
toxicita
Z
výšenie východiskovej hladiny** AST a/alebo ALT, bez zvýšenia hladiny celkového bilirubínu nad
2 x ULN
Stupeň 1*
(
> ULN –
3 x ULN) Nie je potrebná žiadna úprava dávky.
Stupeň 2*
(>3 až 5 x ULN)
Východiskový stupeň <2: Prerušenie dávkovania až do zotavenia na
< východiskový stupeň, potom pokračujte v liečbe
liekom Kisqali v rovnakej dávke. Ak sa stupeň 2
znovu objaví, pokračujte
v liečbe liekom Kisqali v najbližšej nižšej dávke. Východiskový
stupeň = 2:
Žiadne prerušenie
dávkovania.
Stupeň 3*
(>5 až 20 x ULN)
Prerušenie dávkovania liekom Kisqali až do zotavenia na
≤ východiskový stupeň, potom
pokračujte najbližšou nižšou dávkou.
Ak sa stupeň 3 znovu
objaví, ukončite liečbu
liekom Kisqali.
Stupeň 4*
(>20 x ULN)
Ukončite liečbu liekom Kisqali.
K
ombinované zvýšenie hladiny AST a/alebo ALT spolu so zvýšením hladiny celkového bilirubínu, pri absencii cholestázy
Ak sa u pacientky vyvinie hladina ALT a/alebo AST >3x ULN spolu s hladinou celkového bilirubínu >2x ULN bez ohľadu na východiskový stupeň, ukončite liečbu liekom Kisqali.

* Klasifikácia podľa CTCAE verzia 4.03 (CTCAE = Všeobecné kritéria pre terminológiu nežiaducich
udalostí)
** Východisková hladina = pred začatím liečby
ULN = horná hranica normy
Pred začatím liečby liekom Kisqali sa má urobiť EKG vyšetrenie. Po začatí liečby sa má EKG
opakovať približne na 14. deň prvého cyklu a na začiatku druhého cyklu a potom podľa klinickej
potreby. V prípade predĺženia QTcF počas liečby sa odporúča častejšie sledovanie EKG.
T
abuľka 4 Úprava dávkovania a liečba – predĺženie QT
E
K
G
s QTcF >480 ms
E
K
G
s QTcF >500 ms
1. Dávkovanie sa má prerušiť.
2. Ak sa predĺženie QTcF upraví na <481 ms, pokračujte v liečbe
rovnakou dávkou.
3. Ak sa znovu objaví QTcF ≥481 ms, prerušte dávkovanie až do úpravy
QTcF na <481 ms a potom pokračujte v liečbe liekom Kisqali v najbližšej nižšej dávke.
Ak je QTcF väčšie ako 500 ms aspoň na 2 samostatných vyšetreniach EKG,
prerušte liečbu liekom Kisqali až kým je QTcF <481 ms, potom pokračujte
v liečbe liekom Kisqali v najbližšej nižšej dávke.
Ak sa vyskytne predĺženie QTcF intervalu väčšie ako 500 ms alebo zmena oproti východiskovej hodnote väčšia ako 60 ms v kombinácii s torsade de pointes alebo polymorfnou ventrikulárnou tachykardiou alebo prejavmi/symptómami závažnej arytmie, natrvalo ukončite liečbu liekom Kisqali.
T
abuľka 5 Úprava dávkovania a liečba – iné toxicity*
I
né toxicity Stupeň 1 alebo 2** Stupeň 3** Stupeň 4**
Nie je potrebná žiadna úprava dávky. Začnite vhodnú liečbu
a sledujte podľa
klinickej potreby
Prerušenie dávkovania až do zotavenia na stupeň ≤1, potom pokračujte v liečbe liekom Kisqali rovnakej dávke.
Ak sa stupeň 3 znovu
objaví, pokračujte
v liečbe liekom Kisqali v najbližšej nižšej
dávke.
Ukončite liečbu liekom
Kisqali.


* Okrem neutropénie, hepatotoxicity a predĺženia QT intervalu.
** Klasifikácia podľa CTCAE verzia 4.03 (CTCAE = Všeobecné kritéria pre terminológiu
nežiaducich udalostí)
Pre pokyny na úpravu dávky a ďalšie relevantné informácie o bezpečnosti v prípade prejavov toxicity
pozri tiež SmPC súbežne podávaného inhibítora aromatázy.
Úprav a dávk y pre použi ti e lieku Kisqali so silnými inhibítormi CYP3A4Je potrebné vyhnúť sa súbežnej liečbe silnými inhibítormi CYP3A4 a má sa zvážiť výber alternatívnych, súbežne podávaných liekov s menším potenciálom inhibície CYP3A4. Ak sa pacientke
musí podať silný inhibítor CYP3A súbežne s ribociklibom, dávka Kisqali sa má znížiť na 400 mg raz denne (pozri časť 4.5).
U pacientok, ktorým bola dávka ribociklibu znížená na 400 mg denne a u ktorých je nevyhnutné začať súbežné podávanie silných inhibítorov CYP3A4, sa má dávka znížiť na 200 mg.
U pacientok, ktorým bola dávka ribociklibu znížená na 200 mg denne a u ktorých je nevyhnutné začať súbežné podávanie silných inhibítorov CYP3A4, sa má liečba liekom Kisqali prerušiť.
Vzhľadom na variabilitu medzi pacientkami odporúčaná úprava dávky nemusí byť optimálna
u všetkých pacientok, a preto sa odporúča starostlivé sledovanie priznakov toxicity. Keď sa liečba silným inhibítorom ukončí, po uplynutí minimálne 5 polčasov silného inhibítora CYP3A4 sa má dávka Kisqali zmeniť na dávku používanú pred začatím liečby silným inhibítorom CYP3A4 (pozri časti 4.4,
4.5 a 5.2).
O
sobitné skupiny pacientok
Porucha funkcie obličiek
U pacientok s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná úprava dávky
(pozri časť 5.2). Opatrnosť je potrebná u pacientok s ťažkou poruchou funkcie obličiek so starostlivým
sledovaním príznakov toxicity, pretože u tejto populácie pacientok neexistujú žiadne skúsenosti
s liekom Kisqali (pozri časť 5.2).
Porucha funkcie pečene
Na základe farmakokinetickej štúdie u zdravých jedincov a jedincov bez rakoviny s poruchou funkcie pečene nie je potrebná úprava dávky u pacientok s miernou poruchou funkcie pečene (trieda A podľa Childa-Pugha). U pacientok so stredne ťažkou (trieda B podľa Childa-Pugha) a ťažkou poruchou funkcie pečene (trieda C podľa Childa-Pugha) sa môže zvýšiť (menej ako 2-násobne) expozícia ribociklibu a odporúča sa začiatočná dávka 400 mg Kisqali raz denne. Ribociklib sa neskúmal
u pacientok s karcinómom prsníka so stredne ťažkou a ťažkou poruchou funkcie pečene (pozri
časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť lieku Kisqali u detí a dospievajúcich mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Staršie pacientky
U pacientok starších ako 65 rokov nie je potrebná úprava dávky (pozri časť 5.2).
Spôsob podávania
Kisqali sa má užívať perorálne raz denne s jedlom alebo bez jedla. Tablety sa majú prehltnúť celé
a nemajú sa žuvať, drviť alebo deliť pred prehltnutím. Žiadna tableta sa nemá užiť ak je zlomená, prasknutá alebo iným spôsobom porušená.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na arašidy, sóju alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Kritické viscerálne ochorenie
Účinnosť a bezpečnosť ribociklibu sa neskúmala u pacientov s kritickým viscerálnym ochorením.
