profilov toxicity vyššie riziko vzniku nežiaducich udalostí súvisiacich so svalmi. Ak sa vyvinú svalové príznaky, je potrebné pacientov dôsledne sledovať a zvážiť úpravu dávky.
V pivotnom skúšaní fázy II užívalo 12 (15,2 %) pacientov liečených Odomzom 200 mg súbežne aj inhibítory HMG-CoA-reduktázy (9 užívalo pravastatín, 3 užívali iné inhibítory HMG-CoA-reduktázy vrátane rosuvastatínu a simvastatínu). Z týchto pacientov malo 7 (58,3 %) svalové príznaky do
1. stupňa, zatiaľ čo 43 (64,1 %) pacientov, ktorí neužívali inhibítory HMG-CoA-reduktázy, malo príznaky až do 3. stupňa. U žiadneho z pacientov užívajúcich inhibítory HMG-CoA-reduktázy nedošlo
k zvýšeniu CK 3./4. stupňa, na rozdiel od 6 (9,0 %) pacientov, ktorí neužívali inhibítory HMG-CoA- reduktázy.
Interakcie s jedlom
Biologická dostupnosť sonidegibu sa zvyšuje v prítomnosti jedla (pozri časť 5.2). Odomzo sa musí
užívať najmenej dve hodiny po jedle a najmenej jednu hodinu pred nasledujúcim jedlom.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Vzhľadom na riziko embryofetálnej smrti alebo ťažkých vrodených defektov spôsobených
sonidegibom ženy užívajúce Odomzo nesmú byť gravidné alebo otehotnieť počas liečby
a počas 20 mesiacov od skončenia liečby (pozri časť 4.4).
Odomzo je kontraindikované u žien vo fertilnom veku, ktoré nekonajú v súlade s Programom prevencie gravidity pri Odomze (pozri časť 4.3).
V prí pade gravidit y al ebo vyne chani a menštruáci e
Ak pacientka otehotnie, vynechá jej menštruácia alebo má z akéhokoľvek dôvodu podozrenie, že môže byť gravidná, musí to ihneď oznámiť svojmu ošetrujúcemu lekárovi.
Pretrvávajúca neprítomnosť menštruácie počas liečby Odomzom sa až do medicínskeho vyhodnotenia
a potvrdenia má považovať za prejav gravidity.
A
ntikoncepcia pre mužov a ženy
Ženy v o fertilnom veku
Ženy vo fertilnom veku musia byť schopné dodržiavať účinné opatrenia brániace otehotneniu. Počas užívania Odomza a počas 20 mesiacov od poslednej dávky musia používať dve metódy odporúčanej
antikoncepcie, vrátane jednej vysoko účinnej metódy a jednej bariérovej metódy. Ženy vo fertilnom
veku, ktoré majú nepravidelnú menštruáciu alebo nemajú menštruáciu, musia dodržiavať všetky odporúčania týkajúce sa účinnej antikoncepcie.
M uži
Nie je známe, či spermie obsahujú sonidegib. Muži nemajú splodiť dieťa alebo darovať spermie počas užívania Odomza a počas najmenej 6 mesiacov od skončenia liečby. Pacienti, vrátane tých, ktorí podstúpili vazektómiu, musia vždy používať počas pohlavného styku s partnerkou kondóm (so spermicídnou látkou, ak je dostupný) počas užívania Odomza a počas 6 mesiacov od poslednej dávky, aby sa predišlo prípadnej expozícii u plodu počas gravidity.
Odporúč ajú sa nasledujúce formy vy soko úči nných metód:
· Sterilizácia vajíčkovodov
· Vazektómia
· Intrauterinné teliesko (IUD)
Odporúč ajú sa nasledujúce bariérové metódy:
· Akýkoľvek mužský kondóm (so spermicídnou látkou, ak je dostupný)
· Pesar (so spermicídnou látkou, ak je dostupný)
Gravidita
Nie sú k dispozícii údaje o použití sonidegibu u gravidných žien. Štúdie na zvieratách preukázali
teratogenitu a fetotoxicitu (pozri časť 5.3). Odomzo je kontraindikované počas gravidity.
