
edna dávka
každý druhý deň
ekvivalent)
0,5 mg/kg/deň
(najviac 20 mg/deň)
Prednizón (alebo jeho ekvivalent)
0,5 mg/kg každý druhý deň
(najviac 20 mg/deň)
 O
sobitné skupiny pacientov
Starší pacienti
O
sobitné skupiny pacientov
Starší pacienti
Bezpečnosť a účinnosť voretigén neparvoveku u pacientov vo veku ≥ 65 rokov neboli doteraz stanovené. Úprava dávkovania u starších pacientov však nie je potrebná.
Poruchy funkcie obličiek a pečeneBezpečnosť a účinnosť voretigén neparvoveku u pacientov s poruchou funkcie pečene a obličiek
neboli doteraz stanovené. U týchto pacientov nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Pediatrická populáciaBezpečnosť a účinnosť voretigén neparvoveku u detí vo veku do 4 rokov neboli doteraz stanovené.
K dispozícii nie sú žiadne údaje. U pediatrických pacientov nie je potrebná žiadna úprava dávkovania.
Spôsob podávaniaSubretinálne použitie.
Luxturna je sterilný roztok koncentrátu na subretinálnu injekciu, ktorý pred podaním vyžaduje
rozmrazenie a riedenie (pozri časť 6.6).
Tento liek sa nesmie podávať intravitreálnou injekciou.
Luxturna je jednorazová injekčná liekovka, iba na jedno podanie a do jedného oka. Liek sa podáva po vitrektómii ako subretinálna injekcia do každého oka. Nemá sa podávať v bezprostrednej blízkosti fovey, aby fovea ostala neporušená (pozri časť 4.4).
Podávanie voretigén neparvoveku sa má vykonávať na operačnej sále za kontrolovaných aseptických podmienok. Pred týmto úkonom sa má pacientovi podať vhodná anestézia. Zrenica oka, do ktorej sa má podať injekcia, musí byť rozšírená a pred operáciou sa má v súlade so štandardnou lekárskou praxou podať lokálny širokospektrálny mikrobicíd.
Opatrenia pred zaobchádzaním alebo podaním liekuTento liek obsahuje geneticky modifikované organizmy. Počas prípravy alebo podávania voretigén neparvoveku je potrebné používať osobné ochranné prostriedky (zahŕňajúce laboratórny plášť,
ochranné okuliare a rukavice) (pozri časť 6.6).
Pokyny na prípravu, opatrenia pri náhodnej expozícii a likvidáciu Luxturny, pozri časť 6.6.
PodávaniePri podávaní voretigén neparvoveku pacientom dodržiavajte nižšie uvedené kroky:
· Zriedená Luxturna sa má pre podaním vizuálne skontrolovať. Ak sú v lieku viditeľné častice, zakalenie alebo zmena farby, liek sa nesmie použiť.
· Pripojte injekčnú striekačku obsahujúcu zriedený liek k hadičke a mikrokanyle. Liek sa podáva
cez hadičku a mikrokanylu pomaly, aby sa zo systému odstránili všetky vzduchové bubliny.
· Objem, ktorý sa má podať, je v injekčnej striekačke nastavený tak, že koniec piestu je zarovno s
čiarou označujúcou 0,3 ml.
· Po ukončení vitrektómie sa Luxturna podáva subretinálnou injekciou pomocou kanyly subretinálnej injekcie zavedenej cez pars plana (obrázok č. 1A).
· Pri priamej vizualizácii sa hrot kanyly subretinálnej injekcie dostane do kontaktu s povrchom sietnice. Odporúčané miesto podania injekcie sa má nachádzať pri hornej cievnej arkáde
najmenej 2 mm distálne od stredu fovey (obrázok č. 1B). Malé množstvo lieku sa pomaly podáva dovtedy, kým sa neobjaví prvá subretinálna bublina a potom sa pomaly podáva zvyšné
množstvo lieku, až kým sa nepodá celková dávka0,3 ml.
Obrázok č. 1A Kanyla subretinálnej injekcie zavedená cez pars planavitrektomický
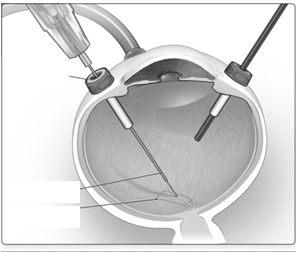
trokár
kanyla subretinálnej
injekcie
odporúčaná oblasť
podania injekcie
O
brázok č. 1B Hrot kanyly subretinálnej injekcie umiestnený v odporúčanej oblasti podania injekcie (z pohľadu chirurga)
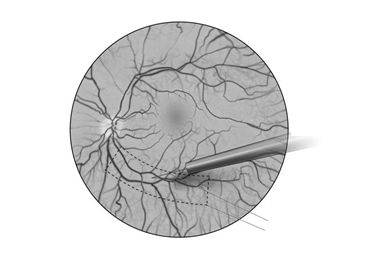
dolná časť
časť smerom k nosu
spánková časť
horná časť
kanyla subretinálnej
injekcie
odporúčaná oblasť
podania injekcie
· Po ukončení podania injekcie sa kanyla subretinálnej injekcie z oka odstráni.
