
Odporúča sa, aby sa akékoľvek ďalšie perorálne lieky podávali s odstupom najmenej 3 hodín od
podania MAVENCLADU, počas vymedzeného počtu dní, kedy sa podáva kladribín. (pozri časť 4.5).
Osobitné skupiny pacientov
Poškodeniefunkcieobličiek
U pacientov s poškodením funkcie obličiek neboli vykonané žiadne cielené štúdie.
U pacientov s miernym poškodením funkcie obličiek (klírens kreatinínu 60 až 89 ml/min) nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Bezpečnosť a účinnosť u pacientov so stredne závažným alebo závažným poškodením funkcie obličiek
neboli stanovené. Preto je MAVENCLAD u týchto pacientov kontraindikovaný (pozri časť 4.3).
Poškodenie funkcie pečene
U pacientov s poškodením funkcie pečene neboli vykonané žiadne štúdie.
Aj keď je význam funkcie pečene pri eliminácii kladribínu považovaný za zanedbateľný (pozri
časť 5.2), používanie MAVENCLADU sa kvôli nedostatku údajov u pacientov so stredne závažným alebo závažným poškodením funkcie pečene (Child-Pughovo skóre > 6) neodporúča.
Starší pacienti
Klinické štúdie s perorálne podávaným kladribínom u pacientov so SM nezahŕňali pacientov vo veku
viac ako 65 rokov, preto nie je známe, či odpovedajú na liečbu odlišne od mladších pacientov.
Pri používaní MAVENCLADU u starších pacientov sa odporúča postupovať opatrne a vziať do úvahy možný častejší výskyt zníženej funkcie pečene alebo obličiek, súbežné ochorenia a ďalšie typy farmakologickej liečby.
Pediatrická populácia
Bezpečnosť a účinnosť MAVENCLADU u pacientov mladších ako 18 rokov neboli stanovené.
K dispozícii nie sú žiadne údaje.
Spôsob podávania
MAVENCLAD je určený na perorálne použitie. Tablety sa musia zapiť vodou a prehĺtnúť celé, bez
rozhryznutia. Tablety sa môžu užívať nezávisle od jedla.
Keďže tablety nie sú obalené, musia sa prehltnúť okamžite po vybratí z blistra, nesmú sa ponechať voľne ležiace na žiadnom povrchu a nesmú sa držať v ruke dlhšie, ako je potrebné na užitie dávky lieku. Ak bola tableta ponechaná na akomkoľvek povrchu alebo ak z blistra vypadla zlomená alebo
rozdrobená tableta, toto miesto sa musí dôkladne umyť.
Pri manipulácii s tabletami, musí mať pacient suché ruky, a potom si ich musí dôkladne umyť.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Infekcia vírusom ľudskej imunodeficiencie (HIV).
Aktívna chronická infekcia (tuberkulóza alebo hepatitída).
Začatie liečby kladribínom u imunokompromitovaných pacientov vrátane pacientov v súčasnosti
užívajúcich imunosupresívnu alebo myelosupresívnu liečbu (pozri časť 4.5). Aktívna malignita.
Stredne závažné alebo závažné poškodenie funkcie obličiek (klírens kreatinínu < 60 ml/min) (pozri
časť 5.2).
Gravidita a dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hematologické monitorovanie
Mechanizmus účinku kladribínu je úzko spojený so znížením počtu lymfocytov. Účinok na počet
lymfocytov je závislý od dávky. V klinických štúdiách bol zaznamenaný aj pokles počtu neutrofilov, erytrocytov, hematokritu, hemoglobínu alebo trombocytov v porovnaní so vstupnými hodnotami, aj keď tieto parametre zvyčajne zostávajú v rámci normálneho rozsahu.
Ak sa kladribín podáva pred alebo súbežne s inými látkami, ktoré ovplyvňujú hematologický profil, je
možné očakávať aditívne hematologické nežiaduce reakcie (pozri časť 4.5).
Počet lymfocytov sa musí stanoviť:
• pred začatím liečby MAVENCLADOM v 1. roku,
• pred začatím liečby MAVENCLADOM v 2. roku,
• 2 mesiace a 6 mesiacov po začatí liečby v každom liečebnom roku. Ak je počet lymfocytov nižší
ako 500 buniek/mm³, má sa aktívne monitorovať pokým sa hodnoty znovu nezvýšia.
Pre rozhodovanie sa o liečbe na základe počtu lymfocytov u pacienta, pozri časť 4.2 a časť „Infekcie“
nižšie.
Infekcie
Kladribín môže znížiť obranyschopnosť imunitného systému organizmu a zvýšiť pravdepodobnosť
výskytu infekcií. Pred začatím liečby kladribínom sa musí vylúčiť prítomnosť infekcie HIV, aktívnej tuberkulózy a aktívnej hepatitídy (pozri časť 4.3).
Môže dôjsť k aktivácii latentných infekcií vrátane tuberkulózy alebo hepatitídy. Preto sa pred začatím liečby v 1. a 2. roku musí previesť skríning na latentné infekcie, a to hlavne na tuberkulózu
a hepatitídu B a C. Začatie liečby MAVENCLADOM sa má odložiť, až kým sa infekcia riadne
nevylieči.
Aj u pacientov s akútnou infekciou sa má zvážiť odklad začiatku liečby kladribínom, až kým sa infekcia úplne nevylieči.
Osobitná pozornosť sa odporúča u pacientov bez predchádzajúcej expozície vírusu varicella zoster.
U pacientov negatívnych na protilátky sa pred začatím liečby kladribínom odporúča vakcinácia. Liečba MAVENCLADOM sa musí odložiť o 4 až 6 týždňov, aby sa umožnilo dosiahnuť úplný účinok vakcinácie.
U pacientov užívajúcich kladribín bol zvýšený výskyt infekcie vírusom herpes zoster. Ak počet lymfocytov klesne pod 200 buniek/mm³, má sa počas obdobia lymfopénie 4. stupňa zvážiť podanie preventívnej antiherpesovej liečby podľa miestnej štandardnej praxe (pozri časť 4.8).
Pacienti s počtom lymfocytov nižším ako 500 buniek/mm³ sa majú aktívne monitorovať na prejavy a príznaky svedčiace o infekciách, hlavne o infekcii herpes zoster. Ak sa vyskytnú takéto prejavy
a príznaky, má sa začať klinicky indikovaná antiinfekčná liečba. Môže sa zvážiť prerušenie alebo
odklad liečby MAVENCLADOM až do úplného zvládnutia infekcie.
