
e a imunogenicita
Pacienti, ktorí sú precitlivení na G-CSF alebo jeho deriváty, sú ohrození aj precitlivenosťou na
lipegfilgrastim z dôvodu možnej skríženej reaktivity. U týchto pacientov sa nemá začínať žiadna liečba lipegfilgrastimom z dôvodu rizika skríženej reakcie.
Väčšina biologických liekov vyvoláva určitú úroveň odpovede protilátok proti lieku. Táto protilátková odpoveď môže v niektorých prípadoch viesť k nežiaducim účinkom alebo k strate účinnosti. Ak pacient nereaguje na liečbu, má podstúpiť ďalšie vyhodnotenie.
Ak dôjde k výskytu závažnej alergickej reakcie, má sa podávať vhodná liečba s dôkladným sledovaním pacienta po dobu niekoľkých dní.
Hematopoetický systém
Liečba lipegfilgrastimom nevylučuje výskyt trombocytopénie a anémie v dôsledku myelosupresívnej
chemoterapie. Lipegfilgrastim môže spôsobovať aj vratnú trombocytopéniu (pozri časť 4.8). Odporúča sa pravidelné sledovanie počtu trombocytov a hematokritu. Počas podávania jedného chemoterapeutického lieku alebo kombinácie chemoterapeutických liekov, o ktorých je známe, že spôsobujú závažnú trombocytopéniu, sa musí postupovať obzvlášť opatrne.
Môže sa vyskytnúť leukocytóza (pozri časť 4.8). Neboli hlásené žiadne nežiaduce udalosti spôsobené priamo leukocytózou. Zvýšenie počtu bielych krviniek (WBC, white blood cells) je v súlade
s farmakodynamickými účinkami lipegfilgrastimu. Počet WBC sa má analyzovať v pravidelných intervaloch počas liečby z dôvodu klinických účinkov lipegfilgrastimu a potenciálu pre vznik leukocytózy. Ak počty WBC prekročia 50 x 109/l po očakávanej najnižšej hodnote, lipegfilgrastim sa má okamžite prestať podávať.
Zvýšená hematopoetická aktivita kostnej drene v odpovedi na liečbu rastovým faktorom bola spojená s prechodne pozitívnymi nálezmi na röntgenových snímkach kostí. To treba zvážiť pri interpretácii výsledkov röntgenových snímok kostí.
Pacienti s myeloidnouleukémioualebomyelodysplastickýmisyndrómami
Faktor stimulujúci kolónie granulocytov môže podporovať rast myeloidných buniek a niektorých
nemyeloidných buniek in vitro.
Bezpečnosť a účinnosť Lonquexu sa neskúmali u pacientov s chronickou myeloidnou leukémiou, myelodysplastickými syndrómami alebo sekundárnou akútnou myeloidnou leukémiou, preto sa nemá používať u týchto pacientov. Zvýšenú pozornosť treba venovať odlíšeniu diagnózy transformácie blastov pri chronickej myeloidnej leukémii od akútnej myeloidnej leukémie.
Slezinové nežiaduce reakcie
Po podaní lipegfilgrastimu boli hlásené všeobecne asymptomatické prípady splenomegálie (pozri
časť 4.8) a po podaní G-CSF alebo jeho derivátov boli hlásené menej časté prípady ruptúry sleziny vrátane fatálnych prípadov (pozri časť 4.8). Z tohto dôvodu sa má dôkladne sledovať veľkosť sleziny (napr. klinickým vyšetrením, ultrazvukom). Diagnóza ruptúry sleziny sa má zvážiť u pacientov, ktorí uvádzajú bolesť v ľavej hornej časti brucha alebo na konci pleca.
Pľúcne nežiaduce reakcie
Po podaní lipegfilgrastimu boli hlásené pľúcne nežiaduce reakcie, najmä intersticiálna pneumónia
(pozri časť 4.8). U pacientov s výskytom pľúcnych infiltrátov alebo pneumóniou v nedávnej anamnéze môže byť toto riziko vyššie.
Nástup pľúcnych príznakov, ako sú kašeľ, horúčka a dyspnoe, v spojení s rádiologicky zistenými príznakmi pľúcnych infiltrátov a zhoršením funkcie pľúc spolu so zvýšeným počtom neutrofilov môžu byť počiatočnými príznakmi syndrómu akútnej respiračnej tiesne (Acute Respiratory Distress Syndrome, ARDS) (pozri časť 4.8). V takýchto prípadoch sa má podávanie Lonquexu podľa uváženia lekára ukončiť a podať vhodná liečba.
