
nických skúšaniach. Nie sú žiadne skúsenosti u pacientov so stredne ťažkou a ťažkou poruchou funkcie obličiek. Ocrevus je monoklonálna protilátka, ktorá podlieha katabolizmu (t. j. rozpadu na peptidy a aminokyseliny), a preto sa nepredpokladá, že u pacientov s poruchou funkcie obličiek je potrebná zmena dávky (pozri časť 5.2).
Porucha funkcie pečene
Bezpečnosť a účinnosť Ocrevusu u pacientov s poruchou funkcie pečene neboli oficiálne skúmané. Pacienti s miernou poruchou funkcie pečene boli zahrnutí v klinických skúšaniach. Nie sú žiadne skúsenosti u pacientov so stredne ťažkou a ťažkou poruchou funkcie pečene. Ocrevus je monoklonálna protilátka, ktorá podlieha katabolizmu (a nie hepatálnemu metabolizmu), a preto sa nepredpokladá, že
u pacientov s poruchou funkcie pečene je potrebná zmena dávky (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť Ocrevusu u detí a dospievajúcich vo veku od 0 do 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Po nariedení sa Ocrevus podáva intravenóznou infúziou cez osobitnú infúznu hadičku. Infúzny roztok
Ocrevusu sa nemá podávať formou pretlakovej infúzie (tzv. i.v. push) ani intravenóznej bolusovej injekcie.
T
abuľka 1: Dávka a schéma podávania Ocrevusu
Množstvo
O
crevusu, ktoré sa
m
á podať
P
okyny na podávanie
i
n
f
úzie
Ú
vodná dávka
(
600 mg)
1. infúzia 300 mg v 250 ml • Infúziu začnite
podávať rýchlosťou
rozdelená do 2 infúzií
2. infúzia (o 2 týždne neskôr)
300 mg v 250 ml
30 ml/hodinu počas
30 minút
• Rýchlosť sa môže zvyšovať v prírastkoch o 30 ml/hodinu každých 30 minút do maximálnej rýchlosti
180 ml/hodinu
• Každá infúzia sa má podávať počas približne 2,5 hodiny
Ď
alšie dávky*
(
600 mg)
raz za 6 mesiacov
Jedna infúzia 600 mg v 500 ml • Infúziu začnite podávať rýchlosťou
40 ml/hodinu počas
30 minút
• Rýchlosť sa môže zvyšovať v prírastkoch o 40 ml/hodinu každých 30 minút do maximálnej rýchlosti
200 ml/hodinu
• Každá infúzia sa má podávať počas približne 3,5 hodiny
Roztok Ocrevusu na intravenóznu infúziu sa pripraví nariedením lieku v infúznom vaku, ktorý
obsahuje 0,9 % roztok chloridu sodného, na konečnú koncentráciu približne 1,2 mg/ml. Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
Pacienti majú byť sledovaní počas podávania infúzie a aspoň jednu hodinu po ukončení infúzie (pozri časť 4.4).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Aktívna infekcia v súčasnosti (pozri časť 4.4).
• Pacienti so závažne oslabeným imunitným systémom (pozri časť 4.4).
• Známe aktívne malignity (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Na zlepšenie sledovateľnosti biologických liekov sa má jasne zaznamenať názov a číslo šarže
podaného lieku.
Reakcie súvisiace sinfúziou(IRR)
K Ocrevusu sú pridružené IRR, ktoré môžu súvisieť s uvoľňovaním cytokínov a/alebo iných
chemických mediátorov.
Príznaky IRR sa môžu vyskytnúť počas ktorejkoľvek infúzie, ale častejšie boli hlásené počas prvej infúzie. IRR sa môžu vyskytnúť do 24 hodín od podania infúzie. Tieto reakcie sa môžu prejavovať ako pruritus, vyrážka, urtikária, erytém, podráždenie hrdla, orofaryngeálna bolesť, dyspnoe, faryngeálny alebo laryngeálny edém, návaly tepla, hypotenzia, pyrexia, únava, bolesť hlavy, závraty, nauzea
a tachykardia (pozri časť 4.8).
Pred infúziou:
• Zvládnutie ťažkých reakcií: K dispozícii má byť vhodné vybavenie na zvládnutie ťažkých reakcií, akými sú závažné IRR, reakcie z precitlivenosti a/alebo anafylaktické reakcie.
• Počas podávania infúzií Ocrevusu sa môže vyskytnúť hypotenzia ako príznak IRR. Preto sa má zvážiť vysadenie antihypertenzív 12 hodín pred podaním každej infúzie Ocrevusu a počas jej podávania. Pacienti s kongestívnym srdcovým zlyhávaním (triedy III a IV podľa New York Heart Association) v anamnéze neboli sledovaní.
• Premedikácia: Pacientom sa musí podať premedikácia na zníženie výskytu a závažnosti IRR
(pozri časť 4.2).
Počas infúzie:
• Nasledujúce opatrenia je potrebné urobiť u pacientov, u ktorých sa vyskytnú závažné pľúcne príznaky, napríklad bronchospazmus alebo exacerbácia astmy:
- podávanie infúzie sa musí ihneď a natrvalo ukončiť
- musí sa podať symptomatická liečba
- pacient musí byť sledovaný, až kým pľúcne príznaky neodznejú, pretože po úvodnom zlepšení klinických príznakov môže nasledovať ich zhoršenie.
• Môže byť ťažké rozlíšiť príznaky precitlivenosti a IRR. Ak počas podávania infúzie vznikne podozrenie na reakciu z precitlivenosti, podávanie infúzie sa musí ihneď a natrvalo ukončiť (pozri nižšie „Reakcie z precitlivenosti“).
Po infúzii:
• Pacienti liečení Ocrevusom majú byť sledovaní aspoň jednu hodinu po ukončení infúzie kvôli akémukoľvek možnému príznaku IRR.
• Lekári majú upozorniť pacientov, že IRR sa môže vyskytnúť do 24 hodín od podania infúzie. Odporúčania týkajúce sa dávkovania u pacientov, u ktorých sa vyskytnú príznaky IRR, pozri časť 4.2.
R
eakcie z precitlivenosti
Môže sa vyskytnúť aj reakcia z precitlivenosti (akútna alergická reakcia na liek). Akútne reakcie
z precitlivenosti 1. typu (sprostredkované IgE) môžu byť klinicky neodlíšiteľné od príznakov IRR.
Reakcia z precitlivenosti sa môže vyskytnúť počas ktorejkoľvek infúzie, hoci sa zvyčajne nevyskytuje počas prvej infúzie. Ak sa pri ďalších infúziách vyskytnú závažnejšie príznaky ako tie, ktoré sa vyskytli predtým, alebo nové závažné príznaky, je nutné zvážiť možnosť reakcie z precitlivenosti. Pacienti so známou IgE sprostredkovanou precitlivenosťou na okrelizumab sa nesmú liečiť (pozri
časť 4.3).
Infekcia
Podávanie Ocrevusu sa musí odložiť u pacientov s aktívnou infekciou, až kým infekcia neustúpi.
Odporúča sa zistiť stav imunitného systému pacienta pred podaním lieku, pretože pacienti so závažne oslabeným imunitným systémom (napr. s lymfopéniou, neutropéniou, hypogamaglobulinémiou) sa nemajú liečiť (pozri časti 4.3 a 4.8).
Celkové percento pacientov, u ktorých sa vyskytla závažná infekcia, bolo podobné ako
pri komparátoroch (pozri časť 4.8). Frekvencia výskytu infekcií 4. stupňa (život ohrozujúcich)
a 5. stupňa (smrteľných) bola nízka vo všetkých liečebných skupinách, ale pri PPMS bola vyššia
pri Ocrevuse v porovnaní s placebom v prípade život ohrozujúcich (1,6 % oproti 0,4 %) a smrteľných
(0,6 % oproti 0 %) infekcií. Všetky život ohrozujúce infekcie ustúpili bez potreby ukončenia liečby okrelizumabom.