Pri parenterálnom kladribíne boli u pacientov liečených na vlasatobunkovú leukémiu iným liečebným
režimom hlásené prípady progresívnej multifokálnej leukoencefalopatie (PML).
V databáze klinickej štúdie kladribínu pri SM (1 976 pacientov, 8 650 pacientorokov) nebol hlásený žiadny prípad PML. Pred začatím liečby MAVENCLADOM (zvyčajne do 3 mesiacov) sa však má urobiť vstupné vyšetrenie magnetickou rezonanciou (MRI).
Malignity
V klinických štúdiách sa u pacientov liečených kladribínom častejšie pozorovali prípady malignity
v porovnaní s pacientmi, ktorí dostávali placebo (pozri časť 4.8).
MAVENCLAD je u pacientov so SM s aktívnymi malignitami kontraindikovaný (pozri časť 4.3). Pred začatím liečby MAVENCLADOM sa má u pacientov s predchádzajúcim výskytom malignity vykonať individuálne posúdenie prínosu a rizika. Pacientov liečených MAVENCLADOM treba upozorniť, aby dodržiavali štandardné pokyny na skríning rakoviny.
Antikoncepcia
Pred začatím liečby v 1. aj 2. roku sa pre ženy vo fertilnom veku a pre mužov, ktorí môžu splodiť dieťa,
vyžaduje poradenstvo týkajúce sa možného závažného rizika pre plod a potreby používania účinnej antikoncepcie (pozri časť 4.6).
Ženy vo fertilnom veku musia počas liečby kladribínom a najmenej 6 mesiacov po jeho poslednej dávke
zabrániť otehotneniu používaním účinnej antikoncepcie (pozri časť 4.5).
Počas liečby kladribínom a najmenej 6 mesiacov po jeho poslednej dávke musia pacienti mužského pohlavia dodržiavať preventívne opatrenia, ktoré zabránia vzniku gravidity u ich partnerky.
Transfúzia krvi
U pacientov, ktorí vyžadujú transfúziu krvi, sa pred jej podaním odporúča ožiarenie krvných zložiek,
aby sa zabránilo reakcii štepu proti hostiteľovi. Odporúča sa konzultácia s hematológom.
Prechod na liečbu kladribínom a z liečbykladribínom
U pacientov, ktorí sa predtým liečili imunomodulačnými alebo imunosupresívnymi liekmi sa má pred
začatím liečby MAVENCLADOM zvážiť spôsob a trvanie účinku týchto liekov (pozri časť 4.2). Pri používaní takýchto liekov po liečbe MAVENCLADOM sa má tiež vziať do úvahy možný aditívny účinok na imunitný systém (pozri časť 4.5).
Pri prechode z iného lieku na liečbu SM sa má urobiť vstupné vyšetrenie MRI (pozri časť „Infekcie“
vyššie).
P
o
š
kod
e
n
i
e funkcie pečene
Aj keď je význam funkcie pečene pri eliminácii kladribínu považovaný za zanedbateľný (pozri
časť 5.2), používanie MAVENCLADU sa kvôli nedostatku údajov u pacientov so stredne závažným
alebo závažným poškodením funkcie pečene (Child-Pughovo skóre > 6) neodporúča (pozri časť 4.2).
Intolerancia fruktózy
MAVENCLAD obsahuje sorbitol. Pacienti s dedičnými problémami intolerancie fruktózy nesmú užívať
tento liek.
4.5 Liekové a iné interakcie
MAVENCLAD obsahuje hydroxypropylbetadex, ktorý môže tvoriť komplexy s inými liekmi, čo môže viesť k zvýšeniu biologickej dostupnosti týchto liekov (najmä liekov s nízkou rozpustnosťou, pozri
časť 5.2). Preto sa podávanie akýchkoľvek iných perorálnych liekov odporúča s odstupom najmenej
3 hodín od podania MAVENCLADU počas daných dní, keď sa podáva kladribín.
Imunosupresívne lieky
U imunokompromitovaných pacientov vrátane pacientov súčasne dostávajúcich imunosupresívnu alebo
myelosupresívnu liečbu látkami ako napr. metotrexát, cyklofosfamid, cyklosporín alebo azatioprín, alebo v prípade chronického užívania kortikosteroidov z dôvodu rizika aditívnych účinkov na imunitný systém, je začatie liečby kladribínom kontraindikované (pozri časť 4.3).
Akútna krátkodobá liečba systémovými kortikosteroidmi je počas liečby kladribínom dovolená. Iné lieky modifikujúce ochorenie
Použitie MAVENCLADU s interferónom beta vedie k zvýšenému riziku lymfopénie. Bezpečnosť
a účinnosť MAVENCLADU v kombinácii s inými liekmi modifikujúcimi ochorenie SM neboli
stanovené. Súbežná liečba sa neodporúča.
Hematotoxické lieky
Z dôvodu kladribínom indukovaného zníženia počtu lymfocytov je možné očakávať aditívne
hematologické nežiaduce reakcie, ak sa kladribín podáva pred alebo súbežne s inými látkami ovplyvňujúcimi hematologický profil (napr. karbamazepín). V takýchto prípadoch sa odporúča dôkladné monitorovanie hematologických parametrov.
Živé alebo živé atenuované vakcíny
Liečba MAVENCLADOM sa nemá začať v priebehu 4 až 6 týždňov po vakcinácii živými alebo živými
atenuovanými vakcínami z dôvodu rizika infekcie živou vakcínou. Vakcinácii živými alebo živými atenuovanými vakcínami sa treba vyhnúť počas liečby a po ukončení liečby kladribínom, kým nie je počet pacientových bielych krviniek v rámci normálneho rozsahu.
Silné inhibítory transportných proteínov ENT1, CNT3 a BCRP
Na úrovni absorpcie kladribínu sa jedinou možnou interakciou s klinickým významom zdá byť
interakcia s proteínom rezistencie rakoviny prsníka (BCRP alebo ABCG2). Inhibícia BCRP
v gastrointestinálnom trakte môže zvýšiť perorálnu biologickú dostupnosť a systémovú expozíciu kladribínu. Známe inhibítory BCRP, ktoré môžu zmeniť farmakokinetické vlastnosti substrátov BCRP o 20 % in vivo, zahŕňajú eltrombopag.
