
ého podávania sildenafilu u pacientov
s erektilnou dysfunkciou.
vardenafil Zvýšené koncentrácie vardenafilu v plazme
(pozri časti 4.4 a 4.5).
Sedatíva/hypnotiká perorálne podávaný midazolam, triazolam
Zvýšené koncentrácie perorálne podávaného midazolamu a triazolamu. Tým sa zvyšuje riziko nadmernej sedácie a útlmu dýchania spôsobené týmito liečivami. Upozornenia týkajúce sa parenterálne podávaného midazolamu, pozri časť 4.5.
 Z
nížená koncentrácia lopinaviru/ritonaviru v plazme
Z
nížená koncentrácia lopinaviru/ritonaviru v plazme
Rastlinné produkty ľubovník bodkovaný Rastlinné prípravky obsahujúce ľubovník bodkovaný (
Hypericum perforatum) spôsobujú riziko znížených koncentrácií lopinaviru a ritonaviru v plazme a zníženie klinických účinkov (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pacienti s pri druž enými ochoreniami
H
epatálna porucha: Bezpečnosť a účinnosť lopinaviru/ritonaviru neboli stanovené u pacientov
so signifikantnou poruchou funkcie pečene. Lopinavir/ritonavir je kontraindikovaný u pacientov s ťažkou poruchou funkcie pečene (pozri časť 4.3). Pacienti s chronickou hepatitídou B alebo C a pacienti liečení kombinovanou antiretrovírusovou terapiou majú zvýšené riziko závažných
a potenciálne fatálnych hepatálnych nežiaducich reakcií. V prípade súbežnej antivirálnej terapie
hepatitídy B alebo C si pozrite príslušné informácie o týchto liekoch.
Pacienti s poruchou funkcie pečene vrátane chronickej hepatitídy majú zvýšený výskyt abnormalít
funkcie pečene v priebehu kombinovanej antiretrovírusovej terapie a majú byť sledovaní v súlade
so štandardnou praxou. Ak u týchto pacientov dôjde k zhoršeniu pečeňového ochorenia, má sa zvážiť prerušenie alebo ukončenie liečby.
U pacientov s monoinfekciou HIV-1 a jedincov užívajúcich liek ako postexpozičnú profylaktickú liečbu boli hlásené zvýšené hodnoty transamináz so zvýšením hodnôt bilirubínu alebo bez nich, a to už po 7 dňoch liečby lopinavirom/ritonavirom súčasne s ďalšími antiretrovírusovými liečivami.
V niektorých prípadoch bola hepatálna dysfunkcia závažná.
Pred začiatkom liečby lopinavirom/ritonavirom je potrebné vykonať príslušné laboratórne vyšetrenia
a počas liečby treba pacienta starostlivo sledovať.
Porucha obličiek: Keďže renálny klírens lopinaviru a ritonaviru je nepatrný, nepredpokladajú sa zvýšené plazmatické hladiny u pacientov s poruchou funkcie obličiek. Keďže lopinavir a ritonavir sú pevne viazané na proteíny, nie je pravdepodobné, že by mohli byť významne eliminované hemodialýzou alebo peritoneálnou dialýzou.
Hemofília: U pacientov s hemofíliou A a B, liečených inhibítormi proteáz, bolo hlásené zvýšené krvácanie vrátane spontánnych kožných hematómov a hemartrózy. Niektorým pacientom bol dodatočne podaný faktor VIII. Vo viac ako polovici hlásených prípadov liečba inhibítormi proteáz pokračovala alebo bola po prerušení opäť začatá. Predpokladá sa kauzálny vzťah, aj keď mechanizmus účinku nebol zatiaľ objasnený. Pacienti s hemofíliou sa majú upozorniť na možnosť zvýšeného krvácania.
Zvýš ená hladi na li pidov
Liečba lopinavirom/ritonavirom zvyšuje, niekedy výrazne, koncentráciu celkového cholesterolu a triglyceridov. Vyšetrenie hladiny triglyceridov a cholesterolu sa má urobiť pred začatím liečby lopinavirom/ritonavirom a počas liečby v pravidelných intervaloch. Zvýšená opatrnosť má byť
u pacientov so zvýšenými hodnotami pred začatím liečby a s anamnézou poruchy metabolizmu
lipidov. Poruchy lipidov sa majú liečiť klinicky primeraným spôsobom (pozri aj časť 4.5 ohľadom ďalších informácií o potenciálnych interakciách s inhibítormi HMG-CoA reduktázy).
Pankreatitída
U pacientov užívajúcich lopinavir/ritonavir vrátane tých, u ktorých sa vyvinula hypertriglyceridémia, boli hlásené prípady pankreatitídy. Vo väčšine prípadov išlo o pacientov s predchádzajúcou pankreatitídou v anamnéze a/alebo pacientov so súbežnou liečbou s inými liekmi, ktoré môžu spôsobovať pankreatitídu. Výrazné zvýšenie hladiny triglyceridov je rizikovým faktorom pre vznik pankreatitídy. Pacienti s pokročilým HIV ochorením majú riziko zvýšenej hladiny triglyceridov
a pankreatitídy.
O pankreatitíde sa má uvažovať, ak sa objavia klinické príznaky (nauzea, vracanie, bolesť brucha) alebo abnormality laboratórnych hodnôt, typické pre pankreatitídu (ako zvýšená hladina lipázy alebo amylázy v sére). Pacienti s týmito prejavmi alebo príznakmi majú byť vyšetrení a liečba lopinavirom/ritonavirom sa má ukončiť, ak sa potvrdí diagnóza pankreatitídy (pozri časť 4.8).
Hyperglykémia
U pacientov užívajúcich inhibítory proteáz bol hlásený novovzniknutý diabetes mellitus,
hyperglykémia alebo exacerbácia existujúceho diabetes mellitus. V niektorých prípadoch išlo
o závažnú hyperglykémiu, niekedy spojenú s ketoacidózou. Mnohí pacienti mali pridružené ochorenia, ktoré vyžadovali podávanie liečiv, ktoré sa spájajú so vznikom diabetes mellitus alebo hyperglykémiou.
Redistribúcia telesného tuku a metabolické poruchy
Kombinovaná antiretrovírusová liečba spôsobila u niektorých pacientov s HIV redistribúciu telesného tuku (lipodystrofiu). Dlhodobé následky týchto účinkov v súčasnosti nie sú známe a rovnako nie je známy ani presný mechanizmus vzniku. Predpokladá sa súvislosť medzi viscerálnou lipomatózou
a inhibítormi proteáz (PI) a lipoatrofiou a inhibítormi nukleozidovej reverznej transkriptázy (NRTI).
Zvýšené riziko lipodystrofie bolo spojené s individuálnymi faktormi ako vyšší vek a faktormi týkajúcimi sa liekov, ako dlhšie trvanie antiretrovírusovej liečby a pridružené metabolické poruchy. Klinické vyšetrenie má zahŕňať aj zhodnotenie fyzikálnych znakov redistribúcie tuku. Má sa venovať pozornosť aj meraniu hladín sérových lipidov a glukózy v krvi nalačno. Dyslipidémie majú byť primerane riešené (pozri časť 4.8).
Syndróm imunitnej reaktivácie
U HIV-infikovaných pacientov s ťažkou imunodeficienciou môže v čase začatia kombinovanej antiretrovírusovej terapie (“Combination Antiretroviral Therapy”, CART) vzniknúť zápalová reakcia na asymptomatické alebo reziduálne oportúnne patogény a spôsobiť závažné klinické stavy alebo zhoršenie symptómov. Takéto reakcie sú zvyčajne pozorované počas prvých niekoľkých týždňov alebo mesiacov po začatí CART. Relevantnými príkladmi sú cytomegalovírusová retinitída, generalizované a/alebo fokálne mykobakteriálne infekcie a pneumónia spôsobená Pneumocystis jiroveci pneumonia. Akékoľvek zápalové symptómy sa musia zhodnotiť a v prípade potreby sa musí začať liečba.
Pri imunitnej reaktivácii boli hlásené aj autoimunitné poruchy (ako napr. Gravesova choroba), ale hlásená doba nástupu je variabilnejšia a môže sa vyskytnúť mnoho mesiacov po začatí liečby.
Osteonekróza
Aj keď sa etiológia považuje za multifaktoriálnu (vrátane používania kortikosteroidov, konzumácie alkoholu, ťažkej imunosupresie, vyššieho indexu telesnej hmotnosti), boli hlásené prípady osteonekrózy, najmä u pacientov s pokročilým HIV ochorením a/alebo dlhodobou expozíciou kombinovanej antiretrovírusovej terapii (“Combination Antiretroviral Therapy”, CART). Pacientom sa má odporučiť, aby vyhľadali lekársku pomoc, ak budú mať bolesť kĺbov, stuhnutosť kĺbov alebo ťažkosti s pohybom.
PredĺženiePRintervalu
Preukázalo sa, že lopinavir/ritonavir spôsobuje u niektorých zdravých dospelých jedincov mierne asymptomatické predĺženie PR intervalu. U pacientov, ktorí užívali lopinavir/ritonavir, a mali štrukturálnu chorobu srdca a už existujúce abnormality prevodového systému alebo dostávali lieky, o ktorých je známe, že predlžujú PR interval (také ako verapamil alebo atazanavir), boli zriedkavé hlásenia o výskyte atrioventrikulárnej blokády II. alebo III. stupňa. U takýchto pacientov sa má lopinavir/ritonavir používať s opatrnosťou (pozri časť 5.1).
Interakcie s liekmi
Lopinavir/Ritonavir Mylan tablety obsahujú lopinavir a ritonavir, ktoré sú inhibítormi izoformy CYP3A cytochrómu P450. Lopinavir/ritonavir pravdepodobne zvyšuje plazmatické koncentrácie liečiv, ktoré sú primárne metabolizované CYP3A. Toto zvýšenie plazmatických koncentrácií súbežne
podávaných liečiv môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce účinky (pozri časti
4.3 a 4.5).
Silné inhibítory CYP3A4, ako sú inhibítory proteázy, môžu zvýšiť expozíciu bedachilínu, čo môže potenciálne zvýšiť riziko nežiaducich reakcií súvisiacich s bedachilínom. Preto je potrebné vyhnúť sa kombinácii bedachilínu s lopinavirom/ritonavirom. Avšak ak prínos prevažuje nad rizikami, súbežné podávanie bedachilínu s lopinavirom/ritonavirom je nutné vykonávať s opatrnosťou. Odporúča sa častejšie monitorovanie EKG a transamináz (pozri časť 4.5 a SmPC bedachilínu).
Je potrebné vyhnúť sa súbežnému podávaniu s kolchicínom, najmä u pacientov so zhoršenou funkciou obličiek alebo pečene (pozri časť 4.5).
Kombinácia lopinaviru/ritonaviru:
- s tadalafilom, indikovaným na liečbu pulmonálnej artériovej hypertenzie sa neodporúča (pozri časť 4.5);
- s kyselinou fusidovou podávanou na liečbu osteoartikulárnych infekcií sa neodporúča (pozri časť 4.5);
- so salmeterolom sa neodporúča (pozri časť 4.5);
- s rivaroxabanom sa neodporúča (pozri časť 4.5).
Kombinácia lopinaviru/ritonaviru s atorvastatínom sa neodporúča. Ak je užívanie atorvastatínu nevyhnutné, má sa podávať v najnižšej možnej dávke za starostlivého monitorovania bezpečnosti. Súbežné použitie lopinaviru/ritonaviru s rosuvastatínom si tiež vyžaduje opatrnosť a majú sa zvážiť nižšie dávky. Ak je indikovaná liečba s inhibítormi HMG-CoA reduktázy, odporúča sa použiť pravastatín alebo fluvastatín (pozri časť 4.5).
Inhibítory PDE5: Zvýšená opatrnosť je potrebná pri predpisovaní sildenafilu alebo tadalafilu na liečbu erektilnej dysfunkcie pacientom, užívajúcim lopinavir/ritonavir. Predpokladá sa, že pri súbežnom užívaní lopinaviru/ritonaviru s týmito liečivami sa výrazne zvýšia ich koncentrácie a môže dôjsť
k výskytu nežiaducich účinkov, ako sú hypotenzia, synkopa, zmeny zraku a predĺžená erekcia (pozri časť 4.5). Súbežné užívanie avanafilu alebo vardenafilu a lopinaviru/ritonaviru je kontraindikované (pozri časť 4.3). Súbežné užívanie sildenafilu, predpísaného na liečbu pľúcnej arteriálnej hypertenzie, a lopinaviru/ritonaviru je kontraindikované (pozri časť 4.3).
Zvýšená opatrnosť je nutná, ak sa užíva lopinavir/ritonavir spolu s liekmi, o ktorých je známe, že predlžujú QT interval ako: chlórfenamín, chinidín, erytromycín, klaritromycín. Lopinavir/ritonavir môže zvýšiť koncentráciu týchto liekov, čo môže viesť k zvýšeniu výskytu ich kardiálnych nežiaducich reakcií. V predklinických štúdiách s lopinavirom/ritonavirom boli hlásené kardiálne príhody; takže nie je možné vylúčiť potencionálne kardiálne účinky lopinaviru/ritonaviru (pozri časti
4.8 a 5.3).