· Po podaní injekcie sa musí všetok nepoužitý liek zlikvidovať. Rezervná injekčná striekačka sa
tiež nesmie uchovávať. Pri likvidácii lieku sa riaďte národnými požiadavkami pre biologickú
bezpečnosť.
· Počas výmeny vzduchu a tekutiny sa treba starostlivo vyhýbať odtoku tekutiny v blízkosti retinotómie vykonávanej kvôli subretinálnej injekcii.
· S polohovaním hlavy v supinačnej polohe sa začína okamžite v pooperačnom období a po prepustení z nemocnice ju pacient musí dodržiavať počas 24 hodín.
4.3 Kontraindikácie
Precitlivenosť na liečivo (liečivá) alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Očná alebo periokulárna infekcia. Aktívny vnútroočný zápal.
4.4 Osobitné upozornenia a opatrenia pri používaní
Na prípravu a podávanie Luxturny sa majú vždy použiť vhodné aseptické postupy.
Počas postupu pri podávaní lieku boli pozorované nasledujúce nežiaduce reakcie:
· Zápal oka (vrátane endoftalmitídy), trhlina v sietnici a odlúčenie sietnice. Pacientov je potrebné poučiť, aby bezodkladne hlásili akékoľvek symptómy pripomínajúce endofthalmitídu alebo odlúčenie sietnice a má im byť podaná vhodná liečba.
· Porucha sietnice (stenčenie fovey, strata foveálnej funkcie), makulárna diera, makulopatia
(epiretinálna membrána, makulárne zvrásnenie) a porucha oka (dehiscencia fovey).
· Zvýšenie vnútroočného tlaku. Vnútroočný tlak sa má sledovať pred aj po podaní lieku a má sa vhodne liečiť. Pacienti majú byť poučení, aby sa vyhýbali cestovaniu lietadlom alebo inému cestovaniu vo výškach dovtedy, kým sa vzduchová bublina, ktorá sa vytvorila ako následok podania Luxturny, úplne z oka vytratí. Vymiznutie vzduchovej bubliny si môže vyžadovať časový úsek v dĺžke až jedného týždňa alebo viac od podania injekcie, čo sa musí overiť oftalmologickým vyšetrením. Rýchly nárast výšky v čase, keď je ešte vzduchová bublina prítomná, môže spôsobiť zvýšenie očného tlaku a nezvratnú stratu videnia.
V priebehu týždňov po liečbe sa môžu vyskytnúť dočasné poruchy videnia, ako napríklad rozmazané videnie a fotofóbia. Pacienti majú byť poučení, aby sa v prípade pretrvávania zrakových porúch obrátili na svojho lekára. Pacienti sa majú vyhýbať plávaniu kvôli zvýšenému riziku infekcie v oku. Pacienti sa majú vyhýbať namáhavej fyzickej aktivite kvôli zvýšenému riziku poranenia oka. Po porade s lekárom, po minimálne jednom až dvoch týždňoch, môžu pacienti opäť začať s plávaním a namáhavou aktivitou.
Vylučovanie vektora (shedding)
V slzách pacienta sa môže vyskytnúť prechodné vylučovanie nízkej hladiny vektora (pozri časť 5.2).
Pacienti/opatrovatelia majú byť poučení o správnej manipulácii s odpadovým materiálom, ktorý bol vytvorený z obväzov, sĺz a nosových sekrétov, čo môže zahŕňať skladovanie odpadového materiálu v hermeticky uzavretých vreciach pred likvidáciou. Tieto preventívne opatrenia na manipuláciu sa majú dodržiavať 14 dní po podaní voretigén neparvoveku. Odporúča sa, aby pacienti/opatrovatelia používali pri výmene obväzov a likvidácii odpadu rukavice, obzvlášť v prípade možnej gravidity, dojčenia a imunodeficiencie opatrovateľov.
Pacienti liečení Luxturnou nesmú darovať krv, orgány, tkanivá ani bunky na transplantáciu. Imunogenita
Na zníženie možnej imunogenity sa majú pacientom podať systémové kortikosteroidy pred a po
subretinálnej injekcii voretigén neparvoveku do každého oka (pozri časť 4.2). Kortikosteroidy môžu znižovať potenciálnu imunitnú reakciu na vektorový kapsid (adeno-asociovaný vírusový vektor
sérotypu 2 [AAV2]) alebo transgénny liek (proteín retinálneho pigmentového epitelu 65 kDa
[RPE65]).