I
n vitro štúdie naznačujú, že kladribín je substrátom ekvilibračných nukleozidových (ENT1) a koncentračných nukleozidových (CNT3) transportných proteínov. Biologická dostupnosť, vnútrobunková distribúcia a vylučovanie kladribínu obličkami môžu byť preto teoreticky zmenené silnými inhibítormi transportných proteínov ENT1 a CNT3, ako sú dilazep, nifedipín, nimodipín, cilostazol, sulindak alebo rezerpín. Avšak čisté účinky, pokiaľ ide o možné zmeny v expozícii kladribínu, sú ťažko predpovedateľné.
Aj keď klinický význam týchto interakcií nie je známy, počas 4 až 5 dní liečby kladribínom sa odporúča vyhnúť súbežnému podávaniu silných inhibítorov ENT1, CNT3 alebo BCRP. Ak to nie je možné, má sa zvážiť výber alternatívnych súbežne podávaných liekov so žiadnymi alebo s minimálnymi vlastnosťami inhibítorov transportných proteínov ENT1, CNT3 alebo BCRP. Ak to nie je možné, odporúča sa zníženie dávky liekov obsahujúcich tieto zlúčeniny na minimálnu účinnú dávku, časový odstup medzi
ich podávaním a dôkladné monitorovanie pacientov.
Silné induktory transportných proteínov BCRP a P-gp
Účinky silných induktorov efluxných transportných proteínov BCRP a P-glykoproteínu (P-gp) na
biologickú dostupnosť a dispozíciu kladribínu sa formálne neskúmali. Pri súbežnom podávaní silných induktorov transportných proteínov BCRP (napr. kortikosteroidy) alebo P-gp (napr. rifampicín, ľubovník bodkovaný) sa má zvážiť možné zníženie expozície kladribínu.
Hormonálna antikoncepcia
V súčasnosti nie je známe, či kladribín môže znižovať účinnosť systémovo pôsobiacej hormonálnej
antikoncepcie. Preto ženy používajúce systémovo pôsobiacu hormonálnu antikoncepciu majú počas liečby kladribínom a po dobu najmenej 4 týždňov po jeho poslednej dávke v každom liečebnom roku používať aj bariérovú metódu (pozri časť 4.6).
4.6 Fertilita, gravidita a laktácia
Antikoncepcia u mužov a žien
Pred začatím liečby v 1. aj 2. roku sa pre ženy vo fertilnom veku a pre mužov, ktorí môžu splodiť dieťa,
vyžaduje poradenstvo týkajúce sa možného závažného rizika pre plod a potreby používania účinnej
antikoncepcie.
U žien vo fertilnom veku sa musí pred začatím liečby MAVENCLADOM v 1. aj 2. roku vylúčiť tehotenstvo a počas liečby kladribínom a najmenej 6 mesiacov po jeho poslednej dávke sa musí zabrániť otehotneniu používaním účinnej antikoncepcie. Preto ženy používajúce systémovo pôsobiacu
hormonálnu antikoncepciu majú počas liečby kladribínom a po dobu najmenej 4 týždňov po jeho poslednej dávke v každom liečebnom roku používať aj bariérovú metódu (pozri časť 4.5). Ženy, ktoré počas liečby MAVENCLADOM otehotnejú, majú prerušiť liečbu.
Vzhľadom na to, že kladribín narúša syntézu DNA, možno očakávať nepriaznivé účinky na ľudskú gametogenézu (pozri časť 5.3). Preto počas liečby kladribínom a najmenej 6 mesiacov po jeho poslednej dávke musia pacienti mužského pohlavia dodržiavať preventívne opatrenia, ktoré zabránia vzniku gravidity u ich partnerky.
Gravidita
Na základe skúsenosti u ľudí s inými látkami inhibujúcimi syntézu DNA môže kladribín pri podávaní
počas gravidity spôsobiť vrodené malformácie. Štúdie na zvieratách preukázali reprodukčnú toxicitu
(pozri časť 5.3).
MAVENCLAD je kontraindikovaný u gravidných žien (pozri časť 4.3).
D
o
j
č
e
n
i
e
Nie je známe, či sa kladribín vylučuje do ľudského mlieka. Z dôvodu možných závažných nežiaducich
reakcií u dojčiat je dojčenie počas liečby MAVENCLADOM a 1 týždeň po poslednej dávke kontraindikované (pozri časť 4.3).
Fertilita
U myší sa nevyskytli žiadne účinky na fertilitu alebo reprodukčnú funkciu potomkov. Pozorovali sa
však testikulárne účinky u myší a opíc (pozri časť 5.3).
Keďže kladribín narúša syntézu DNA, môžu sa očakávať nežiaduce účinky na ľudskú gametogenézu. Preto počas liečby kladribínom a najmenej 6 mesiacov po jeho poslednej dávke musia pacienti mužského pohlavia dodržiavať preventívne opatrenia, ktoré zabránia vzniku gravidity u ich partnerky (pozri vyššie).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
MAVENCLAD nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať
stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Klinicky najvýznamnejšími nežiaducimi reakciami hlásenými u pacientov so SM, ktorí v klinických
štúdiách dostávali odporúčanú kumulatívnu dávku kladribínu 3,5 mg/kg počas 2 rokov boli lymfopénia a herpes zoster. Výskyt herpesu zoster bol vyšší v období lymfopénie 3. a 4. stupňa (< 500 až
200 buniek/mm³ alebo < 200 buniek/mm³) v porovnaní s obdobím, keď sa u pacientov nevyskytovala lymfopénia 3. alebo 4. stupňa (pozri časť 4.4).
Zoznam nežiaducich reakcií
Nežiaduce reakcie uvedené v zozname nižšie sú odvodené zo združených údajov z klinických štúdií so
SM, v ktorých sa perorálne podávaný kladribín používal ako monoterapia v kumulatívnej dávke
3,5 mg/kg. Databáza údajov o bezpečnosti z týchto štúdií zahŕňa 923 pacientov.
