
tatočné zníženie hladiny ALP a/alebo celkového bilirubínu po 3 mesiacoch liečby a pacient znáša kyselinu obeticholovú, titrujte až do
5 mg dvakrát týždenne (s odstupom minimálne 3 dni) a následne do
10 mg dvakrát týždenne (s odstupom minimálne 3 dni) podľa odpovede a znášanlivosti
 Maximálna dávka 10 mg raz denne 10 mg dvakrát týždenne
Maximálna dávka 10 mg raz denne 10 mg dvakrát týždenne (s odstupom minimálne 3 dni)
U pacientov, ktorí dostávajú kyselinu obeticholovú, sa nevyžaduje úprava dávky súbežnej UDCA.
Manažment a úprava dávky pre silné svrbenieStratégie manažmentu zahŕňajú pridanie živíc alebo antihistaminík, ktoré sa viažu na žlčové kyseliny.
U pacientov trpiacich ťažkou neznášanlivosťou kvôli svrbeniu je potrebné zvážiť jednu alebo viaceré z nasledujúcich možností:
PrepacientovbezcirhózyalebosChild-PughovoutriedouA• Zníženie dávky kyseliny obeticholovej na:
§ 5 mg každý druhý deň u pacientov netolerujúcich 5 mg raz denne
§ 5 mg raz denne u pacientov netolerujúcich 10 mg raz denne
• Dočasné vysadenie dávky kyseliny obeticholovej na maximálne 2 týždne s opakovaným nasadením v zníženej dávke.
• Ďalšie zvyšovanie dávky na 10 mg raz denne, podľa znášanlivosti, na dosiahnutie optimálnej odpovede.
Pre pacientov s Child-Pughovou triedou B alebo C alebo s dekompenzovanou cirhózou:• Zníženie dávky kyseliny obeticholovej na:
§ 5 mg raz týždenne u pacientov netolerujúcich 5 mg dvakrát týždenne
§ 10 mg raz týždenne u pacientov netolerujúcich 10 mg dvakrát týždenne
• Dočasné vysadenie dávky kyseliny obeticholovej na maximálne 2 týždne s opakovaným nasadením v zníženej dávke, ak je to relevantné.
• Ďalšie zvyšovanie dávky na 10 mg dvakrát týždenne, podľa znášanlivosti, na dosiahnutie optimálnej odpovede.
Zvážte vysadenie liečby kyselinou obeticholovou u pacientov, u ktorých sa naďalej vyskytuje trvalé neznesiteľné svrbenie.
OsobitnépopuláciePacienti s poruchou funkcie pečenePre odporúčané dávkovanie pozri tabuľku 1. Ďalej pozri časti 4.4 a 5.2.
Starší pacienti (≥ 65 rokov)U starších pacientov existujú obmedzené údaje. U starších pacientov sa nevyžaduje úprava dávky
(pozri časť 5.2).
P
acienti s poruchou funkcie obličiek
U pacientov s miernou a stredne ťažkou poruchou funkcie obličiek existujú obmedzené údaje, a pre ťažkú poruchu funkcie obličiek neexistujú žiadne údaje. U pacientov s poruchou funkcie obličiek sa nevyžaduje úprava dávky (pozri časť 5.2).
Pediatrická populácia
Použitie kyseliny obeticholovej sa netýka pediatrickej populácie pri liečbe primárnej biliárnej cholangitídy (PBC).
Spôsobpodávania
Tableta sa musí užívať perorálne s jedlom alebo bez jedla.
Pacienti užívajúci živice viažuce sa na žlčové kyseliny by mali užiť kyselinu obeticholovú najmenej
4-6 hodín pred alebo 4-6 hodín po užití živice viažucej sa na žlčové kyseliny alebo v čo najväčšom možnom intervale (pozri časť 4.5).
4.3 Kontraindikácie
- Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
- Úplná biliárna obštrukcia
4.4 Osobitné upozornenia a opatrenia pri používaní
Nežiaduceudalostisúvisiacespečeňou
U pacientov užívajúcich kyselinu obeticholovú boli pozorované zvýšené hladiny alanínaminotransferázy (ALT) a aspartátaminotransferázy (AST). Boli tiež pozorované klinické
príznaky a prejavy dekompenzácie pečene. Tieto udalosti sa vyskytli už počas prvého mesiaca liečby.