Dojčenie
Nie je známe, či sa sonidegib vylučuje do ľudského mlieka. Pretože sú možné závažné nežiaduce
reakcie na sonidegib u dojčených novorodencov/detí, napr. závažné vývinové defekty, ženy nesmú
dojčiť počas užívania Odomza a počas 20 mesiacov od ukončenia liečby (pozri časť 5.3).
Fertilita
Údaje zo štúdií na potkanoch a psoch ukazujú, že liečba Odomzom môže ireverzibilne oslabiť mužskú
a ženskú fertilitu (pozri časť 5.3). V klinických skúšaniach sa okrem toho u žien vo fertilnom veku pozorovala amenorea (pozri časť 4.8). Pred začiatkom liečby Odomzom je potrebné prediskutovať so ženami vo fertilnom veku stratégie na zachovanie fertility.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Odomzo nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Pivotná štúdia fázy II vyhodnotila bezpečnosť Odomza u celkovo 229 dospelých pacientov s lokálne
pokročilým alebo metastatickým BCC. Pacienti boli liečení Odomzom 200 mg denne (n=79) alebo Odomzom 800 mg denne (n=150). Medián trvania liečby bol 11,0 mesiacov u pacientov liečených Odomzom v odporúčanej dávke 200 mg (rozmedzie 1,3 až 33,5 mesiacov). Jedno úmrtie sa vyskytlo počas liečby alebo počas 30 dní od poslednej užitej dávky u pacientov s metastatickým BCC alebo lokálne pokročilým BCC, ktorí užívali Odomzo 200 mg.
Najčastejšími nežiaducimi reakciami na liek, ktoré sa vyskytli u ≥10 % pacientov liečených Odomzom
200 mg, boli svalové spazmy, alopécia, dysgeúzia, únava, nauzea, bolesť svalov a kostí, hnačka,
pokles telesnej hmotnosti, znížená chuť do jedenia, myalgia, bolesť brucha, bolesť hlavy, bolesť, vracanie a pruritus.
Najčastejšie nežiaduce účinky 3./4. stupňa, ktoré sa vyskytli u ≥2 % pacientov liečených Odomzom
200 mg, boli únava, pokles telesnej hmotnosti a svalové spazmy.
Pri hlásených nežiaducich reakciách na liek (tabuľka 2) bola frekvencia vyššia u pacientov užívajúcich Odomzo 800 mg ako u pacientov užívajúcich Odomzo 200 mg, okrem bolesti svalov a kostí, hnačky, bolesti brucha, bolesti hlavy a pruritu. Toto platilo aj pre nežiaduce reakcie 3./4. stupňa, okrem únavy.
Tabuľkový zoznam nežiaducichreakcií na liekNežiaduce reakcie na liek pri odporúčanej dávke v pivotnom klinickom skúšaní fázy II (tabuľka 2) sú
uvedené podľa triedy orgánových systémov v Medical Dictionary for Regulatory Activities
(MedDRA), verzia 17.1. V každej triede orgánových systémov sú nežiaduce reakcie na liek zoradené podľa frekvencie, pričom najčastejšie reakcie sú uvedené ako prvé. V každej skupine frekvencií sú nežiaduce reakcie na liek zoradené podľa klesajúcej závažnosti. Okrem toho zodpovedajúca kategória frekvencie pre každú nežiaducu reakciu na liek vychádza z nasledujúcej konvencie (CIOMS III): veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000); neznáme (z dostupných údajov).