Pri PPMS sú pacienti, ktorí majú problémy s prehĺtaním, vystavení vyššiemu riziku vzniku aspiračnej pneumónie. U týchto pacientov môže liečba Ocrevusom ešte viac zvýšiť riziko vzniku závažnej pneumónie. U pacientov s prejavmi pneumónie majú lekári prijať okamžité opatrenia.
Progresívna multifokálna leukoencefalopatia (PML)
Riziko vzniku PML nie je možné vylúčiť, pretože u pacientov, ktorí boli liečení
anti-CD20 protilátkami a inými liekmi na SM, bola hlásená infekcia spôsobená vírusom JC
(pomenovaným podľa Johna Cunninghama), ktorá viedla k PML a dávala sa do súvislosti s rizikovými faktormi (napr. populácia pacientov, liečba viacerými imunosupresívami).
Lekári musia byť ostražití ohľadom včasných prejavov a príznakov PML, ktoré môžu zahŕňať
akékoľvek novovzniknuté alebo zhoršujúce sa neurologické prejavy alebo príznaky, pretože môžu byť podobné SM.
Ak je podozrenie na PML, podávanie Ocrevusu sa musí prerušiť. Je potrebné zvážiť zhodnotenie stavu pacienta zahŕňajúce vyšetrenie magnetickou rezonanciou (MR) najlepšie s použitím kontrastnej látky
(a porovnať s MR snímkou vyhotovenou pred začiatkom liečby), vyšetrenie mozgovomiechového moku (CSF) na potvrdenie prítomnosti kyseliny deoxyribonukleovej (DNA) vírusu JC (pomenovaným podľa Johna Cunninghama) a zopakovanie neurologických vyšetrení. Ak sa PML potvrdí, liečba sa
musí natrvalo ukončiť.
Reaktivácia vírusu hepatitídy B
U pacientov liečených inými anti-CD20 protilátkami bola hlásená reaktivácia vírusu hepatitídy B (HBV), ktorá v niektorých prípadoch viedla k fulminantnej hepatitíde, k zlyhaniu pečene a k smrti.
Pred začiatkom liečby Ocrevusom sa má u všetkých pacientov vykonať skríning na HBV v súlade
s lokálnymi odporúčaniami. Pacienti s aktívnym HBV (t. j. aktívna infekcia potvrdená pozitívnymi výsledkami vyšetrení na prítomnosť HBsAg a anti-HB) sa nemajú liečiť Ocrevusom. Liečba pacientov so sérologickou pozitivitou (t. j. negativita HBsAg a pozitivita protilátok proti jadrového antigénu HB (HBcAb+); nosiči HBV (pozitivita povrchového antigénu, HBsAg+) má byť pred začiatkom konzultovaná s hepatológom a títo pacienti majú byť sledovaní a liečení v súlade s lokálnymi
postupmi, aby sa predišlo reaktivácii hepatitídy B.
Malignity
V klinických skúšaniach sa u pacientov liečených okrelizumabom v porovnaní s kontrolnými
skupinami pozoroval zvýšený počet malignít (vrátane karcinómov prsníka). Ich výskyt bol však v rozmedzí prirodzeného (referenčného) výskytu očakávaného v populácii so SM. U pacientov
so známymi rizikovými faktormi vzniku malignít a u pacientov, ktorí sú aktívne sledovaní kvôli možnej recidíve malignity, sa má individuálne zvážiť prínos a riziko liečby. Pacienti so známou
aktívnou malignitou sa nemajú liečiť Ocrevusom (pozri časť 4.3). Pacienti majú podstúpiť štandardný skríning rakoviny prsníka v súlade s lokálnymi odporúčaniami. Populácie, ktoré neboli sledované, pozri časť 4.2.
Počas kontrolovaného obdobia klinických skúšaní bol výskyt nemelanómových karcinómov kože nízky a medzi liečebnými skupinami nebola nerovnováha v ich výskyte. Zvýšenie výskytu sa pozorovalo medzi 3. a 4. rokom liečby, a to v dôsledku výskytu bazocelulárneho karcinómu, ktorý sa
v nasledujúcich rokoch nepozoroval. Výskyt zostáva v rozmedzí prirodzeného (referenčného) výskytu očakávaného v populácii so SM.
Liečba pacientovsozávažneoslabenýmimunitnýmsystémom
Pacienti so závažne oslabeným imunitným systémom sa nesmú liečiť, až kým sa ich stav neupraví
(pozri časť 4.3).
Pri iných autoimunitných ochoreniach viedlo použitie Ocrevusu súbežne s imunosupresívnymi liekmi (napr. s dlhodobo podávanými kortikosteroidmi, nebiologickými a biologickými antireumatikami modifikujúcimi priebeh ochorenia [disease-modifying antirheumatic drugs, DMARDS], mofetilmykofenolátom, cyklofosfamidom, azatioprínom) k zvýšenému výskytu závažných infekcií vrátane oportúnnych infekcií. Infekcie zahŕňali, ale neobmedzovali sa iba na atypickú pneumóniu
a pneumóniu spôsobenú Pneumocystis jirovecii, pneumóniu spôsobenú vírusom varicella-zoster, tuberkulózu, histoplazmózu. V zriedkavých prípadoch boli niektoré z týchto infekcií smrteľné.
V exploračnej analýze sa identifikovali nasledujúce faktory súvisiace s rizikom vzniku závažných infekcií: vyššie dávky Ocrevusu ako sú dávky odporúčané pri SM, iné komorbidity a dlhodobé používanie imunosupresív/kortikosteroidov.
Neodporúča sa používať iné imunosupresíva súbežne s Ocrevusom okrem kortikosteroidov na symptomatickú liečbu relapsov. K dispozícii sú obmedzené poznatky o tom, či je súbežné používanie kortikosteroidov na symptomatickú liečbu relapsov spájané so zvýšeným rizikom vznikom infekcií v klinickej praxi. V pivotných štúdiách s okrelizumabom zameraných na SM sa podávanie kortikosteroidov na liečbu relapse nespájalo so zvýšeným rizikom vzniku závažnej infekcie.
Keď sa liečba Ocrevusom začína po imunosupresívnej liečbe alebo keď sa imunosupresívna liečba začína po liečbe Ocrevusom, má sa zvážiť možnosť prekrývajúcich sa farmakodynamických účinkov (pozri časť 5.1 Farmakodynamické účinky). Pri predpisovaní Ocrevusu je potrebná obozretnosť a má sa vziať do úvahy farmakodynamika iných liekov modifikujúcich priebeh SM.
O
čkovanie
Bezpečnosť imunizácie živými alebo živými oslabenými očkovacími látkami po liečbe Ocrevusom sa
nesledovala a očkovanie živými oslabenými alebo živými očkovacími látkami sa neodporúča počas liečby a až do obnovy počtu B-lymfocytov (v klinických skúšaniach bol medián času do obnovy počtu B-lymfocytov 72 týždňov). Pozri časť 5.1.
K dispozícii nie sú žiadne údaje o účinkoch očkovania u pacientov liečených Ocrevusom. Lekári majú posúdiť stav imunizácie pacientov, u ktorých sa uvažuje o liečbe Ocrevusom. U pacientov, ktorí potrebujú očkovanie, má byť imunizácia dokončená aspoň 6 týždňov pred začiatkom liečby Ocrevusom.
Ďalšie informácie o očkovaní, pozri časť 4.5.
Expozícia okrelizumabu in utero a očkovanie dojčiat živými a živými oslabenými očkovacími látkami
Vzhľadom na možnú depléciu B-lymfocytov u dojčiat matiek, ktoré boli vystavené pôsobeniu Ocrevusu počas gravidity, majú byť dojčatá sledované kvôli deplécii B-lymfocytov a očkovanie živými a živými oslabenými očkovacími látkami sa má odložiť, až kým sa u dojčaťa počet
B-lymfocytov neobnoví. Bezpečnosť a termín očkovania sa má prediskutovať s lekárom dojčaťa
(pozri časť 4.6).
