
cho
v ústache, bolesť
bruchae
 P
oruchy kože a podkožného tkaniva
P
oruchy reprodukčnéh o systému a prsníkov Celkové poruchy
a reakcie v mieste podania
P
oruchy kože a podkožného tkaniva
P
oruchy reprodukčnéh o systému a prsníkov Celkové poruchy
a reakcie v mieste podania
reakcie
v mieste aplikácie a instiláciea, (vrátane erytému, svrbenia,
pruritus erytéme, hyperhidrózae,
generalizovaný prurituse, podráždenie kožee, kontaktná dermatitídae, generalizovaná vyrážkae
erektilná dysfunkciae
zvýšená dráždivosť, periférny edém
L
aboratórne a funkčné vyšetrenia
Ú
r
azy, otravy a komplikácie liečebného postupu
iritácie, vyrážok, dermatitídy, vezikúl, bolesti, ekzému,
zápalu, opuchu, zmeny farby,
pupencov,
exfoliácie, urtikárie,
precitlivenosti),
astenické
stavya (vrátane únavy, asténie,

mdlôb)
zníženie hmotnostie, zvýšenie pečeňových
enzýmove (vr. AST, ALT, GGT),
zvýšenie hmotnostie, zrýchlenie srdcového rytmue, zvýšenie CPKd,e (pozri Osobitné populácie)
páde
a Nadradený všeobecný názov
b Pozorované v nezaslepených štúdiách
c Pozorované po uvedení lieku na trh
d Pozorované v roku 2011 v súhrnných údajoch z dvojito zaslepených placebom kontrolovaných štúdií
e Pozorované v štúdiách u pacientov s Parkinsonovou chorobou
Popis vybraných nežiaducich reakcií
Náhly nástup spánku a ospalosť
Rotigotín bol spájaný s ospalosťou vrátane nadmernej ospalosti počas dňa a prípadov náhleho zaspania. V zriedkavých prípadoch sa vyskytlo „náhle zaspanie“ počas jazdy, čo následne spôsobilo dopravnú nehodu (pozri aj časť 4.4 a 4.7).
Poruchy kontroly impulzov
Patologické hranie hazardných hier, zvýšené libido, hypersexualita, kompulzívne utrácanie alebo nakupovanie, prejedanie sa a závislosť na jedle sa môžu vyskytnúť u pacientov liečených agonistami dopamínu vrátane rotigotínu (pozri časť 4.4 ).
Osobitné populácie
Nežiaduce účinky zvýšenej kreatínfosfokinázy (CPK) sa pozorovali v klinických štúdiách
s rotigotínom uskutočnených v Japonsku. Vyskytovali sa u 3,4 % Japoncov liečených rotigotínom
v porovnaní s 1,9 % osôb užívajúcich placebo v dvojito zaslepených štúdiách Parkinsonovej choroby
a RLS. Väčšina nežiaducich účinkov zvýšenej CPK pozorovaných vo všetkých dvojito zaslepených a nezaslepených štúdiách ustúpila a boli považované za mierne závažné. V ostatných populáciách sa hladiny CPK bežne nemerali.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieSymptómyNajpravdepodobnejšie nežiaduce reakcie sú tie, ktoré súvisia s farmakodynamickým profilom agonistu
dopamínu, vrátane nevoľnosti, vracania, hypotenzie, mimovoľných pohybov, halucinácií, zmätenosti, kŕčov a ďalších známok centrálnej dopamínergickej stimulácie.
LiečbaNa predávkovanie agonistami dopamínu nie je známy žiadny protiliek. V prípade podozrenia na
predávkovanie sa má zvážiť odstránenie náplaste (náplastí), pretože po odstránení náplaste (náplastí) sa prívod liečiva zastaví a plazmatická koncentrácia rotigotínu rýchlo klesá. Pacient má byť dôkladne sledovaný, vrátane srdcovej frekvencie, srdcového rytmu a tlaku krvi.
Liečba predávkovania môže vyžadovať celkové podporné opatrenia na zachovanie životných funkcií. Dialýza by pravdepodobne nebola prospešná, pretože rotigotín nie je možné eliminovať dialýzou.
Ak je potrebné rotigotín vysadiť, vysadenie má prebehnúť postupne, aby sa zabránilo vzniku
neuroleptického malígneho syndrómu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antiparkinsoniká, agonisty dopamínu; ATC kód: N04BC09
Rotigotín je agonista dopamínu, ktorý nie je odvodený od ergotamínu, na liečbu prejavov
a symptómov Parkinsonovej choroby a syndrómu nepokojných nôh.
Mechanizmus účinkuPredpokladá sa, že rotigotín vyvoláva priaznivé účinky na Parkinsonovu chorobu aktiváciou
receptorov D3, D2 a D1 v
caudate-putamen mozgu.
Presný mechanizmus účinku rotigotínu v liečbe RLS nie je známy. Predpokladá sa, že rotigotín môže vykazovať aktivitu predovšetkým prostredníctvom dopamínových receptorov.
Farmakodynamické účinkyČo sa týka funkčnej aktivity na rôznych podtypoch receptorov a ich distribúcie v mozgu, rotigotín je
agonista receptorov D2 a D3, ktorý pôsobí aj na receptory D1, D4 a D5. V prípade iných receptorov ako
dopaminergných preukázal rotigotín antagonizmus na alfa2B receptoroch a agonizmus na 5HT1A
receptoroch, žiadnu aktivitu však nepreukázal na receptoroch 5HT2B.
Klinická účinnosť
Účinnosť rotigotínu bola hodnotená v 5- tich placebom kontrolovaných štúdiách s viac ako
1400 pacientmi so syndrómom nepokojných nôh (RLS). Účinnosť bola preukázaná v kontrolovaných štúdiách u pacientov liečených až do 29 týždňov. Účinok pretrvával počas 6- mesačného obdobia.
Zmeny oproti východiskovej hodnote boli hodnotené podľa International RLS Rating Scale (IRLS) a CGI-item 1 (závažnosť ochorenia), ktoré predstavovali primárne parametre účinnosti. Pre oba primárne ciele boli zistené štatisticky významné rozdiely oproti placebu v dávkach 1 mg/24 h,
2 mg/24 h a 3 mg/24 h. Po 6- tich mesiacoch udržiavacej liečby u pacientov so stredne závažným až
závažným RLS došlo k zlepšeniu IRLS skóre z 30,7 na 20,7 u placeba a z 30,2 na 13,8 u rotigotínu. Upravená priemerná hodnota rozdielu bola –6,5 bodov (CI95% -8,7; -4,4, p < 0,0001). Počty pacientov, ktorí odpovedali na liečbu podľa CGI-I (veľké zlepšenie, veľmi veľké zlepšenie) zodpovedali 43,0 %
u placeba a 67,5 % u rotigotínu (rozdiel 24,5 % CI 95%: 14,2 %; 34,8 %, p< 0,0001).
V placebom kontrolovanom klinickom skúšaní boli 7 týždňov hodnotené polysomnografické
parametre. Rotigotín významne znížil index periodického pohybu končatín (periodic limb movement
index, PLMI) z 50,9 na 7,7 verzus 37,4 na 32,7 oproti placebu (p< 0,0001).
Augmentácia
V dvoch 6-mesačných, dvojito zaslepených, placebom kontrolovaných štúdiách bola pozorovaná klinicky relevantná augmentácia u 1,5 % pacientov liečených rotigotínom oproti 0,5 % pacientov liečených placebom. V dvoch otvorených nadväzujúcich štúdiách v nasledujúcich 12 mesiacoch bol výskyt relevantnej augmentácie 2,9 %. Žiadny z týchto pacientov neprerušil liečbu z dôvodu augmentácie. V 5-ročnej , otvorenej štúdii sa augmentácia vyskytla u 11,9 % pacientov liečených dávkami schválenými pre RLS (1 – 3 mg/24 h) a u 5,1 % bola považovaná za klinicky významnú.
V tejto štúdii sa väčšina prípadov augmentácie vyskytla v prvom a druhom roku liečby. Navyše v tejto
štúdii bola tiež použitá vyššia dávka 4 mg/24 h, ktorá nie je schválená pre RLS a viedla vo vyššej miere k augmentácii.
Priľnavosť na kožu
V multicentrickej, dvojito zaslepenej, randomizovanej, dvojito skríženej štúdii bola u 52 ambulantných pacientov porovnávaná priľnavosť na kožu rotigotín náplasti 8 mg/24 h, s vylepšeným uchovávaním pri izbovej teplote, s náplasťou s uchovávaním v chlade. Priľnavosť na kožu bola meraná počas 2 po sebe nasledujúcich dní pri 24-hodinovej aplikácii náplasti.
Vylepšená náplasť s uchovávaním pri izbovej teplote preukázala lepšiu priľnavosť na kožu ako náplasť s uchovávaním v chlade, > 90 % náplastí preukazuje dostatočnú priľnavosť (t.j. > 70 %
priliehajúcej plochy náplasti) v porovnaní s <83 %. Pri obidvoch formách bola hlásená porovnateľná
znášanlivosť kože. Väčšina pozorovaných erytémov bolo miernych a žiadny nebol závažný.
5.2 Farmakokinetické vlastnosti
Absorpcia
Rotigotín sa po aplikácii plynule uvoľňuje z transdermálnej náplasti a je absorbovaný cez pokožku.
Ustálené koncentrácie sa dosiahnu za jeden až dva dni po aplikácii náplasti a udržiavajú sa na stabilnej
úrovni aplikáciou náplasti jedenkrát denne, pričom táto sa nosí po dobu 24 hodín. Koncentrácie
rotigotínu v plazme sa zvyšujú proporcionálne v závislosti od dávky v rozsahu dávok od 1 mg/24 h po
24 mg/24 h.
Za 24 hodín sa uvoľní do pokožky približne 45 % liečiva v náplasti. Absolútna biologická dostupnosť
po transdermálnej aplikácii je približne 37 %.
Striedanie miesta aplikácie náplasti môže spôsobiť rozdiely medzi jednotlivými dennými hladinami v plazme. Rozdiely v biologickej dostupnosti rotigotínu siahali od 2 % (nadlaktie v porovnaní s bokom) po 46 % (rameno v porovnaní so stehnom). Neexistuje žiaden náznak relevantného vplyvu na klinické výsledky.
Distribúcia
Približne 92 % rotigotínu sa viaže in vitro na plazmatické proteíny.
Zdanlivý distribučný objem u ľudí je približne 84 l/kg.
Biotransformácia
Rotigotín je z veľkej časti metabolizovaný. Rotigotín je metabolizovaný N-dealkyláciou ako aj
priamou a sekundárnou konjugáciou. Výsledky vykonávané in vitro naznačujú, že rôzne izoformy CYP dokážu katalyzovať N-dealkyláciu rotigotínu. Hlavnými metabolitmi sú sulfáty a glukuronidové konjugáty pôvodnej látky ako aj metabolity N-dealkylácie, ktoré sú biologicky neaktívne.
Informácie o metabolitoch sú neúplné.
Eliminácia
Približne 71 % dávky rotigotínu sa vylučuje močom a menšia časť približne 23 % sa vylučuje stolicou.
Klírens rotigotínu po transdermálnom podaní je približne 10 l/minútu a jeho celkový polčas eliminácie je 5 až 7 hodín. Farmakokinetický profil preukazuje bifázickú elimináciu s počiatočným polčasom
približne 2 až 3 hodiny.
Keďže náplasť sa aplikuje transdermálne, neočakáva sa žiadny vplyv jedál alebo ochorení
gastrointestinálneho traktu.
Osobitné skupiny pacientov
Keďže liečba Neuprom začína nízkou dávkou a táto sa postupne titruje podľa klinickej znášanlivosti,
aby sa získal optimálny terapeutický účinok, úprava dávky na základe pohlavia, hmotnosti alebo veku
nie je potrebná.
Porucha funkcie pečene a obličiek
U pacientov so stredne závažnou poruchou funkcie pečene alebo miernou až závažnou poruchou funkcie obličiek neboli pozorované žiadne relevantné zvýšenia hladiny rotigotínu v plazme. Neupro nebolo skúmané u pacientov so závažnou poruchou funkcie pečene.
V prípade poruchy funkcie obličiek sa zvyšujú hladiny konjugátov rotigotínu a metabolitov N- dealkylácie rotigotínu v plazme. Vplyv týchto metabolitov na klinické účinky je však
nepravdepodobný.
Pediatrická populácia
Obmedzené farmakokinetické údaje získané u dospievajúcich pacientov s RLS (13-17 rokov, n=24) po liečbe viacnásobnými dávkami v rozsahu 0,5 mg až 3 mg/24 h ukázali, že systémová expozícia rotigotínu bola podobná expozícii u dospelých. Údaje o účinnosti/bezpečnosti nie sú dostatočné na stanovenie pomeru medzi expozíciou a odpoveďou (pozri tiež informácie o použití v pediatrickej populácii v časti 4.2).
5.3 Predklinické údaje o bezpečnosti
V štúdiách zameraných na opakované dávky a dlhodobú toxicitu boli hlavné účinky spájané s farmakodynamickými účinkami súvisiacimi s agonistom dopamínu a následným znížením vylučovania prolaktínu.
Po jednorazovej dávke rotigotínu bolo u pigmentovaných potkanov a opíc evidentné viazanie sa rotigotínu na tkanivo obsahujúce melanín (t. j. oči), ktoré však počas 14-dňového pozorovacieho intervalu vymizlo.
V trojmesačnej štúdii bola u bielych potkanov pozorovaná pomocou transmisnej elektrónovej mikroskopie degenerácia sietnice pri dávke ekvivalentnej 2,8-násobku maximálnej odporúčanej dávky pre ľudí (prepočítanej na mg/m2). Účinky boli výraznejšie u samíc potkanov. Dodatočné štúdie na ďalšie vyhodnotenie špecifickej patológie neboli vykonané. Degenerácia sietnice sa nepozorovala počas rutinného histopatologického vyhodnocovania očí skúmaných druhov zvierat v žiadnej
z toxikologických štúdií. Význam týchto zistení pre ľudí nie je známy.
V štúdii karcinogenicity sa u samcov potkanov rozvinuli nádory a hyperplázia Leydigových buniek. Zhubné nádory boli pozorované najmä v maternici samíc, ktorým sa podávali stredné až vysoké
dávky. Tieto zmeny sú dobre známymi účinkami agonistov dopamínu u potkanov po celoživotnej liečbe a boli zhodnotené ako nerelevantné pre ľudí.
Účinky rotigotínu na reprodukciu sa skúmali u potkanov, králikov a myší. Na žiadny z uvedených troch druhov nemal rotigotín teratogénne účinky, ale u potkanov a myší vykazoval embryotoxické
účinky pri dávkach toxických pre matky. Rotigotín neovplyvnil plodnosť samcov u potkanov, avšak jednoznačne znížil plodnosť samíc potkanov a myší z dôvodu účinkov na hladiny prolaktínu, ktoré sú
u hlodavcov obzvlášť významné.
Rotigotín nevyvolal génové mutácie v Amesovom teste, preukázal však účinky v in vitro teste lymfómu u myší s metabolickou aktiváciou a slabšie účinky bez metabolickej aktivácie. Mutagénny účinok je možné pripísať klastogénnemu účinku rotigotínu. Tento účinok nebol potvrdený in vivo
v mikronukleovom teste u myší a v teste na neplánovanú syntézu DNA (UDS, Unsheduled DNA Synthesis) u potkanov. Keďže prebiehal viac-menej súbežne so zníženým relatívnym celkovým rastom
buniek, môže súvisieť s cytotoxickým účinkom látky. Významnosť tohto jedného pozitívneho testu
mutagenicity in vitro preto nie je známa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Krycia vrstva
Polyesterový film s vrstvou silikónu a hliníka
farebný povlak pigmentovej vrstvy (oxid titaničitý (E171), žltý pigment 95, červený pigment 166)
a potlač (červený pigment 144, žltý pigment 95, čierny pigment 7).
Samolepiaca matricová vrstva
Poly(dimetylsiloxán, trimetylsilylsilikát)-kopolymerizát,
Povidón K90,
Metasiričitan sodný (E223), Askorbylpalmitát (E304) a DL-α-tokoferol (E307).
Snímateľná fólia
Priesvitný polyesterový film s fluoropolymérovým povlakom.
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
30 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
6.5 Druh obalu a obsah balenia
Rozlepovacie vrecko v lepenkovej škatuli: Jednu stranu tvorí kopolymér etylénu (najvnútornejšia vrstva), hliníková fólia, polyetylénový film s nízkou hustotou a papier; druhú stranu tvorí polyetylén (najvnútornejšia vrstva), hliník, kopolymér etylénu a papier.
Škatuľa obsahuje 7, 14, 28, 30 alebo 84 (multibalenie obsahujúce 2 balenia po 42) transdermálnych
náplastí, ktoré sú jednotlivo zatavené vo vreckách.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Náplasť po použití ešte stále obsahuje liečivo. Po odstránení sa má použitá náplasť preložiť napoly lepiacou stranou smerom dovnútra tak, aby matricová vrstva nezostala odkrytá, umiestniť do pôvodného vrecka a odložiť. Všetky použité aj nepoužité náplasti majú byť zlikvidované v súlade
s národnými požiadavkami alebo vrátené do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
UCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLA
Neupro1mg/24htransdermálnanáplasť
EU/1/05/331/038
EU/1/05/331/040
EU/1/05/331/041
EU/1/05/331/044
EU/1/05/331/056
Neupro3mg/24htransdermálnanáplasť
EU/1/05/331/047
EU/1/05/331/049
EU/1/05/331/050
EU/1/05/331/053
EU/1/05/331/058
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 15. február 2006
Dátum posledného predĺženia registrácie: 22. január 2016
10. DÁTUM REVÍZIE TEXTU
{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Neupro 2 mg/24 h transdermálna náplasť
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá náplasť uvoľní 2 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 10 cm2 obsahuje 4,5 mg rotigotínu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Transdermálna náplasť.
Tenká štvorcová náplasť matricového typu so zaoblenými rohmi, skladajúca sa z troch vrstiev. Vonkajšia strana krycej vrstvy je béžová s potlačou „Neupro 2 mg/24 h“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Syndróm nepokojných nôh
Neupro sa indikuje na symptomatickú liečbu stredne závažného až závažného idiopatického syndrómu
nepokojných nôh (RLS) u dospelých.
Parkinsonova choroba
Neupro sa indikuje na liečbu známok a symptómov idiopatickej Parkinsonovej choroby v počiatočnom
štádiu v monoterapii (t. j. bez L-dopy) alebo v kombinácii s L-dopou, čiže počas trvania ochorenia až do neskorých štádií, keď účinok L-dopy zoslabne alebo sa stane premenlivým a dochádza
k fluktuáciám terapeutického účinku (fluktuácie ku koncu dávkovacieho intervalu alebo „on-off“
fluktuácie zo stavu dobrej hybnosti tzv. „on“ do stavu zlej hybnosti tzv. „off“).
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Odporúčané dávky predstavujú nominálne dávky.
Syndróm nepokojných nôh
Jednorazová denná dávka sa má začať na 1 mg/24 h. V závislosti od individuálnej odpovede pacienta sa dávka môže zvýšiť v týždňových intervaloch o 1 mg/24 h až po dosiahnutie maximálnej dávky
3 mg/24 h. Potreba pokračovať v liečbe sa má prehodnotiť každých 6 mesiacov.
Parkinsonova choroba
Dávkovanie u pacientov v počiatočnom štádiu Parkinsonovej choroby:
Jednorazová denná dávka má začať na 2 mg/24 h a následne byť zvyšovaná v týždňových intervaloch o 2 mg/24 h až po dosiahnutie účinnej dávky, ktorá je maximálne 8 mg/24 h.
Dávka 4 mg/24 h môže byť v prípade niektorých pacientov účinnou dávkou. U väčšiny pacientov je účinná dávka dosiahnutá v priebehu 3 alebo 4 týždňov pri dávkach 6 mg/24 h alebo 8 mg/24 h,
v uvedenom poradí.
Maximálna dávka je 8 mg/24 h.
Dávkovanie u pacientov v pokročilom štádiu Parkinsonovej choroby s fluktuáciami:
Jednorazová denná dávka má začať na 4 mg/24 h a následne byť zvyšovaná v týždňových intervaloch o 2 mg/24 h až po dosiahnutie účinnej dávky, ktorá je maximálne 16 mg/24 h.
Dávky 4 mg/24 h alebo 6 mg/24 h môžu byť v prípade niektorých pacientov účinnými dávkami.
U väčšiny pacientov je účinná dávka dosiahnutá v priebehu 3 až 7 týždňov pri dávkach 8 mg/24 h až
po maximálnu dávku 16 mg/24 h.
Pre dávky vyššie ako 8 mg/24 h je možné použiť na dosiahnutie konečnej dávky viacero náplastí, napríklad dávka 10 mg/24 h sa môže dosiahnuť kombináciou náplastí 6 mg/24 h a 4 mg/24 h.
Neupro sa aplikuje raz denne. Náplasť sa má aplikovať každý deň približne v rovnakom čase. Náplasť sa ponecháva na koži 24 hodín, a potom sa vymieňa za novú náplasť aplikovanú na inom mieste.
Ak si pacient zabudne náplasť aplikovať v obvyklom čase počas dňa alebo ak sa náplasť odlepí, na zvyšok dňa sa má aplikovať nová náplasť.
Ukončenie liečby
Syndróm nepokojných nôh
Liečba Neuprom má byť ukončená postupne. Denná dávka sa má znižovať o 1 mg/24 h znižovaním dávky podľa možnosti každý druhý deň až do úplného vysadenia Neupra (pozri časť 4.4). Po uplatnení tohto postupu sa nepozoroval tzv. rebound fenomén (zhoršenie pôvodných príznakov so zvýšenou intenzitou po vynechaní liečby).
Parkinsonova choroba
Liečba Neuprom má byť ukončená postupne. Denná dávka sa má znižovať o 2 mg/24 h znižovaním dávky podľa možnosti každý druhý deň až do úplného vysadenia Neupra (pozri časť 4.4).
Osobitné populácie
Porucha funkcie pečene
U pacientov s mierne až stredne závažnými poruchami funkcie pečene nie je potrebná úprava dávky. Opatrnosť sa odporúča pri liečbe pacientov so závažnými poruchami funkcie pečene, u ktorých môže dôjsť k zníženiu klírensu rotigotínu. Rotigotín nebol skúmaný u tejto skupiny pacientov. V prípade zhoršovania poruchy funkcie pečene môže byť potrebná redukcia dávky.
Porucha funkcie obličiek
U pacientov s miernou až závažnou poruchou funkcie obličiek, vrátane dialyzovaných pacientov, nie je potrebná úprava dávky. Neočakávaný nárast hladiny rotigotínu sa môže objaviť tiež v prípade akútneho zhoršenia funkcie obličiek (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť rotigotínu u detí a dospievajúcich neboli doteraz stanovené. V súčasnosti dostupné údaje sú popísané v časti 5.2, avšak na ich základe nie je možné urobiť žiadne odporúčanie pre dávkovanie rotigotínu u detí s RLS.
Neexistuje žiadne relevantné použitie Neupra v pediatrickej populácii pri Parkinsonovej chorobe.
Spôsob podávania
Neupro je určené na transdermálne použitie.
Náplasť sa má aplikovať na čistú, suchú, neporušenú zdravú pokožku v oblasti brucha, stehna, bedier, bokov trupu, ramien alebo nadlaktia. Náplasť sa nemá aplikovať na rovnaké miesto skôr než po
14 dňoch. Neupro sa nesmie aplikovať na červenú, podráždenú alebo poškodenú pokožku (pozri časť 4.4).
Použitie a zaobchádzanie
Každá náplasť je zabalená vo vrecku a aplikuje sa ihneď po otvorení vrecka. Odstráni sa jedna polovica snímateľnej fólie a lepiaca strana sa aplikuje pevným pritlačením na pokožku. Potom sa náplasť prehne dozadu a odstráni sa druhá časť prilepenej fólie. Nedotýkajte sa lepiacej strany náplasti. Dlaňou sa náplasť pevne zatlačí po dobu približne 20 až 30 sekúnd, aby sa dobre prilepila.
Náplasť sa nesmie strihať na kusy.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Zobrazovanie magnetickou rezonanciou alebo kardioverzia (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Ak je odozva pacienta s Parkinsonovou chorobou na liečbu rotigotínom nedostatočná, môžu sa prestupom na liečbu iným agonistom dopamínu dosiahnuť dodatočné pozitívne účinky (pozri časť
5.1).
Obidve indikácie:
Zobrazovanie magnetickou rezonanciou a kardioverzia
Krycia vrstva náplasti Neupro obsahuje hliník. Ak bude pacient podstupovať vyšetrenie zobrazovaním
magnetickou rezonanciou (MRI) alebo kardioverziu, musí sa Neupro odstrániť, aby sa zabránilo
popáleninám pokožky.
Ortostatická hypotenzia
Je známe, že agonisty dopamínu zhoršujú systémovú reguláciu tlaku krvi, v dôsledku čoho vzniká
posturálna/ortostatická hypotenzia. Tieto prípady sa pozorovali aj počas liečby rotigotínom, ich výskyt bol však podobný ako výskyt pozorovaný u pacientov liečených placebom.
Z dôvodu všeobecného rizika ortostatickej hypotenzie spojenej s dopamínergickou liečbou sa odporúča sledovať krvný tlak, a to najmä na začiatku liečby.
Synkopa
V klinických štúdiách s rotigotínom sa pozorovala synkopa s podobnou frekvenciou výskytu, ako bola
pozorovaná u pacientov liečených placebom. Keďže pacienti s klinicky relevantným kardiovaskulárnym ochorením boli z týchto štúdií vylúčení, je potrebné sa pacientov so závažným
kardiovaskulárnym ochorením opýtať na príznaky synkopy a presynkopy.
Náhle zaspanie a ospalosť
Rotigotín bol spájaný s ospalosťou a prípadmi náhleho zaspania. Hlásené boli prípady náhleho
zaspania počas denných aktivít, v niektorých prípadoch bez spozorovania akýchkoľvek varovných známok. Lekári predpisujúci Neupro majú u pacientov nepretržite prehodnocovať príznaky driemot
alebo ospalosti, pretože samotní pacienti si ich nemusia uvedomiť, až kým sa ich na to priamo neopýtajú. Starostlivo sa má zvážiť zníženie dávky alebo ukončenie liečby.
Poruchy kontroly impulzov
Pacientov je treba pravidelne monitorovať pre vývoj porúch kontroly impulzov. Pacienti
a opatrovatelia si majú byť vedomí, že u pacientov liečených agonistami receptorov dopamínu vrátane rotigotínu sa môžu vyskytnúť behaviorálne príznaky porúch kontroly impulzov ako patologické hranie
hazardných hier, zvýšené libido, hypersexualita, kompulzívne utrácanie alebo nakupovanie, prejedanie
sa a závislosť na jedle. Ak sa tieto symptómy objavia, je potrebné zvážiť zníženie dávky/postupné
vysadenie.
Neuroleptický malígny syndróm
Pri náhlom ukončení dopamínergickej liečby boli hlásené symptómy naznačujúce prítomnosť
neuroleptického malígneho syndrómu. Preto sa odporúča liečbu obmedzovať postupne (pozri časť 4.2).
Abnormálne myslenie a správanie
Zaznamenalo sa abnormálne myslenie a správanie a môže pozostávať z rôznych prejavov vrátane
paranoidných myšlienok, preludov, halucinácií, zmätenosti, správania podobného psychotickému, dezorientácie, agresívneho správania, agitácie a delíria.
Fibrotické komplikácie
U niektorých pacientov liečených dopamínergickými látkami odvodenými od námeľu boli hlásené
prípady retroperitoneálnej fibrózy, pľúcnych infiltrátov, pleurálnych výpotkov, pleurálneho zhrubnutia, perikarditídy a srdcovej valvulopatie. Aj keď tieto komplikácie môžu po vysadení liečby ustúpiť, nemusia vždy ustúpiť úplne.
Aj keď sa predpokladá, že tieto nežiaduce reakcie súvisia s ergotamínovou štruktúrou týchto látok, nie je známe, či ich môžu spôsobovať aj agonisty dopamínu, ktoré nie sú odvodené od námeľu.
Neuroleptiká
Neuroleptiká podávané ako antiemetiká sa nemajú podávať pacientom užívajúcim agonistov
dopamínu (pozri aj časť 4.5).
Oftalmologické sledovanie
Oftalmologické sledovanie sa odporúča v pravidelných intervaloch alebo v prípade porúch videnia.
Vystavenie zdroju tepla
Miesto s náplasťou nemá byť vystavované vonkajším zdrojom tepla (nadmerné slnečné žiarenie,
ohrievacie podušky a iné zdroje tepla, napríklad sauna, horúci kúpeľ).
Reakcie v mieste aplikácie
V mieste aplikácie sa môžu vyskytnúť kožné reakcie, zvyčajne s miernou alebo stredne závažnou
intenzitou. Miesto aplikácie sa odporúča denne striedať (napríklad z pravej strany na ľavú stranu
a z hornej časti tela na dolnú časť tela). Rovnaké miesto sa nemá použiť znova skôr než po 14 dňoch. V prípade niekoľkodňového alebo trvalého výskytu reakcií v mieste aplikácie, pri zvýšení závažnosti
alebo pri rozšírení kožnej reakcie mimo miesto aplikácie, sa má zhodnotiť pomer rizík a výhod pre
daného pacienta.
V prípade výskytu vyrážok alebo podráždenia pokožky transdermálnym systémom sa má zabrániť pôsobeniu priameho slnečného žiarenia na dané miesto, kým sa pokožka nezahojí, pretože expozícia môže spôsobiť zmeny sfarbenia pokožky.
V prípade spozorovania celkovej reakcie pokožky (napríklad alergické vyrážky, vrátane erytematóznych, škvrnitých a papulárnych vyrážok alebo svrbenia) spojenej s používaním Neupra, musí sa Neupro prestať používať.
Periférny edém
V klinických štúdiách u pacientov s Parkinsonovou chorobou bol počas 6 mesiacov špecifický výskyt
periférneho edému okolo 4 % a zostal nezmenený počas ďalšieho pozorovania až do 36 mesiacov. V klinických štúdiách uskutočnených u pacientov s RLS sa tiež pozoroval periférny edém.
Citlivosť nasiričitan
Neupro obsahuje metasiričitan sodný, siričitan, ktorý môže vyvolať reakcie alergického typu vrátane
anafylaktických symptómov a život ohrozujúcich alebo menej závažných astmatických epizód u niektorých citlivých osôb.
Pozorované u pacientov s Parkinsonovou chorobou
Dopamínergické nežiaduce reakcie
Výskyt niektorých dopamínergických nežiaducich reakcií, ako sú napríklad halucinácie, dyskinéza a periférny edém, je vo všeobecnosti vyšší pri podávaní v kombinácii s L-dopou u pacientov
s Parkinsonovou chorobou. Treba to zvážiť pri predpisovaní rotigotínu.
Pozorované u pacientov so syndrómom nepokojných nôh
Augmentácia
U pacientov so syndrómom nepokojných nôh sa môže objaviť zintenzívnenie príznakov
(augmentácia). Toto zintenzívnenie sa môže prejaviť ako skorší nástup príznakov večer (alebo dokonca poobede), zvýšenie stupňa závažnosti príznakov a rozšírenie príznakov na iné časti tela.
V dlhodobých klinických štúdiách s rotigotínom sa vo väčšine prípadov augmentácia pozorovala
v prvom a druhom roku liečby. Dávky vyššie ako schválené rozmedzie pre RLS sa nemajú používať, pretože môžu viesť k vyššiemu výskytu augmentácie (pozri časť 5.1).
4.5 Liekové a iné interakcie
Keďže rotigotín je agonista dopamínu, predpokladá sa, že antagonisty dopamínu, ako napríklad neuroleptiká (napríklad fenotiazíny, butyrofenóny, tioxantény) alebo metoklopramid, môžu oslabovať účinnosť Neupra, a preto sa nemajú podávať súbežne s Neuprom. Z dôvodu možných aditívnych účinkov sa má postupovať opatrne, ak pacienti užívajú sedatívne lieky alebo iné lieky utlmujúce činnosť CNS (centrálneho nervového systému) (napríklad benzodiazepíny, antipsychotiká, antidepresíva) alebo alkohol v kombinácii s rotigotínom.
Súbežné podávanie L-dopy a karbidopy s rotigotínom nemalo žiadny účinok na farmakokinetiku rotigotínu a rotigotín nemal žiadny účinok na farmakokinetiku L-dopy a karbidopy.
Súbežné podávanie domperidonu s rotigotínom nemalo žiadny účinok na farmakokinetiku rotigotínu. Súbežné podávanie omeprazolu (inhibítora CYP2C19) v dávkach 40 mg/deň nemalo žiadny účinok na
farmakokinetiku a metabolizmus rotigotínu u zdravých dobrovoľníkov.
Neupro môže zosilniť dopamínergické nežiaduce reakcie L-dopy a môže spôsobiť a/alebo zhoršiť už
existujúcu dyskinézu, podobne ako v prípade ostatných agonistov dopamínu.
Súbežné podávanie rotigotínu (3 mg/24 h) neovplyvňovalo farmakodynamiku a farmakokinetiku
perorálnych kontraceptív (0,03 mg etinylestradiol, 0,15 mg levonorgestrel). Interakcie s inými formami hormonálnej antikoncepcie neboli skúmané.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku, antikoncepcia u žien
Ženy vo fertilnom veku majú používať účinnú antikoncepciu na prevenciu gravidity počas liečby
rotigotínom.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití rotigotínu u gravidných žien. Štúdie na zvieratách
nepreukázali žiadne teratogénne účinky u potkanov a králikov, avšak u potkanov a myší bola pozorovaná embryotoxicita pri dávkach toxických pre matky (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí. Rotigotín sa nemá používať počas gravidity.
Laktácia
Keďže rotigotín znižuje vylučovanie prolaktínu u ľudí, očakáva sa inhibícia tvorby mlieka. Štúdie
vykonávané na potkanoch ukázali, že rotigotín a/alebo jeho metabolit(y) sú vylučované do materského mlieka. Z dôvodu chýbajúcich údajov o účinkoch na ľudský organizmus sa má dojčenie prerušiť.
Fertilita
Pre informácie o štúdiách fertility, prosím, pozri časť 5.3.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Rotigotín môže mať značný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Pacienti liečení rotigotínom, ktorí trpia ospalosťou a/alebo prípadmi náhleho zaspania, musia byť informovaní o zákaze riadenia vozidiel alebo vykonávania aktivít (napríklad obsluhovanie strojov), pri ktorých môže zhoršené vnímanie ohroziť ich samotných alebo ostatných a spôsobiť vážne zranenia alebo smrť. To platí, až kým tieto opakujúce sa prípady náhleho zaspania a ospalosť neustúpia (pozri
aj časti 4.4 a 4.5).
4.8 Nežiaduce účinky
Syndróm nepokojných nôh
Súhrnbezpečnostnéhoprofilu
Podľa analýzy skupiny placebom kontrolovaných klinických štúdií, zahŕňajúcich celkovo
748 pacientov liečených Neuprom a 214 pacientov užívajúcich placebo, 65,5 % pacientov liečených
Neuprom a 33,2 % pacientov liečených placebom hlásilo aspoň jednu nežiaducu reakciu.
Na začiatku liečby sa môžu vyskytovať dopamínergické nežiaduce reakcie ako nevoľnosť a vracanie. Tieto sú zvyčajne mierne alebo stredne závažné a dočasné, a to aj v prípade pokračovania v liečbe.
Medzi nežiaduce liekové reakcie hlásené u viac ako 10 % pacientov liečených Neuprom patria nevoľnosť, reakcie v mieste aplikácie, astenické stavy a bolesť hlavy.
V štúdiách, pri ktorých sa miesto aplikácie striedalo podľa pokynov v SmPC (Súhrn charakteristických
vlastností lieku, Summary of Product Characteristics) a písomnej informácii pre používateľa, sa u 34,2 % z 748 pacientov používajúcich Neupro vyskytovali reakcie v mieste aplikácie. Väčšina
reakcií v mieste aplikácie bola mierna alebo stredne závažná, obmedzená na miesto aplikácie a v ich
dôsledku bola liečba Neuprom ukončená iba u 7,2 % pacientov.
Frekvencia prerušenia liečby
Frekvencia prerušenia liečby sa hodnotila v 3 klinických štúdiách s trvaním až do 3 rokov. Percento
subjektov, ktorí prerušili liečbu, bolo 25 - 38 % v priebehu prvého roka, 10 % v druhom roku a
11 % v treťom roku. Pravidelne sa má hodnotiť účinnosť spolu s hodnotením bezpečnosti, vrátane
augmentácie.
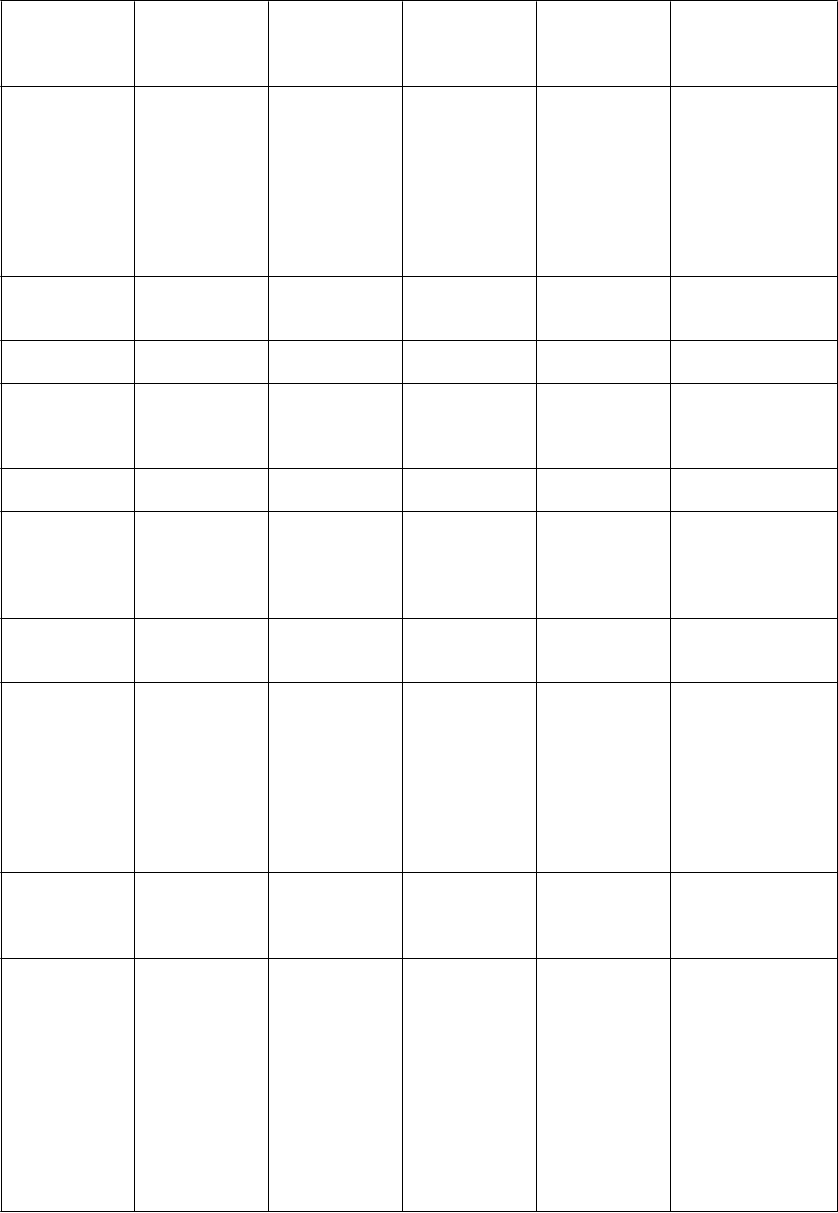
Zoznam nežiaducich reakcií zoradených dotabuľky
V nasledujúcej tabuľke sú uvedené nežiaduce liekové reakcie zo združených štúdií uvedených vyššie
u pacientov so syndrómom nepokojných nôh. V rámci tried orgánových systémov sú nežiaduce reakcie vymenované pod názvami jednotlivých frekvencií (počet pacientov, u ktorých sa predpokladá výskyt reakcie) použitím nasledujúcich kategórií: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé
(< 1/10 000); neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce
účinky usporiadané v poradí klesajúcej závažnosti.
T
ri
edy
orgánových systémov
podľa
MedDRA
P
oruchy imunitného systému
V
e
ľmi časté Časté Menej časté Zriedkavé Neznáme
hypersenzitivit a, ktorá môže zahŕňať angioedém, edém jazyka
a edém pier
 P
sychické poruchy
P
sychické poruchy
náhle zaspávanie/náh ly nástup spánku, poruchy sexuálnej
túžbya (vr. hypersexuality,
zvýšeného libida),
insomnia, porucha spánku,
neobvyklé sny, poruchy
kontroly impulzova,d (vr. patologického hrania hazardných hier, stereotypného/ nutkavého správania, prejedania sa/ porúch
obsedantno- kompulzívna porucha, agitáciad
agresívne správanie/ agresivitab, dezorientácia
syndróm dopamínovej dysreguláciec, poruchy vnímaniae (vr. halucinácie, vizuálnej halucinácie, sluchovej halucinácie, vidín), nočné morye, paranojae, stav zmätenostie, psychotické poruchye, prelude, delíriume
P
oruchy nervového systému
prijímania potravyb, kompulzívneho nakupovaniac)
bolesť hlavy somnolencia závrate, poruchy vedomia NECe
(vr. synkopy,
vazovagálnej synkopy, straty vedomia), dyskinézae, posturálny závrate, letargiae, kŕče
P
oruchy oka rozmazané videniee, poruchy videniae, fotopsiae
P
oruchy ucha a labyrintu Poruchy srdca a srdcovej činnosti
P
oruchy ciev hypertenzia ortostatická hypotenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
vertigoe
palpitáciee,
fibrilácie predsieníe, supraventrikulárna
tachykardiae
hypotenziae
štikútaniee
P
oruchy gastrointestin álneho traktu
nevoľnosť vracanie, dyspepsia
zápchae, sucho
v ústache, bolesť
bruchae
 P
oruchy kože a podkožného tkaniva
P
oruchy reprodukčného systému a prsníkov Celkové poruchy
a reakcie
v mieste podania
P
oruchy kože a podkožného tkaniva
P
oruchy reprodukčného systému a prsníkov Celkové poruchy
a reakcie
v mieste podania
reakcie
v mieste aplikácie a instiláciea, (vr. erytému, svrbenia, iritácie, vyrážok, dermatitídy, vezikúl, bolesti, ekzému,
pruritus erytéme, hyperhidrózae,
generalizovaný prurituse, podráždenie kožee, kontaktná dermatitídae, generalizovaná vyrážkae
erektilná dysfunkciae
zvýšená dráždivosť, periférny edém
L
aboratórne a funkčné vyšetrenia
Ú
r
azy, otravy a komplikácie liečebného postupu
zápalu, opuchu, zmeny farby, pupencov, exfoliácie, urtikárie, precitlivenosti), astenické
stavya (vr. únavy, asténie, mdlôb)
zníženie hmotnostie, zvýšenie pečeňových
enzýmove (vr. AST, ALT, GGT), zvýšenie
hmotnostie, zrýchlenie
srdcového rytmue, zvýšenie CPKd,e (pozri Osobitné
populácie)
páde
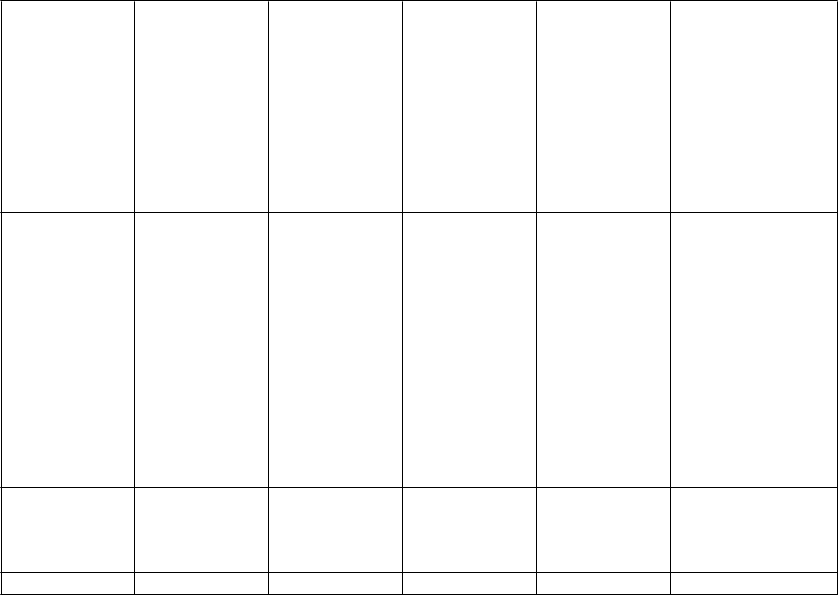
a Nadradený všeobecný názov
b Pozorované v nezaslepených štúdiách
c Pozorované po uvedení lieku na trh
d Pozorované v roku 2011 v súhrnných údajoch z dvojito zaslepených placebom kontrolovaných štúdií
e Pozorované v štúdiách u pacientov s Parkinsonovou chorobou
Parkinsonova chorobaSúhrnbezpečnostnéhoprofiluPodľa analýzy skupinových, placebom kontrolovaných klinických štúdií, zahŕňajúcich celkovo
1 307 pacientov liečených Neuprom a 607 pacientov liečených placebom, 72,5 % pacientov liečených
Neuprom a 58,0 % pacientov liečených placebom hlásilo aspoň jednu nežiaducu reakciu.
Na začiatku liečby sa môžu vyskytovať dopamínergické nežiaduce reakcie ako nevoľnosť a vracanie. Tieto sú zvyčajne mierne alebo stredne závažné a dočasné, a to aj v prípade pokračovania v liečbe.
Medzi nežiaduce liekové reakcie hlásené u viac ako 10 % pacientov liečených transdermálnymi náplasťami Neupro patria nevoľnosť, vracanie, reakcie v mieste aplikácie, ospalosť, závrat a bolesť hlavy.
V štúdiách, pri ktorých sa miesto aplikácie striedalo podľa pokynov v SmPC (Súhrn charakteristických
vlastností lieku,
Summary of Product Characteristics) a písomnej informácii pre používateľa, sa
u 35,7 % z 830 pacientov používajúcich transdermálne náplasti Neupro vyskytovali reakcie v mieste aplikácie. Väčšina reakcií v mieste aplikácie bola mierna alebo stredne závažná, obmedzená na miesto aplikácie a v ich dôsledku bola liečba Neuprom ukončená iba u 4,3 % všetkých pacientov používajúcich Neupro.
Z
o
z
nam nežiaducich reakcií zoradených dotabuľky
V nasledujúcej tabuľke sú uvedené nežiaduce liekové reakcie zo združených štúdií uvedených vyššie
u pacientov s Parkinsonovou chorobou. V rámci tried orgánových systémov sú nežiaduce reakcie vymenované pod názvami jednotlivých frekvencií (počet pacientov, u ktorých sa predpokladá výskyt reakcie) použitím nasledujúcich kategórií: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v
poradí klesajúcej závažnosti.
T
ri
edy
orgánových systémov
podľa
MedDRA
P
oruchy imunitného systému
V
e
ľmi časté Časté Menej časté Zriedkavé Neznáme
hypersenzitivi ta, ktorá môže zahŕňať angioedém, edém jazyka
a edém pier
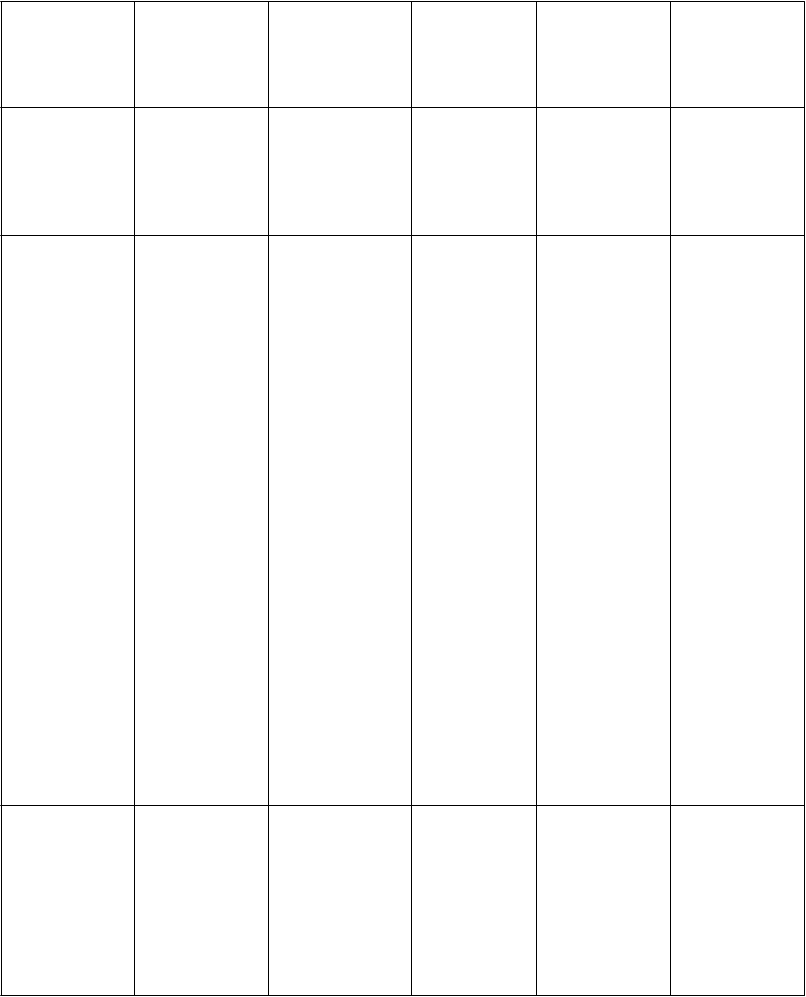 P
sychické poruchy
P
oruchy nervového systému
P
sychické poruchy
P
oruchy nervového systému
ospalosť, závrat, bolesť hlavy
poruchy vnímaniaa (vr. halucinácie, zrakovej halucinácie, sluchovej halucinácie, vidiny), insomnia, porucha spánku, nočné mory, neobvyklé sny, poruchy
kontroly impulzova,d (vr. patologického hrania hazardných hier, stereotypného/ nutkavého správania, prejedania sa/porúch prijímania potravyb, kompulzívneho nakupovaniac) poruchy
vedomia NECa
(vr. synkopy, vazovagálnej
synkopy, straty
vedomia), dyskinéza, posturálny závrat, letargia
náhle zaspávanie/ náhly nástup spánku, paranoja, poruchy sexuálnej túžbya (vrátane hypersexualit y, zvýšeného libida), stav zmätenosti,
dezorientáciad
, agitáciad
psychotická porucha, obsedantno- kompulzívna porucha, agresívne správanie/ agresivitab, preludd, delíriumd
kŕč
syndróm dopamínovej dysreguláciec
P
oruchy oka rozmazané videnie, poruchy videnia, fotopsia
P
oruchy ucha a labyrintu Poruchy srdca a srdcovej činnosti
vertigo
palpitácie atriálna fibrilácia
supraventri- kulárna tachykardia
P
oruchy ciev ortostatická hypotenzia, hypertenzia
hypotenzia
 P
oruchy dýchacej sústavy, hrudníka
a mediastína
P
oruchy gastrointestin álneho traktu Poruchy kože a podkožného tkaniva
P
oruchy reprodukčnéh o systému
a prsníkov
C
el
kové poruchy a reakcie v mieste podania
L
aboratórne a funkčné vyšetrenia
P
oruchy dýchacej sústavy, hrudníka
a mediastína
P
oruchy gastrointestin álneho traktu Poruchy kože a podkožného tkaniva
P
oruchy reprodukčnéh o systému
a prsníkov
C
el
kové poruchy a reakcie v mieste podania
L
aboratórne a funkčné vyšetrenia
nevoľnosť,
vracanie
reakcie
v mieste aplikácie a instiláciea, (napríklad erytém, svrbenie, podráždenie, vyrážka, dermatitída, vezikuly, bolesť, ekzém, zápal, opuch, zmena sfarbenia, pupence, exfoliácie, urtikária, precitlivenosť)
štikútanie
zápcha, sucho v ústach, dyspepsia erytém, hyperhidróza, svrbenie
periférny edém, astenické stavya, (vr. únavy, asténie, malátnosti)
zníženie hmotnosti
bolesť brucha
celkové svrbenie, podráždenie pokožky, kontaktná dermatitída erektilná dysfunkcia
zvýšenie hladiny pečeňového enzýmu (vr. AST, ALT, GGT), zvýšenie
generalizova- ná vyrážka
podráždenosť
Ú
r
azy, otravy a komplikácie liečebného postupu
a Nadradený všeobecný názov
b Pozorované v nezaslepených štúdiách
c Pozorované po uvedení lieku na trh
pád
hmotnosti, zrýchlenie srdcového rytmu, zvýšenie CPKd (pozri Osobitné populácie)
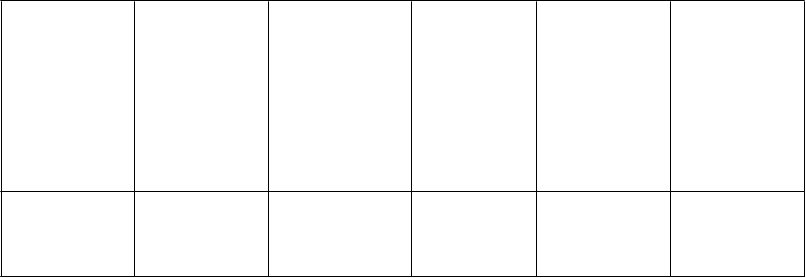
d Pozorované v roku 2011 v súhrnných údajoch z dvojito zaslepených placebom kontrolovaných štúdií
Obidve indikáciePopis vybraných nežiaducich reakciíNáhly nástup spánku a ospalosťRotigotín bol spájaný s ospalosťou vrátane nadmernej ospalosti počas dňa a prípadov náhleho zaspania. V zriedkavých prípadoch sa vyskytlo „náhle zaspanie“ počas jazdy, čo následne spôsobilo dopravnú nehodu (pozri aj časť 4.4 a 4.7).
Poruchy kontroly impulzovPatologické hranie hazardných hier, zvýšené libido, hypersexualita, kompulzívne utrácanie alebo nakupovanie, prejedanie sa a závislosť na jedle sa môžu vyskytnúť u pacientov liečených agonistami dopamínu vrátane rotigotínu (pozri časť 4.4 ).
Osobitné populácieNežiaduce účinky zvýšenej kreatínfosfokinázy (CPK) sa pozorovali v klinických štúdiách
s rotigotínom uskutočnených v Japonsku. Vyskytovali sa u 3,4 % Japoncov liečených rotigotínom
v porovnaní s 1,9 % osôb užívajúcich placebo v dvojito zaslepených štúdiách Parkinsonovej choroby
a RLS. Väčšina nežiaducich účinkov zvýšenej CPK pozorovaných vo všetkých dvojito zaslepených
a nezaslepených štúdiách ustúpila a boli považované za mierne závažné. V ostatných populáciách sa hladiny CPK bežne nemerali.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieSymptómy
Najpravdepodobnejšie nežiaduce reakcie sú tie, ktoré súvisia s farmakodynamickým profilom agonistu dopamínu, vrátane nevoľnosti, vracania, hypotenzie, mimovoľných pohybov, halucinácií, zmätenosti, kŕčov a ďalších známok centrálnej dopamínergickej stimulácie.
Liečba
Na predávkovanie agonistami dopamínu nie je známy žiadny protiliek. V prípade podozrenia na
predávkovanie sa má zvážiť odstránenie náplaste (náplastí), pretože po odstránení náplaste (náplastí)
sa prívod liečiva zastaví a plazmatická koncentrácia rotigotínu rýchlo klesá. Pacient má byť dôkladne
sledovaný, vrátane srdcovej frekvencie, srdcového rytmu a tlaku krvi.
Liečba predávkovania môže vyžadovať celkové podporné opatrenia na zachovanie životných funkcií. Dialýza by pravdepodobne nebola prospešná, pretože rotigotín nie je možné eliminovať dialýzou.
Ak je potrebné rotigotín vysadiť, vysadenie má prebehnúť postupne, aby sa zabránilo vzniku
neuroleptického malígneho syndrómu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiparkinsoniká, agonisty dopamínu; ATC kód: N04BC09
Rotigotín je agonista dopamínu, ktorý nie je odvodený od ergotamínu, na liečbu prejavov
a symptómov Parkinsonovej choroby a syndrómu nepokojných nôh.
Mechanizmus účinku
Predpokladá sa, že rotigotín vyvoláva priaznivé účinky na Parkinsonovu chorobu aktiváciou
receptorov D3, D2 a D1 v caudate-putamen mozgu.
Presný mechanizmus účinku rotigotínu v liečbe RLS nie je známy. Predpokladá sa, že rotigotín môže vykazovať aktivitu predovšetkým prostredníctvom dopamínových receptorov.
Farmakodynamické účinky
Čo sa týka funkčnej aktivity na rôznych podtypoch receptorov a ich distribúcie v mozgu, rotigotín je
agonista receptorov D2 a D3, ktorý pôsobí aj na receptory D1, D4 a D5. V prípade iných receptorov ako dopaminergných preukázal rotigotín antagonizmus na alfa2B receptoroch a agonizmus na 5HT1A receptoroch, žiadnu aktivitu však nepreukázal na receptoroch 5HT2B.
Klinická účinnosť a bezpečnosť
Klinické štúdie u syndrómu nepokojných nôh
Účinnosť rotigotínu bola hodnotená v 5-tich placebom kontrolovaných štúdiách s viac ako
1400 pacientmi so syndrómom nepokojných nôh (RLS). Účinnosť bola preukázaná v kontrolovaných štúdiách u pacientov liečených až do 29 týždňov. Účinok pretrvával počas 6- mesačného obdobia.
Zmeny oproti východiskovej hodnote boli hodnotené podľa International RLS Rating Scale (IRLS) a CGI-item 1 (závažnosť ochorenia), ktoré predstavovali primárne parametre účinnosti. Pre oba primárne ciele boli zistené štatisticky významné rozdiely oproti placebu v dávkach 1 mg/24 h,
2 mg/24 h a 3 mg/24 h. Po 6-tich mesiacoch udržiavacej liečby u pacientov so stredne závažným až závažným RLS došlo k zlepšeniu IRLS skóre z 30,7 na 20,7 u placeba a z 30,2 na 13,8 u rotigotínu. Upravená priemerná hodnota rozdielu bola –6,5 bodov (CI95% -8,7; -4,4, p < 0,0001). Počty pacientov, ktorí odpovedali na liečbu podľa CGI-I (veľké zlepšenie, veľmi veľké zlepšenie) zodpovedali 43,0 %
u placeba a 67,5 % u rotigotínu (rozdiel 24,5 % CI 95%: 14,2 %; 34,8 %, p< 0,0001).
V placebom kontrolovanom klinickom skúšaní boli 7 týždňov hodnotené polysomnografické parametre. Rotigotín významne znížil index periodického pohybu končatín (periodic limb movement index, PLMI) z 50,9 na 7,7 verzus 37,4 na 32,7 oproti placebu (p< 0,0001).
Augmentácia
V dvoch 6-mesačných, dvojito zaslepených, placebom kontrolovaných štúdiách bola pozorovaná klinicky relevantná augmentácia u 1,5 % pacientov liečených rotigotínom oproti 0,5 % pacientov liečených placebom. V dvoch otvorených nadväzujúcich štúdiách v nasledujúcich 12 mesiacoch bol výskyt relevantnej augmentácie 2,9 %. Žiadny z týchto pacientov neprerušil liečbu z dôvodu augmentácie. V 5-ročnej , otvorenej štúdii sa augmentácia vyskytla u 11,9 % pacientov liečených dávkami schválenými pre RLS (1 – 3 mg/24 h) a u 5,1 % bola považovaná za klinicky významnú.
V tejto štúdii sa väčšina prípadov augmentácie vyskytla v prvom a druhom roku liečby. Navyše v tejto
štúdii bola tiež použitá vyššia dávka 4 mg/24 h, ktorá nie je schválená pre RLS a viedla vo vyššej miere k augmentácii.
Klinické štúdie Parkinsonovej choroby
Účinnosť rotigotínu v liečbe známok a symptómov idiopatickej Parkinsonovej choroby bola vyhodnocovaná v multinárodnom programe vývoja liekov, ktorý pozostával zo štyroch kľúčových, paralelných, randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdií a v troch štúdiách skúmajúcich špecifické aspekty Parkinsonovej choroby.
Dve pivotné štúdie (SP512 časť I a SP513 časť I) skúmajúce účinnosť rotigotínu v liečbe známok a
symptómov idiopatickej Parkinsonovej choroby boli vykonané u pacientov, ktorí neboli súbežne
liečení agonistami dopamínu a ešte neboli liečení L-dopou alebo predchádzajúca liečba L-dopou trvala
≤ 6 mesiacov. Hlavným výsledným hodnotením bolo skóre pre komponent aktivity denného života
(časť II) (ADL, Activities of Daily Living) a komponent motorického vyšetrenia (časť III) jednotnej škály pre hodnotenie Parkinsonovej choroby (UPDRS, United Parkinson´s Disease Rating Scale). Účinnosť sa zisťovala na základe odozvy pacienta na liečbu, pričom sa merala ako zlepšenie odozvy pacienta a absolútneho počtu bodov v rámci skóre ADL v kombinácii s motorickým vyšetrením (UPDRS časť II+III).
V dvojito zaslepenej štúdii SP512 časť I dostávalo 177 pacientov rotigotín a 96 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu alebo placeba týždenným zvyšovaním
o 2 mg/24 h od počiatočnej dávky 2 mg/24 h po maximálnu dávku 6 mg/24 h. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 6 mesiacov.
Na konci udržiavacej liečby bola u 91 % pacientov v rotigotínovej vetve, optimálna dávka zhodná s maximálnou povolenou dávkou, t. j. 6 mg/24 h. Zlepšenie o 20 % bolo pozorované u 48 % pacientov, ktorým sa podával rotigotín a u 19 % pacientov, ktorým sa podávalo placebo (rozdiel 29 %, CI95 %
18 %; 39 %, p< 0,0001). V prípade rotigotínu bolo priemerné zlepšenie vyjadrené v skóre UPDRS
(časti II+III) –3,98 bodov (počiatočná hodnota 29,9 bodov), zatiaľ čo u pacientov vo vetve
s placebom, bolo pozorované zhoršenie o 1,31 bodov (počiatočná hodnota 30,0 bodov). Rozdiel bol
5,28 bodov a bol štatisticky významný (p< 0,0001).
V dvojito zaslepenej štúdii SP513 časť I dostávalo 213 pacientov rotigotín, 227 pacientov ropinirol a 117 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 2 mg/24 h po maximálnu dávku 8 mg/24 h počas
4 týždňov. V skupine, v ktorej sa podával ropinirol, bola u pacientov titrovaná optimálna dávka až do maximálnej hodnoty 24 mg/deň počas 13 týždňov. U pacientov v každej liečebnej skupine bola dávka
udržiavaná po dobu 6 mesiacov.
Na konci udržiavacej liečby bola u 92 % pacientov v rotigotínovej vetve optimálna dávka zhodná s
maximálnou povolenou dávkou, t. j. 8 mg/24 h. Zlepšenie o 20 % bolo pozorované u 52 % pacientov, ktorým sa podával rotigotín, u 68 % pacientov, ktorým sa podával ropinirol, a u 30 % pacientov, ktorým sa podávalo placebo. (Rozdiel medzi rotigotínom a placebom bol 21,7 %, CI95 % 11,1 %;
32,4 %, rozdiel medzi ropinirolom a placebom bol 38,4 %, CI95 % 28,1 %; 48,6 %, rozdiel medzi
ropinirolom a rotigotínom bol 16,6 %, CI95 %, 7,6 %; 25,7 %). V rotigotínovej vetve sa dosiahlo
priemerné zlepšenie skóre UPDRS (časti II+III) 6,83 bodov (počiatočná hodnota 33,2 bodov),
v ropinirolovej vetve 10,78 bodov (počiatočná hodnota 32,2 bodov) a vo vetve s placebom 2,33 bodov (počiatočná hodnota 31,3 bodov). Všetky rozdiely medzi aktívnymi liečbami a placebom boli štatisticky významné. Táto štúdia nepreukázala noninferioritu rotigotínu voči ropinirolu.
V následnej nezaslepenej štúdii (SP824), multicentrickej, medzinárodnej štúdii sa skúmala znášanlivosť náhlej zmeny z liečby ropinirolom, pramipexolom alebo kabergolínom na transdermálnu náplasť rotigotínu a jeho účinok na symptómy u osôb s idiopatickou Parkinsonovou chorobou. 116 pacientov prešlo z predchádzajúcej perorálnej liečby na liečbu až 8 mg/24 h rotigotínu, medzi nimi bolo 47 pacientov, ktorí boli liečení až 9 mg/deň ropinirolu, 47 pacientov, ktorí boli liečení až
2 mg/deň pramipexolu a 22 pacientov, ktorí boli liečení až 3 mg/deň kabergolínu. Prechod na rotigotín bolo uskutočniteľný s nevyhnutnou malou úpravou dávky (medián 2 mg/24 h) len u 2 pacientov, ktorí prešli z liečby ropinirolom, u 5 pacientov, ktorí prešli z liečby pramipexolom a u 4 pacientov, ktorí prešli z liečby kabergolínom. Pozorovali sa zlepšenia skóre UPDRS časti I – IV. Bezpečnostný profil bol porovnateľný s profilom v predchádzajúcich štúdiách.
V randomizovanej, nezaslepenej štúdii (SP825) u pacientov s včasných štádiom Parkinsonovej choroby bolo 25 pacientov randomizovaných na liečbu rotigotínom a 26 na ropinirol. V oboch liečebných skupinách bola dávka titrovaná na optimálnu alebo maximálnu dávku 8 mg/24 h alebo
9 mg/24 h, v uvedenom poradí. Obidve liečby preukázali zlepšenia skorej rannej motorickej funkcie
a spánku. Motorické symptómy (UPDRS časť III) sa zlepšili o 6,3 ± 1,3 bodov u pacientov liečených rotigotínom a o 5,9 ± 1,3 bodov v skupine s ropinirolom po 4 týždňoch udržiavacej liečby. Spánok (PDSS) sa zlepšil o 4,1 ± 13,8 bodov u pacientov liečených rotigotínom a o 2,5 ± 13,5 bodov u pacientov liečených ropinirolom. Bezpečnostný profil bol porovnateľný s výnimkou reakcií v mieste aplikácie.
V štúdiách SP824 a SP825 uskutočnených od úvodného komparatívneho klinického skúšania sa preukázalo, že rotigotín a ropinirol v ekvivalentných dávkach majú porovnateľnú účinnosť.
Dve ďalšie pivotné štúdie (SP650DB a SP515) boli vykonané u pacientov súčasne liečených
L-dopou. Hlavným zisteným výsledkom bolo skrátenie trvania tzv. stavu „off“ (v hodinách). Účinnosť sa zisťovala na základe odozvy pacienta na liečbu, pričom sa merala ako zlepšenie stavu pacienta a absolútne zlepšenie v stave „off“.
V dvojito zaslepenej štúdii SP650DB dostávalo 113 pacientov rotigotín až po maximálnu dávku
8 mg/24 h, 109 pacientov dostávalo rotigotín až po maximálnu dávku 12 mg/24 h a 119 pacientov dostávalo placebo. U pacientov bola titrovaná optimálna dávka rotigotínu alebo placeba týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 4 mg/24 h. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 6 mesiacov. Na konci udržiavacej liečby sa pozorovalo zlepšenie o minimálne 30 % u 57 % a u 55 % pacientov dostávajúcich rotigotín 8 mg/24 h a 12 mg/24 h
v uvedenom poradí a u 34 % pacientov dostávajúcich placebo (rozdiely 22 % a 21 % v uvedenom
poradí CI95% 10 %; 35 % a 8 %; 33 % v uvedenom poradí, p< 0,001 pre obe skupiny liečené
rotigotínom). V prípade rotigotínu sa trvanie stavu „off“ skrátilo o priemerne“ 2,7 a 2,1 hodiny
v uvedenom poradí, zatiaľ čo u skupiny dostávajúcej placebo, sa pozorovalo skrátenie o 0,9 hodiny.
Rozdiely boli štatisticky významné (p< 0,001 a p=0,003 v uvedenom poradí).
V dvojito zaslepenej štúdii SP515 dostávalo 201 pacientov rotigotín, 200 pacientov pramipexol a 100 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 4 mg/24 h po maximálnu dávku 16 mg/24 h. V skupine dostávajúcej pramipexol sa pacientom podávala dávka 0,375 mg počas prvého týždňa, 0,75 mg počas druhého týždňa a bola u nich titrovaná optimálna dávka ďalším týždenným zvyšovaním o 0,75 mg až po maximálnu dávku 4,5 mg/deň. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 4 mesiacov.
Na konci udržiavacej liečby sa pozorovalo zlepšenie o minimálne 30 % u 60 % pacientov
dostávajúcich rotigotín, 67 % pacientov dostávajúcich pramipexol a u 35 % pacientov dostávajúcich placebo (rozdiel medzi rotigotínom a placebom bol 25 %, CI95% 13 %; 36 %, rozdiel medzi
pramipexolom a placebom bol 32 %, CI95% 21 %; 43 %, rozdiel medzi pramipexolom a rotigotínom bol 7 %, CI95% -2 %; 17 %). V skupine dostávajúcej rotigotín sa trvanie stavu „off“ skrátilo
o priemerne 2,5 hodiny, v skupine s pramipexolom to bolo 2,8 hodiny a v skupine s placebom
0,9 hodiny. Všetky rozdiely medzi aktívnymi liečbami a placebom boli štatisticky významné.
Ďalšia medzinárodná dvojito zaslepená štúdia (SP889) sa uskutočnila u 287 pacientov vo včasných alebo pokročilých štádiách Parkinsonovej choroby s nedostatočnou kontrolou skorých ranných motorických symptómov. 81,5 % týchto pacientov dostávalo súbežnú liečbu levodopou. 190 pacientov dostávalo rotigotín a 97 placebo. Pacienti boli titrovaní na optimálnu dávku rotigotínu alebo placeba
v týždňových prírastkoch o 2 mg/24h s počiatočnou dávkou 2 mg/24 h až po maximálnu dávku
16 mg/24 h po dobu 8 týždňov, po ktorom nasledovalo udržiavacie obdobie 4 týždňov. Skoré ranné motorické funkcie, hodnotené podľa časti III UPDRS a poruchy nočného spánku, hodnotené podľa upravenej škály spánku pri Parkinsonovej chorobe (Parkinson’s Disease Sleep Scale - PDSS-2), boli ko-primárnymi výslednými meraniami. Na konci udržiavacieho obdobia sa priemerné skóre podľa časti III UPDRS zlepšilo o 7,0 bodov u pacientov liečených rotigotínom (východisková hodnota 29,6) a o 3,9 bodov v placebovej skupine (východisková hodnota 32,0). Zlepšenia v priemernom celkovom skóre PDSS-2 boli 5,9 (rotigotín, východisková hodnota 19,3) a 1,9 bodov (placebo, východisková hodnota 20,5). Liečebné rozdiely v koprimárnych premenných boli štatisticky významné (p=0,0002 a p<0,0001).
Priľnavosť na kožu
V multicentrickej, dvojito zaslepenej, randomizovanej, dvojito skríženej štúdii bola u 52 ambulantných pacientov porovnávaná priľnavosť na kožu rotigotín náplasti 8 mg/24 h, s vylepšeným uchovávaním pri izbovej teplote, s náplasťou s uchovávaním v chlade. Priľnavosť na kožu bola meraná počas 2 po sebe nasledujúcich dní pri 24-hodinovej aplikácii náplasti.
Vylepšená náplasť s uchovávaním pri izbovej teplote preukázala lepšiu priľnavosť na kožu ako náplasť s uchovávaním v chlade, > 90 % náplastí preukazuje dostatočnú priľnavosť (t.j. > 70 %
priliehajúcej plochy náplasti) v porovnaní s <83 %. Pri obidvoch formách bola hlásená porovnateľná
znášanlivosť kože. Väčšina pozorovaných erytémov bolo miernych a žiadny nebol závažný.
5.2 Farmakokinetické vlastnosti
Absorpcia
Rotigotín sa po aplikácii plynule uvoľňuje z transdermálnej náplasti a je absorbovaný cez pokožku.
Ustálené koncentrácie sa dosiahnu za jeden až dva dni po aplikácii náplasti a udržiavajú sa na stabilnej
úrovni aplikáciou náplasti jedenkrát denne, pričom táto sa nosí po dobu 24 hodín. Koncentrácie
rotigotínu v plazme sa zvyšujú v závislosti od dávky v rozsahu dávok od 1 mg/24 h po 24 mg/24 h.
Za 24 hodín sa uvoľní do pokožky približne 45 % liečiva v náplasti. Absolútna biologická dostupnosť
po transdermálnej aplikácii je približne 37 %.
Striedanie miesta aplikácie náplasti môže spôsobiť rozdiely medzi jednotlivými dennými hladinami v plazme. Rozdiely v biologickej dostupnosti rotigotínu siahali od 2 % (nadlaktie v porovnaní s bokom) po 46 % (rameno v porovnaní so stehnom). Neexistuje žiaden náznak relevantného vplyvu na klinické výsledky.
Distribúcia
Približne 92 % rotigotínu sa viaže in vitro na plazmatické proteíny.
Zdanlivý distribučný objem u ľudí je približne 84 l/kg.
Biotransformácia
Rotigotín je z veľkej časti metabolizovaný. Rotigotín je metabolizovaný N-dealkyláciou ako aj
priamou a sekundárnou konjugáciou. Výsledky vykonávané in vitro naznačujú, že rôzne izoformy
CYP dokážu katalyzovať N-dealkyláciu rotigotínu. Hlavnými metabolitmi sú sulfáty a glukuronidové
konjugáty pôvodnej látky ako aj metabolity N-dealkylácie, ktoré sú biologicky neaktívne. Informácie o metabolitoch sú neúplné.
Eliminácia
Približne 71 % dávky rotigotínu sa vylučuje močom a menšia časť približne 23 % sa vylučuje stolicou.
Klírens rotigotínu po transdermálnom podaní je približne 10 l/minútu a jeho celkový polčas eliminácie je 5 až 7 hodín. Farmakokinetický profil preukazuje bifázickú elimináciu s počiatočným polčasom
približne 2 až 3 hodiny.
Keďže náplasť sa aplikuje transdermálne, neočakáva sa žiadny vplyv jedál alebo ochorení
gastrointestinálneho traktu.
Osobitné skupiny pacientov
Keďže liečba Neuprom začína nízkou dávkou a táto sa postupne titruje podľa klinickej znášanlivosti,
aby sa získal optimálny terapeutický účinok, úprava dávky na základe pohlavia, hmotnosti alebo veku
nie je potrebná.
Porucha funkcie pečene a obličiek
U pacientov so stredne závažnou poruchou funkcie pečene alebo miernou až závažnou poruchou funkcie obličiek neboli pozorované žiadne relevantné zvýšenia hladiny rotigotínu v plazme. Neupro nebolo skúmané u pacientov so závažnou poruchou funkcie pečene.
V prípade poruchy funkcie obličiek sa zvyšujú hladiny konjugátov rotigotínu a metabolitov N- dealkylácie rotigotínu v plazme. Vplyv týchto metabolitov na klinické účinky je však nepravdepodobný.
Pediatrická populácia
Obmedzené farmakokinetické údaje získané u dospievajúcich pacientov s RLS (13-17 rokov, n = 24) po liečbe viacnásobnými dávkami v rozsahu 0,5 mg až 3 mg / 24 h ukázali, že systémová expozícia rotigotínu bola podobná expozícii u dospelých. Údaje o účinnosti/bezpečnosti nie sú dostatočné na stanovenie pomeru medzi expozíciou a odpoveďou (pozri tiež informácie o použití v pediatrickej populácii v časti 4.2).
5.3 Predklinické údaje o bezpečnosti
V štúdiách zameraných na opakované dávky a dlhodobú toxicitu boli hlavné účinky spájané s farmakodynamickými účinkami súvisiacimi s agonistom dopamínu a následným znížením vylučovania prolaktínu.
Po jednorazovej dávke rotigotínu bolo u pigmentovaných potkanov a opíc evidentné viazanie sa
rotigotínu na tkanivo obsahujúce melanín (t. j. oči), ktoré však počas 14-dňového pozorovacieho
intervalu vymizlo.
V trojmesačnej štúdii bola u bielych potkanov pozorovaná pomocou transmisnej elektrónovej mikroskopie degenerácia sietnice pri dávke ekvivalentnej 2,8-násobku maximálnej odporúčanej dávky pre ľudí (prepočítanej na mg/m2). Účinky boli výraznejšie u samíc potkanov. Dodatočné štúdie na ďalšie vyhodnotenie špecifickej patológie neboli vykonané. Degenerácia sietnice sa nepozorovala počas rutinného histopatologického vyhodnocovania očí skúmaných druhov zvierat v žiadnej
z toxikologických štúdií. Význam týchto zistení pre ľudí nie je známy.
V štúdii karcinogenicity sa u samcov potkanov rozvinuli nádory a hyperplázia Leydigových buniek. Zhubné nádory boli pozorované najmä v maternici samíc, ktorým sa podávali stredné až vysoké dávky. Tieto zmeny sú dobre známymi účinkami agonistov dopamínu u potkanov po celoživotnej liečbe a boli zhodnotené ako nerelevantné pre ľudí.
Účinky rotigotínu na reprodukciu sa skúmali u potkanov, králikov a myší. Na žiadny z uvedených troch druhov nemal rotigotín teratogénne účinky, ale u potkanov a myší vykazoval embryotoxické účinky pri dávkach toxických pre matky. Rotigotín neovplyvnil plodnosť samcov u potkanov, avšak jednoznačne znížil plodnosť samíc potkanov a myší z dôvodu účinkov na hladiny prolaktínu, ktoré sú u hlodavcov obzvlášť významné.
Rotigotín nevyvolal génové mutácie v Amesovom teste, preukázal však účinky v in vitro teste lymfómu u myší s metabolickou aktiváciou a slabšie účinky bez metabolickej aktivácie. Mutagénny účinok je možné pripísať klastogénnemu účinku rotigotínu. Tento účinok nebol potvrdený in vivo
v mikronukleovom teste u myší a v teste na neplánovanú syntézu DNA (UDS, Unsheduled DNA Synthesis) u potkanov. Keďže prebiehal viac-menej súbežne so zníženým relatívnym celkovým rastom
buniek, môže súvisieť s cytotoxickým účinkom látky. Významnosť tohto jedného pozitívneho testu
mutagenicity in vitro preto nie je známa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Krycia vrstva
Polyesterový film s vrstvou silikónu a hliníka
farebný povlak pigmentovej vrstvy (oxid titaničitý (E171), žltý pigment 95, červený pigment 166)
a potlač (červený pigment 144, žltý pigment 95, čierny pigment 7).
Samolepiaca matricová vrstva
Poly(dimetylsiloxán, trimetylsilylsilikát)-kopolymerizát,
Povidón K90,
Metasiričitan sodný (E223),
Askorbylpalmitát (E304) a
DL-α-tokoferol (E307).
Snímateľná fólia
Priesvitný polyesterový film s fluoropolymérovým povlakom.
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
30 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
6.5 Druh obalu a obsah balenia
Rozlepovacie vrecko v lepenkovej škatuli: Jednu stranu tvorí kopolymér etylénu (najvnútornejšia vrstva), hliníková fólia, polyetylénový film s nízkou hustotou a papier; druhú stranu tvorí polyetylén (najvnútornejšia vrstva), hliník, kopolymér etylénu a papier.
Škatuľa obsahuje 7, 14, 28, 30 alebo 84 (multibalenie obsahujúce 2 balenia po 42) transdermálnych
náplastí, ktoré sú jednotlivo zatavené vo vreckách.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuNáplasť po použití ešte stále obsahuje liečivo. Po odstránení sa má použitá náplasť preložiť napoly lepiacou stranou smerom dovnútra tak, aby matricová vrstva nezostala odkrytá, umiestniť do pôvodného vrecka a odložiť. Všetky použité aj nepoužité náplasti majú byť zlikvidované v súlade
s národnými požiadavkami alebo vrátené do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIUCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLAEU/1/05/331/001
EU/1/05/331/002
EU/1/05/331/015
EU/1/05/331/018
EU/1/05/331/057
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15. február 2006
Dátum posledného predĺženia registrácie: 22. január 2016
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Neupro 4 mg/24 h transdermálna náplasť Neupro 6 mg/24 h transdermálna náplasť Neupro 8 mg/24 h transdermálna náplasť
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Neupro 4 mg/24 h transdermálna náplasť
Každá náplasť uvoľní 4 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 20 cm2 obsahuje 9,0 mg rotigotínu.
Neupro 6 mg/24 h transdermálna náplasť
Každá náplasť uvoľní 6 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 30 cm2 obsahuje
13,5 mg rotigotínu.
Neupro 8 mg/24 h transdermálna náplasť
Každá náplasť uvoľní 8 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 40 cm2 obsahuje
18,0 mg rotigotínu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Transdermálna náplasť.
Tenká štvorcová náplasť matricového typu so zaoblenými rohmi, skladajúca sa z troch vrstiev.
Neupro 4 mg/24 h transdermálna náplasť
Vonkajšia strana krycej vrstvy je béžová s potlačou „Neupro 4 mg/24 h“.
Neupro 6 mg/24 h transdermálna náplasť
Vonkajšia strana krycej vrstvy je béžová s potlačou „Neupro 6 mg/24 h“.
Neupro 8 mg/24 h transdermálna náplasť
Vonkajšia strana krycej vrstvy je béžová s potlačou „Neupro 8 mg/24 h“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Neupro sa indikuje na liečbu známok a symptómov idiopatickej Parkinsonovej choroby v počiatočnom štádiu v monoterapii (t. j. bez L-dopy) alebo v kombinácii s L-dopou, čiže počas trvania ochorenia až do neskorých štádií, keď účinok L-dopy zoslabne alebo sa stane premenlivým a dochádza
k fluktuáciám terapeutického účinku (fluktuácie ku koncu dávkovacieho intervalu alebo „on-off“
fluktuácie zo stavu dobrej hybnosti tzv. „on“ do stavu zlej hybnosti tzv. „off“).
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Odporúčané dávky predstavujú nominálne dávky.
Dávkovanie u pacientov v počiatočnom štádiu Parkinsonovej choroby:
Jednorazová denná dávka má začať na 2 mg/24 h a následne byť zvyšovaná v týždňových intervaloch o 2 mg/24 h až po dosiahnutie účinnej dávky, ktorá je maximálne 8 mg/24 h.
Dávka 4 mg/24 h môže byť v prípade niektorých pacientov účinnou dávkou. U väčšiny pacientov je účinná dávka dosiahnutá v priebehu 3 alebo 4 týždňov pri dávkach 6 mg/24 h alebo 8 mg/24 h,
v uvedenom poradí.
Maximálna dávka je 8 mg/24 h.
Dávkovanie u pacientov v pokročilom štádiu Parkinsonovej choroby s fluktuáciami:
Jednorazová denná dávka má začať na 4 mg/24 h a následne byť zvyšovaná v týždňových intervaloch o 2 mg/24 h až po dosiahnutie účinnej dávky, ktorá je maximálne 16 mg/24 h.
Dávky 4 mg/24 h alebo 6 mg/24 h môžu byť v prípade niektorých pacientov účinnými dávkami.
U väčšiny pacientov je účinná dávka dosiahnutá v priebehu 3 až 7 týždňov pri dávkach 8 mg/24 h až
po maximálnu dávku 16 mg/24 h.
Pre dávky vyššie ako 8 mg/24 h je možné použiť na dosiahnutie konečnej dávky viacero náplastí, napríklad dávka 10 mg/24 h sa môže dosiahnuť kombináciou náplastí 6 mg/24 h a 4 mg/24 h.
Neupro sa aplikuje raz denne. Náplasť sa má aplikovať každý deň približne v rovnakom čase. Náplasť sa ponecháva na koži 24 hodín, a potom sa vymieňa za novú náplasť aplikovanú na inom mieste.
Ak si pacient zabudne náplasť aplikovať v obvyklom čase počas dňa alebo ak sa náplasť odlepí, na zvyšok dňa sa má aplikovať nová náplasť.
Ukončenie liečby
Liečba Neuprom má byť ukončená postupne. Denná dávka sa má znižovať o 2 mg/24 h znižovaním dávky podľa možnosti každý druhý deň až do úplného vysadenia Neupra (pozri časť 4.4).
Osobitné populácie
Porucha funkcie pečene
U pacientov s mierne až stredne závažnými poruchami funkcie pečene nie je potrebná úprava dávky. Opatrnosť sa odporúča pri liečbe pacientov so závažnými poruchami funkcie pečene, u ktorých môže dôjsť k zníženiu klírensu rotigotínu. Rotigotín nebol skúmaný u tejto skupiny pacientov. V prípade zhoršovania poruchy funkcie pečene môže byť potrebná redukcia dávky.
Porucha funkcie obličiek
U pacientov s miernou až závažnou poruchou funkcie obličiek, vrátane dialyzovaných pacientov, nie je potrebná úprava dávky. Neočakávaný nárast hladiny rotigotínu sa môže objaviť tiež v prípade akútneho zhoršenia funkcie obličiek (pozri časť 5.2).
Pediatrická populácia
Neexistuje žiadne relevantné použitie Neupra v pediatrickej populácii pri Parkinsonovej chorobe. Spôsob podávania
Neupro je určené na transdermálne použitie.
Náplasť sa má aplikovať na čistú, suchú, neporušenú zdravú pokožku v oblasti brucha, stehna, bedier, bokov trupu, ramien alebo nadlaktia. Náplasť sa nemá aplikovať na rovnaké miesto skôr než po
14 dňoch. Neupro sa nesmie aplikovať na červenú, podráždenú alebo poškodenú pokožku (pozri časť 4.4).
Použitie a zaobchádzanie
Každá náplasť je zabalená vo vrecku a aplikuje sa ihneď po otvorení vrecka. Odstráni sa jedna polovica snímateľnej fólie a lepiaca strana sa aplikuje pevným pritlačením na pokožku. Potom sa
náplasť prehne dozadu a odstráni sa druhá časť prilepenej fólie. Nedotýkajte sa lepiacej strany náplasti. Dlaňou sa náplasť pevne zatlačí po dobu približne 20 až 30 sekúnd, aby sa dobre prilepila.
Náplasť sa nesmie strihať na kusy.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Zobrazovanie magnetickou rezonanciou alebo kardioverzia (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Ak je odozva pacienta s Parkinsonovou chorobou na liečbu rotigotínom nedostatočná, môžu sa prestupom na liečbu iným agonistom dopamínu dosiahnuť dodatočné pozitívne účinky (pozri časť
5.1).
Zobrazovanie magnetickou rezonanciou a kardioverzia
Krycia vrstva náplasti Neupro obsahuje hliník. Ak bude pacient podstupovať vyšetrenie zobrazovaním
magnetickou rezonanciou (MRI) alebo kardioverziu, musí sa Neupro odstrániť, aby sa zabránilo
popáleninám pokožky.
Ortostatická hypotenzia
Je známe, že agonisty dopamínu zhoršujú systémovú reguláciu tlaku krvi, v dôsledku čoho vzniká
posturálna/ortostatická hypotenzia. Tieto prípady sa pozorovali aj počas liečby rotigotínom, ich výskyt bol však podobný ako výskyt pozorovaný u pacientov liečených placebom.
Z dôvodu všeobecného rizika ortostatickej hypotenzie spojenej s dopamínergickou liečbou sa odporúča sledovať krvný tlak, a to najmä na začiatku liečby.
Synkopa
V klinických štúdiách s rotigotínom sa pozorovala synkopa s podobnou frekvenciou výskytu, ako bola
pozorovaná u pacientov liečených placebom. Keďže pacienti s klinicky relevantným kardiovaskulárnym ochorením boli z týchto štúdií vylúčení, je potrebné sa pacientov so závažným
kardiovaskulárnym ochorením opýtať na príznaky synkopy a presynkopy.
Náhle zaspanie a ospalosť
Rotigotín bol spájaný s ospalosťou a prípadmi náhleho zaspania. Hlásené boli prípady náhleho
zaspania počas denných aktivít, v niektorých prípadoch bez spozorovania akýchkoľvek varovných známok. Lekári predpisujúci Neupro majú u pacientov nepretržite prehodnocovať príznaky driemot alebo ospalosti, pretože samotní pacienti si ich nemusia uvedomiť, až kým sa ich na to priamo neopýtajú. Starostlivo sa má zvážiť zníženie dávky alebo ukončenie liečby.
Poruchy kontroly impulzov
Pacientov je treba pravidelne monitorovať pre vývoj porúch kontroly impulzov. Pacienti
a opatrovatelia si majú byť vedomí, že u pacientov liečených agonistami receptorov dopamínu vrátane rotigotínu sa môžu vyskytnúť behaviorálne príznaky porúch kontroly impulzov ako patologické hranie
hazardných hier, zvýšené libido, hypersexualita, kompulzívne utrácanie alebo nakupovanie, prejedanie
sa a závislosť na jedle. Ak sa tieto symptómy objavia, je potrebné zvážiť zníženie dávky/postupné
vysadenie.
N
e
uroleptický malígny syndróm
Pri náhlom ukončení dopamínergickej liečby boli hlásené symptómy naznačujúce prítomnosť
neuroleptického malígneho syndrómu. Preto sa odporúča liečbu obmedzovať postupne (pozri časť 4.2).
Abnormálne myslenie a správanie
Zaznamenalo sa abnormálne myslenie a správanie a môže pozostávať z rôznych prejavov vrátane
paranoidných myšlienok, preludov, halucinácií, zmätenosti, správania podobného psychotickému, dezorientácie, agresívneho správania, agitácie a delíria.
Fibrotické komplikácie
U niektorých pacientov liečených dopamínergickými látkami odvodenými od námeľu boli hlásené
prípady retroperitoneálnej fibrózy, pľúcnych infiltrátov, pleurálnych výpotkov, pleurálneho zhrubnutia, perikarditídy a srdcovej valvulopatie. Aj keď tieto komplikácie môžu po vysadení liečby ustúpiť, nemusia vždy ustúpiť úplne.
Aj keď sa predpokladá, že tieto nežiaduce reakcie súvisia s ergotamínovou štruktúrou týchto látok, nie je známe, či ich môžu spôsobovať aj agonisty dopamínu, ktoré nie sú odvodené od námeľu.
Neuroleptiká
Neuroleptiká podávané ako antiemetiká sa nemajú podávať pacientom užívajúcim agonistov
dopamínu (pozri aj časť 4.5).
Oftalmologické sledovanie
Oftalmologické sledovanie sa odporúča v pravidelných intervaloch alebo v prípade porúch videnia.
Vystavenie zdroju tepla
Miesto s náplasťou nemá byť vystavované vonkajším zdrojom tepla (nadmerné slnečné žiarenie,
ohrievacie podušky a iné zdroje tepla, napríklad sauna, horúci kúpeľ).
Reakcie v mieste aplikácie
V mieste aplikácie sa môžu vyskytnúť kožné reakcie, zvyčajne s miernou alebo stredne závažnou
intenzitou. Miesto aplikácie sa odporúča denne striedať (napríklad z pravej strany na ľavú stranu
a z hornej časti tela na dolnú časť tela). Rovnaké miesto sa nemá použiť znova skôr než po 14 dňoch. V prípade niekoľkodňového alebo trvalého výskytu reakcií v mieste aplikácie, pri zvýšení závažnosti
alebo pri rozšírení kožnej reakcie mimo miesto aplikácie, sa má zhodnotiť pomer rizík a výhod pre
daného pacienta.
V prípade výskytu vyrážok alebo podráždenia pokožky transdermálnym systémom sa má zabrániť pôsobeniu priameho slnečného žiarenia na dané miesto, kým sa pokožka nezahojí, pretože expozícia
môže spôsobiť zmeny sfarbenia pokožky.
V prípade spozorovania celkovej reakcie pokožky (napríklad alergické vyrážky, vrátane erytematóznych, škvrnitých a papulárnych vyrážok alebo svrbenia) spojenej s používaním Neupra,
musí sa Neupro prestať používať.
Periférny edém
V klinických štúdiách bol počas 6 mesiacov špecifický výskyt periférneho edému okolo 4 % a zostal
nezmenený počas ďalšieho pozorovania až do 36 mesiacov.
Dopamínergické nežiaduce reakcie
Výskyt niektorých dopamínergických nežiaducich reakcií, ako sú napríklad halucinácie, dyskinéza a periférny edém, je vo všeobecnosti vyšší pri podávaní v kombinácii s L-dopou u pacientov
s Parkinsonovou chorobou. Treba to zvážiť pri predpisovaní rotigotínu.
Citlivosť nasiričitan
Neupro obsahuje metasiričitan sodný, siričitan, ktorý môže vyvolať reakcie alergického typu vrátane
anafylaktických symptómov a život ohrozujúcich alebo menej závažných astmatických epizód u niektorých citlivých osôb.
4.5 Liekové a iné interakcie
Keďže rotigotín je agonista dopamínu, predpokladá sa, že antagonisty dopamínu, ako napríklad neuroleptiká (napríklad fenotiazíny, butyrofenóny, tioxantény) alebo metoklopramid, môžu oslabovať účinnosť Neupra, a preto sa nemajú podávať súbežne s Neuprom. Z dôvodu možných aditívnych účinkov sa má postupovať opatrne, ak pacienti užívajú sedatívne lieky alebo iné lieky utlmujúce činnosť CNS (centrálneho nervového systému) (napríklad benzodiazepíny, antipsychotiká, antidepresíva) alebo alkohol v kombinácii s rotigotínom.
Súbežné podávanie L-dopy a karbidopy s rotigotínom nemalo žiadny účinok na farmakokinetiku rotigotínu a rotigotín nemal žiadny účinok na farmakokinetiku L-dopy a karbidopy.
Súbežné podávanie domperidonu s rotigotínom nemalo žiadny účinok na farmakokinetiku rotigotínu. Súbežné podávanie omeprazolu (inhibítora CYP2C19) v dávkach 40 mg/deň nemalo žiadny účinok na
farmakokinetiku a metabolizmus rotigotínu u zdravých dobrovoľníkov.
Neupro môže zosilniť dopamínergické nežiaduce reakcie L-dopy a môže spôsobiť a/alebo zhoršiť už
existujúcu dyskinézu, podobne ako v prípade ostatných agonistov dopamínu.
Súbežné podávanie rotigotínu (3 mg/24 h) neovplyvňovalo farmakodynamiku a farmakokinetiku
perorálnych kontraceptív (0,03 mg etinylestradiol, 0,15 mg levonorgestrel). Interakcie s inými formami hormonálnej antikoncepcie neboli skúmané.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku, antikoncepcia u žien
Ženy vo fertilnom veku majú používať účinnú antikoncepciu na prevenciu gravidity počas liečby
rotigotínom.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití rotigotínu u gravidných žien. Štúdie na zvieratách
nepreukázali žiadne teratogénne účinky u potkanov a králikov, avšak u potkanov a myší bola pozorovaná embryotoxicita pri dávkach toxických pre matky (pozri časť 5.3). Nie je známe
potenciálne riziko u ľudí. Rotigotín sa nemá používať počas gravidity.
Laktácia
Keďže rotigotín znižuje vylučovanie prolaktínu u ľudí, očakáva sa inhibícia tvorby mlieka. Štúdie
vykonávané na potkanoch ukázali, že rotigotín a/alebo jeho metabolit(y) sú vylučované do materského mlieka. Z dôvodu chýbajúcich údajov o účinkoch na ľudský organizmus sa má dojčenie prerušiť.
Fertilita
Pre informácie o štúdiách fertility, prosím, pozri časť 5.3.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Rotigotín môže mať značný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Pacienti liečení rotigotínom, ktorí trpia ospalosťou a/alebo prípadmi náhleho zaspania, musia byť
informovaní o zákaze riadenia vozidiel alebo vykonávania aktivít (napríklad obsluhovanie strojov), pri ktorých môže zhoršené vnímanie ohroziť ich samotných alebo ostatných a spôsobiť vážne zranenia alebo smrť. To platí, až kým tieto opakujúce sa prípady náhleho zaspania a ospalosť neustúpia (pozri
aj časti 4.4 a 4.5).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Podľa analýzy skupinových, placebom kontrolovaných klinických štúdií, zahŕňajúcich celkovo
1 307 pacientov liečených Neuprom a 607 pacientov liečených placebom, 72,5 % pacientov liečených
Neuprom a 58,0 % pacientov liečených placebom hlásilo aspoň jednu nežiaducu reakciu.
Na začiatku liečby sa môžu vyskytovať dopamínergické nežiaduce reakcie ako nevoľnosť a vracanie. Tieto sú zvyčajne mierne alebo stredne závažné a dočasné, a to aj v prípade pokračovania v liečbe.
Medzi nežiaduce liekové reakcie hlásené u viac ako 10 % pacientov liečených transdermálnymi náplasťami Neupro patria nevoľnosť, vracanie, reakcie v mieste aplikácie, ospalosť, závrat a bolesť hlavy.
V štúdiách, pri ktorých sa miesto aplikácie striedalo podľa pokynov v SmPC (Súhrn charakteristických
vlastností lieku, Summary of Product Characteristics) a písomnej informácii pre používateľa, sa
u 35,7 % z 830 pacientov používajúcich transdermálne náplasti Neupro vyskytovali reakcie v mieste aplikácie. Väčšina reakcií v mieste aplikácie bola mierna alebo stredne závažná, obmedzená na miesto aplikácie a v ich dôsledku bola liečba Neuprom ukončená iba u 4,3 % všetkých pacientov používajúcich Neupro.
Zoznam nežiaducich reakcií zoradených dotabuľky
V nasledujúcej tabuľke sú uvedené nežiaduce liekové reakcie zo združených štúdií uvedených vyššie
u pacientov s Parkinsonovou chorobou. V rámci tried orgánových systémov sú nežiaduce reakcie vymenované pod názvami jednotlivých frekvencií (počet pacientov, u ktorých sa predpokladá výskyt reakcie) použitím nasledujúcich kategórií: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v
poradí klesajúcej závažnosti.
T
ri
edy orgánových systémov podľa MedDRA Poruchy imunitného systému
V
e
ľmi časté Časté Menej časté Zriedkavé Neznáme
hypersenzitivita, ktorá môže zahŕňať angioedém,
edém jazyka a edém pier
P
sychické poruchy
P
oruchy nervového systému
ospalosť, závrat, bolesť hlavy
poruchy vnímaniaa (vr. halucinácie, zrakovej halucinácie, sluchovej halucinácie, vidiny), nespavosť, porucha spánku, nočné mory, neobvyklé sny, poruchy
kontroly impulzova,d (vr. patologického hrania hazardných hier, stereotypného/ nutkavého správania, prejedania sa/porúch prijímania potravyb, kompulzívneho nakupovaniac) poruchy
vedomia NECa
(vr. synkopy, vazovagálnej
synkopy, straty
vedomia), dyskinéza, posturálny závrat, letargia
náhle zaspávanie/ náhly nástup spánku, paranoja, poruchy sexuálnej túžbya (vr. hypersexuality, zvýšeného libida), stav zmätenosti, dezorientáciad, agitáciad
psychotická porucha, obsedantno- kompulzívna porucha, agresívne správanie/ agresivitab, preludd, delíriumd
kŕč
syndróm dopamínovej dysreguláciec
P
oruchy oka rozmazané videnie, poruchy videnia, fotopsia
P
oruchy ucha a labyrintu Poruchy srdca a srdcovej činnosti
vertigo
palpitácie atriálna fibrilácia
supraventrik ulárna tachykardia
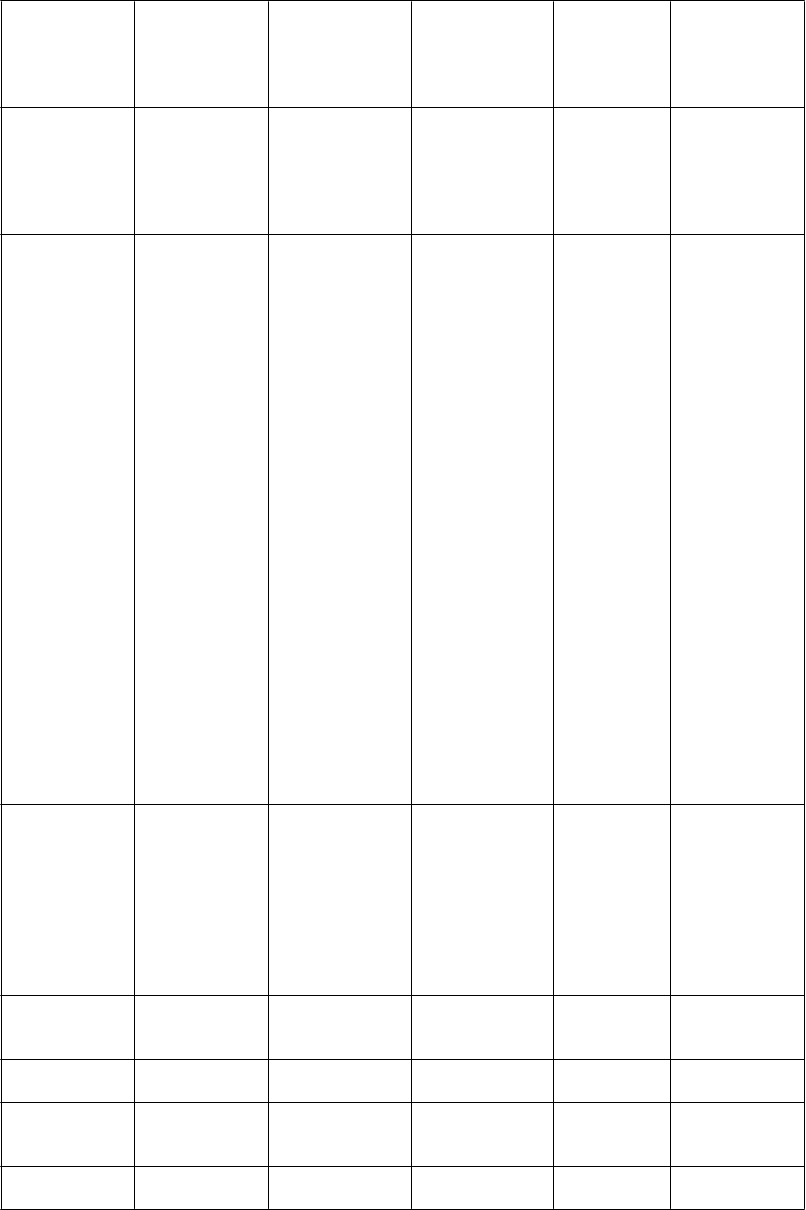 P
oruchy ciev
P
oruchy ciev ortostatická hypotenzia,
hypotenzia
 P
oruchy dýchacej sústavy, hrudníka
a mediastína Poruchy gastrointestin álneho traktu Poruchy kože a podkožného tkaniva
P
oruchy reprodukčnéh o systému
a prsníkov Celkové poruchy
a reakcie
v mieste podania
L
aboratórne a funkčné vyšetrenia
Ú
r
azy, otravy a komplikácie liečebného postupu
P
oruchy dýchacej sústavy, hrudníka
a mediastína Poruchy gastrointestin álneho traktu Poruchy kože a podkožného tkaniva
P
oruchy reprodukčnéh o systému
a prsníkov Celkové poruchy
a reakcie
v mieste podania
L
aboratórne a funkčné vyšetrenia
Ú
r
azy, otravy a komplikácie liečebného postupu
nevoľnosť,
vracanie
reakcie
v mieste aplikácie a instiláciea (napríklad erytém, svrbenie, podráždenie, vyrážka, dermatitída, vezikuly, bolesť, ekzém, zápal, opuch, zmena sfarbenia, pupence, exfoliácie, urtikária, precitlivenosť)
hypertenzia
štikútanie
zápcha, sucho v ústach, dyspepsia erytém, hyperhidróza, svrbenie
periférny edém, astenické stavya, (vr. únavy, asténie, malátnosti)
zníženie hmotnosti
pád
bolesť brucha
celkové svrbenie, podráždenie pokožky, kontaktná dermatitída erektilná dysfunkcia
zvýšenie hladiny pečeňového enzýmu (vr. AST, ALT, GGT), zvýšenie hmotnosti, zrýchlenie srdcového
rytmu, zvýšenie CPKd (pozri Osobitné populácie)
generalizova ná vyrážka
podráždenos
ť
a Nadradený všeobecný názov
b Pozorované v nezaslepených štúdiách
c Pozorované po uvedení lieku na trh
d Pozorované v roku 2011 v súhrnných údajoch z dvojito zaslepených placebom kontrolovaných štúdií
Popis vybraných nežiaducich reakciíNáhly nástup spánku a ospalosťRotigotín bol spájaný s ospalosťou vrátane nadmernej ospalosti počas dňa a prípadov náhleho zaspania. V zriedkavých prípadoch sa vyskytlo „náhle zaspanie“ počas jazdy, čo následne spôsobilo dopravnú nehodu (pozri aj časť 4.4 a 4.7).
Poruchy kontroly impulzovPatologické hranie hazardných hier, zvýšené libido, hypersexualita, kompulzívne utrácanie alebo
nakupovanie, prejedanie sa a závislosť na jedle sa môžu vyskytnúť u pacientov liečených agonistami dopamínu vrátane rotigotínu (pozri časť 4.4 ).
Osobitné populácieNežiaduce účinky zvýšenej kreatínfosfokinázy (CPK) sa pozorovali v klinických štúdiách
s rotigotínom uskutočnených v Japonsku. Vyskytovali sa u 3,4 % Japoncov liečených rotigotínom
v porovnaní s 1,9 % osôb užívajúcich placebo v dvojito zaslepených štúdiách Parkinsonovej choroby
a RLS.. Väčšina nežiaducich účinkov zvýšenej CPK pozorovaných vo všetkých dvojito zaslepených
a nezaslepených štúdiách ustúpila a boli považované za mierne závažné. V ostatných populáciách sa hladiny CPK bežne nemerali.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieSymptómyNajpravdepodobnejšie nežiaduce reakcie sú tie, ktoré súvisia s farmakodynamickým profilom agonistu
dopamínu, vrátane nevoľnosti, vracania, hypotenzie, mimovoľných pohybov, halucinácií, zmätenosti, kŕčov a ďalších známok centrálnej dopamínergickej stimulácie.
LiečbaNa predávkovanie agonistami dopamínu nie je známy žiadny protiliek. V prípade podozrenia na
predávkovanie sa má zvážiť odstránenie náplaste (náplastí), pretože po odstránení náplaste (náplastí)
sa prívod liečiva zastaví a plazmatická koncentrácia rotigotínu rýchlo klesá. Pacient má byť dôkladne
sledovaný, vrátane srdcovej frekvencie, srdcového rytmu a tlaku krvi.
Liečba predávkovania môže vyžadovať celkové podporné opatrenia na zachovanie životných funkcií. Dialýza by pravdepodobne nebola prospešná, pretože rotigotín nie je možné eliminovať dialýzou.
Ak je potrebné rotigotín vysadiť, vysadenie má prebehnúť postupne, aby sa zabránilo vzniku
neuroleptického malígneho syndrómu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiparkinsoniká, agonisty dopamínu; ATC kód: N04BC09
Rotigotín je agonista dopamínu, ktorý nie je odvodený od ergotamínu, na liečbu prejavov
a symptómov Parkinsonovej choroby a syndrómu nepokojných nôh.
Mechanizmus účinku
Predpokladá sa, že rotigotín vyvoláva priaznivé účinky na Parkinsonovu chorobu aktiváciou
receptorov D3, D2 a D1 v caudate-putamen mozgu.
Presný mechanizmus účinku rotigotínu v liečbe RLS nie je známy. Predpokladá sa, že rotigotín môže vykazovať aktivitu predovšetkým prostredníctvom dopamínových receptorov.
Farmakodynamické účinky
Čo sa týka funkčnej aktivity na rôznych podtypoch receptorov a ich distribúcie v mozgu, rotigotín je
agonista receptorov D2 a D3, ktorý pôsobí aj na receptory D1, D4 a D5. V prípade iných receptorov ako dopaminergných preukázal rotigotín antagonizmus na alfa2B receptoroch a agonizmus na 5HT1A receptoroch, žiadnu aktivitu však nepreukázal na receptoroch 5HT2B.
Klinická účinnosť a bezpečnosť
Účinnosť rotigotínu v liečbe známok a symptómov idiopatickej Parkinsonovej choroby bola
vyhodnocovaná v multinárodnom programe vývoja liekov, ktorý pozostával zo štyroch kľúčových,
paralelných, randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdií a v troch
štúdiách skúmajúcich špecifické aspekty Parkinsonovej choroby.
Dve pivotné štúdie (SP512 časť I a SP513 časť I) skúmajúce účinnosť rotigotínu v liečbe známok a
symptómov idiopatickej Parkinsonovej choroby boli vykonané u pacientov, ktorí neboli súbežne
liečení agonistami dopamínu a ešte neboli liečení L-dopou alebo predchádzajúca liečba L-dopou trvala
≤ 6 mesiacov. Hlavným výsledným hodnotením bolo skóre pre komponent aktivity denného života
(časť II) (ADL, Activities of Daily Living) a komponent motorického vyšetrenia (časť III) jednotnej
škály pre hodnotenie Parkinsonovej choroby (UPDRS, United Parkinson´s Disease Rating Scale). Účinnosť sa zisťovala na základe odozvy pacienta na liečbu, pričom sa merala ako zlepšenie odozvy pacienta a absolútneho počtu bodov v rámci skóre ADL v kombinácii s motorickým vyšetrením (UPDRS časť II+III).
V dvojito zaslepenej štúdii SP512 časť I dostávalo 177 pacientov rotigotín a 96 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu alebo placeba týždenným zvyšovaním
o 2 mg/24 h od počiatočnej dávky 2 mg/24 h po maximálnu dávku 6 mg/24 h. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 6 mesiacov.
Na konci udržiavacej liečby bola u 91 % pacientov v rotigotínovej vetve, optimálna dávka zhodná s
maximálnou povolenou dávkou, t. j. 6 mg/24 h. Zlepšenie o 20 % bolo pozorované u 48 % pacientov, ktorým sa podával rotigotín a u 19 % pacientov, ktorým sa podávalo placebo (rozdiel 29 %, CI95 %
18 %; 39 %, p< 0,0001). V prípade rotigotínu bolo priemerné zlepšenie vyjadrené v skóre UPDRS
(časti II+III) –3,98 bodov (počiatočná hodnota 29,9 bodov), zatiaľ čo u pacientov vo vetve
s placebom, bolo pozorované zhoršenie o 1,31 bodov (počiatočná hodnota 30,0 bodov). Rozdiel bol
5,28 bodov a bol štatisticky významný (p< 0,0001).
V dvojito zaslepenej štúdii SP513 časť I dostávalo 213 pacientov rotigotín, 227 pacientov ropinirol a 117 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 2 mg/24 h po maximálnu dávku 8 mg/24 h počas
4 týždňov. V skupine, v ktorej sa podával ropinirol, bola u pacientov titrovaná optimálna dávka až do
maximálnej hodnoty 24 mg/deň počas 13 týždňov. U pacientov v každej liečebnej skupine bola dávka
udržiavaná po dobu 6 mesiacov.
Na konci udržiavacej liečby bola u 92 % pacientov v rotigotínovej vetve optimálna dávka zhodná s
maximálnou povolenou dávkou, t. j. 8 mg/24 h. Zlepšenie o 20 % bolo pozorované u 52 % pacientov, ktorým sa podával rotigotín, u 68 % pacientov, ktorým sa podával ropinirol, a u 30 % pacientov, ktorým sa podávalo placebo. (Rozdiel medzi rotigotínom a placebom bol 21,7 %, CI95 % 11,1 %;
32,4 %, rozdiel medzi ropinirolom a placebom bol 38,4 %, CI95 % 28,1 %; 48,6 %, rozdiel medzi
ropinirolom a rotigotínom bol 16,6 %, CI95 % 7,6 %; 25,7 %). V rotigotínovej vetve sa dosiahlo
priemerné zlepšenie skóre UPDRS (časti II+III) 6,83 bodov (počiatočná hodnota 33,2 bodov),
v ropinirolovej vetve 10,78 bodov (počiatočná hodnota 32,2 bodov) a vo vetve s placebom 2,33 bodov
(počiatočná hodnota 31,3 bodov). Všetky rozdiely medzi aktívnymi liečbami a placebom boli štatisticky významné. Táto štúdia nepreukázala noninferioritu rotigotínu voči ropinirolu.
V následnej nezaslepenej štúdii (SP824), multicentrickej, medzinárodnej štúdii sa skúmala znášanlivosť náhlej zmeny z liečby ropinirolom, pramipexolom alebo kabergolínom na transdermálnu náplasť rotigotínu a jeho účinok na symptómy u osôb s idiopatickou Parkinsonovou chorobou. 116 pacientov prešlo z predchádzajúcej perorálnej liečby na liečbu až 8 mg/24 h rotigotínu, medzi nimi bolo 47 pacientov, ktorí boli liečení až 9 mg/deň ropinirolu, 47 pacientov, ktorí boli liečení až
2 mg/deň pramipexolu a 22 pacientov, ktorí boli liečení až 3 mg/deň kabergolínu. Prechod na rotigotín bolo uskutočniteľný s nevyhnutnou malou úpravou dávky (medián 2 mg/24 h) len u 2 pacientov, ktorí prešli z liečby ropinirolom, u 5 pacientov, ktorí prešli z liečby pramipexolom a u 4 pacientov, ktorí prešli z liečby kabergolínom. Pozorovali sa zlepšenia skóre UPDRS časti I – IV. Bezpečnostný profil bol porovnateľný s profilom v predchádzajúcich štúdiách.
V randomizovanej, nezaslepenej štúdii (SP825) u pacientov s včasných štádiom Parkinsonovej choroby bolo 25 pacientov randomizovaných na liečbu rotigotínom a 26 na ropinirol. V oboch liečebných skupinách bola dávka titrovaná na optimálnu alebo maximálnu dávku 8 mg/24 h alebo
9 mg/24 h, v uvedenom poradí. Obidve liečby preukázali zlepšenia skorej rannej motorickej funkcie
a spánku. Motorické symptómy (UPDRS časť III) sa zlepšili o 6,3 ± 1,3 bodov u pacientov liečených rotigotínom a o 5,9 ± 1,3 bodov v skupine s ropinirolom po 4 týždňoch udržiavacej liečby. Spánok
(PDSS) sa zlepšil o 4,1 ± 13,8 bodov u pacientov liečených rotigotínom a o 2,5 ± 13,5 bodov u pacientov liečených ropinirolom. Bezpečnostný profil bol porovnateľný s výnimkou reakcií v mieste
aplikácie.
V štúdiách SP824 a SP825 uskutočnených od úvodného komparatívneho klinického skúšania sa preukázalo, že rotigotín a ropinirol v ekvivalentných dávkach majú porovnateľnú účinnosť.
Dve ďalšie pivotné štúdie (SP650DB a SP515) boli vykonané u pacientov súčasne liečených
L-dopou. Hlavným zisteným výsledkom bolo skrátenie trvania tzv. stavu „off“ (v hodinách). Účinnosť sa zisťovala na základe odozvy pacienta na liečbu, pričom sa merala ako zlepšenie stavu pacienta a absolútne zlepšenie v stave „off“.
V dvojito zaslepenej štúdii SP650DB dostávalo 113 pacientov rotigotín až po maximálnu dávku
8 mg/24 h, 109 pacientov dostávalo rotigotín až po maximálnu dávku 12 mg/24 h a 119 pacientov dostávalo placebo. U pacientov bola titrovaná optimálna dávka rotigotínu alebo placeba týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 4 mg/24 h. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 6 mesiacov. Na konci udržiavacej liečby sa pozorovalo zlepšenie o minimálne 30 % u 57 % a u 55 % pacientov dostávajúcich rotigotín 8 mg/24 h a 12 mg/24 h
v uvedenom poradí a u 34 % pacientov dostávajúcich placebo (rozdiely 22 % a 21 % v uvedenom
poradí CI95% 10 %; 35 % a 8 %; 33 % v uvedenom poradí, p< 0,001 pre obe skupiny liečené
rotigotínom). V prípade rotigotínu sa trvanie stavu „off“ skrátilo o priemerne“ 2,7 a 2,1 hodiny
v uvedenom poradí, zatiaľ čo u skupiny dostávajúcej placebo sa pozorovalo skrátenie o 0,9 hodiny.
Rozdiely boli štatisticky významné (p< 0,001 a p=0,003 v uvedenom poradí).
V dvojito zaslepenej štúdii SP515 dostávalo 201 pacientov rotigotín, 200 pacientov pramipexol a 100 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 4 mg/24 h po maximálnu dávku 16 mg/24 h. V skupine
dostávajúcej pramipexol sa pacientom podávala dávka 0,375 mg počas prvého týždňa, 0,75 mg počas druhého týždňa a bola u nich titrovaná optimálna dávka ďalším týždenným zvyšovaním o 0,75 mg až po maximálnu dávku 4,5 mg/deň. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 4 mesiacov.
Na konci udržiavacej liečby sa pozorovalo zlepšenie o minimálne 30 % u 60 % pacientov
dostávajúcich rotigotín, 67 % pacientov dostávajúcich pramipexol a u 35 % pacientov dostávajúcich placebo (rozdiel medzi rotigotínom a placebom bol 25 %, CI95% 13 %; 36 %, rozdiel medzi pramipexolom a placebom bol 32 %, CI95% 21 %; 43 %, rozdiel medzi pramipexolom a rotigotínom bol 7 %, CI95% -2 %; 17 %). V skupine dostávajúcej rotigotín sa trvanie stavu „off“ skrátilo
o priemerne 2,5 hodiny, v skupine s pramipexolom to bolo 2,8 hodiny a v skupine s placebom
0,9 hodiny. Všetky rozdiely medzi aktívnymi liečbami a placebom boli štatisticky významné.
Ďalšia medzinárodná dvojito zaslepená štúdia (SP889) sa uskutočnila u 287 pacientov vo včasných alebo pokročilých štádiách Parkinsonovej choroby s nedostatočnou kontrolou skorých ranných motorických symptómov. 81,5 % týchto pacientov dostávalo súbežnú liečbu levodopou. 190 pacientov dostávalo rotigotín a 97 placebo. Pacienti boli titrovaní na optimálnu dávku rotigotínu alebo placeba
v týždňových prírastkoch o 2 mg/24h s počiatočnou dávkou 2 mg/24 h až po maximálnu dávku
16 mg/24 h po dobu 8 týždňov, po ktorom nasledovalo udržiavacie obdobie 4 týždňov. Skoré ranné motorické funkcie, hodnotené podľa časti III UPDRS a poruchy nočného spánku, hodnotené podľa
upravenej škály spánku pri Parkinsonovej chorobe (Parkinson’s Disease Sleep Scale - PDSS-2), boli'
ko-primárnymi výslednými meraniami. Na konci udržiavacieho obdobia sa priemerné skóre podľa časti III UPDRS zlepšilo o 7,0 bodov u pacientov liečených rotigotínom (východisková hodnota 29,6)
a o 3,9 bodov v placebovej skupine (východisková hodnota 32,0). Zlepšenia v priemernom celkovom
skóre PDSS-2 boli 5,9 (rotigotín, východisková hodnota 19,3) a 1,9 bodov (placebo, východisková hodnota 20,5). Liečebné rozdiely v koprimárnych premenných boli štatisticky významné (p=0,0002 a p<0,0001).
Priľnavosť na kožu
V multicentrickej, dvojito zaslepenej, randomizovanej, dvojito skríženej štúdii bola u 52 ambulantných pacientov porovnávaná priľnavosť na kožu rotigotín náplasti 8 mg/24 h, s vylepšeným uchovávaním pri izbovej teplote, s náplasťou s uchovávaním v chlade. Priľnavosť na kožu bola meraná počas 2 po sebe nasledujúcich dní pri 24-hodinovej aplikácii náplasti.
Vylepšená náplasť s uchovávaním pri izbovej teplote preukázala lepšiu priľnavosť na kožu ako náplasť s uchovávaním v chlade, > 90 % náplastí preukazuje dostatočnú priľnavosť (t.j. > 70 % priliehajúcej plochy náplasti) v porovnaní s <83 %. Pri obidvoch formách bola hlásená porovnateľná znášanlivosť kože. Väčšina pozorovaných erytémov bolo miernych a žiadny nebol závažný.
5.2 Farmakokinetické vlastnosti
Absorpcia
Rotigotín sa po aplikácii plynule uvoľňuje z transdermálnej náplasti a je absorbovaný cez pokožku.
Ustálené koncentrácie sa dosiahnu za jeden až dva dni po aplikácii náplasti a udržiavajú sa na stabilnej úrovni aplikáciou náplasti jedenkrát denne, pričom táto sa nosí po dobu 24 hodín. Koncentrácie rotigotínu v plazme sa zvyšujú v závislosti od dávky v rozsahu dávok od 1 mg/24 h po 24 mg/24 h.
Za 24 hodín sa uvoľní do pokožky približne 45 % liečiva v náplasti. Absolútna biologická dostupnosť
po transdermálnej aplikácii je približne 37 %.
Striedanie miesta aplikácie náplasti môže spôsobiť rozdiely medzi jednotlivými dennými hladinami
v plazme. Rozdiely v biologickej dostupnosti rotigotínu siahali od 2 % (nadlaktie v porovnaní
s bokom) po 46 % (rameno v porovnaní so stehnom). Neexistuje žiaden náznak relevantného vplyvu na klinické výsledky.
D
is
t
ri
b
úcia
Približne 92 % rotigotínu sa viaže in vitro na plazmatické proteíny.
Zdanlivý distribučný objem u ľudí je približne 84 l/kg.
Biotransformácia
Rotigotín je z veľkej časti metabolizovaný. Rotigotín je metabolizovaný N-dealkyláciou ako aj
priamou a sekundárnou konjugáciou. Výsledky vykonávané in vitro naznačujú, že rôzne izoformy CYP dokážu katalyzovať N-dealkyláciu rotigotínu. Hlavnými metabolitmi sú sulfáty a glukuronidové konjugáty pôvodnej látky ako aj metabolity N-dealkylácie, ktoré sú biologicky neaktívne.
Informácie o metabolitoch sú neúplné.
Eliminácia
Približne 71 % dávky rotigotínu sa vylučuje močom a menšia časť približne 23 % sa vylučuje stolicou.
Klírens rotigotínu po transdermálnom podaní je približne 10 l/minútu a jeho celkový polčas eliminácie je 5 až 7 hodín. Farmakokinetický profil preukazuje bifázickú elimináciu s počiatočným polčasom
približne 2 až 3 hodiny.
Keďže náplasť sa aplikuje transdermálne, neočakáva sa žiadny vplyv jedál alebo ochorení
gastrointestinálneho traktu.
Osobitné skupiny pacientov
Keďže liečba Neuprom začína nízkou dávkou a táto sa postupne titruje podľa klinickej znášanlivosti,
aby sa získal optimálny terapeutický účinok, úprava dávky na základe pohlavia, hmotnosti alebo veku
nie je potrebná.
Porucha funkcie pečene a obličiek
U pacientov so stredne závažnou poruchou funkcie pečene alebo miernou až závažnou poruchou funkcie obličiek neboli pozorované žiadne relevantné zvýšenia hladiny rotigotínu v plazme. Neupro nebolo skúmané u pacientov so závažnou poruchou funkcie pečene.
V prípade poruchy funkcie obličiek sa zvyšujú hladiny konjugátov rotigotínu a metabolitov N- dealkylácie rotigotínu v plazme. Vplyv týchto metabolitov na klinické účinky je však
nepravdepodobný.
5.3 Predklinické údaje o bezpečnosti
V štúdiách zameraných na opakované dávky a dlhodobú toxicitu boli hlavné účinky spájané s farmakodynamickými účinkami súvisiacimi s agonistom dopamínu a následným znížením vylučovania prolaktínu.
Po jednorazovej dávke rotigotínu bolo u pigmentovaných potkanov a opíc evidentné viazanie sa
rotigotínu na tkanivo obsahujúce melanín (t. j. oči), ktoré však počas 14-dňového pozorovacieho
intervalu vymizlo.
V trojmesačnej štúdii bola u bielych potkanov pozorovaná pomocou transmisnej elektrónovej mikroskopie degenerácia sietnice pri dávke ekvivalentnej 2,8-násobku maximálnej odporúčanej dávky pre ľudí (prepočítanej na mg/m2). Účinky boli výraznejšie u samíc potkanov. Dodatočné štúdie na ďalšie vyhodnotenie špecifickej patológie neboli vykonané. Degenerácia sietnice sa nepozorovala počas rutinného histopatologického vyhodnocovania očí skúmaných druhov zvierat v žiadnej
z toxikologických štúdií. Význam týchto zistení pre ľudí nie je známy.
V štúdii karcinogenicity sa u samcov potkanov rozvinuli nádory a hyperplázia Leydigových buniek. Zhubné nádory boli pozorované najmä v maternici samíc, ktorým sa podávali stredné až vysoké dávky. Tieto zmeny sú dobre známymi účinkami agonistov dopamínu u potkanov po celoživotnej liečbe a boli zhodnotené ako nerelevantné pre ľudí.
Účinky rotigotínu na reprodukciu sa skúmali u potkanov, králikov a myší. Na žiadny z uvedených troch druhov nemal rotigotín teratogénne účinky, ale u potkanov a myší vykazoval embryotoxické účinky pri dávkach toxických pre matky. Rotigotín neovplyvnil plodnosť samcov u potkanov, avšak jednoznačne znížil plodnosť samíc potkanov a myší z dôvodu účinkov na hladiny prolaktínu, ktoré sú u hlodavcov obzvlášť významné.
Rotigotín nevyvolal génové mutácie v Amesovom teste, preukázal však účinky v in vitro teste lymfómu u myší s metabolickou aktiváciou a slabšie účinky bez metabolickej aktivácie. Mutagénny účinok je možné pripísať klastogénnemu účinku rotigotínu. Tento účinok nebol potvrdený in vivo
v mikronukleovom teste u myší a v teste na neplánovanú syntézu DNA (UDS, Unsheduled DNA Synthesis) u potkanov. Keďže prebiehal viac-menej súbežne so zníženým relatívnym celkovým rastom
buniek, môže súvisieť s cytotoxickým účinkom látky. Významnosť tohto jedného pozitívneho testu
mutagenicity in vitro preto nie je známa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Krycia vrstva
Polyesterový film s vrstvou silikónu a hliníka
farebný povlak pigmentovej vrstvy (oxid titaničitý (E171), žltý pigment 95, červený pigment 166)
a potlač (červený pigment 144, žltý pigment 95, čierny pigment 7).
Samolepiaca matricová vrstva
Poly(dimetylsiloxán, trimetylsilylsilikát)-kopolymerizát,
Povidón K90,
Metasiričitan sodný (E223),
Askorbylpalmitát (E304) a
DL-α-tokoferol (E307).
Snímateľná fólia
Priesvitný polyesterový film s fluoropolymérovým povlakom.
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
30 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
6.5 Druh obalu a obsah balenia
Rozlepovacie vrecko v lepenkovej škatuli: Jednu stranu tvorí kopolymér etylénu (najvnútornejšia vrstva), hliníková fólia, polyetylénový film s nízkou hustotou a papier; druhú stranu tvorí polyetylén (najvnútornejšia vrstva), hliník, kopolymér etylénu a papier.
Škatuľa obsahuje 7, 14, 28, 30, alebo 84 (multibalenie obsahujúce 2 balenia po 42) transdermálnych
náplastí, ktoré sú jednotlivo zatavené vo vreckách.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuNáplasť po použití ešte stále obsahuje liečivo. Po odstránení sa má použitá náplasť preložiť napoly lepiacou stranou smerom dovnútra tak, aby matricová vrstva nezostala odkrytá, umiestniť do pôvodného vrecka a odložiť. Všetky použité aj nepoužité náplasti majú byť zlikvidované v súlade
s národnými požiadavkami alebo vrátené do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIUCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLANeupro4mg/24htransdermálnanáplasťEU/1/05/331/004
EU/1/05/331/005
EU/1/05/331/021
EU/1/05/331/024
EU/1/05/331/059
Neupro6mg/24htransdermálnanáplasťEU/1/05/331/007
EU/1/05/331/008
EU/1/05/331/027
EU/1/05/331/030
EU/1/05/331/060
Neupro8mg/24htransdermálnanáplasťEU/1/05/331/010
EU/1/05/331/011
EU/1/05/331/033
EU/1/05/331/036
EU/1/05/331/061
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15. február 2006
Dátum posledného predĺženia registrácie: 22. január 2016
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Neupro
2 mg/24 h
4 mg/24 h
6 mg/24 h
8 mg/24 h
Transdermálna náplasť
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Neupro 2 mg/24 h transdermálna náplasť
Každá náplasť uvoľní 2 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 10 cm2 obsahuje 4,5 mg rotigotínu.
Neupro 4 mg/24 h transdermálna náplasť
Každá náplasť uvoľní 4 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 20 cm2 obsahuje 9,0 mg rotigotínu.
Neupro 6 mg/24 h transdermálna náplasť
Každá náplasť uvoľní 6 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 30 cm2 obsahuje
13,5 mg rotigotínu.
Neupro 8 mg/24 h transdermálna náplasť
Každá náplasť uvoľní 8 mg rotigotínu za 24 hodín. Každá náplasť s povrchom 40 cm2 obsahuje
18,0 mg rotigotínu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Transdermálna náplasť.
Tenká štvorcová náplasť matricového typu so zaoblenými rohmi, skladajúca sa z troch vrstiev. Vonkajšia strana krycej vrstvy je béžová s potlačou „Neupro 2 mg/24 h, 4 mg/24 h, 6 mg/24 h alebo
8 mg/24 h“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Neupro sa indikuje na liečbu známok a symptómov idiopatickej Parkinsonovej choroby v počiatočnom štádiu v monoterapii (t. j. bez L-dopy) alebo v kombinácii s L-dopou, čiže počas trvania ochorenia až do neskorých štádií, keď účinok L-dopy zoslabne alebo sa stane premenlivým a dochádza
k fluktuáciám terapeutického účinku (fluktuácie ku koncu dávkovacieho intervalu alebo „on-off“
fluktuácie zo stavu dobrej hybnosti tzv. „on“ do stavu zlej hybnosti tzv. „off“).
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Odporúčané dávky predstavujú nominálne dávky.
Dávkovanie u pacientov v počiatočnom štádiu Parkinsonovej choroby:
Jednorazová denná dávka má začať na 2 mg/24 h a následne byť zvyšovaná v týždňových intervaloch o 2 mg/24 h až po dosiahnutie účinnej dávky, ktorá je maximálne 8 mg/24 h.
Dávka 4 mg/24 h môže byť v prípade niektorých pacientov účinnou dávkou. U väčšiny pacientov je účinná dávka dosiahnutá v priebehu 3 alebo 4 týždňov pri dávkach 6 mg/24 h alebo 8 mg/24 h,
v uvedenom poradí.
Maximálna dávka je 8 mg/24 h.
Dávkovanie u pacientov v pokročilom štádiu Parkinsonovej choroby s fluktuáciami:
Jednorazová denná dávka má začať na 4 mg/24 h a následne byť zvyšovaná v týždňových intervaloch o 2 mg/24 h až po dosiahnutie účinnej dávky, ktorá je maximálne 16 mg/24 h.
Dávky 4 mg/24 h alebo 6 mg/24 h môžu byť v prípade niektorých pacientov účinnými dávkami.
U väčšiny pacientov je účinná dávka dosiahnutá v priebehu 3 až 7 týždňov pri dávkach 8 mg/24 h až
po maximálnu dávku 16 mg/24 h.
Súprava náplastí Neupro na zahájenie liečby určená na prvé štyri týždne liečby obsahuje 4 rôzne
balenia (jedno pre každú silu), z ktorých každé obsahuje 7 náplastí.
V závislosti od odozvy pacienta na liečbu nemusia byť nutné všetky nasledujúce dávkovacie kroky, prípadne môžu byť po 4. týždni potrebné prídavné vyššie dávky, ktoré nie sú súčasťou tejto súpravy.
Prvý deň liečby začne pacient Neuprom 2 mg/24 h. Počas druhého týždňa používa pacient Neupro
4 mg/24 h. Počas tretieho týždňa používa pacient Neupro 6 mg/24 h a počas štvrtého týždňa Neupro
8 mg/24 h. Balenia sú označené ako „1. (2., 3. alebo 4.) týždeň“.
Neupro sa aplikuje raz denne. Náplasť sa má aplikovať každý deň približne v rovnakom čase. Náplasť sa ponecháva na koži 24 hodín, a potom sa vymieňa za novú náplasť aplikovanú na inom mieste.
Ak si pacient zabudne náplasť aplikovať v obvyklom čase počas dňa alebo ak sa náplasť odlepí, na zvyšok dňa sa má aplikovať nová náplasť.
Ukončenie liečby
Liečba Neuprom má byť ukončená postupne. Denná dávka sa má znižovať o 2 mg/24 h znižovaním dávky podľa možnosti každý druhý deň až do úplného vysadenia Neupra (pozri časť 4.4).
Osobitné populácie
Porucha funkcie pečene
U pacientov s mierne až stredne závažnými poruchami funkcie pečene nie je potrebná úprava dávky. Opatrnosť sa odporúča pri liečbe pacientov so závažnými poruchami funkcie pečene, u ktorých môže dôjsť k zníženiu klírensu rotigotínu. Rotigotín nebol skúmaný u tejto skupiny pacientov. V prípade zhoršovania poruchy funkcie pečene môže byť potrebná redukcia dávky.
Porucha funkcie obličiek
U pacientov s miernou až závažnou poruchou funkcie obličiek, vrátane dialyzovaných pacientov, nie je potrebná úprava dávky. Neočakávaný nárast hladiny rotigotínu sa môže objaviť tiež v prípade akútneho zhoršenia funkcie obličiek (pozri časť 5.2).
Pediatrická populácia
Neexistuje žiadne relevantné použitie Neupra v pediatrickej populácii pri Parkinsonovej chorobe.
Spôsob podávania
Neupro je určené na transdermálne použitie.
Náplasť sa má aplikovať na čistú, suchú, neporušenú zdravú pokožku v oblasti brucha, stehna, bedier, bokov trupu, ramien alebo nadlaktia. Náplasť sa nemá aplikovať na rovnaké miesto skôr než po
14 dňoch. Neupro sa nesmie aplikovať na červenú, podráždenú alebo poškodenú pokožku (pozri časť 4.4).
Použitie a zaobchádzanie:
Každá náplasť je zabalená vo vrecku a aplikuje sa ihneď po otvorení vrecka. Odstráni sa jedna polovica snímateľnej fólie a lepiaca strana sa aplikuje pevným pritlačením na pokožku. Potom sa náplasť prehne dozadu a odstráni sa druhá časť prilepenej fólie. Nedotýkajte sa lepiacej strany náplasti. Dlaňou sa náplasť pevne zatlačí po dobu približne 20 až 30 sekúnd, aby sa dobre prilepila.
Náplasť sa nesmie strihať na kusy.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Zobrazovanie magnetickou rezonanciou alebo kardioverzia (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Ak je odozva pacienta s Parkinsonovou chorobou na liečbu rotigotínom nedostatočná, môžu sa prestupom na liečbu iným agonistom dopamínu dosiahnuť dodatočné pozitívne účinky (pozri časť
5.1).
Zobrazovanie magnetickou rezonanciou a kardioverzia
Krycia vrstva náplasti Neupro obsahuje hliník. Ak bude pacient podstupovať vyšetrenie zobrazovaním
magnetickou rezonanciou (MRI) alebo kardioverziu, musí sa Neupro odstrániť, aby sa zabránilo
popáleninám pokožky.
Ortostatická hypotenzia
Je známe, že agonisty dopamínu zhoršujú systémovú reguláciu tlaku krvi, v dôsledku čoho vzniká
posturálna/ortostatická hypotenzia. Tieto prípady sa pozorovali aj počas liečby rotigotínom, ich výskyt bol však podobný ako výskyt pozorovaný u pacientov liečených placebom.
Z dôvodu všeobecného rizika ortostatickej hypotenzie spojenej s dopamínergickou liečbou sa odporúča sledovať krvný tlak, a to najmä na začiatku liečby.
Synkopa
V klinických štúdiách s rotigotínom sa pozorovala synkopa s podobnou frekvenciou výskytu, ako bola
pozorovaná u pacientov liečených placebom. Keďže pacienti s klinicky relevantným kardiovaskulárnym ochorením boli z týchto štúdií vylúčení, je potrebné sa pacientov so závažným
kardiovaskulárnym ochorením opýtať na príznaky synkopy a presynkopy.
Náhle zaspanie a ospalosť
Rotigotín bol spájaný s ospalosťou a prípadmi náhleho zaspania, najmä u pacientov s Parkinsonovou
chorobou. Hlásené boli prípady náhleho zaspania počas denných aktivít, v niektorých prípadoch bez spozorovania akýchkoľvek varovných známok. Lekári predpisujúci Neupro majú u pacientov
nepretržite prehodnocovať príznaky driemot alebo ospalosti, pretože samotní pacienti si ich nemusia
uvedomiť, až kým sa ich na to priamo neopýtajú. Starostlivo sa má zvážiť zníženie dávky alebo ukončenie liečby.
Poruchy kontroly impulzov
Pacientov je treba pravidelne monitorovať pre vývoj porúch kontroly impulzov. Pacienti
a opatrovatelia si majú byť vedomí, že u pacientov liečených agonistami receptorov dopamínu vrátane rotigotínu sa môžu vyskytnúť behaviorálne príznaky porúch kontroly impulzov ako patologické hranie
hazardných hier, zvýšené libido, hypersexualita, kompulzívne utrácanie alebo nakupovanie, prejedanie
sa a závislosť na jedle. Ak sa tieto symptómy objavia, je potrebné zvážiť zníženie dávky/postupné
vysadenie.
Neuroleptický malígny syndróm
Pri náhlom ukončení dopamínergickej liečby boli hlásené symptómy naznačujúce prítomnosť
neuroleptického malígneho syndrómu. Preto sa odporúča liečbu obmedzovať postupne (pozri časť 4.2).
Abnormálne myslenie a správanie
Zaznamenalo sa abnormálne myslenie a správanie a môže pozostávať z rôznych prejavov vrátane
paranoidných myšlienok, preludov, halucinácií, zmätenosti, správania podobného psychotickému, dezorientácie, agresívneho správania, agitácie a delíria.
Fibrotické komplikácie
U niektorých pacientov liečených dopamínergickými látkami odvodenými od námeľu boli hlásené
prípady retroperitoneálnej fibrózy, pľúcnych infiltrátov, pleurálnych výpotkov, pleurálneho zhrubnutia, perikarditídy a srdcovej valvulopatie. Aj keď tieto komplikácie môžu po vysadení liečby ustúpiť, nemusia vždy ustúpiť úplne.
Aj keď sa predpokladá, že tieto nežiaduce reakcie súvisia s ergotamínovou štruktúrou týchto látok, nie je známe, či ich môžu spôsobovať aj agonisty dopamínu, ktoré nie sú odvodené od námeľu.
Neuroleptiká
Neuroleptiká podávané ako antiemetiká sa nemajú podávať pacientom užívajúcim agonistov
dopamínu (pozri aj časť 4.5).
Oftalmologické sledovanie
Oftalmologické sledovanie sa odporúča v pravidelných intervaloch alebo v prípade porúch videnia.
Vystavenie zdroju tepla
Miesto s náplasťou nemá byť vystavované vonkajším zdrojom tepla (nadmerné slnečné žiarenie,
ohrievacie podušky a iné zdroje tepla, napríklad sauna, horúci kúpeľ).
Reakcie v mieste aplikácie
V mieste aplikácie sa môžu vyskytnúť kožné reakcie, zvyčajne s miernou alebo stredne závažnou
intenzitou. Miesto aplikácie sa odporúča denne striedať (napríklad z pravej strany na ľavú stranu
a z hornej časti tela na dolnú časť tela). Rovnaké miesto sa nemá použiť znova skôr než po 14 dňoch. V prípade niekoľkodňového alebo trvalého výskytu reakcií v mieste aplikácie, pri zvýšení závažnosti
alebo pri rozšírení kožnej reakcie mimo miesto aplikácie, sa má zhodnotiť pomer rizík a výhod pre
daného pacienta.
V prípade výskytu vyrážok alebo podráždenia pokožky transdermálnym systémom sa má zabrániť pôsobeniu priameho slnečného žiarenia na dané miesto, kým sa pokožka nezahojí, pretože expozícia môže spôsobiť zmeny sfarbenia pokožky.
V prípade spozorovania celkovej reakcie pokožky (napríklad alergické vyrážky, vrátane erytematóznych, škvrnitých a papulárnych vyrážok alebo svrbenia) spojenej s používaním Neupra, musí sa Neupro prestať používať.
Periférny edém
V klinických štúdiách u pacientov s Parkinsonovou chorobou bol počas 6 mesiacov špecifický výskyt
periférneho edému okolo 4 % a zostal nezmenený počas ďalšieho pozorovania až do 36 mesiacov.
Dopamínergické nežiaduce reakcie
Výskyt niektorých dopamínergických nežiaducich reakcií, ako sú napríklad halucinácie, dyskinéza a
periférny edém, je vo všeobecnosti vyšší pri podávaní v kombinácii s L-dopou u pacientov
s Parkinsonovou chorobou. Treba to zvážiť pri predpisovaní rotigotínu.
Citlivosť nasiričitan
Neupro obsahuje metasiričitan sodný, siričitan, ktorý môže vyvolať reakcie alergického typu vrátane
anafylaktických symptómov a život ohrozujúcich alebo menej závažných astmatických epizód u niektorých citlivých osôb.
4.5 Liekové a iné interakcie
Keďže rotigotín je agonista dopamínu, predpokladá sa, že antagonisty dopamínu, ako napríklad neuroleptiká (napríklad fenotiazíny, butyrofenóny, tioxantény) alebo metoklopramid, môžu oslabovať účinnosť Neupra, a preto sa nemajú podávať súbežne s Neuprom. Z dôvodu možných aditívnych účinkov sa má postupovať opatrne, ak pacienti užívajú sedatívne lieky alebo iné lieky utlmujúce činnosť CNS (centrálneho nervového systému) (napríklad benzodiazepíny, antipsychotiká, antidepresíva) alebo alkohol v kombinácii s rotigotínom.
Súbežné podávanie L-dopy a karbidopy s rotigotínom nemalo žiadny účinok na farmakokinetiku rotigotínu a rotigotín nemal žiadny účinok na farmakokinetiku L-dopy a karbidopy.
Súbežné podávanie domperidonu s rotigotínom nemalo žiadny účinok na farmakokinetiku rotigotínu. Súbežné podávanie omeprazolu (inhibítora CYP2C19) v dávkach 40 mg/deň nemalo žiadny účinok na
farmakokinetiku a metabolizmus rotigotínu u zdravých dobrovoľníkov.
Neupro môže zosilniť dopamínergické nežiaduce reakcie L-dopy a môže spôsobiť a/alebo zhoršiť už
existujúcu dyskinézu, podobne ako v prípade ostatných agonistov dopamínu.
Súbežné podávanie rotigotínu (3 mg/24 h) neovplyvňovalo farmakodynamiku a farmakokinetiku
perorálnych kontraceptív (0,03 mg etinylestradiol, 0,15 mg levonorgestrel). Interakcie s inými formami hormonálnej antikoncepcie neboli skúmané.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku, antikoncepcia u žien
Ženy vo fertilnom veku majú používať účinnú antikoncepciu na prevenciu gravidity počas liečby
rotigotínom.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití rotigotínu u gravidných žien. Štúdie na zvieratách nepreukázali žiadne teratogénne účinky u potkanov a králikov, avšak u potkanov a myší bola pozorovaná embryotoxicita pri dávkach toxických pre matky (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí. Rotigotín sa nemá používať počas gravidity.
Laktácia
Keďže rotigotín znižuje vylučovanie prolaktínu u ľudí, očakáva sa inhibícia tvorby mlieka. Štúdie
vykonávané na potkanoch ukázali, že rotigotín a/alebo jeho metabolit(y) sú vylučované do materského mlieka. Z dôvodu chýbajúcich údajov o účinkoch na ľudský organizmus sa má dojčenie prerušiť.
Fertilita
Pre informácie o štúdiách fertility, prosím, pozri časť 5.3.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Rotigotín môže mať značný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Pacienti liečení rotigotínom, ktorí trpia ospalosťou a/alebo prípadmi náhleho zaspania, musia byť informovaní o zákaze riadenia vozidiel alebo vykonávania aktivít (napríklad obsluhovanie strojov), pri ktorých môže zhoršené vnímanie ohroziť ich samotných alebo ostatných a spôsobiť vážne zranenia alebo smrť. To platí, až kým tieto opakujúce sa prípady náhleho zaspania a ospalosť neustúpia (pozri
aj časti 4.4 a 4.5).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Podľa analýzy skupinových, placebom kontrolovaných klinických štúdií, zahŕňajúcich celkovo
1 307 pacientov liečených Neuprom a 607 pacientov liečených placebom, 72,5 % pacientov liečených
Neuprom a 58,0 % pacientov liečených placebom hlásilo aspoň jednu nežiaducu reakciu.
Na začiatku liečby sa môžu vyskytovať dopamínergické nežiaduce reakcie ako nevoľnosť a vracanie. Tieto sú zvyčajne mierne alebo stredne závažné a dočasné, a to aj v prípade pokračovania v liečbe.
Medzi nežiaduce liekové reakcie hlásené u viac ako 10 % pacientov liečených transdermálnymi náplasťami Neupro patria nevoľnosť, vracanie, reakcie v mieste aplikácie, ospalosť, závrat a bolesť hlavy.
V štúdiách, pri ktorých sa miesto aplikácie striedalo podľa pokynov v SmPC (Súhrn charakteristických
vlastností lieku, Summary of Product Characteristics) a písomnej informácii pre používateľa, sa
u 35,7 % z 830 pacientov používajúcich transdermálne náplasti Neupro vyskytovali reakcie v mieste
aplikácie. Väčšina reakcií v mieste aplikácie bola mierna alebo stredne závažná, obmedzená na miesto aplikácie a v ich dôsledku bola liečba Neuprom ukončená iba u 4,3 % všetkých pacientov používajúcich Neupro.
Zoznam nežiaducich reakcií zoradených dotabuľky
V nasledujúcej tabuľke sú uvedené nežiaduce liekové reakcie zo združených štúdií uvedených vyššie
u pacientov s Parkinsonovou chorobou. V rámci tried orgánových systémov sú nežiaduce reakcie
vymenované pod názvami jednotlivých frekvencií (počet pacientov, u ktorých sa predpokladá výskyt reakcie) použitím nasledujúcich kategórií: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v
poradí klesajúcej závažnosti.
T
ri
edy orgánových systémov podľa MedDRA Poruchy imunitného systému
V
e
ľmi časté Časté Menej časté Zriedkavé Neznáme
hypersenzitivita, ktorá môže zahŕňať angioedém, edém jazyka a edém pier
P
sychické poruchy
P
oruchy nervového systému
ospalosť, závrat, bolesť hlavy
poruchy vnímaniaa (vr. halucinácie, zrakovej halucinácie, sluchovej halucinácie, vidiny), nespavosť, porucha spánku, nočné mory, neobvyklé sny, poruchy
kontroly impulzova,d (vr. patologického hrania hazardných hier, stereotypného/ nutkavého správania, prejedania sa/porúch prijímania potravyb, kompulzívneho nakupovaniac) poruchy
vedomia NECa
(vr. synkopy, vazovagálnej
synkopy, straty vedomia),
dyskinéza, posturálny
závrat, letargia
náhle zaspávanie/ náhly nástup spánku, paranoja, poruchy sexuálnej túžbya (vr. hypersexuality, zvýšeného libida), stav zmätenosti, dezorientáciad, agitáciad
psychotická porucha, obsedantno- kompulzívna porucha, agresívne správanie/ agresivitab, preludd, delíriumd
kŕč
syndróm dopamínovej dysreguláciec
P
oruchy oka rozmazané videnie, poruchy videnia, fotopsia
P
oruchy ucha a labyrintu Poruchy srdca a srdcovej činnosti
vertigo
palpitácie atriálna fibrilácia supraventrikulá rna tachykardia
 P
oruchy ciev
P
oruchy ciev ortostatická hypotenzia, hypertenzia
hypotenzia
P
oruchy dýchacej sústavy, hrudníka
a mediastína
P
oruchy gastrointestinál neho traktu Poruchy kože
a podkožného tkaniva
P
oruchy reprodukčného systému
a prsníkov Celkové poruchy
a reakcie
v mieste podania
L
aboratórne a funkčné vyšetrenia
Ú
r
azy, otravy a komplikácie liečebného postupu
nevoľnosť,
vracanie
reakcie
v mieste aplikácie a instiláciea, (napríklad erytém, svrbenie, podráždenie, vyrážka, dermatitída, vezikuly, bolesť, ekzém, zápal, opuch, zmena sfarbenia, pupence, exfoliácie, urtikária, precitlivenosť)
štikútanie
zápcha, sucho v ústach, dyspepsia erytém, hyperhidróza, svrbenie
periférny edém, astenické stavya, (vr. únavy, asténie, malátnosti)
zníženie hmotnosti
pád
bolesť brucha
celkové svrbenie, podráždenie pokožky, kontaktná dermatitída erektilná dysfunkcia
zvýšenie hladiny pečeňového enzýmu (vr. AST, ALT, GGT), zvýšenie hmotnosti, zrýchlenie srdcového rytmu, zvýšenie CPKd (pozri Osobitné populácie)
generalizovaná vyrážka
podráždenosť
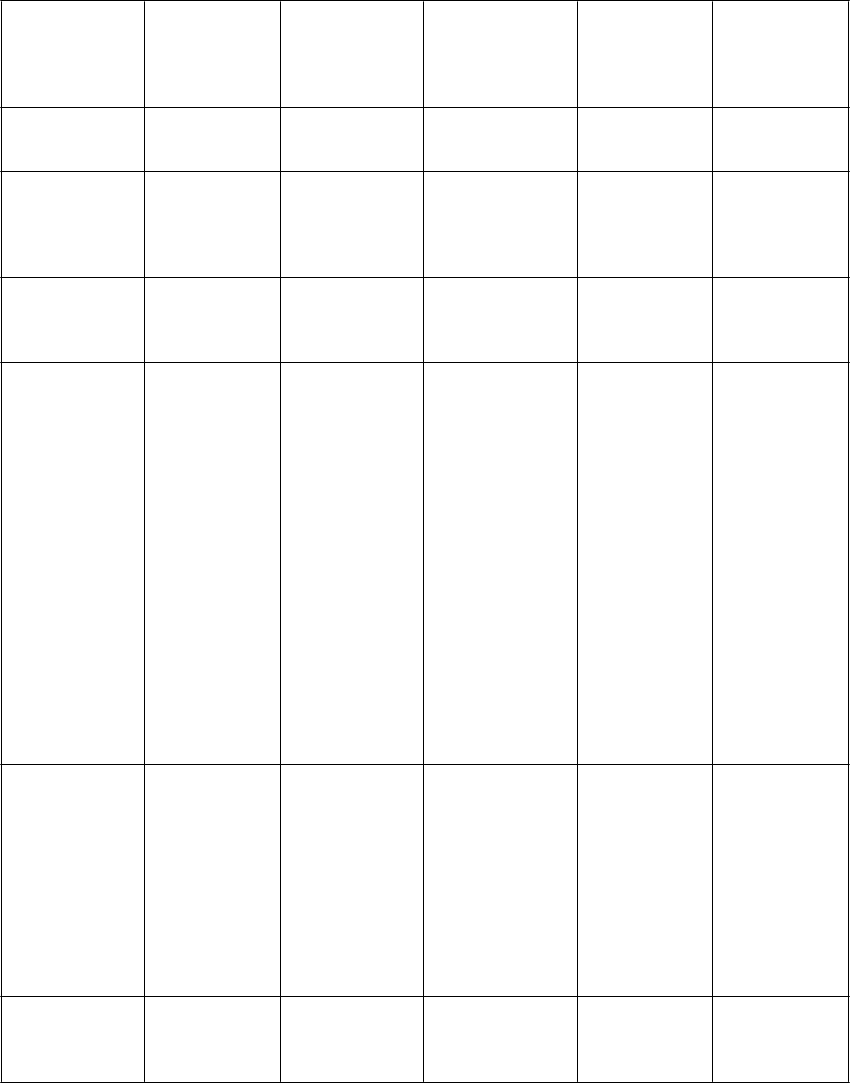
a Nadradený všeobecný názov
b Pozorované v nezaslepených štúdiách
c Pozorované po uvedení lieku na trh
d Pozorované v roku 2011 v súhrnných údajoch z dvojito zaslepených placebom kontrolovaných štúdií
Popis vybraných nežiaducich reakcií
N
áhly nástup spánku a ospalosť
Rotigotín bol spájaný s ospalosťou vrátane nadmernej ospalosti počas dňa a prípadov náhleho zaspania. V zriedkavých prípadoch sa vyskytlo „náhle zaspanie“ počas jazdy, čo následne spôsobilo dopravnú nehodu (pozri aj časť 4.4 a 4.7).
Poruchy kontroly impulzovPatologické hranie hazardných hier, zvýšené libido, hypersexualita, kompulzívne utrácanie alebo nakupovanie, prejedanie sa a závislosť na jedle sa môžu vyskytnúť u pacientov liečených agonistami dopamínu vrátane rotigotínu (pozri časť 4.4 ).
Osobitné populácieNežiaduce účinky kreatínfosfokinázy (CPK) sa pozorovali v klinických štúdiách s rotigotínom
uskutočnených v Japonsku. Vyskytovali sa u 3,4 % Japoncov liečených rotigotínom v porovnaní
s 1,9 % osôb užívajúcich placebo v dvojito zaslepených štúdiách Parkinsonovej choroby
a RLS. Väčšina nežiaducich účinkov zvýšenej CPK pozorovaných vo všetkých dvojito zaslepených
a nezaslepených štúdiách ustúpila a boli považované za mierne závažné. V ostatných populáciách sa hladiny CPK bežne nemerali.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieSymptómyNajpravdepodobnejšie nežiaduce reakcie sú tie, ktoré súvisia s farmakodynamickým profilom agonistu
dopamínu, vrátane nevoľnosti, vracania, hypotenzie, mimovoľných pohybov, halucinácií, zmätenosti, kŕčov a ďalších známok centrálnej dopamínergickej stimulácie.
LiečbaNa predávkovanie agonistami dopamínu nie je známy žiadny protiliek. V prípade podozrenia na
predávkovanie sa má zvážiť odstránenie náplaste (náplastí), pretože po odstránení náplaste (náplastí)
sa prívod liečiva zastaví a plazmatická koncentrácia rotigotínu rýchlo klesá. Pacient má byť dôkladne
sledovaný, vrátane srdcovej frekvencie, srdcového rytmu a tlaku krvi.
Liečba predávkovania môže vyžadovať celkové podporné opatrenia na zachovanie životných funkcií. Dialýza by pravdepodobne nebola prospešná, pretože rotigotín nie je možné eliminovať dialýzou.
Ak je potrebné rotigotín vysadiť, vysadenie má prebehnúť postupne, aby sa zabránilo vzniku
neuroleptického malígneho syndrómu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antiparkinsoniká, agonisty dopamínu; ATC kód: N04BC09
Rotigotín je agonista dopamínu, ktorý nie je odvodený od ergotamínu, na liečbu prejavov
a symptómov Parkinsonovej choroby a syndrómu nepokojných nôh.
Mechanizmus účinku
Predpokladá sa, že rotigotín vyvoláva priaznivé účinky na Parkinsonovu chorobu aktiváciou
receptorov D3, D2 a D1 v caudate-putamen mozgu.
Presný mechanizmus účinku rotigotínu v liečbe RLS nie je známy. Predpokladá sa, že rotigotín môže vykazovať aktivitu predovšetkým prostredníctvom dopamínových receptorov.
Farmakodynamické účinky
Čo sa týka funkčnej aktivity na rôznych podtypoch receptorov a ich distribúcie v mozgu, rotigotín je
agonista receptorov D2 a D3, ktorý pôsobí aj na receptory D1, D4 a D5. V prípade iných receptorov ako dopaminergných preukázal rotigotín antagonizmus na alfa2B receptoroch a agonizmus na 5HT1A receptoroch, žiadnu aktivitu však nepreukázal na receptoroch 5HT2B.
Klinická účinnosť a bezpečnosť
Účinnosť rotigotínu v liečbe známok a symptómov idiopatickej Parkinsonovej choroby bola
vyhodnocovaná v multinárodnom programe vývoja liekov, ktorý pozostával zo štyroch kľúčových,
paralelných, randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdií a v troch
štúdiách skúmajúcich špecifické aspekty Parkinsonovej choroby.
Dve pivotné štúdie (SP512 časť I a SP513 časť I) skúmajúce účinnosť rotigotínu v liečbe známok a
symptómov idiopatickej Parkinsonovej choroby boli vykonané u pacientov, ktorí neboli súbežne
liečení agonistami dopamínu a ešte neboli liečení L-dopou alebo predchádzajúca liečba L-dopou trvala
≤ 6 mesiacov. Hlavným výsledným hodnotením bolo skóre pre komponent aktivity denného života
(časť II) (ADL, Activities of Daily Living) a komponent motorického vyšetrenia (časť III) jednotnej
škály pre hodnotenie Parkinsonovej choroby (UPDRS, United Parkinson´s Disease Rating Scale). Účinnosť sa zisťovala na základe odozvy pacienta na liečbu, pričom sa merala ako zlepšenie odozvy pacienta a absolútneho počtu bodov v rámci skóre ADL v kombinácii s motorickým vyšetrením (UPDRS časť II+III).
V dvojito zaslepenej štúdii SP512 časť I dostávalo 177 pacientov rotigotín a 96 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu alebo placeba týždenným zvyšovaním
o 2 mg/24 h od počiatočnej dávky 2 mg/24 h po maximálnu dávku 6 mg/24 h. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 6 mesiacov.
Na konci udržiavacej liečby bola u 91 % pacientov v rotigotínovej vetve, optimálna dávka zhodná s
maximálnou povolenou dávkou, t. j. 6 mg/24 h. Zlepšenie o 20 % bolo pozorované u 48 % pacientov, ktorým sa podával rotigotín a u 19 % pacientov, ktorým sa podávalo placebo (rozdiel 29 %, CI95 %
18 %; 39 %, p< 0,0001). V prípade rotigotínu bolo priemerné zlepšenie vyjadrené v skóre UPDRS
(časti II+III) –3,98 bodov (počiatočná hodnota 29,9 bodov), zatiaľ čo u pacientov vo vetve
s placebom, bolo pozorované zhoršenie o 1,31 bodov (počiatočná hodnota 30,0 bodov). Rozdiel bol
5,28 bodov a bol štatisticky významný (p< 0,0001).
V dvojito zaslepenej štúdii SP513 časť I dostávalo 213 pacientov rotigotín, 227 pacientov ropinirol a 117 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 2 mg/24 h po maximálnu dávku 8 mg/24 h počas
4 týždňov. V skupine, v ktorej sa podával ropinirol, bola u pacientov titrovaná optimálna dávka až do maximálnej hodnoty 24 mg/deň počas 13 týždňov. U pacientov v každej liečebnej skupine bola dávka udržiavaná po dobu 6 mesiacov.
Na konci udržiavacej liečby bola u 92 % pacientov v rotigotínovej vetve optimálna dávka zhodná s
maximálnou povolenou dávkou, t. j. 8 mg/24 h. Zlepšenie o 20 % bolo pozorované u 52 % pacientov, ktorým sa podával rotigotín, u 68 % pacientov, ktorým sa podával ropinirol, a u 30 % pacientov, ktorým sa podávalo placebo. (Rozdiel medzi rotigotínom a placebom bol 21,7 %, CI95 % 11,1 %;
32,4 %, rozdiel medzi ropinirolom a placebom bol 38,4 %, CI95 % 28,1 %; 48,6 %, rozdiel medzi ropinirolom a rotigotínom bol 16,6 %, CI95 %, 7,6 %; 25,7 %). V rotigotínovej vetve sa dosiahlo priemerné zlepšenie skóre UPDRS (časti II+III) 6,83 bodov (počiatočná hodnota 33,2 bodov),
v ropinirolovej vetve 10,78 bodov (počiatočná hodnota 32,2 bodov) a vo vetve s placebom 2,33 bodov (počiatočná hodnota 31,3 bodov). Všetky rozdiely medzi aktívnymi liečbami a placebom boli štatisticky významné. Táto štúdia nepreukázala noninferioritu rotigotínu voči ropinirolu.
V následnej nezaslepenej štúdii (SP824), multicentrickej, medzinárodnej štúdii sa skúmala znášanlivosť náhlej zmeny z liečby ropinirolom, pramipexolom alebo kabergolínom na transdermálnu náplasť rotigotínu a jeho účinok na symptómy u osôb s idiopatickou Parkinsonovou chorobou. 116 pacientov prešlo z predchádzajúcej perorálnej liečby na liečbu až 8 mg/24 h rotigotínu, medzi nimi bolo 47 pacientov, ktorí boli liečení až 9 mg/deň ropinirolu, 47 pacientov, ktorí boli liečení až
2 mg/deň pramipexolu a 22 pacientov, ktorí boli liečení až 3 mg/deň kabergolínu. Prechod na rotigotín bolo uskutočniteľný s nevyhnutnou malou úpravou dávky (medián 2 mg/24 h) len u 2 pacientov, ktorí
prešli z liečby ropinirolom, u 5 pacientov, ktorí prešli z liečby pramipexolom a u 4 pacientov, ktorí
prešli z liečby kabergolínom. Pozorovali sa zlepšenia skóre UPDRS časti I – IV. Bezpečnostný profil bol porovnateľný s profilom v predchádzajúcich štúdiách.
V randomizovanej, nezaslepenej štúdii (SP825) u pacientov s včasných štádiom Parkinsonovej choroby bolo 25 pacientov randomizovaných na liečbu rotigotínom a 26 na ropinirol. V oboch liečebných skupinách bola dávka titrovaná na optimálnu alebo maximálnu dávku 8 mg/24 h alebo
9 mg/24 h, v uvedenom poradí. Obidve liečby preukázali zlepšenia skorej rannej motorickej funkcie
a spánku. Motorické symptómy (UPDRS časť III) sa zlepšili o 6,3 ± 1,3 bodov u pacientov liečených rotigotínom a o 5,9 ± 1,3 bodov v skupine s ropinirolom po 4 týždňoch udržiavacej liečby. Spánok
(PDSS) sa zlepšil o 4,1 ± 13,8 bodov u pacientov liečených rotigotínom a o 2,5 ± 13,5 bodov u
pacientov liečených ropinirolom. Bezpečnostný profil bol porovnateľný s výnimkou reakcií v mieste
aplikácie.
V štúdiách SP824 a SP825 uskutočnených od úvodného komparatívneho klinického skúšania sa preukázalo, že rotigotín a ropinirol v ekvivalentných dávkach majú porovnateľnú účinnosť.
Dve ďalšie pivotné štúdie (SP650DB a SP515) boli vykonané u pacientov súčasne liečených
L-dopou. Hlavným zisteným výsledkom bolo skrátenie trvania tzv. stavu „off“ (v hodinách). Účinnosť sa zisťovala na základe odozvy pacienta na liečbu, pričom sa merala ako zlepšenie stavu pacienta a absolútne zlepšenie v stave „off“.
V dvojito zaslepenej štúdii SP650DB dostávalo 113 pacientov rotigotín až po maximálnu dávku
8 mg/24 h, 109 pacientov dostávalo rotigotín až po maximálnu dávku 12 mg/24 h a 119 pacientov dostávalo placebo. U pacientov bola titrovaná optimálna dávka rotigotínu alebo placeba týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 4 mg/24 h. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 6 mesiacov. Na konci udržiavacej liečby sa pozorovalo zlepšenie o minimálne 30 % u 57 % a u 55 % pacientov dostávajúcich rotigotín 8 mg/24 h a 12 mg/24 h
v uvedenom poradí a u 34 % pacientov dostávajúcich placebo (rozdiely 22 % a 21 % v uvedenom poradí CI95% 10 %; 35 % a 8 %; 33 % v uvedenom poradí, p< 0,001 pre obe skupiny liečené rotigotínom). V prípade rotigotínu sa trvanie stavu „off“ skrátilo o priemerne“ 2,7 a 2,1 hodiny
v uvedenom poradí, zatiaľ čo u skupiny dostávajúcej placebo sa pozorovalo skrátenie o 0,9 hodiny.
Rozdiely boli štatisticky významné (p< 0,001 a p=0,003 v uvedenom poradí).
V dvojito zaslepenej štúdii SP515 dostávalo 201 pacientov rotigotín, 200 pacientov pramipexol a 100 pacientov placebo. U pacientov bola titrovaná optimálna dávka rotigotínu týždenným zvyšovaním o 2 mg/24 h od počiatočnej dávky 4 mg/24 h po maximálnu dávku 16 mg/24 h. V skupine dostávajúcej pramipexol sa pacientom podávala dávka 0,375 mg počas prvého týždňa, 0,75 mg počas druhého týždňa a bola u nich titrovaná optimálna dávka ďalším týždenným zvyšovaním o 0,75 mg až po maximálnu dávku 4,5 mg/deň. U pacientov v každej liečebnej skupine bola optimálna dávka udržiavaná po dobu 4 mesiacov.
Na konci udržiavacej liečby sa pozorovalo zlepšenie o minimálne 30 % u 60 % pacientov dostávajúcich rotigotín, 67 % pacientov dostávajúcich pramipexol a u 35 % pacientov dostávajúcich placebo (rozdiel medzi rotigotínom a placebom bol 25 %, CI95% 13 %; 36 %, rozdiel medzi pramipexolom a placebom bol 32 %, CI95% 21 %; 43 %, rozdiel medzi pramipexolom a rotigotínom bol 7 %, CI95% -2 %; 17 %). V skupine dostávajúcej rotigotín sa trvanie stavu „off“ skrátilo
o priemerne 2,5 hodiny, v skupine s pramipexolom to bolo 2,8 hodiny a v skupine s placebom
0,9 hodiny. Všetky rozdiely medzi aktívnymi liečbami a placebom boli štatisticky významné.
Ďalšia medzinárodná dvojito zaslepená štúdia (SP889) sa uskutočnila u 287 pacientov vo včasných alebo pokročilých štádiách Parkinsonovej choroby s nedostatočnou kontrolou skorých ranných motorických symptómov. 81,5 % týchto pacientov dostávalo súbežnú liečbu levodopou. 190 pacientov dostávalo rotigotín a 97 placebo. Pacienti boli titrovaní na optimálnu dávku rotigotínu alebo placeba
v týždňových prírastkoch o 2 mg/24h s počiatočnou dávkou 2 mg/24 h až po maximálnu dávku
16 mg/24 h po dobu 8 týždňov, po ktorom nasledovalo udržiavacie obdobie 4 týždňov. Skoré ranné motorické funkcie, hodnotené podľa časti III UPDRS a poruchy nočného spánku, hodnotené podľa upravenej škály spánku pri Parkinsonovej chorobe (Parkinson’s Disease Sleep Scale - PDSS-2), boli ko-primárnymi výslednými meraniami. Na konci udržiavacieho obdobia sa priemerné skóre podľa časti III UPDRS zlepšilo o 7,0 bodov u pacientov liečených rotigotínom (východisková hodnota 29,6) a o 3,9 bodov v placebovej skupine (východisková hodnota 32,0). Zlepšenia v priemernom celkovom skóre PDSS-2 boli 5,9 (rotigotín, východisková hodnota 19,3) a 1,9 bodov (placebo, východisková hodnota 20,5). Liečebné rozdiely v koprimárnych premenných boli štatisticky významné (p=0,0002 a p<0,0001).
Priľnavosť na kožu
V multicentrickej, dvojito zaslepenej, randomizovanej, dvojito skríženej štúdii bola u 52 ambulantných pacientov porovnávaná priľnavosť na kožu rotigotín náplasti 8 mg/24 h, s vylepšeným uchovávaním pri izbovej teplote, s náplasťou s uchovávaním v chlade. Priľnavosť na kožu bola meraná počas 2 po sebe nasledujúcich dní pri 24-hodinovej aplikácii náplasti.
Vylepšená náplasť s uchovávaním pri izbovej teplote preukázala lepšiu priľnavosť na kožu ako náplasť s uchovávaním v chlade, > 90 % náplastí preukazuje dostatočnú priľnavosť (t.j. > 70 %
priliehajúcej plochy náplasti) v porovnaní s <83 %. Pri obidvoch formách bola hlásená porovnateľná
znášanlivosť kože. Väčšina pozorovaných erytémov bolo miernych a žiadny nebol závažný.
5.2 Farmakokinetické vlastnosti
Absorpcia
Rotigotín sa po aplikácii plynule uvoľňuje z transdermálnej náplasti a je absorbovaný cez pokožku.
Ustálené koncentrácie sa dosiahnu za jeden až dva dni po aplikácii náplasti a udržiavajú sa na stabilnej
úrovni aplikáciou náplasti jedenkrát denne, pričom táto sa nosí po dobu 24 hodín. Koncentrácie
rotigotínu v plazme sa zvyšujú v závislosti od dávky v rozsahu dávok od 1 mg/24 h po 24 mg/24 h.
Za 24 hodín sa uvoľní do pokožky približne 45 % liečiva v náplasti. Absolútna biologická dostupnosť
po transdermálnej aplikácii je približne 37 %.
Striedanie miesta aplikácie náplasti môže spôsobiť rozdiely medzi jednotlivými dennými hladinami
v plazme. Rozdiely v biologickej dostupnosti rotigotínu siahali od 2 % (nadlaktie v porovnaní
s bokom) po 46 % (rameno v porovnaní so stehnom). Neexistuje žiaden náznak relevantného vplyvu na klinické výsledky.
Distribúcia
Približne 92 % rotigotínu sa viaže in vitro na plazmatické proteíny.
Zdanlivý distribučný objem u ľudí je približne 84 l/kg.
Biotransformácia
Rotigotín je z veľkej časti metabolizovaný. Rotigotín je metabolizovaný N-dealkyláciou ako aj priamou a sekundárnou konjugáciou. Výsledky vykonávané in vitro naznačujú, že rôzne izoformy CYP dokážu katalyzovať N-dealkyláciu rotigotínu. Hlavnými metabolitmi sú sulfáty a glukuronidové konjugáty pôvodnej látky ako aj metabolity N-dealkylácie, ktoré sú biologicky neaktívne.
Informácie o metabolitoch sú neúplné.
Eliminácia
Približne 71 % dávky rotigotínu sa vylučuje močom a menšia časť približne 23 % sa vylučuje stolicou.
Klírens rotigotínu po transdermálnom podaní je približne 10 l/minútu a jeho celkový polčas eliminácie je 5 až 7 hodín. Farmakokinetický profil preukazuje bifázickú elimináciu s počiatočným polčasom približne 2 až 3 hodiny.
Keďže náplasť sa aplikuje transdermálne, neočakáva sa žiadny vplyv jedál alebo ochorení
gastrointestinálneho traktu.
Osobitné skupiny pacientov
Keďže liečba Neuprom začína nízkou dávkou a táto sa postupne titruje podľa klinickej znášanlivosti,
aby sa získal optimálny terapeutický účinok, úprava dávky na základe pohlavia, hmotnosti alebo veku
nie je potrebná.
Porucha funkcie pečene a obličiek
U pacientov so stredne závažnou poruchou funkcie pečene alebo miernou až závažnou poruchou funkcie obličiek neboli pozorované žiadne relevantné zvýšenia hladiny rotigotínu v plazme. Neupro nebolo skúmané u pacientov so závažnou poruchou funkcie pečene.
V prípade poruchy funkcie obličiek sa zvyšujú hladiny konjugátov rotigotínu a metabolitov N- dealkylácie rotigotínu v plazme. Vplyv týchto metabolitov na klinické účinky je však
nepravdepodobný.
5.3 Predklinické údaje o bezpečnosti
V štúdiách zameraných na opakované dávky a dlhodobú toxicitu boli hlavné účinky spájané s farmakodynamickými účinkami súvisiacimi s agonistom dopamínu a následným znížením vylučovania prolaktínu.
Po jednorazovej dávke rotigotínu bolo u pigmentovaných potkanov a opíc evidentné viazanie sa rotigotínu na tkanivo obsahujúce melanín (t. j. oči), ktoré však počas 14-dňového pozorovacieho intervalu vymizlo.
V trojmesačnej štúdii bola u bielych potkanov pozorovaná pomocou transmisnej elektrónovej mikroskopie degenerácia sietnice pri dávke ekvivalentnej 2,8-násobku maximálnej odporúčanej dávky pre ľudí (prepočítanej na mg/m2). Účinky boli výraznejšie u samíc potkanov. Dodatočné štúdie na ďalšie vyhodnotenie špecifickej patológie neboli vykonané. Degenerácia sietnice sa nepozorovala počas rutinného histopatologického vyhodnocovania očí skúmaných druhov zvierat v žiadnej
z toxikologických štúdií. Význam týchto zistení pre ľudí nie je známy.
V štúdii karcinogenicity sa u samcov potkanov rozvinuli nádory a hyperplázia Leydigových buniek. Zhubné nádory boli pozorované najmä v maternici samíc, ktorým sa podávali stredné až vysoké
dávky. Tieto zmeny sú dobre známymi účinkami agonistov dopamínu u potkanov po celoživotnej liečbe a boli zhodnotené ako nerelevantné pre ľudí.
Účinky rotigotínu na reprodukciu sa skúmali u potkanov, králikov a myší. Na žiadny z uvedených troch druhov nemal rotigotín teratogénne účinky, ale u potkanov a myší vykazoval embryotoxické
účinky pri dávkach toxických pre matky. Rotigotín neovplyvnil plodnosť samcov u potkanov, avšak jednoznačne znížil plodnosť samíc potkanov a myší z dôvodu účinkov na hladiny prolaktínu, ktoré sú
u hlodavcov obzvlášť významné.
Rotigotín nevyvolal génové mutácie v Amesovom teste, preukázal však účinky v in vitro teste lymfómu u myší s metabolickou aktiváciou a slabšie účinky bez metabolickej aktivácie. Mutagénny účinok je možné pripísať klastogénnemu účinku rotigotínu. Tento účinok nebol potvrdený in vivo
v mikronukleovom teste u myší a v teste na neplánovanú syntézu DNA (UDS, Unsheduled DNA Synthesis) u potkanov. Keďže prebiehal viac-menej súbežne so zníženým relatívnym celkovým rastom buniek, môže súvisieť s cytotoxickým účinkom látky. Významnosť tohto jedného pozitívneho testu mutagenicity in vitro preto nie je známa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Krycia vrstva
Polyesterový film s vrstvou silikónu a hliníka
farebný povlak pigmentovej vrstvy (oxid titaničitý (E171), žltý pigment 95, červený pigment 166)
a potlač (červený pigment 144, žltý pigment 95, čierny pigment 7).
Samolepiaca matricová vrstva
Poly(dimetylsiloxán, trimetylsilylsilikát)-kopolymerizát,
Povidón K90,
Metasiričitan sodný (E223), Askorbylpalmitát (E304) a DL-α-tokoferol (E307).
Snímateľná fólia
Priesvitný polyesterový film s fluoropolymérovým povlakom.
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
30 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C.
6.5 Druh obalu a obsah balenia
Rozlepovacie vrecko v lepenkovej škatuli: Jednu stranu tvorí kopolymér etylénu (najvnútornejšia vrstva), hliníková fólia, polyetylénový film s nízkou hustotou a papier; druhú stranu tvorí polyetylén (najvnútornejšia vrstva), hliník, kopolymér etylénu a papier.
Súprava na zahájenie liečby obsahuje 28 transdermálnych náplastí, ktoré sú rozdelené v 4 škatuľkách po 7 náplastiach s obsahom liečiva 2 mg, 4 mg, 6 mg a 8 mg a jednotlivo zatavené vo vreckách.
6.6 Špeciálne opatrenia na likvidáciu
Náplasť po použití ešte stále obsahuje liečivo. Po odstránení sa má použitá náplasť preložiť napoly lepiacou stranou smerom dovnútra tak, aby matricová vrstva nezostala odkrytá, umiestniť do
pôvodného vrecka a odložiť. Všetky použité aj nepoužité náplasti majú byť zlikvidované v súlade
s národnými požiadavkami alebo vrátené do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIUCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLAEU/1/05/331/013
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15. február 2006
Dátum posledného predĺženia registrácie: 22. január 2016
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.