Nežiaduce udalosti súvisiace s pečeňou boli primárne pozorované pri vyšších dávkach, než je maximálna odporúčaná dávka 10 mg raz denne (pozri časť 4.9). V období po uvedení na trh boli hlásené závažné poškodenie pečene a úmrtie pri častejšom dávkovaní kyseliny obeticholovej, ako sa odporúča u pacientov so stredným až závažným zhoršením funkcie pečene.
Po začatí liečby je potrebné všetkých pacientov monitorovať na postup ochorenia PBC
prostredníctvom laboratórnych a klinických vyšetrení na účely stanovenia, či je potrebná úprava
dávky. Pacienti so zvýšeným rizikom hepatickej dekompenzácie vrátané tých s laboratórne potvrdenou zhoršenou funkciou pečene a/alebo progresiou do cirhózy musia byť dôkladne sledovaní. Frekvenciu
dávkovania je potrebné znížiť u pacientov, u ktorých sa rozvinie pokročilé štádium ochorenia (napr. z Child-Pughovej triedy A sa vyvinie Child-Pughovu triedu B alebo C) (pozri časti 4.2 a 5.2).
Silnésvrbenie
Silné svrbenie bolo hlásené u 23 % pacientov liečených v skupine s OCALIVOU 10 mg, u 19 %
pacientov v skupine s titráciou OCALIVY, a u 7 % pacientov v skupine s placebom. Stredný čas nástupu silného svrbenia bolo 11, 158 a 75 dní u pacientov v skupine s OCALIVA 10 mg, s titráciou OCALIVY a s placebom, v uvedenom poradí. Stratégie liečby zahŕňajú pridanie živíc alebo antihistamínov, ktoré sa viažu na žlčové kyseliny, zníženie dávky, zníženie frekvencie dávkovania a/alebo dočasné vysadenie dávky (pozri časť 4.2 a 4.8).
4.5 Liekové a iné interakcie
Lieky ovplyvnené kyselinou obeticholovou
Warfarín
Medzinárodný normalizovaný pomer (INR) sa zníži po súčasnom podávaní warfarínu a kyseliny obeticholovej. Je potrebné monitorovať INR a podľa potreby upraviť dávku warfarínu, aby sa udržal
cieľový rozsah INR pri súčasnom podávaní kyseliny obeticholovej a warfarínu.
InterakciesosubstrátmiCYP1A2súzkymterapeutickýmindexom
Kyselina obeticholová môže zvýšiť expozíciu súbežným liekom, ktoré sú substrátmi CYP1A2. Odporúča sa terapeutické sledovanie substrátov CYP1A2 s úzkym terapeutickým indexom (ako napríklad teofylín a tizanidín).
Lieky, ktoré ovplyvňujú kyselinuobeticholovú
Živiceviažucesanažlčovékyseliny
Živice viažuce sa na žlčové kyseliny ako cholestyramín, kolestipol alebo kolesevelam adsorbujú a znižujú absorpciu žlčových kyselín a môžu znížiť účinnosť kyseliny obeticholovej. Keď sa podávajú
živice viažuce sa na žlčové kyseliny, kyselina obeticholová sa má užiť najmenej 4-6 hodín pred alebo
4-6 hodín po užití živice viažucej sa na žlčové kyseliny alebo v čo najväčšom možnom intervale.
4.6 Fertilita, gravidita a laktácia
Tehotenstvo
O použití kyseliny obeticholovej u tehotných žien neexistujú žiadne údaje. Štúdie na zvieratách neukazujú priame ani nepriame škodlivé účinky na reprodukčnú toxicitu (pozri časť 5.3). Ako bezpečnostné opatrenie je vhodnejšie vyhýbať sa použitiu kyseliny obeticholovej počas tehotenstva.
Dojčenie
Nie je známe, či sa kyselina obeticholová vylučuje do ľudského mlieka. Na základe štúdií na zvieratách a určenej farmakológie sa neočakáva, že by kyselina obeticholová narušovala dojčenie alebo rast alebo vývoj dojčeného dieťaťa. Musí sa urobiť rozhodnutie, či prerušiť dojčenie alebo prerušiť/neužívať terapiu s kyselinou obeticholovou vzhľadom na prínos dojčenia pre dieťa a prínos terapie pre ženu (pozri časť 5.3).
Fertilita
Žiadne údaje o fertilite u ľudí nie sú k dispozícii. Zvieracie štúdie neukazujú žiadne priame ani nepriame účinky na fertilitu alebo reprodukciu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kyselina obeticholová nemá žiadny alebo má len zanedbateľný vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
Najčastejšie hlásenými nežiaducimi reakciami boli svrbenie (63 %) a únava (22 %). Nežiaducich reakcií vedúcich k vysadeniu bolo 1% v skupine s titráciou OCALIVY a 11 % v skupine s
OCALIVOU 10 mg. Najčastejšou nežiaducou udalosťou, ktorá viedla k vysadeniu, bolo svrbenie. Väčšina svrbenia sa vyskytla v prvom mesiaci liečby a časom sa vyriešila pri pokračovaní dávkovania.
Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie spojené s OCALIVOU hlásené vo fáze III klinickej štúdie sú uvedené v tabuľke nižšie a sú zoradené podľa triedy orgánových systémov MedDRA a frekvencie. Frekvencie sú
definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až <1/10), menej časté (≥ 1/1 000 až <1/100),
zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (nemožno odhadnúť na základe dostupných údajov).
Tabuľka 2. Frekvencia nežiaducich reakcií u pacientov s PBC*
Trieda systémových orgánov Veľmi časté Časté

Poruchy endokrinného systému
abnormalita funkcie štítnej žľazy
Poruchy nervového systému závrat
T
rieda systémových orgánov Veľmi časté Časté
Poruchy srdca a srdcovej činnosti
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
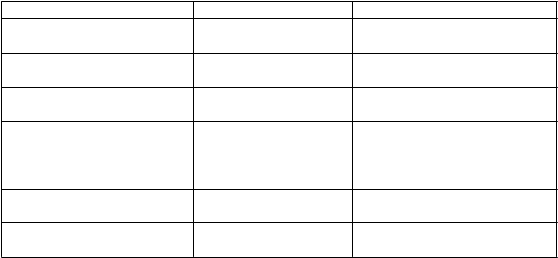
bolesť a nepríjemné pocity v bruchu
palpitácie orofaryngeálna bolesť zápcha
Poruchy kože a podkožného tkaniva
svrbenie ekzém, vyrážka
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
Celkové poruchy a reakcie v mieste podania
artralgia
únava periférny edém, pyrexia
* Nežiaduce reakcie sú definované ako udalosti, ktoré sa vyskytujú v miere vyššej alebo rovnajúcej sa
5 % pacientov v liečebnej skupine s kyselinou obeticholovou a s výskytom vyšším alebo rovným o
1 % viac než v liečebnej skupine s placebom.
Popis vybraných nežiaducichreakciíSvrbeniePribližne 60 % pacientov malo anamnézu svrbenia pri zaradení do štúdie fázy III. Akútne svrbenie obvykle začalo počas prvého mesiaca po začiatku liečby.
V porovnaní s pacientmi, ktorí začali s 10 mg raz denne v skupine s OCALIVOU 10 mg, pacienti v skupine s titráciou OCALIVY mali nižší výskyt svrbenia (70 % a 56 %, v uvedenom poradí) a nižšiu mieru vysadenia kvôli svrbeniu (10 % a 1 %, v uvedenom poradí).
Percento pacientov, ktorí si vyžadovali zásah (t. j. úpravu dávky, prerušenie liečby alebo nasadenie antihistaminík alebo živíc viažucich sa na žlčové kyseliny) bolo 41 % v skupine s OCALIVOU 10 mg,
34 % v skupine s titráciou OCALIVY a 19 % v skupine s placebom.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNajvyššia jednorazová expozícia dávke kyseliny obeticholovej u zdravých dobrovoľníkov bola dávka
500 mg. Opakované dávky 250 mg boli podávané 12 dní po sebe a u niektorých účastníkov sa
vyskytlo svrbenie a reverzibilné zvýšenie pečeňových transamináz. U pacientov s PBC, ktorí dostávali
OCALIVU 25 mg raz denne (2,5-násobok najvyššej odporúčanej dávky) alebo 50 mg raz denne (5- násobok najvyššej odporúčanej dávky), bol hlásený zvýšený výskyt nežiaducich udalostí súvisiacich s pečeňou v závislosti na dávke (napríklad ascites, vzplanutie primárnej biliárnej cholangitídy, nový nástup žltačky) a zvýšenie transamináz a bilirubínu (až viac než 3-násobok hornej hranice normálu
[ULN]). V prípade predávkovania je potrebné pacientov pozorne sledovať a podľa potreby poskytnúť podpornú starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: liečivá na žlčové cesty a pečeň, lieky obsahujúce žlčové kyseliny, kód
ATC: A05AA04
Mechanizmus
účinku
Kyselina obeticholová je selektívny a silný agonista farnezoidového X receptora (FXR), čo je jadrový receptor vysoko exprimovaný v pečeni a čreve. FXR je považovaný za kľúčového regulátora žlčových
kyselín, zápalových, fibrotických a metabolických dráh. Aktivácia FXR znižuje vnútrobunkové
koncentrácie žlčových kyselín v hepatocytoch potlačením syntézy z cholesterolu de novo, ako aj zvýšením prenosu žlčových kyselín z hepatocytov. Tieto mechanizmy limitujú celkový objem žlčových kyselín v obehu a zároveň podporujú cholerézu, čím znižujú expozíciu pečene žlčovým kyselinám.
Farmakodynamické účinky
Klinickáúčinnosťabezpečnosť
Randomizovaná, dvojito zaslepená, placebom kontrolovaná 12-mesačná štúdia fázy III s paralelnými skupinami (POISE) na vyhodnotenie bezpečnosti a účinnosti OCALIVY u 216 pacientov s PBC, ktorí užívali UDCA najmenej 12 mesiacov (stabilná dávka ≥3 mesiace), alebo ktorí boli neschopní
tolerovať UDCA a nedostávali UDCA ≥3 mesiace. Pacienti boli zahrnutí do skúšania, ak bola alkalická fosfatáza (ALP) vyššia alebo rovná 1,67-násobku hornej hranice normálu (ULN) a/alebo ak
bol celkový bilirubín vyšší než 1 x ULN, no nižší než 2 x ULN. Pacienti boli randomizovaní (1:1:1) do skupín, ktorým bolo podávané raz denne placebo, OCALIVA 10 mg alebo titrácia OCALIVY (5 mg
titrovaných na 10 mg po 6 mesiacoch v závislosti od terapeutickej odpovede/znášanlivosti). Väčšina (93 %) pacientov dostávala liečbu v kombinácii s UDCA a malý počet pacientov (7 %) neschopných tolerovať UDCA dostal placebo, OCALIVA (10 mg) alebo titráciu OCALIVY (5 mg na 10 mg) v
monoterapii. ALP a celkový bilirubín boli hodnotené ako kategorické premenné v primárnom kompozitnom koncovom ukazovateli ako aj kontinuálne premenné v priebehu času.
Populáciu v štúdii tvorili prevažne ženy (91 %) a belosi (94 %). Priemerný vek bol 56 rokov, pričom väčšina pacientov mala menej než 65 rokov. Priemerné základné hodnoty ALP sa pohybovali od
316 U/l do 327 U/l. Priemerné základné hodnoty celkového bilirubínu sa pohybovali od 10 μmol/l do
12 μmol/l vo všetkých liečebných skupinách, pričom 92 % pacientov bolo v normálnom rozsahu.
Liečba liekom OCALIVA 10 mg alebo titráciou OCALIVY (5 mg na 10 mg) mala za následok klinicky a štatisticky signifikantné zvýšenia (p < 0,0001) v porovnaní s placebom v počte pacientov, ktorí dosiahli primárny kompozitný koncový ukazovateľ vo všetkých časových bodoch štúdie (pozri tabuľku 3). Odpovede sa vyskytli už po 2 týždňoch a boli závislé na dávke (OCALIVA 5 mg v porovnaní s 10 mg po 6 mesiacoch, p=0,0358).
T
abuľka 3 Percento pacientov s PBC, ktorí dosiahli primárny kompozitný koncový ukazovateľ
a
v 6. mesiaci a 12. mesiaci s alebo bez UDCA
b
6. mesiac
Respondenti, n (%)
OCALIVA
10 mgc
(N = 73)
37 (51)
OCALIVA
Titráciac
(N = 70)
24 (34)
5 (7)
Placebo
(N = 73)
Zodpovedajúci 95 % CI
39 %, 62 %
23 %, 45 %
1 %, 13 %
p-hodnotad <0,0001 <0,0001 neaplikovateľné
12. mesiac
Respondenti, n (%) Zodpovedajúci 95 % CI
35 (48)
36 %, 60 %

32 (46)
34 %, 58 %
7 (10)
4 %, 19 %
p-hodnotad <0,0001 <0,0001 neaplikovateľné
Komponenty primárneho koncového ukazovateľae
ALP menej než 1,67-
násobok ULN, n (%) 40 (55) 33 (47) 12 (16)
Zníženie ALP najmenej o
15 %, n (%) 57 (78) 54 (77) 21 (29)
Celkový bilirubín nižší
alebo rovný 1-násobku
ULNf, n (%)
60 (82) 62 (89) 57 (78)
a Percento účastníkov, ktorí dosiahli odpoveď, definované ako ALP nižšia než 1,67-násobok ULN, celkový bilirubín v normálnom rozsahu, a zníženie ALP aspoň o 15 %. Chýbajúce hodnoty sa považovali za neprítomnosť odpovede. Na výpočet 95 % intervalu spoľahlivosti (CI) sa použil Fisherov exaktný test.
b V skúšaní bolo 16 pacientov (7 %), ktorí neznášali a nedostávali súbežnú liečbu UDCA: 6 pacientov (8 %) v
skupine s OCALIVOU 10 mg, 5 pacientov (7 %) v skupine s titráciou OCALIVY a 5 pacientov (7 %) v skupine s placebom.
c Pacienti boli randomizovaní (1:1:1) do skupín, ktorým bola podávaná OCALIVA 10 mg raz denne po celých 12
mesiacov skúšania, alebo titrácia OCALIVY (5 mg raz denne úvodných 6 mesiacov s možnosťou zvýšenia na
10 mg raz denne posledných 6 mesiacov, ak pacient znášal liek OCALIVA, no mal ALP 1,67-násobok ULN
alebo vyššiu a/alebo celkový bilirubín vyšší než ULN, alebo menej než 15 % zníženie ALP) alebo placebo.
d Skupina s titráciou OCALIVA a OCALIVA 10 mg oproti placebu. P-hodnoty sa získali pomocou
Cochranovho-Mantelovho-Haenszelovho všeobecného asociačného testu stratifikovaného podľa neznášanlivosti UDCA a ALP pred liečbou vyššou než 3-násobok ULN a/alebo AST vyššou než 2-násobok ULN a/alebo celkovým bilirubínom vyšším než ULN.
e Miery odpovedí boli vypočítané na základe analýzy pozorovaných prípadov (t. j. [n=pozorované u respondentov]/[N=populácia s úmyslom liečiť (ITT)]); percento pacientov s hodnotami v 12. mesiaci 86 %,
91 % a 96 % pre skupiny s OCALIVA 10 mg, titráciou OCALIVY a placebom, v uvedenom poradí.
f Priemerná základná hodnota celkového bilirubínu bola 0,65 mg/dl a bola v normálnom rozsahu (t. j. menej než alebo rovná ULN) u 92 % zaradených pacientov.'
PriemernézníženieALP
Priemerné zníženie ALP sa pozorovalo už v 2. týždni a udržalo sa do 12 mesiaca u pacientov, ktorí boli udržiavaní na tej istej dávke celých 12 mesiacov. U pacientov v skupine s titráciou OCALIVY, ktorých dávka OCALIVY bola zvýšená z 5 mg raz denne na 10 mg raz denne, sa pozorovalo ďalšie zníženie ALP v 12. mesiaci u väčšiny pacientov.
Priemernézníženiegamaglutamyltransferázy(GGT)
Priemerné (95 % CI) zníženie GGT bolo 178 (137, 219) U/l v skupine s OCALIVOU 10 mg, 138 (102, 174) U/l v skupine s titráciou OCALIVY, a 8 (-48, 32) U/l v skupine s placebom.
Monoterapia
51 pacientov s PBC so základnou hodnotou ALP 1,67-násobok ULN alebo vyššou a/alebo celkovým bilirubínom vyšším než ULN boli vyhodnotení z hľadiska biochemickej odpovede na OCALIVU v monoterapii (24 pacientov dostávalo OCALIVU 10 mg raz denne a 27 pacientov dostávalo placebo) v súhrnnej analýze údajov z randomizovanej, dvojito zaslepenej, placebom kontrolovanej 12-mesačnej štúdie fázy III (POISE) a z randomizovanej, dvojito zaslepenej, placebom kontrolovanej 3-mesačnej štúdie. V 3. mesiaci 9 (38 %) pacientov liečených OCALIVOU dosiahlo odpoveď na kompozitný koncový ukazovateľ v porovnaní s 1 (4 %) pacientom liečeným placebom. Priemerné (95 % CI) zníženie ALP u pacientov liečených OCALIVOU bolo 246 (165, 327) U/l v porovnaní so zvýšením 17 (-7, 42) U/l u pacientov liečených placebom.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s kyselinou obeticholou vo všetkých podskupinách pediatrickej populácie s PBC (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto
lieku.
Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Absorpcia
Kyselina obeticholová je absorbovaná s nástupom maximálnych plazmatických koncentrácií (Cmax) ku ktorému dochádza v priemernom čase (tmax) približne 2 hodín. Spoločné podanie s jedlom nemení rozsah vstrebávania kyseliny obeticholovej.
Distribúcia
Väzba kyseliny obeticholovej a jej konjugátov na ľudské plazmatické proteíny je vyššia než 99 %. Distribučný objem kyseliny obeticholovej je 618 l. Distribučný objem kyseliny glyko- a
tauro-obeticholovej neboli stanovené.
Biotransformácia
Kyselina obeticholová sa konjuguje s glycínom alebo taurínom v pečeni a vylučuje do žlče. Tieto
glycínové a taurínové konjugáty kyseliny obeticholovej sa vstrebávajú do tenkého červa, čo vedie k enterohepatickej recirkulácii. Konjugáty môže v ileu a hrubom čreve dekonjugovať črevná mikroflóra, čo vedie ku konverzii na kyselinu obeticholovú, ktorá môže byť reabsorbovaná alebo vylúčená v stolici, hlavnou cestou eliminácie.
Po dennom podávaní kyseliny obeticholovej došlo k akumulácii glycínových a taurínových konjugátov kyseliny obeticholovej, ktoré majú farmakologické aktivity in vitro podobné pôvodnému liečivu. Pomery metabolitu k pôvodnému liečivu po dennom podávaní boli 13,8 u glycínových a 12,3 u taurínových konjugátov kyseliny obeticholovej. Vytvára sa aj ďalší, tretí metabolit kyseliny obeticholovej 3-glukuronid, no jeho farmakologická aktivita sa považuje za minimálnu.
Eliminácia
Po podaní rádiologicky označenej kyseliny obeticholovej sa viac než 87 % vylučuje stolicou. Vylučovanie močom je menej než 3 %.
Úmernosťdávky/času
Po podávaní viacerých dávok 5, 10 a 25 mg raz denne po dobu 14 dní sa systémové expozície kyseline obeticholovej proporcionálne zvyšujú s dávkou. Expozície kyseliny glyko- a tauro-obeticholovej a
celkovej kyseliny obeticholovej sa zvyšujú viac než proporčne s dávkou.
Osobitnépopulácie
Starší pacienti
U starších pacientov (≥ 65 rokov) existujú obmedzené farmakokinetické údaje. Farmakokinetická analýza populácie, vypracovaná s použitím údajov od pacientov do veku 65 rokov indikuje, že sa neočakáva, že by vek významne ovplyvnil klírens kyseliny obeticholovej z obehu.
Pediatrická populácia
Žiadne farmakokinetické štúdie neboli vykonané s kyselinou obeticholovou u pacientov do 18 rokov.
Pohlavie
Farmakokinetická analýza populácie ukazuje, že pohlavie neovplyvňuje farmakokinetiku kyseliny obeticholovej.
Rasa
Farmakokinetická analýza populácie ukazuje, že rasa by nemala mať vplyv na farmakokinetiku kyseliny obeticholovej.
Porucha funkcie obličiek
Kyselina obeticholová má minimálnu elimináciu obličkami, menej než 3 % dávky sa vylúči v moči. Na základe farmakokinetickej analýzy populácie funkcia obličiek nemá významný účinok na
farmakokinetiku kyseliny obeticholovej.
Porucha funkcie pečene
Kyselina obeticholová sa metabolizuje v pečeni a črevách. Systémová expozícia kyseline obeticholovej, jej aktívnym konjugátom a endogénnym žlčovým kyselinám sa zvyšuje u pacientov so
strednou a ťažkou poruchou funkcie pečene (Child-Pughova trieda B a C) v porovnaní so zdravými kontrolnými účastníkmi. Z toho dôvodu sa u pacientov so strednou a ťažkou poruchou funkcie pečene
vyžaduje upravený dávkovací režim na dosiahnutie plazmatických hladín expozície podobných pacientom bez poruchy funkcie pečene (pozri časť 4.2).
Účinok miernej poruchy funkcie pečene (Child-Pughova trieda A) na farmakokinetiku kyseliny obeticholovej bol zanedbateľný, preto u pacientov s miernou poruchou funkcie pečene nie je potrebná úprava dávky.
U pacientov s miernou, strednou a ťažkou poruchou funkcie pečene (Child-Pughova trieda A, B a C) sa priemerná AUC celkovej kyseliny obeticholovej, teda súčtu kyseliny obeticholovej a jej dvoch aktívnych konjugátov, zvýšila 1,13-, 4- a 17-násobne, v uvedenom poradí v porovnaní s účastníkmi s normálnou funkciou pečene po jednorazovom podaní dávky 10 mg kyseliny obeticholovej.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podaní, genotoxicity, karcinogénneho potenciálu a toxicity pre fertilitu, reprodukciu a vývoj neodhalili žiadne osobitné riziko pre ľudí.
Perorálne podávanie kyseliny obeticholovej nad NOAEL myšiam, potkanom a psom v pivotných štúdiách toxicity s opakovanými dávkami malo za následok hlavne účinky na hepatobiliárny systém. Medzi ne patrilo zvýšenie hmotnosti pečene, zmeny v parametroch chémie séra (ALT, AST, LDH, ALP, GGT a/alebo bilirubín) a makroskopické/mikroskopické zmeny. Všetky zmeny boli reverzibilné po vysadení dávok, sú konzistentné a predpovedajú dávkami limitovanú toxicitu u ľudí (systémová expozícia pri NOAEL bola až 24-násobne vyššia, než bolo pozorované pri maximálnej odporúčanej ľudskej dávke). V prenatálnej a postnatálnej štúdii toxicity u potkanov sa zistil taurokonjugát kyseliny obeticholovej u mláďat potkanov dojčených samicami, ktorým bola podaná kyselina obeticholová.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
J
adro
t
a
blety
mikrokryštalická celulóza (E460) karboxymetylškrob A, sodná soľ stearan horečnatý
Obaltablety
poly(vinyl alkohol), čiastočne hydrolyzovaný (E1203)
oxid titaničitý (E171)
makrogol 3350 (E1521)
mastenec (E553b)
žltý oxid železitý E172
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek si nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Fľaše z polyetylénu s vysokou hustotou (HDPE) s detským bezpečnostným polypropylénovým uzáverom a indukčným tesnením z hliníkovej fólie.
Veľkosť balenia: 30 alebo 100 filmom obalených tabliet. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Intercept Pharma Ltd.
2 Pancras Square London, N1C 4AG Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/16/1139/001
EU/1/16/1139/002
EU/1/16/1139/003
EU/1/16/1139/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 12. decembra 2016
Dátum posledného predĺženia registrácie: 1. decembera 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.