Súbežné podávanie lopinaviru/ritonaviru s rifampicínom sa neodporúča. Rifampicín v kombinácii
s lopinavirom/ritonavirom zapríčiňuje výrazné zníženie hladín lopinaviru a tým môže významne znížiť terapeutický účinok lopinaviru. Primeraná expozícia lopinaviru/ritonaviru sa môže dosiahnuť vtedy, keď sa použije vyššia dávka lopinaviru/ritonaviru, ale to je spojené s vyšším rizikom pečeňovej
a gastrointestinálnej toxicity. Preto je potrebné sa vyhnúť ich súbežnému podaniu, pokiaľ sa to
nepovažuje za striktne nevyhnutné (pozri časť 4.5).
Súbežné použitie lopinaviru/ritonaviru a flutikazónu alebo iných glukokortikoidov, ktoré sú metabolizované CYP3A4, ako je budezonid, sa neodporúča, pokiaľ potenciálny prínos liečby neprevýši riziko systémových účinkov kortikosteroidov vrátane Cushingovho syndrómu a supresie nadobličiek (pozri časť 4.5).
Iné
Lopinavir/ritonavir nevylieči HIV infekciu alebo AIDS. Hoci sa preukázalo, že účinná vírusová
supresia dosiahnutá pri antiretrovírusovej terapii značne znižuje riziko prenosu HIV pohlavným
stykom, reziduálne riziko nie je možné vylúčiť. Je potrebné prijať opatrenia na zabránenie prenosu HIV v súlade s národnými odporúčaniami. Aj u ľudí, užívajúcich lopinavir/ritonavir, môžu vzniknúť infekcie alebo iné ochorenia spojené s ochorením HIV alebo AIDS.
4.5 Liekové a iné interakcie
Lopinavir/Ritonavir Mylan tablety obsahujú lopinavir a ritonavir, ktoré sú inhibítormi izoformy CYP3A cytochrómu P450 in vitro. Súbežné podávanie lopinaviru/ritonaviru s iným liekmi, primárne metabolizovanými CYP3A, môže viesť k zvýšeným plazmatickým koncentráciám týchto liekov, čo môže zvýšiť alebo predĺžiť jej terapeutické alebo nežiaduce reakcie. Lopinavir/ritonavir neinhibuje CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 ani CYP1A2 v klinicky používaných koncentráciách (pozri časť 4.3).
Ukázalo sa, že lopinavir/ritonavir in vivo indukuje svoj vlastný metabolizmus a zvyšuje biotransformáciu niektorých liekov metabolizovaných enzýmami cytochrómu P450 (vrátane CYP2C9 a CYP2C19) a glukuronidáciou. To môže viesť k zníženej plazmatickej koncentrácii a prípadne znižovať účinnosť súbežne podávaných liekov.
Lieky, ktorých podávanie je špeciálne kontraindikované vzhľadom na očakávanú intenzitu interakcií a riziko závažných nežiaducich účinkov, sú uvedené v časti 4.3.
Všetky interakčné štúdie, pokiaľ nie je uvedené inak, sa vykonali s kapsulami lopinaviru/ritonaviru, pričom dochádza k približne o 20% nižšej expozícii lopinaviru ako u 200/50 mgtabliet.
Známe a teoreticky možné interakcie s vybranými antiretrovirotikami a ne-antiretrovírusovými liekmi
sú uvedené nižšie v tabuľke.
Tabuľka i nt erak ci í
Interakcie medzi lopinavirom/ritonavirom a súbežne podávanými liekmi sú uvedené nižšie v tabuľke
(zvýšenie je označené ako “↑”, zníženie ako “↓”, bez zmeny sa označuje ako “↔”, jedenkrát denne
ako “QD”, dvakrát denne ako “BID” a trikrát denne ako “TID”).
Ak nie je uvedené inak, v nižšie popísaných skúšaniach bolo použité odporúčané dávkovanie lopinaviru/ritonaviru (t.j. 400/100 mg dvakrát denne).
Súbežne podávané
li
e
čivá podľa
t
erapeutických oblastí
A
ntiretrovírusové látky
Ú
činky na hladiny liečiva
G
eometrický priemer zmien
(
%
) AUC, C
m
a
x
, C
m
i
n
Mechanizmus interakcie
K
l
i
nické odporúčanie týkajúce
sa súbežného podávania
s lopinavirom/ritonavirom
I
nhibítory nukleozidovej/nukleotidovej reverznej transkriptázy (NRTI)
Stavudín, lamivudín Lopinavir: ↔ Úprava dávky nie je potrebná.
Abakavir, zidovudín Abakavir, zidovudín:
koncentrácie sa môžu znížiť kvôli lopinavirom/ritonavirom zvýšenej glukuronidácii.
Tenofovir, 300 mg QD Tenofovir: AUC: ↑ 32% Cmax : ↔ Cmin: ↑ 51%
Lopinavir: ↔
Klinický význam znížených
koncentrácií abakaviru
a zidovudínu nie je známy.
Úprava dávky nie je potrebná. Vyššie koncentrácie tenofoviru môžu zosilniť nežiaduce účinky spojené s tenofovirom vrátane porúch obličiek.
I
nhibítory nenukleozidovej reverznej transkriptázy (NNRTI)
Efavirenz, 600 mg QD Lopinavir: AUC: ↓ 20% Cmax : ↓ 13% Cmin: ↓ 42%
Pri súbežnom podávaní
s efavirenzom sa má dávkovanie tabliet lopinaviru/ritonaviru zvýšiť na 500/125 mg dvakrát
Efavirenz, 600 mg QD
(Lopinavir/ritonavir
500/125 mg BID)
Lopinavir: ↔
(v porovnaní s dávkou
400/100 mg BID podávanou samostatne)
denne.
Lopinavir/ritonavir sa nesmie
podávať jedenkrát denne
v kombinácii s efavirenzom.
Nevirapín, 200 mg BID Lopinavir: AUC: ↓ 27% Cmax: ↓ 19% Cmin: ↓ 51%
Pri súbežnom podávaní
s nevirapínom sa má dávkovanie tabliet lopinaviru/ritonaviru
zvýšiť na 500/125 mg dvakrát
denne.
Etravirín (Lopinavir/ritonavir tableta 400/100 mg BID)
Rilpivirín (Lopinavir/ritonavir kapsula 400/100 mg BID)
Antagonista HIV CCR5
Etravirín: AUC: ↓ 35% Cmin: ↓ 45% Cmax: ↓ 30%
Lopinavir: AUC: ↔ Cmin : ↓ 20% Cmax: ↔ Rilpivirín:
AUC: ↑ 52%
Cmin: ↑ 74% Cmax: ↑ 29%
Lopinavir: AUC: ↔ Cmin: ↓ 11% Cmax: ↔
(inhibícia enzýmov CYP3A)
Úprava dávky nie je potrebná.
Súbežné používanie
lopinaviru/ritonaviru
s rilpivirínom spôsobuje zvýšenie
plazmatických koncentrácií
rilpivirínu, ale úprava dávky nie je
potrebná.
Maravirok Maravirok: AUC: ↑ 295% Cmax: ↑ 97%
Spôsobené inhibíciou CYP3A
lopinavirom/ritonavirom.
Pri súbežnom podávaní
s lopinavirom/ritonavirom
400/100 mg dvakrát denne sa má
dávka maraviroku znížiť na
150 mg dvakrát denne.
 I
nhibítor integrázy
I
nhibítor integrázy
Raltegravir Raltegravir: AUC: ↔ Cmax: ↔
C12: ↓ 30%
Lopinavir: ↔
Úprava dávky nie je potrebná.
Súbežné podávanie s inými inhibítormi HIV proteázy (PI)
Na základe aktuálnych liečebných smerníc sa duálna liečba inhibítormi proteáz vo všeobecnosti neodporúča.
Fosamprenavir/ritonavir (700/100 mg BID) (Lopinavir/ritonavir
400/100 mg BID)
alebo
Fosamprenavir
(1400 mg BID)
(Lopinavir/ritonavir
533/133 mg BID)
Fosamprenavir:
koncentrácie amprenaviru sa
signifikantne znižujú.
Súbežné podávanie zvýšených dávok fosamprenaviru (1400 mg BID) s lopinavirom/ritonavirom (533/133 mg BID) pacientom, ktorí už boli liečení inhibítorom proteázy, malo pri tomto kombinovanom režime
v porovnaní so štandardnými dávkami fosamprenaviru/ritonaviru za následok vyšší výskyt gastrointestinálnych nežiaducich účinkov a zvýšenie triacylglycerolov bez zvýšenia virologickej účinnosti. Preto sa súbežné podávanie týchto liekov neodporúča.
Indinavir, 600 mg BID Indinavir:
AUC: ↔
Cmin: ↑ 3,5-násobne
Cmax: ↓
(v porovnaní so samotným indinavirom 800 mg TID)
Vhodné dávky pre túto kombináciu, s ohľadom na účinnosť a bezpečnosť, neboli stanovené.
Sachinavir
1000 mg BID
Lopinavir: ↔
(na základe predchádzajúcich porovnaní)
Sachinavir: ↔ Úprava dávky nie je potrebná.
Tipranavir/ritonavir
(500/100 mg BID)
Antacidá
Lopinavir: AUC: ↓ 55% Cmin: ↓ -70% Cmax: ↓ 47%
Súbežné podávanie týchto liekov sa neodporúča.
Omeprazol (40 mg QD) Omeprazol: ↔ Lopinavir: ↔
Úprava dávky nie je potrebná.
Ranitidín (150 mg
v jednorazovej dávke)
Ranitidín: ↔ Úprava dávky nie je potrebná.
 Antagonisty alfa
1
adrenoreceptoru
Antagonisty alfa
1
adrenoreceptoru
Alfuzosín Alfuzosín:
kvôli inhibícii CYP3A
lopinavirom/ritonavirom sa predpokladajú zvýšené koncentrácie alfuzosínu.
AnalgetikáFentanyl Fentanyl:
zvýšené riziko nežiaducich účinkov (respiračná depresia, sedácia) kvôli vyšším plazmatickým koncentráciám z dôvodu inhibície CYP3A4 lopinavirom/ritonavirom.
Súbežné podávanie lopinaviru/ritonaviru a alfuzosínu je kontraindikované (pozri časť
4.3), pretože sa môže zvýšiť
toxicita súvisiaca s alfuzosínom vrátane hypotenzie.
Pri súbežnom podávaní fentanylu s lopinavirom/ritonavirom sa odporúča starostlivé monitorovanie nežiaducich účinkov (najmä respiračnej depresie, ale aj sedácie).
Antiarytmiká
Digoxín Digoxín:
plazmatické koncentrácie sa môžu zvýšiť z dôvodu inhibície P- glykoproteínu lopinavirom/ritonavirom.
Zvýšená hladina digoxínu sa môže časom znížiť, pretože sa
vyvinie indukcia Pgp.
V prípade súbežného podávania lopinaviru/ritonaviru a digoxínu je potrebná opatrnosť a pokiaľ je to možné, odporúča sa terapeutické monitorovanie koncentrácií digoxínu. Osobitná pozornosť je nutná pri predpisovaní lopinaviru/ritonaviru pacientom užívajúcim digoxín, pretože je možné očakávať, že akútny inhibičný účinok ritonaviru na Pgp výrazne zvýši hladiny digoxínu. Predpokladá sa, že začatie liečby digoxínom
u pacientov, ktorí už užívajú
lopinavir/ritonavir, má za následok nižší vzostup koncentrácií digoxínu.
Bepridil, systémovo podaný lidokaín
a chinidín
Antibiotiká
Bepridil, systémovo podaný lidokaín a chinidín:
pri súbežnom podávaní
s lopinavirom/ritonavirom môžu byť ich koncentrácie zvýšené.
Je nutná zvýšená opatrnosť
a odporúča sa monitorovanie terapeutických koncentrácií, ak je to možné.
Klaritromycín Klaritromycín:
Mierne zvýšenie AUC
klaritromycínu sa očakáva na základe inhibície CYP3A lopinavirom/ritonavirom.
Cytostatiká
U pacientov s poruchou funkcie obličiek (CrCL < 30 ml/min) sa má zvážiť zníženie dávky klaritromycínu (pozri časť 4.4). Pri podávaní klaritromycínu
s lopinavirom/ritonavirom pacientom s poruchou funkcie pečene alebo obličiek je potrebná zvýšená opatrnosť.
Väčšina inhibítorov tyrozínkinázy, ako sú dasatinib a nilotinib, vinkristín, vinblastín
Antikoagulanciá
Väčšina inhibítorov tyrozínkinázy, ako sú dasatinib a nilotinib a tiež vinkristín
a vinblastín:
riziko zvýšenia výskytu nežiaducich účinkov kvôli vyšším sérovým koncentráciám z dôvodu inhibície CYP3A4 lopinavirom/ritonavirom.
Starostlivé monitorovanie tolerancie týchto cytostatík.
Warfarín Warfarín:
pri súbežnom podávaní
s lopinavirom/ritonavirom môžu
byť koncentrácie ovplyvnené na
základe indukcie CYP2C9.
Odporúča sa monitorovanie INR
(international normalised ratio).