Tabuľka 2 Nežiaduce reakcie na liek pozorované v pivotnom klinickom skúšaní fázy IIPrimárna trieda orgánových systémov
Preferovaný názov
| Frekvencia všetkých stupňov
200 mg
|
Poruchy metabolizmu a výživy
|
Znížená chuť do jedenia
| Veľmi časté
|
Dehydratácia
| Časté
|
Poruchy nervového systému
|
Dysgeúzia
| Veľmi časté
|
Bolesť hlavy
| Veľmi časté
|
Poruchy gastrointestinálneho traktu
|
Nauzea
| Veľmi časté
|
Hnačka
| Veľmi časté
|
Bolesť brucha
| Veľmi časté
|
Vracanie
| Veľmi časté
|
Dyspepsia
| Časté
|
Zápcha
| Časté
|
Gastroezofágová refluxná porucha
| Časté
|
P
oruchy kože a podkožného tkaniva
|
Alopécia
|
Veľmi časté
|
Pruritus
|
Veľmi časté
|
Exantém
|
Časté
|
Abnormálny rast vlasov
|
Časté
|
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Svalové spazmy
|
Veľmi časté
|
Bolesť svalov a kostí
|
Veľmi časté
|
Myalgia
|
Veľmi časté
|
Myopatia
[svalová únava a slabosť]
|
Časté
|
P
oruchy reprodukčného systému a prsníkov
|
Amenorea*
|
Veľmi časté
|
C
elkové poruchy a reakcie v mieste podania
|
Únava
|
Veľmi časté
|
Bolesť
|
Veľmi časté
|
L
aboratórne a funkčné vyšetrenia
|
Pokles telesnej hmotnosti
|
Veľmi časté
|
* Zo 79 pacientov, ktorí dostávali Odomzo 200 mg, bolo 5 žien vo fertilnom veku. Medzi týmito
ženami sa amenorea pozorovala u 1 pacientky (20 %).
|
K
l
i
n
i
cky významné laboratórne abnormality
Najčastejšie hlásené laboratórne abnormality 3./4. stupňa s incidenciou ≥5 % vyskytujúce sa u
pacientov liečených Odomzom 200 mg boli zvýšenie lipázy a zvýšenie CK v krvi (tabuľka 3).
Tabuľka 3 Laboratórne abnormality*
Laboratórny test
| Frekvencia všetkých stupňov
200 mg
|
Hematologické ukazovatele
|
Znížený hemoglobín
| Veľmi časté
|
Znížený počet lymfocytov
| Veľmi časté
|
Biochemické ukazovatele
|
|
Zvýšený sérový kreatinín
| Veľmi časté
|
Zvýšená sérová kreatínfosfokináza (CK)
| Veľmi časté
|
Zvýšená glykémia
| Veľmi časté
|
Zvýšená lipáza
| Veľmi časté
|
Zvýšená alanínaminotransferáza (ALT)
| Veľmi časté
|
Zvýšená aspartátaminotransferáza (AST)
| Veľmi časté
|
Zvýšená amyláza
| Veľmi časté
|
* Na základe najhoršej laboratórnej hodnoty po liečbe bez ohľadu na východiskovú hodnotu, klasifikácia
podľa CTCAE verzia 4.03
|
Opisvybranýchnežiaducich reakcií na liekUdalosti súvi siace so sval mi vr át ane z výš eni a CK U pacientov liečených sonidegibom je svalová toxicita klinicky najvýznamnejším hláseným vedľajším účinkom a predpokladá sa, že je to skupinový účinok inhibítorov signálnej dráhy Hedgehog (Hh).
Svalové spazmy boli v pivotnej štúdii fázy II najčastejšími nežiaducimi udalosťami „súvisiacimi so
svalmi“ a boli hlásené u menej pacientov v skupine Odomza 200 mg (54 %) ako v skupine Odomza
800 mg (69 %).
Zvýšenie CK v krvi 3./4. stupňa bolo hlásené u 8 % pacientov užívajúcich Odomzo 200 mg. U väčšiny
pacientov, ktorí mali zvýšenie CK 2. alebo vyššieho stupňa, sa svalové príznaky objavili pred zvýšením CK. U týchto pacientov bol medián času do zvýšenia laboratórnych hodnôt CK na 2. a vyšší stupeň závažnosti 12,9 týždňov (rozmedzie 2 až 39 týždňov) od začiatku liečby Odomzom a medián času do úpravy (na normu alebo 1. stupeň) bol 12 dní (95 % IS, 8 až 14 dní).
Svalové príznaky a zvýšenie CK nad 10x ULN, vyžadujúce intravenózne podanie tekutín, sa vyskytli u jedného pacienta užívajúceho Odomzo 200 mg v porovnaní so 6 pacientmi užívajúcimi Odomzo
800 mg.