Cievne nežiaduce reakcie
Po podaní G-CSF alebo derivátov sa zaznamenal syndróm kapilárneho presakovania, ktorý je
charakterizovaný hypotenziou, hypoalbuminémiou, edémom a hemokoncentráciou. Pacienti, u ktorých sa vyvinú príznaky syndrómu kapilárneho presakovania, sa majú starostlivo sledovať a majú dostať štandardnú symptomatickú liečbu, ktorá môže zahŕňať potrebu intenzívnej starostlivosti (pozri
časť 4.8).
Aortitída bola hlásená po podaní faktora stimulujúceho kolónie granulocytov (granulocyte-colony stimulating factor, G-CSF) u zdravých pacientov a u pacientov s rakovinou. Medzi príznaky patrí horúčka, abdominálna bolesť, nevoľnosť, bolesť chrbta a zvýšená hladina zápalových markerov (napr. C-reaktívny proteín a počet bielych krviniek). Vo väčšine prípadov bola aortitída diagnostikovaná pomocou snímky počítačovej tomografie (computed tomography, CT) a vo všeobecnosti ustúpila po vysadení G-CSF. Pozri tiež časť 4.8.
Pacienti s kosáčikovitou anémiou
Kríza kosáčikovitej anémie sa u pacientov s kosáčikovitou anémiou spája s používaním G-CSF alebo
jeho derivátov (pozri časť 4.8). Lekári majú preto pri podávaní Lonquexu u pacientov s kosáčikovitou anémiou postupovať opatrne, sledovať príslušné klinické parametre a laboratórne výsledky, a venovať pozornosť možnej súvislosti medzi lipegfilgrastimom a zväčšením sleziny a vazookluzívnou krízou.
Hypokaliémia
Môže sa vyskytnúť hypokaliémia (pozri časť 4.8). U pacientov so zvýšeným rizikom hypokaliémie
z dôvodu základného ochorenia alebo súbežne podávaných liekov sa odporúča dôkladne sledovať hladinu sérového draslíka a v prípade potreby ho dopĺňať.
Glomerulonefritída
U pacientov, ktorí dostávali filgrastim, lenograstim alebo pegfilgrastim, bola hlásená
glomerulonefritída. Vo všeobecnosti sa udalosti glomerulonefritídy vyriešili po znížení dávky alebo prerušení podávania filgrastimu, lenograstimu alebo pegfilgrastimu. Odporúča sa sledovanie rozboru moču (pozri časť 4.8).
Pomocné látky so známym účinkom
Tento liek obsahuje sorbitol. Pacienti so zriedkavými dedičnými problémami intolerancie fruktózy
nesmú používať tento liek.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v naplnenej injekčnej striekačke, t. j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Z dôvodu potenciálnej citlivosti rýchlo sa deliacich myeloidných buniek na cytotoxickú chemoterapiu sa má Lonquex podávať približne 24 hodín po podaní cytotoxickej chemoterapie. Súbežné použitie lipegfilgrastimu s akýmkoľvek chemoterapeutickým liekom sa u pacientov nevyhodnocovalo.
Na zvieracích modeloch sa ukázalo, že súbežné podávanie G-CSF a 5-fluórouracilu (5-FU) alebo iných antimetabolitov posilňuje myelosupresiu.
Bezpečnosť a účinnosť Lonquexu sa u pacientov dostávajúcich chemoterapiu, spojenú s oneskorenou myelosupresiou, ako sú napríklad nitrózomočoviny, nevyhodnocovali.
Potenciál pre vznik interakcie s lítiom, ktorý tiež podporuje uvoľňovanie neutrofilov, sa špecificky neskúmal. Neexistuje žiadny dôkaz o tom, že by takáto interakcia bola škodlivá.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii sú iba veľmi obmedzené údaje (menej ako 300 ukončených gravidít) o použití
lipegfilgrastimu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri
časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Lonquexu počas gravidity.
Dojčenie
Nie je známe, či sa lipegfilgrastim/metabolity vylučuje/vylučujú do ľudského mlieka. Riziko
u dojčeného dieťaťa nemôže byť vylúčené. Dojčenie má byť počas liečby Lonquexom ukončené.
