
detí vo veku do 18 rokov nebola stanovená. Nie sú k dispozícii žiadne údaje.
Spôsobpodávania
Subkutánne použitie.
Celý obsah (1,14 ml) naplnenej injekčnej striekačky/naplneného injekčného pera sa má podať vo forme subkutánnej injekcie. Miesta vpichu injekcie (brucho, stehno a rameno) sa majú po každej injekcii meniť. Kevzara sa nemá pichať do kože, ktorá je citlivá, poškodená alebo má modriny alebo jazvy.
Po náležitom posúdení odborným zdravotníckym pracovníkom, si môže pacient vpichnúť injekciu Kevzary sám alebo mu ju môže podať ošetrovateľ. Pred použitím majú byť pacienti a/alebo ošetrovatelia primerane zaškolení o príprave a podávaní Kevzary.
Ďalšie podrobnosti o podávaní tohto lieku, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Aktívne, závažné infekcie (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
SledovateľnosťKevzary
V súlade so zlepšením sledovateľnosti biologických liekov sa má obchodný názov a číslo šarže podávaného lieku jasne zaznamenať.
Závažnéinfekcie
Pacienti majú byť starostlivo monitorovaní na rozvoj prejavov a symptómov infekcie počas liečby
Kevzarou (pozri časti 4.2 a 4.8). Keďže vo všeobecnosti existuje vysoký výskyt infekcií u staršej populácie, pri liečbe starších pacientov je potrebná opatrnosť.
Kevzara sa nemá podávať pacientom s aktívnou infekciou, vrátane lokálnych infekcií. Pred začatím liečby Kevzarou zvážte riziká a výhody liečby u pacientov, ktorí majú:
· chronickú alebo opakovanú infekciu;
· v anamnéze závažné alebo oportúnne infekcie;
· HIV infekciu;
· základné ochorenie, ktoré ich môže robiť náchylnými na infekciu;
· boli vystavení tuberkulóze alebo
· žili alebo cestovali do oblastí s endemickou tuberkulózou alebo endemickou mykózou.
Liečba Kevzarou sa má prerušiť, ak sa u pacienta vyskytne závažná alebo oportúnna infekcia. Pacient, u ktorého sa vyskytne infekcia počas liečby Kevzarou, má tiež podstúpiť okamžité
a kompletné diagnostické vyšetrenie vhodné pre pacientov s oslabeným imunitným systémom; má sa iniciovať vhodná antimikrobiálna liečba a pacient má byť starostlivo monitorovaný.
U pacientov liečených imunosupresívami, vrátane Kevzary pri RA boli hlásené závažné a niekedy fatálne infekcie spôsobené baktériami, mykobaktériami, invazívnymi hubami, vírusovými alebo inými oportúnnymi patogénmi. Najčastejšími pozorovanými závažnými infekciami súvisiacimi s Kevzarou boli pneumónia a celulitída (pozri časť 4.8). V súvislosti s Kevzarou boli medzi hlásenými oportúnnymi infekciami tuberkulóza, kandidóza a pneumocystitída. V ojedinelých prípadoch, skôr s rozšírenými ako lokalizovanými infekciami, boli pozorované u pacientov často súbežne liečených imunosupresívami, napr. metotrexátom (MTX) alebo kortikoidmi, ktoré pri prebiehajúcej RA môžu zvyšovať náchylnosť k infekciám.
Tuberkulóza
Pred začatím liečby s Kevzarou sa majú u pacientov zhodnotiť rizikové faktory tuberkulózy a majú sa vyšetriť na latentnú infekciu. Pacienti s latentnou alebo aktívnou tuberkulózou sa majú pred začatím
liečby s Kevzarou liečiť štandardnou antimykobakteriálnou liečbou. Pred začatím liečby s Kevzarou
zvážte antituberkulóznu liečbu u pacientov s anamnézou latentnej alebo aktívnej tuberkulózy,
u ktorých nemožno zaručiť adekvátny priebeh liečby. Pri uvažovaní o antituberkulóznej liečbe, je vhodná konzultácia s lekárom, ktorý má skúsenosti s tuberkulózou.
U pacientov sa má starostlivo monitorovať rozvoj prejavov a príznakov tuberkulózy vrátane tých pacientov, ktorí pred začatím liečby mali negatívny test na latentnú tuberkulóznu infekciu.
Reaktivácia vírusov
V súvislosti s imunosupresívnou biologickou liečbou bola hlásená reaktivácia vírusov. V klinických štúdiach s Kevzarou boli pozorované prípady herpes zoster. V klinických štúdiach neboli hlásené žiadne prípady hepatitídy B; napriek tomu pacienti s rizikom reaktivácie boli vylúčení.
Laboratórneparametre
Počet neutrofilov
Liečba s Kevzarou bola spojená s vyšším výskytom zníženého absolútneho počtu neutrofilov (absolute neutrophil count, ANC). Znížený ANC nebol spojený s častejším výskytom infekcií, vrátane
závažných infekcií.
· Začatie liečby s Kevzarou sa neodporúča u pacientov s nízkym počtom neutrofilov, t.j. ANC
menší ako 2 x 109/l. U pacientov, u ktorých ANC klesol na menej ako 0,5 x 109/l, liečba s Kevzarou sa má ukončiť.
· Má sa sledovať počet neutrofilov 4 až 8 týždňov od začatia liečby a potom podľa zhodnotenia klinického stavu. Odporúčané úpravy dávky vychádzajú z výsledkov ANC, pozri časť 4.2.
· Vychádzajúc z farmakodynamických zmien ANC, použite pri zvažovaní úpravy dávky výsledky získané na konci dávkovacieho intervalu (pozri časť 5.1).
Počet krvných doštičiek
Liečba s Kevzarou bola spojená so znížením počtu krvných doštičiek v klinických štúdiach. Zníženie počtu krvných doštičiek sa nespájalo s krvácavými príhodami (pozri časť 4.8).
· Začatie liečby s Kevzarou sa neodporúča u pacientov s počtom krvných doštičiek nižším ako
150 x 103/µl. U pacientov, u ktorých klesol počet krvných doštičiek na menej ako 50 x 103/µl, liečba s Kevzarou sa má ukončiť.
· Má sa sledovať počet krvných doštičiek 4 až 8 týždňov po začatí liečby a potom podľa zhodnotenia klinického stavu. Odporúčané úpravy dávky vychádzajú z výsledkov počtu
krvných doštičiek, pozri časť 4.2.
Pečeňové enzýmy
Liečba s Kevzarou bola spojená s vyšším výskytom zvýšených transamináz. Tieto zvýšenia boli prechodné a v klinických štúdiach nevyústili do žiadneho klinicky zjavného poškodenia pečene (pozri
časť 4.8). Zvýšená frekvencia a rozsah týchto zvýšení boli pozorované, keď sa potenciálne
hepatotoxické lieky (napr. MTX) použili v kombinácii s Kevzarou.
Začatie liečby s Kevzarou sa neodporúča u pacientov so zvýšenými transaminázami, ALT alebo AST
väčším ako 1,5 x ULN. U pacientov, u ktorých sa zvýšila ALT na viac ako 5 x ULN, sa má liečba s Kevzarou prerušiť (pozri časť 4.2).
Hladiny ALT a AST sa majú sledovať 4 až 8 týždňov po začatí liečby a potom každé 3 týždne. Keď je to klinicky indikované, zvážte iné vyšetrenia funkcie pečene, ako napr. bilirubín. Odporúčané úpravy dávky vychádzajú z výsledkov zvýšenia transamináz, pozri časť 4.2.