V pivotnej štúdii fázy II sa nepotvrdili žiadne hlásené prípady rabdomyolýzy (definovanej ako hladiny CK >10-násobné v porovnaní s hodnotou pred liečbou alebo východiskovou hodnotou, alebo >10x ULN, ak nebola udaná východisková hodnota, plus 1,5-násobné zvýšenie kreatinínu v sére
v porovnaní s hodnotou pred liečbou alebo východiskovou hodnotou). Potvrdil sa však jeden hlásený prípad u pacienta liečeného Odomzom 800 mg v nepivotnej štúdii.
AmenoreaV pivotnej štúdii fázy II sa počas liečby Odomzom 200 mg alebo 800 mg raz denne vyskytla amenorea u 2 (14,3 %) zo 14 žien vo fertilnom veku alebo vo fertilnom veku po sterilizácii
podviazaním vajíčkovodov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieOdomzo sa v štúdiách so stupňovaním dávky podávalo v dávkach až do 3 000 mg perorálne raz denne. Vo všetkých prípadoch predávkovania majú byť pacienti starostlivo sledovaní pre nežiaduce udalosti a má sa im poskytnúť primeraná podporná starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, iné antineoplastiká, ATC kód: L01XX48
Mechanizmus účinkuSonidegib je perorálne biologicky dostupný inhibítor signálnej dráhy Hh. Viaže sa na Smoothened
(Smo), molekulu podobnú receptoru viazanú na proteín G, ktorá pozitívne reguluje dráhu Hh
a napokon aktivuje a uvoľňuje transkripčné faktory onkogénu spojeného s gliómom (GLI), ktorý indukuje transkripciu cieľových génov Hh podieľajúcich sa na proliferácii, diferenciácii a prežívaní. Aberantná signalizácia Hh sa dávala do súvislosti s patogenézou viacerých typov rakoviny vrátane bazocelulárneho karcinómu (BCC). Sonidegib viažuci sa na Smo bude inhibovať signalizáciu Hh
a následne blokovať transdukciu signálu.
Farmakodynamické účinky
Analýza QTc a plazmatickej koncentrácie sonidegibu ukázala, že horná hranica jednostranného 95 %
intervalu spoľahlivosti pre nárast QTc bola nižšia ako 5 msek pri Cmax v rovnovážnom stave pri denných dávkach 800 mg, ktoré vedú k 2,3-násobnej plazmatickej expozícii v porovnaní
s odporúčanou dávkou 200 mg. Preto sa pri terapeutických dávkach Odomza neočakáva klinicky významné predĺženie QTc. Okrem toho sa plazmatické koncentrácie sonidegibu vyššie ako tie, ktoré sa dosahujú pri terapeutických dávkach, nespájali s arytmiami alebo torsades de pointes ohrozujúcimi
život.
Odpoveď nádoru nezávisela od dávky Odomza alebo od plazmatickej koncentrácie v rozmedzí dávok
200 mg až 800 mg.
Klinická účinnosťabezpečnosť
Randomizovaná, dvojito zaslepená štúdia fázy II s dvoma veľkosťami dávky Odomza (200 mg alebo
800 mg raz denne) sa vykonala s 230 pacientmi, ktorí mali buď lokálne pokročilý bazocelulárny
karcinóm (laBCC) (n=194), alebo metastatický bazocelulárny karcinóm (mBCC) (n=36). Z týchto
230 pacientov malo 16 diagnózu Gorlinovho syndrómu (15 laBCC a 1 mBCC). Dospelí (vo veku
≥18 rokov) pacienti s laBCC alebo mBCC, u ktorých nebola vhodná rádioterapia, chirurgický zákrok
alebo iná lokálna liečba, boli randomizovaní na podávanie Odomza buď 200 mg, alebo 800 mg denne
až do progresie choroby alebo neprijateľnej toxicity.
Primárnym cieľovým ukazovateľom účinnosti v štúdii bol podiel objektívnych odpovedí podľa modifikovaných kritérií pre hodnotenie odpovede pri solídnych nádoroch (modified Response Evaluation Criteria in Solid Tumours, mRECIST) u pacientov s laBCC a RECIST 1.1 u pacientov'
s mBCC, ktoré sa stanovili centrálnym posúdením. Sekundárne cieľové ukazovatele zahŕňali trvanie odpovede, čas do odpovede nádoru a prežívanie bez progresie (PFS) podľa mRECIST u pacientov s
laBCC a RECIST 1.1 u pacientov s mBCC, stanovené centrálnym posúdením.