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t.j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Nie sú známe žiadne klinicky významné interakcie. Neuskutočnili sa žiadne interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
Na základe neklinických štúdií a klinických údajov z klinických skúšaní AAV2 vektorov a vzhľadom na subretinálnu cestu podávania Luxturny je neúmyselný prenos v zárodočnej línii prostredníctvom AAV vektorov vysoko nepravdepodobný.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov (menej ako 300 ukončených gravidít)
o použití voretigén neparvoveku u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu voretigén neparvoveka počas gravidity.
Dojčenie
Luxturna nebola skúmaná u dojčiacich žien. Nie je známe, či sa voretigén neparvovek vylučuje do
ľudského mlieka. Riziko u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu voretigén neparvovekom sa má urobiť po zvážení prínosu
dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne klinické údaje o účinku tohto lieku na fertilitu. V štúdiách na zvieratách
neboli hodnotené účinky na mužskú a ženskú fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Voretigén neparvovek má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U pacientov sa po podaní subretinálnej injekcie Luxturny môžu vyskytnúť dočasné poruchy videnia. Pacienti nesmú viesť vozidlá ani obsluhovať ťažké stroje dovtedy, kým sa ich vizuálna funkcia dostatočne nezlepší, ako to odporúča ich oftalmológ.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
U troch zo 41 (7 %) pacientov sa vyskytli tri nezávažné nežiaduce reakcie vo forme retinálnych
usadenín, ktoré pravdepodobne súviseli s voretigén neparvovekom. Všetky tieto tri prípady boli iba prechodný výskyt asymptomatického subretinálneho precipitátu pod miestom retinálnej injekcie
1-6 dní po podaní injekcie a vymizli bez následkov.
Závažné nežiaduce reakcie súvisiace s postupom pri podávaní injekcie boli v priebehu klinického programu hlásené u troch pacientov. Jeden zo 41 (2 %) pacientov hlásil závažný výskyt zvýšeného vnútroočného tlaku (po podaní depozitného steroidu), ktorý súvisel s liečbou endoftalmitídy spojenej s postupom pri podávaní lieku a mal za následok atrofiu zrakového nervu a jeden zo 41 (2 %)
pacientov hlásil závažný výskyt retinálnej poruchy (stratu funkcie fovey), ktorá bola vyhodnotená ako súvisiaca s postupom pri podávaní injekcie. Jeden zo 41 (2 %) pacientov hlásil závažný prípad odlúčenia sietnice, ktorý bol vyhodnotený ako súvisiaci s postupom pri podávaní lieku.
Najčastejšími nežiaducimi reakciami (výskyt ≥ 5 %) súvisiacimi s postupom pri podávaní lieku boli hyperémia spojoviek, katarakta, zvýšený vnútroočný tlak, trhlina v sietnici, stenčenie rohovky dellen, makulárna diera, subretinálne usadeniny, zápal oka, podráždenie oka, bolesť oka a makulopatia (zvrásnenie na povrchu makuly).
Tabuľkový zoznam nežiaducichreakcií
Nežiaduce reakcie sú zatriedené podľa triedy orgánových systémov a frekvencie v súlade s
nasledujúcou konvenciou: veľmi časté (≥1/10), časté ≥1/100 až <1/10), menej časté (≥1/1 000 až
<1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov).
Tabuľka č. 2 Nežiaduce reakcie súvisiace s voretigén neparvovekom
T
rieda orgánových
systémov
/
f
rekvencia
P
oruchy oka
 N
ežiaduce reakcie
N
ežiaduce reakcie
Časté retinálne usadeniny
T
abuľka č. 3 Nežiaduce reakcie súvisiace s postupom pri podávaní
T
rieda orgánových
systémov
/
f
rekvencia
P
sychické poruchy
N
ežiaduce reakcie
Časté úzkosť
Poruchy nervového systému
Časté bolesť hlavy, závrat
Poruchy oka
Veľmi časté hyperémia spojovky, katarakta
trhlina v sietnici, stenčenie rohovky dellen, makulárna diera, zápal oka,
podráždenie oka, bolesť oka, makulopatia, choroidálne krvácanie, spojovková
Časté
cysta, porucha oka, opuch oka, pocit cudzieho telieska v očiach, degenerácia makuly, endoftalmitída, odlúčenie sietnice, porucha sietnice, retinálne krvácanie
P
oruchy gastrointestinálneho traktu
Časté nauzea, vracanie, bolesť v hornej časti brucha, bolesť pery
Poruchy kože a podkožného tkanivaČasté vyrážka, opuch tváre
Laboratórne a funkčné vyšetreniaVeľmi časté zvýšený vnútroočný tlak Časté inverzia T vlny EKG
Úrazy, otravy a komplikácie liečebného postupuČasté komplikácie pri endotrachálnej intubácii, dehiscencia rany
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNie sú žiadne klinické skúsenosti s predávkovaním voretigén neparvovekom. V prípade predávkovania sa odporúča symptomatická a podporná liečba, ak ju ošetrujúci lekár považuje za potrebnú.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: { zatiaľ nepridelená}, ATC kód: { zatiaľ nepridelený }.