Poruchy lipidov
Hladiny lipidov môžu byť znížené u pacientov s chronickým zápalom. Liečba s Kevzarou bola spojená so zvýšením parametrov lipidov ako napr. LDL cholesterol, HDL cholesterol a/alebo triglyceridy
(pozri časť 4.8).
Parametre lipidov sa majú posúdiť približne 4 až 8 týždňov po začatí liečby s Kevzarou, potom približne v 6 mesačnom intervale.
Pacienti sa majú liečiť podľa klinických odporúčaní pre liečbu hyperlipidémie. Gastrointestinálneperforácie
Z klinických štúdií boli hlásené prípady gastrointestinálnych perforácií, najmä ako komplikácia
divertikulitídy. U pacientov s anamnézou intestinálnej ulcerácie alebo divertikulitídy používajte
Kevzaru s opatrnosťou. Pacienti s prítomnými novovzniknutými brušnými príznakmi, napr. s pretrvávajúcou bolesťou s horúčkou, sa majú urýchlene vyšetriť (pozri časť 4.8).
Malígneochorenia
Liečba imunosupresívami môže vyústiť do zvýšeného rizika malígnych ochorení. Vplyv liečby
s Kevzarou na rozvoj malígnych ochorení nie je známy, ale v klinických štúdiach boli hlásené malígne ochorenia (pozri časť 4.8).
Reakciezprecitlivenosti
V súvislosti s Kevzarou boli hlásené reakcie z precitlivenosti (pozri časť 4.8). Najčastejšími reakciami z precitlivenosti boli vyrážka v mieste podania injekcie, vyrážka a urtikária. Pacientov treba poučiť, aby vyhľadali okamžitú lekársku pomoc, ak sa objavia akékoľvek príznaky reakcie z precitlivenosti. Ak sa objaví anafylaxia alebo iná reakcia z precitlivenosti, podávanie Kevzary sa má okamžite zastaviť. Kevzara sa nemá podávať pacientom so známou precitlivenosťou na sarilumab (pozri časť
4.3).
Poruchafunkciepečene
Liečba s Kevzarou sa neodporúča u pacientov s aktívnym ochorením pečene alebo poruchou funkcie pečene (pozri časti 4.2 a 4.8).
Vakcinácie
Počas liečby s Kevzarou sa vyhnite súbežnému používaniu živých vakcín, rovnako ako aj živých oslabených vakcín, keďže klinická bezpečnosť nebola stanovená. Nie sú k dispozícii žiadne údaje o
sekundárnom prenose infekcie z osôb dostávajúcich živé očkovacie látky na pacientov, ktorým je
podávaná Kevzara. Odporúča sa, aby pred začatím liečby s Kevzarou všetci pacienti absolvovali všetky očkovania v súlade s aktuálnymi odbornými smernicami o imunizácii. Interval medzi očkovaním živými vakcínami a začatím liečby s Kevzarou má byť v súlade s aktuálnymi smernicami o očkovaní týkajúcimi sa imunosupresív (pozri časť 4.5).
K
a
r
diovaskulárne
ri
z
i
k
o
Pacienti s RA majú zvýšené riziko kardiovaskulárnych porúch, a rizikové faktory (napr. hypertenzia, hyperlipidémia) sa majú korigovať vrámci bežnej štandardnej zdravotnej starostlivosti.
4.5 Liekové a iné interakcie
Vystavenie sarilumabu nebolo ovplyvnené súbežným podávaním metotrexátu (MTX) na základe farmakokinetických analýz populácie a porovnaní naprieč štúdiou. Neočakáva sa, že by sa súbežným podávaním sarilumabu zmenilo vystavenie MTX; hoci neboli zozberané žiadne klinické údaje. Kevzara nebola skúšaná v kombinácii s inhibítormi Janus kinázy (JAK), ani s biologickými DMARDs, ako napr. antagonisty tumor nekrotizujúceho faktora (Tumor Necrosis Factor, TNF).
Rôzne in vitro a obmedzené in vivo štúdie u ľudí preukázali, že cytokíny a modulátory cytokínov môžu ovplyvňovať expresiu a aktivitu špecifických enzýmov cytochrómu P450 (CYP) (CYP1A2, CYP2C9, CYP2C19, and CYP3A4) a preto majú potenciál zmeniť farmakokinetiku súčasne užívaných liekov, ktoré sú substrátmi pre tieto enzýmy. Zvýšené hladiny interleukínu-6 (IL-6) môžu potláčať aktivitu CYP, napr. u pacientov s RA, a tým zvyšovať hladiny lieku v porovnaní s pacientmi bez RA. Blokáda IL-6 signalizovaná antagonistami IL-6Rα, napr. sarilumabom, môže zvrátiť inhibičný účinok IL-6 a znovu obnoviť CYP aktivitu, čo vedie k zmeneným koncentráciam liekov.
Modulovanie účinku IL-6 na CYP enzýmy sarilumabom môže byť klinicky relevantné pre substráty CYP s úzkym terapeutickým indexom, pri ktorom sa dávka upravuje individuálne. Po začatí alebo prerušení liečby Kevzarou u pacientov liečených liekmi, ktoré sú substrátmi CYP, sa má sledovať terapeutický účinok (napr. warfarín) alebo koncentrácia lieku (napr. teofylín) a ak je to potrebné, individuálna dávka lieku sa má upraviť.
U pacientov, ktorí začali liečbu s Kevzarou je potrebná opatrnosť počas liečby so substrátmi CYP3A4 (napr. perorálne kontraceptíva alebo statíny), pretože Kevzara môže zvrátiť inhibičný účinok IL-6
a obnoviť aktivitu CYP3A4, čo vedie k poklesu vystavenia a aktivity substrátu CYP3A4 (pozri časť
5.2).
Interakcia sarilumabu s inými substrátmi CYP (CYP2C9, CYP 2C19, CYP2D6) nebola študovaná.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku
Ženy vo fertilnom veku majú používať účinnú antikoncepciu až 3 mesiace po ukončení liečby.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití sarilumabu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity
(pozri časť 5.3).
Kevzara sa nemá používať v tehotenstve, kým si klinický stav ženy nevyžiada liečbu sarilumabom. Dojčenie
Nie je známe, či sa sarilumab vylučuje do ľudského mlieka alebo sa vstrebáva systémovo po požití. Vylučovanie sarilumabu do mlieka nebolo u zvierat skúmané (pozri časť 5.3).
Pretože sa IgG1 vylučuje do ľudského mlieka, rozhodnutie, či ukončiť dojčenie alebo ukončiť liečbu
sarilumabom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne údaje o vplyve sarilumabu na ľudskú plodnosť. Štúdie na zvieratách nepreukázali žiadnu poruchu plodnosti u samcov ani u samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kevzara nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšími nežiaducimi reakciami pozorovanými v klinických štúdiach s Kevzarou bola neutropénia, zvýšené ALT, erytém v mieste podania injekcie, infekcie horných dýchacích ciest
a infekcie močových ciest. Najčastejšími závažnými nežiaducimi účinkami boli infekcie (pozri
časť4.4).
Zoznamnežiaducichúčinkovvtabuľke
Bezpečnosť Kevzary v kombinácii s DMARDs bola hodnotená na základe údajov zo siedmych klinických štúdií, z ktorých dve boli kontrolované placebom, pozostávajúcich z 2887 pacientov (populácia pre hodnotenie dlhodobej bezpečnosti). Z týchto, 2170 pacientov dostávalo Kevzaru aspoň počas 24 týždňov, 1546 aspoň počas 48 týždňov, 1020 aspoň počas 96 týždňov a 624 aspoň počas 144 týždňov.
Frekvencia výskytu nežiaducich účinkov uvedených nižšie je definovaná použitím nasledovných konvencií: veľmi časté (≥ 1/10); časté (≥ 1/100 to < 1/10); menej časté (≥ 1/1,000 to < 1/100); zriedkavé (≥ 1/10,000 to < 1/1,000); veľmi zriedkavé (< 1/10,000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky zoradené podľa klesajúcej závažnosti.
Tabuľka 1: ADRs (adverse drug reactions) v kontrolovaných klinických štúdiach
Trieda orgánových systémov Frekvencia Nežiaduca reakcia
Infekcie a nákazy Časté Infekcia horných dýchacích ciest Infekcia močových ciest Nazofaryngitída
Orálny herpes
Poruchy krvi a lymfatického systému
Veľmi časté Neutropénia

Časté Trombocytopénia
Poruchy metabolizmu a výživy Časté Hypercholesterolémia
Hypertriglyceridémia
Poruchy pečene a žlčových ciest Časté Zvýšené transaminázy
Celkové poruchy a reakcie v
mieste podania
Časté Erytém v mieste podania injekcie
Pruritus v mieste podania injekcie
Popis
vy
braných
nežiaducich
reakcií
I
nfekcie
V placebom kontrolovanej populácii bol výskyt infekcií 84,5; 81,0 a 75,1 prípadov na 100 pacientov ročne, v skupine s 200 mg a 150 mg dávkou Kevzara + DMARDs a placebo + DMARDs, v uvedenom
poradí. Najčastejšie zaznamenanými infekciami (5 % až 7 % pacientov) boli infekcie horných
dýchacích ciest, infekcie močového traktu a nazofaryngitída. Výskyt závažných infekcií bol 4,3; 3,0
a 3,1 prípadov na 100 pacientov ročne, v skupine s 200 mg a 150 mg dávkou Kevzara + DMARDs a placebo + DMARDs, v uvedenom poradí.
V populácii pre hodnotenie dlhodobej bezpečnosti s Kevzarou + DMARDs bol výskyt infekcií 57,3
prípadov na 100 pacientov ročne a 3,4 prípradov závažných infekcií na 100 pacientov ročne.
Najčastejšie pozorované závažné infekcie zahŕňali pneumóniu a celulitídu. Boli hlásené prípady oportúnnej infekcie (pozri časť 4.4).
Celkový výskyt infekcií a závažných infekcií v populácii liečenej monoterapiou s Kevzarou bol konzistentný s výskytom v populácii s Kevzarou + DMARDs.
Gastrointestinálna perforácia
V placebom kontrolovanej populácii sa počas liečby Kevzarou u jedného pacienta vyskytla gastrointestinálna (GI) perforácia (0,11 prípadov na 100 pacientov ročne). V populácii pre hodnotenie
dlhodobej bezpečnosti s Kevzarou + DMARDs bol výskyt GI perforácií 0,14 prípadov na 100
pacientov ročne.
Hlásenia gastrointestinálnych perforácií boli zaznamenané predovšetkým ako komplikácie divertikulitídy, vrátane perforácií dolnej časti gastrointestinálneho traktu a abscesu. Väčšina pacientov, u ktorých sa vyskytli gastrointestinálne perforácie súbežne užívali nesteroidové protizápalové antiflogistiká (NSAIDs), kortikosteroidy alebo metotrexát. Príspevok týchto súbežne užívaných liekov vo vzťahu ku Kevzare pri vzniku gastroinstestinálnych perforácií nie je známy (pozri časť 4.4).
V populácii s monoterapiou Kevzary neboli žiadne hlásenia gastrointestinálnych perforácií.
Reakcie z precitlivenosti
V placebom kontrolovanej populácii bol podiel pacientov, ktorí prerušili liečbu kvôli reakciám
z precitlivenosti vyšší u tých, ktorí boli liečení s Kevzarou (0,9 % v 200 mg skupine, 0,5 % v 150 mg skupine) oproti placebu (0,2 %). Počet prípadov ukončenia liečby z dôvodu precitlivenosti v populácii
pre hodnotenie dlhodobej bezpečnosti s Kevzarou + DMARDs a v populácii s monoterapiou Kevzary
bol konzistentný s populáciou kontrolovanou placebom. V placebom kontrolovanej populácii sa u
0,2 % pacientov liečených Kevzarou 200 mg každé dva týždne + DMARD zaznamenali závažné nežiaduce udalosti reakcií z precitlivenosti, a žiadne v skupine Kevzary 150 mg každé dva týždne +
DMARD.
Reakcie v mieste podania injekcie
V placebom kontrolovanej populácii boli hásené reakcie v mieste podania injekcie u 9,5 % pacientov dostávajúcich Kevzaru 200 mg, u 8 % pacientov dostávajúcich Kevzaru 150 mg a u 1,4 % pacientov
dostávajúcich placebo. Tieto reakcie v mieste podania injekcie (vrátane erytému a pruritu) boli pre
väčšinu týchto pacientov miernej závažnosti. Dvaja pacienti na Kevzare (0,2 %) prerušili liečbu kvôli reakciám v mieste podania injekcie.
Laboratórne abnormality
Aby bolo možné priamo porovnať frekvencie laboratórnych abnormalít medzi placebom a aktívnou liečbou, použili sa údaje z 0. – 12. týždňa, pretože to bolo predtým, ako sa pacientom umožnilo
vymeniť placebo za Kevzaru.
Počet neutrofilov
Pokles počtu neutrofilov pod 1 x 109/l sa vysktol u 6,4 % pacientov v skupine s 200 mg Kevzarou
+ DMARDs a u 3,6 % pacientov v skupine so 150 mg Kevzary + DMARDs v porovnaní so žiadnymi pacientmi v skupine s placebom + DMARDs. Pokles počtu neutrofilov pod 0,5 x 109/l sa vyskytol
u 0,8 % pacientov v skupine s 200 mg Kevzarou + DMARDs a u 0,6 % pacientov v skupine so 150
mg Kevzarou + DMARDs. U pacientov, u ktorých došlo k poklesu v absolútnom počte neutrofilov (ANC), úprava liečebného režimu, napr. prerušenie liečby Kevzarou alebo zníženie dávky vyústilo do zvýšenia alebo znormalizovania ANC (pozri časť 4.2). Pokles ANC nebol spojený s vyšším výskytom infekcií, vrátane závažných infekcií.
V populácii pre hodnotenie dlhodobej bezpečnosti s Kevzarou + DMARDs a v populácii
s monoterapiou Kevzary, pozorované počty neutrofilov zodpovedali tým, ktoré boli v placebom kontrolovanej skupine (pozri časť 4.4).
Počet krvných doštičiek
Pokles počtu krvných doštičiek pod 100 x 103/µl sa vyskytol u 1,2 % pacientov v skupine s 200 mg Kevzarou + DMARDs a u 0,6 % pacientov v skupine so 150 mg Kevzary + DMARDs v porovnaní so žiadnymi pacientmi v skupine s placebom + DMARDs.
V populácii pre hodnotenie dlhodobej bezpečnosti s Kevzarou + DMARDs a v populácii
s monoterapiou Kevzary, pozorované počty krvných doštičiek zodpovedali tým, ktoré boli v placebom kontrolovanej skupine (pozri časť 4.4).
Zníženie počtu krvných doštičiek sa nespájalo so žiadnymi krvácavými príhodami.
Pečeňové enzýmy
Abnormality pečeňových enzýmov sú zosumarizované v Tabuľke 2. U pacientov, u ktorých sa vyskytli zvýšenia pečeňových enzýmov, úprava liečebného režimu, napr. prerušenie liečby Kevzarou
alebo zníženie dávky, vyústilo do poklesu alebo znormalizovania pečeňových enzýmov (pozri časť
4.2). Tieto zvýšenia sa nespájali s klinicky významnými zvýšeniami priameho bilirubínu, ani s klinickým dôkazom hepatitídy alebo hepatálnej nedostatočnosti (pozri časť 4.4).
Tabuľka 2: Výskyt abnormalít pečeňových enzýmov v kontrolovaných klinických štúdiach
A
ST
Placebo +
D
MARD N = 661
K
evzara 150 mg
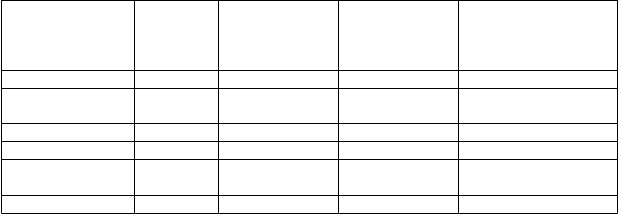 + DMARD N = 660
K
evzara 200 mg
+ DMARD N = 661
Monoterapia
K
evzarou Akákoľvek dávka N = 467
+ DMARD N = 660
K
evzara 200 mg
+ DMARD N = 661
Monoterapia
K
evzarou Akákoľvek dávka N = 467
>3 x ULN –
5 x ULN
0 % 1,2 % 1,1 % 1,1 %
>5 x ULN 0 % 0,6 % 0,2 % 0 %
ALT
>3 x ULN –
5 x ULN
0,6 % 3,2 % 2,4 % 1,9 %
>5 x ULN 0 % 1,1 % 0,8 % 0,2 %
Lipidy
Parametre lipidov (LDL, HDL a triglyceridy) boli prvýkrát hodnotené v 4. týždni od začatia liečby
Kevzarou + DMARDs v placebom kontrolovanej populácii. V 4. týždni vzrástla priemerná hodnota LDL o 14 mg/dl; priemerná hodnota triglyceridov vzrástla o 23 mg/dl a priemerná hodnota HDL vzrástla o 3 mg/dl. Po 4. týždni sa nepozorovali žiadne ďalšie zvýšenia. Medzi dávkami neboli žiadne významné rozdiely.
V populácii pre hodnotenie dlhodobej bezpečnosti s Kevzarou + DMARDs a v populácii
s monoterapiou Kevzary, pozorované parametre lipidov zodpovedali tým, ktoré boli zistené v placebom kontrolovanej populácii.
Imunogenicita
Rovnako ako u všetkých terapeutických proteínov, existuje možnosť imunogenicity pri Kevzare.
V placebom kontrolovanej populácii, 4,0 % pacientov liečených Kevzarou 200 mg + DMARDs, 5,6 %
pacientov liečených Kevzarou 150 mg + DMARDs a 2,0 % pacientov na placebe + DMARDs dosiahlo pozitívnu odpoveď v teste na protilátky proti lieku (anti-drug antibody, ADA). Pozitívne odpovede v teste na neutralizujúce protilátky (neutralizing antibody, NAb) boli zistené u 1,0 % pacientov na Kevzare 200 mg, 1,6 % pacientov na Kevzare 150 mg a u 0,2 % pacientov na placebe.
V populácii s monoterapiou Kevzary zodpovedali zistenia tým, ktoré boli v populácii s Kevzarou + DMARDs.
Tvorba protilátok proti lieku (Anti Drug Antibody, ADA) môže mať vplyv na farmakokinetiku Kevzary. Medzi vznikom ADA a stratou účinnosti alebo nežiaducimi účinkami sa nepozoroval žiaden vzájomný vzťah.
Preukázanie imunitnej odpovede závisí vo vysokej miere od citlivosti a špecificity použitých testov a od skúšobných podmienok. Z týchto dôvodov porovnanie výskytu protilátok proti Kevzare
s výskytom protilátok proti iným produktom môže byť zavádzajúce.
MalignityV placebom kontrolovanej populácii sa vyskytli malignity s rovnakým stupňom výskytu u pacientov dostávajúcich Kevzaru + DMARDs alebo placebo + DMARDs (1,0 udalostí na 100 pacientov ročne).
V populácii pre hodnotenie dlhodobej bezpečnosti s Kevzarou + DMARDs a v populácii
s monoterapiou Kevzary výskyt malignít zodpovedal stupňu pozorovanému v placebom kontrolovanej populácii.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.*
4.9 PredávkovanieK dispozícii sú obmedzené údaje o predávkovaní Kevzarou. Pri predávkovaní Kevzarou neexistuje špecifická liečba. V prípade predávkovania má byť pacient starostlivo monitorovaný, liečený symptomaticky a podľa potreby sa majú vykonať podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, Inhibítory interleukínov. ATC kód: L04AC14
MechanizmusúčinkuSarilumab je ľudská monoklonálna protilátka (podtyp IgG1), ktorá sa špecificky viaže na obidva
rozpustné a na membránu sa viažuce IL-6 receptory (IL-6Ra) a inhibuje IL-6 sprostredkované signály, ktoré zahŕňajú všadeprítomný glykoproteín 130 transudkujúci signál a signálny transduktor a aktivátor transkripcie-3 (Signal Transducer and Activator of Transcription-3, STAT-3).
Vo funkčných testoch na báze ľudských buniek bol sarilumab schopný blokovať IL-6 signálnu dráhu meranú ako inhibíciu STAT-3, iba za prítomnosti IL-6.
IL-6 je pleiotropný cytokín, ktorý stimuluje rôzne bunkové odpovede, napr. proliferáciu, diferenciáciu, prežívanie a apoptózu, a môže aktivovať hepatocyty, čo spôsobí uvoľňovanie proteínov v akútnej fáze, vrátane C-reaktívneho proteínu (CRP) a sérového amyloidu A. Zvýšené hladiny IL-6 sa nachádzajú
v synoviálnej tekutine pacientov s reumatoidnou artritídou a hrajú dôležitú úlohu pri patologických zápaloch a poškodení kĺbov, ktoré sú charakteristickými znakmi RA. U pacientov s RA sa IL-6 podieľa na rôznych fyziologických procesoch, ako napr. migrácia a aktivácia T-buniek, B-buniek, monocytov a osteoklastov vedúca k systémovému zápalu, synoviálnemu zápalu a kostnej erózii.
Aktivita sarilumabu pri zmierňovaní zápalu je spojená s laboratórnymi zmenami, ako napr. pokles
ANC a zvýšenie hladín lipidov (pozri časť 4.4).
Farmakodynamickéúčinky Po jednorazovom subkutánnom podaní sarilumabu 200 mg a 150 mg u pacientov s RA sa pozorovalo rýchle zníženie hladín CRP. Hladiny boli redukované na normálne už po 4 dňoch od začatia liečby. Po jednorazovom podaní sarilumabu u pacientov s RA klesol ANC na úplné dno medzi 3. a 4. dňom
a potom sa vrátil späť na východiskovú hodnotu (pozri časť 4.4). Liečba sarilumabom vyústila do poklesu fibrinogénu a sérového amyloidu A a zvýšenia hemoglobínu a sérového albumínu.
Klinickáúčinnosť
Účinnosť a bezpečnosť Kevzary bola hodnotená v troch randomizovaných, dvojito zaslepených, kontrolovaných multicentrických štúdiach (MOBILITY a TARGET boli placebom kontrolované
štúdie a MONARCH bola aktívna kontrolovaná porovnávacia štúdia) u pacientov starších ako 18
rokov s miernou až závažnou aktívnou reumatoidnou artritídou diagnostikovanou podľa kritérií Americkej reumatologickej spoločnosti (American College of Rheumatology, ACR). Pacienti mali pred začiatkom liečby aspoň 8 bolestivých a 6 opuchnutých kĺbov.
Placebom kontrolované štúdie
MOBILITY hodnotila 1197 pacientov s RA, ktorí mali neadekvátnu klinickú odpoveď na MTX. Pacienti dostávali Kevzaru 200 mg, Kevzaru 150 mg alebo placebo každé 2 týždne so súbežným MTX. Primárne koncové body boli podiel pacientov, ktorí dosiahli ACR20 odpoveď v 24. týždni, zmeny v skóre indexu invalidity v Dotazníku hodnotenia zdravia (Health Assessment Questionnaire – Disability Index, HAQ-DI) v 16. týždni oproti stavu pred liečbou a zmeny vo van der Heijde modifikovanom celkovom Sharp skóre (van der Heijde-modified Total Sharp Score, mTSS) v 52. týždni oproti stavu pred liečbou.
TARGET hodnotila 546 pacientov s RA, ktorí mali neadekvátnu klinickú odpoveď alebo netolerovali jeden alebo viac antagonistov TNF-α. Pacienti dostávali Kevzaru 200 mg, Kevzaru 150 mg alebo placebo každé 2 týždne so súbežnými konvenčnými DMARDs (cDMARDs). Primárne koncové body boli podiel pacientov, ktorí dosiahli ACR20 odpoveď v 24. týždni a zmeny v skóre HAQ-DI v 12. týždni oproti stavu pred liečbou.
Klinická odpoveď
Percentuálny podiel pacientov liečených Kevzarou + DMARDs dosahujúcich odpovede ACR20, ACR50 a ACR70 v MOBILITY a TARGET je zobrazený v Tabuľke 3. V obidvoch štúdiach mali pacienti liečení buď 200 mg alebo 150 mg Kevzary + DMARDs každé dva týždne vyššiu mieru ACR20, ACR50 a ACR70 odpovede oproti placebom liečeným pacientom v 24. týždni. Tieto odpovede pretrvávali počas 3 rokov liečby v otvorenej rozšírenej štúdii.
V MOBILITY, väčší pomer pacientov liečených Kevzarou 200 mg alebo 150 mg každé dva týždne plus MTX dosiahol remisiu, definovanú ako skóre aktivity ochorenia 28-C-reaktívny proteín (DAS28- CRP) < 2,6 v porovnaní s placebom + MTX v 52. týždni. Výsledky v 24. týždni v TARGET boli podobné výsledkom v 52. týždni v MOBILITY (pozri Tabuľku 3).
T
abuľka 3: Klinická odpoveď v 12, 24. a 52. týždni v placebom kontrolovaných štúdiach
MOBILITY a TARGET
MOBILITY
P
e
rcento pacientov
TARGE
T
P
acienti s nedostatočnou odpoveďou na MTX
P
acienti s nedostatočnou odpoveďou na
TN
F inhibítor
12. týžden
DA
S28-CRP
Placebo
+ MTX N = 398
K
evzara
150 mg
+ MTX
N = 400
†††
Kevzara
200 mg
+ MTX N = 399
†††
Placebo
+ cDMA RDs*
N = 181
Kevzara
150 mg
+ cDMARD
s*
N = 181
†††
Kevzara
200 mg
+ cDMARD
s*
N = 184
†††
remisia (< 2,6) 4,8 % 18,0 %
†††
23,1 %
†††
3,9 % 17,1 %
†
17,9 %
†††
ACR
20 34,7 % 54,0 %
†††
ACR50 12,3 % 26,5 %
††
ACR70 4,0 % 11,0 %
24. týždeň
DAS28-CRP
64,9 %
†††
36,3 %
†††
17,5 %
37,6 % 54,1 %
†††
13,3 % 30,4 %
†††
2,2 % 13,8 %
62,5 %
†††
33,2 %
†††
14,7 %
remisia (< 2,6) 10,1 % 27,8 %††† 34,1 %††† 7,2 % 24,9 %††† 28,8 %†††
†††
ACR20 33,4 % 58,0 %
†††
ACR50 16,6 % 37,0 %
†††
ACR70 7,3 % 19,8 %
52. týždeňDAS28-CRP†††
66,4 %
†††
45,6 %
†††
24,8 %
†††
33,7 % 55,8 %
†††
18,2 % 37,0 %
††
7,2 % 19,9 %
†††
60,9 %
†††
40,8 %
†
16,3 %
remisia (< 2,6) 8,5 % 31,0 %††† 34,1 %††† NA§ NA§ NA§
†††
ACR20 31,7 % 53,5 %
†††
ACR50 18,1 % 40,0 %
†††
58,6 %
†††
42,9 %
ACR
70 9,0 % 24,8 % 26,8 %
Významná
†††
klinická odpoveď 3,0 % 12,8 %
†††
14,8 %

* cDMARDs v TARGET vrátane MTX, sulfasalazínu, leflunomidu a hydroxychlorochínu
†
p-hodnota <0,01 rozdiel oproti placebu
††p-hodnota <0,001 rozdiel oproti placebu
†††p-hodnota <0,0001 rozdiel oproti placebu
‡ Primárny koncový bod
§ NA = neaplikovateľné, keďže TARGET bola 24-týždňová štúdia
¶Významná klinická odpoveď = ACR70 pre 24 po sebe nasledujúcich týždňov počas 52 týždňového obdobia
Aj v MOBILITY aj v TARGET sa pozorovala vyššia miera ACR20 odpovede počas 2 týždňov v
porovnaní s placebom a pretrvávala počas trvania štúdií (pozri Obrázok 1 a 2).
 O
brázok 1:
O
brázok 1: Percento ACR20 odpovedí pri návšteve pre MOBILITY
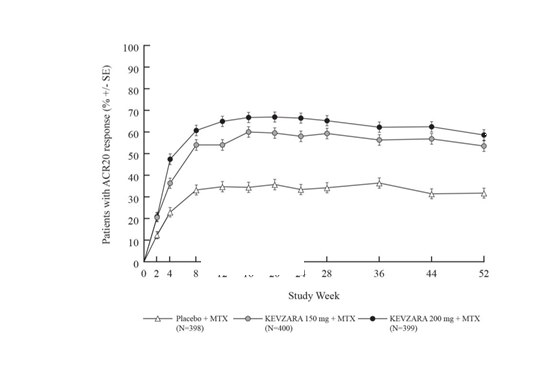
Týždeň štúdie
 Obrázok 2:
Obrázok 2: Percento ACR20 odpovedí pri návšteve pre TARGET
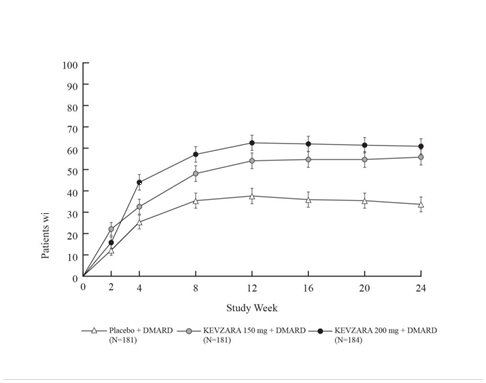
Týždeň štúdie
Výsledky jednotlivých kritérií ACR20 odpovedí v 24. týždni pre MOBILITY a TARGET sú
zobrazené v Tabuľke 4. Výsledky v 52. týždni v MOBILITY boli podobné výsledkom z 24. týždňa pre
TARGET.
T
abuľka 4: Priemerné zníženie zložiek ACR skóre oproti východiskovej hodnote v 24. týždni.
MOBILITY TARGET
Z
l
ožky
(
rozsah)
B
o
l
estivé
Placebo
+ MTX (N =
398)
K
EV
Z
AR
A
150 mg q2w* + MTX
(
N = 400)
K
EV
Z
AR
A
200 mg q2w* + MTX
(
N = 399)
Placebo
+ cDMARDs (N = 181)
K
EV
Z
AR
A
150 mg q2w* + cDMARDs (N = 181)
K
EV
Z
AR
A
200 mg q2w* + cDMARDs (N = 184)
kĺby (0-68) Opuchnuté kĺby
(
0-66)
-14,38 -19,25††† -19,00††† -17,18 -17,30† -20,58†††
-8,70 -11,84††† -12,43††† -12,12 -13,04†† -14,03†††
B
o
l
esť VAS
†
(
0-100 mm) -19,43 -30,75
Celkové zhodnotenie
†††
-34,35†††
-27,65 -36,28††
-39,60
†††
l
ekárom VAS
‡
(
0
-
100 mm)
C
elkové zhodnotenie pacientom VAS
‡
(
0
-
100 mm)
-32,04 -40,69††† -42,65††† -39,44 -45,09††† -48,08†††
-19,55 -30,41††† -35,07††† -28,06 -33,88†† -37,36†††
H
AQ
-
D
I
(
0-3) -0,43 -0,62
†††
-0,64
†††
-0,52 -0,60†
-0,69††
 CR
P
CR
P -0,14 -13,63
††† -18,04
††† -5,21 -13,11
††† -29,06
†††* q2w = každé dva týždne
‡ Vizuálna analógová stupnica'
†p-hodnota <0,01 rozdiel oproti placebu
††p-hodnota <0,001 rozdiel oproti placebu
†††p-hodnota <0,0001 rozdiel oproti placebu
Rádiografická odpoveďV MOBILITY sa hodnotilo štrukturálne poškodenie kĺbov rádiograficky a bolo vyjadrené ako zmena van der Heijde modifikovanom celkovom Sharp skóre (modified Total Sharp Score, mTSS) a jeho
komponentov, skóre erózie a skóre zúženia kĺbovej štrbiny v 52. týždni. Röntgenové snímky rúk a nôh boli urobené na začiatku liečby, v 24. týždni a v 52. týždni a boli nezávisle obodované najmenej
dvomi kvalitne vyškolenými posudzovateľmi, ktorí boli zaslepení voči liečebnej skupine a číslu návštevy.
Obidve dávky Kevzara + MTX boli nadradené placebu + MTX pri zmene z východiskovej hodnoty v mTSS v 24. a 52. týždni (pozri Tabuľku 5). V skupinách liečených sarilumabom sa dosiahol menší pokrok skóra erózie a skóra zúženia kĺbovej štrbiny v 24. a 52. týždni v porovnaní s placebo skupinou.
Liečba s Kevzarou + MTX bola spojená s výrazne menšou rádiografickou progresiou štrukturálneho poškodenia v porovnaní s placebom. V 52. týždni 55,6 % pacientov dostávajúcich Kevzaru 200 mg a
47,8 % pacientov dostávajúcich Kevzaru 150 mg nemalo progresiu štrukturálnych poškodení (ako je
to definované nulovou alebo nižšou zmenou TSS) v porovnaní s 38,7 % pacientmi dostávajúcimi placebo.
Liečba Kevzarou 200 mg + MTX inhibovala progresiu štrukturálneho poškodenia v 52. týždni o 91 %, liečba Kevzarou 150 mg + MTX o 68 % v porovnaní s placebom + MTX.
Účinnosť sarulimabu so súbežnými DMARDs na inhibíciu rádiografickej progresie, ktorá bola hodnotená ako súčasť primárnych koncových bodov v 52. týždni v MOBILITY bola udržaná počas troch rokov od začiatku liečby.
Tabuľka 5: Priemerná rádiografická zmena oproti východiskovej hodnote v 24. týždni a v 52. týždni v MOBILITY
MOBILITY
Pacienti nedostatočne reagujúci na MTX
P
r
i
e
m
erná zmena v 24. týždni
Modifikované celkové Sharp skóre
Placebo
+ MTX (N = 398)
K
evzara
150 mg q2w*
+ MTX (N = 400)
†
Kevzara
200 mg q2w*
+ MTX (N = 399)
††
(m
T
SS) 1,22 0,54
0,13
Skóre erózie (0-280) 0,68 0,26† 0,02††
Skóre zúženia kĺbovej štrbiny 0,54 0,28 0,12†
Priemerná zmena v 52. týždni
Modifikované celkové Sharp skóre
(m
T
SS) ‡
2,78 0,90†† 0,25††
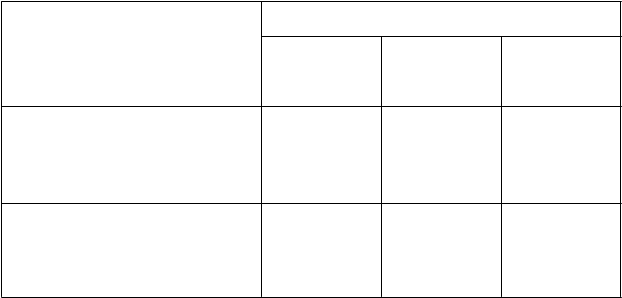 Skóre erózie (0-280)
Skóre erózie (0-280) 1,46 0,42†† 0,05††
Skóre zúženia kĺbovej štrbiny 1,32 0,47† 0,20††
* q2w=každé dva týždne
† p-hodnota <0,001
†† p-hodnota <0,0001
‡ Primárny koncový bod
Odpoveď podľa telesných funkciíV MOBILITY a TARGET boli hodnotené telesné funkcie a poruchy Dotazníkom hodnotiacim zdravie a index porúch (Health Assessment Questionnaire Disability Index, HAQ-DI). Pacienti dostávajúci
Kevzaru 200 mg + DMARDs každé dva týždne preukázali v MOBILITY v 16. týždni väčšie zlepšenie
telesných funkcií oproti východiskovej hodnote, v porovnaní s placebom a pacienti dostávajúci
Kevzaru 150 mg + DMARDs každé dva týždne preukázali v TARGET v 12. týždni väčšie zlepšenie telesných funkcií oproti východiskovej hodnote, v porovnaní s placebom.
MOBILITY preukázala významné zlepšenie telesných funkcií hodnotených podľa HAQ-DI v 16. týždni v porovnaní s placebom (-0,58 pre Kevzaru 200 mg + MTX, -0,54 pre Kevzaru 150 mg + MTX a –0,30 pre placebo + MTX, každé dva týždne). TARGET preukázal významné zlepšenie v skóre
HAQ-DI v 12. týždni v porovnaní s placebom (-0,49 pre Kevzaru 200 mg + DMARDs, -0,50 pre
Kevzaru 150 mg + DMARDs a –0,29 pre placebo + DMARDs, každé dva týždne).
V MOBILITY bolo zlepšenie telesných funkcií hodnotených podľa HAQ-DI udržiavané do 52. týždňa (-0,75 pre skupinu liečenú Kevzarou 200 mg + MTX, -0,71 pre skupinu liečenú Kevzarou 150 mg + MTX a –0,46 pre skupinu liečenú placebom + MTX).
Pacienti liečení s Kevzarou + MTX (47,6 % v skupine liečenej 200 mg a 47,0 % v skupine liečenej
150 mg) dosiahli klinicky významné zlepšenie v HAQ-DI (zmena oproti východiskovej hodnote ≥ 0,3
jednotiek) v 52. týždni v porovnaní s 26,1 % v skupine liečenej placebom + MTX.
Výsledky zaznamenané pacientmi
Celkový zdravotný stav bol hodnotený v Skrátenej forme dotazníka zdravia (SF-36). V MOBILITY a TARGET, pacienti dostávajúci Kevzaru 200 mg + DMARDs každé dva týždne alebo Kevzaru 150 mg + DMARDs každé dva týždne preukázali výraznejšie zlepšenie oproti východiskovej hodnote
v porovnaní s placebom + DMARDs každé dva týždne v súhrne zložiek telesného zdravia (physical component summary, PCS) a žiadne zhoršenie v súhrne zložiek duševného zdravia (mental component summary, MCS) v 24. týždni. Pacienti dostávajúci Kevzaru 200 mg + DMARDs zaznamenali výraznejšie zlepšenie v porovnaní s placebom v oblastiach Telesných funkcií, Obmedzenia kvôli fyzickým problémom, Fyzickej bolesti, Celkového vnímania zdravotného stavu, Vitality, Spoločenskej funkcie a Duševného zdravia.
Únava bola posúdená podľa stupnice FACIT-Fatigue. V MOBILITY a TARGET preukázali pacienti dostávajúci sarilumab 200 mg + DMARDs každé dva týždne alebo sarilumab 150 mg + DMARDs každé dva týždne výraznejšie zlepšenie oproti východiskovej hodnote, v porovnaní s placebom + DMARDs.
Aktívnym komparátorom kontrolovaná štúdia
MONARCH bola 24 týždňová randomizovaná dvojito zaslepená, dvojito placebom kontrolovaná štúdia, ktorá porovnávala monoterapiu Kevzarou 200 mg s monoterapiou adalimumabom 40 mg
podávaným subkutánne každé dva týždne 369 pacientom s miernou až závažnou aktívnou RA, ktorá bola nevhodne liečená s MTX vrátane tých, ktorí netolerovali alebo nedostatočne odpovedali na MTX.
Kevzara 200 mg bola účinnejšia oproti adalimumabu 40 mg pri znižovaní aktivity ochorenia
a zlepšovaní telesných funkcií, s viacerými pacientmi dosahujúcimi klinickú remisiu počas 24 týždňov
(pozri Tabuľku 6).
Tabuľka 6: Výsledky účinnosti v štúdii MONARCH
A
dalimumab
 40 mg q2w* (N=185)
K
evzara
200 mg q2w
(
N=
184)
40 mg q2w* (N=185)
K
evzara
200 mg q2w
(
N=
184)
DA
S28-ESR (primárny koncový bod)
p-hodnota oproti adalimumabu
-2,20 (0,106) -3,28 (0,105)
< 0,0001
DA
S28-ESR remisia (< 2,6), n (%)
p-hodnota oproti adalimumabu
ACR20 odpoveď, n (%)
p-hodnota oproti adalimumabu
ACR50 odpoveď, n (%)
p-hodnota oproti adalimumabu
ACR70 odpoveď, n (%)
p-hodnota oproti adalimumabu
HAQ-DI
p-hodnota oproti adalimumabu
13 (7,0 %) 49 (26,6 %)
< 0,0001
108 (58,4 %) 132 (71,7 %)
0,0074
55 (29,7 %) 84 (45,7 %)
0,0017
22 (11,9 %) 43 (23,4 %)
0,0036
-0,43 (0,045) -0,61(0,045)
0,0037
*Zahŕňa pacientov, ktorí kvôli nedostatočnej odpovedi zvýšili frekvenciu dávkovania adalimumabu
40 mg na každé dva týždne.
Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Kevzarou
(sarilumab) v jednej alebo vo viacerých podskupinách pediatrickej populácie pri chronickej idiopatickej artritíde (vrátane reumatoidnej artritídy, spondylartritídy, psoriatickej artritídy a juvenilnej
idiopatickej artritídy) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika sarilumabu bola charakterizovaná u 2186 pacientov s RA liečených sarilumabom, ktorí zahŕňali 751 pacientov liečených 150 mg a 891 pacientov liečených 200 mg subkutánnymi dávkami každé dva týždne počas 52 týždňov.
Absorpcia
AbsolútnabiodostupnosťpresarilumabpoSCinjekciisaPKanalýzoupopulácieodhadovalana80%. Medián tmax po jednorazovej subkutánnej dávke bol pozorovaný 2 až 4 dni. Po viacnásobnej dávke 150 až 200 mg každé dva týždne sa dosiahol rovnovážny stav v 12. a 16. týždni s 2 až 3 násobnou akumuláciou v porovnaní s jednorazovou dávkou.
Pri dávkovacom režime 150 mg každé dva týždne bola odhadovaná stredná hodnota (± smerodajná odchýlka, SD) plochy pod krivkou (area under curve, AUC) sarilumabu v rovnovážnom stave
210±115 mg.day/l, Cmin bola 6,95±7,60 mg/l a Cmax bola 20,4±8,27 mg/l.
Pri dávkovacom režime 200 mg každé dva týždne bola odhadovaná stredná hodnota (± SD) plochy pod krivkou (area under curve, AUC) sarilumabu v rovnovážnom stave 396±194 mg.day/l, Cmin bola
16,7±13,5 mg/l a Cmax bola 35,4±13,9 mg/l.
V štúdii účinnosti sarilumabu bolo vystavenie po 200 mg každé dva týždne mierne vyššie (Cmax + 24-
34%, AUC(0-2w) +7-21%) po použití naplneného injekčného pera v porovnaní s naplnenou injekčnou
striekačkou.
Distribúcia
U pacientov s RA bol zdanlivý distribučný objem v rovnovážnom stave 8,3 l.
Biotransformácia
Metabolická dráha sarilumabu nebola charakterizovaná. Predpokladá sa, že sarilumab ako monoklonálna protilátka je degradovaný na malé peptidy a aminokyseliny katabolickou cestou,
rovnakým spôsobom ako endogénne IgG.
Eliminácia
Sarilumab sa eliminuje súbežnými lineárnymi a nelineárnymi cestami. Pri vyšších koncentráciach je eliminácia prevažne lineárna nesaturovateľná proteolytická cesta, kým pri nižších koncentráciach
prevláda nelineárna saturovateľná sprostredkovaná eliminácia. Tieto súbežné eliminačné cesty majú za následok úvodný polčas od 8 do 10 dní a odhadovaný účinný polčas v rovnovážnom stave 21 dní.
Po poslednej 150 mg dávke sarilumabu v rovnovážnom stave bol medián času na dosiahnutie nedetekovateľnej koncentrácie 30 dní a po 200 mg dávke bol 49 dní.
Monoklonálne protilátky sa neeliminujú obličkami ani pečeňou.
Linearita/nelinearita
U pacientov s RA sa pozorovalo viac ako od dávky závislé zvýšenie farmakokinetickej expozície. V rovnovážnom stave sa zvýšilo vystavenie počas dávkovacieho intervalu merané AUC približne
dvojnásobne pri 1,33 násobnom zvýšení dávky od 150 do 200 mg každé dva týždne.
InterakciasCYP450substrátmi
Simvastatín je substrátom CYP3A4 a OATP1B1. U 17 pacientov s RA pokleslo vystavenie simvastatínu a kyseliny simvastatínovej jeden týždeň po jednorazovom 200 mg podaní sarilumabu o
45 % a 36 %, v uvedenom poradí (pozri časť 4.5).
Osobitnéskupinypacientov
Vek, pohlavie, etnická príslušnosť a telesná hmotnosť
Farmakokinetické analýzy populácie u dospelých pacientov s RA (vo veku od 18 do 88 rokov so 14 % nad 65 rokov) preukázali, že vek, pohlavie a rasa nemajú významný vplyv na farmakokinetiku sarilumabu.
Farmakokinetiku sarilumabu ovplyvnila telesná hmotnosť. 150 mg a 200 mg dávky preukázali účinok u pacientov s vyššou telesnou hmotnosťou (> 100 kg), ale pacienti vážiaci > 100 kg mali väčší terapeutický prínos z 200 mg dávky.
Porucha funkcie obličiek
Nebola vykonaná žiadna oficiálna štúdia o vplyve poruchy funkcie obličiek na farmakokinetiku sarilumabu. Mierna až stredná porucha funkcie obličiek neovplyvnila farmakokinetiku sarilumabu. U pacientov s miernou až strednou poruchou funkcie obličiek sa nevyžaduje žiadna úprava dávkovania. Pacienti so závažnou poruchou funkcie obličiek neboli skúmaní.
Porucha funkcie pečene
Nebola vykonaná žiadna oficiálna štúdia o vplyve poruchy funkcie pečene na farmakokinetiku sarilumabu (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií toxicity po opakovanom podaní, posúdenia karcinogénneho rizika a reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Neboli vykonané žiadne dlhodobé štúdie na zvieratách na stanovenie karcinogénneho potenciálu sarilumabu. Závažné dôkazy pre inhibíciu IL-6Rα väčšinou indikujú protinádorové účinky sarilumabu sprostredkované viacnásobnými mechanizmami prevažne vyžadujúcimi inhibíciu STAT-3. In vitro
a in vivo štúdie so sarilumabom používajúce ľudské nádorové bunkové línie ukázali inhibíciu STAT-3
aktivácie a inhibíciu rastu tumoru na zvieracích modeloch s ľudskými xenotransplantátmi tumoru.
Štúdie fertility uskutočnené u samcov a samíc myší, v ktorých sa použili myšie náhradné protilátky IL-
6Ra nepreukázali žiadne zhoršenie plodnosti.
V rozšírenej štúdii toxicity prenatálneho/postnatálneho vývoja sa gravidným opiciam Cynomolgus raz týždenne podával sarilumab intravenózne, od skorej gestácie až do prirodzeného pôrodu (približne 21 týždňov). Expozícia matiek približne až 83 krát väčšia ako expozícia u ľudí založená na AUC po subkutánnych dávkach 200 mg každé 2 týždne nepreukázala žiadne účinky na matku, zárodok ani plod. Sarilumab nemá vplyv na udržanie gravidity ani na novorodencov, u ktorých sa počas 1 mesiaca po narodení vyhodnocovalo váženie telesnej hmotnosti, parametre funkčného alebo morfologického vývinu, vrátane ohodnotenia kostí, imunofenotypizácia periférnych krvných lymfocytov
a mikroskopické zhodnotenia. Sarilumab bol detekovaný v sére novorodencov až do 1 mesiaca. Vylučovanie sarilumabu do mlieka opíc Cynomolgus nebolo skúmané.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Histidín Arginín Polysorbát 20
Sacharóza
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
Po vybratí z chladničky sa má Kevzara podať do 14 dní, a nemá sa skladovať pri teplote nad 25 ºC.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke.
Naplnenú injekčnú striekačku/naplnené injekčné pero uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Všetky balenia obsahujú 1,14 ml roztoku v injekčnej striekačke (sklo typu 1) vybavenej vsadenou ihlou z nehrdzavejúcej ocele a piestovou elastomérovou zátkou.
Naplnenáinjekčnástriekačka150mg:
Naplnená injekčná striekačka na jednorazové použitie má styréno-butadiénový elastomérový kryt ihly
a pozostáva z bieleho polystyrénového piestu a svetlo oranžovej polypropylénovej prstencovej obruby.
Naplnenáinjekčnástriekačka200mg:
Naplnená injekčná striekačka na jednorazové použitie má styréno-butadiénový elastomérový kryt ihly a pozostáva z bieleho polystyrénového piestu a tmavo oranžovej polypropylénovej prstencovej
obruby.
Naplnenéinjekčnépero150mg:
Jednotlivé časti pera sú zostavené do jednorazového naplneného pera so žltým krytom ihly a svetlo oranžovým uzáverom.
Naplnenéinjekčnépero200mg:
Jednotlivé časti pera sú zostavené do jednorazového naplneného pera so žltým krytom ihly a tmavo oranžovým uzáverom.
Veľkosti balenia:
· 1 naplnená injekčná striekačka
· 2 naplnené injekčné striekačky
· viacnásobné balenie obsahujúce 6 naplnených injekčných striekačiek (3 balenia po 2)
· 1 naplnené injekčné pero
· 2 naplnené injekčné perá
· viacnásobné balenie obsahujúce 6 naplnených injekčných pier (3 balenia po 2)
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Naplnená injekčná striekačka/naplnené injekčné pero sa majú pred použitím vizuálne skontrolovať. Roztok sa nemá použiť, ak je zakalený, má zmenenú farbu alebo obsahuje častice alebo ak je akákoľvek časť pomôcky poškodená.
Po vybratí naplnenej injekčnej striekačky/naplneného pera z chladničky, nechajte pred podaním injekcie Kevzary striekačku/pero zohriať na izbovú teplotu (<25°C).
Podrobný návod na používanie Kevzary v naplnenej injekčnej striekačke/naplnenom pere je uvedený v písomnej informácii.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami. Po použití vložte injekčnú striekačku/pero do nádoby odolnej proti prepichnutiu
a zlikvidujte v súlade s lokálnymi požiadavkami. Nádobu nerecyklujte. Nádobu uchovávajte mimo dohľadu a dosahu detí.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
sanofi-aventis groupe
54, rue La Boétie
75008 Paríž
Francúzsko
8. REGISTRAČNÉ ČÍSLOEU/1/17/1196/001
EU/1/17/1196/002
EU/1/17/1196/003
EU/1/17/1196/004
EU/1/17/1196/005
EU/1/17/1196/006
EU/1/17/1196/007
EU/1/17/1196/008
EU/1/17/1196/009
EU/1/17/1196/010
EU/1/17/1196/011
EU/1/17/1196/012
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu <a na internetovej stránke Štátneho ústavu pre kontrolu liečiv
http://www.sukl.sk>.