Rivaroxaban (Ritonavir 600 mg dvakrát denne)
Rivaroxaban: AUC: ↑ 153% Cmax: ↑ 55%
Spôsobené inhibíciou CYP3A
a P-gp lopinavirom/ritonavirom.
Súčasné podávanie rivaroxabanu a lopinaviru/ritonaviru môže zvýšiť vystavenie rivaroxabanu, čo môže zvýšiť riziko krvácania. Použitie rivaroxabanu sa neodporúča u pacientov, ktorí
Antikonvulzíva
Fenytoín Fenytoín:
rovnovážne koncentrácie boli mierne znížené z dôvodu indukcie
CYP2C9 a CYP2C19
lopinavirom/ritonavirom.
Lopinavir:
koncentrácie sú znížené z dôvodu indukcie CYP3A fenytoínom.
súbežne užívajú
lopinavir/ritonavir (pozri časť
4.4).
Pri súbežnom užívaní fenytoínu a lopinaviru/ritonaviru je potrebná zvýšená opatrnosť. Keď sa fenytoín podáva spolu
s lopinavirom/ritonavirom, majú
sa monitorovať jeho hladiny. Pri súbežnom podávaní
s fenytoínom možno uvažovať
o zvýšení dávky lopinaviru/ritonaviru. Úprava dávky nebola hodnotená
v klinickej praxi. Lopinavir/ritonavir sa
v kombinácii s fenytoínom
nesmie podávať jedenkrát denne.
Karbamazepín a fenobarbital
Karbamazepín:
sérové koncentrácie sa môžu zvýšiť na základe inhibície
CYP3A lopinavirom/ritonavirom.
Lopinavir:
koncentrácie sa môžu znížiť na
základe indukcie CYP3A
karbamazepínom a fenobarbitalom.
Pri podávaní karbamazepínu alebo fenobarbitalu
s lopinavirom/ritonavirom je potrebná zvýšená opatrnosť. Hladiny karbamazepínu
a fenobarbitalu sa majú
monitorovať ak podáva spolu s lopinavirom/ritonavirom.
Pri súbežnom podávaní
s karbamazepínom alebo fenobarbitalom treba zvážiť zvýšenie dávky lopinaviru/ritonaviru. Úprava dávky nebola hodnotená
v klinickej praxi. Lopinavir/ritonavir sa nesmie podávať jedenkrát denne
v kombinácii s karbamazepínom a fenobarbitalom

Lamotrigín a valproát Lamotrigín: AUC: ↓ 50% Cmax: ↓ 46% Cmin: ↓ 56%
Spôsobené indukciou lamotrigínu glukuronidáciou.
Valproát: ↓
Keď sa lopinavir/ritonavir a kyselina valproová alebo valproát podávajú súbežne, je potrebné pacientov starostlivo monitorovať na znížený účinok VPA.
Pacienti,ktorízačínajúaleboukončujúliečbulopinavirom/ritonavirom počassúbežnéhoužívaniaudržiavacejdávky lamotrigínu:
Môže byť potrebné zvýšiť dávku lamotrigínu, keď sa pridáva lopinavir/ritonavir, alebo znížiť, keď sa ukončuje liečba lopinavirom/ritonavirom, a preto sa má monitorovať plazmatický lamotrigín, najmä pred a počas
Antidepresíva a anxiolytiká
2 týždňov po začatí alebo ukončení liečby lopinavirom/ritonavirom, aby sa zistilo, či je potrebná úprava dávky lamotrigínu.
Pacienti, ktorí užívajú v súčasnej
dobe lopinavir/ritonavir
a začínajúliečbulamotrigínom: Nie je potrebná žiadna úprava odporúčaného zvyšovania dávky lamotrigínu.
Trazodon jednorazová dávka
(Ritonavir, 200 mg
BID)
Antimykotiká Ketokonazol a itrakonazol
Trazodon:
AUC: ↑ 2,4-násobne
Pri súbežnom podaní trazodonu a ritonaviru sa zaznamenali nežiaduce účinky ako nauzea, závrat, hypotenzia a synkopa.
Ketokonazol, itrakonazol: sérové koncentrácie sa môžu zvýšiť na základe inhibície CYP3A lopinavirom/ritonavirom.
Nie je známe, či kombinácia lopinaviru/ritonaviru spôsobuje podobné zvýšenie expozície trazodonu. Kombinácia sa má používať s opatrnosťou a má sa zvážiť nižšia dávka trazodonu.
Neodporúča sa podávať vysoké
dávky ketokonazolu
a itrakonazolu (> 200 mg/deň).
Vorikonazol Vorikonazol:
koncentrácie sa môžu znížiť.
Liečivá proti dne
Je potrebné vyhnúť sa súbežnému užívaniu vorikonazolu v nízkej dávke ritonaviru (100 mg BID) obsiahnutej v tabletách lopinaviru/ritonaviru, pokiaľ zhodnotenie pomeru
prínosu/rizika pre pacienta neodôvodní použitie vorikonazolu.
Kolchicín jednorazová dávka
(Ritonavir 200 mg 2x denne )
Antiinfektíva
Kolchicín:
AUC: ↑ 3-násobne
Cmax: ↑ 1,8-násobne
Z dôvodu inhibície P-gp a/alebo
CYP3A4 ritonavirom.
Súbežné podávanie
lopinaviru/ritonaviru
s kolchicínom sa neodporúča kvôli možnému zvýšeniu
neuromuskulárnej toxicity súvisiacej s kolchicínom (vrátane
rabdomyolýzy), najmä
u pacientov so zhoršenou funkciou obličiek alebo pečene
(pozri časť 4.4).

Kyselina fusidová Kyselina fusidová:
Môžu sa zvýšiť koncentrácie v dôsledku inhibície CYP3A lopinavirom/ritonavirom.
Súbežné podávanie lopinaviru/ritonaviru s kyselinou fusidovou je kontraindikované pri dermatologických indikáciách kvôli zvýšenému riziku nežiaducich účinkov súvisiacich
s kyselinou fusidovou, najmä
rabdomyolýzy (pozri časť 4.3). Pri použití v liečbe
osteoartikulárnych infekcií, kde
sa súbežnému podávaniu nedá
Antimykobakteriálne lieky
Bedachilín
(jednorazová dávka)
(Lopinavir/ritonavir
400 mg/100 mg 2x denne, opakovaná dávka)
Bedachilín: AUC: ↑ 22% Cmax: ↔
Výraznejší vplyv na plazmatické expozície bedachilínu sa môže pozorovať pri dlhšom súbežnom podávaní
s lopinavirom/ritonavirom.
Inhibícia CYP3A4 pravdepodobne spôsobená lopinavirom/ritonavirom.
vyhnúť, sa dôrazne odporúča starostlivé monitorovanie nežiaducich účinkov týkajúcich sa svalov.
Vzhľadom na riziko nežiaducich účinkov súvisiacich
s bedachilínom je potrebné
vyhnúť sa kombinácii bedachilínu a lopinaviru/ritonaviru. Ak prínos
prevažuje nad rizikami, súbežné
podávanie bedachilínu
s lopinavirom/ritonavirom je nutné vykonávať s opatrnosťou. Odporúča sa častejšie monitorovanie EKG
a transamináz (pozri časť 4.4
a SmPC bedachilínu).
Rifabutín, 150 mg QD Rifabutín (materské liečivo
a aktívny 25-O-desacetylový metabolit):
AUC: ↑ 5,7-násobne
Cmax: ↑ 3,5-násobne
Rifampicín Lopinavir:
môže sa pozorovať výrazné zníženie koncentrácií lopinaviru
z dôvodu indukcie CYP3A
rifampicínom.
Pri súbežnom podávaní
s lopinavirom/ritonavirom je
odporúčaná dávka rifabutínu
150 mg 3-krát týždenne v určených dňoch (napr. pondelok - streda - piatok). Starostlivé monitorovanie
s rifabutínom súvisiacich
nežiaducich reakcií vrátane neutropénie a uveitídy sa vyžaduje kvôli očakávanému zvýšeniu expozície rifabutínu. Ďalšia redukcia dávky rifabutínu na 150 mg 2-krát týždenne
v určených dňoch sa odporúča
u pacientov, ktorí netolerovali dávku 150 mg 3-krát týždenne. Treba brať do úvahy, že dávkovanie 150 mg 2-krát denne nemusí poskytovať optimálnu expozíciu rifabutínu, čo môže viesť k riziku vzniku rezistencie na rifamycín a k zlyhaniu liečby.
Dávku lopinaviru/ritonaviru nie je
potrebné upravovať.
Súbežné podávanie
lopinaviru/ritonaviru
s rifampicínom sa neodporúča.
Rifampicín podávaný spolu s lopinavirom/ritonavirom
zapríčiňuje veľké zníženie
koncentrácií lopinaviru, čo môže významne znížiť terapeutický
účinok lopinaviru. Úprava dávky
lopinaviru/ritonaviru na
400 mg/400 mg (t.j. lopinavir/ritonavir 400/100 mg + ritonavir 300 mg) dvakrát denne umožnila vykompenzovať
 Antipsychotiká
Antipsychotiká
Kvetiapín Kvôli inhibícii CYP3A lopinavirom/ritonavirom sa predpokladajú zvýšené koncentrácie kvetiapínu.
BenzodiazepínyMidazolam Perorálny midazolam: AUC: ↑ 13-násobne Parenterálny midazolam: AUC: ↑ 4-násobne
Z dôvodu inhibície CYP3A
lopinavirom/ritonavirom.
Agonisty beta2 adrenoreceptoru (s dlhodobým účinkom)Salmeterol Salmeterol:
predpokladá sa zvýšenie
koncentrácií kvôli inhibícii
CYP3A lopinavirom/ritonavirom.
indukčný účinok rifampicínu na CYP 3A4. Takáto úprava dávky však môže byť spojená so vzostupmi hladín ALT/AST a so zvýšením gastrointestinálnych porúch. Z tohto dôvodu sa takému súbežnému podaniu treba vyhnúť, pokiaľ nie je striktne potrebné.
Ak je súbežné podanie
nevyhnutné, dávka lopinaviru/ritonaviru zvýšená na
400 mg/400 mg dvakrát denne
môže byť podávaná
s rifampicínom pri pozornom
monitorovaní bezpečnosti
a terapeutického účinku. Dávka lopinaviru/ritonaviru sa má titrovať nahor len potom, keď sa začal podávať rifampicín (pozri časť 4.4).
Súbežné podávanie lopinaviru/ritonaviru a kvetiapínu je kontraindikované, pretože
môže zvyšovať toxicitu
kvetiapínu.
Lopinavirom/ritonavir sa nesmie podávať súbežne s perorálnym midazolamom (pozri časť 4.3), zatiaľ čo pri súbežnom podávaní lopinaviru/ritonaviru
a parenterálneho midazolamu je
potrebná obozretnosť. Ak je lopinavir/ritonavir podávaný spolu s parenterálnym midazolamom, má sa podávať na
jednotke intenzívnej starostlivosti
(JIS) alebo v podobnom zariadení, kde je pre prípad respiračnej depresie a/alebo predĺženej sedácie zabezpečené podrobné klinické monitorovanie a primeraný lekársky manažment. Má sa zvážiť úprava dávkovania midazolamu, najmä ak sa podáva viac dávok midazolamu.
Kombinácia môže mať za následok zvýšené riziko nežiaducich účinkov na srdce a cievy súvisiacich so
salmeterolom vrátane predĺženia
QT intervalu, palpitácií
a sínusovej tachykardie. Preto sa
súbežné podávanie
lopinaviru/ritonaviru
Blokátory kalciového kanála
a salmeterolu neodporúča (pozri časť 4.4).
Felodipín, nifedipín, nikardipín
Kortikosteroidy
Felodipín, nifedipín, nikardipín: koncentrácie sa môžu zvýšiť na základe inhibície CYP3A lopinavirom/ritonavirom.
Pri súbežnom podávaní týchto liekov s lopinavirom/ritonavirom sa odporúča klinické monitorovanie terapeutických
a nežiaducich účinkov.
Dexametazón Lopinavir:
Koncentrácie sa môžu znížiť na
základe indukcie CYP3A
dexametazónom.
Pri súbežnom podávaní týchto liekov s lopinavirom/ritonavirom sa odporúča klinické monitorovanie antivírusovej účinnosti.
Flutikazónpropionát,
50 mg intranazálne 4 – krát denne (100 mg ritonaviru BID)
Flutikazónpropionát: plazmatické koncentrácie ↑ hladiny kortizolu ↓ 86%
Väčšie účinky sa dajú očakávať, ak je flutikazónpropionát inhalovaný. Systémové účinky kortikosteroidov vrátane Cushingovho syndrómu
a supresie nadobličiek boli
hlásené u pacientov, ktorí užívali ritonavir a ktorým sa inhalačne alebo intranazálne podával flutikazónpropionát; tieto účinky sa môžu vyskytnúť aj pri iných kortikosteroidoch, ktoré sú metabolizované P450 3A, napr. budesonid. V dôsledku toho sa neodporúča súbežné podávanie lopinaviru/ritonaviru a týchto glukokortikoidov, pokiaľ potenciálny prínos liečby neprevýši riziko systémových účinkov kortikosteroidov (pozri časť 4.4). Je potrebné zvážiť zníženie dávky glukokortikoidu
s dôkladným monitorovaním lokálnych a systémových účinkov
alebo prejsť na glukokortikoid,
ktorý nie je substrátom pre
CYP3A4 (napr. beklometazón). Navyše, v prípade vysadzovania glukokortikoidov sa môže počas dlhšej doby postupne znižovať dávka.