Neutropénia
Na základe závažnosti neutropénie možno bude nutné liečbu liekom Kisqali prerušiť, znížiť dávku
alebo liečbu ukončiť, ako je opísané v Tabuľke 2 (pozri časti 4.2 a 4.8).
Hepatobiliárna toxicita
Pred začatím liečby liekom Kisqali majú pacientky podstúpiť funkčné vyšetrenie pečene. Po začatí
liečby sa má sledovať funkcia pečene (pozri časti 4.2 a 4.8).
Na základe závažnosti zvýšenia hladín transamináz možno bude nutné liečbu liekom Kisqali prerušiť, znížiť dávku alebo liečbu ukončiť, ako je opísané v Tabuľke 3 (pozri časti 4.2 a 4.8). Odporúčania pre pacientky so zvýšenou hladinou AST/ALT s východiskovým stupňom ≥ 3 neboli stanovené.
Predĺženie intervalu QT
Pred začatím liečby sa má urobiť EKG vyšetrenie. Liečba liekom Kisqali sa má začať len u pacientok
s hodnotami QTcF menej ako 450 ms. EKG sa má opakovať približne na 14. deň prvého cyklu a na
začiatku druhého cyklu a potom podľa klinickej potreby (pozri časti 4.2 a 4.8).
Vhodné sledovanie sérových elektrolytov (vrátane draslíka, vápnika, fosforu a horčíka) sa má vykonať pred začatím liečby, na začiatku prvých 6 cyklov a potom podľa klinickej potreby. Akákoľvek abnormalita sa má upraviť pred začatím liečby liekom Kisqali.
Použitiu lieku Kisqali je potrebné sa vyhnúť u pacientok, ktoré už majú predĺžený QTc alebo majú signifikantné riziko vzniku jeho predĺženia. Patria sem pacientky:
· so syndrómom dlhého QT;
· s nekompenzovaným alebo signifikantným ochorením srdca, vrátane nedávneho infarktu myokardu, kongestívneho srdcového zlyhávania, nestabilnej angíny pektoris a bradyarytmie;
· s abnormalitami elektrolytov.
Je potrebné sa vyhnúť použitiu lieku Kisqali s liekmi, o ktorých je známe, že predlžujú interval QTc, a/alebo so silnými inhibítormi CYP3A4, pretože to môže viesť ku klinicky významnému predĺženiu intervalu QTcF (pozri časti 4.2, 4.5 a 5.1). Ak nie je možné vyhnúť sa liečbe silným inhibítorom CYP3A4, dávka sa má znížiť na 400 mg raz denne (pozri časti 4.2 a 4.5).
Na základe pozorovaného predĺženia QT počas liečby, možno bude nutné liečbu liekom Kisqali prerušiť, znížiť dávku alebo liečbu ukončiť, ako je opísané v Tabuľke 4 (pozri časti 4.2, 4.8 a 5.2).
Substráty CYP3A4
Ribociklib je pri dávke 600 mg silným inhibítorom CYP3A4 a pri dávke 400 mg stredne silným
inhibítorom CYP3A4. Medzi ribociklibom a liekmi metabolizovanými prostredníctvom CYP3A4 preto môže dochádzať k interakciám vedúcim k zvýšenej sérovej koncentrácii substrátov CYP3A4 (pozri časť 4.5). Pri súbežnom užívaní senzitívnych CYP3A4 substrátov s úzkym terapeutickým indexom sa odporúča opatrnosť a je potrebné preveriť odporúčania uvedené v SmPC týchto liekov týkajúce sa ich súbežného užívania s inhibítormi CYP3A4.
Sójový lecitín
Kisqali obsahuje sójový lecitín. Pacienti, ktorí sú precitlivení na arašidy alebo sóju, nesmú užívať
Kisqali (pozri časť 4.3).
4.5 Liekové a iné interakcie
Liečivá, ktorémôžuzvýšiťplazmatickékoncentrácie ribociklibu
Ribociklib je primárne metabolizovaný prostredníctvom CYP3A4. Preto lieky, ktoré môžu ovplyvniť
aktivitu enzýmu CYP3A4, môžu zmeniť farmakokinetiku ribociklibu. Súbežné podanie silného
inhibítora CYP3A4 ritonaviru (100 mg dvakrát denne po dobu 14 dní) s jednorazovou 400 mg dávkou ribociklibu zvýšilo expozíciu ribociklibu (AUCinf) a maximálnu koncentráciu (Cmax) u zdravých jedincov 3,2- a 1,7-násobne, v uvedenom poradí, v porovnaní s jednorazovou dávkou 400 mg ribociklibu podanou samostatne. Cmax a AUClast pre LEQ803 (hlavný metabolit ribociklibu predstavujúci menej ako 10 % expozície pôvodnej zlúčeniny) sa znížili o 96 % a 98 %, v uvedenom poradí.
Musí sa vyhnúť súbežnému použitiu silných inhibítorov CYP3A4, zahŕňajúcich okrem iného: klaritromycín, indinavir, itrakonazol, ketokonazol, lopinavir, ritonavir, nefazodón, nelfinavir, posakonazol, sachinavir, telaprevir, telitromycín, verapamil a vorikonazol (pozri časť 4.4). Je potrebné zvážiť súbežné podávanie náhradných liekov s menším potenciálom inhibovať CYP3A4 a pacientky
sa majú sledovať kvôli výskytu nežiaducich účinkov súvisiacich s ribociklibom (pozri časti 4.2, 4.4
a 5.2).
Ak nie je možné vyhnúť sa súbežnému podaniu lieku Kisqali so silným inhibítorom CYP3A4, dávka Kisqali sa má znížiť tak, ako je uvedené v časti 4.2. Pre takúto úpravu dávky však neexistujú žiadne klinické údaje. Vzhľadom na variabilitu medzi pacientkami nemusí byť odporúčaná úprava dávky optimálna u všetkých pacientok, a preto sa odporúča starostlivé sledovanie nežiaducich účinkov súvisiacich s ribociklibom. V prípade toxicity súvisiacej s ribociklibom sa má upraviť dávka alebo sa má prerušiť liečba až do ustúpenia toxicity (pozri časti 4.2 a 5.2). Po ukončení liečby silným inhibítorom CYP3A4 a uplynutí minimálne 5 polčasov inhibítora CYP3A4 (pozri SmPC príslušného CYP3A4 inhibítora) sa má pokračovať dávkou Kisqali, ktorá sa použila pred začatím liečby silným inhibítorom CYP3A4.
Fyziologicky založené farmakokinetické simulácie naznačili, že pri 600 mg dávke ribociklibu môže stredne silný inhibítor CYP3A4 (erytromycín) zvýšiť v rovnovážnom stave Cmax a AUC ribociklibu
1,2-násobne a 1,3-násobne, v uvedenom poradí. U pacientov užívajúcich zníženú dávku ribociklibu
400 mg raz denne sa odhaduje v rovnovážnom stave zvýšenie Cmax a AUC 1,4-násobne a 2,1-násobne, v uvedenom poradí. Pri dávke 200 mg raz denne sa predpokladá 1,7-násobné a 2,8-násobné zvýšenie,
v uvedenom poradí. Pri začatí liečby miernymi a stredne silnými inhibítormi CYP3A4 nie je potrebná
žiadna úprava dávky ribociklibu. Odporúča sa však sledovať nežiaduce účinky súvisiace s ribociklibom.
Pacientky je potrebné informovať, aby sa vyhli granátovým jablkám alebo šťave z granátových jabĺk a grapefruitu alebo grapefruitovému džúsu. Je o nich známe, že inhibujú enzýmy cytochrómu CYP3A4 a môžu zvýšiť expozíciu ribociklibu.