Nasledujúce definície určujú terminológiu frekvencií výskytu používanú nižšie:
veľmi časté (≥ 1/10)
časté (≥ 1/100 až < 1/10)
menej časté (≥ 1/1 000 až < 1/100) zriedkavé (≥ 1/10 000 až < 1/1 000) veľmi zriedkavé (< 1/10 000)
neznáma frekvencia (z dostupných údajov)
Infekcie a nákazy
Časté: perorálny herpes , dermatomálny herpes zoster
Veľmi zriedkavé: tuberkulóza (pozri časť 4.4)
Poruchy krvi a lymfatického systému
Veľmi časté: lymfopénia
Časté: pokles počtu neutrofilov
Poruchy kože a podkožného tkanivaČasté: vyrážka, alopécia
Opis vybraných nežiaducich reakciíLymfopéniaV klinických štúdiách sa u 20 % až 25 % pacientov liečených kumulatívnou dávkou kladribínu
3,5 mg/kg počas 2 rokov monoterapie vyvinula prechodná lymfopénia 3. alebo 4. stupňa. Lymfopénia
4. stupňa sa pozorovala u menej než 1 % pacientov. Najväčší podiel pacientov s lymfopéniou 3. alebo
4. stupňa sa pozoroval 2 mesiace po prvej dávke kladribínu v každom roku (4,0 % a 11,3 % pacientov
s lymfopéniou 3. stupňa v 1. roku a v 2. roku, 0 % a 0,4 % pacientov s lymfopéniou 4. stupňa v 1. roku a v 2. roku). Očakáva sa, že u väčšiny pacientov sa počet lymfocytov upraví na normálnu hodnotu
alebo sa lymfopénia zmení na stupeň 1 do 9 mesiacov.
Aby sa znížilo riziko vzniku závažnej lymfopénie, počet lymfocytov sa musí stanoviť pred liečbou, počas liečby a po liečbe kladribínom (pozri časť 4.4) a musia sa dodržiavať prísne kritériá pre začatie a pokračovanie v liečbe kladribínom (pozri časť 4.2).
MalignityV klinických štúdiách a pri následnom dlhodobom sledovaní pacientov liečených kumulatívnou dávkou
3,5 mg/kg perorálneho kladribínu sa u pacientov liečených kladribínom pozorovali častejšie prípady
výskytu malignít (10 prípadov za 3 414 pacientorokov [0,29 prípadu na 100 pacientorokov])
v porovnaní s pacientmi, ktorí dostávali placebo (3 prípady za 2 022 pacientorokov [0,15 prípadu na
100 pacientorokov]) (pozri časť 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieS predávkovaním perorálne podávaného kladribínu sú malé skúsenosti. Je známe, že lymfopénia je
závislá od dávky (pozri časti 4.4 a 4.8).
U pacientov, ktorí boli vystavení predávkovaniu kladribínom, sa odporúča mimoriadne dôkladné monitorovanie hematologických parametrov.
Pri predávkovaní kladribínom nie je známe žiadne špecifické antidotum. Liečba pozostáva z dôkladného sledovania a zavedenia príslušných podporných opatrení. Môže byť potrebné zvážiť prerušenie podávania MAVENCLADU. Z dôvodu rýchlej a rozsiahlej vnútrobunkovej a tkanivovej distribúcii sa nepredpokladá, že by hemodialýza eliminovala kladribín vo významnom rozsahu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, antimetabolity, analógy purínov, ATC kód: L01BB04
M
e
c
h
a
n
i
z
m
us účinku
Kladribín je nukleozidový analóg deoxyadenozínu. Substitúcia chlóru v purínovom kruhovom reťazci
chráni kladribín pred odbúravaním adenozíndeaminázou, čím sa predlžuje čas, počas ktorého sa prekurzor kladribínu nachádza vo vnútrobunkovom priestore. Nasledujúca fosforylácia kladribínu na jeho aktívnu trifosfátovú formu, 2-chlorodeoxyadenozín trifosfát (Cd-ATP) sa veľmi účinne dosahuje
v lymfocytoch vďaka ich konštitučne vysokému obsahu deoxycitidínkinázy (DCK) a relatívne nízkemu obsahu 5'-nukleotidázy (5'-NTáza). Vysoký pomer DCK ku 5'-NTáze je priaznivý pre akumuláciu
Cd-ATP, čo robí lymfocyty obzvlášť citlivými na bunkovú smrť. Ako výsledok nižšieho pomeru DCK/5'-NTázy sú ostatné bunky pochádzajúce z kostnej drene v porovnaní s lymfocytmi menej postihnuté. DCK je enzým limitujúci mieru konverzie prekurzora kladribínu na jeho aktívnu trifosfátovú formu, čo vedie k selektívnemu vyčerpaniu deliacich sa a nedeliacich sa T a B buniek.
Primárny mechanizmus účinku Cd-ATP indukujúci apoptózu má priamy a nepriamy účinok na syntézu DNA a mitochondriálnu funkciu. V deliacich sa bunkách narušuje Cd-ATP syntézu DNA inhibíciou ribonukleotidovej reduktázy a konkuruje s deoxyadenozíntrifosfátom pri začleňovaní do DNA pomocou DNA polymeráz. V nedeliacich sa bunkách spôsobuje kladribín prerušenia v jednotlivých reťazcoch DNA, rýchlu spotrebu nikotínamidadenín dinukleotidov, vyčerpanie ATP a bunkovú smrť. Existujú dôkazy, že kladribín môže spôsobiť aj priamu apoptózu, závislú aj nezávislú od kaspázy, a to uvoľňovaním cytochrómu c a faktora indukujúceho apoptózu do cytosolu nedeliacich sa buniek.
Patológia SM zahŕňa komplexné reťazové udalosti, v ktorých hrajú úlohu rôzne typy imunitných buniek vrátane autoreaktívnych T a B buniek. Mechanizmus, ktorým kladribín prejavuje svoj liečebný účinok pri SM nie je úplne objasnený, avšak jeho prevládajúci účinok na B a T lymfocyty sa považuje za prerušujúci kaskádu centrálnych imunitných udalostí pri SM.
Variácie v hladinách expresie DCK a 5'-NTázy medzi podtypmi imunitných buniek môžu vysvetľovať rozdiely v citlivosti imunitných buniek voči kladribínu. Kvôli týmto hladinám expresie sú bunky vrodeného imunitného systému postihnuté menej ako bunky adaptívneho imunitného systému.
Farmakodynamické účinky
Kladribín vykazuje dlhodobý účinok preferenčne cielený na lymfocyty a autoimunitné procesy
zúčastňujúce sa na patofyziológii SM.