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne oficiálne štúdie liekových interakcií, pretože sa neočakávajú žiadne liekové interakcie sprostredkované enzýmami cytochrómu P450, inými enzýmami metabolizujúcimi lieky alebo transportérmi.
Očkovanie
Bezpečnosť imunizácie živými alebo živými oslabenými vírusovými očkovacími látkami po liečbe
Ocrevusom sa nesledovala.
K dispozícii nie sú žiadne údaje o účinkoch očkovania u pacientov liečených Ocrevusom. Pozri časť 4.4.
Po liečbe Ocrevusom trvajúcej 2 roky bol podiel pacientov s pozitívnymi titrami protilátok proti
S. pneumoniae, príušniciam, ružienke a ovčím kiahňam vo všeobecnosti podobný podielu zistenému
pred začiatkom liečby.
Imunosupresíva
Neodporúča sa používať iné imunosupresíva súbežne s Ocrevusom okrem kortikosteroidov
na symptomatickú liečbu relapsov.
Informáciu o používaní imunosupresív pred, počas alebo po liečbe Ocrevusom, pozri časť 4.4 pod
„Liečba pacientov so závažne oslabeným imunitným systémom“.
4.6 Fertilita, gravidita a laktácia
Ž
eny vofertilnomveku
Ženy vo fertilnom veku musia používať antikoncepciu počas liečby Ocrevusom a počas 12 mesiacov
po poslednej infúzii Ocrevusu (pozri nižšie a časti 5.1 a 5.2).
Gravidita
Ocrevus je humanizovaná monoklonálna protilátka podtriedy imunoglobulínu G1 a je známe, že
imunoglobulíny prechádzajú placentárnou bariérou.
K dispozícii je obmedzené množstvo údajov o použití Ocrevusu u gravidných žien. Nezozbierali sa žiadne údaje o počte B-lymfocytov u dojčiat vystavených pôsobeniu Ocrevusu a možná dĺžka trvania deplécie B-lymfocytov nie je známa (pozri časť 4.4).
U dojčiat narodených matkám, ktoré boli vystavené pôsobeniu iných anti-CD20 protilátok počas gravidity, bola hlásená prechodná deplécia B-lymfocytov v periférnej krvi a lymfocytopénia.
Štúdie (embryofetálnej toxicity) na zvieratách nepreukázali teratogénne účinky. Zistila sa deplécia
B-lymfocytov in utero. V štúdiách prenatálneho a postnatálneho vývoja bola pozorovaná reprodukčná toxicita (pozri časť 5.3).
Je potrebné vyhnúť sa liečbe Ocrevusom počas gravidity, pokiaľ možný prínos pre matku neprevažuje možné riziko pre plod.
Dojčenie
Nie je známe, či sa okrelizumab/metabolity vylučujú do ľudského mlieka. Dostupné
farmakodynamické/toxikologické údaje získané u zvierat preukázali vylučovanie okrelizumabu
do mlieka (podrobnosti, pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť vylúčené. Ženám sa má odporučiť, aby počas liečby Ocrevusom nedojčili.
Fertilita
Predklinické údaje získané na základe štúdií samčej a samičej fertility vykonaných na opiciach rodu
Cynomolgus neodhalili žiadne osobitné riziká pre ľudí.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ocrevus nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Najvýznamnejšie a najčastejšie hlásené nežiaduce reakcie na liek (adverse drug reactions, ADR) boli
IRR a infekcie. Ďalšie podrobnosti, pozri časť 4.4 a časť 4.8 (podčasť „Opis vybraných nežiaducich reakcií“).
Tabuľkový zoznamnežiaducichreakcií
Celkový bezpečnostný profil Ocrevusu pri skleróze multiplex je založený na údajoch od pacientov
z pivotných klinických skúšaní zameraných na SM (RSM a PPSM).
V tabuľke 2 sú zhrnuté ADR, ktoré boli hlásené v súvislosti s použitím Ocrevusu u 1 311 pacientov
(3 054 pacientorokov) počas kontrolovaných období liečby v klinických skúšaniach zameraných na SM.
Frekvencie výskytu sú definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) a veľmi zriedkavé (< 1/10 000). V rámci každej triedy orgánových systémov (SOC) sú nežiaduce reakcie zoradené podľa klesajúcej frekvencie.
Tabuľka 2 ADR hlásené pri Ocrevuse (pri RSM alebo PPSM)
MedDRA
V
eľmi časté Časté
T
rieda orgánových
systémov (SOC)
Infekcie a nákazy Infekcia horných dýchacích
ciest, nazofaryngitída, chrípka
Poruchy dýchacej sústavy, hrudníka a mediastína
Sinusitída, bronchitída,
orálny herpes, gastroenteritída, infekcia dýchacích ciest, vírusová infekcia, herpes zoster, konjunktivitída, celulitída Kašeľ, katar
L
aboratórne a funkčné vyšetrenia
Znížená koncentrácia imunoglobulínu M v krvi
Znížená koncentrácia imunoglobulínu G v krvi
P
oruchy krvi
a lymfatického systému
Neutropénia
Ú
razy, otravy a komplikácie liečebného postupu
Reakcie súvisiace s infúziou1

1 Príznaky hlásené ako IRR do 24 hodín od podania infúzie sú opísané nižšie pod „Reakcie súvisiace s infúziou“.
Opis vybraných nežiaducichreakciíReakcie súvisiace s infúziouNaprieč klinickými skúšaniami zameranými na RSM a PPSM príznaky súvisiace s IRR zahŕňali,
ale neobmedzovali sa iba na: pruritus, vyrážku, urtikáriu, erytém, návaly tepla, hypotenziu, pyrexiu, únavu, bolesť hlavy, závraty, podráždenie hrdla, orofaryngeálnu bolesť, dyspnoe, faryngeálny
alebo laryngeálny edém, nauzeu, tachykardiu. V kontrolovaných klinických skúšaniach sa nevyskytli
žiadne smrteľné IRR.
V klinických skúšaniach kontrolovaných aktívnym komparátorom (zameraných na RSM) bola IRR
najčastejšia nežiaduca udalosť u pacientov liečených Ocrevusom, s celkovým výskytom 34,3 % v porovnaní s výskytom 9,9 % v liečebnej skupine s interferónom beta-1a (infúzia s placebom). Výskyt IRR bol najvyšší počas podávania 1. dávky, 1. infúzie (27,5 %) a v priebehu času sa znížil na < 10 % pri podávaní 4. dávky. IRR boli v obidvoch liečebných skupinách väčšinou mierne
až stredne ťažké. Mierne IRR sa vyskytli u 21,7 % a stredne ťažké IRR u 10,1 % pacientov liečených
Ocrevusom, u 2,4 % sa vyskytli ťažké IRR a u 0,1 % sa vyskytli život ohrozujúce IRR. Pozri časť 4.4.
V klinickom skúšaní kontrolovanom placebom (zameranom na PPSM) bola IRR najčastejšia nežiaduca udalosť u pacientov liečených Ocrevusom, s celkovým výskytom 40,1 % v porovnaní s výskytom 25,5 % v skupine s placebom. Výskyt IRR bol najvyšší počas podávania 1. dávky,
1. infúzie (27,4 %) a pri ďalších dávkach sa znižoval, a to až na < 10 % pri podávaní 4. dávky. U vyššieho percenta pacientov v každej skupine sa IRR vyskytla pri prvej infúzii každej dávky
v porovnaní s druhou infúziou danej dávky. IRR boli väčšinou mierne až stredne ťažké. Mierne IRR sa vyskytli u 26,7 % a stredne ťažké IRR u 11,9 % pacientov liečených Ocrevusom, u 1,4 % sa vyskytli ťažké IRR. Nevyskytli sa žiadne život ohrozujúce IRR. Pozri časť 4.4.
