
ch tumorov (GEP-NET) stupňa 1 a podskupiny stupňa 2 (index proliferácie Ki67 do 10 %) stredného čreva („midgut“), tumorov pankreasu alebo tumorov s neznámou lokalizáciou, u ktorých bol vylúčený pôvod v zadnom čreve („hindgut“), u dospelých pacientov s neresekovateľným lokálne pokročilým alebo metastatickým ochorením
Odporúčaná dávka je 1 injekcia Mytolente 120 mg podávaná každých 28 dní. Liečba liekom Mytolente má pokračovať tak dlho, ako je to potrebné na kontrolu nádoru.
Liečba príznakov spojených s neuroendokrinnými tumormi
Odporúčaná začiatočná dávka je 60 až 120 mg podávaná každých 28 dní.
Dávka sa má upraviť podľa toho, do akej miery sa dosiahne úľava od príznakov.
Porucha funkcie obličiek a/alebo pečene
U pacientov s poruchou funkcie obličiek alebo pečene nie je potrebná žiadna úprava dávkovania z dôvodu širokého terapeutického okna lanreotidu (pozri časť 5.2).
Starší pacienti
U starších pacientov nie je potrebná žiadna úprava dávkovania z dôvodu širokého terapeutického okna
lanreotidu (pozri časť 5.2).
Pediatrická populácia
Mytolente sa neodporúča používať u detí a dospievajúcich vzhľadom na chýbajúce údaje o bezpečnosti a účinnosti.
Spôsob podávania
Mytolente sa podáva hlbokou subkutánnou injekciou do horného vonkajšieho kvadrantu sedacieho svalu
alebo do hornej vonkajšej časti stehna.
U pacientov, ktorí dostávajú stabilnú dávku Mytolente, môže byť liek po primeranom zaškolení podávaný buď samotným pacientom, alebo zaškolenou osobou. V prípade samopodania injekcie sa má injekcia aplikovať do hornej vonkajšej časti stehna.
Rozhodnutie o podávaní samotným pacientom alebo zaškolenou osobou má urobiť zdravotnícky pracovník. Bez ohľadu na miesto vpichu injekcie sa na koži nemá vytvárať kožná riasa a ihla sa má vpichnúť rýchlo
celou svojou dĺžkou, kolmo do kože.
Miesto vpichu sa má striedať medzi pravou a ľavou stranou.
4.3 Kontraindikácie
Precitlivenosť na lanreotid, somatostatín alebo príbuzné peptidy alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1
4.4 Osobitné upozornenia a opatrenia pri používaní
Lanreotid môže znížiť motilitu žlčníka a viesť k tvorbe žlčových kameňov. Preto je potrebné pacientov pravidelne sledovať. U pacientov užívajúcich lanreotid boli po uvedení na trh hlásené komplikácie spôsobené žlčovými kameňmi, vrátane cholecystitídy, cholangitídy a pankreatitídy, vyžadujúce cholecystektómiu. Ak máte podozrenie na komplikácie cholelitiázy, liečbu lanreotidom ukončite a začnite s príslušnou liečbou komplikácie.
Farmakologické štúdie u zvierat a u ľudí dokazujú, že lanreotid, rovnako ako somatostatín a jeho analógy, inhibuje sekréciu inzulínu a glukagónu. Z tohto dôvodu sa môže u pacientov liečených lanreotidom objaviť hypoglykémia alebo hyperglykémia. Na začiatku liečby lanreotidom alebo pri úprave dávky je potrebné kontrolovať hladiny glukózy v krvi a podľa toho upraviť antidiabetickú liečbu.
Počas liečby lanreotidom sa u pacientov s akromegáliou pozorovalo mierne zníženie funkcie štítnej žľazy, hoci klinická hypotyreóza je zriedkavá (<1 %). Tam, kde je to klinicky indikované, sa má vykonať vyšetrenie funkcie štítnej žľazy.
U pacientov bez prítomných srdcových problémov môže lanreotid viesť k zníženiu srdcového rytmu, pričom nemusí dosiahnuť hranicu bradykardie. U pacientov, ktorí pred liečbou lanreotidom mali srdcové poruchy, sa môže objaviť sínusová bradykardia. Na začiatku liečby lanreotidom u pacientov s bradykardiou je potrebná opatrnosť (pozri časť 4.5).
