
th="170" valign="top"> Stupeň 4
Vysadenie
| Nenasadzovať znova
|
Tabuľka 3 Nežiaduce reakcie vyžadujúce úpravu dávky lenvatinibu pri DTC a HCC
|
Nežiaduca reakcia
|
Z
ávažnosť
|
Akcia
|
Redukcia dávky a obnovenie lenvatinibu
|
Proteinúria
|
≥ 2 g / 24 hodín
|
Prerušenie
|
Odznenie na menej ako 2 g /
24 hodín.
|
Nefrotický syndróm
|
-------
|
Vysadenie
|
Nenasadzovať znova
|
Porucha funkcie obličiek alebo renálne zlyhanie
|
Stupeň 3
|
Prerušenie
|
Odznenie na stupeň 0-1 alebo na stav pred začatím liečby.
|
Stupeň 4*
|
Vysadenie
|
Nenasadzovať znova
|
Porucha srdca
a srdcovej činnosti
|
Stupeň 3
|
Prerušenie
|
Odznenie na stupeň 0-1 alebo na stav pred začatím liečby.
|
Stupeň 4
|
Vysadenie
|
Nenasadzovať znova
|
PRES/RPLS
|
Akýkoľvek stupeň
|
Prerušenie
|
Zváženie nasadenia pri zníženej dávke, ak sa zmierni na stupeň 0-1.
|
Hepatotoxicita
|
Stupeň 3
|
Prerušenie
|
Odznenie na stupeň 0-1 alebo na stav pred začatím liečby.
|
Stupeň 4*
|
Vysadenie
|
Nenasadzovať znova
|
Artériová tromboembolizácia
|
Akýkoľvek stupeň
|
Vysadenie
|
Nenasadzovať znova
|
Krvácanie
|
Stupeň 3
|
Prerušenie
|
Odznenie na stupeň 0-1.
|
Stupeň 4
|
Vysadenie
|
Nenasadzovať znova
|
Perforácia GI alebo fistula
|
Stupeň 3
|
Prerušenie
|
Odznenie na stupeň 0-1 alebo na stav pred začatím liečby
|
Stupeň 4
|
Vysadenie
|
Nenasadzovať znova
|
Non-GI fistula
|
Stupeň 4
|
Vysadenie
|
Nenasadzovať znova
|
Predĺženie QT
intervalu
|
>500 ms
|
Prerušenie
|
Odznenie na <480 ms alebo na stav
pred začatím liečby
|
Hnačka
|
Stupeň 3
|
Prerušenie
|
Odznenie na stupeň 0-1 alebo na stav pred začatím liečby
|
Stupeň 4 (napriek liečbe)
|
Vysadenie
|
Nenasadzovať znova
|
*Stupeň 4 laboratórnych abnormalít, ktoré sa nepovažujú za život ohrozujúce, sa môžu liečiť ako
závažné reakcie (napr. stupeň 3).
Osobitné skupiny pacientovStarší pacientiDTCU pacientov vo veku ≥ 75 rokov, ázijskej rasy, so sprievodnými ochoreniami (ako je hypertenzia a porucha funkcie pečene alebo obličiek), alebo s telesnou hmotnosťou pod 60 kg sa pozoruje nižšia tolerancia lenvatinibu (pozri časť 4.8, Ostatné osobitné skupiny). Všetci pacienti okrem tých s ťažkou poruchou funkcie pečene alebo obličiek (pozri nižšie) majú začať liečbu s odporúčanou dávkou 24 mg a tá sa má následne upravovať podľa individuálnej znášanlivosti.
HCCU pacientov vo veku ≥75 rokov, bielej rasy alebo ženského pohlavia, prípadne u tých, ktorí mali na začiatku liečby výraznejšie poškodenie funkcie pečene (skóre Child-Pugh A 6 v porovnaní so skóre 5) sa pozoruje nižšia tolerancia lenvatinibu.
Pacienti s HCC, okrem tých s miernym až stredne ťažkým poškodením funkcie pečene by mali začať liečbu na odporúčanej úvodnej dávke 8 mg (dve 4 mg kapsuly) pri telesnej hmotnosti <60 kg a 12 mg (tri 4 mg kapsuly) pri telesnej hmotnosti ≥60 kg. Môže byť potrebná aj ďalšia úprava dávky podľa individuálnej znášanlivosti.
Pacienti s hypertenziou
Pred liečbou lenvatinibom má byť krvný tlak dostatočne kontrolovaný a počas liečby sa má pravidelne sledovať (pozri časť 4.4). Pozri tiež časť 4.8, Ostatné osobitné skupiny.
Pacienti s poruchou funkcie pečene
DTC
U pacientov s miernou poruchou (Child-Pugh A) alebo stredne ťažkou poruchou (Child-Pugh B) funkcie pečene nie je úprava úvodnej dávky potrebná. U pacientov s ťažkou poruchou funkcie pečene (Child-Pugh C) sa odporúča úvodná dávka 14 mg jedenkrát denne. Môže byť potrebná aj ďalšia úprava dávky podľa individuálnej znášanlivosti. Pozri tiež časť 4.8, Ostatné osobitné skupiny.
HCC
U pacientov zaradených do štúdie HCC neboli potrebné žiadne úpravy dávkovania u tých pacientov, ktorí mali miernu poruchu funkcie pečene (Child-Pugh A). Dostupné veľmi obmedzené údaje nie sú dostatočné na vypracovanie odporúčania na dávkovanie u pacientov HCC s miernou poruchou funkcie pečene (Child-Pugh B). U týchto pacientov sa odporúča dôkladné monitorovanie celkovej bezpečnosti (pozri časti 4.4 a 5.2). Lenvatinib nebol skúšaný u pacientov s ťažkou poruchou funkcie pečene
(Child-Pugh C) a u týchto pacientov sa užívanie neodporúča.
Pacienti s poruchou funkcie obličiek
DTC
U pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je úprava úvodnej dávky potrebná. U pacientov s ťažkou poruchou funkcie obličiek sa odporúča úvodná dávka 14 mg jedenkrát denne. Môže byť potrebná aj ďalšia úprava dávky podľa individuálnej znášanlivosti. U pacientov
v konečnom štádiu ochorenia obličiek sa klinické štúdie s lenvatinibom nerobilia preto sa podávanie lenvatinibu u týchto pacientov neodporúča. Pozri tiež časť 4.8, Ostatné osobitné skupiny.
HCC
U pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná úprava dávkovania. Dostupné údaje nie sú dostatočné na vypracovanie odporúčania na dávkovanie u pacientov s HCC a ťažkou poruchou funkcie obličiek.
Starší pacienti
V súvislosti s vekom nie je potrebná žiadna úprava úvodnej dávky. O použití lenvatinibu u pacientov vo veku ≥ 75 rokov sú obmedzené údaje (pozri tiež časť 4.8, Ostatné osobitné skupiny).
Pediatrická populácia
Lenvatinib sa nemá podávať deťom mladším ako 2 roky kvôli možným bezpečnostným rizikám, ktoré sa zistili v štúdiách na zvieratách (pozri časť 5.3). Bezpečnosť a účinnosť lenvatinibu u detí vo veku
2 až < 18 rokov sa ešte nestanovila (pozri časť 5.1). Nie sú dostupné žiadne údaje.
Rasa
V súvislosti s rasou nie je potrebná žiadna úprava úvodnej dávky (pozri časť 5.2). O použití lenvatinibu u pacientov iných rás ako kaukazskej alebo ázijskej sú obmedzené údaje (pozri tiež časť
4.8, Ostatné osobitné skupiny).
Spôsob podávania
Lenvatinib je určený na perorálne použitie. Kapsuly sa majú užívať každý deň v rovnaký čas, s jedlom
alebo bez jedla (pozri časť 5.2). Kapsuly sa majú prehĺtať celé a zapiť vodou. Opatrovatelia by nemali
kapsuly otvárať, aby nedošlo k opakovanej expozícii obsahom kapsuly.
Alternatívne sa kapsuly levanitibu môžu pridávať celé alebo rozdrvené v polievkovej lyžici vody alebo jablkového džúsu do malého pohára, aby vznikol roztok. Kapsuly musia ostať v tekutine aspoň
10 minút a musia sa aspoň 3 minúty miešať, aby sa obal kapsuly rozpustil. Roztok sa musí prehltnúť. Po vypití je potrebné pridať do pohára rovnaké množstvo vody alebo jablkového džúsu (jedna polievková lyžica) a pohár niekoľkokrát premiešať. Aj zvyšná tekutina sa musí prehltnúť.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Laktácia (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaníHypertenziaU pacientov liečených lenvatinibom sa zvyčajne krátko po začatí liečby pozorovala hypertenzia (pozri
časť 4.8, Popis vybraných nežiaducich reakcií). Krvný tlak (TK) by mal byť pred začatím liečby lenvatinibom dostatočne kontrolovaný a pacienti so známou hypertenziou by mali užívať stabilnú dávku antihypertenzív najmenej 1 týždeň pred začatím liečby lenvatinibom. Boli hlásené závažné komplikácie zle liečenej hypertenzie vrátane disekcie aorty. Včasné zistenie a účinné zvládnutie hypertenzie je dôležité na predchádzanie potreby prerušiť alebo redukovať dávku lenvatinibu. Liečba antihypertenzívami sa má začať hneď, ako sa potvrdí zvýšený TK. TK sa má sledovať po jednom týždni liečby lenvatinibom, potom každé dva týždne prvé 2 mesiace a následne v mesačnom intervale. Výber antihypertenzívnej liečby by mal zohľadniť klinický stav každého pacienta a mal by byť
v súlade so štandardnými liečebnými postupmi. U pacientov s normálnym tlakom krvi by sa pred liečbou lenvatinibom malo po zistení zvýšeného tlaku krvi začať s monoterapiou liekom z jednej z tried antihypertenzív. U pacientov užívajúcich antihypertenzívum sa môže, ak je to vhodné, pristúpiť k zvýšeniu dávky aktuálnej medikácie alebo sa môže k liečbe pridať jedno alebo viac antihypertenzív z rozdielnej triedy. V prípade potreby hypertenziu liečte podľa odporučenia v tabuľke 4.
Tabuľka 4 Odporúčané postupy pre zvládnutie hypertenzie
|
Hodnota tlaku krvi (TK)
|
Odporučený postup
|
Systolický TK od ≥140 mmHg do <160 mmHg alebo diastolický TK od ≥90 mmHg
do <100 mmHg
|
Pokračovať s lenvatinibom a začať antihypertenzívnu
liečbu, ak ešte nebola začatá
ALEBO
Pokračovať s lenvatinibom a zvýšiť dávku súčasnej antihypertenzívnej liečby alebo pridať ďalšie antihypertenzíva
|
Tabuľka 4 Odporúčané postupy pre zvládnutie hypertenzie
|
Hodnota tlaku krvi (TK)
|
Odporučený postup
|
Systolický TK od ≥160 mmHg alebo diastolický TK od ≥100 mmHg
napriek optimálnej antihypertenzívnej liečbe
|
1. Nepodať lenvatinib
2. Keď systolický TK klesne ≤150 mmHg, diastolický TK ≤95 mmHg a pacient je nastavený na stabilnú dávku antihypertenzív najmenej 48 hodín, opäť podať lenvatinib
v redukovanej dávke (pozri časť 4.2)
|
Život ohrozujúce následky
(malígna hypertenzia, neurologické poruchy alebo hypertenzná kríza)
|
Je nutná urgentná intervencia. Vysadiť lenvatinib
a začať patričnú liečbu.
|
Aneuryzmy a arteriálne disekcie
Používanie inhibítorov dráhy vaskulárneho endotelového rastového faktora (vascular endothelial
growth factor, VEGF) u pacientov s hypertenziou alebo bez hypertenzie môže podporovať tvorbu aneuryziem a/alebo arteriálnych disekcií. Pred začatím liečby lenvatinibom je potrebné toto riziko dôkladne zvážiť u pacientov s rizikovými faktormi, ako je hypertenzia alebo aneuryzma v anamnéze.
ProteinúriaU pacientov liečených lenvatinibom sa zvyčajne krátko po začatí liečby pozorovala proteinúria (pozri
časť 4.8, Popis vybraných nežiaducich reakcií). Bielkoviny v moči je treba pravidelne sledovať. Ak je proteinúria papierikovým testom ≥ 2+, môže byť potrebné prerušiť podávanie, upraviť dávku alebo liečbu ukončiť (pozri časť 4.2). Prípady nefrotického syndrómu boli hlásené u pacientov užívajúcich lenvatinib. V prípade nefrotického syndrómu sa má liečba lenvatinibom ukončiť.
HepatotoxicitaPri DTC, hlavne u pacientov liečených lenvatinibom boli hlásené nežiaduce reakciespojené
s poruchami pečeňových funkcií so zvýšením alanínaminotransferázy (ALT),
aspartátaminotransferázy (AST) a sérového bilirubínu. U pacientov s DTC užívajúcich lenvatinib bolo hlásené hepatálne zlyhanie a akútna hepatitída (<1 %; pozri časť 4.8, Popis vybraných nežiaducich reakcií). Prípady hepatálneho zlyhania boli hlásené u pacientov s progresívnymi pečeňovými metastázami.
U pacientov s HCC liečených lenvatinibom v rámci skúšania REFLECT, boli s vyššou frekvenciou hlásené nežiaduce reakcie spojené s pečeňou, vrátane hepatálnej encefalopatie a hepatálneho zlyhania (vrátane fatálnych reakcií) (pozri časť 4.8) v porovnaní s pacientmi liečenými sorafenibom. Pacienti s ťažším poškodením funkcie pečene a/alebo silnejšou záťažou tumorov pečene na začiatku liečby mali vyššie riziko rozvoja hepatálnej encefalopatie a hepatálneho zlyhania. Hepatálna encefalopatia sa tak isto objavovala častejšie u pacientov vo veku nad 75 rokov. Približne polovica prípadov hepatálneho zlyhania a jedna tretina prípadov hepatálnej encefalopatie bola hlásená u pacientov s progresívnym ochorením.
Údaje pacientov s HCC so stredne ťažkou poruchou funkcie pečene (Child-Pugh B) sú veľmi obmedzené a v súčasnosti nie sú k dispozícii žiadne údaje pacientov s ťažkou poruchou funkcie pečene (Child-Pugh C). Keďže lenvatinib sa eliminuje najmä hepatálnym metabolizmom, očakáva sa nárast vystavenia pacientov so stredne ťažkou až ťažkou poruchou funkcie pečene.
U pacientov s miernou až stredne ťažkou poruchou funkcie pečene sa odporúča dôkladné monitorovanie celkovej bezpečnosti (pozri aj časti 4.2 a 5.2).Testy hepatálnych funkcií sa majú vyšetriť pred začatím liečby, potom každé dva týždne prvé 2 mesiace a počas liečby v mesačnom intervale. Pacienti s HCC by sa mali monitorovať vo vzťahu k zhoršovaniu funkcie pečene, vrátane hepatálnej encefalopatie. V prípade hepatotoxicity môže byť potrebné prerušenie podávania, úprava dávky alebo ukončenie liečby (pozri časť 4.2).
Renálne zlyhanie alebo porucha funkcie obličiek
U pacientov liečených lenvatinibom sa pozorovali prípady poruchy funkcie obličiek a renálne zlyhanie
(pozri časť 4.8, Popis vybraných nežiaducich reakcií). Primárnym rizikovým faktorom bola dehydratácia a/alebo hypovolémia v dôsledku gastrointestinálnej toxicity. Gastrointestinálnu toxicitu treba aktívne liečiť, aby sa znížilo riziko rozvoja poruchy funkcie obličiek alebo renálneho zlyhania. Ďalej môže byť potrebné prerušenie podávania, úprava dávky alebo ukončenie liečby (pozri časť 4.2).
U pacientov s ťažkou poruchou funkcie obličiek je potrebné upraviť úvodnú dávku lenvatinibu (pozri časti 4.2. a 5.2).
Hnačka
U pacientov liečených lenvatinibom bola často hlásená hnačka, zvyčajne sa vyskytovala včasne
v priebehu liečby (pozri časť 4.8, Popis vybraných nežiaducich reakcií). Aby sa predišlo dehydratácii, je potrebné začaťokamžitú liečbu hnačky. Lenvatinib sa má vysadiť v prípade perzistencie hnačky stupňa 4 napriek poskytnutej liečbe.
Srdcová dysfunkcia
U pacientov liečených lenvatinibom sa pozorovali srdcové zlyhanie (< 1 %) a znížená ejekčná frakcia
ľavej komory (pozri časť 4.8, Popis vybraných nežiaducich reakcií). U pacientov treba sledovať klinické príznaky alebo známky kardiálnej dekompenzácie. V takom prípade môže byť potrebné prerušenie podávania, úprava dávky alebo ukončenie liečby (pozri časť 4.2).
Reverzibilný posteriórny encefalopatický syndróm (PRES) / Reverzibilný posteriórnyleukoencefalopatický syndróm (RPLS)
U pacientov liečených lenvatinibom sa pozoroval PRES, tiež známy ako RPLS, (< 1 %; pozri časť 4.8,
Popis vybraných nežiaducich reakcií). PRES je neurologická porucha, ktorá sa môže prejavovať bolesťami hlavy, záchvatmi, letargiou, zmätenosťou, zmenenými mentálnymi funkciami, slepotou a inými zrakovými alebo neurologickými poruchami. Môže byť prítomná aj mierna alebo závažná hypertenzia. Na potvrdenie diagnózy PRES je potrebná magnetická rezonancia. Na kontrolu tlaku krvi sa majú prijať primerané opatrenia (pozri časť 4.4 Hypertenzia). U pacientov so známkami alebo
symptómami PRES môže byť potrebné prerušenie podávania, úprava dávky alebo ukončenie liečby
(pozri časť 4.2).
Artériová tromboembolizácia
U pacientov liečených lenvatinibom boli hlásené prípady artériovej tromboembolizácie (náhle cievne
mozgové príhody, tranzientné ischemické ataky a infarkt myokardu) (pozri časť 4.8, Popis vybraných nežiaducich reakcií). Používanie lenvatinibu sa netestovalo u pacientov s anamnézou artériovej
tromboembolizácie v predchádzajúcich 6 mesiacoch. Preto sa má lenvatinib používať u týchto
pacientov s opatrnosťou. Rozhodnutie o liečbe má byť na základe pomeru prínosu/rizika
u jednotlivého pacienta. V prípade artériovej trombotickej príhody sa má liečba lenvatinibom ukončiť.
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia počas liečby lenvatinibom a mesiac po skončení liečby používať
vysokoúčinnú antikoncepciu (pozri časť 4.6). Zatiaľ nie je známe, či lenvatinib zvyšuje riziko
tromboembolických príhod pri kombinácii s perorálnou antikoncepciou.
Krvácanie
V klinických skúšaniach sa vyskytli a po uvedení na trh boli hlásené závažné krvácania spojené
s nádormi, vrátane fatálnych prípadov krvácania (pozri časť 4.8, Popis vybraných nežiaducich reakcií). Počas sledovania po uvedení na trh sa u pacientov s anaplastickým karcinómom štítnej žľazy (anaplastic thyroid cancer- ATC) pozorovali závažné a fatálne krvácania z karotídy častejšie ako
u pacientov s DTC alebo s inými typmi nádorov. Kvôli potenciálnemu riziku závažného krvácania spojeného so zmenšovaním nádoru/nekrózou nasledujúcou po liečbe lenvatinibom sa má zohľadniť
stupeň invázie/infiltrácie nádoru do veľkých ciev (napr. karotídy). Niektoré prípady krvácania sa
vyskytli sekundárne po zmenšení nádoru a formácii fistuly, napr. tracheo-ezofageálnej fistuly. Boli hlásené prípady fatálneho intrakraniálneho krvácania u niektorých pacientov s alebo bez metastáz do mozgu. Boli tiež hlásené krvácania na iných miestach ako je mozog (napr. trachea, brucho, pľúca). Bol hlásený jeden fatálny prípad krvácania spojeného s nádorom u pacienta s HCC.
V rámci štandardnej starostlivosti pred nasadením liečby lenvatinibom by sa mal vykonať skríning a
následná liečba ezofagiálnych varixov u pacientov s cirhózou pečene.
V prípade krvácania môže byť potrebné prerušenie, úprava alebo vysadenie liečby (pozri časť
4.2,tabuľka 3).
Gastrointestinálna perforácia a tvorba fistúl
U pacientov liečených lenvatinibom boli hlásené prípady gastrointestinálnych perforácií alebo fistúl
(pozri časť 4.8). Vo väčšine prípadov sa gastrointestinálne perforácie a fistuly vyskytli u rizikových pacientov ako napríklad po chirurgických zákrokoch alebo rádioterapii. V prípade gastrointestinálnej perforácie alebo fistuly môže byť potrebné prerušenie podávania, úprava dávky alebo ukončenie liečby (pozri časť 4.2).
Non-gastrointestinálna fistula
U pacientov liečených lenvatinibom môže byť zvýšené riziko vzniku fistuly. V klinických skúšaniach
a po uvedení na trh sa pozorovali prípady tvorby fistuly alebo zväčšenia fistuly, ktoré postihujú oblasti iné ako žalúdok alebo črevá (napr. tracheálna, tracheo-ezofageálna, ezofageálna, kožná, fistula ženského pohlavného traktu). Okrem toho bol pneumotorax hlásený tak s jasnými známkami bronchopleurálnej fistuly, ako aj bez nich. Niektoré prípady fistuly a pneumotoraxu boli spojené s regresiou tumoru alebo nekrózou. Podieľajúcimi sa faktormi môžu byť predchádzajúci chirurgický výkon alebo rádioterapia. Pľúcne metastázy môžu tiež zvýšiť riziko pneumotoraxu. Liečba lenvatinibom sa nemá začať u pacientov s fistulou, aby sa vyhlo zhoršeniu a lenvatinib sa má vysadiť
u pacientov s ezofageálnym alebo tracheobronchiálnym postihnutím alebo s akoukoľvek fistulou stupňa 4 (pozri časť 4.2). K dispozícii sú iba obmedzené informácie ohľadom použitia prerušenia liečby alebo zníženia dávky pri riešení iných príhod, ale v niektorých prípadoch sa pozorovalo zhoršenie a odporúča sa opatrnosť. Lenvatinib môže nepriaznivo ovplyvňovať proces hojenia rán, tak ako ďalšie lieky rovnakej skupiny.
Predĺženie QT intervalu
Predĺženie QT/QTc intervalu bolo hlásené vo vyššej miere u pacientov liečených lenvatinbom než
u pacientov na placebe (pozri časť 4.8, Popis vybraných nežiaducich reakcií). Elektrokardiogram sa má sledovať pri nasadení liečby a pravidelne počas liečby u všetkých pacientov a mimoriadnu pozornosť treba venovať pacientom s vrodeným syndrómom predĺženého QT intervalu, kongestívnym srdcovým zlyhaním, bradyarytmiami a tých, čo užívajú lieky predlžujúce QT interval (vrátane
antiarytmík triedy Ia a III). Lenvatinib sa má vysadiť v prípade rozvoja predĺženia QT intervalu viac ako 500 ms. Lenvatinib sa má nasadiť v zníženej dávke, ak QTc predĺženie odznie na < 480 ms alebo na hladinu rovnakú ako pred začiatkom liečby.
Poruchy elektrolytov ako hypokaliémia, hypokalciémia a hypomagneziémia zvyšujú riziko predĺženia QT intervalu. Abnormality elektrolytov je preto potrebné sledovať a upraviť u všetkých pacientov pred začatím liečby. Počas liečby treba pravidelne monitorovať elektrolyty (magnézia, kália
a kalcia).V priebehu liečby lenvatinibom sa má hladina kalcia v krvi monitorovať najmenej jedenkrát mesačne a ak je to potrebné, kalcium sa má doplňovať. Dávka lenvatinibu sa má upraviť alebo prerušiť ako je potrebné, v závislosti na závažnosti, prítomnosti zmien na EKG a pretrvávaní hypokalciémie.
Porucha supresie tyreotropného hormónu (TSH)/ Dysfunkcia štítnej žľazy
U pacientov liečených lenvatinibom bol hlásený hypotyreoidizmus (pozri časť 4.8, Popis vybraných
nežiaducich reakcií). Funkcia štítnej žľazy sa má monitorovať pred liečbou a pravidelne v priebehu liečby lenvatinibom. Hypotyreoidizmus má byť liečený v súlade so štandardnými postupmi na udržanie eutyreoidného stavu.
Lenvatinib narušuje exogénnu tyreoidnú supresiu (pozri časť 4.8, Popis vybraných nežiaducich reakcií). Hodnoty tyreotropného hormónu sa majú pravidelne sledovať a dávkovanie hormónov štítnej žľazy sa má upraviť tak, aby sa dosiahla taká hladina TSH, ktorá je v súlade s terapeutickým cieľom
u jednotlivého pacienta.
Komplikácie pri hojení rán
Nevykonali sa žiadne formálne štúdie účinku lenvatinibu na hojenie rán. U pacientov dostávajúcich
lenvatinib bolo hlásené zhoršené hojenie rán. U pacientov podstupujúcich veľké chirurgické zákroky sa má zvážiť dočasné prerušenie liečby lenvatinibom. Klinické skúsenosti týkajúce sa načasovania obnovenia podávania lenvatinibu po veľkom chirurgickom zákroku sú obmedzené. Preto sa má rozhodnutie o opätovnom začatí podávania lenvatinibu po veľkom chirurgickom zákroku zakladať na klinickom posúdení adekvátneho zahojenia rany.
Osobitné skupiny pacientov
U pacientov iného ako kaukazského a ázijského etnického pôvodu a u pacientov vo veku ≥75 rokov
nie sú k dispozícii dostatočné údaje. Preto sa má u týchto pacientov lenvatinib používať s opatrnosťou, s ohľadom na zníženú znášanlivosť lenvatinibu u ázijských a starších pacientov (pozri časť 4.8,
Ostatné osobitné skupiny).
K dispozícii nie sú žiadne údaje o použití lenvatinibu hneď po liečbe sorafenibom alebo inou protinádorovou terapiou. V prípade nedostatočnej vymývacej periódy medzi terapiami existuje potenciálne riziko prídavnej toxicity. Minimálna dĺžka vymývacej periódy v klinických skúšaniach bola 4 týždne.
4.5 Liekové a iné interakcie
Účinok iných liekov na lenvatinib
Chemoterapeutiká
Súčasné podávanie lenvatinibu, karboplatiny a paklitaxelu nemalo signifikantný vplyv na farmakokinetiku ktoréhokoľvek z týchto 3 liekov.
Účinok lenvatinibu na iné liečivá
Klinická štúdia liekových interakcií (drug-drug interaction, DDI) u pacientov s rakovinou preukázala,
že plazmatické koncentrácie midazolamu (citlivého na substrát CYP3A a Pgp) sa prítomnosťou lenvatinibu nezmenili. Medzi lenvatinibom a ostatnými CYP3A4/Pgp substrátmi sa preto neočakáva žiadna významná lieková interakcia.
Perorálna antikoncepcia
Zatiaľ nie je známe, či môže lenvatinib znižovať účinnosť hormonálnej antikoncepcie. Preto by mali ženy užívajúce hormonálnu antikoncepciu pridať aj bariérovú metódu (pozri časť 4.6).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Počas liečby lenvatinibom a aspoň 1 mesiac po ukončení liečby sa majú ženy vo fertilnom veku
vyhýbať otehotneniu a používať vysokoúčinnú antikoncepciu. Zatiaľ nie je známe, či môže lenvatinib znižovať účinnosť hormonálnej antikoncepcie. Preto by mali ženy užívajúce hormonálnu antikoncepciu pridať aj bariérovú metódu.
Gravidita
Nie sú k dispozícii žiadne údaje o použití lenvatinibu u gravidných žien. Pri podaní potkanom a
králikom bol lenvatinib embryotoxický a teratogénny(pozri časť 5.3).
Lenvatinib sa má používať počas gravidity iba pokiaľ to je jednoznačne nevyhnutné a až po dôkladnom zvážení potrieb matky a rizika pre plod.
Dojčenie
Nie je známe, či sa lenvatinib vylučuje do ľudského mlieka. Lenvatinib a jeho metabolity sa vylučujú
do mlieka potkanov (pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť vylúčené, a preto je
použitie lenvatinibu počas dojčenia kontraindikované (pozri časť 4.3).
Fertilita
Vplyv u ľudí nie je známy. Avšak u potkanov, psov a opíc sa pozorovala testikulárna a ovariálna
toxicita (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Lenvatinib má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje, vzhľadom na nežiaduce účinky ako sú únava a závraty. Pacienti, u ktorých sa vyskytli tieto symptómy by mali dbať na zvýšenú opatrnosť pri vedení vozidiel alebo pri obsluhe strojov.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnostný profil lenvatinibu vychádza z údajov 452 pacientov s DTC a 496 pacientov s HCC; čo
umožňuje len charakterizovanie častých nežiaducich reakcií na liečivo u pacientov s DTC a HCC. Nežiaduce reakcie uvedené v tejto časti vychádzajú z bezpečnostných údajov pacientov s DTC a HCC (pozri časť 5.1).
DTC
Najčastejšie zaznamenané nežiaduce reakcie (s výskytom u ≥ 30 % pacientov) sú
hypertenzia (68,6 %), hnačka (62,8 %), zníženie chute do jedla (51,5 %), hmotnostný úbytok (49,1 %), únava (45,8 %), nauzea (44,5 %), proteinúria (36,9 %), stomatitída (35,8 %), vracanie (34,5 %), dysfónia (34,1 %), bolesť hlavy (34,1 %) a syndróm palmárno-plantárnej erytrodysestézie
(PPE) (32,7 %). Hypertenzia a proteinúria sa zvyčajne vyskytujú včasne po začatí liečby lenvatinibom (pozri časti 4.4 a 4.8, Popis vybraných nežiaducich reakcií). Väčšina nežiaducich reakcií stupňa 3 a 4 sa vyskytla počas prvých 6 mesiacov liečby, ale hnačka sa vyskytovala v priebehu celej liečby a
hmotnostný úbytok sa kumuloval s trvaním liečby.
Najdôležitejšie závažné nežiaduce reakcie boli renálne zlyhanie a porucha funkcie obličiek (2,4 %), artériová tromboembolizácia (3,9 %), zlyhanie srdca (0,7 %), krvácanie intrakraniálnych
tumorov (0,7 %), PRES/RPLS (0,2 %), hepatálne zlyhanie (0,2 %) a artériové tromboembolizácie (náhla cievna mozgová príhoda (1,1 %), prechodné ischemické príhody (0,7 %), a infarkt myokardu (0,9 %).
U 452 pacientov s RAI-refraktérnym DTC bola kvôli nežiaducim reakciám znížená dávka u 63,1 % pacientov a u 19,5 % pacientov došlo k vysadeniu dávky. Nežiaduce reakcie, ktoré viedli k redukcii dávky (u ≥5 % pacientov) boli najčastejšie hypertenzia, proteinúria, hnačka, únava, PPE, hmotnostný úbytok a zníženie chute do jedla. Nežiaduce reakcie, ktoré najčastejšie viedli k ukončeniu liečby lenvatinibom boli proteinúria, asténia, hypertenzia, náhla cievna mozgová príhoda, hnačka
a pľúcna embolizácia.
HCC
Najčastejšie zaznamenané nežiaduce reakcie (s výskytom u ≥30 % pacientov) sú hypertenzia (44,0 %),
hnačka (38,1 %), zníženie chute do jedla (34,9 %), únava (30,6 %) a hmotnostný úbytok (30,4 %).
Najdôležitejšie závažné nežiaduce reakcie boli zlyhanie pečene (2,8 %), hepatálna encefalopatia (4.6 %), krvácanie z varixov pažeráka (1,4 %), krvácanie do mozgu (0,6 %), arteriálne tromboembolicképríhody (2,0 %) vrátane infarktov myokardu (0,8 %), mozgový infarkt (0,4 %) a náhla cievna príhoda (0,4 %) a renálne zlyhanie/poruchy (1,4 %). U pacientov s HCC bol zaznamenaný vyšší výskyt zníženého počtu neutrofilov (8,7 % na lenvatinibeoproti iným typom tumoru než HCC (1,4 %)), ktorý nebol spojený s infekciou, sepsou alebo bakteriálnym zápalom pobrušnice.
U 496 pacientov s HCC bola kvôli nežiaducim reakciám upravená dávka (prerušenie alebo zníženie) u 62,3 % pacientov a u 20,2 % pacientov došlo k vysadeniu dávky. Nežiaduce reakcie, ktoré viedli k redukcii dávky (u ≥5 % pacientov) boli najčastejšie zníženie chute do jedla, hnačka, proteinúria, hypertenzia, únava, PPE a znížený počet krvných doštičiek. Nežiaduce reakcie, ktoré najčastejšie
viedli k ukončeniu liečby lenvatinibom boli hepatálna encefalopatia, únava, zvýšenie bilirubínu v krvi, proteinúria, a zlyhanie pečene.
Tabuľkový zoznam nežiaducich reakcií
V klinických skúšaniach DTC aj HCC boli pozorované podobné nežiaduce reakcie.
Nežiaduce reakcie, ktoré sa pozorovali v klinických skúšaniach pri DTC a HCC, a ktoré boli hlásené z
používania lenvatinibu po uvedení na trh, sú uvedené v tabuľke 5. Kategória frekvencie výskytu nežiaducich reakcií predstavuje tie najkonzervatívnejšie odhady frekvencie dvoch samostatných skupín pacientov.
Frekvencia je definovaná nasledovne:
·
| veľmi časté
| (≥1/10)
|
·
| časté
| (≥1/100 až <1/10)
|
·
| menej časté
| (≥1/1 000 až <1/100)
|
·
| neznáme
| (z dostupných údajov)
|
V každej kategórii frekvencie výskytu sú nežiaduce účinky zoradené podľa klesajúcej závažnosti.
Tabuľka 5 Nežiaduce reakcie hlásené u pacientov liečených lenvatinibom
|
Trieda orgánových systémov
(Terminológia
MedDRA*)
|
Veľmi časté
|
Časté
|
Menej časté
|
Neznáme
|
Infekcie a nákazy
| Infekcie močového
traktu
|
| Perineálny absces
|
|
Poruchy krvi a lymfatického
systému
| Trombocytopéniaa
Leukopéniaa
Neutropéniaa
| Lymfocytopéniaa
| Slezinový infarkt
|
|
Poruchy
endokrinného systému
| Hypotyreóza
| Zvýšenie
tyreotropného
hormónu v krvi‡
|
|
|
Poruchy
metabolizmu a výživy
| Hypokalciémia‡
Hypokaliémia Hmotnostný úbytok Znížená chuť do jedla
| Dehydratácia Hypomagneziémiab Hyper- cholesterolémiab
|
|
|
Psychické
poruchy
| Nespavosť
|
|
|
|
Poruchy
nervového systému
| Závraty
Bolesti hlavy
Dysgeúzia
| Cievne mozgové
príhody
| Reverzibilný
posteriórny encefalopatický syndróm Monoparéza Tranzientný ischemický atak
|
|
Poruchy srdca a
srdcovej činnosti
|
| Infarkt myokarduc,†
Srdcové zlyhanie Predĺženie QT intervalu (na EKG) Zníženie ejekčnej frakcie
|
|
|
Poruchy ciev
| Krvácanied, †,‡
Hypertenziae,‡
Hypotenzia
|
|
| Aneuryzmy
a arteriálne disekcie
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
| Dysfónia
| Pľúcna embólia†,
| Pneumotorax
|
|
Tabuľka 5 Nežiaduce reakcie hlásené u pacientov liečených lenvatinibom
|
Trieda orgánových systémov
(Terminológia
MedDRA*)
|
Veľmi časté
|
Časté
|
Menej časté
|
Neznáme
|
Poruchy gastrointestinálne
ho traktu
|
Hnačka
Bolesti gastrointestinálneho traktu a brucha f Vracanie
Nauzea
Zápal ústnej dutinyg
Bolesť v ústnej dutineh Konstipácia Dyspepsia Sucho v ústach
|
Análna fistula
Flatulencia Zvýšenie lipáz Zvýšenie amyláz
|
Pankreatitída i,†
|
|
Poruchy pečene a žlčových ciest
|
Zvýšenie bilirubínu v krvij, ‡ Hypoalbuminémiaj,‡ Zvýšenie alanínamino- transferázy‡ Zvýšenie aspartátamino- transferázy‡
|
Zlyhanie pečenek,‡,†
Hepatálna encefalopatial,‡,† Zvýšenie alkalickej fosfatázy v krvi Abnormálne hepatálne funkcie Zvýšenie gamaglutamyl- transferázy Cholecystitída
|
Hepatocelulárne poškodenie/hepatitíd am
|
|
Poruchy kože a
podkožného
tkaniva
|
Syndróm palmárno-
plantárnej erytrodysestésie Vyrážka Alopécia
|
Hyperkeratóza
|
|
|
Poruchy kostrovej a
svalovej sústavy a spojivového tkaniva
|
Bolesť chrbta
Artralgia
Myalgia Bolesť v končatine Muskuloskeletárna bolesť
|
|
|
|
Poruchy obličiek
a močových ciest
|
Proteinúria‡
|
Prípady renálneho zlyhanian, †
Porucha renálnych
funkcií Zvýšenie kreatinínu v krvi
Zvýšenie urey v krvi
|
Nefrotický syndróm
|
|
Celkové poruchy
a reakcie v mieste podania
|
Únava
Asténia
Periférny edém
|
Nevoľnosť
|
Zhoršené hojenie
rán*
|
Non-gastro- intestinálna fistulao
|
|
|
*: Zistené na základe používania lenvatinibu po uvedení na trh.
†: Zahŕňa prípady s fatálnym následkom.
‡: Pre ďalší popis pozri časť 4.8, Popis vybraných nežiaducich reakcií.
Následné pojmy boli zlúčené:
a: Trombocytopénia zahŕňa trombocytopéniu a znížený počet krvných doštičiek. Neutropénia
zahŕňa neutropéniu a znížený počet neutrofilov. Leukopénia zahŕňa leukopéniu a znížený počet
bielych krviniek. Lymfocytopénia zahŕňa lymfocytopéniu a pokles počtu lymfocytov.
b: Hypomagneziémia zahŕňa hypomagneziémiu a zníženie horčíku v krvi.
Hypercholesterolémia zahŕňa hypercholesterolémiu a zvýšenie cholesterolu v krvi.
c: Infarkt myokardu zahŕňa infarkt myokardu a akútny infarkt myokardu.
d: Zahŕňa všetky prípady krvácania.
Krvácanie, ktoré sa vyskytlo u 5 alebo viacerých pacientov s DTC bolo: epistaxa, hemoptýza, hematúria, kontúzia, hematochézia, krvácanie z ďasien, petéchia, pľúcne krvácanie, rektálne krvácanie, prítomnosť krvi v moči, hematómy a vaginálne krvácanie.
Krvácanie, ktoré sa vyskytlo u 5 alebo viacerých pacientov s HCC bolo: epistaxa, hematúria, krvácanie z ďasien, hemoptýza, krvácanie z varixovpažeráka, krvácanie z hemoroidov, krvácanie z ústnej dutiny, rektálne krvácanie a krvácanie z horného tráviaceho traktu.
e: Hypertenzia zahŕňa: hypertenziu, hypertenznú krízu, zvýšenie diastolického krvného tlaku, ortostatickú hypertenziu a zvýšenie krvného tlaku.
f: Bolesť gastrointestinálneho traktu a bolesť brucha zahŕňajú: abdominálny dyskomfort, abdominálnu bolesť, bolesť dolnej časti brucha, bolesť hornej časti brucha, citlivosť brucha, dyskomfort v epigastriu a bolesti gastrointestinálneho traktu.
g: Zápal v ústnej dutine zahŕňa: aftóznu stomatitídu, aftózny vred, gingiválnu eróziu, gingiválne
vredy, pľuzgiere ústnej sliznice, stomatitídu, glositídu, ulceráciu v ústnej dutine a zápal slizníc. h: Bolesť v ústnej dutine zahŕňa: bolesť v ústnej dutine, gingiválnu bolesť, orofaryngálne ťažkosti,
bolesť jazyka, bolesť v ústnej dutine a faryngu.
i: Pankreatitída zahŕňa: pankreatitídu a akútnu pankreatitídu.
j: Hyperbilirubinémia zahŕňa: hyperbilirubinémiu, zvýšenie bilirubínu v krvi, žltačku a zvýšenie konjugovaného bilirubínu. Hypoalbuminémia zahŕňa hypoalbuminémiu a zníženie albumínu v krvi.
k: Zlyhanie pečene zahŕňa: zlyhanie pečene, akútne zlyhanie pečene a chronické zlyhanie pečene. l: Hepatálna encefalopatia zahŕňa: hepatálnu encefalopatiu, pečeňová kóma, metabolická
encefalopatia a encefalopatia.
m: Hepatocelulárne poškodenie a hepatitída zahŕňajú: liekmi-indukované hepatálne poškodenie, steatózu pečene a cholestatické poškodenie pečene.
n: Prípady renálneho zlyhania zahŕňajú: akútne pre-renálne zlyhanie, renálne zlyhanie, akútne renálne zlyhanie, akútne poranenie obličiek a renálnu tubulonekrózu.¨
o: Non-gastrointestinálna fistula zahŕňa prípady fistuly vyskytujúcej sa mimo oblasť žalúdka
a čriev ako je tracheálna, tracheo-ezofageálna, ezofageálna, fistula ženského pohlavného traktu
a kožná fistula.
Popis vybraných nežiaducich reakcií
Hypertenzia (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) sa zaznamenala hypertenzia (vrátane hypertenzie,
hypertenznej krízy, zvýšenia diastolického krvného tlaku a zvýšenia krvného tlaku) u 72,8 %
pacientov liečených lenvatinibom a 16,0 % pacientov liečených placebom.
Medián času do nástupu u pacientov liečených lenvatinbom bol 16 dní. Reakcie stupňa 3 alebo vyššieho (vrátane 1 reakcie stupňa 4) sa vyskytli u 44,4 % pacientov liečených lenvatinbom
v porovnaní s 3 % pacientov liečených placebom.Vo väčšine prípadov došlo k uzdraveniu (u 13 % pacientov) alebo zlepšeniu (u 13.4 % pacientov) po prerušení alebo znížení dávky. V dôsledku hypertenzie došlo k trvalému prerušeniu liečby u 1,1 % pacientov.
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1) sa zaznamenala hypertenzia (vrátane hypertenzie,
zvýšenia krvného tlaku, zvýšenia diastolického krvného tlaku a ortostatickej hypertenzie)
u 44,5 %pacientov liečených lenvatinibom a u 23,5 % pacientov sa zaznamenala hypertenzia 3.
stupňa.Medián času do nástupubol 26 dní. Vo väčšine prípadov došlo k uzdraveniupo prerušení
(3,6 %) alebo znížení dávky (3,4 %). V dôsledku hypertenzie došlo k prerušeniu liečby lenvatinibom
u jedného pacienta (0,2 %).
Proteinúria (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) sa proteinúria zaznamenala u 33,7 % pacientov
liečených lenvatinibom a u 3,1 % pacientov na placebe. Medián času do nástupu bol 6,7 týždňa. Reakcie 3.stupňa sa vyskytli u 10,7 % pacientov liečených lenvatinibom a nevyskytli sa u žiadneho pacienta na placebe. Vo väčšine prípadov došlo k uzdraveniu alebo zlepšeniu po prerušení (16,9 %) alebo znížení (10,7 %) dávky. Kvôli proteinúrii došlo k trvalému prerušeniu liečby u 0,8 % pacientov.
HCC
Vo fáze3 skúšania REFLECT (pozri časť 5.1)sa proteinúria zaznamenala u 26,3 % pacientov
liečených lenvatinibom a reakcie 3. stupňa sa zaznamenali u 5,9 %. Medián času do nástupubol
6,1 týždňa. Vo väčšine prípadov došlo k uzdraveniupo prerušení (6,9 %) alebo znížení (2,5 %) dávky.
Kvôli proteínúrii došlo k trvalému prerušeniu liečby u 0,6 % pacientov.
Renálne zlyhanie alebo porucha funkcie obličiek (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) sa u 5,0 % pacientov vyvinulo zlyhanie obličiek
a u 1,9 % porucha funkcie obličiek (3,1 % pacientov malo stupeň ≥ 3 renálneho zlyhania alebo poruchy funkcie obličiek). V skupine s placebom sa vyvinulo renálne zlyhanie alebo porucha funkcie obličiek u 0,8 % pacientov (0,8 % malo stupeň ≥ 3).
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1), sa u 7,1 % pacientov liečených lenvatinibomvyvinulo
zlyhanie/porucha funkcie obličiek. Reakcie 3. alebo vyššieho stupňa sa zaznamenali u 1,9 %pacientov
liečených lenvatinibom.
Kardiálna dysfunkcia (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1), klesla ejekčná frakcia/bolo hlásené zlyhanie
srdca u 6,5 % pacientov (1,5 % mali stupeň ≥ 3) v skupine s lenvatinibom a 2,3 % v skupine s placebom (nikto nemal stupeň ≥ 3).
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1), bola v skupine liečenej lenvatinibom hlásená kardiálna
dysfunkcia (vrátane kongestívneho srdcového zlyhania, kardiogénneho šoku a kardiopulmonárneho zlyhania) u 0,6 % pacientov (0,4 % boli stupeň ≥3).
Reverzibilný posteriórny encefalopatický syndróm (PRES) / Reverzibilný posteriórny leukoencefalopatický syndróm (RPLS) (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) bol 1 prípad PRES (stupeň 2) v skupine
s lenvatinibom a žiaden prípad v skupine s placebom.
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1) bol 1 prípad PRES (stupeň 2) v skupine s lenvatinibom.
Medzi 1 823 pacientmi liečenými lenvatinibom v rámci monoterapie v klinických skúšaniachbolo5 prípadov (0,3 %) PRES (0,2 % boli stupeň 3 alebo 4), všetky odzneli po liečbe a/alebo prerušení dávky alebo po jej trvalom vysadení.
Hepatotoxicita (pozri časť 4.4)
DTC
Najčastejšie zaznamenané nežiaduce reakcie spojené s pečeňou v pivotnej fáze 3 skúšania SELECT
(pozri časť 5.1) boli hypoalbuminémia (9,6 % pri lenvatinibe vs. 1,5 % pri placebe) a elevácia hepatálnych enzýmov, vrátane vzostupu hladiny alanínaminotransferázy (7,7 % pri lenvatinibe vs.
0 pri placebe), aspartátaminotranferázy (6,9 % pri lenvatinibe vs. 1,5 % pri placebe) a bilirubínu v krvi (1,9 % pri lenvatinibe vs. 0 pri placebe). Medián času do nástupu pečeňových reakcií u pacientov liečených lenvatinibom bol 12,1 týždňa. Nežiaduce reakcie spojené s pečeňou 3. stupňa alebo vyššie
(vrátane jedného prípadu hepatálneho zlyhania – stupeň 5) sa vyskytli v 5,4 % pacientov liečených
lenvatinibom oproti 0,8 % pacientov na placebe. Nežiaduce reakcie spojené s pečeňou viedli k prerušeniu alebo zníženiu dávky v 4,6 % a 2,7 %, v tomto poradí a k trvalému prerušeniu v 0,4 %.
Medzi 1 166 pacientmi liečenými lenvatinibom boli 3 prípady (0,3 %) hepatálneho zlyhania, ktoré viedli k úmrtiu. Jeden prípad bol u pacienta bez hepatálnych metastáz. Taktiež sa vyskytol prípad akútnej hepatitídy u pacienta bez hepatálnych metastáz.
HCC
Najčastejšie zaznamenané nežiaduce reakcie spojené s hepatotoxicitou vo fáze 3 skúšania REFLECT
(pozri časť 5.1) boli zvýšenie bilirubínu v krvi (14,9 %), zvýšenie hladiny aspartátaminotranferázy (13,7 %), zvýšenie hladiny alanínaminotransferázy (11,1 %), hypoalbuminémia (9,2 %), hepatálna encefalopatia (8,0 %), zvýšenie hladiny gamaglutamyltransferázy (7,8 %) a zvýšenie alkalickej fosfatázy v krvi (6,7 %). Medián času do nástupunežiaducich reakcií hepatotoxocity bol 6,4 týždňa. Reakcie hepatotoxicity≥3. stupňa sa objavili u 26,1 %pacientov liečených lenvatinibom. Zlyhanie pečene (vrátane fatálnych prípadov u 12 pacientov) sa objavilo u 3,6 %pacientov (všetci mali
≥3. stupeň). Hepatálna encefalopatia (vrátane fatálnych prípadov u 4 pacientov) sa objavila u 8,4 %
pacientov (5,5 % bolo ≥3. stupeň). Vyskytlo sa 17 úmrtí (3,6 %) v dôsledku prípadov hepatotoxicity v skupine pacientov liečených lenvatinibom a 4 úmrtia (0,8 %) v skupine liečenej sorafenibom. Nežiaduce reakcie spojené s hepatotoxicitou viedli k prerušeniu dávky u 12,2 % alebo zníženiu dávky u 7,4 % pacientov liečených lenvatiniboma v 5,5 % k trvalému vysadeniu.
V rámci klinických skúšaní, pri ktorých sa 1 327 pacientom podávala monoterapia lenvatinibom na iné indikácie ako HCC, bolo hlásené zlyhanie pečene (vrátane fatálnych prípadov) u 4 pacientov (0,3 %), poškodenie pečene u 2 pacientov (0,2 %), akútna hepatitída u 2 pacientov (0,2 %) a hepatocelulárne poškodenie u 1 pacienta (0,1 %).
Artériová tromboembolizácia (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) boli hlásené prípady artériovej
tromboembolizácie u 5,4 % pacientov liečených lenvatinibom a u 2,3 % pacientov v skupine s placebom.
Vo fáze 3 skúšania REFLECT (pozri časť 5.1) boli hlásené prípady artériovej tromboembolizácie u 2,3 %pacientovliečených lenvatinibom.
Medzi 1 823 pacientmi liečenými lenvatinibom v rámci monoterapie v klinických skúšaniach bolo
10 prípadov (0,5 %) artériovej tromboembolizácie (5 prípadov infarktu myokardu a 5 prípadov cerebrovaskulárnej príhody) s fatálnym výsledkom.
Krvácanie (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) sa krvácanie zaznamenalo u 34,9 % (1,9 % mali
stupeň ≥ 3) pacientov liečených lenvatinibom oproti 18,3 % (3,1 % mali stupeň ≥ 3) pacientov na placebe. Reakcie, ktoré sa vyskytli s incidenciou ≥ 0,75 % oproti placebu boli: epistaxa (11,9 %), hematúria (6,5 %), kontúzia (4,6 %), krvácanie z ďasien (2,3 %), hematochézia (2,3 %), rektálne krvácanie (1,5 %), hematóm (1,1 %), krvácanie z hemoroidov (1,1 %), laryngeálne krvácanie (1,1 %), petéchie (1,1 %) a krvácanie z intrakraniálneho nádoru (0,8 %). V tejto štúdii bol 1 prípad fatálneho
intrakraniálneho krvácania zo 16 pacientov, ktorí užívali lenvatinib a mali metastázy do CNS na
začiatku liečby.
Medián času do nástupu u pacientov liečených lenvatinibom bol 10,1 týždňa. Medzi pacientmi na lenvatinibe a pacientmi na placebe sa nepozorovali žiadne rozdiely vo výskyte závažných reakcií (3,4 % vs. 3,8 %), reakcií vedúcich k predčasnému ukončeniu liečby (1,1 % vs. 1,5 %) alebo reakcií vedúcich k prerušeniu podávania (3,4 % vs. 3,8 %) alebo redukcii dávky (0,4 % vs. 0).
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1) sa krvácanie zaznamenalo u 24,6 % pacientov(5,0 %
mali≥3. stupeň.Reakcie stupňa 3 sa objavili u 3,4 %, reakcie stupňa 4 u 0,2 % a7 pacientov (1,5 %) malo reakciu stupňa 5 vrátane krvácania do mozgu, krvácania horného tráviaceho traktu, krvácania v črevách a krvácania z nádoru. Medián času do prvého nástupu bol 11,9 týždňa. Krvácanie viedlo k prerušeniu dávky u 3,2 % a k zníženiu dávky u 0,8 % pacientov, pričom liečba bola ukončená
u 1,7 %pacientov.
V rámci klinických skúšaní, pri ktorých bolo monoterapiou lenvatinibom liečených 1 327
pacientovs inou indikáciou ako HCC, boli u 2 % pacientov hlásené krvácanie stupňa 3 alebo väčšie. U 3 pacientov (0,2 %) sa vyskytlo krvácanie stupňa 4 a u 8 pacientov (0,6 %) sa vyskytlo krvácanie stupňa 5, zahŕňajúce arteriálne krvácanie, hemoragickú mozgovú príhodu, intrakraniálne krvácanie, krvácanie intrakraniálneho nádoru, hematemézu, melénu, hemoptýzu a krvácanie z nádoru.
Hypokalciémia (pozri časť 4.4 Predĺženie QT intervalu)
DTC
V pivotnejfáze 3 skúšania SELECT (pozri časť 5.1) sa hypokalciémia zaznamenala u 12,6 %
pacientov liečených lenvatinibom a u žiadneho pacienta na placebe. Medián času do nástupu u pacientov liečených lenvatinibom bol 11,1 týždňa. Reakcie 3. alebo 4.stupňa závažnosti sa vyskytli u
5,0 % pacientov liečených lenvatinibom a u žiadneho pacienta na placebe. Väčšina prípadov sa zlepšila po podpornej liečbe, bez prerušenia alebo zníženia dávky, čo sa vyskytlo u 1,5 % a 1,1 % pacientov, v tomto poradí. K trvalému prerušeniu liečby došlo u jedného pacienta.
Vo fáze 3 skúšania REFLECT (pozri časť 5.1)sa hypokalciémia zaznamenala u 1,1 % pacientov, pričom reakcie stupňa 3 sa objavili v 0,4 %. Prerušenie dávok lenvatinibu v dôsledku hypokalciémie nastalo u jedného pacienta (0,2 %) a nevyskytli sa žiadne prípady zníženia alebo vysadenia dávok.
Gastrointestinálna perforácia a tvorba fistúl (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) boli hlásené prípady gastrointestinálnej perforácie
alebo fistuly u 1,9 % lenvatinibom liečených pacientov a u 0,8 % pacientov v skupine s placebom.
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1)boli hlásené prípady gastrointestinálnej perforácie alebo
fistuly u 1,9 % pacientov liečených lenvatinibom.
Non-gastrointestinálne fistuly (pozri časť 4.4)
Používanie lenvatinibu bolo asociované s prípadmi fistúl vrátane reakcií končiacich smrťou. Prípady fistúl, ktoré postihujú oblasti tela iné ako žalúdok a črevá sa pozorovali pri rôznych indikáciách. Reakcie boli hlásené v rôznom čase v priebehu liečby, v rozmedzí od dvoch týždňov do viac ako
1 roka od začiatku užívania lenvatinibu, s mediánom latencie okolo 3 mesiacov.
Predĺženie QT intervalu (pozri časť 4.4)
DTC
V pivotnejfáze 3 skúšania SELECT (pozri časť 5.1) bolo hlásené predĺženie QT/QTc intervalu u 8,8 %
pacientov liečených lenvatinibom a u 1,5 % pacientov v skupine s placebom. Incidencia predĺženia QT intervalu viac ako 500 ms bola 2 % u pacientov liečených lenvatinibom v porovnaní so žiadnym prípadom v skupine s placebom.
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1) bolo hlásené predĺženie QT/QTc intervalu u 6,9 %
pacientov liečených lenvatinibom. Incidencia predĺženia intervalu QTcF viac ako 500 ms bola 2,4 %.
Zvýšenie hladiny tyreotropného hormónu v krvi (pozri časť 4.4 Porucha supresie tyreotropného hormónu/ Dysfunkcia štítnej žľazy)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1), malo 88 % všetkých pacientov hodnotu TSH pri
vstupe do štúdie menej ako alebo rovné 0,5 mU/l. U pacientov s normálnou hodnotou TSH pri vstupe do štúdie sa pozorovalo zvýšenie TSH hodnoty nad 0,5 mU/l v priebehu štúdie u 57 % pacientov liečených lenvatinibom v porovnaní so 14 % pacientov na placebe.
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1)malo 89,6 %pacientovhodnotu TSH pri vstupe do štúdie
menej ako alebo rovné hornému limitu normy. Zvýšenie hladiny TSH nad horný limit normy bolo
pozorované po pri vstupe do štúdie u 69,6 % pacientov liečených lenvatinibom.
Hnačka (pozri časť 4.4)
DTC
V pivotnej fáze 3 skúšania SELECT (pozri časť 5.1) bola hnačka hlásená u 67,4 % pacientov
liečených lenvatinibom (9,2 % malo stupeň ≥ 3) a u 16,8 % pacientov v skupine s placebom (nikto
nemal stupeň ≥ 3).
HCC
Vo fáze 3 skúšania REFLECT (pozri časť 5.1) bola hnačka hlásená u 38,7 % pacientov liečených
lenvatinibom (4,2 %malo ≥ 3. stupeň).
Pediatrická populácia
Pre túto populáciu nie sú dostupné žiadne klinické údaje (pozri časť 4.2).
Ostatné osobitné skupiny
Starší pacienti
DTC
Pacienti vo veku ≥75 rokov mali väčšiu pravdepodobnosť rozvoja hypertenzie stupňa 3 alebo 4,
proteinúrie, zníženej chute do jedla a dehydratácie.
HCC
Pacienti vo veku ≥75 rokov mali vyššiu pravdepodobnosť rozvoja hypertenzie, proteinúrie, zníženej
chute do jedla, asténie, dehydratácie, závratu, malátnosti, periférneho edému, svrbenia a hepatálnej encefalopatie. Hepatálna encefalopatia sa objavila viac ako dvojnásobne u pacientov nad ≥75 rokov (17,2 %) oproti pacientom do <75 rokov (7,1 %). Hepatálna encefalopatia zvykla byť spojená
s nežiaducou charakteristikou ochorenia pri vstupe do štúdie alebo pri použití sprievodných liekov. V tejto vekovej skupine sa tak isto objavili častejšie arteriálne tromboembolické príhody.
Pohlavie
DTC
Ženy mali vyšší výskyt hypertenzie (vrátane hypertenzie stupňa 3 až 4), proteinúrie a PPE, zatiaľ čo
u mužov sa častejšie vyskytla znížená ejekčná frakcia, perforácia gastrointestinálneho traktu a tvorba
fistúl.
HCC
Ženy mali vyšší výskyt hypertenzie, únavy, predĺženia ECG QT a alopécie. U mužov bol vyšší výskyt
(26,5 %) dysfónie ako u žien (12,3 %), úbytok telesnej hmotnosti a znížený počet krvných doštičiek.
Zlyhanie funkcie pečene bolo pozorované výlučne u mužov.
Etnický pôvod
DTC
U ázijských pacientov bol oproti pacientom kaukazského pôvodu vyšší výskyt periférneho edému,
hypertenzie, únavy, PPE, proteinúrie, trombocytopénie a zvýšenia tyreotropného hormónu.
HCC
U ázijských pacientov bol oproti pacientom kaukazského pôvodu vyšší výskyt proteinúrie, zníženého
počtu neutrofilov, zníženého počtu krvných doštičiek, zníženého počtu bielych krviniek a syndróm PPE, pričom u pacientov kaukazského pôvodu bol vyšší výskyt únavy, hepatálnej encefalopatie, akútneho poškodenia obličiek, úzkosti, asténie, nevoľnosti, trombocytopénie a vracania.
Hypertenzia pri vstupe do štúdie
DTC
Pacienti s hypertenziou pri vstupe do štúdie mali vyšší výskyt hypertenzie, proteinúrie, hnačky
a dehydratácie stupňa 3 alebo 4 a tiež mali závažnejšie prípady dehydratácie, hypotenzie, pľúcnej embolizácie, malígneho pleurálneho výpotku, atriálnej fibrilácie a GI príznakov (bolesť brucha, hnačka, vracanie).
Porucha funkcie pečene
DTC
Pacienti s poruchou funkcie pečene pri vstupe do štúdie mali vyšší výskyt hypertenzie a PPE a vyšší
výskyt hypertenzie, asténie, únavy a hypokalciémie stupňa 3 až 4, v porovnaní s pacientmi s
normálnou funkciou pečene.
HCC
Pacienti so skóre Child-Pugh (CP) 6 pri vstupe do štúdie (okolo 20 % pacientov v štúdii REFLECT)
mali vyšší výskyt zníženej chute do jedla, únavy, proteinúrie, hepálnej encefalopatie a zlyhania pečene v porovnaní s pacientmi, ktorí mali pri vstupe do štúdie skóre CP 5. Prípady hepatotoxicity a krvácania sa tak isto objavovali častejšie u pacientov so skóre CP 6 oproti pacientom so skóre CP 5.
Porucha funkcie obličiek
DTC
Pacienti s poruchou funkcie obličiek pri vstupe do štúdie mali vyšší výskyt hypertenzie proteinúrie,
únavy, stomatitídy, periférneho edému, trombocytopénie, dehydratácie, predĺženého intervalu QT, hypotyreózy, hyponatriémie, zvýšenej hodnoty tyreotropného hormónu v krvi a pneumónie stupňa 3 alebo 4, v porovnaní s pacientmi s normálnou funkciou obličiek. Títo pacienti mali tiež vyšší výskyt renálnych nežiaducich reakcií a sklon k vyššiemu výskytu hepatálnych nežiaducich reakcií.
HCC
Pacienti s poruchou funkcie obličiek pri vstupe do štúdie mali vyšší výskytúnavy, hypotyreózy,
dehydratácie, hnačky, zníženej chute do jedla, proteinúrie a hepatálnej encefalopatie. U týchto
pacientov sa tak isto vo vyššej miere vyskytli renálne reakcie a arteriálne tromboembolické príhody.
Pacienti s telesnou hmotnosťou < 60 kg
DTC
Pacienti s nízkou telesnou hmotnosťou (< 60 kg) mali vyšší výskyt PPE, proteinúrie, hypokalciémie
a hyponatriémie stupňa 3 alebo 4 a mali tendenciu k zvýšenému výskytu zníženej chute do jedla
stupňa 3 alebo 4.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNajvyššie dávky lenvatinibu, ktoré sa klinicky testovali, boli 32 mg a 40 mg denne.
V štúdiách sa vyskytli prípady náhodných medikačných chýb, čo viedlo k jednotlivým dávkam
40 až 48 mg. Najčastejšie pozorované nežiaduce liekové účinky pri týchto dávkach boli hypertenzia, nauzea, hnačka, únava, stomatitída, proteinúria, bolesť hlavy a zhoršenie PPE. Tiež boli hlásené prípady predávkovania lenvantinibom zahŕňajúce jednotlivé podania dávky 6-10 násobne vyššej ako je odporučená denná dávka. V týchto prípadoch sa vyskytli nežiaduce reakcie, ktoré sú konzistentné so známym bezpečnostným profilom lenvatinibu (napr. renálne alebo kardiálne zlyhanie) alebo neboli prítomné žiadne nežiaduce reakcie.
Symptómy a odporúčané postupyPri predávkovaní lenvatinibom nie je k dispozícii žiadne špecifické antidotum. V prípade podozrenia
na predávkovanie sa má lenvatinib vysadiť a má sa poskytnúť potrebná podporná starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE29
Lenvatinib je multikinázový inhibítor, u ktorého sa
in vitro a
in vivo preukázali najmä antiangiogénne vlastnosti a tiež sa pozorovala priama inhibícia rastu tumoru v
in vitro modeloch.
Mechanizmus účinkuLenvatinib je inhibítor receptora tyrozínkinázy, ktorý selektívne inhibuje aktivity kinázy receptorov
cievneho endoteliálneho rastového faktoru (VGEF) VGEF1 (FLT1), VEGFR2 (KDR) a VEGFR3 (FLT4), spolu s ďalšími RTK súvisiacich s proangiogénnymi a onkogénnymi dráhami, vrátane receptorov rastového faktora fibroblastov (FGF) FGFR1, 2, 3 a 4, receptorov rastového faktora odvodeného od krvných doštičiek (PDGF) PDGFRα, KIT a RET.
Okrem toho,lenvatinib pôsobí selektívne a priamoantiproliferatívne na hepatocelulárne bunkové línie v závislosti od aktivovanej signalizácie FGFR, čo sa pripisuje inhibícii signalizácie FGFR pôsobením lenvatinibu.
Hoci sa nerealizovali štúdie priamo s lenvatinibom, predpokladá sa, že mechanizmus vzniku (
mechanism of action, MOA) hypertenzie je sprostredkovaný inhibíciou VEGFR2 vo vaskulárnych endoteliálnych bunkách. Podobne, hoci neprebehla priama štúdia, sa predpokladá, že MOA proteinúrie je sprostredkovaný down reguláciou VEGFR1 a VEGFR2 v podocytoch glomerulov.
Mechanizmus vzniku hypotyreózy nie je úplne objasnený.
KlinickáúčinnosťDiferencovaný karcinóm štítnej žľazy refraktérny na rádiojód'
Štúdia SELECT bola multicentrická, randomizovaná, dvojito zaslepená, placebom kontrolovaná klinická štúdia, ktorá sa uskutočnila u 392 pacientov s diferencovaným karcinómom štítnej žľazy refraktérnym na rádiojód, s nezávislým centrálnym hodnotením rádiologického nálezu progresie
ochorenia počas 12 mesiacov (+1 mesiac) pred zaradením do štúdie. Refraktérnosť na rádiojód bola definovaná ako nález jednej alebo viacerých merateľných lézií buď s nedostatočným vychytávaním rádiojódu alebo s progresiou napriek liečbe rádiojódom alebo s kumulatívnou aktivitou rádiojódu
> 600 mCi alebo 22 GBq po poslednej dávke najmenej 6 mesiacov pred zaradením do štúdie. Randomizácia bola stratifikovaná podľa geografických oblastí (Európa, Severná Amerika a ostatné), podľa predchádzajúcej liečby cielenej na VEGF/VEGFR (pacienti nepodstúpili žiadnu alebo
podstúpili 1 liečbu cielenú na VEGF/VEGFR) a veku (≤ 65 rokov alebo > 65 rokov). Hlavné kritérium miery účinnosti bol interval dĺžky prežitia bez progresie (PFS), určený zaslepeným nezávislým rádiologickým hodnotením s použitím kritérií na hodnotenie odpovede u solídnych tumorov (Response
Evaluation Criteria in Solid Tumours RECIST) 1.1. Druhotné kritérium miery účinnosti zahŕňalo
celkovú mieru odpovede a celkové prežívanie. Pacienti v placebovom ramene mali možnosť si zvoliť liečbu lenvatinibom po potvrdení progresie ochorenia.
Vhodní pacienti s merateľným ochorením podľa RECIST 1.1 boli randomizovaní v pomere 2:1
na liečbu lenvatinibom 24 mg jedenkrát denne (n=261) alebo placebom (n=131). Demografické údaje
a charakteristiky ochorenia pri vstupe do štúdie boli vyhovujúco vyvážené medzi oboma skupinami. Z 392 randomizovaných pacientov 76,3 % nedostalo predchádzajúcu liečby cielenú na VEGF/VEGFR, 49,0 % boli ženy, 49,7 % boli Európania a medián veku bol 63 rokov. Histologicky malo 66,1 % potvrdený nález papilárneho karcinómu štítnej žľazy a 33,9 % malo nález folikulárneho karcinómu štítnej žľazy, čo zahŕňalo variantu z Hürthleho buniek u 14,8 % a svetlobunkovú variantu u 3,8 %. Metastázy boli prítomné u 99 % pacientov: v pľúcach u 89,3 %, v lymfatických uzlinách
u 51,5 %, v kostiach u 38,8 %, v pečeni u 18,1 %, v pleure u 16,3 %, a v mozgu u 4,1 %. Väčšina pacientov mala výsledok stavu výkonnosti hodnotený pomocou ECOG v hodnote 0; 42,1 % pacientov malo výsledok 1; 3,9 % malo výsledok viac ako 1. Medián kumulatívnej aktivity rádiojódu, ktorý bol podaný pred vstupom do štúdie bol 350 mCi (12,95 GBq).
Štatisticky významné predĺženie PFS sa preukázalo u pacientov liečených lenvatinibom oproti pacientom, ktorí dostávali placebo (p<0,0001) (pozri schému 1). Bol pozorovaný pozitívny účinok na PFS vo všetkých podskupinách podľa veku (nad a pod 65 rokov), pohlavia, rasy, histologického subtypu, geografickej oblasti a u pacientov, ktorí nedostávali žiadnu alebo dostávali 1 terapiu cielenú na VEGF/VEGFR (pozri tabuľku 4). Následne po potvrdení progresie ochorenia nezávislým hodnotením prešlo 109 (83,2 %) pacientov zaradených do ramena s placebom v čase primárnej analýzy účinnosti na otvorenú liečbu lenvatinibom.
Objektívna miera odpovede (kompletná odpoveď [CR] plus čiastočná odpoveď [PR]) na základe nezávislého rádiologického hodnotenia bola významne (p<0,0001) vyššia u skupiny liečenej lenvatinibom ako u skupiny placeba (1,5 %). Štyria (1,5 %) pacienti liečení lenvatinibom dosiahli kompletnú odpoveď a 165 pacientov (63,2 %) čiastočnú odpoveď. V skupine placeba nedosiahol kompletnú odpoveď žiaden pacient a 2 (1,5 %) pacienti dosiahli čiastočnú odpoveď.
Medián času do prvej redukcie dávky bol 2,8 mesiaca. Medián doby do objektívnej odpovede bol 2,0 (95 % CI: 1,9, 3,5) mesiace, ale u pacientov, ktorí dosiahli kompletnú alebo čiastočnú odpoveď na lenvatinibe, sa u 70,4 % pozorovalo dosiahnutie odpovede v 30. deň alebo v priebehu 30 dní od začiatku užívania dávky 24 mg.
Fakt, že pacienti na placebe mohli po potvrdení progresie ochorenia prejsť na otvorenú liečbu lenvatinibom, neumožnil uskutočniť celkovú analýzu prežívania. V čase primárnej analýzy nebol žiaden štatisticky významný rozdiel v celkovom prežívaní medzi liečebnými skupinami (HR=0,73;
95 % CI: 0,50, 1,07; p=0,1032). Medián celkového prežívania (OS) sa nedosiahol ani u skupiny na lenvatinibe, ani u prekríženej placebovej skupiny.
Tabuľka 6 Výsledky účinnosti u pacientov s DTC
|
|
Lenvatinib
N=261
|
Placebo
N=131
|
Dĺžka prežitia bez progresie (PFS)
a
|
Počet prípadov progresie alebo úmrtia (%)
|
107 (41,0)
|
113 (86,3)
|
Medián PFS v mesiacoch (95 % CI)
|
18,3 (15,1; NE)
|
3,6 (2,2; 3,7)
|
Miera rizika(99 % CI)b,c
|
0,21 (0,14; 0,31)
|
p-hodnotab
|
<0,0001
|
Pacienti, ktorí nedostali žiadnu predchádzajúcu liečbu
cielenú na VEGF/VEGFR (%)
|
195 (74,7)
|
104 (79,4)
|
Počet prípadov progresie alebo úmrtia
|
76
|
88
|
Medián PFS v mesiacoch (95 % CI)
|
18,7 (16,4; NE)
|
3,6 (2,1; 5,3)
|
Miera rizika (95 % CI)b,c
|
0,20 (0,14; 0,27)
|
Pacienti, ktorí dostali 1 predchádzajúcu liečbu cielenú
na VEGF/VEGFR (%)
|
66 (25,3)
|
27 (20,6)
|
Počet prípadov progresie alebo úmrtia
|
31
|
25
|
Medián PFS v mesiacoch (95 % CI)
|
15,1 (8,8; NE)
|
3,6 (1,9; 3,7)
|
Miera rizika (95 % CI)b,c
|
0,22 (0,12; 0,41)
|
Objektívna miera odpovede
a
|
Počet pacientov s objektívnou odpoveďou (%)
|
169 (64,8)
|
2 (1,5)
|
(95 % CI)
|
(59,0; 70,5)
|
(0,0; 3,6)
|
p-hodnotab
|
<0,0001
|
Počet pacientov s kompletnou odpoveďou
|
4
|
0
|
Počet pacientov s čiastočnou odpoveďou
|
165
|
2
|
Medián času do objektívnej odpovede,d v mesiacoch
(95 % CI)
|
2,0 (1,9; 3,5)
|
5,6 (1,8; 9,4)
|
Trvanie odpovede,d v mesiacoch, medián (95 % CI)
|
NE (16,8; NE)
|
NE (NE; NE)
|
Celkové prežívanie
|
Počet prípadov úmrtia (%)
|
71 (27,2)
|
47 (35,9)
|
Medián OS v mesiacoch (95 % CI)
|
NE (22,0; NE)
|
NE (20,3; NE)
|
Miera rizika (95 % CI) b, e
|
0,73 (0,50; 1,07)
|
p-hodnotab,e
|
0,1032
|
CI, interval spoľahlivosti; NE, nevypočítané; OS, celkové prežívanie; PFS, prežitie bez progresie; RPSFT,
rank preserving structural failure time model; VEGF/VEGFR, cievny endoteliálny rastový faktor /
receptor cievneho endoteliálneho rastového faktora. a: Nezávislé rádiologické hodnotenie.
b: Stratifikované podľa regiónov (Európa vs. Severná Amerika vs. Ostatné), vekové skupiny
(≤65 rokov vs. >65 rokov), a predchádzajúca liečba proti VEGF/VEGFR (0 vs. 1). c: Odhadované pomocou Coxovho modelu proporcionálneho rizika.
d: Odhadovanépoužitím Kaplan-Meierovej metódy; 95 % CI bola vytvorená pomocou generalizovanej metódy Brookmeyer a Crowley u pacientov s najlepšou celkovou odpoveďou kompletnej alebo čiastočnej odpovede.
e: Bez zohľadnenia efektu prekríženia.
 Schéma 1 Kaplan-Meierova krivka prežitia bez progresie – DTC
Medián (mesiace) (95 % CI)
Schéma 1 Kaplan-Meierova krivka prežitia bez progresie – DTC
Medián (mesiace) (95 % CI)
Lenvatinib 18,3 (15,1; NE)
Placebo 3,6 (2,2; 3,7)
HR (99 % CI): 0,21 (0,14; 0,31)
Log-rank test: P<0,0001
Počet rizikových pacientov:
Čas (mesiace)
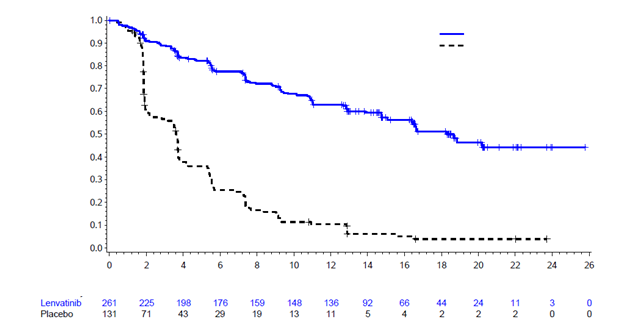
CI, interval spoľahlivosti; NE, neodhadnuteľné.
Hepatocelulárny karcinómKlinická účinnosť a bezpečnosť lenvatinibu sa vyhodnocovala v medzinárodnej, multicentrickej,
otvorenej, randomizovanej štúdii fázy 3 (REFLECT) u pacientov s neresekovateľným
hepatocelulárnym karcinómom (HCC).
Celkovo bolo 954 pacientov randomizovaných 1:1 na podávanie buď lenvatinibu (12 mg [telesná hmotnosť pri vstupe ≥60 kg] alebo 8 mg [telesná hmotnosť pri vstupe <60 kg]), podávanom perorálne raz denne, alebo sorafenibu 400 mg podávaného perorálne dvakrát denne.
Pacienti boli spôsobilí na účasť, ak mali stav funkčnosti pečene Child-Pugh triedy A a status Eastern Cooperative Oncology Group Performance (ECOG PS) 0 alebo 1. Boli vylúčení pacienti, ktorí predtým užívali systémovú protirakovinovú liečbu pokročilého/neresekovateľného HCC alebo akúkoľvek predchádzajúcu liečbu anti-VEGF. Cieľové lézie, ktoré sa predtým liečili rádioterapiou alebo lokoregionálnou terapiou, museli vykazovať rádiografický dôkaz o progresii ochorenia. Pacienti s obsadením pečene ≥50 %, jasnou inváziou do žlčovodu alebo hlavnej vetvy vrátnice (Vp4) pri zobrazení boli tak isto vylúčení.
· Demografická a úvodná charakteristika ochorenia bola podobná medzi skupinami
s lenvatinibom a sorafenibom a je uvedená nižšie pre všetkých 954 randomizovaných pacientov:
· Priemerný vek: 62 rokov
· Mužov: 84 %
· Bielych: 29 %, ázijských: 69 %, čiernych alebo afroameričanov: 1,4 %
· Telesná hmotnosť: <60 kg -31 %, 60-80 kg – 50 %, >80 kg– 19 %
· Status Eastern Cooperative Oncology Group Performance (ECOG PS) 0: 63 %, ECOG PS
1: 37 %
· Child-Pugh A: 99 %, Child-Pugh B: 1 %
· Etiológia: Hepatitída B (50 %), Hepatitída C (23 %), alkohol (6 %)
· Absencia makroskopického zasiahnutia vrátnice (MPVI): 79 %
· Absencia MPVI, rozšírenie tumoru mimo pečene (EHS) alebo obidvoje: 30 %
· Základná cirhóza (podľa nezávislého zobrazenia): 75 %
· Barcelona Clinic Liver Cancer (BCLC) stupňaB: 20 %;BCLCstupňaC: 80 %
· Predchádzajúca liečba: hepatektómia (28 %), rádioterapia (11 %), lokoregionálne terapie vrátane transarteriálnej (chemo)embolizácie (52 %), rádiofrekvenčná ablácia (21 %)
a perkutánna etanolinjekcia (4 %)
Primárnym koncovým bodom účinnosti bolo celkové prežívanie (OS). Lenvatinib nebol nižší vo vzťahu k OS oproti sorafenibu s HR = 0,92 [95 % CI z (0,79, 1,06)] a priemerným OS 13,6 mesiaca oproti 12,3 mesiacom (pozri tabuľku 7 a obrázok 2). Výsledky pre náhradné koncové body (PFS a ORR) sú uvedené v tabuľke 7 nižšie.
Tabuľka 7: Výsledky účinnosti zo štúdie REFLECT u pacientov s HCC
|
Parameter účinnosti
|
a, b
(95 %CI)
|
P-hodnotad
|
Medián(95 %CI)e
| Lenvatinib
(N= 478)
| Sorafenib
(N=476)
|
OS
|
0,92 (0,79,1,06)
|
Nedostupné
|
13,6 (12,1, 14,9)
| 12,3 (10,4,
13,9)
| PFSg(mRECIST)
| 0,64 (0,55, 0,75)
| <0,00001
| 7,3 (5,6, 7,5)
| 3,6 (3,6, 3,7)
|
|
|
| Percentá (95 % CI)
| ORRc, f, g (mRECIST)
| Nedostupné
| <0,00001
| 41 % (36 %, 45 %)
| 12 % (9 %,
15 %)
|
|
|
Miera rizikaDátum uzavretia údajov: 13. November 2016.
a Miera rizika je pre lenvatinib vs. sorafenib, za základe modelu Cox vrátane liečebnej skupiny
ako faktora.
b Stratifikované podľa regiónov (Región 1: Ázia-Tichomorie; Región 2: Západný), makroskopické zasiahnutie vrátnice alebo rozšírenie mimo pečene alebo obidvoje (áno, nie), ECOG PS (0, 1) a telesná hmotnosť (<60 kg, ≥60 kg).
c Výsledky sú založené na potvrdených aj nepotvrdených reakciách. d P-hodnota pre test superiority lenvatinibu versus sorafenibu.
e Kvartily sa odhadujú použitím Kaplan-Meierovej metódy; 95 % CI bola vytvorená pomocou
generalizovanej metódy Brookmeyer a Crowley
f Miera reakcií (úplných alebo čiastočných reakcií)
g Podľa nezávislej retrospektívnej rádiologickej analýzy. Medián trvania objektívnej reakcie bol
7,3 (95 %CI 5,6, 7,4) mesiaca pre skupinu lenvatinibu a 6,2 (95 %CI 3,7, 11,2) mesiaca pre skupinu sorafenibu.
 Schéma 2 Kaplan-Meierova krivka celkového prežitia – HCC
Medián (mesiace) (95 % CI)
Schéma 2 Kaplan-Meierova krivka celkového prežitia – HCC
Medián (mesiace) (95 % CI) Lenvatinib: 13,6 (12,1, 14,9) Sorafenib: 12,3 (10,4, 13,9)
HR (95 % CI): 0,92 (0,79, 1,06)
Počet rizikových pacientov:
Čas (mesiace)
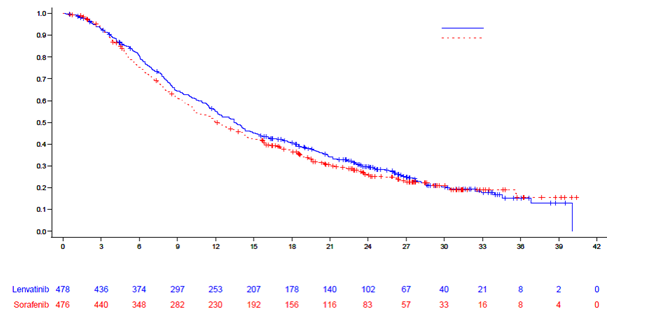
1. Dátum uzavretia údajov: 13. November 2016.
2. Krajná hodnota pre nepodradenosť miery rizika (HR: lenvatinib vs sorafenib = 1,08).
3. Medián bol odhadovaný použitím Kaplan-Meierovej metódy; 95 % CI bola vytvorená pomocou generalizovanej metódy Brookmeyer a Crowley
4. HR bol odhadovaný podľa Coxovho modelu proporcionálneho rizika s liečbou ako nezávislou
premennou a stratifikované podľa stratifikačných faktorov IxRS. Na väzby bola použitá
Efronova metóda.
5. + = cenzurované pozorovania.
V analýzach podskupín stratifikačnými faktormi (prítomnosť alebo absencia MPVI alebo EHS alebo obidvoch, ECOG PS 0 alebo 1, telesná hmostnosť<60 kg alebo ≥60 kg a región) HR výsledky súhlasne hovorili v prospech lenvatinibupred sorafenibom, s výnimkou západného regiónu [HR 1,08 (95 % CI 0,82, 1,42], pacientov bez EHS [HR 1,01 (95 % CI 0,78, 1,30)] a pacientov bez MPVI, EHS alebo obidve [HR 1,05 (0,79, 1,40)].Výsledky analýz podskupín by sa mali interpretovať opatrne. Priemerné trvanie liečby bolo 5,7 mesiacov (Q1: 2,9, Q3: 11,1) pre časť lenvatinibu a 3,7 mesiacov (Q1: 1,8, Q3: 7,4) pre časť sorafenibu.
V obidvoch častiach štúdie REFLECT bolo priemerné OS približne o 9 mesiacov dlhšie u pacientov, ktorí dostávali následnú protirakovinovú terapiu oproti pacientom bez nej. V časti lenvatinibu bolo priemerné OS 19,5 mesiaca (95 % CI: 15,7, 23,0) u pacientov, ktorí dostávali následnú protirakovinovú terapiu (43 %) a 10,5 mesiaca (95 % CI: 8,6, 12,2) u pacientov bez nej. V časti sorafenib bolo priemerné OS 17,0 mesiaca (95 % CI: 14,2, 18,8) u pacientov, ktorí dostávali následnú protirakovinovú terapiu (51 %) a 7,9 mesiaca (95 % CI: 6,6, 9,7)u pacientov bez nej. Priemerné OS bolo dlhšie približne o 2,5 mesiaca v skupine lenvatinibu v porovnaní so sorafenibom u obidvoch podskupín pacientov (s následnou protirakovinovou terapiou alebo bez nej).
Predĺženie QT intervaluPodľa výsledkov dôkladných QT štúdií u zdravých dobrovoľníkov jednotlivá dávka lenvatinibu 32 mg
nepredlžovala QT/QTc interval. Avšak, u pacientov liečených lenvatinibom sa pozorovala zvýšená incidencia predĺženia QT/QTc intervalu oproti pacientom na placebe (pozri časti 4.4 a 4.8).
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s lenvatinibom
v jednej alebo vo viacerých podskupinách pediatrickej populácie liečených pre diferencovaný karcinóm štítnej žľazy refraktérny na rádiojód a zrušila povinnosť predkladať výsledky štúdie pre jednu alebo viacero podskupín pediatrickej populácie liečenej na hepatocelulárny karcinóm (HCC).
5.2 Farmakokinetické vlastnosti
Farmakokinetické parametre lenvatinibu sa skúmali u zdravých dospelých, dospelých s poruchou
funkcie pečene, poruchou funkcie obličiek a solídnymi nádormi.
Absorpcia
Lenvatinib sa rýchlo absorbuje po perorálnom podaní s typicky pozorovaným tmax od 1 do 4 hodín
po podaní dávky. Jedlo neovplyvňovalo rozsah absorpcie, ale spomaľovalo rýchlosť absorpcie. Keď sa
podal s jedlom zdravým osobám, maximálna plazmatická koncentrácia sa oneskorila o 2 hodiny. Absolútna biologická dostupnosť sa u ľudí nehodnotila, ale údaje zo štúdie hmotnostnej bilancie naznačujú, že je to rádovo 85 %. Lenvatinib vykazoval dobrú perorálnu biologickú dostupnosť u psov (70,4 %) a opíc (78,4 %).
Distribúcia
Väzba lenvatinibu na plazmatické bielkoviny in vitro je vysoká a pohybovala sa v rozmedzí od 98 %
do 99 % (0,3 - 30 μg/ml, mesilát). Viazal sa hlavne na albumín a v malej miere na kyslý α1- glykoproteín a γ-globulin.
V in vitro sledovaní sa pohyboval pomer koncentrácie v krvi a v plazme od 0,589 do 0,608 (0,1 –
10 μg/ml, mesilát).
Lenvatinib je substrát pre P-gp a BCRP. Lenvatinib nie je substrát pre OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2,MATE1, MATE2-K alebo pre pumpu na export žlčovej soli(BSEP).