Mechanizmus účinku
Proteín 65 kDa (RPE65) špecifický pre retinálny pigmentový epitel sa nachádza v retinálnych
pigmentových epitelovýh bunkách a mení all-trans-retinol na 11-cis-retinol, ktorý následne v priebehu zrakového (retinoidného) cyklu vytvára chromofór, 11-cis-retinálny. Tieto kroky sú kľúčové pri
biologickej konverzii fotónu svetla na elektrický signál v sietnici. Mutácie v géne RPE65 vedú
k zníženiu alebo absencii aktivity RPE65 all-trans-retinyl izomerázy, pričom blokujú zrakový cyklus a majú za následok stratu videnia. V priebehu času hromadenie toxických prekuzorov vedie k smrti
buniek retinálneho pigmentového epitelu a následne k smrti progresívnej fotoreceptorovej bunky.
U osôb s bialelickou RPE65 mutáciou súvisiacou s retinálnou dystrofiou sa často v detstve a počas dospievania prejavuje strata videnia vrátane zhoršenia funkčných zrakových parametrov ako sú zraková ostrosť a zrakové pole. Táto strata zraku nakoniec prechádza do úplnej slepoty.
Injekcia voretigén neparvoveku do subretinánej oblasti má za následok transdukciu buniek retinálneho pigmentového epitelu s cDNA kódujúcou normálny ľudský RPE65 proteín (génová augmentačná
liečba), čo poskytuje možnosť obnovenia zrakového cyklu.
Klinická účinnosťabezpečnosť
Dlhodobá bezpečnosť a účinnosť Luxturny sa hodnotili v štúdii fázy 1 skúmajúcej bezpečnosť
a zvyšovanie dávky (101), v ktorej boli 12 pacientom unilaterálne podané subretinálne injekcie voretigén neparvoveku; v kontrolnej štúdii (102), v ktorej bol voretigén neparvovek podaný do kontralaterálneho oka u 11 z 12 pacientov, ktorí sa zúčastnili štúdie so zvyšovaním dávky;
v jednoročnej, otvorenej kontrolovanej štúdii fázy 3 (301), v ktorej bolo v dvoch centrách výskumu randomizovaných 31 pacientov a v pokračovaní štúdie fázy 3, v ktorej 9 kontrolných pacientov prešlo
na inú liečbu a podstúpilo zákrok. V klinickom programe sa zúčastnilo celkom 41 pacientov (injekcia bola podaná do 81 očí [jeden pacient z fázy 1 nesplnil kritérium spôsobilosti pre druhú injekciu]).
Všetci účastníci štúdií mali klinickú diagnózu Leberova vrodená amauróza a niektorí mohli mať aj predchádzajúce alebo ďalšie klinické diagnózy, vrátane retinitis pigmentosa. Potvrdené bialelické mutácie RPE65 a prítomnosť dostatočne životaschopných retinálnych buniek (oblasť sietnice na
zadnom póle s hrúbkou > 100 mikrónov, určených pomocou optickej koherentnej tomografie [OCT]), boli preukázané u všetkých účastníkov.
Št údi a f ázy 3
Štúdia 301 bola otvorená, randomizovaná, kontrolná štúdia. Bolo do nej zaradených 31 pacientov,
13 mužov a 18 žien. Priemerný vek bol 15 rokov (rozpätie od 4 do 44 rokov) vrátane 64 % pediatrických pacientov (n=20, vek od 4 do 17 rokov) a 36 % dospelých (n=11). Všetci pacienti mali diagnózu Leberova vrodená amauróza, ako následok RPE65 mutácií, potvrdených genetickou analýzou v certifikovanom laboratóriu.
21 pacientov bolo randomizovaných na podávanie subretinálnej injekcie voretigén neparvoveku. Zraková ostrosť (LogMAR) prvého oka týchto pacientov na začiatku liečby bola 1,18 (0,14), priemer (SE). Účasť jedného pacienta v štúdii bola ukončená ešte pred začiatkom liečby. 10 pacientov bolo randomizovaných do kontrolnej (neintervenčnej) skupiny. Zraková ostrosť (LogMAR) prvého oka týchto pacientov na začiatku liečby bola 1,29 (0,21), priemer (SE). Jeden pacient v kontrolnej skupine odvolal svoj súhlas a jeho účasť v štúdii bola ukončená. Deväť pacientov, ktorí boli randomizovaní do kontrolnej skupiny, prešlo po jednom roku pozorovania na liečbu subretinálnou injekciou voretigén neparvoveku. Do každého oka bola podaná jedna subretinálna injekcia s 1,5 x 1011 vg voretigén neparvoveku v celkovom objeme 300 μl. Interval medzi injekciami do očí bol u každého pacienta od 6 do 18 dní.
Primárny koncový ukazovateľ v štúdii fázy 3 meral priemernú zmenu od začiatku liečby do jedného roka u intervenčnej a kontrolnej skupiny v testovaní binokulárnej multiluminančnej mobility (MLMT). MLMT bolo určené na meranie zmien vo funkčnom videní, konkrétne schopnosti pacienta nájsť cestu presne a v rozumnom tempe pri rôznych úrovniach osvetlenia okolitého prostredia. Táto schopnosť závisí od zrakovej ostrosti pacienta, zrakového poľa a rozsahu nyktalopie (znížená schopnosť vnímať a/alebo vidieť pri tlmenom svetle), pričom každá z týchto funkcií je jasne ovplyvnená retinálnym ochorením súvisiacim s mutáciami RPE65. V štúdii fázy 3 MLMT testovaní sa použilo sedem hladín osvetlenia v rozsahu 400 luxov až 1 lux (čo napríklad zodpovedá jasne osvetlenej kancelárii až bezmesačnej letnej noci). Vyšetrovanie každého pacienta sa nahrávalo na videokazetu a hodnotili ho nezávislí klasifikátori. Pozitívna zmena skóre vyjadruje zvládnutie testu MLMT pri nižšej hladine svetla a lux skóre 6 vyjadruje najvyššie možné zlepšenie MLMT. Skúmali sa aj tri sekundárne
koncové ukazovatele: testovanie prahu citlivosti na plné svetlo (FST) s použitím bieleho svetla, zmena v skóre MLMT pri prvom určenom oku a vyšetrenie zrakovej ostrosti (VA).
Na začiatku liečby dosahovali pacienti v teste mobility úspešné hodnotenie medzi 4 a 400 luxov okolitého prostredia.
Tabuľka č. 4 Zmeny v skóre MLMT: po jednom roku, v porovnaní so začiatkom liečby
(populácia ITT: n=21 intervenčná skupina, n=10 kontrolná skupina)
Z
m
ena v skóre MLMT
R
ozdiel
 (
95 % CI) intervenčná skupina – kontrolná skupina
p-hodnota
(
95 % CI) intervenčná skupina – kontrolná skupina
p-hodnota
použitie binokulárneho videnia 1,6 (0,72; 2,41) 0,001 použitie iba prvého určeného oka 1,7 (0,89; 2,52) 0,001 použitie iba druhého určeného oka 2,0 (1,14; 2,85) < 0,001
Skóre zmeny monokulárnej MLMT sa v liečenej skupine významne zlepšilo a bolo podobné
výsledkom binokulárnej MLMT (pozri tabuľku č. 4).
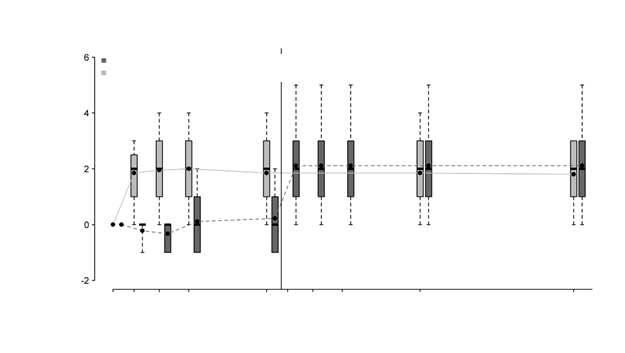
Obrázok č. 2 ukazuje účinok lieku v priebehu trojročného obdobia v skupine liečenej s voretigén neparvovekom, ako aj účinok v kontrolnej skupine po prechode na podávanie subretinálnej injekcie voretigén neparvoveku. Významné rozdiely vo výsledku binokulárnej MLMT boli v skupine liečenej voretigén neparvovekom zaznamenané v 30. dni a udržali sa počas zostávajúcich kontrolných návštev v priebehu trojročného obdobia, v porovnaní so žiadnou zmenou v kontrolnej skupine. Po prechode na podávanie subretinálnej injekcie voretigén neparvoveku však pacienti v kontrolnej skupine vykazovali podobnú odpoveď na voretigén neparvovek, ako pacienti v skupine liečenej voretigén neparvovekom.