I
nhibítory fosfodiesterázy (PDE5)
Avanafil
(ritonavir 600 mg BID)
Avanafil:
AUC: ↑ 13-násobne kvôli inhibícii CYP3A4 lopinavirom/ritonavirom.
Podávanie avanafilu
s lopinavirom/ritonavirom je
kontraindikované (pozri časť
4.3).
Tadalafil Tadalafil:
AUC: ↑ 2-násobne
kvôli inhibícii CYP3A4
lopinavirom/ritonavirom.
V liečbepulmonálnejartériovejhypertenzie: súbežné podávanie lopinaviru/ritonaviru so sildenafilom je kontraindikované
Sildenafil Sildenafil:
AUC: ↑ 11-násobne kvôli inhibícii CYP3A
lopinavirom/ritonavirom.
Vardenafil Vardenafil:
AUC: ↑ 49-násobne kvôli inhibícii CYP3A
lopinavirom/ritonavirom
(pozri časť 4.3). Súbežné podávanie lopinaviru/ritonaviru
s tadalafilom sa neodporúča.
V liečbeerektilnejdysfunkcie: Predpisovanie sildenafilu alebo tadalafilu pacientom, ktorí užívajú lopinavir/ritonavir si vyžaduje zvýšenú opatrnosť
a starostlivé sledovanie nežiaducich účinkov vrátane hypotenzie, synkopy, porúch videnia a pretrvávajúcej erekcie (pozri časť 4.4).
Pri súbežnom podávaní
s lopinavirom/ritonavirom nesmie
byť dávka sildenafilu vyššia ako
25 mg v priebehu každých
48 hodín a dávka tadalafilu
nesmie presiahnuť 10 mg počas každých 72 hodín.
Užívanie vardenafilu
s lopinavirom/ritonavirom je
kontraindikované (pozri časť 4.3).
 I
nhibítory HCV proteázy
I
nhibítory HCV proteázy Boceprevir 800 mg trikrát denne
Simeprevir 200 mg denne (ritonavir 100 mg BID)
Telaprevir 750 mg trikrát denne
Rastlinné prípravky Ľubovník bodkovaný (Hypericum perforatum)
Boceprevir: AUC: ↓ 45% Cmax: ↓ 50% Cmin: ↓ 57%
Lopinavir: AUC: ↓ 34% Cmax: ↓ 30% Cmin: ↓ 43% Simeprevir:
AUC: ↑ 7,2-násobne
Cmax: ↑ 4,7-násobne Cmin: ↑ 14,4-násobne Telaprevir:
AUC: ↓ 54%
Cmax: ↓ 53%
Cmin: ↓ 52%
Lopinavir: ↔
Lopinavir:
koncentrácie sa môžu znížiť
z dôvodu indukcie CYP3A rastlinným prípravkom ľubovníkom bodkovaným.
Neodporúča sa súbežné podávanie lopinaviru/ritonaviru a bocepreviru.
Neodporúča sa súbežné podávanie lopinaviru/ritonaviru a simepreviru.
Neodporúča sa súbežné podávanie lopinaviru/ritonaviru a telapreviru.
Rastlinné prípravky obsahujúce ľubovník bodkovaný sa nesmú kombinovať s lopinavirom
a ritonavirom. Ak pacient už užíva ľubovník bodkovaný, treba
ukončiť jeho užívanie a ak je to
možné, zistiť hladinu vírusu.
Hladiny lopinaviru a ritonaviru sa po ukončení užívania ľubovníka bodkovaného môžu zvýšiť. Môže
I
m
unosupresíva Cyklosporín, sirolimus (rapamycín)
a takrolimus
Hypolipidemiká
Cyklosporín, sirolimus (rapamycín) a takrolimus: koncentrácie sa môžu zvýšiť na základe inhibície CYP3A lopinavirom/ritonavirom.
byť potrebné upraviť dávku lopinaviru/ritonaviru. Účinok indukcie pretrváva minimálne
2 týždne od ukončenia podávania ľubovníka bodkovaného (pozri
časť 4.3). Bezpečné podávanie
lopinaviru/ritonaviru sa preto
môže začať 2 týždne po ukončení liečby ľubovníkom bodkovaným.
Odporúča sa častejšie monitorovanie terapeutických koncentrácií, až kým nedôjde k stabilizácii plazmatických hladín týchto liekov.
Lovastatín a simvastatín Lovastatín, simvastatín:
výrazné zvýšenie plazmatických
koncentrácií z dôvodu inhibície
CYP3A lopinavirom/ritonavirom.
Atorvastatín Atorvastatín:
AUC: ↑ 5,9-násobne
Cmax: ↑ 4,7-násobne
z dôvodu inhibície CYP3A
lopinavirom/ritonavirom.
Rosuvastatín, 20 mg QD Rosuvastatín: AUC: ↑ 2-násobne Cmax: ↑ 5-násobne
Rosuvastatín sa len slabo metabolizuje CYP3A4, napriek
tomu však bolo pozorované zvýšenie jeho plazmatickej
koncentrácie. Mechanizmus tejto
interakcie môže vyplývať
z inhibície transportných proteínov.
Pretože zvýšené koncentrácie inhibítorov HMG-CoA reduktázy môžu spôsobovať myopatiu vrátane rabdomyolýzy, kombinácia týchto liekov
s lopinavirom/ritonavirom je kontraindikovaná (pozri časť 4.3). Kombinácia lopinaviru/ritonaviru s atorvastatínom sa neodporúča. Ak je užívanie atorvastatínu nevyhnutné, má sa podávať
v najnižších možných dávkach za
starostlivého monitorovania bezpečnosti (pozri časť 4.4). Pri súbežnom podávaní lopinaviru/ritonaviru
s rosuvastatínom je potrebná opatrnosť a má sa zvážiť zníženie dávky (pozri časť 4.4).
Fluvastatín alebo pravastatín
Ópioidy
Fluvastatín, pravastatín: neočakáva sa žiadna klinicky relevantná interakcia. Pravastatín sa nemetabolizuje CYP450.
Fluvastatín sa čiastočne
metabolizuje CYP2C9.
Ak je indikovaná liečba
inhibítormi HMG-CoA
reduktázy, odporúča sa fluvastatín
alebo pravastatín.
Buprenorfín, 16 mg QD Buprenorfín: ↔ Úprava dávky nie je potrebná. Metadón Metadón: ↓ Odporúča sa monitorovanie
plazmatických koncentrácií
metadónu.
Perorálne kontraceptívaEtinylestradiol Etinylestradiol: ↓ V prípade súbežného podávania
lopinavir/ritonaviru
Lieky na pomoc pri odvykaní od fajčenia
Bupropión Bupropión a jeho aktívny metabolit hydroxybupropión: AUC a Cmax ↓ ~50%
Tento účinok môže byť spôsobený indukciou metabolizmu bupropiónu.
 Vazodilatanciá
VazodilatanciáBosentan Lopinavir - ritonavir: Plazmatické koncentrácie lopinaviru/ritonaviru sa môžu znížiť z dôvodu indukcie CYP3A4 bosentanom.
Bosentan:
AUC: ↑ 5- násobne
Cmax: ↑ 6-násobne
Na začiatku, bosentan Cmin:
↑ približne 48-násobne
Z dôvodu inhibície CYP3A4
lopinavirom/ritonavirom.
s kontraceptívami obsahujúcimi etinylestradiol (akýkoľvek typ kontraceptív, napr. perorálne alebo náplasť) sa majú použiť ďalšie metódy antikoncepcie.
Ak je súbežné podávanie lopinaviru/ritonaviru
s bupropiónom nevyhnutné, má
sa podávať za podrobného klinického monitorovania účinnosti bupropiónu a, napriek pozorovanej indukcii, bez prekročenia odporúčaného dávkovania.
Podávanie lopinaviru/ritonaviru s bosentanom si vyžaduje opatrnosť.
Ak sa lopinavir/ritonavir podáva
súbežne s bosentanom, má sa
monitorovať liečba HIV
a pacienti majú byť starostlivo
sledovaní na toxicitu bosentanu, najmä v priebehu 1. týždňa
súbežného podávania.
I
né lieky
Na základe známych metabolických profilov sa neočakávajú klinicky signifikantné interakcie medzi lopinavirom/ritonavirom a dapsonom, trimetoprimom/sulfametoxazolom, azitromycínom alebo flukonazolom.
4.6 Fertilita, gravidita a laktácia
Gravidita
Pri rozhodovaní o podávaní antiretrovírusových látok na liečbu infekcie HIV tehotným ženám
a zároveň pri znížení rizika vertikálneho prenosu HIV na novorodenca je všeobecne potrebné brať do úvahy tak údaje pochádzajúce zo skúšaní na zvieratách, ako aj klinické skúsenosti z podávania tehotným ženám, a to za účelom charakterizovania bezpečnosti pre plod.
Lopinavir/ritonavir bol hodnotený u viac ako 3000 žien počas gravidity vrátane viac ako 1000 počas
prvého trimestra.
V rámci postmarketingového sledovania prostredníctvom registra “Antiretroviral Pregnancy
Registry“, zavedeného v januári 1989, nebolo hlásené zvýšené riziko vrodených chýb pri
sledovaní viac ako 1000 žien užívajúcich liek počas prvého trimestra gravidity. Prevalencia vrodených
chýb po expozícii lopinaviru v ktoromkoľvek trimestri gravidity je porovnateľná s prevalenciou
u bežnej populácie. Nebol pozorovaný žiaden súbor príznakov vrodených chýb s možnou spoločnou etiológiou. Pri štúdiách na zvieratách sa preukázala reprodukčná toxicita (pozri časť 5.3). Na základe týchto údajov je riziko malformácií u ľudí nepravdepodobné. Lopinavir sa môže používať v gravidite, ak je klinicky potrebný.
 D
ojčenie
D
ojčenie
Štúdie na potkanoch ukázali, že lopinavir sa vylučuje do mlieka. Nie je známe, či sa toto liečivo vylučuje do materského mlieka u ľudí. Všeobecne sa odporúča, aby matky infikované HIV za žiadnych okolností nedojčili svoje deti, aby sa zabránilo prenosu HIV.
FertilitaŠtúdie na zvieratách nepreukázali žiadny vplyv na fertilitu. Údaje o vplyve lopinaviru/ritonaviru na
plodnosť u ľudí nie sú dostupné.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeNeuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti majú byť informovaní, že počas liečby lopinavirom/ritonavirom bola hlásená nauzea (pozri časť 4.8).
4.8 Nežiaduce účinkya. Súhrn bezpečnostnéhoprofiluBezpečnosť lopinaviru/ritonaviru bola sledovaná u viac ako 2600 pacientov v II.-IV. fáze klinických
skúšok, viac ako 700 z týchto pacientov užívalo dávku 800/200 mg (6 kapsúl alebo 4 tablety) raz denne. Okrem nukleozidových inhibítorov reverznej transkriptázy (NRTI) bol lopinavir/ritonavir v niektorých štúdiách používaný v kombinácii s efavirenzom alebo nevirapínom.
Najčastejšími nežiaducimi reakciami, súvisiacimi s liečbou lopinaviru/ritonaviru, boli počas klinických skúšaní hnačka, nevoľnosť, vracanie, hypertriglyceridémia a hypercholesterolémia. Riziko hnačky môže byť vyššie pri dávkovaní lopinaviru/ritonaviru jedenkrát denne. Hnačka, nevoľnosť
a vracanie sa môžu objaviť na začiatku liečby, kým hypertriglyceridémia a hypercholesterolémia sa
môžu vyskytnúť neskôr. Liečbu si vyžadujúce nežiaduce udalosti viedli k predčasnému ukončeniu liečby u 7% pacientov, zaradených do klinických skúšaní fázy II-IV.
Je dôležité uviesť, že boli hlásené prípady pankreatitídy u pacientov užívajúcich lopinavir/ritonavir vrátane tých, u ktorých sa vyvinula hypertriglyceridémia. Okrem toho bolo zriedkavo hlásené predĺženie PR intervalu počas liečby lopinavirom/ritonavirom (pozri časť 4.4).
b. Tabuľkový zoznamnežiaducichreakciíNežiaduce reakcie z klinických skúšaní a postmarketingového sledovania u dospelých a pediatrickýchpacientov:Nasledovné udalosti boli identifikované ako nežiaduce reakcie. Frekvenčná skupina zahŕňa všetky hlásené nežiaduce udalosti strednej až ťažkej intenzity, bez ohľadu na individuálne posúdenie kauzality. Nežiaduce reakcie sú uvedené podľa tried orgánových systémov. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti: veľmi časté
(≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100) a neznáme (nemožno odhadnúť
z dostupných údajov).
Nežiaduce účinky s neznámou frekvenciou boli identifikované počas postmarketingových sledovaní.