Fertilita
K dispozícii nie sú žiadne údaje. Štúdie na zvieratách s G-CSF a derivátmi nepreukázali škodlivé
účinky z hľadiska plodnosti (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Lonquex nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšími nežiaducimi účinkami sú bolesť kostrovej a svalovej sústavy a nevoľnosť.
Syndróm kapilárneho presakovania, ktorý môže ohrozovať život, ak sa oneskorí liečba, sa po podaní G-CSF alebo derivátov zaznamenal väčšinou u pacientov s nádorovým ochorením, podstupujúcich chemoterapiu (pozri časť 4.4 a časť 4.8).
Tabuľkový zoznam nežiaducich reakcií
Bezpečnosť lipegfilgrastimu sa vyhodnocovala na základe výsledkov z klinických štúdií, vrátane
506 pacientov a 76 zdravých dobrovoľníkov, liečených lipegfilgrastimom najmenej jedenkrát.
Nežiaduce reakcie uvedené nižšie v tabuľke 1 sú klasifikované podľa triedy orgánových systémov. Skupiny frekvencií výskytu sú definované podľa nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka
1
:
N
ežiaduce
reakcie
|
Trieda
o
rgánových
s
y
stémov
|
Frekvencia
|
Nežiaduca
re
akcia
|
Poruchy krvi a lymfatického systému
|
Časté
|
Trombocytopénia*
|
Menej časté
|
Leukocytóza*, splenomegália*
|
Poruchy imunitného systému
|
Menej časté
|
Reakcie z precitlivenosti*
|
Poruchy metabolizmu a výživy
|
Časté
|
Hypokaliémia*
|
Poruchy nervového systému
|
Časté
|
Bolesť hlavy
|
Poruchy ciev
|
Neznáme
|
Syndróm kapilárneho presakovania*, aortitída*
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Časté
|
Hemoptýza
|
Menej časté
|
Pľúcne nežiaduce reakcie*, pľúcne krvácanie
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
|
Nevoľnosť*
|
Poruchy kože a podkožného
tkaniva
|
Časté
|
Kožné reakcie*
|
Menej časté
|
Reakcie v mieste vpichu*
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Veľmi časté
|
Bolesť kostrovej a svalovej sústavy*
|
Celkové poruchy a reakcie v mieste podania
|
Časté
|
Bolesť v hrudi
|
Laboratórne a funkčné
vyšetrenia
|
Menej časté
|
Zvýšená hladina alkalickej fosfatázy v krvi*, zvýšená hladina laktátovej dehydrogenázy v krvi*
|
*Pozri časť „Popis vybraných nežiaducich reakcií“ nižšie
|
Popis
v
yb
raných
n
ežiaducich
reakcií
Boli hlásené trombocytopénia a leukocytóza (pozri časť 4.4).
Bola hlásená splenomegália, vo všeobecnosti asymptomatická (pozri časť 4.4).
Môžu sa vyskytnúť reakcie z precitlivenosti, ako napríklad alergické kožné reakcie, urtikária, angioedém a závažné alergické reakcie.
Bola hlásená hypokaliémia (pozri časť 4.4).
Boli hlásené pľúcne nežiaduce reakcie, najmä intersticiálna pneumónia (pozri časť 4.4). Tieto pľúcne nežiaduce reakcie môžu zahŕňať aj edém pľúc, pľúcne infiltráty, fibrózu pľúc, zlyhávanie respiračných funkcií alebo ARDS (pozri časť 4.4).
Nevoľnosť bola veľmi často pozorovaná u pacientov dostávajúcich chemoterapiu. Môžu sa vyskytnúť kožné reakcie, ako napríklad erytém a vyrážka.
Môžu sa vyskytnúť reakcie v mieste vpichu, ako napríklad stvrdnutie miesta vpichu a bolesť v mieste vpichu.
Najčastejšie nežiaduce reakcie zahŕňajú bolesti kostrovej a svalovej sústavy, ako napríklad bolesť kostí a myalgia. Bolesť kostrovej a svalovej sústavy má všeobecne miernu až strednú závažnosť, je prechodná a u väčšiny pacientov ju možno tlmiť štandardnými analgetikami. Hlásili sa však prípady závažnej bolesti kostrovej a svalovej sústavy (hlavne bolesť kostí a bolesť chrbta), vrátane prípadov, ktoré viedli k hospitalizácii.