V rámci všetkých štúdií sa najväčší podiel pacientov s lymfopéniou 3. alebo 4. stupňa (< 500 až
200 buniek/mm³ alebo < 200 buniek/mm³) pozoroval 2 mesiace po prvej dávke kladribínu v každom roku, čo naznačuje časový odstup medzi plazmatickými koncentráciami a maximálnym hematologickým účinkom.
V rámci všetkých klinických štúdií ukazujú údaje s navrhovanou kumulatívnou dávkou 3,5 mg/kg
telesnej hmotnosti postupné zlepšovanie mediánu počtu lymfocytov späť do normálneho rozsahu do
84 týždňov po prvej dávke kladribínu (približne 30 týždňov po poslednej dávke kladribínu). U viac ako
75 % pacientov sa počty lymfocytov vrátili do normálneho rozsahu do 144 týždňov od prvej dávky
kladribínu (približne 90 týždňov po poslednej dávke kladribínu).
Liečba perorálne podávaným kladribínom vedie k rýchlemu zníženiu cirkulujúcich CD4+ a CD8+
T buniek. V prípade CD8+ T buniek je zníženie menej výrazné a obnova ich počtov je rýchlejšia než
v prípade CD4+ T buniek, čo vedie k dočasnému zníženému pomeru CD4 ku CD8. Kladribín znižuje počet CD19+ B buniek a počet CD16+/CD56+ prirodzených zabíjačov, ktoré sa tiež obnovujú rýchlejšie než CD4+ T bunky.
K
li
n
i
c
ká
účinnosť a bezpečnosť
Relaps-remitujúcaSM
Účinnosť a bezpečnosť perorálne podávaného kladribínu sa vyhodnocovali v randomizovanej, dvojito
zaslepenej, placebom kontrolovanej klinickej štúdii (CLARITY) u 1 326 pacientov s relaps-remitujúcou SM. Cieľmi štúdie boli vyhodnotenie účinnosti kladribínu v porovnaní s placebom v znižovaní priemerného ročného výskytu relapsov (primárny koncový bod), spomalenie progresie postihnutia
a zníženie aktívnych lézií meraných pomocou vyšetrenia MRI.
Pacienti dostávali buď placebo (n = 437) alebo kumulatívnu dávku kladribínu 3,5 mg/kg (n = 433)
alebo 5,25 mg/kg telesnej hmotnosti (n = 456) počas 96-týždňového (2-ročného) obdobia štúdie
v 2 liečebných cykloch. Pacienti randomizovaní na kumulatívnu dávku 3,5 mg/kg dostali prvý liečebný cyklus v 1. a 5. týždni prvého roku a druhý liečebný cyklus v 1. a 5. týždni druhého roku. Pacienti randomizovaní na kumulatívnu dávku 5,25 mg/kg dostali dodatočnú liečbu v 9. a 13. týždni prvého roku. Väčšina pacientov v liečebných skupinách s placebom (87,0 %) a s kladribínom 3,5 mg/kg
(91,9 %) a 5,25 mg/kg (89,0 %) dokončila celých 96 týždňov liečby.
U pacientov sa vyžadoval výskyt aspoň 1 relapsu v predchádzajúcich 12 mesiacoch. V celkovej populácii štúdie bol priemerný vek pacientov 39 rokov (18 až 65 rokov) a pomer žien a mužov bol približne 2:1. Priemerné trvanie SM pred zaradením do štúdie bolo 8,7 roku a medián počiatočného neurologického postihnutia na základe skóre Kurtzkého rozšírenej škály postihnutia (EDSS, Expanded Disability Status Scale) v rámci všetkých skupín pacientov bol 3,0 (rozsah 0 až 6,0). Viac ako dve tretiny pacientov v štúdii predtým neužívalo lieky modifikujúce priebeh SM. Ostatní pacienti boli predtým liečení buď interferónom beta-1a, interferónom beta-1b, glatirameracetátom alebo natalizumabom.
U pacientov s relaps-remitujúcou SM dostávajúcich 3,5 mg/kg kladribínu sa preukázali štatisticky významné zlepšenia hodnoty priemerného ročného výskytu relapsov, podielu pacientov bez relapsu počas 96 týždňov, podielu pacientov bez trvalého postihnutia počas 96 týždňov a času do 3-mesačnej progresie EDSS v porovnaní s pacientmi užívajúcimi placebo (pozri tabuľku 3).
T
a
b
u
ľ
k
a 3 Klinické výsledky štúdie CLARITY (96 týždňov)
K
u
m
u
l
a
t
í
vna dávka kladribínu
Pa
r
a
m
ete
r
P
l
a
ce
b
o
(
n = 437)
3,5 mg/kg
(
n = 433)
5,25 mg/kg
(
n = 456)
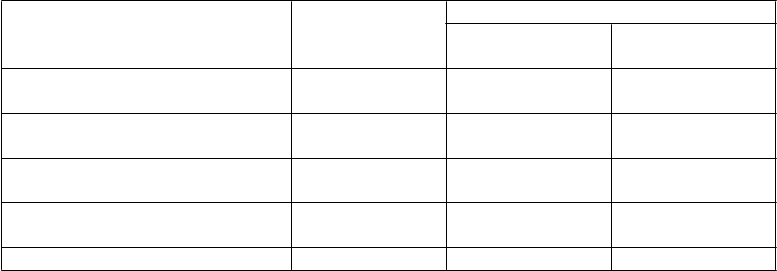
Priemerný ročný výskyt relapsov
(95 % CI) 0,33 (0,29; 0,38) 0,14* (0,12; 0,17) 0,15* (0,12; 0,17)
Relatívne zníženie (kladribín oproti
placebu) 57,6 % 54,5 %
Podiel pacientov bez relapsu počas
96 týždňov 60,9 % 79,7 % 78,9 %
Čas do 3-mesačnej progresie EDSS,
10. percentil (v mesiacoch) 10,8 13,6 13,6
Pomer rizík (95 % CI) 0,67* (0,48; 0,93) 0,69* (0,49; 0,96)
* p < 0,001 v porovnaní s placebom
Okrem toho skupina pacientov užívajúca kladribín 3,5 mg/kg dosahovala počas celých 96 týždňov
štúdie štatisticky významne lepšie výsledky v porovnaní s placebom čo sa týka počtu a relatívneho zníženia T1 Gd+ lézií, aktívnych T2 lézií a kombinovaných špecifických lézií, ako bolo potvrdené pri vyšetrení mozgu pomocou vyšetrenia MRI. Pacienti užívajúci kladribín v porovnaní s liečebnou skupinou s placebom dosiahli relatívne zníženie priemerného počtu T1 Gd+ lézií o 86 % (upravený priemerný počet pre skupinu s 3,5 mg/kg kladribínu bol 0,12 a pre skupinu s placebom 0,91), 73 % relatívne zníženie priemerného počtu aktívnych T2 lézií (upravený priemerný počet pre skupinu
s 3,5 mg/kg kladribínu bol 0,38 a pre skupinu s placebom 1,43) a 74 % relatívne zníženie priemerného
počtu kombinovaných špecifických lézií na pacienta a sken (upravený priemerný počet pre skupinu s 3,5 mg/kg kladribínu bol 0,43 a pre skupinu s placebom 1,72) (p < 0,001 v rámci všetkých
troch cieľových ukazovateľov na MRI).