4.5 Liekové a iné interakcie
Farmakologické gastrointestinálne účinky lanreotidu môžu spôsobiť zníženie intestinálnej absorpcie súbežne podávaných liečiv vrátane cyklosporínu. Súčasné podanie cyklosporínu s lanreotidom môže znížiť relatívnu biologickú dostupnosť cyklosporínu, preto môže byť nevyhnutné dávku cyklosporínu upraviť a udržovať jeho terapeutické hladiny.
Interakcie s liekmi s vysokou väzbou na plazmatické bielkoviny sú nepravdepodobné vzhľadom na miernu
väzbu lanreotidu na sérové proteíny.
Obmedzené publikované údaje naznačujú, že súbežné podávanie analógov somatostatínu a bromokryptínu môže zvyšovať dostupnosť bromokryptínu.
Súbežné podávanie liečiv indukujúcich bradykardiu (napr. betablokátory) môže mať aditívny účinok na mierne zníženie srdcového rytmu v súvislosti s lanreotidom. Môže byť nevyhnutné upraviť dávky takýchto súčasne podávaných liekov.
Obmedzené publikované údaje naznačujú, že analógy somatostatínu môžu znižovať metabolický klírens zlúčenín, o ktorých je známe, že sú metabolizované enzýmami cytochrómu P450, čo môže byť dôsledkom supresie rastového hormónu. Pretože nemožno vylúčiť, že lanreotid môže mať takýto účinok, iné liečivá, predovšetkým tie, ktoré sú metabolizované CYP3A4 a ktoré majú úzky terapeutický index (napr. chinidín, terfenadín), sa majú používať opatrne.
4.6 Fertilita, gravidita a laktáciaGraviditaV štúdiách na zvieratách sa nepreukázali teratogénne účinky v súvislosti s lanreotidom počas organogenézy.
Údaje o obmedzenom počte gravidných žien vystavených lanreotidu nepreukázali žiadne nežiaduce účinky na graviditu alebo na zdravie plodu/novorodenca. V súčasnosti nie sú dostupné žiadne ďalšie relevantné epidemiologické údaje.
Pretože zo štúdií u zvierat sa nedá vždy predpovedať, aká bude odpoveď u ľudí, lanreotid sa má podávať gravidným ženám len v prípade, keď to je jednoznačne potrebné.
DojčenieNie je známe, či sa toto liečivo vylučuje do materského mlieka u ľudí.
Keďže veľa liečiv sa vylučuje do materského mlieka u ľudí, pri podávaní lanreotidu počas laktácie je
potrebná opatrnosť.
FertilitaU samíc potkanov sa pozorovala znížená fertilita z dôvodu inhibície sekrécie GH pri dávkach oveľa vyšších,
ako sa dosahujú u ľudí pri terapeutických dávkach.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeMytolente má malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Nevykonali sa
žiadne štúdie účinku tohto lieku na schopnosť viesť vozidlá a obsluhovať stroje.
V súvislosti s liekom Mytolente bol však zaznamenaný závrat (pozri časť 4.8). Ak sa u pacienta objaví závrat, nemá viesť motorové vozidlo ani obsluhovať stroje.
4.8 Nežiaduce účinkyNežiaduce účinky hlásené u pacientov s akromegáliou a GEP-NET liečených lanreotidom v klinických štúdiách sú vymenované pod príslušnou triedou orgánových systémov podľa nasledujúcej klasifikácie:
veľmi časté (
>1/10); časté (
>1/100 až <1/10); menej časté (
>1/1 000 až <1/100).
Najčastejšie očakávané nežiaduce reakcie na liek po liečbe lanreotidom sú gastrointestinálne poruchy
(najčastejšie sú hlásené hnačka a bolesti brucha, zvyčajne mierne alebo stredne závažné a prechodné), cholelitiáza (často asymptomatická) a reakcie v mieste podania injekcie (bolesť, uzliny a indurácie).
Profil nežiaducich účinkov je pri všetkých indikáciách podobný.
Trieda orgánového systému
| Veľmi časté
(≥1/10)
| Časté (≥1/100 až
<1/10)
| Menej časté
(≥1/1 000 až
<1/100)
| Informácie
o bezpečnosti po uvedení na
trh (frekvencia
nie je známa)
|
Infekcie a nákazy
|
|
|
| Absces v mieste
podania injekcie
|
P
oruchy metabolizmu a
výživy
|
|
Hypoglykémia,
znížená chuť do
jedla**, hyperglykémia, diabetes mellitus
|
|
|
P
sychické poruchy
|
|
|
Insomnia*
|
|
P
oruchy nervového systému
|
|
Závrat, bolesť
hlavy, letargia**
|
|
|
P
oruchy srdca
a srdcovej činnosti
|
|
Sínusová
bradykardia*
|
|
|
P
oruchy ciev
|
|
|
Návaly horúčavy*
|
|
P
oruchy
gastrointestinálneho
t
raktu
|
Hnačka,
riedka stolica*, bolesť brucha
|
Nauzea, vracanie,
zápcha, flatulencia, brušná distenzia, nepríjemné pocity
v bruchu, dyspepsia,
steatorea**
|
Zmena sfarbenia
stolice*
|
Pankreatitída
|
P
oruchy pečene
a žlčových ciest
|
Cholelitiáza
|
Dilatácia žlčníka*
|
|
Cholecystitída,
cholangitída
|
P
oruchy kostrovej
a svalovej sústavy a spojivového tkaniva
|
|
Muskuloskeletálna
bolesť**,
myalgia**
|
|
|
P
oruchy kože
a podkožného
t
kaniva
|
|
Alopécia,
hypotrichóza*
|
|
|
C
elkové poruchy a reakcie v mieste podania
|
|
Asténia,
vyčerpanosť,
reakcie v mieste podania injekcie
(bolesť, opuch,
indurácia, uzlík,
pruritus)
|
|
|
L
aboratórne a funkčné vyšetrenia
|
|
Zvýšené ALT*,
abnormálne AST*,
abnormálne ALT*, zvýšená hladina
bilirubínu v krvi*,
zvýšená hladina glukózy v krvi*, zvýšená hladina glykozylovaného hemoglobínu*, úbytok telesnej hmotnosti, znížená hladina pankreatických enzýmov**
|
Zvýšené AST*,
zvýšená hladina alkalickej fosfatázy
v krvi*,
abnormálna hladina bilirubínu
v krvi*, znížená
hladina sodíka
v krvi*
|
|
P
oruchy
im
unitného systému
|
|
|
|
Alergické
reakcie (vrátane angioedému,
anafylaxie,
hypersenzitivity)
|
* založené na súbore štúdií uskutočnených s pacientmi s akromegáliou
** založené na súbore štúdií uskutočnených s pacientmi s GEP-NET
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek
podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieAk dôjde k predávkovaniu, je indikovaná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Hormóny hypofýzy a hypotalamu a analógy, somatostatín a analógy,
ATC kód: H01CB03.
Lanreotid je oktapeptidový analóg prirodzeného somatostatínu. Rovnako ako somatostatín, lanreotid je inhibítorom rôznych endokrinných, neuroendokrinných, exokrinných a parakrinných funkcií. Lanreotid má vysokú afinitu k ľudským receptorom pre somatostatín (SSTR) 2 a 5 a redukovanú väzobnú afinitu
k ľudským SSTR 1, 3 a 4. Zdá sa, že hlavným mechanizmom zodpovedným za inhibíciu GH je účinok na ľudské SSTR 2 a 5. Lanreotid je účinnejší ako prirodzený somatostatín a vykazuje dlhšie trvanie účinku.
Lanreotid, rovnako ako somatostatín, preukazuje všeobecný exokrinný antisekretorický účinok. Inhibuje bazálnu sekréciu motilínu, žalúdočného inhibičného peptidu a pankreatického polypeptidu, ale nemá signifikantný účinok na sekretín na lačno alebo na sekréciu gastrínu. Okrem toho, znižuje hladinu plazmatického chromogranínu A a kyseliny 5-HIAA v moči (5-hydroxyindoloctová kyselina) u pacientov
s GEP-NET a zvýšené hladiny týchto nádorových markerov. Lanreotid výrazne inhibuje jedlom indukovaný zvýšený prietok krvi v arteria mesenterica superior a vo vena portae. Lanreotid významne znižuje
prostaglandínom E1 stimulovanú jejunálnu sekréciu vody, sodíka, draslíka a chloridov. Lanreotid znižuje
hladiny prolaktínu u dlhodobo liečených pacientov s akromegáliou.