Liečivá, ktorémôžuznížiťplazmatickékoncentrácie ribociklibu
Súbežné podanie silného induktora CYP3A4 rifampicínu (600 mg denne po dobu 14 dní)
s jednorazovou 600 mg dávkou ribociklibu znížilo AUCinf a Cmax ribociklibu o 89 % a 81 %,
v uvedenom poradí, v porovnaní s jednorazovou dávkou 600 mg ribociklibu podanou samostatne
zdravým jedincom. Cmax LEQ803 sa zvýšila 1,7-násobne a AUCinf sa znížila o 27 %, v uvedenom poradí. Súbežné použitie silných induktorov CYP3A4 môže preto viesť k zníženej expozícii
a následne riziku nedostatočnej účinnosti. Je potrebné sa vyhnúť súbežnému použitiu silných induktorov CYP3A4, zahŕňajúcich okrem iného fenytoín, rifampicín, karbamazepín a ľubovník bodkovaný (Hypericum perforatum). Má sa zvážiť náhradný súbežne podávaný liek bez potenciálu
alebo s minimálnym potenciálom indukovať CYP3A4.
Účinok stredne silných induktorov CYP3A4 na expozíciu ribociklibu sa neskúmal. Fyziologicky založené farmakokinetické simulácie naznačili, že stredne silný induktor CYP3A4 (efavirenz) môže v rovnovážnom stave znížiť Cmax a AUC ribociklibu o 51 % a 70 %, v uvedenom poradí. Súbežné použitie stredne silných induktorov CYP3A4 preto môže viesť k zníženiu expozície a následne k riziku zníženia účinnosti, najmä u pacientov liečených dávkou 400 mg alebo
200 mg ribociklibu raz denne.
Liečivá, ktorých plazmatickékoncentráciemôžubyťzmenené liekom Kisqali
Ribociklib je stredne silný až silný inhibítor CYP3A4 a môže ovplyvňovať lieky, ktoré sú
metabolizované prostredníctvom CYP3A4, čo môže viesť k zvýšeniu sérových koncentrácií súbežne použitých liekov.
Súbežné podanie midazolamu (substrát CYP3A4) s viacnásobnými dávkami Kisqali (400 mg) zvýšilo u zdravých jedincov expozíciu midazolamu o 280 % (3,80-násobne), v porovnaní s podaním samotného midazolamu. Simulácie použitím fyziologicky založených farmakokinetických modelov naznačili, že po podaní lieku Kisqali v klinicky relevantnej dávke 600 mg sa očakáva 5,2-násobné zvýšenie AUC midazolamu. Preto sa zakaždým, keď sa ribociklib podáva súbežne s inými liekmi, musí preveriť SmPC iných liekov kvôli odporúčaniam týkajúcim sa súbežného podania s inhibítormi
CYP3A4. Ak sa Kisqali podáva so senzitívnymi substrátmi CYP3A4 s úzkym terapeutickým indexom, odporúča sa opatrnosť (pozri časť 4.4). Dávku citlivého substrátu CYP3A4 s úzkym terapeutickým
indexom, zahŕňajúceho okrem iného alfentanil, cyklosporín, everolimus, fentanyl, sirolimus
a takrolimus, môže byť potrebné znížiť, pretože ribociklib môže zvýšiť ich expozíciu.
Je potrebné sa vyhnúť súbežnému podaniu ribociklibu v dávke 600 mg s nasledujúcimi substrátmi CYP3A4: alfuzosín, amiodarón, cisaprid, pimozid, chinidín, ergotamín, dihydroergotamín, kvetiapín, lovastatín, simvastatín, sildenafil, midazolam, triazolam.
Súbežné podanie kofeínu (substrát CYP1A2) s viacnásobnými dávkami Kisqali (400 mg) zvýšilo
v porovnaní s podaním samotného kofeínu u zdravých jedincov expozíciu kofeínu o 20 %
(1,20-násobne). V klinicky relevantnej dávke 600 mg predpovedali simulácie použitím fyziologicky založených farmakokinetických (physiologically-based pharmacokinetic, PBPK) modelov len slabé
inhibičné účinky ribociklibu na substráty CYP1A2 (<2-násobné zvýšenie AUC).
V súčasnosti nie je známe, či Kisqali môže znížiť účinnosť systémovo pôsobiacej hormonálnej
antikoncepcie.
Liečivá, ktoré sú substrátmi transportérov
In vitro hodnotenia naznačili, že ribociklib má potenciál inhibovať aktivitu transportérov liekov P-gp,
BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 a BSEP. Pri súbežnej liečbe so senzitívnymi substrátmi týchto transportérov s úzkym terapeutickým indexom, zahŕňajúcimi okrem iného digoxín, pitavastatín, pravastatín, rosuvastatín a metformín sa odporúča opatrnosť a sledovanie toxicity.
Interakcie liek - jedlo
Kisqali sa môže podávať s jedlom alebo bez jedla (pozri časti 4.2 a 5.2).
Lieky, ktoré zvyšujúpHžalúdka
Ribociklib vykazuje vysokú solubilitu pri alebo pod pH 4,5 a v bio relevantnom médiu (pri pH 5,0
a 6,5). Súbežné podanie ribociklibu s liekmi, ktoré zvyšujú pH v žalúdku, sa nehodnotilo v klinickej štúdii; v populačnej farmakokinetickej analýze a nekompartmentovej farmakokinetickej analýze sa však nepozorovala zmenená absorpcia ribociklibu.
Liekové interakcie medzi ribociklibom a letrozolom
Údaje z klinickej štúdie u pacientok s karcinómom prsníka a populačná farmakokinetická analýza
ukázali, že medzi ribociklibom a letrozolom nie je žiadna lieková interakcia po súbežnom podaní
týchto liekov.
Očakávané interakcie
Antiarytmiká a i né li eky, ktoré môžu pre dĺ ži ť i nt erval QT
Je potrebné sa vyhnúť súbežnému podaniu Kisqali s liekmi so známym potenciálom predĺžiť interval
QT, ako sú antiarytmiká (zahŕňajúce okrem iného amiodarón, dizopyramid, prokaínamid, chinidín a sotalol), a liekmi, o ktorých je známe, že predlžujú interval QT (zahŕňajúcimi okrem iného
chlorochín, halofantrín, klaritromycín, haloperidol, metadón, moxifloxacín, bepridil, pimozid
a intravenózny ondansetrón) (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Pred začatím liečby s liekom Kisqali sa má overiť neprítomnosť gravidity.
Na základe nálezov u zvierat môže ribociklib spôsobiť poškodenie plodu, ak sa podá gravidným
ženám (pozri časť 5.3).
Pre ďalšie informácie týkajúce sa gravidity, laktácie a fertility pozri časť 5.3.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kisqali môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientky je potrebné informovať, aby boli opatrné pri vedení vozidiel alebo obsluhe strojov v prípade, že počas liečby liekom Kisqali pocítia únavu (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Celkové vyhodnotenie bezpečnosti Kisqali je založené na údajoch od 898 pacientok; 568 z nich bolo
vystavených ribociklibu v odporúčanej 600 mg dávke, s použitím navrhovaného liečebného režimu
600 mg ribociklibu (1.-21. deň 28-dňového cyklu), a vrátane 381, ktoré dostávali ribociklib v kombinácii s letrozolom v dávke 2,5 mg raz denne.
Údaje o bezpečnosti uvedené nižšie sú založené na údajoch z klinickej štúdie fázy III s mediánom trvania expozície ribociklibu a letrozolu 13 mesiacov (58,1 % pacientok vystavených po dobu
≥12 mesiacov).
K zníženiu dávky v dôsledku nežiaducich udalostí, bez ohľadu na príčinu, došlo u 44,6 % pacientok, ktorým boli podávané Kisqali a letrozol a trvalé ukončenie liečby bolo hlásené u 7,5 % pacientok.
Najčastejšími nežiaducimi liekovými reakciami a najčastejšími nežiaducimi liekovými reakciami stupňa 3/4 (hlásené s frekvenciou ≥20 % a ≥2 %, v uvedenom poradí), u ktorých frekvencia pre Kisqali a letrozol presahuje frekvenciu pre placebo a letrozol, boli neutropénia, leukopénia, bolesť hlavy, bolesť chrbta, nauzea, únava, hnačka, vracanie, zápcha, alopécia, vyrážka a neutropénia,
leukopénia, abnormálne hodnoty testov funkcie pečene, lymfopénia, hypofosfatémia, vracanie, nauzea, únava a bolesť chrbta, v uvedenom poradí.
Tabuľkový zoznam nežiaducichreakcií
Nežiaduce liekové reakcie z klinickej štúdie fázy III (tabuľka 6) sú uvedené podľa triedy orgánových
systémov MedDRA. V každej triede orgánových systémov sú nežiaduce liekové reakcie zoradené podľa frekvencie, s najčastejšími reakciami ako prvými. V každej skupine frekvencií sú nežiaduce
liekové reakcie uvedené v poradí klesajúcej závažnosti. Okrem toho je zodpovedajúca kategória frekvencie pre každú liekovú reakciu založená na nasledujúcej konvencii (CIOMS III): veľmi časté
(≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až
<1/1 000); veľmi zriedkavé (<1/10 000); a neznáme (z dostupných údajov).
T
abuľka 6 Nežiaduce liekové reakcie pozorované v klinickej štúdii fázy III
N
ežiaduca lieková reakcia Frekvencia
Infekcie a nákazy
Infekcie močových ciest Veľmi časté
Poruchy krvi a lymfatického systémuNeutropénia, leukopénia, anémia, lymfopénia Veľmi časté Trombocytopénia, febrilná neutropénia Časté
Poruchy metabolizmu a výživyZnížená chuť do jedla Veľmi časté Hypokalciémia, hypokaliémia, hypofosfatémia Časté
Poruchy nervového systémuBolesť hlavy, insomnia Veľmi časté
Poruchy okaZvýšená lakrimácia, suché oko Časté
Poruchy srdca a srdcovej činnostiSynkopa Časté
Poruchy dýchacej sústavy, hrudníka a mediastínaDyspnoe Veľmi časté Epistaxa Časté
Poruchy gastrointestinálneho traktu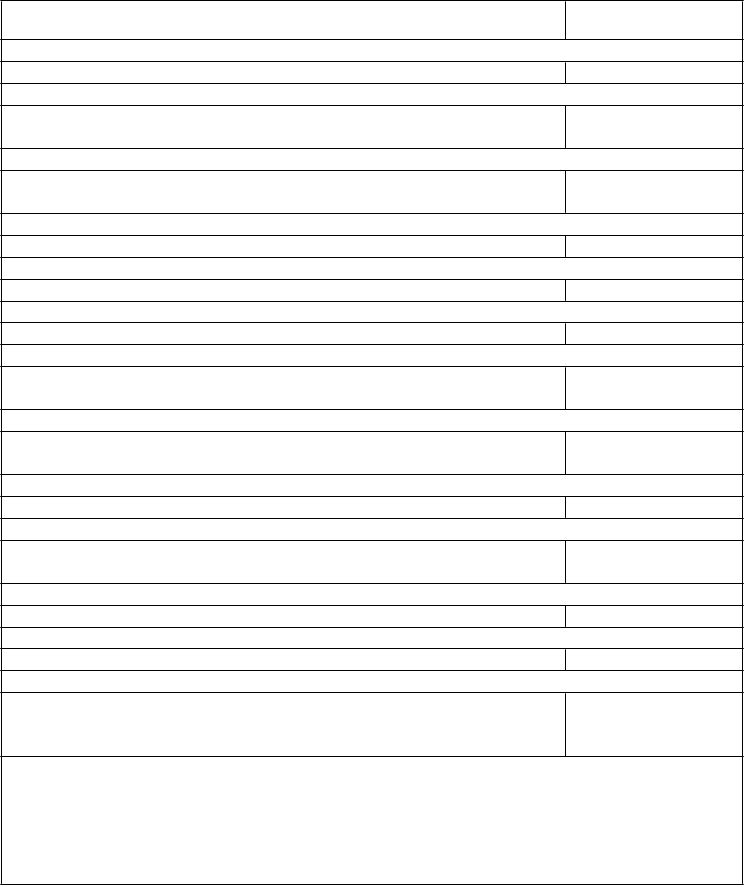
Nauzea, hnačka, vracanie, zápcha, stomatitída, bolesť brucha Veľmi časté
Dysgeúzia, dyspepsia Časté
Poruchy pečene a žlčových ciestHepatotoxicita1 Časté
Poruchy kože a podkožného tkanivaAlopécia, vyrážka2, pruritus Veľmi časté
Erytém Časté
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaBolesť chrbta Veľmi časté
Celkové poruchy a reakcie v mieste podaniaÚnava, periférny edém, asténia, pyrexia Veľmi časté
Laboratórne a funkčné vyšetreniaAbnormálne hodnoty testov funkcie pečene3 Veľmi časté
Zvýšená hladina kreatinínu v krvi, zníženie hmotnosti, predĺženie QT na
elektrokardiograme Časté
1Hepatotoxicita: hepatocelulárne poškodenie, poškodenie pečene spôsobené liekom, hepatotoxicita, zlyhanie pečene (jediný prípad bez úmrtia), autoimunitná hepatitída (jediný
prípad).
2Vyrážka: vyrážka, makulopapulárna vyrážka.
3Abnormálne hodnoty testov funkcie pečene: zvýšená hladina ALT, zvýšená hladina AST,
zvýšená hladina bilirubínu v krvi.
PopisvybranýchnežiaducichliekovýchreakciíNeutropéniaNeutropénia bola najčastejšie hlásenou nežiaducou liekovou reakciou (74,3 %) a pokles počtu neutrofilov stupňa 3 alebo 4 (na základe laboratórnych nálezov) bol hlásený u 59,6 % pacientok
dostávajúcich Kisqali a letrozol v štúdii fázy III.
U pacientok, ktoré mali neutropéniu stupňa 2, 3 alebo 4, bol medián do nástupu 16 dní u tých pacientok, u ktorých sa udalosť vyskytla. Medián času do ústupu stupňa ≥3 (na normalizáciu alebo stupeň <3) bol 15 dní v skupine liečenej ribociklibom a letrozolom po prerušení liečby a/alebo znížení dávky a/alebo ukončení liečby. Febrilná neutropénia bola hlásená približne u 1,5 % pacientok vystavených lieku Kisqali v štúdii fázy III. Pacientky majú byť poučené, aby ihneď hlásili akúkoľvek horúčku.
Neutropénia bola na základe závažnosti kontrolovaná laboratórnym sledovaním, prerušením liečby a/alebo úpravou dávky. Ukončenie liečby v dôsledku neutropénie bolo nízke (0,9 %) (pozri časti 4.2 a 4.4).
Hepatobiliárna toxicitaV klinickej štúdii fázy III sa vyskytli udalosti hepatobiliárnej toxicity u vyššieho podielu pacientok
v ramene s ribociklibom a letrozolom v porovnaní s ramenom placebo a letrozol (24,0 % oproti
13,6 %, v uvedenom poradí), s väčším počtom nežiaducich udalostí stupňa 3/4 hlásených u pacientok
liečených ribociklibom a letrozolom (11,4 % oproti 3,6 %, v uvedenom poradí). Pozorované bolo
zvýšenie hladín transamináz. V ramenách s ribociklibom a placebom boli hlásené zvýšenia hladín
ALT stupňa 3 alebo 4 (10,2 % oproti 1,2 %) a AST (6,9 % oproti 1,5 %) v uvedenom poradí.
U 4 (1,2 %) pacientok sa vyskytli súbežné zvýšenia hladín ALT alebo AST väčšie ako trojnásobok
hornej hranice normálu a celkového bilirubínu väčšie ako dvojnásobok hornej hranice normálu,
s normálnou alkalickou fosfatázou a v neprítomnosti cholestázy, pričom u všetkých pacientok sa
hladiny upravili do normálu v priebehu 154 dní po ukončení liečby liekom Kisqali.
Prerušenie dávkovania a/alebo úprava dávky v dôsledku udalostí hepatobiliárnej toxicity boli hlásené u 8,4 % pacientok liečených ribociklibom a letrozolom, hlavne kvôli zvýšeniu hladiny ALT (5,7 %) a/alebo zvýšeniu hladiny AST (4,5 %). K ukončeniu liečby liekom Kisqali a letrozolom kvôli abnormálnym hodnotám testov funkcie pečene alebo hepatotoxicite došlo u 3,0 % a 0,6 % pacientok v uvedenom poradí (pozri časti 4.2 a 4.4).
V klinickej štúdii fázy III a štúdii fázy Ib v liečbe ribociklibom a letrozolom sa vyskytlo 83,8 % (31/37)
udalostí zvýšenia hladín ALT alebo AST stupňa 3 alebo 4 v priebehu prvých 6 mesiacov liečby.
U pacientok, ktoré mali zvýšenie hladín ALT/AST stupňa 3 alebo 4, bol medián času do nástupu
57 dní v skupine liečenej ribociklibom a letrozolom. Medián času do ústupu (na normalizáciu alebo stupeň ≤ 2) bol 24 dní v skupine liečenej ribociklibom a letrozolom.
Predĺže ni e QTV klinickej štúdii fázy III malo 7,5 % pacientok v ramene s ribociklibom a letrozolom a 2,4 %
v ramene s placebom a letrozolom najmenej jednu udalosť predĺženia intervalu QT (vrátane predĺženia
QT na elektrokardiograme a synkopy). Preskúmanie EKG údajov (priemer troch vyšetrení) ukázalo, že
1 pacientka (0,3 %) mala hodnotu QTcF >500 ms po východiskovej hodnote a 9 pacientok (2,7 %)
malo >60 ms zvýšenie oproti východiskovým intervalom QTcF. Neboli žiadne hlásenia
torsade de pointes. Prerušenia liečby/úpravy dávky z dôvodu predĺženia QT na elektrokardiograme a synkopy
boli hlásené u 0,9 % pacientok liečených ribociklibom a letrozolom.
Centrálna analýza EKG údajov (priemer troch vyšetrení) ukázala 11 pacientok (3,3 %) a 1 pacientku (0,3 %) s aspoň jednou hodnotou QTcF >480 ms po východiskovej hodnote pre rameno s ribociklibom a letrozolom a rameno s placebom a letrozolom v uvedenom poradí. U pacientok, ktoré mali
predĺženie QTcF >480 ms, bol medián času do nástupu 15 dní a tieto zmeny boli reverzibilné pri
prerušení liečby a/alebo znížení dávky (pozri časti 4.2, 4.4 a 5.2).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Nie sú známe žiadne prípady predávkovania liekom Kisqali. V prípade predávkovania sa môžu vyskytnúť symptómy ako nauzea a vracanie. Okrem toho sa môžu vyskytnúť hematologická toxicita (napr. neutropénia, trombocytopénia) a prípadné predĺženie QTc. Vo všetkých prípadoch predávkovania sa má začať podľa potreby všeobecná podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE42
Mechanizmus účinku
Ribociklib je selektívnym inhibítorom cyklín-závislej kinázy (cyclin-dependent kinase, CDK) 4 a 6, čo
vedie u 50 % inhibícií (IC50) k hodnotám 0,01 μM (4,3 ng/ml) a 0,039 μM (16,9 ng/ml)
v biochemických rozboroch, v uvedenom poradí. Tieto kinázy sú aktivované po väzbe na D-cyklíny
a zohrávajú kľúčovú úlohu v signálnych dráhach, ktoré vedú k progresii bunkového cyklu a bunkovej proliferácii. Komplex cyklínu D-CDK4/6 reguluje priebeh bunkového cyklu prostredníctvom
fosforylácie proteínu retinoblastómu (pRb).
V in vitro štúdiách ribociklib znížil fosforyláciu pRb, čo viedlo k zastaveniu bunkového cyklu vo fáze G1, a znížil bunkovú proliferáciu v bunkových líniách karcinómu prsníka. In vivo liečba ribociklibom v monoterapii viedla k regresii tumoru, ktorá korelovala s inhibíciou fosforylácie pRb.
V in vivo štúdiách s použitím xenograftického modelu získaného od pacientky s karcinómom prsníka pozitívneho na estrogén viedlo použitie kombinácie ribociklibu a antiestrogénov (t.j. letrozol)
k inhibícii rastu superiórneho tumoru s udržiavaním regresie tumoru a oneskoreniu opätovného rastu
tumoru po ukončení liečby v porovnaní s podávaním každej látky samostatne.
Pri testovaní v paneli bunkových línií karcinómu prsníka so známym stavom ER ribociklib preukázal,
že je účinnejší v ER+ bunkových líniách karcinómu prsníka než v líniách ER-.
Elektrofyziológia srdca
Po podaní jednorazovej dávky a v rovnovážnom stave boli pre vyhodnotenie účinku ribociklibu na interval QTc u pacientok s pokročilým karcinómom urobené sériové, trojnásobné EKG vyšetrenia. Do
farmakokineticko-farmakodynamickej analýzy bolo zaradených celkovo 267 pacientok liečených ribociklibom v dávkach v rozmedzí od 50 do 1 200 mg, vrátane 193 pacientok liečených ribociklibom v dávke 600 mg. Analýza naznačila, že ribociklib spôsobuje zvýšenie intervalu QTcF závislé na
koncentrácii. Odhadovaná priemerná zmena východiskovej hodnoty QTcF po podaní odporúčanej
600 mg dávky bola 22,87 ms (90% IS: 21,6; 24,1) pri pozorovanej strednej hodnote Cmax
v rovnovážnom stave Cmax (2 237 ng/ml) (pozri časť 4.4).
Klinická účinnosťabezpečnosť
Št údi a CLEE011A2301 (MONALEESA-2)
Kisqali sa hodnotil v randomizovanej, dvojito zaslepenej, placebom kontrolovanej, multicentrickej
klinickej štúdii fázy III v liečbe postmenopauzálnych žien s pokročilým karcinómom prsníka s pozitivitou hormonálnych receptorov, s negativitou HER2, ktoré nedostali žiadnu predchádzajúcu
terapiu pri pokročilom ochorení, v kombinácii s letrozolom oproti samotnému letrozolu.
Celkovo 668 pacientok bolo randomizovaných v pomere 1:1 a dostávalo buď Kisqali v dávke 600 mg a letrozol (n=334) alebo placebo a letrozol (n=334), stratifikovaných podľa prítomnosti metastáz
v pečeni a/alebo pľúcach (áno [n=292 (44 %)]) oproti nie [n=376 (56 %)]). Demografické
a východiskové charakteristiky ochorenia boli vyrovnané a porovnateľné medzi ramenami štúdie.
Kisqali sa podával perorálne v dávke 600 mg denne po dobu 21 po sebe nasledujúcich dní, po ktorých nasledovalo 7 dní bez liečby v kombinácii s letrozolom v dávke 2,5 mg raz denne po dobu 28 dní. Pacientky nesmeli prejsť z placeba na Kisqali počas štúdie alebo po progresii ochorenia.
Pacientky zaradené do tejto štúdie mali medián veku 62 rokov (rozmedzie 23 až 91). 44,2 % pacientok bolo starších ako 65 rokov vrátane 69 pacientok starších ako 75 rokov. Pacientky zahŕňali belošky (82,2 %), aziatky (7,6 %) a černošky (2,5 %). Všetky pacientky mali výkonnostný stav podľa ECOG 0 alebo 1. 43,7 % pacientok dostávalo chemoterapiu v neoadjuvantnom alebo adjuvantnom nastavení
a 52,4 % dostávalo antihormonálnu terapiu v neoadjuvantnom alebo adjuvantnom nastavení pred
vstupom do štúdie. 34,1 % pacientok bolo de novo. 20,7 % pacientok malo iba ochorenie kostí
a 59,0 % pacientok malo viscerálne ochorenie. Pacientky s predchádzajúcou (neo)adjuvantnou liečbou s anastrozolom alebo letrozolom museli ukončiť túto liečbu najneskoršie 12 mesiacov pred randomizáciou.
Primárny cieľ štúdie bol splnený v čase plánovanej predbežnej analýzy uskutočnenej po zaznamenaní
80 % cieľových udalostí prežívania bez progresie (progression-free survival, PFS) za použitia kritérií hodnotenia odpovede solídnych tumorov (Response Evaluation Criteria in Solid Tumors, RECIST
v1.1), na základe hodnotenia skúšajúceho v celej populácii (všetky randomizované pacientky)
a zaslepeného nezávislého centrálneho rádiologického hodnotenia.
Výsledky účinnosti preukázali v analýze celého súboru štatisticky signifikantné zlepšenie PFS u pacientok, ktorým boli podávané Kisqali a letrozol, v porovnaní s pacientkami, ktorým boli podávané placebo a letrozol (pomer rizika 0,556; 95% IS: 0,429; 0,720, p-hodnota jednostranného stratifikovaného log-rank testu 0,00000329), s klinicky významným liečebným účinkom.
Údaje celkového stavu zdravia/QoL nepreukázali žiadny relevantný rozdiel medzi ramenom s
liekom Kisqali a letrozolom a ramenom s placebom a letrozolom.
Novšia aktualizácia údajov účinnosti (ukončenie 2. januára 2017) je uvedená v tabuľkách 7 a 8. Medián PFS bol 25,3 mesiacov (95% IS: 23,0; 30,3) u pacientok liečených ribociklibom a letrozolom
a 16,0 mesiacov (95% IS: 13,4; 18,2) u pacientok, ktorým boli podávané placebo a letrozol. U 54,7 %
pacientok dostávajúcich ribociklib a letrozol sa odhadovalo, že budú PFS v 24. mesiaci v porovnaní s 35,9 % v ramene s placebom a letrozolom.
Medzi ramenom s liekom Kisqali a letrozolom a ramenom s placebom a letrozolom nebol žiadny štatisticky signifikantný rozdiel v celkovom prežívaní (overall survival, OS) (HR 0,746 [95% IS 0,517;
1,078]). Údaje o OS sú predčasné.
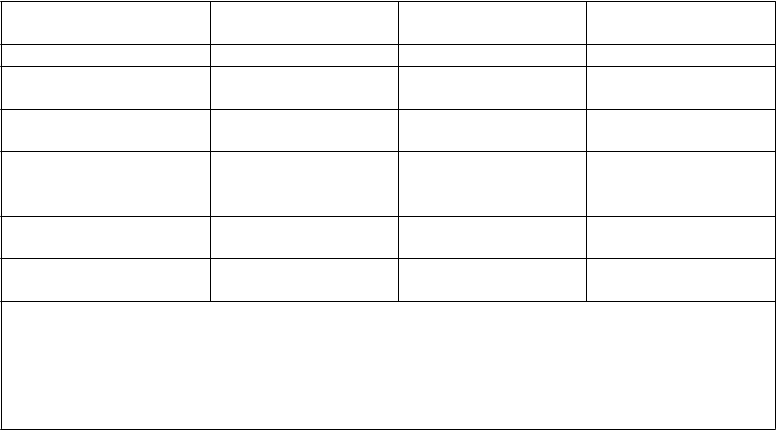
Tabuľka 7 Výsledky účinnosti MONALEESA-2– výsledky primárnej účinnosti (PFS) založené
na rádiologickom hodnotení skúšajúcim (ukončenie 2. januára 2017)
Aktualizovaná analýza (ukončenie 2. januára 2017)
P
r
ežívanie bez progresie
'
K
i
sqali a letrozol
 N=
334
Placebo a letrozol
N=
334
N=
334
Placebo a letrozol
N=
334
Medián PFS [mesiace] (95% IS) 25,3 (23,0-30,3) 16,0 (13,4-18,2)
Stupeň rizika (95% IS) 0,568 (0,457-0,704)
p-hodnotaa 9,63×10-8
IS=interval spoľahlivosti; N=počet pacientok;
ap-hodnota je získaná z jednostranného stratifikovaného log-rank testu.

 O
brázok 1 Kaplanova-Meierova krivka PFS na základe hodnotenia skúšajúcim –
MONALEESA-2 (analýza celého súboru ukončená 2. januára 2017)
O
brázok 1 Kaplanova-Meierova krivka PFS na základe hodnotenia skúšajúcim –
MONALEESA-2 (analýza celého súboru ukončená 2. januára 2017)
100
80
60 Cenzorované časy
ribociklib (N=334)

placebo (N=334)
40
Počet udalostí - ribociklib: 140, placebo: 205
Miera rizika = 0,568; 95% IS [0,475; 0,704]
20
Kaplanov-Meierov medián - ribociklib: 25,3 mesiacov; placebo: 16,0 mesiacov
Log rank p-hodnota = 9,63*10^(-8)
0
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34
Čas (mesiace)
Počet pacientov s pretrvávajúcim rizikom
Čas 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34
ribociklib 334 294 277 257 240 227 207 196 188 176 164 132 97 46 17 11 1 0
placebo 334 279 265 239 219 196 179 156 138 124 110 93 63 34 10 7 2 0
Uskutočnila sa séria analýz s vopred špecifikovanou podskupinou PFS na základe prognostických
faktorov a východiskových charakteristík za účelom skúmania vnútornej konzistencie liečebného účinku. Zníženie rizika progresie ochorenia alebo úmrtia v prospech ramena s ribociklibom
a letrozolom sa pozorovalo vo všetkých individuálnych pacientskych podskupinách z hľadiska veku,
rasy, predchádzajúcej adjuvantnej alebo neoadjuvantnej chemoterapie alebo hormonálnej terapie, ochorením pečene a/alebo pľúc a len metastatického ochorenia kostí. Toto bolo evidentné u pacientok
s metastázami v pečeni a/alebo pľúcach (HR 0,561 [95% IS: 0,424; 0,743], s mediánom prežívania bez
progresie [mPFS] 24,8 mesiacov pre ribociklib s letrozolom oproti 13,4 mesiacom pre samotný letrozol alebo bez metastáz v pečeni a/alebo pľúcach (HR 0,597 [95% IS: 0,426; 0,837], mPFS
27,6 mesiacov oproti 18,2 mesiacom).
Aktualizované výsledky pre celkovú odpoveď a mieru klinického prínosu sú uvedené v tabuľke 8.
T
abuľka 8 Výsledky účinnosti MONALEESA-2 (ORR, CBR) na základe hodnotenia
skúšajúcim (ukončenie 2. januára 2017)
A
nalýza Kisqali + letrozol
(
%
, 95% IS)
Placebo + letrozol
(
%
, 95% IS)
p-hodnota
c
A
nalýza celého súboru N=334 N=334
Miera celkovej odpovede
a
Miera klinického prínosu
b
P
acientky
s merateľným ochorením Miera celkovej odpovede
a
Miera klinického prínosu
b
42,5 (37,2; 47,8) 28,7 (23,9; 33,6) 9,18 × 10-5
79,9 (75,6; 84,2) 73,1 (68,3; 77,8) 0,018
N=257 N=245
54,5 (48,4; 60,6) 38,8 (32,7; 44,9) 2,54 × 10-4
80,2 (75,3; 85,0) 71,8 (66,2; 77,5) 0,018
a ORR (
overall response rate, ORR): Miera celkovej odpovede = podiel pacientok s úplnou
odpoveďou + čiastočnou odpoveďou
b CBR (
clinical benefit rate, CBR): Miera klinického prínosu = podiel pacientok s úplnou odpoveďou
+ čiastočnou odpoveďou (+ stabilné ochorenie alebo neúplná odpoveď/neprogresívne ochorenie
≥24 týždňov)
c p-hodnoty sú získané z jednostranného Cochranovho-Mantelovho-Haenszelovho chí-kvadrát testu
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Kisqali
vo všetkých podskupinách pediatrickej populácie pre karcinóm prsníka (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiFarmakokinetika ribociklibu sa skúmala u pacientok s pokročilým karcinómom po perorálnych denných dávkach 50 mg až 1 200 mg. Zdraví jedinci dostali jednorazové perorálne dávky v rozmedzí od 400 mg do 600 mg alebo opakované denné dávky (8 dní) 400 mg.
AbsorpciaAbsolútna biologická dostupnosť ribociklibu nie je známa.
Čas pre dosiahnutie Cmax (Tmax) po perorálnom podaní ribociklibu bol v rozmedzí 1 a 4 hodín. Ribociklib vykazoval o niečo viac ako dávke úmerné zvýšenie expozície (Cmax a AUC) v celom skúšanom rozmedzí dávkovania (50 až 1 200 mg). Po opakovanom podávaní raz denne sa rovnovážny stav dosiahol zvyčajne po 8 dňoch a ribociklib sa akumuloval s geometrickou strednou hodnotou pomeru akumulácie 2,51 (rozsah: 0,97 až 6,40).
Vplyv jedlaV porovnaní s podaním nalačno nemalo perorálne podanie jednorazovej 600 mg dávky filmom obalených tabliet ribociklibu s vysokokalorickým jedlom s vysokým obsahom tuku žiadny účinok na
rýchlosť a rozsah absorpcie ribociklibu.
D
i
stribúcia
Väzba ribociklibu na plazmatické proteíny u ľudí in vitro bola približne 70 % a bola nezávislá od
koncentrácie (10 až 10 000 ng/ml). Ribociklib bol rovnomerne distribuovaný medzi červené krvinky
a plazmu s priemerným in vivo pomerom krv-plazma 1,04. Zdanlivý objem distribúcie v rovnovážnom stave (Vss/F) bol na základe populačnej farmakokinetickej analýzy 1 090 l.
Biotransformácia
In vitro a in vivo štúdie ukázali, že ribociklib je primárne eliminovaný u ľudí hepatálnym
metabolizmom hlavne prostredníctvom CYP3A4. Po perorálnom podaní jednorazovej 600 mg dávky
[14C] ribociklibu ľuďom zahŕňali primárne metabolické dráhy ribociklibu oxidáciu (dealkylácia, C a/alebo N-oxygenácia, oxidácia (-2H)) a ich kombinácie. Konjugáty ribociklibu fázy II metabolitov fázy I zahŕňali N-acetyláciu, sulfáciu, konjugáciu cystínu, glykozyláciu a glukuronidáciu. Ribociklib
bol hlavnou od lieku odvodenou zlúčeninou cirkulujúcou v plazme. Hlavné cirkulujúce metabolity
zahŕňali metabolit M13 (CCI284, N-hydroxylácia), M4 (LEQ803, N-demetylácia) a M1 (sekundárny glukuronid). Klinická aktivita (farmakologická a bezpečnostná) ribociklibu bola spôsobená
predovšetkým materskou zlúčeninou so zanedbateľným príspevkom cirkulujúcich metabolitov.
Ribociklib bol extenzívne metabolizovaný, nezmenená forma predstavuje 17,3 % a 12,1 % dávky
v stolici a moči, v uvedenom poradí. Metabolit LEQ803 bol signifikantným metabolitom v exkrétoch
a predstavoval približne 13,9 % a 3,74 % podanej dávky v stolici a moči, v uvedenom poradí. Početné
iné metabolity boli zistené v stolici a moči v menších množstvách (≤2,78 % podanej dávky).
Eliminácia
Geometrický priemer plazmatických účinných polčasov (na základe pomeru akumulácie) bol
32,0 hodín (63 % CV) a geometrický priemer zdanlivého perorálneho klírensu (CL/F) bol 25,5 l/hod
(66 % CV) v rovnovážnom stave pri dávke 600 mg u pacientok s pokročilým karcinómom. Geometrický priemer zdanlivého plazmatického terminálneho polčasu (T1/2) ribociklibu bol
v rozmedzí od 29,7 do 54,7 hodín a geometrický priemer CL/F ribociklibu bol v rozmedzí od 39,9 do
77,5 l/hod pri dávke 600 mg vo všetkých štúdiách u zdravých jedincov.
Ribociklib a jeho metabolity sa vylučujú hlavne stolicou, v malej miere renálnou cestou. U 6 zdravých
mužských jedincov bolo po peorálnom podaní jednorazovej dávky [14C] ribociklibu vylúčených
91,7 % celkovej podanej rádioaktívnej dávky v priebehu 22 dní; stolica bola hlavnou cestou vylučovania (69,1 %), s 22,6 % dávky vylúčenej močom.
Linearita/nelinearita
Ribociklib vykazoval o niečo viac ako dávke úmerné zvýšenie expozície (Cmax a AUC) v celom
skúšanom rozmedzí dávkovania 50 mg až 1 200 mg po jednorazovej dávke a opakovaných dávkach.
Táto analýza je obmedzená malou veľkosťou vzoriek pre väčšinu dávkovacích kohort s väčšinou
údajov pochádzajúcich zo skupiny so 600 mg dávkou.
Osobitné skupiny pacientov
Porucha f unkc ie obl i či ek
Na základe populačnej farmakokinetickej analýzy, ktorá zahŕňala 77 pacientok s normálnou funkciou
obličiek (eGFR ≥90 ml/min/1,73 m2), 76 pacientok s miernou poruchou funkcie obličiek (eGFR 60 až
<90 ml/min/1,73 m2) a 35 pacientok so stredne ťažkou poruchou funkcie obličiek (eGFR 30 až
<60 ml/min/1,73 m2), nemala mierna a stredne ťažká porucha funkcie obličiek žiadny vplyv na expozíciu ribociklibu (pozri časť 4.2). Farmakokinetika ribociklibu u pacientok s ťažkou poruchou
funkcie obličiek sa neskúmala.
Porucha f unkc ie peče ne
Na základe farmakokinetickej štúdie u pacientok s poruchou funkcie pečene nemala mierna porucha funkcie pečene žiadny vplyv na expozíciu ribociklibu (pozri časť 4.2). Priemerná expozícia ribociklibu sa zvýšila menej ako 2-násobne u pacientok so stredne ťažkou (geometrický priemer pomeru
[geometric mean ratio, GMR]: 1,50 pre Cmax; 1,32 pre AUCinf) a ťažkou (GMR: 1,34 pre Cmax; 1,29 pre AUCinf) poruchou funkcie pečene. Na základe populačnej farmakokinetickej analýzy, ktorá zahŕňala 160 pacientok s karcinómom prsníka s normálnou funkciou pečene a 47 pacientok s miernou poruchou funkcie pečene, nemala mierna porucha funkcie pečene žiadny vplyv na expozíciu ribociklibu, čo bolo ďalej podporené zisteniami zo štúdie zameranej na poškodenie funkcie pečene (pozri časť 4.2).
Vplyv veku, hmotnosti, pohlavia a rasy
Populačná farmakokinetická analýza ukázala, že neexistuje žiadny klinicky relevantný vplyv veku, telesnej hmotnosti alebo pohlavia na systémovú expozíciu ribociklibu, ktoré by vyžadovali úpravu
dávky. Údaje o rozdieloch vo farmakokinetike kvôli rase sú príliš obmedzené na vyvodenie záverov.
In vitro údaje o interakcii
Úči nok ribociklibu na enzýmy cytochrómu P450
In vitro je ribociklib v klinicky relevantných koncentráciách reverzibilným inhibítorom CYP1A2, CYP2E1 a CYP3A4/5 a časovo závislým inhibítorom CYP3A4/5. In vitro hodnotenia naznačili, že
ribociklib nemá v klinicky relevantných koncentráciách žiadny potenciál inhibovať aktivitu CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP2D6. V klinicky relevantných koncentráciách nemá ribociklib žiadny potenciál pre časovo závislú inhibíciu CYP1A2, CYP2C9 a CYP2D6.
In vitro údaje naznačujú, že ribociklib nemá žiadny potenciál indukovať enzýmy UGT alebo CYP enzýmy CYP2C9, CYP2C19 a CYP3A4 prostredníctvom PXR. Preto je nepravdepodobné, že Kisqali ovplyvňuje substráty týchto enzýmov. Pre nedostačujúce in vitro údaje nemožno vylúčiť indukčný potenciál ribociklibu na CYP2B6 prostredníctvom CAR.
Úči nok t ransport érov na ribociklib
In vitro je ribociklib substrátom P-gp, podľa údajov z hmotnostnej bilancie inhibície P-gp alebo BCRP
je však vplyv na expozíciu ribociklibu v terapeutických dávkach nepravdepodobný. In vitro ribociklib nie je substrátom hepatálnych transportérov OATP1B1, OATP1B3 alebo OCT-1.
Úči nok ribociklibu na transportéry
In vitro hodnotenia naznačili, že ribociklib má potenciál inhibovať aktivitu transportérov liekov P-gp, BCRP, OATP1B1/B3, OCT1, MATE1 a BSEP. In vitro ribociklib nie je v klinicky relevantných
koncentráciách inhibítorom OAT1, OAT3 alebo MRP2.
5.3 Predklinické údaje o bezpečnosti
Farmakologické štúdiebezpečnosti
In vivo štúdie kardiologickej bezpečnosti na psoch preukázali predĺženie intervalu QTc súvisiace s
dávkou a koncentráciou pri očakávanej expozícii, ktorá sa dosiahne u pacientov po odporúčanej dávke
600 mg. Existuje tiež možnosť indukovanej incidencie predčasných ventrikulárnych kontrakcií
(premature ventricular contractions, PVC) pri zvýšených expozíciách (približne 5-násobok
očakávanej klinickej Cmax).
T
oxicita po opakovanom podávaní
Štúdie toxicity po opakovanom podávaní (schéma 3 týždne liečba/1 týždeň bez liečby) v trvaní až do
27 týždňov na potkanoch a až do 39 týždňov na psoch preukázali ako primárny cieľový orgán toxicity ribociklibu hepatobiliárny systém (proliferatívne zmeny, cholestáza, žlčníkové kamene podobné piesku a zhustnutá žlč). Medzi cieľové orgány spojené s farmakologickým účinkom ribociklibu
v štúdiách po opakovanom podávaní patria kostná dreň (hypocelularita), lymfatický systém
(lymfoidná deplécia), črevná sliznica (atrofia), koža (atrofia), kosti (znížená tvorba kostí), obličky
(súbežná degenerácia a regenerácia epitelových buniek tubulu) a semenníky (atrofia). Okrem atrofických zmien pozorovaných v semenníkoch, ktoré preukázali tendenciu k reverzibilite, boli všetky ostatné zmeny úplne reverzibilné po období 4 týždňov bez liečby. Expozícia ribociklibu
v štúdiách toxicity na zvieratách bola všeobecne menšia alebo rovná expozícii pozorovanej
u pacientok dostávajúcich viacnásobné dávky 600 mg/deň (na základe AUC).
Reprodukčná toxicity/fertilita
Ribociklib preukázal fetotoxicitu a teratogenitu pri dávkach, ktoré nepreukázali toxicitu u matiek
potkanov alebo králikov. U potkanov bolo zaznamenané zníženie hmotnosti plodov sprevádzané skeletálnymi zmenami považovanými za prechodné a/alebo súvisiace s nižšou hmotnosťou plodov.
U králikov boli nežiaduce účinky na embryofetálny vývoj, ako dokazuje zvýšená incidencia fetálnych
abnormalít (malformácie a externé, viscerálne a skeletálne varianty) a rast plodu (nižšia hmotnosť plodov). Tieto zistenia zahŕňali zmenšené/malé pľúcne laloky a ďalšiu cievu na aortálnom oblúku a diafragmatickú herniu, chýbajúci vedľajší lalok alebo (čiastočne) spojené pľúcne laloky
a zmenšený/malý vedľajší pľúcny lalok (30 a 60 mg/kg), extra/rudimentárne trináste rebrá
a znetvorená jazylka a znížený počet článkov na palci. Neexistujú žiadne dôkazy o embryofetálnej mortalite.
Ribociklib sa nehodnotil v štúdiách fertility. Avšak štúdie chronickej toxicity na potkanoch a psoch odhalili atrofické zmeny semenníkov po histopatologickom hodnotení. Tieto účinky môžu byť spojené s priamymi antiproliferatívnymi účinkami na testikulárne zárodočné bunky, čo vedie k atrofii semenotvorných kanálikov.
Ribociklib a jeho metabolity prechádzali ľahko do mlieka potkanov. Expozícia ribociklibu bola vyššia
v mlieku ako v plazme.
Genotoxicita
V štúdiách genotoxicity v bakteriálnych systémoch in vitro a v cicavčích systémoch in vitro a in vivo
s metabolickou aktiváciou alebo bez nej sa nenašiel žiadny dôkaz genotoxického potenciálu ribociklibu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
mikrokryštalická celulóza
krospovidón typ A
čiastočne substituovaná hydroxypropylcelulóza
stearan horečnatý
koloidný oxid kremičitý bezvodý
Filmotvorná vrstva
čierny oxid železitý (E172)
červený oxid železitý (E172)
sójový lecitín (E322)
polyvinylalkohol (čiastočne hydrolyzovaný)
mastenec
oxid titaničitý (E171)
xantánová guma
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PCTFE (polyvinylchlorid/polychlórtrifluóretylén) alebo PA/Al/PVC (polyamid/hliník/polyvinylchlorid) blistre obsahujúce 14 alebo 21 filmom obalených tabliet.
Jednotlivé balenia obsahujúce 21, 42 alebo 63 filmom obalených tabliet a multibalenia obsahujúce 63 (3 balenia po 21), 126 (3 balenia po 42) alebo 189 (3 balenia po 63) filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLA
EU/1/17/1221/001-012
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.