Post-hoc analýza času do 6-mesačnej potvrdenej progresie EDSS mala za výsledok 47 % zníženie rizika progresie postihnutia v skupine s 3,5 mg/kg kladribínu v porovnaní s placebom (pomer rizík = 0,53,
95 % CI [0,36; 0,79], p < 0,05); v skupine s placebom sa hodnota 10. percentilu dosiahla po 245 dňoch
a v skupine s 3,5 mg/kg kladribínu sa počas štúdie nedosiahla vôbec.
Ako je ukázané v tabuľke 3 hore, vyššie kumulatívne dávky nemali žiadny klinicky významný prínos, ale boli spojené s vyšším výskytom lymfopénie ≥ 3. stupňa (44,9 % v skupine s 5,25 mg/kg oproti
25,6 % v skupine s 3,5 mg/kg).
Pacienti, ktorí ukončili štúdiu CLARITY, mohli byť zaradení do rozširujúcej štúdie CLARITY (CLARITY Extension). V tejto rozširujúcej štúdii dostávalo počas 96-týždňového obdobia
806 pacientov buď placebo, alebo kumulatívnu dávku 3,5 mg/kg kladribínu (v režime podobnom ako sa použil v štúdii CLARITY). Primárnym cieľom tejto štúdie bola bezpečnosť, zatiaľ čo cieľové ukazovatele účinnosti boli exploratívne.
Rozsah účinku v znížení frekvencie relapsov a spomalení progresie postihnutia u pacientov dostávajúcich dávku 3,5 mg/kg počas 2 rokov sa udržal v 3. aj 4. roku (pozri časť 4.2).
Účinnosť u pacientov s vysokou aktivitou ochorenia'
U pacientov s vysokou aktivitou ochorenia liečených perorálne podávaným kladribínom v odporúčanej
kumulatívnej dávke 3,5 mg/kg sa previedli u podskupín post-hoc analýzy účinnosti. Patrili sem
• pacienti s 1 relapsom v predchádzajúcom roku a aspoň 1 T1 Gd+ léziou alebo 9 alebo viacerými
T2 léziami počas liečby inými ochorenie modifikujúcimi liekmi (DMD),
• pacienti s 2 alebo viacerými relapsmi v prechádzajúcom roku, liečení alebo neliečení inými DMD.
V analýze údajov zo štúdie CLARITY sa pozoroval konzistentný liečebný účinok na výskyt relapsov
s priemerným ročným výskytom relapsov v rozsahu od 0,16 do 0,18 v skupine s kladribínom a 0,47 až
0,50 v skupine s placebom (p < 0,0001). V porovnaní s celkovou populáciou sa pozoroval väčší účinok
v čase do 6-mesačného udržania stavu postihnutia, keď kladribín znížil riziko progresie postihnutia
o 82 % (pomer rizík = 0,18; 95 % CI [0,07; 0,47]). Pre skupinu s placebom sa 10. percentil progresie postihnutia dosiahol medzi 16 a 23 týždňami, zatiaľ čo pre skupiny s kladribínom sa nedosiahol počas celej štúdie.
Sekundárna progresívna SM s relapsmi
Podporná štúdia u pacientov liečených kladribínom ako doplnkovou liečbou k interferónu beta oproti
podávaniu placeba + interferónu-beta tiež zahŕňala obmedzený počet pacientov so sekundárnou progresívnou SM (26 pacientov). U týchto pacientov viedla liečba kladribínom 3,5 mg/kg ku zníženiu priemerného ročného výskytu relapsov v porovnaní s placebom (0,03 oproti 0,30, pomer rizík: 0,11,
p < 0,05). Nevyskytol sa rozdiel v priemernom ročnom výskyte relapsov medzi pacientmi s relaps- remitujúcou SM a pacientmi so sekundárnou progresívnou SM s relapsmi. Účinok na progresiu postihnutia nebolo možné ukázať v žiadnej z podskupín.
Pacienti so sekundárnou progresívnou SM boli vylúčení zo štúdie CLARITY. Avšak post-hoc analýza zmiešaných kohort zahrňujúca pacientov zo štúdií CLARITY a ONWARD, definovaných počiatočným skóre EDSS ≥ 3,5 ako zástupcom sekundárnej progresívnej SM, ukázala podobné zníženie priemerného ročného výskytu relapsov v porovnaní s pacientmi s EDSS skóre pod 3.
P
e
d
i
a
t
r
i
c
k
á populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií
s MAVENCLADOM vo všetkých podskupinách pediatrickej populácie so sklerózou multiplex
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Kladribín je liekový prekurzor, ktorý sa musí fosforylovať vnútrobunkovo, aby sa stal biologicky účinným. Farmakokinetické vlastnosti kladribínu boli sledované po perorálnom a intravenóznom podaní u pacientov so SM, u pacientov s malignitami a v in vitro systémoch.
Absorpcia
Po perorálnom podaní sa kladribín rýchlo absorbuje. Podanie 10 mg kladribínu viedlo k priemernej
koncentrácii kladribínu Cmax v rozsahu od 22 do 29 ng/ml a príslušnej priemernej hodnote AUC
v rozsahu od 80 do 101 ng*h/ml (aritmetické priemery z rôznych štúdií).