Môžu sa vyskytnúť vratné, mierne až stredné zvýšenia alkalickej fosfatázy a laktátovej dehydrogenázy, bez súvisiacich klinických účinkov. Zvýšenia hladín alkalickej fosfatázy a laktátovej dehydrogenázy
sú najpravdepodobnejšie spôsobené zvýšenými hladinami neutrofilov.
Niektoré nežiaduce reakcie sa s lipegfilgrastimom doteraz nepozorovali, sú však všeobecne akceptované ako spôsobované G-CSF a jeho derivátmi:
Poruchy krvi a lymfatického systému- Ruptúra sleziny vrátane niektorých fatálnych prípadov (pozri časť 4.4)
- Kríza kosáčikovitej anémie u pacientov s kosáčikovitou anémiou (pozri časť 4.4)
Poruchy ciev- Syndróm kapilárneho presakovania
Po podaní G-CSF alebo derivátov sa po uvedení na trh zaznamenali prípady syndrómu kapilárneho presakovania. Zvyčajne sa vyskytovali u pacientov s pokročilými nádorovými ochoreniami, sepsou, u pacientov liečených kombinovanou chemoterapiou alebo podstupujúcich aferézu (pozri časť 4.4).
- Aortitída (pozri časť 4.4).
Poruchy kože a podkožného tkaniva- Akútna febrilná neutrofilná dermatóza (Sweetov syndróm)
- Dermálna vaskulitída
Poruchy obličiek a močových ciest- Glomerulonefritída (pozri časť 4.4)
Pediatrická populáciaSkúsenosti s použitím u detí sú obmedzené na štúdiu fázy 1 skúmajúcu podanie jednorazovej dávky
u 21 pediatrických pacientov vo veku od 2 do < 18 rokov (pozri časť 5.1), ktorá nenaznačila žiadny rozdiel v bezpečnostnom profile lipegfilgrastimu u detí v porovnaní s dospelými. Nežiaducimi udalosťami súvisiacimi s liečbou boli bolesť chrbta, bolesť kostí a zvýšený počet neutrofilov
(1 udalosť z každej z nich).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNie sú žiadne skúsenosti s predávkovaním lipegfilgrastimom. V prípade predávkovania sa má pravidelne vykonávať sledovanie počtov WBC a trombocytov a má sa dôkladne sledovať veľkosť sleziny (napr. klinickým vyšetrením, ultrazvukom).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunostimulanciá, faktory stimulujúce kolónie, ATC kód: L03AA14
Mechanizmus účinkuLipegfilgrastim je kovalentný konjugát filgrastimu s jednou molekulou metoxypolyetylénglykolu
(PEG) vytvorený prostredníctvom uhľohydrátového spojiva pozostávajúceho z glycínu, kyseliny
N-acetylneuramínovej a
N-acetylgalaktozamínu. Priemerná molekulárna hmotnosť je približne 39 kDa, z čoho proteínový podiel tvorí približne 48 %. Ľudský G-CSF je glykoproteín, ktorý reguluje tvorbu
a uvoľňovanie funkčných neutrofilov z kostnej drene. Filgrastim je neglykozylovaný rekombinantný metionylový ľudský G-CSF. Lipegfilgrastim je forma filgrastimu s predĺženým účinkom z dôvodu zníženého obličkového klírensu. Lipegfilgrastim sa viaže na ľudský receptor G-CSF ako filgrastim
a pegfilgrastim.
Farmakodynamické účinkyLipegfilgrastim a filgrastim vyvolávali značné zvýšenie počtov neutrofilov v periférnej krvi v priebehu
24 hodín, s malými zvýšeniami hladín monocytov a/alebo lymfocytov. Tieto výsledky naznačujú, že podiel G-CSF v lipegfilgrastime zodpovedá za očakávanú aktivitu tohto rastového faktora: stimuláciu množenia hematopoetických progenitorových buniek, diferenciáciu do zrelých buniek a uvoľňovanie do periférnej krvi. Tento účinok zahŕňa nielen líniu neutrofilných buniek, ale rozširuje sa aj na iné jednolíniové a viaclíniové progenitory a pluripotentné hematopoetické kmeňové bunky. G-CSF tiež zvyšuje antibakteriálne aktivity neutrofilov vrátane fagocytózy.
Klinická účinnosť a bezpečnosťDávkovanie lipegfilgrastimu jedenkrát za cyklus sa skúmalo v dvoch kľúčových randomizovaných,
dvojito zaslepených klinických štúdiách u pacientov podstupujúcich myelosupresívnu chemoterapiu.
Prvá kľúčová klinická štúdia XM22-03 (fázy III) bola aktívne kontrolovaná štúdia u 202 pacientov s rakovinou prsníka v štádiu II-IV, dostávajúcich najviac 4 cykly chemoterapie obsahujúcej doxorubicín a docetaxel. Pacienti boli randomizovaní v pomere 1:1 na podávanie 6 mg lipegfilgrastimu alebo 6 mg pegfilgrastimu. Táto štúdia preukázala nepodradnosť dávky 6 mg lipegfilgrastimu v porovnaní s dávkou 6 mg pegfilgrastimu z hľadiska primárneho koncového ukazovateľa, ktorým bolo trvanie závažnej neutropénie (
Duration of Severe Neutropenia, DSN)
v prvom cykle chemoterapie (pozri tabuľku 2).
Tabuľka2: DSN, závažnáneutropénia(severe neutropenia, SN) a febrilná neutropénia (FN)
v 1. cykle štúdie XM22-03 (ITT)
|
| Pegfilgrastim 6 mg
(n = 101)
| Lipegfilgrastim 6 mg
(n = 101)
|
Trvaniezávažnejneutropénie(DSN)
|
Priemerná hodnota ± SD (d)
| 0,9 ± 0,9
| 0,7 ± 1,0
|
Δ priemernej hodnoty zistenej metódou LS
| -0,186
|
95 % IS
| -0,461 až 0,089
|
Závažnáneutropénia(SN)
|
Miera výskytu (%)
| 51,5
| 43,6
|
Febrilnáneutropénia(FN)
|
Miera výskytu (%)
| 3,0
| 1,0
|
ITT – populácia s liečebným zámerom (všetci randomizovaní pacienti) SD – smerodajná odchýlka
d – dni
IS – interval spoľahlivosti
Δ priemernej hodnoty zistenej metódou LS (rozdiel priemernej hodnoty zistenej metódou najmenších štvorcov medzi lipegfilgrastimom a pegfilgrastimom) a IS z multivariačnej Poissonovej regresnej analýzy
|
Druhá kľúčová klinická štúdia XM22-04 (fázy III) bola placebom kontrolovaná štúdia u 375 pacientov
s nemalobunkovým karcinómom pľúc dostávajúcich najviac 4 cykly chemoterapie obsahujúcej cisplatinu a etopozid. Pacienti boli randomizovaní v pomere 2:1 na podávanie buď 6 mg lipegfilgrastimu, alebo placeba. Výsledky tejto štúdie sú uvedené v tabuľke 3. Po dokončení hlavnej štúdie bola miera výskytu úmrtia na úrovni 7,2 % (placebo) a 12,5 % (6 mg lipegfilgrastim), aj keď po
360-dňovom období sledovania bola celková miera výskytu úmrtia medzi placebom
a lipegfilgrastimom (44,8 % a 44,0 %, populácia na vyhodnotenie bezpečnosti) podobná.
Tabuľka3: DSN, SN a FN v 1. cykle štúdie XM22-04 (ITT)
|
| Placebo
(n = 125)
| Lipegfilgrastim 6 mg
(n = 250)
|
FN
|
Miera výskytu (%)
| 5,6
| 2,4
|
95 % IS
| 0,121 až 1,260
|
Hodnota p
| 0,1151
|
Trvaniezávažnejneutropénie(duration of severe neutropenia, DSN)
|
Priemerná hodnota ± SD (d)
| 2,3 ± 2,5
| 0,6 ± 1,1
|
Δ priemernej hodnoty zistenej metódou LS
| -1,661
|
95 % IS
| -2,089 až -1,232
|
Hodnota p
| < 0,0001
|
Závažnáneutropénia(severe neutropenia, SN)
|
Miera výskytu (%)
| 59,2
| 32,1
|
Miera pravdepodobnosti
| 0,325
|
95 % IS
| 0,206 až 0,512
|
Hodnota p
| < 0,0001'
|
Δ priemernej hodnoty zistenej metódou LS (rozdiel priemernej hodnoty zistenej metódou najmenších štvorcov medzi lipegfilgrastimom a placebom), IS a hodnota p z multivariačnej Poissonovej regresnej
analýzy
Miera pravdepodobnosti (lipegfilgrastim/placebo), IS a hodnota p z multivariačnej logaritmickej regresnej analýzy
|
Po registrácii lieku sa vykonala štúdia bezpečnosti XM22-ONC-40041 na zozbieranie údajov
týkajúcich sa progresie ochorenia a úmrtnosti u pacientov s pokročilým karcinómom pľúc skvamóznych alebo neskvamóznych buniek dostávajúcich lipegfilgrastim dodatočne k chemoterapii na základe platiny. Pre lipegfilgrastim sa nepozorovalo zvýšené riziko progresie ochorenia alebo úmrtnosti.