Kompozitná celková odpoveď nezávislej posudzovacej komisie u pacientov s laBCC zahŕňala centrálne vyhodnotené snímky MRI, digitálne klinické fotografie a histopatológiu podľa mRECIST. Viaceré biopsie sa pri laBCC odoberali vždy, keď hodnotenie odpovede bolo sťažené prítomnosťou ulcerácie lézie, cystou a/alebo zjazvením/fibrózou. Odpoveď nádoru v MRI sa vyhodnocovala podľa RECIST 1.1. Odpoveď na digitálnej klinickej fotografii sa hodnotila podľa upravených kritérií Svetovej zdravotníckej organizácie (WHO) [čiastočná odpoveď (PR): pokles súčtu kolmých priemerov (SPD) lézie o ≥50 %; kompletná odpoveď (CR): zmiznutie všetkých lézií; progredujúca choroba: nárast SPD lézií o ≥25 %]. Pri zloženej kompletnej odpovedi museli všetky metódy použité pri hodnotení preukázať neprítomnosť nádoru.
Z 230 randomizovaných pacientov bolo 79 pacientov zadelených do skupiny Odomza 200 mg.
Z týchto 79 pacientov bolo 66 (83,5 %) pacientov s laBCC (37 [46,8 %] s agresívnym histologickým nálezom a 29 [36,7 %] s neagresívnym histologickým nálezom) a 13 (16,5 %) pacientov s mBCC.
Medián veku všetkých pacientov užívajúcich Odomzo 200 mg bol 67 rokov (59,5 % bolo vo veku
>65 rokov), 60,8 % bolo mužov a 89,9 % belochov.
Väčšina pacientov (laBCC 74 %, mBCC 92 %) podstúpila v minulosti liečbu vrátane chirurgického zákroku (laBCC 73 %, mBCC 85 %), rádioterapie (laBCC 18 %, mBCC 54 %) a antineoplastických terapií (laBCC 23 %, mBCC 23 %).
Kľúčové výsledky účinnosti podľa centrálneho posúdenia a hodnotenia lokálnym skúšajúcim lekárom
sú uvedené v tabuľke 4.
T
abuľka 4 Prehľad účinnosti podľa centrálneho posúdenia a hodnotenia lokálnym skúšajúcim lekárom pre FAS
a
 O
domzo 200 mg
Centrálne Lokálny skúšajúci
O
domzo 200 mg
Centrálne Lokálny skúšajúci
 lekár laBCC laBCC N=66 N=66
Podiel objektívnych odpovedí, n (%) 37 (56,1) 47 (71,2)
lekár laBCC laBCC N=66 N=66
Podiel objektívnych odpovedí, n (%) 37 (56,1) 47 (71,2)
95 % IS (43,3; 68,3) (58,7; 81,7)
Kompletná odpoveď
| 3 (4,5)b
|
6 (9,1)
| Čiastočná odpoveď
| 34 (51,5)
| 41 (62,1)
| Stabilizácia choroby
| 23 (34,8)
| 14 (21,2)
| Progresia choroby
| 1 (1,5)
| 1 (1,5)
| Neznáma
| 5 (7,6)
| 4 (6,1)
|
|
|
Najlepšia celková odpoveď, n (%)
Čas do odpovede nádoru (mesiace)Medián 4,0 2,5
95 % IS (3,8; 5,6) (1,9; 3,7)
Trvanie odpovede
|
|
|
Počet udalostí*
| 10
| 21
|
Počet cenzorovaných
| 27
| 26
|
Medián (mesiace)
| NE
| 14,3
|
95 % IS (NE) (12,0; 20,2)
6 mesiacov
| 86,9 (68,6; 94,9)
| 89,8 (74,8; 96,1)
| 9 mesiacov
| 75,8 (55,7; 87,7)
| 80,7 (63,5; 90,4)
| 12 mesiacov
| 65,6 (43,2; 81,0)
| 70,9 (52,2; 83,3)
|
|
|
Pravdepodobnosť bez udalosti (%), (95 % IS)
Prežívanie bez progresie
|
|
|
Počet udalostí*
| 15
| 26
|
Počet cenzorovaných
| 51
| 40
|
Medián (mesiace)
| 22,1
| 19,4
|
95 % IS (NE) (16,6; 22,6)
Pravdepodobnosť prežívania bez progresie (%),
(95 % IS)
6 mesiacov
| 94,8 (84,6; 98,3)
| 94,8 (84,7; 98,3)
| 12 mesiacov
| 82,2 (67,0; 90,8)
| 76,0 (61,4; 85,7)
|
|
|
a Kompletný súbor pre analýzu zahŕňal všetkých randomizovaných pacientov (populácia, ktorá sa má liečiť).