V otvorenej štúdii sa lanreotid 120 mg podával každých 28 dní počas 48 týždňov u 90 predtým neliečených pacientov s akromegáliou s diagnózou makroadenómu hypofýzy. Pacienti, u ktorých sa očakávalo, že budú počas štúdie potrebovať operáciu hypofýzy alebo rádioterapiu, boli vylúčení.
U 63 % pacientov (95 % IS: 52 %-73 %) bolo pozorované zníženie objemu tumoru o viac ako 20 %. V 48. týždni bolo priemerné percentuálne zníženie objemu tumoru 26,8 %, hladiny GH pod 2,5 μg/l boli u 77,8 % pacientov a hladiny IGF-1 sa normalizovali u 50 % pacientov. Normalizované IGF-1 hladiny a zároveň hladiny GH pod 2,5 μg/l boli pozorované u 43,5 % pacientov. Väčšina pacientov hlásila zreteľnú úľavu od príznakov akromegálie, ako sú únava, nadmerné potenie, bolesť kĺbov a opuch mäkkých tkanív. Včasné
a pretrvávajúce zníženie objemu tumoru, ako aj hladín GH a IGF-1 bolo pozorované od 12. týždňa.
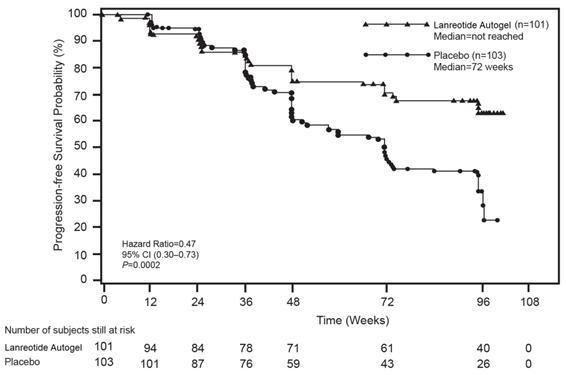
Bola uskutočnená randomizovaná, dvojito zaslepená, multicentrická, placebom kontrolovaná klinická štúdia fázy III s fixným časom trvania 96 týždňov s lanreotidom u pacientov s gastroenteropankreatickými neuroendokrinnými tumormi s cieľom posúdiť antiproliferačný účinok lanreotidu.
Pacienti boli randomizovaní v pomere 1:1 na podanie buď lanreotidu 120 mg každých 28 dní (n=101), alebo placeba (n=103). Randomizácia bola stratifikovaná na základe prítomnosti alebo neprítomnosti predchádzajúcej liečby a na základe prítomnosti alebo neprítomnosti progresie ochorenia pri vstupe, hodnotenej podľa kritérií RECIST 1,0 (Response Evaluation Criteria in Solid Tumours) počas 3 až 6 mesačnej skríningovej fázy.
Pacienti mali metastatické a/alebo lokálne pokročilé inoperabilné ochorenie s histologicky potvrdenými dobre alebo stredne diferencovanými tumormi primárne lokalizovanými v pankrease (44,6 % pacientov), strednom čreve – „midgut“ (35,8 %), zadnom čreve –„hindgut“ (6,9 %) alebo v inej/neznámej primárnej lokalizácii (12,7 %).
69 % pacientov s GEP-NET mali tumor 1. stupňa (G1), definovaný buď indexom proliferácie Ki67 ≤ 2 % (50,5 % z celkového počtu pacientov) alebo mitotickým indexom < 2 mitózy / 10 HPF (18,5 % z celkového počtu pacientov) a 30 % pacientov s GEP-NET mali tumor 2. stupňa v dolnom rozsahu (G2) (definovaný indexom proliferácie Ki67 > 2 % - ≤ 10 %). Stupeň tumoru nebol dostupný pri 1 % pacientov. Štúdia vylúčila pacientov s G2 GEP-NET s vyšším indexom bunkovej proliferácie (Ki67 >10 % - ≤20 %) a s G3
GEP – neuroendokrinnými karcinómami (index proliferácie Ki67 > 20 %).
Celkovo 52,5 % pacientov malo hepatálnu tumoróznu nálož ≤ 10 %, 14,5 % malo hepatálnu tumoróznu nálož > 10 a ≤ 25 % a 33 % malo hepatálnu tumoróznu nálož > 25 %.
Primárnym ukazovateľom bolo prežitie bez progresie (progression-free survival – PFS) merané buď ako čas do progresie ochorenia pomocou RECIST 1,0, alebo ako úmrtie v priebehu 96 týždňov po prvom podaní liečby. Na analýzu PFS bolo využité nezávislé centrálne hodnotené rádiologické posúdenie progresie.
Tabuľka 1: Výsledky účinnosti z klinickej štúdie fázy III
Medián času prežitia bez progresie
(týždne)
|
Pomer rizika
(95% IS)
|
Redukcia rizika progresie alebo úmrtia
|
p-hodnota
|
Lanreotid
(n = 101 )
| Placebo
(n =103 )
|
|
> 96 týždňov
| 72,00 týždňov
(95% IS: 48,57, 96,00)
| 0,470 (0,304,
0,729)
| 53 %
| 0,0002
|
Obr. 1 : Krivky prežitia podľa Kaplan-Meiera
'
Prospešný účinok lanreotidu v znížení rizika progresie ochorenia alebo úmrtia bol jednotný bez ohľadu na lokalizáciu primárneho tumoru, hepatálnu tumoróznu nálož, predchádzajúcu chemoterapiu, počiatočný Ki67, stupeň tumoru alebo ďalšie vopred špecifikované charakteristiky uvedené na obrázku 2.
Klinicky významný prínos liečby lanreotidom bol pozorovaný u pacientov s tumormi pankreasu, midgut
a iného/neznámeho pôvodu v celej sledovanej populácii. Obmedzený počet pacientov s hind-gut tumormi (14/204) spôsobil sťaženú interpretáciu výsledkov tejto podskupiny. Dostupné dáta naznačujú, že lanreotid u týchto pacientov nie je prínosný.
Obr. 2 – Výsledky analýzy z Coxovho modelu proporcionálnych rizík PFS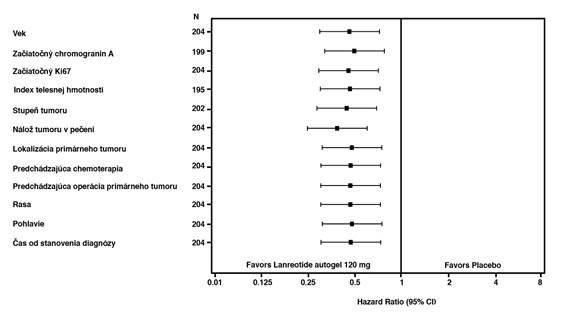
Poznámka: Všetky pomery rizík sú pomery rizík pre lanreotid autogel versus placebo. Výsledky premenných sú odvodené zo samostatných CoX PH modelov s podmienkami pre liečbu, progresiu pri úvodnom vyšetrení, predchádzajúcu liečbu pri vstupe a podmienkami uvedenými na zvislej osi.
Prechod z placeba na otvorené podávanie lanreotidu, v predĺženej štúdii, nastal u 45,6 % (47/103) pacientov.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s referenčným liekom
obsahujúcim lanreotid vo všetkých podskupinách pediatrickej populácie pre akromegálie a hypofyzárny gigantizmus (pozri časť 4.2 – informácie pre pediatrické použitie). Európska agentúra pre lieky zaradila gastroenteropankreatické neuroendokrinné tumory (okrem neuroblastómu, neuroganglioblastómu, feochromocytómu) do zoznamu výnimiek.
5.2 Farmakokinetické vlastnosti
Vlastné farmakokinetické parametre lanreotidu po intravenóznej aplikácii u zdravých dobrovoľníkov ukázali limitovanú extravaskulárnu distribúciu, s ustáleným distribučným objemom 16,1 l. Celkový klírens bol 23,7 l/h, terminálny biologický polčas bol 1,14 hodiny a priemerný čas pretrvávania bol 0,68 hodiny.
V štúdiách hodnotiacich vylučovanie sa menej ako 5 % lanreotidu vylúčilo v moči a menej ako 0,5 %
v nezmenenej forme v stolici, čo svedčí o malom vylučovaní žlčou.
Po hlbokom subkutánnom podaní lanreotidu 60, 90 a 120 mg u zdravých dobrovoľníkov dosiahli koncentrácie lanreotidu priemerné maximálne sérové koncentrácie 4,25; 8,39 a 6,79 ng/ml. Tieto hodnoty Cmax sa dosahujú v priebehu prvého dňa 8, 12 a 7 hodín (stredné hodnoty) po podaní. Koncentrácie lanreotidu klesajú pomaly z maximálnych sérových hladín kinetikou prvého rádu s terminálnym eliminačným polčasom 23,3; 27,4 a 30,1 dní, v uvedenom poradí. 4 týždne po podaní boli priemerné sérové hladiny lanreotidu 0,9; 1,11 a 1,69 ng/ml, v uvedenom poradí. Absolútna biologická dostupnosť bola 73,4;
69,0 a 78,4 %, v uvedenom poradí.
Po hlbokom subkutánnom podaní lanreotidu 60, 90 a 120 mg pacientom s akromegáliou dosiahli koncentrácie lanreotidu priemerné maximálne sérové koncentrácie 1,6; 3,5 a 3,1 ng/ml, v uvedenom poradí. Tieto hodnoty Cmax sa dosahujú v priebehu prvého dňa 6, 6 a 24 hodín po podaní. Koncentrácie lanreotidu pomaly klesajú z maximálnych sérových hladín kinetikou prvého rádu a 4 týždne po podaní boli priemerné sérové hladiny lanreotidu 0,7; 1,0 a 1,4 ng/ml, v uvedenom poradí.
Sérové hladiny lanreotidu v rovnovážnom stave sa dosiahli v priemere po 4 injekciách každé 4 týždne. Po opakovanom podaní dávky každé 4 týždne boli priemerné hodnoty Cmax v rovnovážnom stave 3,8; 5,7
a 7,7 ng/ml pre 60, 90 a 120 mg, v uvedenom poradí. Priemerné hodnoty Cmin boli 1,8; 2,5 a 3,8 ng/ml. Index
kolísania maximálnej a minimálnej hodnoty bol mierny a pohyboval sa v rozmedzí od 81 do 108 %.
U pacientov s akromegáliou sa pozorovali po hlbokom subkutánnom podaní lanreotidu 60, 90 a120 mg lineárne farmakokinetické profily uvoľňovania.
Najnižšie sérové hladiny lanreotidu dosiahnuté po troch hlbokých subkutánnych injekciách lanreotidu 60, 90
alebo 120 mg podaných každých 28 dní boli podobné ako najnižšie sérové hladiny lanreotidu
v rovnovážnom stave u pacientov s akromegáliou, ktorí boli predtým liečení intramuskulárnym podaním
lanreotidu 30 mg vo forme mikročastíc s predĺženým uvoľňovaním každých 14, 10 alebo 7 dní, v uvedenom
poradí.
V populačnej farmakokinetickej analýze s 290 GEP-NET pacientmi liečenými lanreotidom 120 mg bolo
pozorované rýchle uvoľnenie liečiva s priemernými hodnotami Cmax 7,49 ± 7,58 ng/ml, dosiahnutými
v priebehu prvého dňa po jednej injekcii. Koncentrácie v rovnovážnom stave boli dosiahnuté po 5 injekciách
lanreotidu 120 mg podávaných každých 28 dní a udržali sa do posledného hodnotenia (až 96 týždňov po
prvej injekcii). V rovnovážnom stave boli priemerné hodnoty Cmax 13,9 ± 7,44 ng/ml a priemerné minimálne sérové hladiny 6,56 ± 1,99 ng/ml. Zdanlivý terminálny polčas eliminácie bol 49,8 ± 28,0 dní.
Poruchafunkcieobličiek/pečene
U osôb so závažnou poruchou funkcie obličiek klesol celkový sérový klírens lanreotidu približne dvojnásobne, s následným zvýšením polčasu a AUC. U osôb so stredne závažnou až závažnou poruchou funkcie pečene sa pozorovalo zníženie klírensu (30 %). Distribučný objem a stredný čas pretrvávania sa zvýšil u osôb so všetkými stupňami insuficiencie pečene.
Nebol pozorovaný žiaden vplyv na klírens lanreotidu pri populačnej farmakokinetickej analýze GEP-NET pacientov, vrátane 165 pacientov s mierne a stredne závažnou poruchou funkcie obličiek (v poradí 106 a 59) liečených lanreotidom. GEP-NET pacienti so závažným poškodením funkcie obličiek neboli skúmaní.