Keď sa kladribín podal nalačno, medián Tmax bol 0,5 h (rozsah 0,5 až 1,5 h). Pri podávaní s jedlom obsahujúcim vysoké množstvo tukov bola absorpcia kladribínu oneskorená (medián Tmax 1,5 h, rozsah 1 až 3 h) a koncentrácia Cmax bola znížená o 29 % (na základe geometrického priemeru), zatiaľ čo hodnota AUC zostala nezmenená. Biologická dostupnosť 10 mg perorálne podaného kladribínu bola približne
40 %.
Distribúcia
Distribučný objem je rozsiahly, čo naznačuje rozsiahlu distribúciu v tkanivách a vnútrobunkovú
absorpciu. Štúdie ukázali priemerný distribučný objem kladribínu v rozsahu 480 až 490 l. 20 %
kladribínu sa viaže na plazmatické proteíny nezávisle od plazmatickej koncentrácie.
Distribúcia kladribínu v rámci biologických membrán je sprostredkovaná rôznymi transportnými proteínmi vrátane ENT1, CNT3 a BCRP.
In vitro štúdie naznačujú, že eflux kladribínu súvisí len minimálne s P-gp. Klinicky významné interakcie s inhibítormi P-gp sa neočakávajú. Možné dôsledky indukcie P-gp na biologickú dostupnosť kladribínu sa formálne neskúmali.
In vitro štúdie preukázali zanedbateľnú absorpciu kladribínu do ľudských hepatocytov sprostredkovanú
transportnými proteínmi.
Kladribín má potenciál prechádzať cez hematoencefalickú bariéru. Malá štúdia u pacientov
s karcinómom preukázala pomer koncentrácií v mozgovomiechovom moku/plazme približne 0,25.
Kladribín a/alebo jeho fosforylované metabolity sa v podstatnej miere akumulujú a zostávajú
v ľudských lymfocytoch. Pomer vnútrobunkovej a mimobunkovej koncentrácie in vitro bol približne 30
až 40 už 1 hodinu po expozícii kladribínu.
Biotransformácia
Metabolizmus kladribínu bol sledovaný u pacientov so SM po podaní jednej 10 mg tablety a jednej
3 mg intravenóznej dávky. Po perorálnom aj intravenóznom podaní bola pôvodná zlúčenina kladribín hlavnou zložkou prítomnou v plazme a v moči. Metabolit 2-chloroadenín bol menej podstatným metabolitom v plazme a v moči, napr. predstavoval len ≤ 3 % plazmatickej expozície pôvodnej zlúčeniny po perorálnom podaní. V plazme a v moči sa našli iba stopové množstvá iných metabolitov.
V in vitro hepatických systémoch bol pozorovaný zanedbateľný metabolizmus kladribínu (najmenej
90 % tvoril nezmenený kladribín).
Kladribín nie je významným substrátom enzýmov cytochrómu P450 a nepreukazuje významný potenciál pre inhibíciu CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ani CYP3A4. Neočakáva sa, že inhibícia týchto enzýmov alebo genetické polymorfizmy (napr. CYP2D6, CYP2C9 alebo CYP2C19) budú viesť ku klinicky významnému účinku na farmakokinetické vlastnosti alebo expozíciu kladribínu. Kladribín nemá žiadny klinicky významný indukčný účinok na enzýmy CYP1A2, CYP2B6 ani CYP3A4.
Po vniknutí do cieľovej bunky sa kladribín fosforyluje na monofosfát kladribínu (Cd-AMP) prostredníctvom DCK (a tiež prostredníctvom deoxyguanozínkinázy v mitochondriách). Cd-AMP sa ďalej fosforyluje na difosfát kladribínu (Cd-ADP) a trifosfát kladribínu (Cd-ATP). Defosforylácia
a deaktivácia Cd-AMP sa katalyzuje prostredníctvom cytoplazmatickej 5´-NTázy. V štúdii vnútrobunkových farmakokinetických vlastností Cd-AMP a Cd-ATP u pacientov s chronickou myelogénnou leukémiou boli hodnoty Cd-ATP približne polovičné v porovnaní s hodnotami Cd-AMP.
Vnútrobunkový polčas Cd-AMP bol 15 h. Vnútrobunkový polčas Cd-ATP bol 10 h. Eliminácia
Na základe združených farmakokinetických údajov z rôznych štúdií boli mediány eliminácie 22,2 l/h pre
renálny klírens a 23,4 l/h pre iný ako renálny klírens. Renálny klírens prevyšoval rýchlosť glomerulárnej filtrácie, čo naznačuje aktívnu tubulárnu sekréciu kladribínu.
Nerenálna časť eliminácie kladribínu (približne 50 %) pozostáva zo zanedbateľného hepatického
metabolizmu a rozsiahlej vnútrobunkovej distribúcie a odchytu aktívneho kladribínu (Cd-ATP)
v cieľových intracelulárnych kompartmentoch (t. j. lymfocytoch) a následnej eliminácie intracelulárneho
Cd-ATP podľa životného cyklu a eliminačných dráh týchto buniek.
Odhadovaný terminálny polčas pre typického pacienta je na základe populačnej famakokinetickej analýzy približne 1 deň. To však nevedie k akumulácii lieku pri podávaní jedenkrát denne, keďže tento polčas predstavuje len malú časť hodnoty AUC.
Závislosť od dávky a času
Po perorálnom podaní kladribínu v rozsahu dávok od 3 do 20 mg sa hodnoty Cmax a AUC zvýšili
spôsobom úmerným dávke, čo naznačuje, že do perorálnej dávky 20 mg absorpcia nie je ovplyvnená
procesmi obmedzenými rýchlosťou ani kapacitou.
Po opakovanom podávaní sa nepozorovala žiadna významná akumulácia koncentrácií kladribínu v plazme. Neexistuje žiadny dôkaz, že by sa po opakovanom podávaní menili farmakokinetické vlastnosti kladribínu v časovo závislej forme.
Osobitné skupiny pacientov
Nevykonali sa žiadne štúdie na vyhodnotenie farmakokinetických vlastností kladribínu u starších
pacientov ani pediatrických pacientov so SM, ani u pacientov s poškodením obličiek alebo pečene.
Populačná kinetická analýza nepreukázala žiadny vplyv veku (18 až 65 rokov) alebo pohlavia na farmakokinetické vlastnosti kladribínu.
P
oškodenie
f
unkcie
obličiek
Renálny klírens kladribínu je závislý od klírensu kreatinínu. Na základe populačnej farmakokinetickej
analýzy zahŕňajúcej pacientov s normálnou funkciou obličiek a s miernym poškodením funkcie obličiek sa pre celkový klírens u pacientov s miernym poškodením funkcie obličiek (CLCR = 60 ml/min) očakáva mierny pokles, čo vedie k zvýšenej expozícii o 25 %.
Poškodenie funkcie pečene
Úloha hepatálnej funkcie na elimináciu kladribínu sa považuje za zanedbateľnú.
Farmakokinetické interakcie
Štúdia liekových interakcií u pacientov so SM preukázala, že biologická dostupnosť 10 mg perorálne
podaného kladribínu sa pri súbežnom podávaní s pantoprazolom nezmenila.
5.3 Predklinické údaje o bezpečnosti
Predklinické farmakologické a toxikologické hodnotenia bezpečnosti kladribínu vykonané na zvieracích modeloch relevantných pre posúdenie bezpečnosti kladribínu neodhalili žiadne iné významné nálezy okrem tých, ktoré boli predpovedané na základe farmakologického mechanizmu kladribínu. Primárnymi cieľovými orgánmi identifikovanými v štúdiách toxicity po opakovanom podávaní parenterálnymi cestami (intravenózne alebo subkutánne) trvajúcich až 1 rok na myšiach a opiciach boli lymfoidný
a hematopoetický systém. Ďalšími cieľovými orgánmi po dlhšom podávaní (14 cyklov) kladribínu opiciam subkutánnou cestou boli obličky (karyomegália tubulárneho epitelu obličiek), nadobličky (atrofia kôry a znížená vakuolizácia), gastrointestinálny trakt (atrofia sliznice) a semenníky. Účinky na obličky sa pozorovali aj u myší.
Mutagenita
Kladribín sa inkorporuje do DNA reťazcov a inhibuje syntézu a opravu DNA. Kladribín neindukoval
mutáciu génov v baktériách ani bunkách cicavcov, ale bol klastogénny, čo spôsobovalo chromozomálne poškodenie buniek cicavcov in vitro pri koncentrácii, ktorá bola 17-násobne vyššia ako očakávané klinické hodnoty Cmax. In vivo sa pozorovala klastogenita u myší pri 10 mg/kg, čo bola najnižšia testovaná dávka.
Karcinogenita
Karcinogénny potenciál kladribínu bol posudzovaný v rámci dlhodobej 22-mesačnej štúdie so
subkutánnym podávaním myšiam a v rámci krátkodobej 26-týždňovej štúdie s perorálnym podávaním transgénnym myšiam.
• V dlhodobej štúdii karcinogenity u myší bola najvyššia použitá dávka 10 mg/kg denne, ktorá bola v mikronukleárnej štúdii u myší genotoxická (zodpovedá približne 16-násobku očakávanej expozície u ľudí v AUC u pacientov užívajúcich maximálnu dennú dávku 20 mg kladribínu).
U myší sa nepozoroval zvýšený výskyt lymfoproliferatívnych porúch ani iných typov nádorov (okrem karcinómov Harderovej žľazy, hlavne adenómov). Karcinómy Harderovej žľazy sa nepovažujú za klinicky významné, keďže ľudia nemajú porovnateľnú anatomickú štruktúru.
• V krátkodobej štúdii karcinogenity u Tg rasH2 myší sa pri žiadnej testovanej dávke až do
30 mg/kg denne (zodpovedá približne 25-násobku očakávanej expozície u ľudí v AUC
u pacientov užívajúcich maximálnu dennú dávku 20 mg kladribínu) nepozorovalo zvýšenie výskytu lymfoproliferatívnych porúch alebo iných typov nádorov súvisiacich s kladribínom.
Kladribín bol hodnotený aj v 1-ročnej štúdii u opíc pri subkutánnom podávaní. V tejto štúdii sa nezaznamenal zvýšený výskyt lymfoproliferatívnych porúch ani žiadne nádory.
Aj keď kladribín môže mať potenciál vyvolať genotoxicitu, dlhodobé údaje u myší a opíc neposkytli žiadne dôkazy o významnom zvýšení rizika karcinogenity u ľudí.
Reprodukčná toxicita
Aj keď sa nepozorovali žiadne účinky na samičiu fertilitu, reprodukčnú funkciu ani všeobecný
výkonnostný stav potomstva, pri podávaní gravidným myšiam bol kladribín preukázateľne embryoletálny a zlúčenina bola u myší (aj po liečbe iba samotných samcov) a králikov teratogénna. Pozorované embryoletálne a teratogénne účinky sú v súlade s farmakologickým mechanizmom kladribínu. V štúdii fertility u samcov myší sa pozorovali znetvorené plody s malformáciami s agenézou distálnych častí končatín, humeru a/alebo femuru. Frekvencia výskytu postihnutých myších plodov
v tejto štúdii bola v rovnakom rozsahu ako spontánny výskyt amélie alebo fokomélie u tohto kmeňa myší. Avšak pri zohľadnení genotoxicity kladribínu sa nedajú vylúčiť samcami sprostredkované účinky súvisiace s možnými genetickými zmenami diferencujúcich sa spermií.
Kladribín neovplyvnil samčiu fertilitu u myší, pozorovali sa však testikulárne účinky, a to zníženie hmotnosti semenníkov a zvýšenie počtu nepohyblivých spermií. U opíc sa tiež pozorovala testikulárna degenerácia a reverzibilné zníženie počtu spermií s rýchlou progresívnou pohyblivosťou.
Z histologického hľadiska sa testikulárna degenerácia pozorovala len u jedného samca opice v 1-ročnej
štúdii toxicity so subkutánnym podávaním.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
hydroxypropylbetadex (2-hydroxypropyl-ß-cyklodextrín)
sorbitol magnéziumstearát
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Blister z orientovaného polyamidu (OPA)/hliníka (Al)/polyvinylchloridu (PVC) - hliníka (Al), zatavený do kartónového puzdra a upevnený do detskej bezpečnostnej škatule.
Veľkosti balenia: 1, 4, 5, 6, 7 alebo 8 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Serono Europe Limited
56 Marsh Wall London E14 9TP Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/17/1212/001
EU/1/17/1212/002
EU/1/17/1212/003
EU/1/17/1212/004
EU/1/17/1212/005
EU/1/17/1212/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.