Obrázok č. 2 Zmena v skóre MLMT pri používaní binokulárneho videnia oproti časupred/po expozícii voretigén neparvovekukontrolná skupina/intervenčná skupina (N = 9)
pôvodná intervenčná skupina (N = 20)
zmena skóre
binokulárnej
MLMT
B D30 D90 D180 R1 R2 R3L XBLXD30 XD90 XD180 XR1 XR2návšteva u lekára v priebehu štúdie
Každý rámček predstavuje stredných 50 % distribúcie zmeny skóre MLMT. Vertikálne vybodkované čiary
predstavujú ďalších 25 % nad a pod rámčekom. Vodorovný prúžok v každom políčku predstavuje strednú hodnotu. Bodka v každom políčku predstavuje priemer. Plná čiara spája priemerné zmeny MLMT skóre
v priebehu návštev pre každú skupinu. Vybodkovaná čiara spája priemernú zmenu MLMT skóre v priebehu návštev u kontrolnej skupiny vrátane piatich návštev v priebehu prvého roka bez podávania voretigén neparvoveku. Kontrolnej skupine bol podávaný voretigén neparvovek po 1 roku pozorovania.
BL (Baseline): začiatok liečby;
D30, D90, D180: 30, 90 a 180 dní po začiatku štúdie; R1, R2, R3: jeden, dva a tri roky po začiatku štúdie.
XBL; XD30; XD90; XD180: začiatok liečby, 30, 90 a 180 dní po začiatku štúdie u kontrolnej skupiny po'
prechode na inú liečbu;
XR1; XR2: jeden a dva roky po začiatku štúdie u kontrolnej skupiny po prechode na inú liečbu.
Výsledky vyšetrenia celého zorného poľa na citlivosť na svetlo v prvom roku štúdie: biele svetlo
[Log10(cd.s/m2)] sú uvedené v tabuľke č. 5 nižšie.
Tabuľka č. 5 Vyšetrenie celého zorného poľa na citlivosť na svetloVyšetrenie celého zorného poľa na citlivosť na svetlo – prvé určené oko (ITT) Intervenčná skupina, N = 21začiatok liečby 1. rok zmenaN 20 20 19
Priemer (SE) -1,23 (0,10) -3,44 (0,30) -2,21 (0,30)
Kontrolná skupina, N = 10N 9 9 9
Priemer (SE) -1,65 (0,14) -1,54 (0,44) 0,12 (0,45) Rozdiel (95 % CI) (intervenčná skupina-kontrolná skupina)
-2,33 (-3,44; -1,22), p<0,001
Vyšetrenie celého zorného poľa na citlivosť na svetlo – druhé určené oko (ITT) Intervenčná skupina, N = 21začiatok liečby 1. rok zmenaN 20 20 19
Priemer (SE) -1,35 (0,09) -3,28 (0,29) -1,93 (0,31)
Kontrolná skupina, N = 10N 9 9 9
Priemer (SE) -1,64 (0,14) -1,69 (0,44) 0,04 (0,46) Rozdiel (95 % CI) (intervenčná skupina-kontrolná skupina)
-1,89 (-3,03; -0,75), p=0,002
Vyšetrenie celého zorného poľa na citlivosť na svetlo – priemer za obe oči (ITT)Rozdiel (95 % CI) (intervenčná skupina-kontrolná skupina): -2,11 (-3,19; -1,04), p<0,001
Zlepšenie celého zorného poľa na citlivosť na svetlo sa udržalo až 3 roky po expozícii voretigén
neparvoveku.
Po jednom roku od expozície voretigén neparvovekom sa v intervenčnej skupine zaznamenalo zlepšenie zrakovej ostrosti s hodnotou najmenej 0,3 LogMAR u 11/20 (55 %) očí liečených ako prvých a u 4/20 (20 %) očí liečených ako druhých. U nikoho z kontrolnej skupiny sa nezaznamenalo také zlepšenie zrakovej ostrosti, ani v prvom ani v druhom oku.
5.2 Farmakokinetické vlastnostiPredpokladá sa, že voretigén neparvovek bude vstrebaný bunkami prostredníctvom receptorov proteoglykánu heparínsulfátu a že sa bude odbúravať pomocou endogénnych proteínov
a katabolických dráh DNA.
Neklinická biodistribúciaBiodistribúcia Luxturny sa hodnotila u nehumánnych primátov v treťom mesiaci od subretinálneho
podania. Najvyššia hladina sekvencií DNA vektora bola zistená vo vnútroočných tekutinách (tekutina v prednej komore a sklovci) očí, do ktorých bol vektor injekčne podaný. Nízke hladiny sekvencií DNA vektora boli zistené v zrakovom nerve oka, do ktorého bol vektor injekčne podaný, v optickej chiasme, slezine a pečeni a ojedinele v žalúdku a lymfatických uzlinách. U jedného zvieraťa, ktorému bola podaná Luxturna s objemom 7,5 x 1011 vg (5-krát viac ako je odporúčaná dávka pre jedno oko), boli sekvencie DNA vektora zistené v hrubom čreve, dvanástniku a v priedušnici. V gonádach sekvencie DNA vektora zistené neboli.
K
l
i
n
i
cká farmakokinetika a vylučovanie vektora (shedding)
Vylučovanie a biodistribúcia vektora boli hodnotené v slzách oboch očí, v sére a v celej krvi pacientov
v klinickej štúdii fázy 3. U 13/29 (45 %) pacientov ktorým bola Luxturna podávaná bilaterálne, boli vo
vzorkách sĺz zistené sekvencie DNA vektora. U väčšiny týchto pacientov boli vzorky pri návšteve
1 deň po podaní injekcie negatívne, ale u štyroch z týchto pacientov boli vzorky sĺz pozitívne po prvom dni a u jedného pacienta až do 14. dňa po podaní injekcie do oka. Sekvencie vektorovej DNA boli zistené v sére u 3/29 (10 %) pacientov vrátane dvoch s pozitívnymi vzorkami sĺz, a to iba do
3. dňa po každej injekcii. Celkovo boli prechodné a nízke hladiny vektorovej DNA zistené v slzách a príležitostných vzorkách séra u 14/29 (48 %) pacientov v štúdii fázy 3.
Farmakokinetika v osobitných skupinách pacientov
V osobitných skupinách pacientov sa neuskutočnili žiadne farmakokinetické štúdie s voretigén
neparvovekom.
Poruchy f unkci e peče ne a obl iči ek
Luxturna sa injekčne podáva priamo do oka. Nepredpokladá sa, že funkcie pečene a obličiek,
polymorfizmus cytochrómu P450 a starnutie majú vplyv na klinickú účinnosť alebo bezpečnosť lieku. Preto u pacientov s poruchou funkcie pečene alebo obličiek nie je potrebná žiadna úprava dávky.
5.3 Predklinické údaje o bezpečnosti
Očná histopatológia u psov a nehumánnych primátov vystavených pôsobeniu voretigén neparvoveku ukázala iba mierne zmeny, ktoré sa väčšinou týkali uzdravovania sa po chirurgickom poškodení. V predchádzajúcej toxikologickej štúdii mal podobný AAV2 vektor podávaný subretinálne psom v dávke 10-krát vyššej ako odporúčaná dávka za následok fokálnu retinálnu toxicitu a infiltráty zápalových buniek histologicky v oblastiach vystavených pôsobeniu vektora. Ďalšie zistenia
z neklinických štúdií s voretigén neparvovekom zahŕňali príležitostný a izolovaný výskyt zápalových buniek v sietnici bez akejkoľvek zjavnej degenerácie sietnice. Po jednom podaní vektora sa u psov vyvinuli protilátky proti kapsidu vektora AAV2, ktoré u predtým neliečených nehumánnych primátov neboli prítomné.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Koncentrát chlorid sodný
monohydrát dihydrogénfosforečnanu sodného (na úpravu pH)
dihydrát hydrogénfosforečnanu sodného (na úpravu pH)
poloxamér 188
voda na injekcie
Rozpúšťadlo
chlorid sodný
monohydrát dihydrogénfosforečnanu sodného (na úpravu pH)
dihydrát hydrogénfosforečnanu sodného (na úpravu pH)
poloxamér 188
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
N
eotvorené zmrazené injekčné liekovky
21 mesiacov.
Po rozmrazení a zriedení
Po rozmrazení sa liek nesmie opakovane zmraziť a má sa nechať pri izbovej teplote (neprevyšujúcej
25 °C).
Po zriedení pri aseptických podmienkach sa roztok musí ihneď použiť; ak sa nepoužije okamžite, uchovávanie pri izbovej teplote (neprevyšujúcej 25 °C) nemá byť dlhšie ako 4 hodiny.
6.4 Špeciálne upozornenia na uchovávanie
Koncentrát a rozpúšťadlo sa musia uchovávať a prepravovať v mraze pri ≤ -65 ºC. Podmienky na uchovávanie po rozmrazení a riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
0,5 ml extrahovateľného objemu koncentrátu v 2 ml injekčnej liekovke z cyklického olefínového polyméru s chlorobutylovou gumenou zátkou utesnenou odtrhávacím hliníkovým viečkom.
1,7 ml extrahovateľného objemu rozpúšťadla v 2 ml z cyklického olefínového polyméru s chlorobutylovou gumenou zátkou utesnenou odtrhávacím hliníkovým viečkom.
V každom hliníkovom váčiku sa nachádza škatuľa obsahujúca 1 injekčnú liekovku koncentrátu a 2
injekčné liekovky rozpúšťadla.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Každá škatuľa obsahujúca 1 injekčnú liekovku koncentrátu a 2 injekčné liekovky rozpúšťadla je
určená iba na jednorazové použitie.
Luxturna sa má pred podaním vizuálne skontrolovať. Ak sú v nej viditeľné častice, zakalenie alebo zmena farby, injekčná liekovka na jednorazové použitie sa nesmie použiť.
Je potrebné vyhýbať sa náhodnej expozícii. Pri príprave, podávaní a manipulácii s voretigén neparvovekom je potrebné dodržiavať národné predpisy pre biologickú bezpečnosť.
· Počas prípravy a podávania voretigén neparvoveku je potrebné používať osobné ochranné prostriedky (zahŕňajúce laboratórny plášť, ochranné okuliare a rukavice).
· Je potrebné vyhýbať sa náhodnej expozícii voretigén neparvoveku, vrátane jeho styku s pokožkou, očami a sliznicami. Akékoľvek otvorené rany sa majú pred manipuláciou s liekom prikryť.
· Všetky prípady rozliatia voretigén neparvoveku sa musia ošetriť virusocídnym prostriedkom, ako je napr. 1 % chlórnan sodný a osušiť pomocou absorpčných materiálov.
· Všetky materiály, ktoré mohli prísť do styku s voretigén neparvovekom (napr. injekčná liekovka, injekčná striekačka, ihla, bavlnená gáza, rukavice, masky alebo obväzy) sa musia zlikvidovať v súlade s národnými predpismi pre biologickú bezpečnosť.
N
áhodná expozícia
· V prípade náhodnej expozície na pracovisku (napr. postriekania očí alebo slizníc), oplachujte
najmenej 5 minút čistou vodou.
· V prípade expozície poškodenej pokožky alebo poranenia injekčnou ihlou dôkladne omyte zasiahnutú plochu mydlom a vodou a/alebo dezinfekčným prostriedkom.
Tento liek obsahuje geneticky modifikované organizmy. Nepoužitý liek sa musí zlikvidovať v súlade s národnými predpismi pre biologickú bezpečnosť.
Príprava
Príprava Luxturny sa má vykonať do 4 hodín od začiatku postupu jeho prípravy, v súlade
s nasledujúcim odporúčaným postupom vykonávaným za aseptických podmienok.
Pri izbovej teplote rozmrazte jednu injekčnú liekovku koncentrátu na jednorazové použitie a dve
injekčné liekovky rozpúšťadla. Injekčné liekovky päťkrát opatrne obráťte, aby sa obsah premiešal.
Skontrolujte injekčné liekovky na akékoľvek viditeľné častice alebo akékoľvek odlišnosti. Akékoľvek odlišnosti alebo vznik viditeľných častíc sa majú nahlásiť držiteľovi rozhodnutia o registrácii a liek sa nemá používať.
Preneste 2,7 ml rozpúšťadla odobratého z dvoch rozmrazených injekčných liekoviek pomocou 3 ml
injekčnej striekačky do sterilnej 10 ml prázdnej sklenenej injekčnej liekovky.
Na riedenie naberte 0,3 ml rozmrazeného koncentrátu do 1 ml injekčnej striekačky a pridajte ho do
10 ml sterilnej injekčnej liekovky obsahujúcej rozpúšťadlo. Opatrne injekčnú liekovku najmenej päťkrát obráťte, aby sa obsah riadne premiešal. Skontrolujte obsah na akékoľvek viditeľné častice.
Zriedený roztok má byť číry až mierne opalescenčný. Označte 10 ml sklenenú injekčnú liekovku
obsahujúcu zriedený koncentrát nápisom: ‘Zriedená Luxturna’.
Nepripravujte injekčnú striekačku, ak injekčná liekovka vykazuje nejaké poškodenie alebo ak sa v nej dajú pozorovať nejaké viditeľné častice. Pripravte si injekčné striekačky na podanie injekcie tak, že do sterilnej 1 ml injekčnej striekačky naberiete 0,8 ml zriedeného roztoku. Ten istý postup zopakujte pri príprave rezervnej injekčnej striekačky. Injekčné striekačky naplnené liekom sa potom majú
v označenom prepravnom kontajneri preniesť do operačnej sály.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Spark Therapeutics Ireland Ltd. Studio G3, The Tower
Trinity Technology & Enterprise Campus
Pearse Street
Dublin 2
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/18/1331/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.