b Pri použití len negatívneho histologického nálezu na definovanie CR medzi pacientmi, ktorí majú aspoň PR
podľa inej metódy (MRI alebo fotografia), bol podiel CR 22,7 %.
*Udalosť znamená progresiu choroby alebo úmrtie z akejkoľvek príčiny.
FAS: Kompletný súbor pre analýzu
IS: interval spoľahlivosti
NE: nedá sa odhadnúť (medián sa zatiaľ nedosiahol; celkové stredné trvanie ďalšieho sledovania

26,3 mesiacov)

Obrázok 1 zobrazuje najlepšiu zmenu veľkosti cieľovej lézie pre každého pacienta s laBCC pri dávke
200 mg podľa centrálneho posúdenia.
Obrázok 1 Najlepšia zmena cieľových lézií u pacientov s laBCC oproti východiskovej hodnotepodľa centrálneho posúdenia pre FAS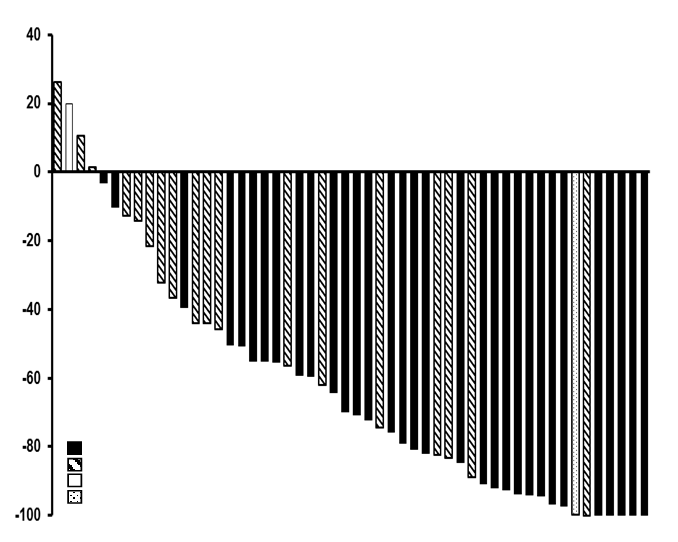
Odpoveď (kompletná odpoveď/čiastočná odpoveď)
Stabilná choroba Progredujúca choroba Neznáme
Výsledky hlásené pacientmi sa vyhodnotili ako výskumný cieľový ukazovateľ podľa dotazníka
o kvalite života Európskej organizácie pre výskum a liečbu rakoviny (European Organisation for
Research and Treatment of Cancer Quality of Life Questionnaire Core 30, EORTC QLQ-C30) a jeho
špecifického modulu pre rakovinu v oblasti hlavy a krku (H&N35).
U väčšiny pacientov došlo k stabilizácii a/alebo zlepšeniu príznakov súvisiacich s chorobou, bežných aktivít a zdravotného stavu. Čas do zhoršenia podľa vopred špecifikovaných škál PRO (zodpovedá zhoršeniu o >10 bodov bez následného zlepšenia) sa v podstate zhodoval s odhadovaným PFS.
V pivotnom klinickom skúšaní 27,8 % pacientov ukončilo účasť pre nežiaduce udalosti, ktoré boli väčšinou nezávažné alebo stredne závažné (pozri časť 4.8).
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Odomzom vo
všetkých podskupinách pediatrickej populácie pre bazocelulárny karcinóm (informácie o použití v
pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
A
bsorpcia
Po podaní jednorazovej dávky Odomza (100 mg až 3 000 mg) bez jedla pacientom s rakovinou bol
medián času do dosiahnutia maximálnej koncentrácie (Tmax) 2 až 4 hodiny. Sonidegib vykazoval zväčšenia AUC a Cmax úmerné dávke v rozmedzí dávok od 100 mg do 400 mg, ale menej ako dávke úmerné zväčšenia nad 400 mg. Podľa analýzy farmakokinetiky populácie sa nepreukázala zmena klírensu pri opakovanom podávaní a odhadovaná akumulácia v rovnovážnom stave predstavovala 19- násobok bez ohľadu na dávku. Rovnovážny stav sa dosahoval približne po 4 mesiacoch od začiatku užívania sonidegibu. Priemerná Ctrough v rovnovážnom stave pri 200 mg bola u pacientov s rakovinou
830 ng/ml (rozmedzie 200 až 2 400 ng/ml). V porovnaní so stavom nalačno sa pri dávke Odomza
800 mg zvýšili Cmax 7,8-násobne a AUC 7,4-násobne, keď sa dávka podala s jedlom s vysokým obsahom tukov.
Distribúcia
Na základe analýzy farmakokinetiky populácie 351 pacientov, ktorí dostali perorálne dávky Odomza
v rozmedzí dávok od 100 mg do 3 000 mg, bol zdanlivý distribučný objem v rovnovážnom stave (Vss/F) 9 170 litrov. Koncentrácia sonidegibu v koži bola v rovnovážnom stave 6-krát vyššia ako v plazme.
Sonidegib sa vo veľkej miere viazal na bielkoviny ľudskej plazmy (ľudský sérový albumín a kyslý alfa-1-glykoproteín) in vitro (>97 %), a väzba nezávisela od koncentrácie od 1 ng/ml do 2 500 ng/ml.
Podľa údajov in vitro nie je sonidegib substrátom P-gp, BCRP alebo proteínu multirezistencie 2 (MRP2). Sonidegib neinhiboval pri klinicky významných koncentráciách apikálne efluxné transportéry, P-gp alebo MRP2, hepatálne transportéry vychytávania OATP1B1 alebo OATP1B3, obličkové transportéry vychytávania organických aniónov OAT1 a OAT3, alebo transportéry vychytávania organických katiónov OCT1 alebo OCT2.
Biotransformácia
Sonidegib sa primárne metabolizuje prostredníctvom CYP3A4. Nezmenený sonidegib predstavoval
36 % cirkulujúcej rádioaktivity a hlavný cirkulujúci metabolit (45 % expozície pôvodnej látke)
identifikovaný v plazme je produktom hydrolýzy sonidegibu a je farmakologicky neaktívny. Všetky metabolity sa považovali za 4- až 90-krát menej účinné ako sonidegib.
Eliminácia
Sonidegib a jeho metabolity sa eliminujú primárne pečeňou, pričom 93,4 % podanej dávky sa našlo
v stolici a 1,95 % v moči. Nezmenený sonidegib v stolici predstavoval 88,7 % podanej dávky a v moči nebol zistiteľný. Polčas eliminácie (t1/2) sonidegibu odhadovaný na základe modelovania farmakokinetiky populácie bol približne 28 dní.
Osobitné skupiny pacientov
Pacienti s poruchou f unkcie pe č ene
Farmakokinetika sonidegibu sa skúmala u osôb s ľahkou (trieda A podľa Childa-Pugha; n=8) alebo
stredne ťažkou (trieda B podľa Childa-Pugha; n=8) poruchou funkcie pečene a u 8 zdravých osôb
s normálnou funkciou pečene. Cmax sonidegibu bola po jednorazovej perorálnej dávke 800 mg o 10 % nižšia pri ľahkej a o 19 % nižšia pri stredne ťažkej poruche funkcie pečene v porovnaní s normálnou funkciou pečene. AUCinf sonidegibu boli nižšie o 39 % a 6 %. AUClast bola o 34 % nižšia pri ľahkej poruche funkcie pečene a o 13 % vyššia pri stredne ťažkej poruche funkcie pečene. U pacientov
s ľahkou až stredne ťažkou poruchou funkcie pečene nie je potrebná úprava dávky. Sonidegib sa neskúmal u pacientov s ťažkou poruchou funkcie pečene (trieda C podľa Childa-Pugha).
Pacienti s poruchou f unkcie obli či ek
Vplyv poruchy funkcie obličiek na systémovú expozíciu sonidegibu sa neskúmal. Keďže sonidegib sa nevylučuje obličkami, neočakáva sa zmena systémovej expozície u pacientov s poruchou funkcie obličiek. Analýza farmakokinetiky populácie nezistila významný vplyv funkcie obličiek (klírens kreatinínu >27 ml/min) na zdanlivý klírens (CL/F) sonidegibu, čo naznačuje, že u pacientov
s poruchou funkcie obličiek nie je potrebná úprava dávky.
Vplyv veku, telesnej hmotnosti a pohlavia
Analýzy farmakokinetiky populácií nepreukázali klinicky významný vplyv veku (testované rozmedzie od 20-93 rokov, priemer 61 rokov), telesnej hmotnosti (testované rozmedzie 42-181 kg, priemer
77 kg), pohlavia, alebo klírensu kreatinínu (testované rozmedzie 27,3-290 ml/min, priemer
92,9 ml/min) na systémovú expozíciu sonidegibu.
Vply v et ni cke j príslušnosti
Pri jednorazovej dávke 200 mg bola u zdravých Japoncov Cmax sonidegibu 1,56-násobne vyššia a
AUCinf 1,68-násobne vyššia ako hodnoty pozorované u zdravých belochov.
5.3 Predklinické údaje o bezpečnosti
Sonidegib sa vyhodnotil u potkanov a psov.
Všeobecná toxikológia
Väčšina nežiaducich účinkov sonidegibu sa môže pripísať jeho farmakologickému mechanizmu
účinku na vývinové dráhy, pričom účinky u potkanov a psov boli podobné. Väčšina účinkov sa
vyskytla blízko plánovaných expozícií u ľudí. Tieto účinky pozorované pri klinicky významných expozíciách zahŕňajú uzavretie kostných rastových štrbín, vplyv na rastúce zuby, účinky na samčí a samičí reprodukčný systém, atrofiu vlasových folikulov s alopéciou, gastrointestinálnu toxicitu
s poklesom telesnej hmotnosti a účinky na lymfatické uzliny. Pri expozíciách podstatne vyšších, ako je klinická expozícia, boli ďalším cieľovým orgánom obličky.
Karcinogenéza a mutagenéza
Štúdie karcinogenity sa so sonidegibom nevykonali, ale sonidegib nebol genotoxický v štúdiách
in vitro a in vivo.
Reprodukčná a vývinová toxicita
Pri sonidegibe sa preukázala fetotoxicita u králikov, čo sa prejavilo abortom a/alebo úplnou resorpciou
plodov, a teratogenita, ktorá spôsobila ťažké malformácie pri veľmi nízkej expozícii. Teratogénne
účinky zahŕňali malformácie stavcov, distálnych častí končatín a prstov, ťažké kraniofaciálne malformácie a iné závažné defekty pozdĺž stredovej línie. Fetotoxicita u králikov sa pozorovala aj pri veľmi nízkej expozícii u matiek. Pri nízkej expozícii bola znížená fertilita u samíc potkana. U samcov potkana, ktorým sa podával sonidegib, expozícia približne 2-násobná oproti klinickej expozícii neovplyvnila samčiu fertilitu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
O
bsah kapsuly
krospovidón (typ A)
monohydrát laktózy magnéziumstearát
poloxamér (188)
koloidný oxid kremičitý
laurylsíran sodný
Telo kapsuly
želatína
červený oxid železitý (E172)
oxid titaničitý (E171)
Atrament na potlač
čierny oxid železitý (E172)
propylénglykol (E1520)
šelak
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
10 x 1 tvrdá kapsula v perforovaných PCTFE/PVC/Al blistroch s jednotlivými dávkami.
Každé balenie obsahuje buď 10, alebo 30 tvrdých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/15/1030/001
EU/1/15/1030/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu