
lóza, pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovanie
S cieľom zlepšiť sledovanie biologických liekov sa má jasne zaznamenať názov a číslo šarže
podaného lieku.
Crohnova choroba
U pacientov s anamnézou Crohnovej choroby sú dostupné iba obmedzené údaje. Pri predpisovaní lieku Kyntheum pacientom s anamnézou Crohnovej choroby je potrebná opatrnosť. U pacientov s
anamnézou Crohnovej choroby je potrebné sledovať prejavy a príznaky aktívnej Crohnovej choroby.
Ak u pacientov dôjde k rozvoju aktívnej Crohnovej choroby, liečba sa má natrvalo ukončiť.
Samovražedné myšlienky a správanie
U pacientov liečených liekom Kyntheum boli hlásené samovražedné myšlienky a správanie vrátane dokonanej samovraždy. Väčšina pacientov so samovražedným správaním mala v anamnéze depresiu a/alebo samovražedné myšlienky či správanie. Nebola zistená príčinná súvislosť medzi liečbou liekom Kyntheum a zvýšeným rizikom výskytu samovražedných myšlienok a správania.
U pacientov s anamnézou depresie a/alebo samovražedných myšlienok či správania alebo u pacientov, u ktorých dôjde k rozvoju takýchto príznakov starostlivo zvážte pomer rizika a prínosu liečby liekom Kyntheum. Pacienti, ošetrovatelia a rodinní príslušníci majú byť poučení o potrebe zvýšenej
pozornosti pri výskyte alebo zhoršení depresie, samovražedných myšlienok, úzkosti alebo iných zmien nálady a v prípade výskytu takýchto udalostí majú kontaktovať poskytovateľa zdravotnej
starostlivosti. Ak má pacient nové alebo zhoršujúce sa symptómy depresie a/alebo sa uňho identifikujú samovražedné myšlienky či samovražedné správanie, liečbu liekom Kyntheum sa odporúča prerušiť.
Infekcie
Kyntheum môže zvyšovať riziko infekcií.
Počas 12-týždňového placebom kontrolovaného klinického skúšania u pacientov so psoriázou boli závažné infekcie pozorované u 0,5 % pacientov liečených liekom Kyntheum (pozri časť 4.8).
Pri zvažovaní použitia lieku Kyntheum u pacientov s chronickou infekciou alebo anamnézou rekurentnej infekcie je potrebná opatrnosť. Pacienti by mali byť poučení, aby vyhľadali lekársku pomoc v prípade, ak sa u nich vyskytnú prejavy alebo príznaky poukazujúce na infekciu. Ak sa u
pacienta rozvinie závažná infekcia, je potrebne pacienta starostlivo sledovať a nesmie sa mu podávať
Kyntheum, až kým infekcia neustúpi.
V klinických skúšaniach neboli hlásené žiadne prípady aktívnej tuberkulózy. Pacientom s aktívnou tuberkulózou sa však Kyntheum nemá podávať. U pacientov s latentnou formou tuberkulózy sa má pred začatím liečby liekom Kyntheum zvážiť antituberkulózna liečba.
Zníženýabsolútnypočetneutrofilov
Počas 12-týždňového placebom kontrolovaného klinického skúšania u pacientov so psoriázou bol pozorovaný pokles počtu neutrofilov (ANC) u 5,6 % pacientov liečených liekom Kyntheum, ktorý bol vo všeobecnosti prechodný a reverzibilný. Zriedkavo bol pozorovaný stupeň 3 a 4. Žiaden z poklesov na stupeň 3 alebo 4 ANC u pacientov so psoriázou nebol spojený s vážnou infekciou (pozri tiež
časť 4.8).
Očkovania
Pred začatím liečby liekom Kyntheum sa pacientom odporúča podstúpiť všetky očkovania v súlade s
národnými smernicami týkajúcimi sa imunizácie. Živé vakcíny sa nemajú podávať súčasne s liekom Kyntheum (pozri časť 4.5). Nie sú k dispozícii žiadne údaje o odpovedi na očkovanie živými vakcínami, o riziku infekcie ani o prenose infekcie po podaní živých vakcín pacientom liečených liekom Kyntheum.
Očkovanie dojčiat
Očkovanie dojčiat živými vakcínami po expozícii lieku Kyntheum počas tretieho trimestra je nutné prekonzultovať s lekárom (pozri tiež časť 4.6).
Súbežná imunosupresívnaliečba
Bezpečnosť a účinnosť lieku Kyntheum v kombinácii s imunosupresívami vrátane biologických liekov alebo fototerapie neboli hodnotené.
4.5 Liekové a iné interakcie
Živé vakcíny sa nemajú podávať súbežne s liekom Kyntheum (pozri časť 4.4).
Tvorba enzýmov CYP450 môže byť zmenená kvôli zvýšeným koncentráciám určitých cytokínov (napr. IL-1, IL-6, IL-10, TNFα, IFN) pri chronickom zápale. Hoci úloha interleukínov (IL)-17A a IL-17RA v regulácii enzýmov CYP450 nebola pozorovaná, v štúdii interakcií medzi liekmi a ochorením bol vyhodnotený účinok brodalumabu na aktivitu enzýmu CYP3A4/3A5.
U pacientov so stredne závažnou až závažnou ložiskovou psoriázou zvýšila jedna subkutánna dávka 210 mg brodalumabu pôsobenie midazolamu (substrátu enzýmu CYP3A4/3A5) o 24 %. Na základe rozsahu zmeny expozície midazolamu nie je pri súbežnom podávaní lieku Kyntheum nutná úprava dávky substrátov enzýmu CYP3A4/3A5.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a až 12 týždňov po liečbe.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití brodalumabu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity
(pozri časť 5.3).
O ľudskom imunoglobulíne IgG2 je známe, že prechádza placentárnou bariérou. Brodalumab je ľudský imunoglobulín IgG2, a preto sa môže preniesť z matky na vyvíjajúci sa plod. Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu lieku Kyntheum počas gravidity.
Pretože metabolizmus brodalumabu u dojčiat nie je známy, pomer prínosu a rizika expozície dojčaťa
živým vakcínam po expozícii lieku Kyntheum počas tretieho trimestra sa má konzultovať s lekárom.
Dojčenie
Nie je známe, či sa brodalumab vylučuje do ľudského mlieka. Brodalumab je monoklonálna protilátka a predpokladá sa, že sa vyskytne v prvom mlieku a následne v nízkych koncentráciách.
Riziko pre novorodencov/dojčatá sa nedá vylúčiť.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu liekom Kyntheum sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku brodalumabu na ľudskú fertilitu. Štúdie na zvieratách
nepreukázali žiadne účinky na mužské a ženské reprodukčné orgány ani na počet, pohyblivosť a
morfológiu spermií (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kyntheum nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie hlásenými nežiaducimi reakciami u všetkých pacientov liečených liekom Kyntheum boli bolesť kĺbov (4,6 %), bolesť hlavy (4,3 %), únava (2,6 %), hnačka (2,2 %) a orofaryngeálna bolesť (2,1 %).
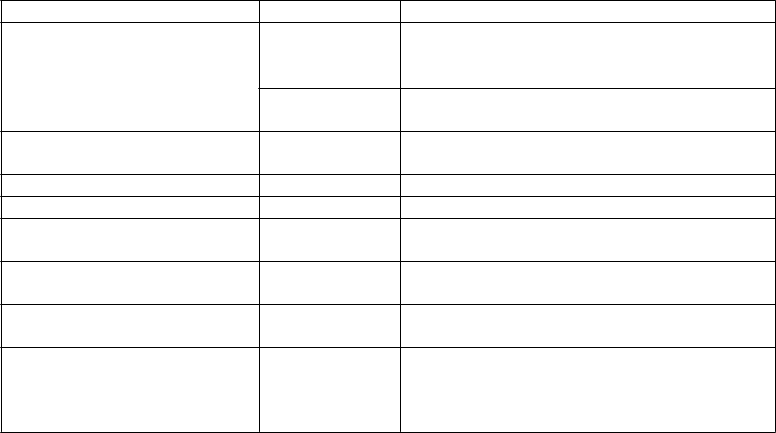
Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie z klinických skúšaní (Tabuľka 1) sú uvedené na základe tried orgánových systémov (SOC) podľa databázy MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, pričom najčastejšie reakcie sú prvé. V rámci jednotlivých skupín
frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. Okrem toho sú zodpovedajúce kategórie frekvencií pre každú nežiaducu reakciu založené na nasledujúcej konvencii:
veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥
1/10 000 až < 1/1 000) a veľmi zriedkavé (< 1/10 000).
T
abuľka 1: Zoznam nežiaducich reakcií v klinických skúšaniach
T
rieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Časté Chrípka
Plesňové infekcie (vrátane tinea pedis, tinea
versicolor, tinea cruris)
Menej časté Kandidové infekcie (vrátane orálnych, genitálnych a ezofágových infekcií)
Poruchy krvi a lymfatického systému
Časté Neutropénia
Poruchy nervového systému Časté Bolesť hlavy
Poruchy oka Menej časté Konjunktivitída
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva Celkové poruchy a reakcie
v mieste podania
Časté Orofaryngeálna bolesť
Časté Hnačka
Nevoľnosť
Časté Artralgia
Myalgia
Časté Únava
Reakcie v mieste podania (vrátane erytému, bolesti, svrbenia, krvnej podliatiny a krvácania v mieste podania)
 O
pis vybraných nežiaducich reakcií
I
nfekcie
O
pis vybraných nežiaducich reakcií
I
nfekcie
Počas 12-týždňového placebom kontrolovaného skúšania u pacientov s ložiskovou psoriázou boli infekcie hlásené u 25,4 % pacientov liečených liekom Kyntheum v porovnaní s 23,4 % pacientov liečených placebom. Väčšinu prípadov infekcií predstavovali nazofaryngitída, infekcia horných dýchacích ciest, faryngitída, infekcie močových ciest, bronchitída a chrípka, ktoré si nevyžadovali ukončenie liečby. Závažné infekcie sa vyskytli u 0,5 % pacientov liečených liekom Kyntheum a
u 0,2 % pacientov s placebom. U pacientov liečených liekom Kyntheum bol v porovnaní s pacientmi s
placebom pozorovaný vyšší výskyt plesňových infekcií, najmä nezávažných kandidových infekcií
kože a slizníc, a to u 1,8 % pacientov liečených liekom Kyntheum verzus 0,9 % pacientov s placebom. V klinických skúšaniach bol pozorovaný jeden závažný prípad kryptokokovej meningitídy a jeden
závažný prípad kokcidioidomykózy (pozri časť 4.4).
V 52. týždni boli výskyty infekcií po úprave na základe expozície (na 100 pacientorokov) 114,6
u pacientov liečených liekom Kyntheum a 118,1 u pacientov liečených ustekinumabom. Výskyty závažných infekcií po úprave na základe expozície (na 100 pacientorokov) boli 1,3 u pacientov liečených liekom Kyntheum a 1,0 u pacientov liečených ustekinumabom.
NeutropéniaPočas 12-týždňového placebom kontrolovaného klinického skúšania bola neutropénia hlásená u 0,8 % pacientov liečených liekom Kyntheum a u 0,5 % pacientov s placebom. Väčšina pozorovaných nežiaducich reakcií spojených s liečbou liekom Kyntheum bola mierna, prechodná a reverzibilná.
Neutropénia stupňa 3 a 4 bola hlásená u 0,4 % pacientov liečených liekom Kyntheum v porovnaní
s 0,2 % pacientov liečených ustekinumabom a žiadnymi pacientmi na placebe. S neutropéniou neboli spojené žiadne závažné infekcie.
ImunogenicitaU 2,7 % pacientov so psoriázou (122/4 461), ktorí boli v klinických skúšaniach liečení liekom
Kyntheum až 52 týždňov, sa vytvorili protilátky proti brodalumabu (0,3 % týchto pacientov malo protilátky proti brodalumabu už na začiatku liečby). Žiaden z týchto pacientov nemal neutralizačné protilátky.
S tvorbou protilátok proti brodalumabu nie je spojená žiadna zmena farmakokinetického profilu,
klinickej odpovede ani bezpečnostného profilu.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinickych skúšaniach sa podávali intravenózne dávky až do 700 mg bez prejavov toxicity, ktorá by limitovala dávku. V prípade predávkovania sa odporúča pacienta sledovať na akékoľvek prejavy a príznaky nežiaducich účinkov a bezodkladne začať primeranú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory interleukínov, ATC kód: L04AC12
MechanizmusúčinkuBrodalumab je rekombinantná plne ľudská monoklonálna imunoglobulínová protilátka IgG2, ktorá sa s vysokou afinitou viaže na ľudský interleukín IL-17RA a blokuje biologické aktivity prozápalových cytokínov IL-17A, IL-17F, IL-17A/F heterodiméru a IL-25, čím inhibuje zápal a klinické príznaky spojené so psoriázou. IL-17RA je proteín exprimovaný na povrchu bunky a je nevyhnutnou súčasťou receptorových komplexov využívaných viacerými cytokínmi z rodiny IL-17. U pacientov so psoriázou boli hlásené zvýšené koncentrácie cytokínov z rodiny IL-17. Cytokíny IL-17A, IL-17F a IL-17A/F heterodimér vykazujú pleiotropné aktivity vrátane indukcie prozápalových mediátorov, ako sú IL-6, GROα a G-CSF, z epiteliálnych buniek, endoteliálnych buniek a fibroblastov podporujúcich zápal v tkanivách. Blokovanie interleukínu IL-17RA inhibuje odpovede vyvolané cytokínom IL-17, čo vedie
k normalizácii zápalu v koži.
FarmakodynamickéúčinkyV psoriatických ložiskách bola zistená zvýšená expresia génov pre cytokíny IL-17A, IL-17C a IL-17F. V psoriatických ložiskách bola tiež zistená zvýšená expresia génov pre IL-12B a IL-23A, čo sú dve podjednotky IL-23, aktivátora „upstream“ expresie IL-17A a IL-17F. U pacientov so psoriázou liečených liekom Kyntheum bol pozorovaný pokles koncentrácií interleukínu IL-17A a markerov bunkovej proliferácie a zníženie hrúbky epidermy pri biopsii kožných lézií na hodnoty pozorované pri biopsii kože bez lézií a to až po dobu 12 týždňov po liečbe.
Klinickáúčinnosťa bezpečnosťÚčinnosť a bezpečnosť lieku Kyntheum bola hodnotená u 4 373 dospelých pacientov s ložiskovou psoriázou v troch medzinárodných, randomizovaných, dvojito zaslepených, placebom kontrolovaných
klinických skúšaniach fázy 3 (AMAGINE-1, AMAGINE-2 a AMAGINE-3). Skúšania AMAGINE-2 a
AMAGINE-3 boli kontrolované aj aktívnou referenčnou vzorkou (ustekinumabom). Všetky tri
skúšania zahŕňali 12-týždňovú placebom kontrolovanú indukčnú fázu, 52-týždňovú dvojito zaslepenú fázu a dlhodobú otvorenú pokračovaciu fázu.
Zaradení pacienti boli vhodní na systémovú liečbu vrátane fototerapie, biologickej a nebiologickej
systémovej liečby. Približne 21 % pacientov malo v anamnéze výskyt psoriatickej artritídy. Približne
30 % pacientov dostávalo v minulosti biologickú liečbu a u 12 % pacientov došlo k zlyhaniu
biologickej liečby.
Prevažná väčšina pacientov boli muži (69 %), bielej rasy (91 %), v priemernom veku 45 rokov
(18 až 86 rokov), z ktorých 6,1 % bolo starších ako 65 rokov a 0,3 % starších ako 75 rokov.
V liečebných skupinách bolo východiskové skóre indexu plochy postihnutia a závažnosti psoriázy PASI (Psoriasis Area Severity Index) v rozsahu od 9,4 do 72 (medián: 17,4) a východisková hodnota postihnutej plochy povrchu tela BSA (body surface area) bola v rozsahu od 10 do 97 (medián: 21). Východiskové skóre sPGA (static Physician Global Assessment) bolo v rozsahu od „3 (stredne závažné)“ (58 %) do „5 (veľmi závažné)” (5 %).
V skúšaní AMAGINE-1 sa zúčastnilo 661 pacientov. Zahŕňalo 12-týždňovú dvojito zaslepenú, placebom kontrolovanú indukčnú fázu a následnú dvojito zaslepenú fázu ukončenia a opätovného začatia liečby v trvaní až do 52 týždňov. Pacienti randomizovaní do skupiny s liekom Kyntheum dostávali dávky 210 mg alebo 140 mg v 0. týždni (deň 1), 1. týždni a 2. týždni a následne im bola podávaná rovnaká dávka každé 2 týždne. V 12. týždni boli pacienti, ktorí boli pôvodne randomizovaní do skupiny s liekom Kyntheum a ktorí dosiahli štatické skóre celkového hodnotenia lekárom sPGA
(0 alebo 1), opätovne randomizovaní do skupiny s placebom alebo do skupiny pokračujúcej v liečbe
liekom Kyntheum v indukčnej dávke. Pacienti pôvodne randomizovaní do skupiny s placebom a pacienti, ktorí nesplnili kritéria na opätovnú randomizáciu, dostávali Kyntheum v dávke 210 mg každé dva týždne od 12. týždňa. Opakovaná liečba bola k dispozícii od 16. týždňa pre pacientov s recidívou choroby a záchranná liečba bola k dispozícii po 12 týždňoch opakovanej liečby.
Skúšania AMAGINE-2 a AMAGINE-3 boli identické placebom a ustekinumabom kontrolované skúšania. V skúšaní AMAGINE-2 sa zúčastnilo 1 831 pacientov a v skúšaní
AMAGINE-3 1 881 pacientov. Obe skúšania zahŕňali 12-týždňovú dvojito zaslepenú, placebom a ustekinumabom kontrolovanú indukčnú fázu a následnú dvojito zaslepenú udržiavaciu fázu v trvaní až 52 týždňov. Pacienti randomizovaní do skupiny s liekom Kyntheum dostávali v indukčnej fáze
dávky 210 mg alebo 140 mg v 0. týždni (deň 1), 1. týždni a 2. týždni a následne im bola podávaná rovnaká dávka každé 2 týždne. Pacienti randomizovaní do skupiny s ustekinumabom dostávali
dávky 45 mg (pacienti s hmotnosťou ≤ 100 kg) a 90 mg (pacienti s hmotnosťou > 100 kg) v 0.
týždni, 4. týždni a 16. týždni a následne im bola podávaná rovnaká dávka každých 12 týždňov. V 12. týždni boli pacienti pôvodne randomizovaní do skupiny s liekom Kyntheum opäť randomizovaní do
skupín pacientov dostávajúcich buď 210 mg každé 2 týždne, alebo 140 mg každé 2 týždne,
alebo 140 mg každé 4 týždne, alebo 140 mg každých 8 týždňov počas udržiavacej fázy. Pacienti pôvodne randomizovaní do skupiny s placebom začali od 12. týždňa dostávať Kyntheum v
dávke 210 mg každé 2 týždne. Pacienti v skupine s ustekinumabom v 12. týždni ďalej dostávali
ustekinumab a potom v 52. týždni prešli na Kyntheum v dávke 210 mg každé 2. týždne. Pre pacientov s nedostatočnou odpoveďou podľa jediného hodnotenia sPGA ≥ 3 alebo pretrvávajúceho hodnotenia sPGA so skóre 2 po dobu minimálne 4 týždňov bola od 16. týždňa k dispozícii záchranná liečba.
T
abuľka 2: Prehľad hlavných výsledkov účinnosti
A
MAGINE-1 AMAGINE-2 a AMAGINE-3
Placebo Kyntheum
210 mg Q2W
Placebo Kyntheum
210 mg
Q2W
Ustekinumab
n-randomizovaní 220 222 624 1 236 613
n-dokončili 12. týždeň 209 212 601 1 205 594
n-v udržiavacej fáze 84 83 Nevzťahuje
sa
n-dokončili 52. týždeň 2 74 Nevzťahuje
sa
339 590
236 300
P
A
SI
PASIVýchodiskové skóre (priemer
±SD)
19,7 ±7,7 19,4 ±6,6 20,2 ±8,4 20,3 ±8,3 20,0 ±8,4
PASI 75 12. týždeň (%) 3 83* 7 86* 70*
PASI 75 52. týždeň (%) 0 87* Nevzťahuje
sa
65 48
s
P
G
A (%)
sPGA 0 alebo 1 12. týždeň 1 76* 4 79* 59*
sPGA 0 alebo 1 52. týždeň 0 83* Nevzťahuje
sa
65 45
 P
SI
P
SI
PSIVýchodiskové skóre (priemer ±SD) 19,0 ±6,7 18,9 ±6,7 18,8 ±6,9 18,7 ±7,0 18,8 ±6,9
PSIrespondér 12. týždeň (%) 4 61* 7 64* 54*
Q2W = každé 2 týždne
PSI = Psoriasis Symptom Inventory. Respondér podľa PSI: celkové skóre ≤ 8 so žiadnou položkou zo skóre
> 1; SD: štandardná odchýlka.
Hodnota NRI (Non-responder imputation) sa používa na prisúdenie chýbajúcich údajov.
Z dôvodu opätovnej randomizácie do skupín s inými skúmanými dávkovacími režimami je skupina n-
v udržiavacej fáze v niekoľkých skupinách výrazne menšia ako skupina n-randomizovaní. Udržiavacia fáza v skúšaniach AMAGINE -2 a -3 nezahŕňala placebo.
* p-hodnota verzus zodpovedajúce placebo, prispôsobené pre stratifikačné faktory < 0,001
Skóre PASI 75 po 2 týždňoch bolo v rozsahu medzi 20 % a 25 % v skúšaniach vo fáze 3 v porovnaní s
placebom (0 % až 0,6 %) a ustekinumabom (3 % až 3,5 %).
 G
raf 1: PASI 100 počas indukčnej a udržiavacej fázy pre Kyntheum a ustekinumab (súhrn údajov z AMAGINE-2 a AMAGINE-3)
G
raf 1: PASI 100 počas indukčnej a udržiavacej fázy pre Kyntheum a ustekinumab (súhrn údajov z AMAGINE-2 a AMAGINE-3)
,
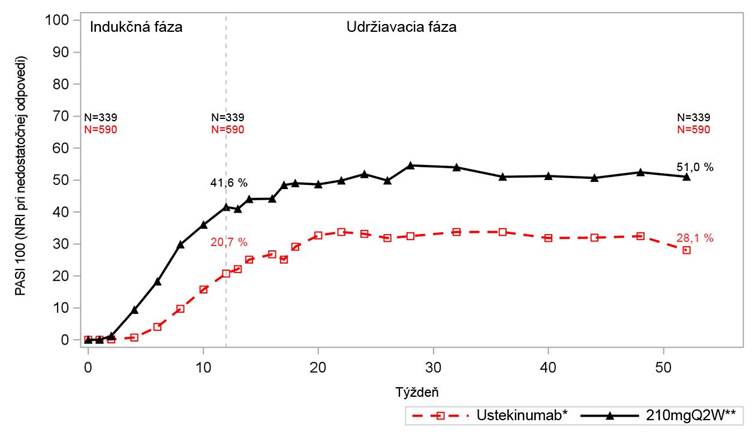
N = počet pacientov na začiatku, v 12. a 52. týždni. Q2W = každé 2 týždne
*Pacientom bol podávaný ustekinumab v indukčnej fáze a pokračovali v jeho užívaní aj v udržiavacej fáze.
**Pacientom bol podávaný Kyntheum 210 mg každé 2 týždne v indukčnej fáze a následne boli opätovne
randomizovaní do skupiny s liekom Kyntheum v dávke 210 mg každé 2 týždne v udržiavacej fáze. NRI= Non-responder imputation
Vo všetkých troch klinických skúšaniach sa nepreukázali žiadne rozdiely v odpovedi na liek
Kyntheum, a to v žiadnom z kľúčových cieľových ukazovateľoch [PASI 75, PASI 100, úspešnosť podľa sPGA (0 alebo 1) a sPGA čisté (0)] v podskupinách podľa veku, pohlavia, rasy, predchádzajúceho užívania systémovej liečby alebo fototerapie, predchádzajúceho užívania biologických liekov a zlyhania biologickej liečby.
Spolu s primárnymi cieľovými ukazovateľmi účinnosti boli tiež pozorované klinicky dôležité zlepšenia v indexe závažnosti psoriázy skalpu (Psoriasis Scalp Severity Index, PSSI) v 12. týždni (AMAGINE-1) a v indexe závažnosti psoriázy nechtov (Nail Psoriasis Severity Index, NAPSI) v 12. a 52. týždni (AMAGINE-1,-2 a -3).
Kvalita života / pacientom hlásené výsledkyPodiel pacientov, ktorí dosiahli v 12. týždni skóre v dotazníku príznakov psoriázy Psoriasis Symptom Inventory (PSI) 0 (čisté) alebo 1 (mierne) pre každý bod (svrbenie, pálenie, štípanie, bolesť, sčervenenie, šupinatosť, praskanie a odlupovanie), je uvedený v Tabuľke 2.'
Percentuálny podiel pacientov, ktorí dosiahli v 12. týždni skóre DLQI (Dermatology Life Quality
Index) 0 alebo 1, bol 56 %, 61 %, 59 % v skupine s 210 mg lieku Kyntheum a 5 %, 5 %, 7 %
v skupine s placebom v skúšaní AMAGINE-1, -2 a -3 v uvedenom poradí (upravená p-hodnota <
0,001) a 44 % v skupinách s ustekinumabom (AMAGINE-2 a -3).
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky skúšaní s liekom Kyntheum na liečbu ložiskovej psoriázy v jednej alebo vo viacerých podskupinách pediatrickej populácie (informácie o použití v pediatrickej populácii nájdete v časti 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Na základe populačných farmakokinetických modelov je odhadovaná akumulácia po 20 týždňoch
podávania 2,5-násobná. U pacientov so stredne závažnou až závažnou ložiskovou psoriázou bola po jednom subkutánnom podaní 210 mg lieku Kyntheum zistená priemerná maximálna sérová koncentrácia (Cmax) 13,4 μg/ml (štandardná odchýlka [SD] = 7,29 μg/ml). Medián času do maximálnej koncentrácie (Tmax) bol 3,0 dní (rozsah: 2,0 až 4,0 dní) a priemerná plocha pod krivkou závislosti koncentrácie na čase bola do poslednej merateľnej koncentrácie (AUCposledný) 111 μg*deň/ml (SD
= 64,4 μg*deň/ml). Subkutánna biologická dostupnosť brodalumabu odhadovaná na základe
populačných farmakokinetických modelov bola 54,7 % (relatívna štandardná chyba [RSE] = 4,25 %.
Pozorované farmakokinetické parametre počas rovnovážneho stavu (10. – 12. týždeň) boli: priemerná plocha pod krivkou času/koncentrácie pri rovnovážnom stave za interval dávkovania (AUCtau) –
227,4 μg*deň/ml (SD = 191,7 μg*deň/ml); odpovedajúca priemernej koncentrácii (Cav,ss) 16,2 μg/ml
priemerná hodnota Cmax – 20,9 μg/ml (SD = 17,0 μg/ml) a minimálna sérová koncentrácia v 12. týždni
(Cmin) – 9,8 μg/ml (SD = 11,2 μg/ml).
Distribúcia
Na základe populačných farmakokinetických modelov bol odhadovaný priemerný distribučný objem
brodalumabu v rovnovážnom stave približne 7,24 l.
Biotransformácia
Brodalumab je ľudská monoklonálna protilátka IgG2 a predpokladá sa, že sa rozkladá na malé peptidy a aminokyseliny prostredníctvom katabolických dráh podobným spôsobom ako endogénny IgG.
Eliminácia
Po subkutánnom podaní 210 mg vykazuje brodalumab nelineárnu farmakokinetiku typickú pre monoklonálnu protilátku, ktorá sa distribuuje podľa charakteru farmakologického cieľa.
So stúpajúcou dávkou brodalumabu sa znižuje klírens a dochádza k väčšiemu nárastu expozície než je úmerné dávke. V prípade 3-násobného zvýšenia subkutánnej dávky brodalumabu zo 70 na 210 mg sa sérová koncentrácia Cmax brodalumabu v rovnovážnom stave zvýšila približne 18-násobne a
AUC0-t 25-násobne v uvedenom poradí.
Po jednom subkutánnom podaní brodalumabu v dávke 210 mg pacientom s ložiskovou psoriázou bol zjavný klírens (CL/F) 2,95 l/deň.
Podľa populačných farmakokinetických modelov sa predpovedalo, že u 95 % pacientov klesnú sérové koncentrácie brodalumabu pod limit merateľnosti (0,05 μg/ml) 63 dní po prerušení podávania brodalumabu v dávke 210 mg v rovnovážnom stave pri podávaní každé 2 týždne. Koncentrácie brodalumabu pod LLOQ (Lower Limit of Quantification) boli však spojené až s 81% obsadenosťou receptorov pre IL-17.
Na základe populačných farmakokinetických modelov bol odhadovaný eliminačný polčas brodalumabu 10,9 dňa v rovnovážnom stave po subkutánnej dávke 210 mg podávanej každý druhý týždeň.
Vplyv hmotnosti na farmakokinetiku
Na populačných farmakokinetických modeloch sa preukázalo, že expozícia klesá s narastajúcou
telesnou hmotnosťou. Neodporúča sa žiadna úprava dávky.
Starší pacienti
Populačné farmakokinetické modely naznačili, že vek nemal vplyv na farmakokinetiku brodalumabu, čo bolo založené na údajoch od 259 (6 %) pacientov vo veku 65 – 74 rokov a od 14 (0,3 %) pacientov vo veku ≥ 75 rokov, ktorí spadali do celkovej FK populácie 4 271 pacientov s ložiskovou psoriázou.
Poruchafunkcieobličiekalebopečene
Nie sú k dispozícii žiadne farmakokinetické údaje týkajúce sa pacientov s poruchou funkcie obličiek alebo pečene. Predpokladá sa, že eliminácia intaktného brodalumabu, monoklonálnej protilátky IgG, obličkami bude nízka a nevýznamná. Predpokladá sa, že brodalumab bude eliminovaný najmä katabolizmom a porucha funkcie pečene zrejme nemá vplyv na jeho klírens.
Iné populácie
Farmakokinetika brodalumabu bola u pacientov so psoriázou japonského a pacientov iného ako japonského pôvodu podobná.
Populačná farmakokinetická analýza preukázala, že pohlavie nemalo vplyv na farmakokinetiku brodalumabu.
Farmakokinetický/farmakodynamickývzťah
Populačný farmakokinetický/farmakodynamický model pripravený na základe všetkých dostupných údajov preukázal, že pri dávke 210 mg každé 2 týždne sa predpokladá, že u 90 % pacientov sa udrží minimálna koncentrácia vyššia ako je odhadovaná hodnota IC90 1,51 μg/ml. Na základe výskumnej popisnej analýzy nebol pozorovaný žiadny vzťah medzi expozíciou a výskytom závažných infekcií a nákaz, kandidových infekcií, vírusových infekcií a samovražedných myšlienok a správania. Analýza vzťahu medzi expozíciou a odpoveďou naznačuje, že vyššie koncentrácie brodalumabu súvisia s lepšími odpoveďami PASI a sPGA.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií toxicity po opakovanom podávaní (vrátane bezpečnostných farmakologických cieľových ukazovateľov a hodnotenia cieľových ukazovateľov súvisiacich s fertilitou) a reprodukčnej toxicity a vývinu neodhalili žiadne osobitne riziko pre ľudí.
Štúdie karcinogenity s brodalumabom sa neuskutočnili. Na makakoch dlhochvostých, ktoré dostávali brodalumab v týždenných subkutánnych dávkach 90 mg/kg počas 6 mesiacov (expozícia
AUC 47-násobne vyššia ako u ľudských pacientov liečených liekom Kyntheum v dávke 210 mg každé 2 týždne), sa však nevyskytli žiadne proliferatívne zmeny. Mutagénny potenciál brodalumabu
nebol hodnotený. Neočakáva sa však, že by monoklonálne protilátky alterovali DNA alebo chromozómy.
Po podaní brodalumabu v dávkach až do 90 mg/kg raz týždenne po dobu 6 mesiacov neboli na makakoch dlhochvostých zistené žiadne účinky na mužské a ženské reprodukčné orgány ani na počet, pohyblivosť a morfológiu spermií (expozícia AUC 47-násobne vyššia ako u ľudských pacientov liečených liekom Kyntheum v dávke 210 mg každé 2 týždne).
Na makakoch dlhochvostých neboli pozorované žiadne účinky na embryonálny/fetálny či postnatálny (až do 6 mesiacov veku) vývin pri subkutánnom podávaní brodalumabu počas gravidity pri expozičných hladinách, ktoré boli až 27-násobne vyššie ako koncentrácie dosiahnuté u ľudských pacientov dostávajúcich Kyntheum v dávke 210 mg každé 2 týždne, na základe plochy pod koncentračnou krivkou (AUC). Sérové koncentrácie u novorodených mláďat opíc a králikov vo fetálnom štádiu naznačujú výrazný prechod brodalumabu z matky na plod na konci gravidity.
Na makakoch dlhochvostých boli účinky súvisiace s brodalumabom po týždennom subkutánnom podávaní brodalumabu v dávke až do 90 mg/kg po dobu 6 mesiacov obmedzené na reakcie v mieste vpichu a zápal kože a slizníc, ktorý vyplynul z farmakologickej modulácie kontrolného systému hostiteľa nad symbiotickou mikroflórou. Nevyskytli sa žiadne účinky na výsledky imunofenotypizácie
na králikoch bol po subkutánnej injekcii lieku s obsahom brodalumabu v klinickej koncentrácii 140 mg/ml pozorovaný stredne závažný až závažný opuch.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prolín Glutamát Polysorbát 20
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
4 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Neuchovávajte v mrazničke.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Kyntheum sa môže uchovávať vo vonkajšom obale pri izbovej teplote (do 25°C) maximálne 14 dní, a to len raz. Po vybratí lieku Kyntheum z chladničky a po dosiahnutí izbovej teploty (do 25°C) sa musí použiť do 14 dní alebo zlikvidovať.
6.5 Druh obalu a obsah balenia
1,5 ml roztok v naplnenej injekčnej striekačke zo skla typu I s ihlou s priemerom 27G x ½” z nehrdzavejúcej ocele zakrytou elastomérovým krytom.
Kyntheum je dostupný v jednotlivých baleniach obsahujúcich 2 naplnené injekčné striekačky a v spoločných baleniach obsahujúcich 6 (3 balenia po 2) naplnených injekčných striekačiek.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Kyntheum je sterilný injekčný roztok naplnený v injekčnej striekačke. Každá naplnená injekčná striekačka je určená len na jedno použitie. Podrobné pokyny na podávanie lieku Kyntheum by ste mali nájsť v časti Pokyny na použitie.
Pred podaním injekcie by sa malo počkať aspoň 30 minút, aby sa naplnená injekčná striekačka zohriala na izbovú teplotu. Predídete tak nepríjemnému pocitu v mieste vpichu. Naplnená injekčná striekačka by nemala byť žiadnym spôsobom zahrievaná. Naplnenou injekčnou striekačkou by sa nemalo triasť. Pri dosahovaní izbovej teploty by sivý kryt ihly naplnenej injekčnej striekačky nemal byť odstránený.
Pred podaním by malo byť pohľadom skontrolované, či Kyntheum neobsahuje častice alebo či nezmenil farbu. Kyntheum je číra až slabo opaleskujúca, bezfarebná až slabožltá tekutina bez častíc. Tento liek by nemal byť používaný, ak spozorujete, že roztok je zakalený, zmenil farbu alebo obsahuje hrudky, vločky alebo častice.
Naplnená injekčná striekačka by nemala byť používaná, ak spadla na tvrdý povrch.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIILEO Pharma A/S Industriparken 55
DK-2750 Ballerup
Dánsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1155/001
EU/1/16/1155/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 17. júla 2017
10. DÁTUM REVÍZIE TEXTUPodrobne informácie o tomto lieku sú dostupne na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.