Infekcia
V klinických skúšaniach kontrolovaných aktívnym komparátorom a zameraných na RSM sa infekcie vyskytli u 58,5 % pacientov liečených Ocrevusom v porovnaní s 52,5 % pacientov liečených interferónom beta-1a. Závažné infekcie sa vyskytli u 1,3 % pacientov liečených Ocrevusom
v porovnaní s 2,9 % pacientov liečených interferónom beta-1a. V placebom kontrolovanom klinickom skúšaní zameranom na PPSM sa infekcie vyskytli u 72,2 % pacientov liečených Ocrevusom
v porovnaní so 69,9 % pacientov, ktorým sa podávalo placebo. Závažné infekcie vyskytli
u 6,2 % pacientov liečených Ocrevusom v porovnaní so 6,7 % pacientov, ktorým sa podávalo placebo. Zvýšenie výskytu závažných infekcií sa pozorovalo pri RMS medzi 2. a 3. rokom, ale nie
v nasledujúcich rokoch. Pri PPMS sa nepozorovalo žiadne zvýšenie ich výskytu.
Infekcie dýchacích ciest
Podiel infekcií dýchacích ciest bol vyšší u pacientov liečených Ocrevusom v porovnaní s pacientmi,
ktorým sa podával interferón beta-1a a placebo.
V klinických skúšaniach zameraných na RSM sa infekcia horných dýchacích ciest vyskytla
u 39,9 % pacientov liečených Ocrevusom a u 33,2 % pacientov liečených interferónom beta-1a a infekcia dolných dýchacích ciest vyskytla u 7,5 % pacientov liečených Ocrevusom
a u 5,2 % pacientov liečených interferónom beta-1a.
V klinických skúšaniach zameraných na PPSM sa infekcia horných dýchacích ciest vyskytla u 48,8 % pacientov liečených Ocrevusom a u 42,7 % pacientov, ktorým sa podávalo placebo
a infekcia dolných dýchacích ciest vyskytla u 9,9 % pacientov liečených Ocrevusom
a u 9,2 % pacientov, ktorým sa podávalo placebo.
Infekcie dýchacích ciest hlásené u pacientov liečených Ocrevusom boli prevažne mierne až stredne ťažké (80 - 90 %).
Herpes
V klinických skúšaniach kontrolovaných aktívnym komparátorom (zameraných na RSM) boli
herpetické infekcie hlásené častejšie u pacientov liečených Ocrevusom ako u pacientov liečených interferónom beta-1a a zahŕňali herpes zoster (2,1 % oproti 1,0 %), herpes simplex (0,7 % oproti
0,1 %), orálny herpes (3,0 % oproti 2,2 %), genitálny herpes (0,1 % oproti 0 %) a infekciu spôsobenú
herpetickým vírusom (0,1 % oproti 0 %). Infekcie boli prevažne miernej až stredne ťažkej závažnosti a pacienti sa uzdravili po liečbe štandardnými liekmi.
V placebom kontrolovanom klinickom skúšaní (zameranom na PPSM) bol pozorovaný vyšší percentuálny podiel pacientov s orálnym herpesom v liečebnej skupine s Ocrevusom (2,7 % oproti
0,8 %).
Laboratórne abnormality
I
m
unoglobulíny
Liečba Ocrevusom viedla k zníženiu koncentrácií celkových imunoglobulínov počas kontrolovaného
obdobia klinických skúšaní, čo bolo podmienené hlavne znížením koncentrácie IgM. Môže existovať súvislosť medzi pretrvávajúcim znížením koncentrácií IgG, IgM alebo IgA a závažnými infekciami, ale z dôvodu obmedzenej expozície a obmedzeného počtu pacientov nie je možné vyvodiť definitívne závery.
V klinických skúšaniach kontrolovaných aktívnym komparátorom (zameraných na RSM) bol percentuálny podiel pacientov, u ktorých sa pri zaradení do štúdie zistili koncentrácie IgG, IgA a IgM < dolná hranica referenčného rozpätia (lower limit of normal, LLN), v liečebnej skupine
s Ocrevusom 0,5 %, 1,5 % a 0,1 % v uvedenom poradí. Po liečbe bol percentuálny podiel pacientov liečených Ocrevusom, u ktorých sa zistili koncentrácie IgG, IgA a IgM < LLN v 96. týždni, 1,5 %,
2,4 % a 16,5 % v uvedenom poradí.
V placebom kontrolovanom klinickom skúšaní (zameranom na PPSM) bol percentuálny podiel pacientov, u ktorých sa pri zaradení do štúdie zistili koncentrácie IgG, IgA a IgM < LLN, v liečebnej skupine s Ocrevusom 0,0 %, 0,2 % a 0,2 % v uvedenom poradí. Po liečbe bol percentuálny podiel pacientov liečených Ocrevusom, u ktorých sa zistili koncentrácie IgG, IgA a IgM < LLN v 120. týždni,
1,1 %, 0,5 % a 15,5 % v uvedenom poradí.
Lymfocyty
Pri RMS bolo zníženie počtu lymfocytov na < LLN pozorované u 20,7 % pacientov liečených
Ocrevusom v porovnaní s 32,6 % pacientov liečených interferónom beta-1a. Pri PPMS bolo zníženie počtu lymfocytov na < LLN pozorované u 26,3 % pacientov liečených Ocrevusom v porovnaní
s 11,7 % pacientov, ktorým sa podávalo placebo.
Zníženia počtu lymfocytov hlásené u pacientov liečených Ocrevusom boli väčšinou 1. stupňa
(< LLN - 800 buniek/mm3) a 2. stupňa (500 až 800 buniek/mm3) závažnosti. Približne u 1 % pacientov v skupine s Ocrevusom sa vyskytla lymfopénia 3. stupňa (200 až 500 buniek/mm3). U žiadneho
z týchto pacientov nebola hlásená lymfopénia 4. stupňa (< 200 buniek/mm3).
Pozoroval sa zvýšený výskyt závažných infekcií počas epizód potvrdeného zníženia celkového počtu lymfocytov u pacientov liečených okrelizumabom. Počet závažných infekcií bol príliš nízky na vyvodenie definitívnych záverov.
Neutrofily
V období liečby (RSM) kontrolovanej aktívnym komparátorom bolo zníženie počtu neutrofilov
na < LLN pozorované u 14,7 % pacientov liečených Ocrevusom v porovnaní so 40,9 % pacientov
liečených interferónom beta-1a. V placebom kontrolovanom klinickom skúšaní (zameranom
na PPSM) bol percentuály podiel pacientov, ktorí mali znížený počet neutrofilov, vyšší v skupine s Ocrevusom (12,9 %) ako v skupine s placebom (10,0 %); u týchto pacientov sa neutropénia
2. alebo vyššieho stupňa vyskytla u vyššieho percenta pacientov v skupine s Ocrevusom (4,3 %) ako
u pacientov v skupine s placebom (1,3 %); neutropénia 4. stupňa sa vyskytla približne u 1 % pacientov v skupine s Ocrevusom v porovnaní s 0 % pacientov v skupine s placebom.
Zníženie počtu neutrofilov bolo väčšinou prechodné (zistené iba jedenkrát u daného pacienta liečeného Ocrevusom) a 1. stupňa (< 1 500 buniek/mm3) a 2. stupňa (1 000 až 1 500 buniek/mm3) závažnosti. U jedného pacienta s neutropéniou 3. stupňa (500 až 1 000 buniek/mm3) a u jedného pacienta s neutropéniou 4. stupňa (< 500 buniek/mm3) bola potrebná špecifická liečba faktorom stimulujúcim kolónie granulocytov a po epizóde pokračovali v liečbe okrelizumabom.
I
né
Jeden pacient, ktorému sa podalo 2 000 mg Ocrevusu, zomrel na syndróm systémovej zápalovej
odpovede (systemic inflammatory response syndrome, SIRS) neznámej etiológie, a to po vyšetrení magnetickou rezonanciou (MR) vykonaným 12 týždňov po poslednej infúzii; anafylaktoidná reakcia na gadolíniovú kontrastnú látku použitú pri MR mohla prispieť k SIRS.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieZ klinických skúšaní sú k dispozícii obmedzené skúsenosti s podávaním dávok vyšších ako je schválená intravenózna dávka Ocrevusu. Najvyššia dávka doteraz testovaná u pacientov so SM je
2 000 mg, podaných vo forme dvoch 1 000 mg intravenóznych infúzií s časovým odstupom 2 týždňov
(štúdia fázy II zisťujúca optimálnu dávku pri RRSM). Nežiaduce reakcie na liek sa zhodovali s bezpečnostným profilom Ocrevusu zisteným v pivotných klinických skúšaniach.
Informáciu o syndróme systémovej zápalovej odpovede (SIRS), ktorá sa vyskytla u jedného pacienta liečeného 2 000 mg Ocrevusu, pozri časť 4.8.
K dispozícii nie je špecifické antidotum pre prípad predávkovania; je potrebné okamžite prerušiť podávanie infúzie a sledovať pacienta kvôli možným IRR (pozri časť 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: selektívne imunosupresíva, ATC kód: L04AA36.
MechanizmusúčinkuOkrelizumab je rekombinantná humanizovaná monoklonálna protilátka, ktorá sa selektívne zameriava
na B-lymfocyty exprimujúce CD20.
CD20 je povrchový antigén nachádzajúci sa na pre-B-lymfocytoch, zrelých a pamäťových
B-lymfocytoch, ale nie je exprimovaný na lymfoidných kmeňových bunkách a plazmatických bunkách.
Presný mechanizmus, ktorým okrelizumab vykazuje terapeutické klinické účinky pri SM,
nie je úplne objasnený, ale predpokladá sa, že zahŕňa imunomoduláciu prostredníctvom zníženia počtu a funkcie CD20-exprimujúcich B-lymfocytov. Po naviazaní sa na bunkový povrch spôsobuje okrelizumab selektívnu depléciu CD20-exprimujúcich B-lymfocytov prostredníctvom fagocytózy závislej
od protilátky (ADCP), cytotoxicity závislej od protilátky (ADCC), cytotoxicity závislej
od komplementu (CDC) a apoptózy. Schopnosť obnovy B-lymfocytov a už existujúca humorálna imunita sú zachované. Okrem toho, vrodená imunita a celkový počet T-lymfocytov nie sú ovplyvnené.
Farmakodynamické účinkyLiečba Ocrevusom vedie k rýchlej deplécii CD19+ B-lymfocytov v krvi do 14 dní po začatí liečby
(prvý časový bod hodnotenia), čo je očakávaný farmakologický účinok. Táto deplécia sa udržala počas celého obdobia liečby. Na meranie počtu B-lymfocytov sa používa CD19, pretože prítomnosť
Ocrevusu interferuje s testom na rozpoznávanie CD20.
V štúdiách fázy III sa preukázalo, že medzi podaním jednotlivých dávok Ocrevusu došlo až
u 5 % pacientov k obnove počtu B-lymfocytov (na > dolná hranica referenčného rozpätia (lower limit of normal, LLN) alebo na východiskový počet) aspoň v jednom hodnotenom časovom bode. Rozsah
a trvanie deplécie B-lymfocytov sa v klinických skúšaniach zameraných na PPSM a RSM zhodovali.
Najdlhšie obdobie sledovania po podaní poslednej infúzie Ocrevusu (štúdia fázy II, WA21493,
N = 51) ukazuje, že medián času do obnovy počtu B-lymfocytov (návrat na východiskový počet/LLN, podľa toho, čo sa vyskytlo ako prvé) bol 72 týždňov (rozmedzie 27 - 175 týždňov). U 90 % všetkých
pacientov sa počet B-lymfocytov vrátil na LLN alebo východiskový počet približne do dva a pol roka po poslednej infúzii.
Klinická účinnosť abezpečnosť
Relapsujúce formy SM
Účinnosť a bezpečnosť Ocrevusu sa hodnotili v dvoch randomizovaných, dvojito zaslepených, dvojito maskovaných (double-dummy) kontrolovaných klinických skúšaniach kontrolovaných aktívnym komparátorom (WA21092 a WA21093), ktoré mali rovnaké usporiadanie (dizajn) a vykonali sa
u pacientov s relapsujúcimi formami SM (v súlade s McDonaldovými kritériámi z roku 2010)
a s dôkazom o aktivite ochorenia (definovanej klinickými znakmi alebo nálezmi zo zobrazovacieho vyšetrenia) v priebehu predchádzajúcich dvoch rokov. Dizajn štúdií a východiskové charakteristiky
populácie v štúdiách sú zhrnuté v tabuľke 3.
Obe liečebné skupiny boli dobre vyvážené v demografických a východiskových charakteristikách. Pacientom liečeným Ocrevusom (skupina A) sa podávalo 600 mg raz za 6 mesiacov (1. dávka vo forme dvoch 300 mg intravenóznych infúzií, ktoré sa podali s časovým odstupom 2 týždňov,
a následné dávky sa podávali vo forme jednej 600 mg intravenóznej infúzie). Pacientom v skupine B
sa podával interferón beta-1a (Rebif) 44 µg subkutánnou injekciou trikrát týždenne.
T
abuľka 3 Dizajn štúdií, demografické údaje a východiskové charakteristiky
Štúdia č. 1
Štúdia č. 2
N
ázov štúdie WA21092 (OPERA I) (n = 821)
D
i
z
ajn štúdií
WA21093 (OPERA II) (n = 835)
Populácia v štúdiách Pacienti s relapsujúcimi formami SM
Anamnéza ochorenia v čase skríningu
Aspoň dva relapsy v priebehu predchádzajúcich dvoch rokov alebo jeden relaps v priebehu predchádzajúceho roka; skóre EDSS* medzi 0 a 5,5 vrátane
Dĺžka trvania štúdií 2 roky
Liečebné skupiny Skupina A: Ocrevus 600 mg
Skupina B: interferón beta-1a 44 µg s.c. (IFN)
V
ýchodiskové charakteristiky Ocrevus
600 mg
(n = 410)
IFN
44 µg
(n = 411)
Ocrevus
600 mg
(n = 417)
IFN
44 µg
(n = 418)
Priemerný vek (roky) 37,1 36,9 37,2 37,4
Vekové rozmedzie (roky) v čase zaradenia do štúdie
Distribúcia podľa pohlavia
(% mužov/% žien)
Priemerné trvanie/medián trvania ochorenia od stanovenia diagnózy (roky)
Pacienti bez predchádzajúcej liečby DMT (%)**
Priemerný počet relapsov v minulom roku
Percentuálny podiel pacientov s gadolíniom (Gd) zvýraznenými T1 léziami
18 - 56 18 - 55 18 - 55 18 - 55
34,1/65,9 33,8/66,2 35,0/65,0 33,0/67,0
3,82/1,53 3,71/1,57 4,15/2,10 4,13/1,84
73,4 71,0 72,7 74,9
1,31 1,33 1,32 1,34
42,5 38,1 39,0 41,4
Priemerné skóre EDSS* 2,82 2,71 2,73 2,79
* Rozšírená škála funkčnej nespôsobilosti, známa aj ako Kurtzkeho škála (Expanded Disability Status
Scale).
** Pacienti, ktorí neboli liečení žiadnym liekom na SM v priebehu 2 rokov pred randomizáciou.
Kľúčové klinické výsledky a výsledky účinnosti z hľadiska MR nálezov sú prezentované v tabuľke 4
a na grafe 1.
Výsledky týchto štúdií ukazujú, že Ocrevus v porovnaní s interferónom beta-1a podávaným subkutánne v dávke 44 µg významne potlačil vznik relapsov, subklinickú aktivitu ochorenia meranú pomocou MR a progresiu ochorenia.