GEP-NET pacienti s poškodením pečene (podľa Child-Pugh skóre) neboli skúmaní.
Nie je potrebné upravovať počiatočnú dávku u pacientov s poruchou funkcie obličiek alebo pečene, pretože sérové koncentrácie lanreotidu u týchto populácií sú pravdepodobne v rámci rozmedzia sérových koncentrácií, ktoré sú bezpečne tolerované u zdravých jedincov.
Staršípacienti
U starších pacientov sa preukázalo zvýšenie biologického polčasu a priemerného času pretrvávania
v porovnaní so zdravými mladými jedincami. Nie je potrebné upravovať počiatočnú dávku u starších pacientov, pretože sérové koncentrácie lanreotidu v tejto populácii sú pravdepodobne v rámci rozmedzia sérových koncentrácií, ktoré sú bezpečne tolerované u zdravých jedincov.
V populačnej farmakokinetickej analýze GEP-NET pacientov, vrátane 122 vo veku 65 až 85 rokov, nebol pozorovaný žiaden vplyv veku na klírens a distribučný objem lanreotidu.
5.3 Predklinické údaje o bezpečnosti
Účinky v predklinických štúdiách sa pozorovali iba pri expozíciách považovaných za dostatočne vyššie, ako je maximálna expozícia u ľudí, čo poukazuje na malý význam týchto zistení pre klinické použitie.
V štúdiách s biologickými testmi na karcinogenitu uskutočnených u potkanov a myší sa nepozorovali žiadne systémové neoplastické zmeny pri dávkach oveľa vyšších, ako sú terapeutické dávky u ľudí. Zvýšená incidencia subkutánnych tumorov sa pozorovala v mieste injekcie pravdepodobne z dôvodu zvýšenej frekvencie podávania dávky u zvierat (denne) v porovnaní s dávkovaním raz mesačne u ľudí, a preto nemusí byť klinicky významná.
V sérii štandardných in vitro a in vivo testov lanreotid nepreukázal žiadny genotoxický potenciál.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Voda na injekcie
Ľadová kyselina octová (na úpravu pH)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Po otvorení ochranného hliníkového vrecka sa má liek podať okamžite.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C) v pôvodnom obale na ochranu pred svetlom.
Ak bol liek v hermeticky uzavretom vrecku vystavený maximálnej teplote 40 °C spolu najviac 24 hodín, môže sa vrátiť do chladničky (počet teplotných výkyvov nesmie byť viac ako trikrát) na ďalšie uchovávanie a neskoršie použitie.
6.5 Druh obalu a obsah balenia
Mytolente je dodávaný v naplnenej injekčnej striekačke (z polypropylénu s gumenou piestovou zátkou
z termoplastického elastoméru s polypropylénovým bezpečnostným uzáverom) umiestnenej v plastovej podložke v hermeticky uzatvorenom hliníkovom vrecku a so samostatne zabaleným automatickým bezpečnostným systémom na jednorazové ihly. Oboje je zabalené v papierovej škatuľke.
Škatuľka s jednou 0,5 ml injekčnou striekačkou a pribalenou bezpečnostnou ihlou (1,2 mm x 20 mm). Multibalenie s 3 škatuľkami, z ktorých každá obsahuje jednu 0,5 ml injekčnú striekačku s pribalenou
bezpečnostnou ihlou (1,2 mm x 20 mm).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Injekčný roztok v naplnenej injekčnej striekačke je pripravený na použitie.
Po prvom otvorení je na okamžité jednorazové použitie. Nepoužívajte, ak je vrecko poškodené alebo otvorené.
Je dôležité liek aplikovať presne podľa pokynov v písomnej informácii pre používateľa.
Použitá injekčná pomôcka sa musí zlikvidovať do nádoby určenej na ostré predmety.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Amdipharm Limited
3 Burlington Road
Dublin 4
Írsko
Amdipharm Limited je súčasťou skupiny ADVANZ PHARMA.
8. REGISTRAČNÉ ČÍSLA
Mytolente 60 mg injekčný roztok v naplnenej injekčnej striekačke: 56/0107/21-S Mytolente 90 mg injekčný roztok v naplnenej injekčnej striekačke: 56/0108/21-S Mytolente 120 mg injekčný roztok v naplnenej injekčnej striekačke: 56/0109/21-S
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIE
10. DÁTUM REVÍZIE TEXTU
04/2021