ImunogenitaVykonala sa analýza protilátok proti lieku u 579 pacientov a zdravých dobrovoľníkov liečených lipegfilgrastimom, 188 pacientov a zdravých dobrovoľníkov liečených pegfilgrastimom
a 121 pacientov liečených placebom. Liekové špecifické protilátky objavujúce sa po začatí liečby boli zistené u 0,86 % pacientov dostávajúcich lipegfilgrastim, u 1,06 % pacientov dostávajúcich pegfilgrastim a u 1,65 % pacientov dostávajúcich placebo. Nepozorovali sa žiadne neutralizačné protilátky proti lipegfilgrastimu.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Lonquexom vo
všetkých podskupinách pediatrickej populácie pri liečbe chemoterapiou vyvolanej neutropénie a prevencii chemoterapiou vyvolanej febrilnej neutropénie (informácie o použití v pediatrickej populácii, pozri časť 4.2). V štúdii fázy 1 vykonávanej u 21 detí vo veku od 2 do 16 rokov
s Ewingovou skupinou nádorov alebo rabdomyosarkómom sa lipegfilgrastim podával ako jednorazová subkutánna dávka 100 μg/kg (do maximálne 6 mg, čo je fixná dávka pre dospelých) 24 hodín po skončení poslednej chemoterapeutickej liečby v 1. týždni režimu liečby. Miera výskytu FN sa líšila podľa veku (od 14,3 % do 71,4 %), pričom najvyššia frekvencia bola v najstaršej vekovej skupine. Použitie troch rôznych režimov chemoterapeutickej liečby s rôznymi myelosupresívnymi účinkami
a vekovými distribúciami komplikovalo porovnanie účinnosti v rámci vekových skupín (pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Všeobecné informácie
Zdraví dobrovoľníci
V 3 štúdiách (XM22-01, XM22-05, XM22-06) u zdravých dobrovoľníkov sa maximálna koncentrácia v krvi dosiahla po mediáne 30 až 36 hodín a priemerný terminálny polčas bol v rozsahu od približne
32 do 62 hodín po jednorazovej subkutánnej injekcii 6 mg lipegfilgrastimu.
Po subkutánnej injekcii 6 mg lipegfilgrastimu do troch rôznych miest (rameno, brucho a stehno)
u zdravých dobrovoľníkov bola biologická dostupnosť (maximálna koncentrácia a plocha pod krivkou
[AUC]) nižšia po subkutánnej injekcii do stehna v porovnaní so subkutánnou injekciou do brucha
a ramena. V tejto obmedzenej štúdii XM22-06 boli biologická dostupnosť lipegfilgrastimu
a pozorované rozdiely medzi miestami vpichu vyššie u mužov než u žien. Napriek tomu boli farmakodynamické účinky podobné a nezávislé od pohlavia a miesta vpichu.
Metabolizmus
Lipegfilgrastim sa metabolizuje prostredníctvom intra- alebo extracelulárnej degradácie
proteolytickými enzýmami. Lipegfilgrastim sa internalizuje neutrofilmi (nelineárny proces) a následne sa rozkladá vnútri bunky endogénnymi proteolytickými enzýmami. Lineárna dráha je pravdepodobná
z dôvodu extracelulárnej proteínovej degradácie neutrofilovou elastázou a inými plazmatickými proteázami.
Liekové interakcie
In vitro údaje ukazujú, že lipegfilgrastim má malé alebo nemá žiadne priame ani imunitným systémom sprostredkované účinky na aktivitu enzýmov CYP1A2, CYP2B6, CYP2C8, CYP2C9,
CYP2C19 a CYP3A4/5. Preto u lipegfilgrastimu nie je pravdepodobné, že by ovplyvňoval metabolizmus prostredníctvom enzýmov ľudského cytochrómu P450.
Osobitné populácie
Pacienti s rakovinou
V 2 štúdiách (XM22-02 a XM22-03) u pacientov s rakovinou prsníka, dostávajúcich chemoterapiu obsahujúcu doxorubicín a docetaxel, sa priemerné maximálne koncentrácie 227 a 262 ng/ml v krvi dosiahli po mediánoch časov do maximálnej koncentrácie (tmax) v trvaní 44 a 48 hodín. Priemerné terminálne polčasy boli približne 29 a 31 hodín po jednorazovej subkutánnej injekcii 6 mg
lipegfilgrastimu počas prvého cyklu chemoterapie. Po jednorazovej subkutánnej injekcii 6 mg lipegfilgrastimu počas štvrtého cyklu boli maximálne koncentrácie v krvi nižšie, než sa pozorovali v prvom cykle (priemerné hodnoty 77 a 111 ng/ml), a dosiahli sa po mediáne tmax v trvaní 8 hodín. Priemerné terminálne polčasy v štvrtom cykle boli približne 39 a 42 hodín.
V štúdii (XM22-04) u pacientov s nemalobunkovým karcinómom pľúc, dostávajúcich chemoterapiu obsahujúcu cisplatinu a etopozid, sa priemerná maximálna koncentrácia v krvi 317 ng/ml dosiahla po mediáne tmax v trvaní 24 hodín a priemerný terminálny polčas bol približne 28 hodín po jednorazovej subkutánnej injekcii 6 mg lipegfilgrastimu počas prvého cyklu chemoterapie. Po jednorazovej subkutánnej injekcii 6 mg lipegfilgrastimu počas štvrtého cyklu sa priemerná maximálna koncentrácia v krvi 149 ng/ml dosiahla po mediáne tmax v trvaní 8 hodín a priemerný terminálny polčas bol približne
34 hodín.
Zdá sa, že lipegfilgrastim sa vylučuje prevažne cez klírens sprostredkovaný neutrofilmi, ktorý pri vyšších dávkach saturuje. Sérová koncentrácia lipegfilgrastimu počas chemoterapiou vyvolanej prechodnej najnižšej hodnoty počtu neutrofilov klesá pomaly a pri nasledujúcom nástupe obnovy neutrofilov klesá rýchlo, v súlade so samoregulačným mechanizmom klírensu (pozri obrázok 1).
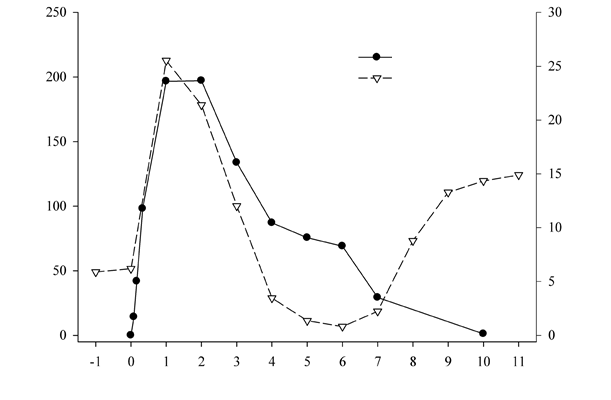 Obrázok 1: Profil mediánu sérovej koncentrácie lipegfilgrastimu a mediánu ANC u pacientov
Obrázok 1: Profil mediánu sérovej koncentrácie lipegfilgrastimu a mediánu ANC u pacientov
 liečených
chemoterapiou
p
o
jednej
6
mg injekcii lipegfilgrastimu
liečených
chemoterapiou
p
o
jednej
6
mg injekcii lipegfilgrastimu
Lipegfilgrastim
ANC
Počet dní štúdie, injekcia lipegfilgrastimu v 0. deň
Pacienti s poruchou funkcie obličiek alebo pečeneZ dôvodu neutrofilmi sprostredkovaného mechanizmu klírensu sa neočakáva ovplyvnenie farmakokinetických vlastností lipegfilgrastimu poškodením obličiek ani pečene.
Starší pacientiObmedzené údaje získané u pacientov ukázali, že farmakokinetické vlastnosti lipegfilgrastimu
u starších pacientov (65-74 rokov) sú podobné ako u mladších pacientov. U pacientov vo veku
≥ 75 rokov nie sú k dispozícii žiadne farmakokinetické údaje.
Pediatrická populáciaV štúdii fázy 1 (pozri časť 5.1), v ktorej sa používal roztok na subkutánnu injekciu s koncentráciou
10 mg/ml špecificky navrhnutý pre pediatrické štúdie, dosahovali priemerné maximálne koncentrácie v krvi (Cmax) po injekčnom podaní jednorazovej subkutánnej dávky 100 μg/kg (maximálne 6 mg) lipegfilgrastimu s prvým cyklom chemoterapie úroveň 243 ng/ml v skupine od 2 do < 6 rokov,
255 ng/ml v skupine od 6 do < 12 rokov a 224 ng/ml v skupine od 12 do < 18 rokov. Maximálne koncentrácie v krvi sa dosahovali po mediáne času (tmax) na úrovni 23,9 hodiny; 30,0 hodín
a 95,8 hodiny, v uvedenom poradí. Pozri časť 4.2.
Pacienti s nadváhouPozoroval sa trend smerujúci k zníženiu expozície lipegfilgrastimu so zvyšovaním hmotnosti. Toto môže spôsobiť znížené farmakodynamické odpovede u ťažkých pacientov (> 95 kg). Na základe aktuálnych údajov nemožno vylúčiť následné zníženie účinnosti u týchto pacientov.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po jednorazovom a opakovanom podávaní a lokálnej znášanlivosti neodhalili žiadne osobitné riziko pre ľudí.
V štúdii reprodukčnej a vývojovej toxicity u králikov sa pri vysokých dávkach lipegfilgrastimu pozorovala zvýšená miera výskytu poimplantačných úmrtí a potratov, pravdepodobne z dôvodu nadmerného farmakodynamického účinku špecifického pre králikov. Nie sú žiadne dôkazy o tom, že je lipegfilgrastim teratogénny. Tieto nálezy sú v súlade s výsledkami získanými s G-CSF a jeho derivátmi. Publikované informácie o G-CSF a jeho derivátoch neodhalili žiadny dôkaz nežiaducich účinkov na fertilitu a embryofetálny vývoj u potkanov ani pre-/postnatálnych účinkov iných než súvisiacich aj s materskou toxicitou. Existuje dôkaz, že filgrastim a pegfilgrastim môžu u potkanov prechádzať pri nízkych hladinách cez placentu, avšak pre lipegfilgrastim nie sú k dispozícii žiadne informácie. Význam týchto zistení pre ľudí nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
ľadová kyselina octová hydroxid sodný (na úpravu pH) sorbitol (E420)
polysorbát 20
voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Lonquex možno vybrať z chladničky a uchovávať pri teplote do 25 °C počas maximálne jedného obdobia trvajúceho najviac 3 dni. Po vybratí z chladničky sa liek musí použiť počas tohto obdobia alebo zlikvidovať.
6.5 Druh obalu a obsah balenia
Naplnená injekčná striekačka (sklo typu I) s piestovou zátkou [brómbutylová guma s povlakom
z poly(etylén-ko-tetrafluóroetylénu)] a pevne nasadenou injekčnou ihlou (z nehrdzavejúcej ocele, 29G
[0,34 mm] alebo 27G [0,4 mm] x 0,5 palca [12,7 mm]). Každá naplnená injekčná striekačka obsahuje 0,6 ml roztoku.
Veľkosti balení s 1 a 4 naplnenými injekčnými striekačkami s bezpečnostným zariadením (ktoré zabraňuje poraneniu pichnutím ihlou a opätovnému použitiu) alebo s 1 injekčnou striekačkou bez bezpečnostného zariadenia.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6
.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Roztok sa má pred použitím vizuálne skontrolovať. Majú sa použiť iba číre, bezfarebné roztoky bez častíc.
Roztok sa má nechať, aby dosiahol príjemnú teplotu (15 °C - 25 °C) na injekciu.
Vyhýbajte sa prudkému traseniu. Nadmerné trasenie môže spôsobiť agregáciu lipegfilgrastimu, čím sa stane biologicky neaktívny.
Lonquex neobsahuje žiadnu konzervačnú látku. S ohľadom na možné riziko mikrobiologickej kontaminácie sú injekčné striekačky Lonquex určené iba na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIITeva B.V. Swensweg 5
2031 GA Haarlem
Holandsko
8. REGISTRAČNÉ ČÍSLAEU/1/13/856/001
EU/1/13/856/002
EU/1/13/856/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 25. júl 2013.
Dátum posledného predĺženia registrácie: 08. mája 2018.
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.