T
abuľka 4 Kľúčové klinické cieľové ukazovatele a ukazovatele hodnotené pomocou MR
v štúdiách WA21092 a WA21093 (RMS)
Štúdia č. 1: WA21092
(OP
E
RA I)
Štúdia č. 2: WA21093
(OP
E
RA II)
Cieľové ukazovatele
K
linické cieľové ukazovatele
Ocrevus
600 mg
(n = 410)
IFN
44 µg
(n = 411)
Ocrevus
600 mg
(n = 417)
IFN
44 µg
(n = 418)
Ročný výskyt relapsov (primárny cieľový ukazovateľ) Relatívne zníženie
Percentuálny podiel pacientov s potvrdenou progresiou funkčného zneschopnenia pretrvávajúcou 12 týždňov3
Zníženie rizika (súhrnná analýza1)
Zníženie rizika (jednotlivé štúdie2)
Percentuálny podiel pacientov s potvrdenou progresiou funkčného zneschopnenia pretrvávajúcou 24 týždňov3
Zníženie rizika (súhrnná analýza1)
Zníženie rizika (jednotlivé štúdie2)
Percentuálny podiel pacientov s potvrdeným zlepšením miery funkčného zneschopnenia pretrvávajúcim aspoň 12 týždňov4
Relatívne zvýšenie (súhrnná analýza1)
Relatívne zvýšenie (jednotlivé štúdie2)
0,156 0,292 0,155 0,290
46 % (p < 0,0001) 47 % (p < 0,0001)
9,8 % pri Ocrevuse vs 15,2 % pri IFN
40 % (p = 0,0006)7
43 % (p = 0,0139)7 37 % (p = 0,0169)7
7,6 % pri Ocrevuse vs 12,0 % pri IFN
40 % (p = 0,0025)7
43 % (p = 0,0278)7 37 % (p = 0,0370)7
20,7 % pri Ocrevuse vs 15,6 % pri IFN
33 % (p = 0,0194)
61 % (p = 0,0106) 14 % (p = 0,4019)
Percentuálny podiel pacientov bez relapsov v 96. týždni2 80,4 % 66,7 % 78,9 % 64,3 % (p < 0,0001) (p < 0,0001)
Percentuálny podiel pacientov bez známok aktivity ochorenia
(No Evidence of Disease Activity, NEDA)5
Relatívne zvýšenie2
Cieľové ukazovatele hodnotené pomocou MR
Priemerný počet Gd zvýraznených T1 lézií na MR snímke
Relatívne zníženie
Priemerný počet nových a/alebo zväčšujúcich sa
T2 hyperintenzívnych lézií na MR snímke
Relatívne zníženie
Percentuálna zmena v objemu mozgu od 24. týždňa
do 96. týždňa
Relatívne zníženie rýchlosti úbytku objemu mozgu
48 % 29 % 48 % 25 %
64 % (p < 0,0001) 89 % (p < 0,0001)
0,016 0,286 0,021 0,416
94 % (p < 0,0001) 95 % (p < 0,0001)
0,323 1,413 0,325 1,904
77 % (p < 0,0001) 83 % (p < 0,0001)
-0,572 -0,741 -0,638 -0,750
22,8 % (p = 0,0042)6 14,9 % (p = 0,0900)
1 Údaje prospektívne skombinované zo štúdie č. 1 a štúdie č. 2.
2 Nekonfirmatórna (t. j. iba deskriptívna) p-hodnota; nie je súčasťou vopred špecifikovaného hierarchického testovania.
3 Definované ako zvýšenie o ≥ 1,0 bodu v porovnaní s východiskovým skóre EDSS (
Expanded DisabilityStatus Scale) u pacientov s východiskovým skóre 5,5 bodu alebo nižším, alebo o ≥ 0,5 bodu, keď je východiskové skóre
> 5,5 bodu; odhad v 96. týždni podľa Kaplana-Meiera.'
4 Definované ako zníženie o ≥ 1,0 bodu v porovnaní s východiskovým skóre EDSS u pacientov
s východiskovým skóre EDSS ≥ 2 a ≤ 5,5 bodu, alebo o ≥ 0,5 bodu, keď je východiskové skóre
> 5,5 bodu. Pacienti s východiskovým skóre < 2 neboli zahrnutí v analýze.
5 NEDA definované ako absencia protokolom definovaných relapsov, potvrdenej progresie funkčného zneschopnenia pretrvávajúcej 12 týždňov (
Confirmed Disability Progression, CDP) a akejkoľvek aktivity preukázanej MR vyšetrením (buď Gd zvýraznené T1 lézie, alebo nové alebo zväčšujúce sa T2 lézie)
počas celej 96-týždňovej liečby. Exploračný výsledok bol založený na kompletnej ITT populácii.
6 Nekonfirmatórna (t. j. iba deskriptívna) p-hodnota; hierarchická testovacia procedúra bola ukončená
pred dosiahnutím cieľového ukazovateľa.
7 Log-rank test.
G
raf 1: Kaplanova-Meierova krivka* času do objavenia sa potvrdenej progresie funkčného zneschopnenia pretrvávajúcej aspoň 12 týždňov, pričom prvou udalosťou bolo zhoršenie neurologických funkcií vyskytujúce sa počas obdobia dvojito zaslepenej liečby (Súhrnná ITT populácia zo štúdií WA21092 a WA21093)*
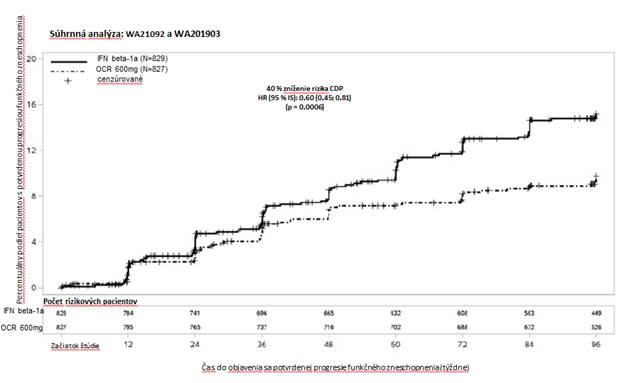
*Vopred špecifikovaná súhrnná analýza štúdií WA21092 a WA21093
Výsledky vopred špecifikovaných súhrnných analýz času do CPD pretrvávajúcej aspoň 12 týždňov (40 % zníženie rizika pri Ocrevuse v porovnaní s interferónom beta-1a (p = 0,0006) sa vo vysokej miere zhodovali s výsledkami analýzy času do CPD pretrvávajúcej aspoň 24 týždňov (40 % zníženie rizika pri Ocrevuse v porovnaní s interferónom beta-1a, p = 0,0025).
Do štúdií boli zaradení pacienti s aktívnym ochorením. Zahŕňali dovtedy neliečených pacientov aj pacientov nedostatočne odpovedajúcich na predchádzajúcu liečbu, pričom aktívne ochorenie bolo definované klinickými znakmi alebo nálezmi zo zobrazovacieho vyšetrenia. Analýza podskupín pacientov s rozličnými východiskovými stupňami aktivity ochorenia vrátane aktívneho a vysoko aktívneho ochorenia ukázala, že účinnosť Ocrevusu z hľadiska ARR a CPD pretrvávajúcej 12 týždňov sa zhodovala s účinnosťou zistenou v celkovej populácii.
Primárne progresívna SMÚčinnosť a bezpečnosť Ocrevusu sa hodnotili aj v randomizovanom, dvojito zaslepenom, placebom kontrolovanom klinickom skúšaní u pacientov s primárne progresívnou SM (štúdia WA25046), ktorí boli vo včasnom štádiu ochorenia podľa hlavných inklúznych kritérií, t. j. vek 18 - 55 rokov vrátane; skóre EDSS v čase skríningu od 3,0 do 6,5 boda; trvanie ochorenia od nástupu príznakov SM kratšie ako 10 rokov u pacientov so skóre EDSS v čase skríningu ≤ 5,0 bodov alebo kratšie ako 15 rokov
u pacientov so skóre EDSS v čase skríningu > 5,0 bodov. Pokiaľ ide o aktivitu ochorenia, nálezy typické pre zápalovú aktivitu môžu byť, dokonca aj pri progresívnej SM, získané zobrazovacím
vyšetrením (t. j. gadolíniom zvýraznené T1 lézie a/alebo aktívne [nové alebo zväčšujúce sa] T2 lézie).
MR dôkaz sa má použiť na potvrdenie zápalovej aktivity u všetkých pacientov. Pacienti vo veku nad
55 rokov neboli sledovaní. Dizajn štúdie a východiskové charakteristiky populácie v štúdii sú prezentované v tabuľke 5.
Obe liečebné skupiny boli dobre vyvážené v demografických a východiskových charakteristikách. MR vyšetrením mozgu sa preukázali nálezy typické pre zápalovú aktivitu, a to buď Gd zvýraznené T1 lézie alebo T2 lézie.
V štúdii fázy 3 zameranej na PPSM sa pacientom podávalo 600 mg Ocrevusu raz za 6 mesiacov
vo forme dvoch 300 mg infúzií, ktoré boli podané s časovým odstupom dvoch týždňov, počas celého obdobia liečby. Preukázalo sa, že 600 mg infúzie podávané pri RSM a dve 300 mg infúzie podávané pri PPSM majú zhodné FK/FD profily. Profily IRR pri jednotlivých infúziách boli tiež podobné,
nezávisle od toho, či sa 600 mg dávka podávala vo forme jednej 600 mg infúzie, alebo vo forme dvoch
300 mg infúzií s časovým odstupom dvoch týždňov (pozri časti 4.8 a 5.2), ale vzhľadom na celkovo vyšší počet infúzií pri schéme s dvomi 300 mg infúziami, celkový počet IRR bol vyšší. Preto sa po
podaní 1. dávky odporúča podávať Ocrevus v jednej 600 mg infúzii (pozri časť 4.2), aby sa znížil
celkový počet infúzií (a súbežná expozícia profylakticky podávanému metylprednizolónu a antihistaminiku) a reakcií súvisiacich s infúziou.
Tabuľka 5 Dizajn štúdie, demografické údaje a východiskové charakteristiky v štúdii WA25046
Názov štúdie Štúdia WA25046 ORATORIO (n = 732)
Dizajn štúdie
Populácia v štúdii Pacienti s primárne progresívnou formou SM
Dĺžka trvania štúdie Závislá od dosiahnutia vopred určeného počtu sledovaných
udalostí (event-driven) (minimálne 120 týždňov a 253 prípadov potvrdenej progresie funkčného zneschopnenia)
(Medián obdobia sledovania:
Ocrevus 3,0 roky, placebo 2,8 roka)
Anamnéza ochorenia v čase skríningu
Vek 18 - 55 rokov, skóre EDSS od 3,0 do 6,5

Liečebné skupiny Skupina A: Ocrevus 600 mg
Skupina B: placebo, pri randomizácii 2:1
V
ýchodiskové charakteristiky
Ocrevus 600 mg (n = 488) Placebo (n = 244)
Priemerný vek (roky) 44,7 44,4
Vekové rozmedzie (roky)
v čase zaradenia do štúdie
Distribúcia podľa pohlavia (% mužov/% žien) Priemerné trvanie/medián
trvania ochorenia od
stanovenia diagnózy PPSM
(roky)
20 - 56 18 - 56
51,4/48,6 49,2/50,8
2,9/1,6 2,8/1,3
Priemerné skóre EDSS 4,7 4,7
Kľúčové klinické výsledky a výsledky účinnosti z hľadiska MR nálezov sú prezentované v tabuľke 6
a na grafe 2.
Výsledky tejto štúdie ukazujú, že Ocrevus v porovnaní s placebom významne oddiaľuje progresiu ochorenia a znižuje mieru zhoršenia rýchlosti chôdze.
T
abuľka 6 Kľúčové klinické cieľové ukazovatele a ukazovatele hodnotené pomocou MR
v štúdii WA25046 (PPSM)
Štúdia č. 3
WA25046 (Oratorio)
C
i
eľové ukazovatele
K
l
i
nické cieľové ukazovatele
P
r
i
m
árny cieľový ukazovateľ účinnosti
Ocrevus 600 mg
(n = 488)
Placebo
(n = 244)
Percentuálny podiel pacientov s potvrdenou progresiou funkčného zneschopnenia pretrvávajúcou
12 týždňov1 (primárny cieľový ukazovateľ)
Zníženie rizika
Percentuálny podiel pacientov s potvrdenou progresiou funkčného zneschopnenia pretrvávajúcou
24 týždňov1
Zníženie rizika
Percentuálna zmena (nárast) v čase potrebnom
na prejdenie 25-stopovej (cca 7,62 m) vzdialenosti
(Timed 25-Foot Walk - test chôdze na čas)
v 120. týždni v porovnaní s východiskovým časom
Relatívne zníženie miery nárastu času chôdze
Cieľové ukazovatele hodnotené pomocou MR
Percentuálna zmena v objeme T2 hyperintenzívnych lézií v 120. týždni v porovnaní s východiskovým stavom
Percentuálna zmena v objeme mozgu od 24. týždňa
do 120. týždňa
Relatívne zníženie rýchlosti úbytku objemu mozgu
30,2 % 34,0 %
24 %
(p = 0,0321)
28,3 % 32,7 %
25 %
(p = 0,0365)
38,9 55,1
29,4 %
(p = 0,0404)
-3,4 7,4
(p < 0,0001)
-0,902 -1,093
17,5 %
(p = 0,0206)

1 Definované ako zvýšenie o ≥ 1,0 bodu v porovnaní s východiskovým skóre EDSS u pacientov
s východiskovým skóre 5,5 bodu alebo nižším, alebo o ≥ 0,5 bodu, keď je východiskové skóre
> 5,5
bodu; odhad v 120. týždni podľa Kaplana-Meiera.
G
raf 2: Kaplanova-Meierova krivka* času do objavenia sa potvrdenej progresie funkčného zneschopnenia pretrvávajúcej aspoň 12 týždňov, pričom prvou udalosťou bolo zhoršenie neurologických funkcií vyskytujúce sa počas obdobia dvojito zaslepenej liečby (ITT populácia zo štúdie WA25046)*
* Všetci pacienti v tejto analýze boli sledovaní počas obdobia minimálne 120 týždňov. Primárna analýza je založená na kumulatívnom výskyte všetkých udalostí.
Vopred špecifikovaná analýza primárneho cieľového ukazovateľa v podskupinách, ktorá však nemala dostatočnú štatistickú silu, naznačuje, že pacienti mladšieho veku alebo pacienti s Gd zvýraznenými T1 léziami prítomnými pred začiatkom liečby majú väčší prínos z liečby ako starší pacienti alebo pacienti bez Gd zvýraznených T1 lézií (≤ 45 rokov: HR 0,64 [0,45; 0,92], > 45 rokov: HR 0,88
[0,62; 1,26], prítomnosť Gd zvýraznených T1 lézií pred začiatkom liečby: HR 0,65 [0,40 - 1,06], neprítomnosť Gd zvýraznených T1 lézií pred začiatkom liečby: HR 0,84 [0,62 - 1,13]).
Post-hoc analýzy navyše naznačili, že u mladších pacientov s Gd zvýraznenými T1 léziami
prítomnými pred začiatkom liečby je efekt liečby lepší (≤ 45 rokov: HR 0,52 [0,27 - 1,00]; ≤ 46 rokov
[medián veku v štúdii WA25046]; HR 0,48 [0,25 - 0,92]; < 51 rokov: HR 0,53 [0,31 - 0,89].
ImunogenicitaPacienti v klinických skúšaniach zameraných na SM (WA21092, WA21093 a WA25046) boli
vo viacerých časových bodoch (pri zaradení do štúdie a každých 6 mesiacov po začatí liečby počas trvania klinického skúšania) testovaní na prítomnosť protilátok proti lieku (
anti-drug antibodies,
ADA). U 12 (~1 %) z 1 311 pacientov liečených Ocrevusom sa zistila pozitivita ADA vytvorených
počas liečby, pričom u 2 z nich sa zistila prítomnosť neutralizujúcich protilátok. Vplyv ADA vytvorených počas liečby na bezpečnosť a účinnosť nie je možné posúdiť vzhľadom na nízky výskyt ADA súvisiacich s Ocrevusom.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Ocrevusom
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe sklerózy multiplex
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika okrelizumabu v štúdiách zameraných na SM bola opísaná pomocou dvojkompartmentového modelu s časovo závislým klírensom a s FK parametrami typickými pre monoklonálnu protilátku typu IgG1.
Celková expozícia (AUC počas 24-týždňového dávkovacieho intervalu) bola identická pri podávaní dvoch 300 mg dávok v štúdiách zameraných na PPSM a jednej 600 mg dávky v štúdiách zameraných
na RSM, ako sa očakávalo vzhľadom na to, že sa podávala identická dávka. Plocha pod krivkou
(AUCτ) po 4. dávke 600 mg okrelizumabu bola 3 510 µg/ml•deň a priemerná maximálna koncentrácia
(Cmax) bola 212 µg/ml pri RSM (600 mg infúzia) a 141 µg/ml pri PPSM (300 mg infúzie).
Absorpcia
Ocrevus sa podáva intravenóznou infúziou. Neuskutočnili sa žiadne štúdie s inými spôsobmi
podávania.
Distribúcia
Na základe populačného farmakokinetického modelu bol distribučný objem v centrálnom
kompartmente odhadnutý na 2,78 l. Distribučný objem v periférnom kompartmente bol odhadnutý na 2,68 l a klírens medzi kompartmentami bol odhadnutý na 0,294 l/deň.
Biotransformácia
Metabolizmus Ocrevusu sa priamo neskúmal, pretože protilátky podliehajú výlučne katabolizmu
(t. j. rozpadu na peptidy a aminokyseliny).
Eliminácia
Konštantný klírens bol odhadnutý na 0,17 l/deň a úvodný časovo závislý klírens na 0,0489 l/deň, ktorý
klesal s polčasom 33 týždňov. Terminálny eliminačný polčas okrelizumabu bol 26 dní.
Farmakokinetika v osobitných skupináchpacientov
Pediatrická populácia
Neuskutočnili sa žiadne štúdie skúmajúce farmakokinetiku okrelizumabu u detí a dospievajúcich vo veku < 18 rokov.
Staršie osoby
K dispozícii nie sú žiadne štúdie s okrelizumabom zamerané na FK u pacientov vo veku ≥ 55 rokov z dôvodu obmedzených klinických skúseností (pozri časť 4.2).
Porucha funkcie obličiek
Neuskutočnila sa žiadna oficiálna farmakokinetická štúdia. Pacienti s miernou poruchou funkcie obličiek boli zahrnutí v klinických skúšaniach a u týchto pacientov sa nepozorovala žiadna zmena vo farmakokinetike Ocrevusu. K dispozícii nie sú žiadne FK údaje týkajúce sa pacientov so stredne ťažkou alebo ťažkou poruchou funkcie obličiek.
Porucha funkcie pečene
Neuskutočnila sa žiadna oficiálna farmakokinetická štúdia. Pacienti s miernou poruchou funkcie pečene boli zahrnutí v klinických skúšaniach a u týchto pacientov sa nepozorovala žiadna zmena
vo farmakokinetike Ocrevusu. K dispozícii nie sú žiadne FK údaje týkajúce sa pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a embryofetálneho vývinu neodhalili žiadne osobitné riziko pre ľudí. S okrelizumabom sa neuskutočnili žiadne štúdie karcinogenity ani mutagenity.
V štúdiách prenatálneho a postnatálneho vývoja na opiciach rodu
Cynomolgus sa podávanie okrelizumabu od 20. dňa gestácie do približne 5 týždňov po pôrode dávalo do súvislosti
s glomerulopatiou, s tvorbou lymfoidných folikulov v kostnej dreni, s lymfoplazmocytovým zápalom obličiek a so zníženou hmotnosťou semenníkov u potomkov. Dávky podávané zvieracím matkám

v tejto štúdii viedli k maximálnym priemerným koncentráciám v sére (Cmax), ktoré boli
4,5- a 21-násobne vyššie ako koncentrácie v sére predpokladané v klinickej praxi.
Vyskytli sa dva prípady moribundných (umierajúcich) mláďat, pričom jeden z nich sa pripisoval slabosti v dôsledku predčasného narodenia sprevádzanej oportúnnou infekciou a druhý infekčnej meningoencefalitíde postihujúcej mozoček novorodeného mláďata zvieracej matky trpiacej aktívnou infekciou (mastitídou). Priebeh obidvoch neonatálnych infekcií mohol byť potenciálne ovplyvnený depléciou B-lymfocytov. U novorodených mláďat zvieracích matiek vystavených okrelizumabu sa
v postnatálnej fáze zistilo zníženie populácií B-lymfocytov. Počas obdobia laktácie sa zistili merateľné hladiny okrelizumabu v mlieku (približne 0,2 % z minimálnych rovnovážnych hladín v sére).
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokTrihydrát octanu sodného Ľadová kyselina octová Dihydrát trehalózy Polysorbát 20
Voda na injekciu
6.2 InkompatibilityNepozorovali sa žiadne inkompatibility medzi Ocrevusom a polyvinylchloridovými (PVC)
alebo polyolefínovými (PO) vakmi a infúznymi súpravami.
Na riedenie Ocrevusu nepoužívajte iné riedidlá ako tie, ktoré sú vymenované v časti 6.6, pretože ich použitie sa netestovalo.
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnostiNeotvorená injekčná liekovka18 mesiacov
Nariedený roztok na intravenóznu infúziuChemická a fyzikálna stabilita pripraveného infúzneho roztoku je preukázaná na 24 hodín pri teplote
2 °C - 8 °C a následne na 8 hodín pri izbovej teplote.
Z mikrobiologického hľadiska sa má pripravený infúzny roztok použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky uchovávania pred použitím je zodpovedný používateľ a za normálnych okolností to nemá byť dlhšie ako 24 hodín pri teplote 2 °C - 8 °C a následne na 8 hodín pri izbovej teplote, pokiaľ sa riedenie nevykonalo za kontrolovaných a validovaných aseptických podmienok.
V prípade, že podanie intravenóznej infúzie nie je možné dokončiť v rovnaký deň, zvyšný roztok sa má zlikvidovať.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Injekčné liekovky uchovávajte vo vonkajšej škatuľke na ochranu pred svetlom. Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
10 ml koncentrátu v sklenenej injekčnej liekovke. Veľkosť balenia po 1 alebo 2 injekčných liekovkách. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pokyny na riedenie
Ocrevus má pripraviť zdravotnícky pracovník s použitím aseptickej techniky. Injekčnou liekovkou
netraste.
Liek je určený len na jednorazové použitie.
Nepoužívajte roztok, ak má zmenenú farbu alebo ak roztok obsahuje cudzorodé častice (opis roztoku, pozri časť 3).
Liek Ocrevus sa pred podaním musí nariediť. Roztoky Ocrevusu na intravenózne podanie sa pripravia nariedením lieku v infúznom vaku, ktorý obsahujúce izotonický 0,9 % roztok chloridu sodného
(300 mg/250 ml alebo 600 mg/500 ml), na konečnú koncentráciu liečiva približne 1,2 mg/ml.
Nariedený infúzny roztok sa musí podať pomocou infúznej súpravy s 0,2- alebo 0,22-mikrónovým
in-line filtrom.
Pred začiatkom podávania intravenóznej infúzie musí obsah infúzneho vaku dosiahnuť izbovú teplotu, aby sa zabránilo reakcii na infúziu spôsobenej podávaním roztoku s nízkou teplotou.
Likvidácia
Likvidácia nepoužitých/exspirovaných liekov
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
Veľká Británia
8. REGISTRAČNÉ ČÍSLO9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTU<{DD mesiac RRRR}>
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.