Nežiaduce účinky v klinických štúdiách a postmarketingových sledovaniach u dospelých pacientovTrieda orgánových systémov Frekvencia Nežiaduca reakciaInfekcie a nákazy Veľmi časté Infekcie horných dýchacích ciest
Časté Infekcie dolných dýchacích ciest, infekcie
kože vrátane celulitídy, folikulitídy
a furunkulózy
Poruchy krvi a lymfatického systému
Časté Anémia, leukopénia, neutropénia, lymfadenopatia
Poruchy imunitného systému Časté
Menej časté
Hypersenzitivita vrátane urtikárie a angioedému
Syndróm imunitnej reaktivácie
Poruchy endokrinného systému Menej časté Hypogonadizmus
Poruchy metabolizmu a výživy Časté Poruchy glykémie vrátane diabetes mellitus, hypertriglyceridémia, hypercholesterolémia, pokles hmotnosti, znížená chuť do jedla.
Menej časté Zvýšenie hmotnosti, zvýšenie chuti do jedla
Psychické poruchy Časté Úzkosť
Menej časté Abnormálne sny, znížené libido
Poruchy nervového systému Časté Bolesť hlavy (vrátane migrény), neuropatia (vrátane periférnej neuropatie), závraty, nespavosť
Menej časté Cerebrovaskulárna príhoda, kŕče, poruchy
chuti, strata chuti, tras Poruchy oka Menej časté Porucha zraku Poruchy ucha a labyrintu Menej časté Tinitus, vertigo
Poruchy srdca a srdcovej
činnosti
Menej časté Ateroskleróza, ako je infarkt myokardu,
atrioventrikulárna blokáda, insuficiencia trikuspidálnej chlopne
Poruchy ciev Časté Hypertenzia
Menej časté Hlboká žilová trombóza
Poruchy gastrointestinálneho
traktu
Veľmi časté Hnačka, nevoľnosť
Časté Pankreatitída1, vracanie, gastroezofageálna refluxná choroba, gastroenteritída a kolitída, bolesť brucha (hornej aj dolnej časti), abdominálna distenzia, dyspepsia, hemoroidy, flatulencia
Menej časté Gastrointestinálna hemorágia vrátane gastrointestinálneho vredu, duodenitída, gastritída a rektálna hemorágia, stomatitída a ulcerácie v ústach, fekálna inkontinencia, zápcha, sucho v ústach
Poruchy pečene a žlčových ciest Časté Hepatitída vrátane zvýšenia AST, ALT
a GMT
Menej časté Steatóza pečene, hepatomegália, cholangitída,
hyperbilirubinémia
Poruchy kože a podkožného
tkaniva
Neznáme Žltačka
Časté Získaná lipodystrofia vrátane atrofie tváre, vyrážka vrátane makulopapulárnej vyrážky, dermatitída/exantém vrátane ekzému
a seboroickej dermatitídy, nočné potenie,
svrbenie
Menej časté Alopécia, kapilaritída, vaskulitída

Neznáme Stevensov-Johnsonov syndróm, multiformný
erytém
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Poruchy obličiek a močových
ciest
Poruchy reprodukčného systému a prsníkov Celkové poruchy a reakcie v mieste podania
Časté Myalgia, muskuloskeletálna bolesť vrátane artralgie a bolesti chrbta, poruchy svalov, ako sú slabosť a spazmy
Menej časté Rabdomyolýza, osteonekróza
Menej časté Zníženie klírensu kreatinínu, nefritída,
hematúria
Časté Erektilná dysfunkcia, poruchy menštruácie - amenorea, menorágia
Časté Únava vrátane asténie
1Pozri časť 4.4: Pankreatitída a lipidy
c. PopisvybranýchnežiaducichreakciíCushingov syndróm bol hlásený u pacientov, užívajúcich ritonavir a inhalačný alebo intranazálny flutikazónpropionát; môže sa tiež vyskytnúť pri užívaní iných kortikosteroidov, metabolizovaných pomocou P450 3A, napr. budezonidu (pozri časti 4.4 a 4.5).
Pri používaní inhibítorov proteáz, najmä v kombinácii s inhibítormi nukleozidovej reverznej transkriptázy, bolo hlásené zvýšenie kreatínfosfokinázy (CPK), myalgia, myozitída a zriedkavo rabdomyolýza.
Kombinovaná antiretrovírusová liečba bola spojená s redistribúciou telesného tuku (lipodystrofia) u HIV pacientov vrátane straty periférneho a tvárového podkožného tuku, zvýšenia množstva intraabdominálneho a viscerálneho tuku, hypertrofiou prsníkov a akumuláciou tuku v dorzocervikálnej oblasti (byvolí hrb).
Kombinovaná antiretrovírusová terapia bola spojená aj s niektorými metabolickými abnormalitami ako sú hypertriglyceridémia, hypercholesterolémia, rezistencia na inzulín, hyperglykémia
a hyperlaktacidémia (pozri časť 4.4).
U HIV-infikovaných pacientov s ťažkou imunodeficienciou môže v čase začatia kombinovanej antiretrovírusovej terapie (CART) vzniknúť zápalová reakcia na asymptomatické alebo reziduálne oportúnne infekcie. Boli hlásené aj autoimunitné poruchy (ako napr. Gravesova choroba), ale hlásená doba nástupu je variabilnejšia a môže sa vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne uznanými rizikovými faktormi, pokročilým HIV ochorením alebo dlhodobou expozíciou kombinovanej antiretrovírusovej terapie (CART). Ich frekvencia nie je známa (pozri časť 4.4).
d. Pediatrická populáciaU 2-ročných a starších detí je bezpečnostný profil podobný ako u dospelých (pozri tabuľku v časti b).
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieV súčasnosti existujú len obmedzené skúsenosti s akútnym predávkovaním lopinavirom/ritonavirom u ľudí.
Nežiaduce klinické prejavy, pozorované u psov boli salivácia, vracanie a hnačka/abnormálna stolica. Prejavy toxicity pozorované u myší, potkanov alebo psov zahŕňali zníženú aktivitu, ataxiu, emaciáciu, dehydratáciu a tremor.
Neexistuje žiadne špecifické antidotum pri predávkovaní lopinavirom/ritonavirom. Liečba predávkovania lopinavirom/ritonavirom zahŕňa všeobecné podporné opatrenia vrátane monitorovania základných životných funkcií a sledovania klinického stavu pacienta. Ak je to potrebné, je možné
na odstránenie neabsorbovanej látky vyvolať vracanie alebo vykonať gastrickú laváž. Na odstránenie neabsorbovaného liečiva sa môže podať živočíšne uhlie. Vzhľadom na to, že lopinavir/ritonavir sa pevne viaže na bielkoviny, je málo pravdepodobné, že by sa dialýzou odstránilo signifikantné množstvo liečiva.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antivirotiká na systémové použitie, antivirotiká na liečbu HIV infekcií,
kombinácie, ATC kód: J05AR10
M ec hanizmus úči nku: Antivírusový účinok lopinaviru/ritonaviru je vyvolaný lopinavirom. Lopinavir je inhibítor HIV-1 a HIV-2 proteáz. Inhibícia HIV proteázy zabraňuje štiepeniu gag-pol polyproteínu, čo vedie k produkcii nezrelých, neinfekčných vírusov.
Úči nky na el ekt rokar di ogram: QTcF interval bol hodnotený 10 meraniami počas 12 hodín na 3. deň v randomizovanej, placebom a liečivom (moxifloxacín 400 mg raz denne) kontrolovanej prekríženej štúdií u 39 zdravých dospelých jedincov. Maximálne priemerné (95% horný limit spoľahlivosti) rozdiely QTcF v porovnaní s placebom boli 3,6 (6,3) pri dávke 400/100 mg LPV/r dvakrát denne
a 13,1 (15,8) pri dávke 800/200 mg dvakrát denne, ktorá je vyššia ako terapeutická dávka. Indukované
predĺženie QRS intervalu zo 6 ms na 9,5 ms vysokými dávkami lopinaviru/ritonaviru (800/200 mg dvakrát denne) prispieva k predĺženiu QT. Tieto dva režimy mali na 3. deň za následok expozície, ktoré boli približne 1,5 a 3-násobne vyššie ako expozície v rovnovážnom stave pri odporúčaných dávkach LPV/r raz denne alebo dvakrát denne. Žiadny jedinec nemal predĺženie QTcF o³ 60 ms oproti počiatočnému stavu alebo QTcF interval prevyšujúci potenciálne klinicky relevantnú prahovú hodnotu 500 ms.
U jedincov, ktorí dostávali lopinavir/ritonavir, bolo v tej istej štúdii na 3. deň pozorované aj mierne predĺženie PR intervalu. Priemerné zmeny oproti počiatočnému stavu PR intervalu boli v rozsahu od
11,6 ms do 24,4 ms v intervale 12 hodín po podaní dávky. Maximálny PR interval bol 286 ms
a nezistila sa žiadna srdcová blokáda II. alebo III. stupňa (pozri časť 4.4).
Antivírusová aktivita in vitro:Antivírusová aktivita lopinaviru in vitro proti laboratórnym a klinickým
HIV kmeňom bola vyhodnotená u akútne infikovaných lymfoblastických bunkových línií
a lymfocytov z periférnej krvi. V neprítomnosti ľudského séra bola priemerná IC50 lopinaviru proti piatim rôznym laboratórnym kmeňom HIV-1 19 nM. V neprítomnosti a v prítomnosti 50% ľudského séra bola priemerná IC50 lopinaviru proti HIV-1IIIB v MT4 bunkách 17 nM a 102 nM. V neprítomnosti ľudského séra bola priemerná IC50 lopinaviru 6,5 nM proti rôznym HIV-1 klinickým izolátom.
Rezistencia
In vitro selekcia rezistencie:
V pokusoch in vitro boli selektované HIV-1 izoláty so zníženou citlivosťou na lopinavir. HIV-1 bol pasážovaný in vitro s lopinavirom samotným a s lopinavirom v kombinácii s ritonavirom v pomeroch koncentrácií, reprezentujúcich rozsah plazmatických pomerov koncentrácií pri terapii lopinavirom/ritonavirom. Genotypová a fenotypová analýza vírusov, selektovaných pri tomto pasážovaní naznačuje, že prítomnosť ritonaviru v použitých pomeroch koncentrácií neovplyvňuje
merateľne selekciu vírusov rezistentných na lopinavir. Celkovo, in vitro charakterizácia fenotypovej skríženej rezistencie medzi lopinavirom a inými inhibítormi proteáz nasvedčuje, že znížená citlivosť na lopinavir tesne korelovala so zníženou citlivosťou na ritonavir a indinavir, ale nekorelovala tesne so zníženou citlivosťou na amprenavir, sachinavir a nelfinavir.
Analýza rezistencie u pacientov bez predchádzajúceho podávania ARV:
V klinických skúšaniach s limitovaným množstvom analyzovaných izolátov nebola selekcia rezistencie na lopinavir pozorovaná u zatiaľ neliečených pacientov bez signifikantnej rezistencie na inhibítor proteázy na začiatku liečby. Detailný popis klinických skúšaní, pozri ďalej.
Analýza rezistencie u pacientov s predchádzajúcim podávaním inhibítora proteázy:
Selekcia rezistencie na lopinavir u pacientov, u ktorých zlyhala predchádzajúca liečba inhibítorom proteázy, sa skúmala analýzou pozdĺžnych izolátov u 19 subjektov s predchádzajúcou liečbou inhibítorom proteázy v dvoch štúdiách fázy II a jednej štúdii fázy III, u ktorých došlo buď k neúplnej virologickej supresii alebo opätovnému namnoženiu vírusu po počiatočnej odpovedi na lopinavir/ritonavir a u ktorých sa prejavil nárast in vitro rezistencie medzi východiskovým stavom
a opätovným namnožením vírusu (definované ako vznik nových mutácií alebo 2-násobná zmena vo fenotypovej citlivosti na lopinavir). Nárast rezistencie bol najčastejší u subjektov, ktorých východiskové izoláty mali niekoľko mutácií spojených s inhibítorom proteázy, ale < 40-násobne zníženú citlivosť na lopinavir na začiatku liečby. Najčastejšie vznikali mutácie V82A, I54V a M46I. Pozorovali sa tiež mutácie L33F, I50V a V32I v kombinácii s I47V/A. V porovnaní s izolátmi na začiatku liečby malo 19 izolátov 4,3-násobné zvýšenie IC50 (od 6,2- do 43-násobného, v porovnaní
s divokým typom vírusu).
Genotypové koreláty zníženej fenotypovej citlivosti na lopinavir pri vírusoch, selektovaných inými inhibítormi proteáz. Bola hodnotená antivírusová aktivita lopinaviru in vitro proti 112 klinickým izolátom, získaným od pacientov, u ktorých zlyhala terapia s jedným alebo viacerými inhibítormi proteáz. Medzi týmito izolátmi boli spojené so zníženou in vitro citlivosťou na lopinavir nasledovné mutácie HIV proteázy: L10F/I/R/V, K20M/R, L24I, M46I/L, F53L, I54L/T/V, L63P, A71I/L/T/V, V82A/F/T, I84V a L90M. Medián EC50 lopinaviru proti izolátom s 0 - 3, 4 - 5, 6 - 7 a 8 - 10 mutáciami vo vyššie uvedených polohách aminokyselín bol 0,8; 2,7; 13,5 a 44,0-krát vyšší ako EC50 proti divokému typu HIV. Všetkých 16 vírusov, ktoré vykazovali > 20-násobnú zmenu v citlivosti, mali mutácie v polohách 10, 54, 63 plus 82 a/alebo 84. Navyše obsahovali medián 3 mutácií aminokyselín v polohách 20, 24, 46, 53, 71 a 90. U pacientov s predchádzajúcou liečbou inhibítorom proteázy, ktorí dostávali lopinavir/ritonavir, sa okrem mutácií opísaných vyššie zaznamenali
v izolátoch po opätovnom namnožení vírusu mutácie V32I a I47A so zníženou citlivosťou na lopinavir a u pacientov liečených lopinavirom/ritonavirom sa zaznamenali v izolátoch poopätovnom namnožení vírusu mutácie I47A a L76V so zníženou citlivosťou na lopinavir.
Závery, týkajúce sa relevancie jednotlivých mutácií alebo mutačných vzorcov sa môžu pri získavaní dodatočných údajov ďalej meniť, pre analýzu výsledkov rezistenčných testov sa preto odporúča zoznámiť sa vždy so súčasnými interpretáciami.
Antivírusová aktivita lopinaviru/ritonaviru u pacientov, u ktorých zlyhala terapia inhibítorom proteázy: U 56 pacientov, u ktorých predtým zlyhala liečba s viacerými inhibítormi proteáz, sa študoval klinický význam zníženej in vitro citlivosti lopinaviru hodnotením virologickej odpovede na terapiu lopinavirom/ritonavirom, s ohľadom na pôvodný vírusový genotyp a fenotyp. EC50 lopinaviru proti 56 pôvodným vírusovým izolátom bola 0,6 až 96-násobne vyššia ako EC50 proti divokému typu HIV. Po 48 týždňoch liečby lopinavirom/ritonavirom, efavirenzom a inhibítormi nukleozidovej reverznej transkriptázy sa pozoroval výskyt plazmatickej HIV RNA £ 400 kópií/ml u 93% (25/27),'
73% (11/15) a 25% (2/8) pacientov s < 10-násobným, 10 až 40-násobným a > 40-násobným znížením citlivosti na lopinavir oproti pôvodnému stavu. Okrem toho bola pozorovaná virologická odpoveď
u 91% (21/23), 71% (15/21) a 33% (2/6) pacientov s 0 - 5, 6 - 7 a 8 - 10 mutáciami z vyššie uvedených mutácií v HIV proteáze, ktoré boli spojené so zníženou in vitro citlivosťou na lopinavir. Keďže títo pacienti neboli predtým vystavení účinku lopinaviru/ritonaviru alebo efavirenzu, za časť z týchto odpovedí môže byť zodpovedná antivírusová aktivita efavirenzu, najmä u pacientov
s vírusom vysoko rezistentným na lopinavir. Táto štúdia neobsahovala kontrolnú skupinu pacientov, ktorí neužívali lopinavir/ritonavir.
Skrížená rezistencia: Účinok iných inhibítorov proteázy proti izolátom, ktoré vyvolali nárast rezistencie na lopinavir po podávaní lopinaviru/ritonaviru u pacientov s predchádzajúcou liečbou inhibítorom proteázy: Prítomnosť skríženej rezistencie na iné inhibítory proteázy sa analyzovala v 18 izolátoch so znova namnoženým vírusom, ktoré preukázali rozvoj rezistencie na lopinavir počas
3 štúdií fázy II a jednej štúdie fázy III s lopinavirom/ritonavirom u pacientov s predchádzajúcou liečbou inhibítorom proteázy. V porovnaní s divokým typom vírusu bolo priemerné zvýšenie IC50 lopinaviru v 18 východiskových izolátoch 6,9-násobné a 63-násobné v izolátoch po namnožení vírusu. Vo všeobecnosti sa izoláty s namnoženým vírusom buď nezmenili (ak boli skrížene rezistentné na začiatku liečby) alebo vyvinuli významnú skríženú rezistenciu na indinavir, sachinavir a atazanavir. Zaznamenali sa mierne zníženia účinku amprenaviru s priemerným zvýšením IC50 od 3,7-násobného
vo východiskových izolátoch do 8-násobného v izolátoch s namnoženým vírusom. V porovnaní
s divokým typom vírusu mali izoláty s nezmenenou citlivosťou na tipranavir priemerné zvýšenie IC50 na začiatku 1,9-násobné a izoláty po namnožení vírusu 1,8–násobné. Dodatočné informácie o užívaní tipranaviru vrátane genotypových prediktorov odpovede pri liečbe infekcie HIV-1 rezistentnej na lopinavir, pozri v súhrne charakteristických vlastností Aptivisu.
Klinické výsledkyÚčinky lopinaviru/ritonaviru (v kombinácii s inými antiretrovírusovými látkami) na biologické
markery (plazmatické hladiny HIV RNA a počet CD4+ T-buniek) boli sledované v kontrolovaných
štúdiách lopinaviru/ritonaviru, trvajúcich 48 až 360 týždňov.
Použitie u dospelýchPacienti bez predchádzajúcej antiretrovírusovej terapieŠtúdia M98-863 bola randomizovaná, dvojito zaslepená štúdia so 653 pacientmi bez predchádzajúcej
antiretrovírusovej liečby, v ktorej sa porovnával lopinavir/ritonavir (400/100 mg dvakrát denne)
s nelfinavirom (750 mg trikrát denne) plus stavudín a lamivudín. Priemerná východisková hodnota počtu CD4+ T-buniek bola 259 buniek/mm3 (rozpätie: 2 až 949 buniek/ mm3) a priemerná východisková hodnota plazmatickej HIV-1 RNA bola 4,9 log10 kópií/ml (rozpätie: 2,6 až 6,8 log10 kópií/ml).
Tabuľka 1
Výsledky v 48. týždni: skúšanie M98-863 Lopinavir/ritonavir (N=326) Nelfinavir (N=327)
Lopinavir/ritonavir (N=326) Nelfinavir (N=327)HIV RNA < 400 kópií/ml* 75% 63% HIV RNA < 50 kópií/ml*† 67% 52% Priemerný nárast počtu CD4+
T-buniek (bunky/mm3) oproti
východiskovej hodnote
207 195
* intent-to-treat analýza, kde sa pacienti s chýbajúcimi hodnotami považujú za pacientov
s virologickým zlyhaním
† p < 0,001
Stotrinásť pacientov liečených nelfinavirom a 74 pacientov liečených lopinavirom/ritonavirom malo počas liečby v týždňoch 24 až 96 viac ako 400 kópií HIV RNA/ml. Z toho sa mohli použiť na testovanie rezistencie izoláty od 96-tich pacientov liečených nelfinavirom a 51 pacientov liečených lopinavirom/ritonavirom. Rezistencia na nelfinavir, definovaná ako prítomnosť D30N alebo L90M mutácií proteázy, bola pozorovaná u 41/96 (43%) pacientov. Rezistencia na lopinavir, definovaná ako prítomnosť akejkoľvek primárnej mutácie alebo mutácie aktívneho miesta proteázy (pozri vyššie), sa
pozorovala u 0/51 (0%) pacientov. Chýbanie rezistencie na lopinavir bolo potvrdené fenotypovými analýzami.
Štúdia M05-730 bola randomizovaná, otvorená, multicentrická skúška porovnávajúca liečbu lopinavirom/ritonavirom 800/200 mg raz denne spolu s tenofovirom DF a emtricitabínom a liečbu lopinavirom/ritonavirom 400/100 mg dvakrát denne spolu s tenofovirom DF a emtricitabínom u 664 pacientov bez predchádzajúcej antiretrovírusovej liečby. Vzhľadom na farmakokinetickú interakciu medzi lopinavirom/ritonavirom a tenofovirom (pozri časť 4.5) nemusia byť výsledky tejto štúdie striktne extrapolovateľné v prípade, že sa používa iný základný režim s lopinavirom/ritonavirom. Pacienti boli randomizovaní v pomere 1:1 a dostávali buď lopinavir/ritonavir 800/200 mg raz denne (n = 333) alebo lopinavir/ritonavir 400/100 mg dvakrát denne (n = 331). Ďalšie rozdelenie v rámci každej skupiny bolo 1:1 (tablety a mäkké kapsuly). Pacienti dostávali buď tablety alebo mäkké
kapsuly počas 8 týždňov, potom bola všetkým pacientom podávaná tabletová lieková forma raz denne
alebo dvakrát denne až do ukončenia štúdie. Pacienti dostávali emtricitabín 200 mg raz denne
a tenofovir DF 300 mg raz denne. V protokole definovaná non-inferiorita dávkovania raz denne v porovnaní s dávkovaním dvakrát denne bola preukázaná, ak dolná hranica 95% intervalu spoľahlivosti pre rozdiel v pomere reagujúcich subjektov (raz denne mínus dvakrát denne) vylúčila -
12% v 48. týždni. Priemerný vek zaradených pacientov bol 39 rokov (rozpätie: 19 až 71); 75% bolo
kaukazskej rasy a 78% bolo mužov. Priemerná východisková hodnota počtu CD4+ T-buniek bola 216
buniek/mm3 (rozpätie: 20 až 775 buniek/mm3) a priemerná východisková hodnota HIV-1 RNA
v plazme bola 5,0 log10 kópií/ml (rozpätie: 1,7 až 7,0 log10 kópií/ml).
Tabuľka 2
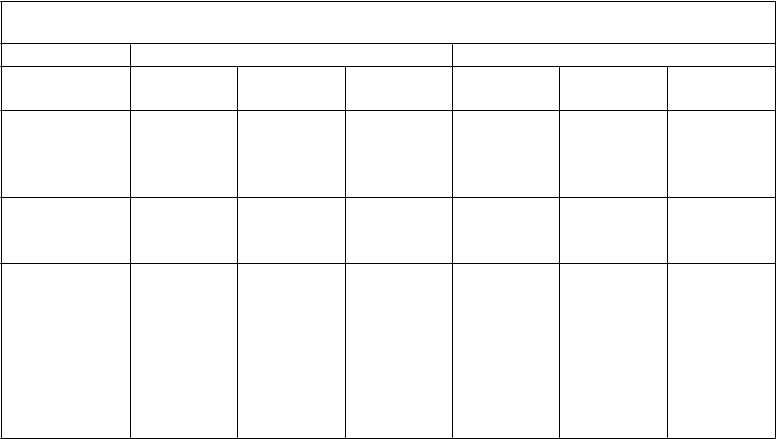
Virologická odpoveď subjektov skúšania v 48. týždni a v 96. týždni
48. týždeň 96. týždeň
R
az denne Dvakrát denne
R
ozdiel
[
95% IS]
R
az denne Dvakrát denne
R
ozdiel
[
95% IS]
NC = zlyhanie 257/333
(77,2%)
251/331
(75,8%)
1,3%
[-5,1; 7,8]
216/333
(64,9%)
229/331
(69,2%)
-4,3%
[-11,5; 2,8]
Pozorované
hodnoty
257/295
(87,1%)
250/280
(89,3%)
-2,2%
[-7,4; 3,1]
216/247
(87,4%)
229/248
(92,3%)
-4,9%
[-10,2; 0,4]
Priemerný nárast počtu CD4+ T- buniek (bunky/mm3) oproti východiskovej hodnote
186 198 238 254
Počas 96. týždňa boli k dispozícii výsledky testov genotypovej rezistencie od 25 pacientov užívajúcich
liek raz denne a 26 pacientov užívajúcich liek 2-krát denne, ktorí mali nekompletnú virologickú odpoveď. V skupine pacientov, užívajúcich liek raz denne sa rezistencia na lopinavir nepozorovala u žiadneho pacienta a v skupine pacientov, užívajúcich liek 2-krát denne sa u 1 pacienta so signifikantnou rezistenciou na inhibítor proteázy na začiatku liečby pozorovala aj rezistencia na lopinavir v skúšaní.
Pretrvávajúca virologická odpoveď na lopinavir/ritonavir (v kombinácii s inhibítormi nukleozidovej/nukleotidovej reverznej transkriptázy) bola pozorovaná aj v malej štúdii fázy II (M97-
720) počas 360 týždňov liečby. V štúdii bolo pôvodne lopinavirom/ritonavirom liečených 101
pacientov (vrátane 51 pacientov, ktorí dostávali 400/100 mg dvakrát denne a 49 pacientov, ktorí
dostávali buď 200/100 mg dvakrát denne alebo 400/200 mg dvakrát denne). Všetci pacienti medzi
48. týždňom a 72. týždňom prešli do otvorenej fázy štúdie s lopinavirom/ritonavirom s dávkou
400/100 mg dvakrát denne. Štúdiu prerušilo 39 pacientov (39%) vrátane 16 (16%) prerušení kvôli nežiaducim udalostiam, z ktorých jedno bolo spojené s úmrtím. Štúdiu ukončilo 61 pacientov (odporúčanú dávku 400/100 mg dvakrát denne dostalo počas štúdie 35 pacientov).
Tabuľka 3
 Výsledky v 360. týždni: skúšanie M97-720Lopinavir/ritonavir(N=100)
Výsledky v 360. týždni: skúšanie M97-720Lopinavir/ritonavir(N=100) HIV RNA < 400 kópií/ml 61% HIV RNA < 50 kópií/ml 59%
Priemerný nárast počtu CD4+ T-buniek (bunky/mm3) oproti
východiskovej hodnote
501
Počas 360 týždňov liečby bola úspešne vykonaná genotypová analýza vírusových izolátov u 19 z 28
pacientov s potvrdenou HIV RNA v počte viac ako 400 kópií/ml, pričom neboli odhalené žiadne
mutácie primárneho alebo aktívneho miesta proteázy (aminokyseliny na pozíciách 8, 30, 32, 46, 47,
48, 50, 82, 84 a 90) alebo fenotypová rezistencia proteázového inhibítora.
Pacienti s predchádzajúcou antiretrovírusovou terapiou
M06-802 bola randomizovaná otvorená štúdia, v ktorej sa porovnávala bezpečnosť, znášanlivosť
a antivírusová aktivita tabliet lopinaviru/ritonaviru v dávkovaní jedenkrát denne a dvakrát denne
u 599 pacientov s detekovateľnou vírusovou záťažou počas užívania ich súčasnej antivírusovej liečby. Pacienti sa predtým neliečili lopinavirom/ritonavirom. Pacienti boli náhodne vybraní v pomere 1:1 na liečbu lopinavirom/ritonavirom v dávke 800/200 mg jedenkrát denne (n = 300) alebo na liečbu lopinavirom/ritonavirom v dávke 400/100 mg dvakrát denne (n = 299). Pacienti dostávali minimálne dva nukleozidové/nukleotidové inhibítory reverznej transkriptázy, ktoré vybral skúšajúci. Zaradená populácia mala stredne veľkú skúsenosť s PI, pričom viac ako polovica pacientov nikdy predtým nedostávala PI a približne 80% pacientov malo vírusový kmeň s menej ako 3 mutáciami PI. Priemerný vek zaradených pacientov bol 41 rokov (rozpätie: 21 až 73); 51% bolo kaukazskej rasy a 66% bolo mužov. Priemerný východiskový počet CD4+ T-buniek bol 254 buniek/mm3 (rozpätie: 4 až
952 buniek/mm3) a priemerná východisková hodnota HIV-1 RNA v plazme bola 4,3 log10 kópií/ml
(rozpätie: 1,7 až 6,6 log10 kópií/ml). Asi 85% pacientov malo vírusovú záťaž < 100 000 kópií/ml.
Tabuľka 4
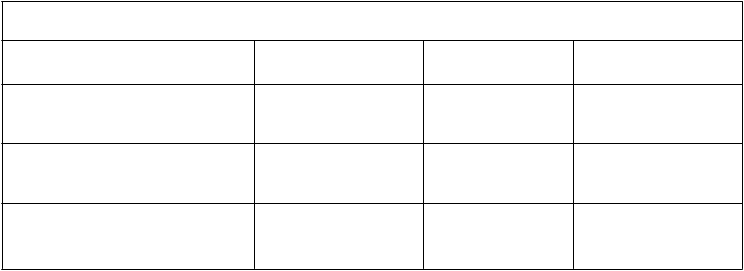 Virologická odpoveď subjektov skúšania v 48. týždni skúšania 802Raz denne Dvakrát denne Rozdiel[95% IS]
Virologická odpoveď subjektov skúšania v 48. týždni skúšania 802Raz denne Dvakrát denne Rozdiel[95% IS] NC = zlyhanie 171/300 (57%) 161/299 (53,8%) 3,2%
[-4,8%, 11,1%]
Pozorované hodnoty 171/225 (76,0%) 161/223 (72,2%) 3,8%
[-4,3%, 11,9%]
Priemerný nárast počtu CD4+ T-
buniek (bunky/mm3) oproti východiskovej hodnote
135 122
Počas 48. týždňa boli k dispozícii výsledky testov genotypovej rezistencie od 75 pacientov užívajúcich
liek raz denne a 75 pacientov užívajúcich liek 2-krát denne, ktorí mali nekompletnú virologickú
odpoveď. V skupine pacientov, užívajúcich liek raz denne sa u 6/75 (8%) pacientov pozorovali
novoobjavené primárne mutácie proteázových inhibítorov (kodóny 30, 32, 48, 50, 82, 84, 90), rovnako ako u 12/77 (16%) pacientov zo skupiny, užívajúcej liek 2-krát denne.
Pediatrické použitie
M98-940 bola otvorená štúdia s tekutou formou lopinavir/ritonaviru u 100 pediatrických pacientov, predtým neliečených (44%) alebo liečených (56%) antiretrovírusovou liečbou. Žiaden z pacientov nebol predtým liečený inhibítorom nenukleozidovej reverznej transkriptázy. Pacienti boli randomizovaní do skupín s 230 mg lopinaviru/57,5 mg ritonaviru na m2 alebo 300 mg lopinaviru/7 mg ritonaviru na m2. Pacienti bez predchádzajúcej liečby užívali aj inhibítory nukleozidovej reverznej transkriptázy. Pacienti s predchádzajúcou liečbou užívali nevirapín s jedným až dvomi inhibítormi nukleozidovej reverznej transkriptázy. Bola hodnotená bezpečnosť, účinnosť a farmakokinetický
profil oboch dávkovacích režimov po 3 týždňoch terapie u každého pacienta. Následne všetci pacienti pokračovali v liečbe s dávkou 300/75 mg na m2. Priemerný vek pacientov bol 5 rokov (rozmedzie od 6 mesiacov do 12 rokov), z čoho malo 14 pacientov menej ako 2 roky a 6 pacientov jeden rok alebo menej. Priemerný počet CD4+ T-buniek pred začiatkom liečby bol 838 buniek/mm3 a priemerný počet HIV-1 RNA v plazme 4,7 log10 kópií/ml.
Tabuľka 5
Výsledky v 48. týždni: skúšanie M98-940
Bez predchádzajúcej
S predchádzajúcou
antiretrovírusovej liečby
 (
N=
44)
antiretrovírusovou liečbou
(
N=
56)
(
N=
44)
antiretrovírusovou liečbou
(
N=
56)
HIV RNA < 400 kópií/ml 84% 75%
Priemerný nárast počtu CD4+
T-buniek (bunky/mm3) oproti východiskovej hodnote
404 284
KONCERT/PENTA 18 je prospektívna, multicentrická, randomizovaná, otvorená štúdia, ktorá
hodnotila farmakokinetický profil, účinnosť a bezpečnosť podávania tabliet lopinaviru/ritonaviru
100 mg/25 mg dávkovaných na základe telesnej hmotnosti dvakrát denne v porovnaní s podávaním jedenkrát denne ako súčasť kombinovanej antiretrovírusovej terapie (cART) u HIV-1 infikovaných detí s virologickou supresiou (n = 173). Deti mohli byť zaradené do štúdie, ak boli vo veku
< 18 rokov, mali telesnú hmotnosť ≥ 15 kg, dostávali terapiu cART, ktorá zahŕňala lopinavir/ritonavir,
mali < 50 kópií HIV-1 ribonukleovej kyseliny (RNA)/ml počas najmenej 24 týždňov a boli schopné
prehĺtať tablety. V 24. týždni bola účinnosť a bezpečnosť dávkovania tabliet lopinaviru/ritonaviru
100 mg/25 mg dvakrát denne u detskej populácie (n = 87) v súlade so zisteniami o účinnosti
a bezpečnosti v predchádzajúcich štúdiách u dospelých a pediatrických pacientoch s podávaním lopinaviru/ritonaviru dvakrát denne. Percento pacientov, ktorí dosiahli < 50 kópií HIV-1 RNA/ml
v 24. týždni bolo nižšie u detských pacientov užívajúcich tablety lopinaviru/ritonaviru jedenkrát denne
(88,2%) ako u pacientov, ktorí dostávali dávku dvakrát denne (96,6%, p = 0,040), najmä z dôvodu nižšieho dodržiavania liečby v skupine s liečbou jedenkrát denne. Údaje o účinnosti v prospech režimu dávkovania dvakrát denne sú potvrdené rozdielom vo farmakokinetických parametroch, ktoré sú významne v prospech režimu dvakrát denne (pozri časť 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti lopinaviru, podávaného súbežne s ritonavirom, boli hodnotené u zdravých dospelých dobrovoľníkov aj u HIV infikovaných pacientov. Neboli pozorované žiadne zásadné rozdiely medzi týmito skupinami. Lopinavir je v podstate kompletne metabolizovaný CYP3A. Ritonavir inhibuje metabolizmus lopinaviru, čím zvyšuje plazmatické hladiny lopinaviru. V rôznych štúdiách viedlo podávanie lopinaviru/ritonaviru v dávke 400/100 mg dvakrát denne k priemernej rovnovážnej plazmatickej koncentrácii lopinaviru 15 až 20-násobne vyššej, ako bola koncentrácia ritonaviru u HIV infikovaných pacientov. Plazmatické hladiny ritonaviru predstavovali menej ako 7% hladín, ktoré sa dosiahli pri dávke ritonaviru 600 mg dvakrát denne. Antivírusová EC50 lopinaviru in
vitro je približne 10-násobne nižšia ako EC50 ritonaviru. Antivírusový účinok lopinaviru/ritonaviru je teda spôsobený lopinavirom.
Absorpcia: Viacnásobné podávanie 400/100 mg lopinaviru/ritonaviru dvakrát denne počas 2 týždňov bez obmedzenia jedla viedlo k priemernej maximálnej plazmatickej koncentrácii (Cmax) lopinaviru ± SD 12,3 ± 5,4 mg/ml, ktorá sa dosahovala približne 4 hodiny po podaní. Priemerná minimálna koncentrácia v rovnovážnom stave pred prvou rannou dávkou bola 8,1 ± 5,7 mg/ml. Priemerná AUC lopinaviru pri 12-hodinovom dávkovacom intervale bola 113,2 ± 60,5 mg·h/ml. Absolútna biologická dostupnosť lopinaviru v kombinovanom prípravku s ritonavirom u ľudí nebola stanovená.
Účinkypotravynaperorálnuabsorpciu: Podanie jednorazovej dávky 400/100 mg tabliet lopinaviru/ritonaviru po jedle (s vysokým obsahom tuku, 872 kcal, z toho 56% tuk) v porovnaní s podaním nalačno sa nespájal so signifikantnými zmenami v Cmax a AUCinf. Preto sa tablety
lopinaviru/ritonaviru môžu užívať s jedlom alebo bez jedla. Ukázalo sa, že bez ohľadu na stav príjmu potravy majú tablety lopinaviru/ritonaviru v porovnaní s mäkkými kapsulami lopinaviru/ritonaviru menšiu farmakokinetickú variabilitu.
Distribúcia: V rovnovážnom stave je väzba lopinaviru na sérové proteíny približne 98 - 99%. Lopinavir sa viaže na alfa-1 kyslý glykoproteín (AAG) aj na albumín, avšak má vyššiu afinitu pre AAG. V rovnovážnom stave zostáva väzba lopinaviru na proteíny konštantná v sledovanom rozsahu koncentrácií po podaní 400/100 mg lopinaviru/ritonaviru dvakrát denne a je podobná
u zdravých dobrovoľníkov aj HIV-pozitívnych pacientov.
Biotransformácia: Pokusy in vitro s ľudskými hepatálnymi mikrozómami naznačujú, že lopinavir je primárne metabolizovaný oxidatívnou cestou. Lopinavir je extenzívne metabolizovaný hepatálnym cytochrómovým systémom P450, takmer výlučne izoenzýmom CYP3A. Ritonavir je silným inhibítorom CYP3A, čím inhibuje metabolizmus lopinaviru a vedie k zvýšeným plazmatickým hladinám lopinaviru. V štúdii s 14C-lopinavirom u ľudí sa ukázalo, že 89% plazmatickej rádioaktivity po jednorazovej 400/100 mg dávke lopinaviru/ritonaviru tvorí materské liečivo. U ľudí bolo identifikovaných minimálne 13 oxidatívnych metabolitov lopinaviru. Epimerický pár 4-oxo a 4- hydroxymetabolitu je hlavným metabolitom s antivírusovou aktivitou, ale predstavuje len nepatrné množstvo z celkovej plazmatickej rádioaktivity. Bolo dokázané, že ritonavir indukuje metabolické enzýmy, čo vedie k indukcii jeho vlastného metabolizmu a pravdepodobne aj k indukcii metabolizmu lopinaviru. Koncentrácie lopinaviru pred podaním ďalšej dávky klesajú počas viacnásobného podávania a stabilizujú sa približne po 10 dňoch až 2 týždňov.
Eliminácia: Po 400/100 mg dávke 14C-lopinaviru/ritonaviru sa dostane približne 10,4 ± 2,3% podanej dávky 14C-lopinaviru do moču a 82,6 ± 2,5% do stolice. Nezmenený lopinavir sa dostane v množstve približne 2,2% podanej dávky do moču a 19,8% do stolice. Po viacnásobnej dávke je menej ako 3% dávky lopinaviru vylučovanej močom v nezmenenej forme. Účinný polčas (pomer maximálnej
a minimálnej hladiny) lopinaviru pri 12-hodinovom dávkovacom intervale je priemerne 5-6 hodín a zdanlivý perorálny klírens (CL/F) lopinaviru je 6 až 7 l/h.
Dávkovanie raz denne: Hodnotila sa farmakokinetika lopinaviru/ritonaviru podávanej raz denne HIV- infikovaným subjektom bez predchádzajúcej antiretrovírusovej liečby. Lopinavir/ritonavir 800/200 mg sa podávala v kombinácii s emtricitabínom 200 mg a tenofovirom DF 300 mg v dávkovacom režime raz denne. Pri viacnásobnom podaní 800/200 mg lopinaviru/ritonaviru jedenkrát denne počas 2
týždňov bez obmedzenia jedla (n = 16) bola priemerná maximálna plazmatická koncentrácia (Cmax)
lopinaviru ± SD 14,8 ± 3,5 μg/ml, ktorá sa dosahovala približne 6 hodín po podaní. Priemerná minimálna koncentrácia v rovnovážnom stave pred rannou dávkou bola 5,5 ± 5,4 μg/ml. AUC lopinaviru pri 24-hodinovom dávkovacom intervale bola v priemere 206,5 ± 89,7 μg h/ml.
V porovnaní s dávkovacím režimom dvakrát denne je podávanie raz denne spojené s redukciou hodnôt
Cmin/Ctrough približne o 50%.
O
sobitné skupiny pacientov
Pediatrickí pacienti:
Existujú len obmedzené farmakokinetické údaje u detí mladších ako 2 roky. Farmakokinetika lopinaviru/ritonaviru perorálneho roztoku 300/75 mg/m2 dvakrát denne a 230/57,5 mg/m2 dvakrát denne bola študovaná celkovo u 53 pediatrických pacientov vo veku od 6 mesiacov do 12 rokov. Priemerná AUC lopinaviru v rovnovážnom stave bola 72,6 ± 31,1 mg h/ml, Cmax 8,2 ± 2,9 mg/ml a Cmin
3,4 ± 2,1 mg/ml po podávaní lopinaviru/ritonaviru perorálneho roztoku 230/57,5 mg/m2 dvakrát denne bez nevirapínu (n = 12) a boli 85,8 ± 36,9 mg h/ml, 10,0 ± 3,3 mg/ml a 3,6 ± 3,5 mg/ml pri dávke
300/75 mg/m2 dvakrát denne s nevirapínom (n = 12). Dávkovací režim 230/57,5 mg/m2 dvakrát denne bez nevirapínu a 300/75 mg/m2 dvakrát denne s nevirapínom viedlo k plazmatickým koncentráciám lopinaviru podobným ako u dospelých pacientov s dávkovacím režimom 400/100 mg dvakrát denne
bez nevirapínu.
Pohlavie, rasa a vek:
Farmakokinetika lopinaviru/ritonaviru nebola študovaná u starších ľudí. U dospelých pacientov neboli pozorované žiadne rozdiely vo farmakokinetike vo vzťahu k veku a pohlaviu. Nezistili sa farmakokinetické rozdiely v závislosti od rasy.
Gravidita a popôrodné obdobie:
V otvorenej farmakokinetickej štúdii dostávalo 12 HIV-infikovaných gravidných žien, ktoré boli na začiatku štúdie v menej ako 20. týždni gravidity a boli na antiretrovírusovej liečbe, dávku lopinaviru/ritonaviru 400 mg/100 mg (dve tablety 200/50 mg) až do 30. týždňa gravidity.
Od 30. týždňa gravidity sa dávka zvýšila na 500/125 mg (dve tablety 200/50 mg a jedna tableta
100/25 mg) dvakrát denne až do 2. týždňa po pôrode. Plazmatické koncentrácie lopinaviru boli merané počas štyroch 12-hodinových časových úsekov, a to v priebehu druhého trimestra (20. – 24. týždeň gravidity), v treťom trimestri pred zvýšením dávky (30. týždeň gravidity), v treťom trimestri po
zvýšení dávky (32. týždeň gravidity) a v 8. týždni po pôrode. Zvýšenie dávky neviedlo
k signifikantnému zvýšeniu plazmatických koncentrácií lopinaviru.
V ďalšej otvorenej farmakokinetickej štúdii dostávalo 19 HIV-infikovaných gravidných žien dávku lopinaviru/ritonaviru 400/100 mg dvakrát denne ako súčasť kombinovanej antiretrovírusovej liečby počas gravidity od doby pred počatím. Séria krvných vzoriek bola odobratá pred podaním dávky
a v intervaloch v priebehu 12 hodín v 2. a 3. trimestri, pri pôrode, a 4 - 6 týždňov po pôrode (u žien,
ktoré pokračovali v liečbe po pôrode) na farmakokinetickú analýzu hladín celkového a voľného
lopinaviru v plazme.
Farmakokinetické údaje u HIV-1 infikovaných gravidných žien, užívajúcich tablety
lopinaviru/ritonaviru 400/100 mg dvakrát denne sú uvedené v tabuľke 6 (pozri časť 4.2).
Tabuľka 6
Priemerné (% CV) farmakokinetické parametre lopinaviru v rovnovážnom stave
u HIV-infikovaných gravidných žien
P
harmakokinetický
parameter
2. trimester n = 17* 3. trimester n = 23
P
opôrodné obdobie n = 17**
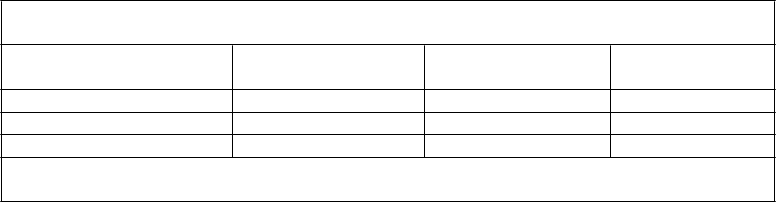
AUC0-12 μg·h/ml 68,7 (20,6) 61,3 (22,7) 94,3 (30,3) Cmax 7,9 (21,1) 7,5 (18,7) 9,8 (24,3) Cpred podaním dávky μg/ml 4,7 (25,2) 4,3 (39,0) 6,5 (40,4)
* n = 18 pre Cmax
** n = 16 pre Cpred podaním dávky
Renálna insuficiencia:Farmakokinetika lopinaviru/ritonaviru nebola študovaná u pacientov s renálnou insuficienciou, avšak keďže renálny klírens lopinaviru je nepatrný, zníženie celkového klírensu u pacientov s renálnou insuficienciou sa neočakáva.
H
epatálna insuficiencia:
Farmakokinetické parametre lopinaviru v rovnovážnom stave u HIV-infikovaných pacientov
s miernou až stredne ťažkou poruchou funkcie pečene boli porovnávané s parametrami zistenými u HIV-infikovaných pacientov s normálnou funkciou pečene, v štúdii s viacnásobnými dávkami lopinaviru/ritonaviru 400/100 mg dvakrát denne. Bol pozorovaný približne 30% nárast v celkových koncentráciách lopinaviru, nepredpokladá sa však, že je klinicky významný (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Štúdie toxicity po opakovanom podávaní u hlodavcov a psov identifikovali ako cieľové orgány pečeň, obličky, štítnu žľazu, slezinu a cirkulujúce erytrocyty. Pečeňové zmeny poukazujú na bunkový opuch
s fokálnou degeneráciou. Zatiaľ čo expozícia vyvolávajúca tieto zmeny bola porovnateľná alebo nižšia
ako klinická expozícia u ľudí, dávkovanie u zvierat bolo viac ako šesťkrát väčšie ako sú odporúčané
klinické dávky. U myší bola preukázaná mierna tubulárna degenerácia obličiek pri minimálne dvojnásobnej expozícii ako je odporúčaná u ľudí; u potkanov a psov neboli obličky ovplyvnené. Znížená hladina sérového tyroxínu viedla k zvýšenému uvoľňovaniu TSH s následnou folikulárnou bunkovou hypertrofiou v štítnej žľaze potkanov. Tieto zmeny boli reverzibilné po ukončení podávania liečiva a neboli prítomné u myší a psov. Coombs negatívna anizocytóza a poikilocytóza boli pozorované u potkanov, nie však u myší alebo u psov. Zväčšenie sleziny s histiocytózou bolo pozorované u potkanov, nie však u ostatných druhov. Zvýšenie sérového cholesterolu bolo zistené
u hlodavcov, ale nie u psov, zatiaľ čo triglyceridy boli zvýšené iba u myší.
Počas štúdií in vitro boli inhibované klonované ľudské srdcové draslíkové kanály (HERG) o 30% pri najvyšších testovaných koncentráciách lopinaviru/ritonaviru, ktoré zodpovedali expozícii 7- násobku celkovej a 15-násobku maximálnych plazmatických koncentrácií lopinaviru, dosiahnutých
u ľudí pri maximálnych odporúčaných terapeutických dávkach. Pre porovnanie podobné koncentrácie lopinaviru/ritonaviru nespôsobovali oneskorenie repolarizácie v srdcových Purkyňových vláknach psa. Nižšie koncentrácie lopinaviru/ritonaviru nespôsobovali významnú blokádu draslíkového vstupu (HERG). Štúdie tkanivovej distribúcie, vykonané na potkanoch, nesvedčia o významnej retencii
liečiva v srdci; 72-hodinová AUC v srdci bola približne 50% nameranej plazmatickej AUC. Preto je opodstatnené sa domnievať, že hladiny lopinaviru v srdci nebudú signifikantne vyššie ako plazmatické
hladiny.
U psov boli na elektrokardiograme pozorované prominujúce vlny U spolu s predĺženým intervalom
PR a bradykardiou. Predpokladá sa, že tieto účinky boli spôsobené elektrolytovou nerovnováhou.
Klinický význam týchto predklinických údajov nie je známy, avšak potenciálne účinky tohto lieku
na srdce u ľudí nie je možné vylúčiť (pozri aj časti 4.4 a 4.8).
U potkanov bola pri podávaní dávok toxických pre matku pozorovaná embryotoxicita (potraty, znížená životnosť plodov, znížená telesná hmotnosť plodov, zvýšená frekvencia odchýlok kostry) a toxicita pri postnatálnom vývoji (znížené prežívanie mláďat). Systémová expozícia lopinaviru/ritonaviru v dávkach toxických pre matku a vývoj plodu bola nižšia ako zamýšľaná terapeutická expozícia u ľudí.
Dlhodobé štúdie karcinogenity lopinaviru/ritonaviru na myšiach ukázali negenotoxickú mitogénnu indukciu tumorov pečene. Toto riziko je všeobecne považované za málo významné pre ľudí.
Štúdie karcinogenity na potkanoch neodhalili žiadne tumorogénne nálezy. V súbore testov in vitro a in vivo vrátane Amesovho testu bakteriálnej reverznej mutácie, testu myšieho lymfómu, myšieho mikrojadrového testu a testu chromozomálnych aberácií ľudských lymfocytov nebolo zistené, že by lopinavir/ritonavir bol mutagénny alebo klastogénny.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Tableta obsahuje:
sorbitánlaurát
koloidný oxid kremičitý bezvodý
kopovidón
nátrium-stearylfumarát
Filmový obal:
hypromelóza
oxid titaničitý (E171)
macrogol hydroxypropylcelulóza mastenec
koloidný oxid kremičitý
polysorbát 80
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
HDPE fľaška: Po prvom otvorení použite do 120 dní.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne požiadavky na uchovávanie. Podmienky uchovávania po prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Lopinavir/Ritonavir Mylan 100 mg/25 mg filmom obalené tablety
OPA/Al/PVC-hliníkové blistrové balenie. Dostupné veľkosti balenia sú:
- Viacpočetné balenie obsahujúce 60, 60x1 (2 škatuľky po 30 alebo 30x1) filmom obalených tabliet.
HDPE fľaška s bielym nepriehľadným polypropylénovým skrutkovacím uzáverom s indukčne tesniacou hliníkovou vložkou a vysúšadlom. Dostupné veľkosti balenia sú:
- 1 fľaška po 60 filmom obalených tabliet.
Lopinavir/Ritonavir Mylan 200 mg/50 mg filmom obalené tablety
OPA/Al/PVC-hliníkové blistrové balenie. Dostupné veľkosti balenia sú:
- Viacpočetné balenie obsahujúce 120, 120x1 (4 škatuľky po 30 alebo 30x1) alebo 360
(12 škatuliek po 30) filmom obalených tabliet.
HDPE fľaška s bielym nepriehľadným polypropylénovým skrutkovacím uzáverom skrutkovacím uzáverom s indukčne tesniacou hliníkovou vložkou a vysúšadlom. Dostupné veľkosti balenia sú:
- 1 fľaška po 120 filmom obalených tabliet.
- Viacpočetné balenie obsahujúce 360 (3 fľašky po 120) filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu <a iné zaobchádzanie s liekom>
Žiadne zvláštne požiadavky.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMylan SAS
117 Allee des Parcs
69800 Saint-Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLAEU/1/15/1067/001
EU/1/15/1067/002
EU/1/15/1067/003
EU/1/15/1067/004
EU/1/15/1067/005
EU/1/15/1067/006
EU/1/15/1067/007
EU/1/15/1067/008
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: {DD. mesiac RRRR}
10. DÁTUM REVÍZIE TEXTU{MM/YYYY}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.