Medián zdanlivého distribučného objemu (Vz/F) po prvej dávke sa u pacientov pohyboval od 50,5 l do
92 l a spravidla bol stály v skupinách s dávkou od 3,2 mg do 32 mg. Analógový medián zdanlivého distribučného objemu v ustálenom stave (Vz/Fss) bol tiež spravidla stály a pohyboval sa od 43,2 l do 121 l.
Biotransformácia
V in vitro sledovaní sa cytochróm P450 3A4 ukazoval ako prevládajúca (> 80 %) izoforma, ktorá je
zapojená do P450-mediovaného metabolizmu lenvatinibu. Ale in vivo údaje naznačujú, že
významný podiel metabolizmu lenvatinibu prebieha aj mimo P450–mediovanej cesty.
V dôsledku tohto mali induktory a inhibítory CYP 3A4 minimálny efekt na expozíciu lenvatinibu
(pozri časť 4.5).
V ľudských hepatálnych mikrozómoch sa ako hlavný metabolit identifikovala demetylovaná forma lenvatinibu (M2). M2’ a M3’, ako hlavné metabolity prítomné v ľudských výkaloch, boli tvorené aldehydoxidázou z M2, resp. z lenvatinibu.
Vo vzorkách plazmy, ktoré sa zbierali počas 24 hodín po podaní, tvoril lenvatinib 97 % rádioaktivity v plazmatických rádiochromatografoch, zatiaľ čo metabolit M2 tvoril zvyšných 2,5 %. Na základe AUC(0 – inf) sa lenvatinib podieľal na rádioaktivite v plazme a v krvi 60 %, resp. 64 %.
Údaje z humánnej štúdiehmotnostnej bilancie vylučovaní naznačujú, že lenvatinib sa u ľudí značne metabolizuje. Hlavná metabolická dráha je oxidácia aldehydoxidázou, demetylácia cez CYP 3A4, konjugácia glutatiónom s elimináciou O-arylovej skupiny (časť chlórfenylu) a kombináciou týchto metabolických dráh s ďalšou biotransformáciou (napr.glukuronidácia, hydrolýza glutationóvej časti, degradácia cysteínovej časti a preusporiadanie cysteínglycínových a cysteínových konjugátov vnútri molekuly s následnou dimerizáciou). Tieto in vivo metabolické cesty sú v súlade s údajmi získanými v in vitro štúdiách s humánnymi biomateriálmi.
Transportné in vitro štúdie
Na základe rozdelenia IC50> 50 ´ Cmax,unbound sa vylúčila klinicky relevantná inhibícia nasledovných prenášačov, OAT1, OAT3, OATP1B1, OCT1, OCT2 a BSEP.
Lenvatinib vykazoval minimálnu alebo žiadnu inhibičnú aktivitu voči transportným aktivitám mediovaným P-gp a proteínu rezistencie rakoviny prsníka (BCRP). Podobne sa nepozorovala žiadna indukcia P-gp mRNA.
Lenvatinib vykazoval minimálny alebo žiadny inhibičný účinok voči OATP1B3a MATE2-K. Lenvatinib slabo inhibuje MATE1. V cytozóle ľudských hepatálnych buniek neinhiboval lenvatinib aktivitu aldehydoxidázy.
Eliminácia
Koncentrácia v plazme klesá bi-exponenciálne po Cmax. Priemerný terminálny exponenciálny polčas
lenvatinibu je približne 28 hodín.
Po podaní rádioaktívne značeného lenvatinibu 6 pacientom so solídnymi nádormi, približne dve tretiny zo značenej látky boli eliminované do stolice a jedna štvrtina do moču. M3 metabolit bol prevládajúcim analytom v exkrétoch (~ 17 % dávky), nasledovaný M2’(~ 11 % dávky) a M2 (~ 4,4 % dávky).
Linearita/nelinearita
Proporcionalita dávky a akumulácia
U pacientov so solídnymi nádormi, ktorým boli podávané jednorazové alebo opakované dávky lenvatinibu raz denne, sa expozícia lenvatinibu (Cmax a AUC) zvyšovala priamo úmerne k podanej dávke v rozsahu 3,2 až 32 mg, podávanom jedenkrát denne.
Lenvatinib vykazuje minimimálnu akumuláciu v rovnovážnom stave. Mimo tento rozsah bol medián
indexu kumulácie (Rac) v rozmedzí od 0,96 (20 mg) do 1,54 (6,4 mg).Rac u pacientov s HCC
s miernym až stredne ťažkým poškodením funkcie pečene bol podobný ako sa pozoruje u iných solídnych tumorov.
Osobitné skupiny
Porucha funkcie pečene
Farmakokinetika lenvatinibu po podaní jednotlivej dávky 10 mg sa hodnotila u 6 jedincov s miernou alebo stredne ťažkou poruchou funkcie pečene (Child-Pugh A a Child-Pugh B, v tomto poradí). Dávka
5 mg sa hodnotila u 6 jedincov s ťažkou poruchu funkcie pečene (Child-Pugh C).
Osem zdravých, demograficky porovnateľných jedincov, ktorí boli priradení ako kontrola, dostávalo dávku 10 mg. Expozícia lenvatinibu na základe údajov AUC0-inf u jedincov s miernou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek bola 101 %, 90 % a 122 % normálu,v tomto poradí. Bolo stanovené, že u pacientov s poruchou funkcie pečene bola väzba na plazmatické proteíny podobná ako u zdravých jedincov a nebola pozorovaná žiadna závislosť na koncentrácii. Pozri časť 4.2
o odporúčanom dávkovaní.
Neexistujú dostatočné údaje pre pacientov s HCC, ktorí majú Child-Pugh B (mierne poškodenie pečene, 3 pacienti liečení v pivotálnej štúdii lenvimou) a neexistujú žiadne údaje pre pacientov
s Child-Pugh C s HCC (ťažké poškodenie funkcie pečene). Lenvatinib sa eliminuje najmä v pečeni
a u týchto skupín pacientov sa expozícia môže zvyšovať.
Medián polčasu bol porovnateľný u jedincov s miernou, stredne ťažkou aj ťažkou poruchou funkcie pečene ako aj u jedincov s normálnou funkciou pečene. Pohyboval sa v rozmedzí 26 až 31 hodín. Percento dávky lenvatinibu vylúčenej do moču bolo nízke vo všetkých kohortách (<2,16 %
vo všetkých liečebných kohortách).
Porucha funkcie obličiek
Farmakokinetika lenvatinibu po podaní jednotlivej dávky 24 mg sa hodnotila u 6 jedincov s miernou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek v porovnaní s 8 zdravými, demograficky
porovnateľnými jedincami. Štúdia sa nerealizovala u pacientov s terminálnym zlyhaním obličiek.
Expozícia lenvatinibu na základe údajov AUC0-inf u jedincov s miernou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek bola 101 %, 90 % a 122 % normálu,v tomto poradí. Bolo stanovené, že
u pacientov s poruchou funkcie obličiek bola väzba na plazmatické proteíny podobná ako u zdravých jedincov a nebola pozorovaná žiadna závislosť na koncentrácii. Pozri časť 4.2 o odporúčanom
dávkovaní.
Vek, pohlavie, hmotnosť, rasa
Na základe populačnej farmakokinetickej analýzy u pacientov s dávkou 24 mg lenvatinibu jedenkrát
denne nemali vek, pohlavie, hmotnosť a rasa (Japonci vs. ostatní, kaukazská rasa vs. ostatní) žiaden významný vplyv na klírens (pozri časť 4.2).
Pediatrická populácia
U pediatrických pacientov sa klinické štúdie nerealizovali.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovanou dávkou (až 39 týždňov) spôsoboval lenvatinib toxické zmeny v rôznych orgánoch a tkanivách, ktoré súviseli s očakávanými farmakologickými účinkmi lenvatinibu. Tieto zahŕňali glomerulopatiu, testikulárnu hypocelularitu, folikulárnu atréziu ovárií, zmeny v gastrointestinálnom trakte, kostné zmeny, zmeny v nadobličkách (potkany a psy) a artériálne lézie (fibrinoidná nekróza artérií, degenerácie médie alebo krvácanie) u potkanov, psov a makakov (Macaca fascicularis). U potkanov, psov a opíc sa tiež pozorovali zvýšené hodnoty transamináz
s príznakmi hepatotoxicity. U všetkých testovaných zvieracích druhov sa na konci 4 týždňového
zotavovacieho obdobia pozorovala reverzibilita toxikológických zmien.
Genotoxicita
Lenvatinib nebol genotoxický.
Štúdie karcinogenicity s lenvatinibom sa nerealizovali.
Reprodukčná a vývojovátoxicita
U lenvatinibu sa nerealizovali žiadne špeciálne štúdie na zhodnotenie vplyvu na fertilitu u zvierat.
Napriek tomu sa u zvierat pozorovali testikulárne (hypocelularita semenotvorného epitelu) a ovariálne (folikulárna atrézia) zmeny počas toxikologických štúdií s opakovanou dávkou u zvierat, pri expozíciách 11 až 15 násobku (u potkanov) alebo 0,6 až 7 násobku (u opíc) očakávanej klinickej expozície (na základe AUC) pri maximálnej tolerovanej dávke u ľudí. Tieto nálezy boli reverzibilné
na konci 4 týždňového zotavovacieho obdobia.
Podanie lenvatinibu počas organogenézy spôsobilo úmrtia embryí a teratogenicitu u potkanov (vonkajšie a kostrové anomálie plodu) pri nižšej ako klinickej expozícii (na základe AUC), pri maximálnej tolerovanej dávke u ľudí a u králikov (vonkajšie, viscerálne a kostrové anomálie plodu)
na základe plochy povrchu tela; mg/m2 v maximálnej tolerovanej dávke u ľudí. Tieto nálezy poukazujú
na to, že lenvatinib má teratogénny potenciál, pravdepodobne vzhľadom k farmakologickej aktivite ako anti-angiogénny činiteľ. Lenvatinib a jeho metabolity sa vylučujú do mlieka potkanov.
Juvenilné toxikologické štúdie na zvieratách
Mortalita bola toxicitou limitujúcou dávku u mladých potkanov, u ktorých sa začalo podávanie dávky
na postnatálny deň (PND)7 alebo PND21. Pozorovala sa pri expozíciách, ktoré boli 125-krát alebo 12-
krát nižšie, v tomto poradí, v porovnaní s expozíciami, pri ktorých bola pozorovaná mortalita
u dospelých potkanov. To naznačuje, že čím je nižší vek, tým je vyššia citlivosť na toxicitu. Z toho dôvodu sa mortalita prisudzuje komplikáciám súvisiacim s primárnymi duodenálnymi léziami
a pôsobeniu možnej prídavnej toxicity na nezrelé cieľové orgány.
Toxicita lenvatinibu bola významnejšia u mladších potkanov (začiatok dávkovania na PND7), ako u potkanov, u ktorých sa dávkovanie začalo na PND21a mortalita a niektoré toxické účinky sa pozorovali skôr u juvenilných potkanov pri 10 mg/kg v porovnaní s dospelými potkanmi s tou istou dávkou. U juvenilných potkanov sa taktiež pozorovalo spomalenie rastu, sekundárne oneskorenie fyzického vývoja a lézie zodpovedajúce farmakologickému pôsobeniu (rezáky, femur [epifýzová rastová platnička], obličky, nadobličky a duodenum).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly Uhličitan vápenatý Manitol
Mikrokryštalická celulóza
Hydroxypropylcelulóza
Nízko substituovaná hydroxypropylcelulóza
Mastenec
Puzdro kapsuly
Hypromelóza
Oxid titaničitý (E171) Žltý oxid železitý (E172) Červený oxid železitý (E172)
Potlačováfarba
Šelak
Čierny oxid železitý (E172) Hydroxid draselný Propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25 °C. Uchovávajte v pôvodnom blistri na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Polyamidové/hliníkové/PVC/hliníkové blistre obsahujúce 10 kapsúl. Každá škatuľa obsahuje 30, 60
alebo 90 tvrdých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Opatrovatelia by nemali kapsuly otvárať, aby nedošlo k opakovanej expozícii obsahu kapsúl.
Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIEisai GmbH Lyoner Straße 36
60528 Frankfurt am Main
Nemecko
E-mail:
medinfo_de@eisai.net8. REGISTRAČNÉ ČÍSLO (ČÍSLA)Lenvima 4 mg tvrdé kapsulyEU/1/15/1002/001
EU/1/15/1002/003
EU/1/15/1002/004
Lenvima 10 mg tvrdé kapsulyEU/1/15/1002/002
EU/1/15/1002/005
EU/1/15/1002/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. máj 2015
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTU