
155 – 164
|
1,6
|
165 – 174
|
1,7
|
175 – 184
|
1,8
|
185 – 194
|
1,9
|
≥ 195
|
2,0
|
1 Ak sa vyžaduje injekčný objem väčší ako 1 ml, môže byť potrebná viac ako 1 injekčná liekovka.
Účinok difelikefalínu pri znižovaní pruritu sa očakáva po 2 – 3 týždňoch liečby.
Vynechané dávky
Ak sa vynechá pravidelne naplánovaná hemodialyzačná liečba, Kapruvia sa má podať pri nasledujúcej hemodialyzačnej liečbe v rovnakej dávke.
Extra liečba
Ak sa za týždeň vykoná 4. hemodialyzačná liečba, Kapruvia sa má podať na konci hemodialýzy podľa odporúčaného dávkovania. Nemajú sa podávať viac ako 4 dávky za týždeň, a to ani v prípade, ak
počet hemodialyzačných terapií za týždeň prekračuje 4. Nie je pravdepodobné, že 4. dávka Kapruvie spôsobí kumuláciu difelikefalínu, ktorá by mohla ohroziť bezpečnosť, lebo väčšina zvyšného difelikefalínu z predchádzajúcej liečby sa odstráni pri hemodialýze (pozri časti 4.9 a 5.2). Bezpečnosť a účinnosť 4. dávky však nebola z dôvodu nedostatočných údajov presne stanovená.
Pacienti s neúplnou hemodialyzačnou liečbou
Pri hemodialýzach trvajúcich kratšie ako 1 hodinu sa má podávanie difelikefalínu vysadiť až do ďalšej aplikácie hemodialýzy.
Po podaní difelikefalínu sa u osôb na hemodialýze z tela eliminuje až 70 % difelikefalínu pred ďalšou hemodialýzou (pozri časti 4.9 a 5.2). Hladina difelikefalínu v plazme zostávajúca v čase ďalšej hemodialýzy sa zníži o približne 40 – 50 % do jednej hodiny hemodialýzy.
Pacienti s poruchou funkcie pečene
U pacientov s miernou alebo stredne závažnou poruchou funkcie pečene sa nevyžaduje žiadna úprava dávkovania (pozri časť 5.2). Difelikefalín sa neštudoval u osôb so závažnou poruchou funkcie pečene (Pracovná skupina orgánovej dysfunkcie Národného onkologického inštitútu (NCI ODWG, National Cancer Institute Organ Dysfunction Working Group)), a preto sa jeho použitie v tejto populácii pacientov neodporúča.
Starší pacienti (≥ 65 rokov)
Odporúčania týkajúce sa dávkovania u starších pacientov sú rovnaké ako u dospelých pacientov.
Pediatrická populácia
Bezpečnosť a účinnosť difelikefalínu u detí vo veku 0 – 17 rokov neboli doteraz stanovené.
K dispozícii nie sú žiadne údaje.
Spôsob podávania
Kapruvia sa nemá riediť ani miešať s inými liekmi.
Difelikefalín sa odstraňuje membránou dialyzátora a musí sa podávať vtedy, keď už krv neprechádza dialyzátorom. Difelikefalín sa podáva 3-krát týždenne intravenóznou bolusovou injekciou do venóznej hadičky dialyzačného okruhu na konci hemodialyzačnej liečby počas návratu do tela alebo po ňom. Keď sa podá po návrate do tela, po injekcii Kapruvie sa má podať aspoň 10 ml návratového objemu injekčného roztoku chloridu sodného 9 mg/ml (0,9 %). Ak sa dávka podá počas návratu do tela, nie je potrebný žiadny ďalší injekčný roztok chloridu sodného 9 mg/ml (0,9 %) na prepláchnutie hadičky.
4
.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Hyperkaliémia
Hyperkaliémia sa často vyskytuje u pacientov s chronickým ochorením obličiek, ktorí sú na
hemodialýze. V placebom kontrolovaných klinických štúdiách sa hlásil číselne vyšší výskyt nežiaducich udalostí hyperkaliémie u pacientov liečených difelikefalínom (4,7 %; 20/424 pacientov) v porovnaní s pacientmi s placebom (3,5 %, 15/424 pacientov). Príčinná súvislosť nebola stanovená. Odporúča sa časté monitorovanie hladiny draslíka.
Zlyhávanie srdca a fibriláciapredsiení
Difelikefalín sa neštudoval u pacientov so zlyhávaním srdca New York Heart Association (NYHA)
triedy IV. V pivotných klinických štúdiách sa u pacientov liečených difelikefalínom v porovnaní s placebom pozoroval malý číselný nepomer udalostí zlyhávania srdca a fibrilácie predsiení, najmä u pacientov s anamnézou fibrilácie predsiení, ktorí prerušili alebo vynechali liečbu fibrilácie predsiení. Príčinná súvislosť nebola stanovená.
Pacienti s poškodenou hematoencefalickou bariérou
Difelikefalín je periférne pôsobiaci agonista kappa opioidného receptora s obmedzeným prístupom do
centrálneho nervového systému (CNS). Na minimalizáciu absorbcie difelikefalínu do CNS je nevyhnutná integrita hematoencefalickej bariéry (pozri časť 5.1). U pacientov s klinicky významnými poškodeniami hematoencefalickej bariéry (napr. primárny zhubný nádor mozgu, metastázy v CNS alebo iné podmienky spôsobujúce zápal, aktívna skleróza multiplex, pokročilá Alzheimerova choroba) môže byť riziko prenosu difelikefalínu do CNS. Kapruvia sa má takýmto pacientom predpisovať s opatrnosťou, berúc do úvahy ich individuálny pomer prínosu a rizika s pozorovaním možných účinkov na CNS.
Závraty a somnolencia
U pacientov užívajúcich difelikefalín sa vyskytli závraty a somnolencia. V čase a s pokračovaním
liečby môžu ustupovať (pozri časť 4.8). Súbežné používanie sedatívnych antihistaminík, opioidných analgetík alebo iných látok tlmiacich CNS môže zvyšovať pravdepodobnosť výskytu týchto nežiaducich reakcií a pri liečbe difelikefalínom sa majú používať opatrne (pozri časť 4.5).
V porovnaní s placebom bola incidencia somnolencie vyššia u osôb vo veku 65 rokov a starších liečených difelikefalínom (7,0 %) ako u osôb mladších ako 65 rokov liečených difelikefalínom (2,8 %).
Pomocné látky so známym účinkom
Tento liek obsahuje menej ako 1 mmol sodíka v injekčnej liekovke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne klinické interakčné štúdie. Difelikefalín neinhibuje ani neindukuje enzýmy CYP450 a nie je substrátom enzýmov CYP450. Nie je ani inhibítorom glukuronidačných enzýmov. Difelikefalín nie je substrátom ani inhibítorom ľudských transportérov (pozri časť 5.2). Preto sú interakcie difelikefalínu s inými liekmi nepravdepodobné.
Súbežné podávanie liekov, ako sú sedatívne antihistaminiká, opioidné analgetiká alebo iné látky tlmiace CNS (napr. klonidín, ondansetrón, gabapentín, pregabalín, zolpidem, alprazolam, sertralín, trazodón) môže zvýšiť pravdepodobnosť závratov a somnolencie (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití difelikefalínu u gravidných
žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity
(pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu Kapruvie počas gravidity. Dojčenie
Nie je známe, či sa difelikefalín vylučuje do ľudského materského mlieka.
Riziko u novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Kapruviou sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Štúdie na zvieratách preukázali vylučovanie difelikefalínu do materského mlieka. Fertilita
K dispozícii nie sú žiadne údaje o vplyve difelikefalínu na fertilitu u ľudí. V štúdiách s difelikefalínom
na potkanoch sa nepozoroval žiadny účinok na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kapruvia má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
U pacientov liečených difelikefalínom sa hlásila somnolencia a/alebo závraty (pozri časť 4.8). Pacienti majú byť obozretní ohľadom vedenia vozidiel alebo obsluhy nebezpečných strojov, pokiaľ nebude známy účinok difelikefalínu na ich schopnosť viesť vozidlá a obsluhovať stroje. Somnolencia sa vyskytovala počas prvých 3 týždňov liečby a s pokračujúcou liečbou mala tendenciu ustupovať. Závraty sa vyskytovali počas prvých 9 týždňov liečby a boli zvyčajne prechodné.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
V placebom kontrolovaných a nekontrolovaných klinických štúdiách vo fáze 3 sa u približne 6,6 %
pacientov vyskytla počas liečby difelikefalínom aspoň jedna nežiaduca reakcia. Najčastejšími nežiaducimi reakciami boli somnolencia (1,1 %), závraty (0,9 %), parestézia (vrátane hypestézie, orálnej parestézie a orálnej hypestézie) (1,1 %), bolesť hlavy (0,6 %), nevoľnosť (0,7 %), vracanie (0,7 %), hnačka (0,2 %) a zmeny duševného stavu (vrátane stavu zmätenosti) (0,3 %). Väčšina týchto udalostí mala miernu alebo strednú závažnosť, neviedli k škodlivým následkom a vymizli
s pokračujúcou liečbou. Žiadna udalosť nebola závažná a výskyt udalostí, ktoré viedli k prerušeniu
liečby, bol ≤ 0,5 % zo všetkých vyššie uvedených nežiaducich reakcií.
Tabuľkový zoznam nežiaducich reakciíNežiaduce reakcie pozorované v placebom kontrolovaných a nekontrolovaných klinických štúdiách vo
fáze 3 u pacientov liečených difelikefalínom (N = 1 306) sú uvedené v tabuľke 1 podľa triedy orgánových systémov MedDRA, preferovaného termínu a frekvencie.
Frekvencia je klasifikovaná ako časté (≥ 1/100 až < 1/10) a menej časté (≥ 1/1 000 až < 1/100).

 Tabuľka 1: Nežiaduce reakciepripisované liečbedifelikefalínomupacientovnahemodialýze
Tabuľka 1: Nežiaduce reakciepripisované liečbedifelikefalínomupacientovnahemodialýze Trieda orgánových systémov
MedDRA
| Časté
| Menej časté
| Psychické poruchy
|
| Zmeny duševného stavu1
| Poruchy nervového systému
| Somnolencia, parestézia2
| Závraty, bolesť hlavy
| Poruchy gastrointestinálneho traktu
|
| Vracanie, nevoľnosť, hnačka
|
|
|
1 Zmeny duševného stavu zahŕňali preferované termíny MedDRA stav zmätenosti a zmeny duševného stavu.
2 Parestézia zahŕňala uprednostňované pojmy databázy MedDRA: parestézia, hypestézia, orálna parestézia a orálna hypestézia.
Opis vybratých nežiaducich reakciíSomnolenciaSomnolencia sa hlásila ako nežiaduca udalosť po začatí liečby u 2,2 % osôb randomizovaných na
používanie difelikefalínu. Prevažná väčšina týchto udalostí bola mierna alebo stredne závažná. U
0,3 % pacientov viedla somnolencia k prerušeniu liečby difelikefalínom. Somnolencia sa hlásila ako závažná nežiaduca udalosť u < 0,1 % osôb liečených difelikefalínom. U 1,1 % pacientov sa somnolencia hlásila ako udalosť, ktorá mala príčinnú súvislosť s liečbou difelikefalínom. Somnolencia sa vyskytovala počas prvých 3 týždňov liečby a s pokračujúcou liečbou mala tendenciu ustupovať. Pravdepodobnosť výskytu somnolencie sa môže zvýšiť, ak sa difelikefalín súbežne používa s inými liekmi (pozri časť 4.4 a 4.5).
ZávratZávrat sa hlásil ako nežiaduca udalosť po začatí liečby u 7,9 % osôb randomizovaných na používanie difelikefalínu. Prevažná väčšina týchto udalostí bola mierna alebo stredne závažná. U 0,5 % pacientov viedol závrat k prerušeniu liečby difelikefalínom. Závrat sa hlásil ako závažná nežiaduca udalosť u
0,5 % osôb liečených difelikefalínom. U 0,9 % pacientov sa závrat hlásil ako udalosť, ktorá mala príčinnú súvislosť s liečbou difelikefalínom. Závrat sa vyskytoval počas prvých 9 týždňov liečby a bol zvyčajne prechodný.
Pravdepodobnosť výskytu závratu sa môže zvýšiť, ak sa difelikefalín súbežne používa s inými liekmi
(pozri časť 4.4 a 4.5).
Zmeny duševného stavuZmena duševného stavu (vrátane stavu zmätenosti) sa hlásila ako nežiaduca udalosť po začatí liečby u
4,4 % osôb randomizovaných na užívanie difelikefalínu.
Prevažná väčšina týchto udalostí bola mierna alebo stredne závažná. U 0,2 % pacientov viedli zmeny duševného stavu k prerušeniu liečby difelikefalínom.
Zmeny duševného stavu sa hlásili ako závažná nežiaduca udalosť u 2,2 % osôb liečených difelikefalínom. U 0,3 % pacientov sa zmeny duševného stavu hlásili ako udalosť, ktorá mala príčinnú súvislosť s liečbou difelikefalínom.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4
.9 Predávkovanie
V klinických štúdiách u pacientov podstupujúcich hemodialýzu sa podávala jedna dávka difelikefalínu až 12-násobne vyššia a opakovane podávané dávky difelikefalínu až 5-násobne vyššie ako klinická dávka 0,5 mikrogramov/kg. Pozoroval sa od dávky závislý nárast nežiaducich udalostí zahŕňajúcich závraty, somnolenciu, zmeny duševného stavu, parestézie, únavu, hypertenziu a vracanie.
V prípade predávkovania je potrebné poskytnúť príslušnú lekársku pomoc na základe klinického stavu pacienta. Hemodialýza počas 4 hodín pomocou vysokoprietokového dialyzátora účinne odstránila približne 70 % – 80 % difelikefalínu z plazmy a difelikefalín nebol detegovateľný v plazme na konci druhého z dvoch dialyzačných cyklov (pozri časť 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: všetky ostatné liečivá, iné liečivá, ATC kód: V03AX04
Mechanizmus účinku
Difelikefalín je selektívny agonista kappa opioidného receptora s nízkou penetráciou do centrálnej
nervovej sústavy.
Fyzikálno-chemické vlastnosti difelikefalínu (hydrofilný, syntetický D-aminokyselinový peptid
s veľkou polárnou povrchovou oblasťou a nábojom pri fyziologickom pH) minimalizujú jeho pasívnu difúziu (permeabilitu) a aktívny transport cez membrány, čím obmedzujú penetráciu do centrálnej nervovej sústavy.
Patofyziológia pruritu spojeného s chronickým ochorením obličiek sa považuje za viac faktorovú vrátane systémového zápalu a nerovnováhy endogénneho opioidného systému (t. j. nadmerná expresia mu opioidných receptorov a súčasná znížená regulácia kappa opioidných receptorov). Je známe, že opioidné receptory modulujú signály svrbenia a zápal, pričom aktivácia kappa opioidných receptorov znižuje svrbenie a má imunomodulačné účinky.
Aktivácia kappa opioidných receptorov na periférnych senzorických neurónoch a imunitných bunkách difelikefalínom sa považuje za mechanicky zodpovednú za antipruritické a protizápalové účinky.
Klinická účinnosť a bezpečnosť
Placebom kontrolované štúdie
V dvoch pivotných klinických štúdiách vo fáze 3 s podobným, dvojito zaslepeným, randomizovaným, placebom kontrolovaným dizajnom (KALM-1 a KALM-2) dostávali pacienti s chronickým ochorením obličiek na hemodialýze, ktorí mali stredne závažný až závažný pruritus, placebo alebo
0,5 mikrogramu/kg difelikefalínu intravenózne 3-krát týždenne po hemodialýze po dobu 12 týždňov. U pacientov podstupujúcich ďalšiu dialýzu počas daného týždňa boli povolené maximálne 4 dávky za týždeň. Primárnym koncovým ukazovateľom v oboch štúdiách bol percentuálny podiel pacientov, ktorí dosiahli zníženie od počiatočnej hodnoty škály číselného hodnotenia najhoršieho svrbenia (WI- NRS) o minimálne 3 body po 12 týždňoch. Hlavnými sekundárnymi koncovými ukazovateľmi
v oboch štúdiách boli percentuálne podiely pacientov so zlepšením vo WI-NRS o aspoň 4 body po
12 týždňoch a zmeny závažnosti svrbenia a kvality života (QoL) súvisiacej so svrbením podľa merania pomocou celkovej škály Skindex-10 a škály svrbenia 5-D. Bola zahrnutá aj analýza odpovedajúcich pacientov založená na celkovom dojme pacienta ohľadom zmeny.
V pivotných štúdiách bolo zahrnutých celkovo 851 pacientov so stredne závažným až závažným pruritom (východisková hodnota WI-NRS > 4). Priemerný vek bol 59 rokov, pričom 33,1 % bolo vo
veku 65 rokov a vyššom a 11,1 % bolo vo veku 75 rokov a vyššom, 60 % pacientov tvorili muži.
Východiskové priemerné skóre WI-NRS bolo 7,18 v oboch skupinách, v skupine s difelikefalínom aj v skupine s placebom, východiskový medián skóre WI-NRS bol 7,13 (rozsah 4,2 až 10) v skupine s difelikefalínom a 7,13 (rozsah 4,1 až 10) v skupine s placebom. Ostatné charakteristiky ochorenia na začiatku boli v skupinách s difelikefalínom a placebom porovnateľné: čas od stanovenia diagnózy chronického ochorenia obličiek (8,22 roka oproti 8,54 roka), trvanie pruritu (3,20 roka oproti
3,31 roka) a používanie liekov určených na zmiernenie pruritu, ako sú antihistaminiká, kortikosteroidy, gabapentín alebo pregabalín (37,5 % oproti 38 %). Naprieč štúdiami difelikefalín za
12 týždňov významne znížil intenzitu svrbenia a zlepšil kvalitu života súvisiacu so svrbením, ako je to
uvedené v tabuľke 2.
Tabuľka 2: Súhrn primárnych a kľúčových sekundárnych výsledkov v štúdiách KALM-1 a KALM-2 v 12. týždni KALM-1 (n = 378) KALM-2 (n = 473)
Koncový ukazovateľ do
| difelikefalín
| Placebo
| difelikefalín
| Placebo
| konca 12. týždňa
| (n = 189)
| (n = 189)
| (n = 237)
| (n = 236)
| Primárny koncový
|
|
|
|
| ukazovateľ
|
|
|
|
| WI-NRS
|
|
|
|
| Pacienti so zlepšením
| 51,0 %
| 27,6 %
| 54,0 %
| 42,2 %
| o ≥ 3 body (%)
| (p < 0,001)
|
| (p = 0,02)
|
| Sekundárne koncové
|
|
|
|
| ukazovatele
|
|
|
|
| WI-NRS
|
|
|
|
| Pacienti so zlepšením
| 38,9 %
| 18,0 %
| 41,2 %
| 28,4 %
| o ≥ 4 body (%)
| (p < 0,001)
|
| (p = 0,01)
|
| Skindex-10
|
|
|
|
| Zmena od počiatočnej
| -17,2
| -12,0
| -16,6
| -14,8
| hodnoty
| (p < 0,001)
| '
| (p = 0,171)
|
| [celkové skóre]
|
|
|
|
| Škála svrbenia 5-D
|
|
|
|
| Zmena od počiatočnej
| -5,0
| -3,7
| -4,9
| -3,8
| hodnoty
| (p < 0,001)
|
| Neaplikovateľné1
|
| [celkové skóre]
|
|
|
|
|
|
|
1 Nebolo testované na základe hierarchického poradia testovania.
Obrázok 1 zobrazuje percentuálne priemerné skóre v štúdiách KALM-1 a KALM-2 s ≥ 3-bodovým zlepšením od počiatočnej hodnoty skóre WI-NRS podľa týždňa štúdie. Na základe pomeru šancí sa pozorovalo štatisticky významné zlepšenie svedčiace v prospech skupiny s difelikefalínom do
3. týždňa v štúdii KALM-1 a do 2. týždňa v štúdii KALM-2, ktoré pokračovalo počas každého nasledujúceho týždňa až do 12. týždňa v oboch štúdiách.
 Obrázok 1: Percentuálny podiel pacientov so zlepšením o ≥ 3 body vzhľadom na skóre WI-NRSpodľa jednotlivých týždňov v štúdii KALM-1 a KALM-2 – (populácia s ITT)KALM-1
Obrázok 1: Percentuálny podiel pacientov so zlepšením o ≥ 3 body vzhľadom na skóre WI-NRSpodľa jednotlivých týždňov v štúdii KALM-1 a KALM-2 – (populácia s ITT)KALM-1Počiatočná 1. týždeň 2. týždeň 3. týždeň 4. týždeň 5. týždeň 6. týždeň 7. týždeň 8. týždeň 9. týždeň 10. týždeň 11. týždeň 12. týždeň
hodnota

Návšteva
Sekvencia liečby Difelikefalín Placebo
KALM-2
Počiatočná 1. týždeň 2. týždeň 3. týždeň 4. týždeň 5. týždeň 6. týždeň 7. týždeň 8. týždeň 9. týždeň 10. týždeň 11. týždeň 12. týždeň
hodnota
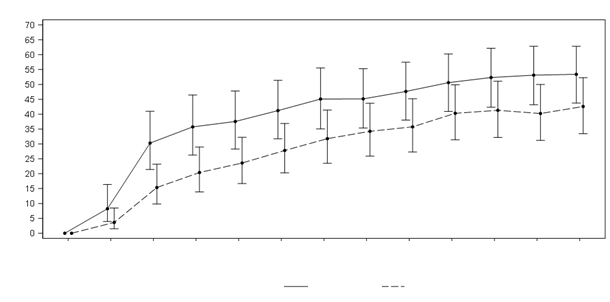
Návšteva
Sekvencia liečby Difelikefalín Placebo
CI = interval spoľahlivosti, ITT = snaha liečiť, LS = najmenšie štvorce, WI-NRS = škála číselného hodnotenia najhoršieho

svrbenia
Otvorené predĺženia štúdiíÚčinok liečby difelikefalínom až do 52 týždňov sa hodnotil pomocou škály svrbenia 5-D, otvorených predĺženiach štúdií KALM-1 a KALM-2 s jednou skupinou zahŕňajúcich 712 pacientov.
U pacientov prechádzajúcich z placeba na difelikefalín na konci dvojito zaslepenej fázy sa pozorovalo zlepšenie v skóre škály svrbenia 5-D po 4 týždňoch liečby s priemernou zmenou LS (SE) od počiatočnej hodnoty porovnateľnou s pacientmi dostávajúcimi difelikefalín od začiatku štúdie: -6,0 (0,22) oproti -5,7 (0,23). Zlepšenie skóre škály svrbenia 5-D sa udržalo v oboch skupinách liečby počas celej 52-týždňovej liečby.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s difelikefalínom
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe pruritu spojeného
s chronickým ochorením obličiek (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiU pacientov so závažnou poruchou funkcie obličiek podstupujúcich hemodialýzu bol celkový telesný klírens difelikefalínu znížený v porovnaní so zdravými osobami a koncentrácie v plazme pomaly klesali, kým počas dialýzy nevymizli. Vzhľadom na to, že počas dialýzy sa odstráni 70 – 80 % difelikefalínu, týmto pacientom sa difelikefalín podáva po každej hemodialýze. Dostupné údaje o variabilite medzi jednotlivými osobami na hemodialýze, ktorým sa podával difelikefalín v množstve
0,5 mikrogramu/kg, poukazujú na to, že variabilita plochy pod krivkou (AUC) môže presiahnuť 30 %.
DistribúciaVäzba difelikefalínu na plazmatické proteíny je nízka až stredná (24 – 32 %) a nie je ovplyvnená
poruchou funkcie obličiek. Priemerný distribučný objem v ustálenom stave bol v rozsahu
145 až 189 ml/kg u zdravých osôb a 214 až 301 ml/kg u pacientov podstupujúcich hemodialýzu so stredne závažným až závažným pruritom. Penetrácia difelikefalínu do centrálnej nervovej sústavy je obmedzená (pod limitom kvantifikácie), ako ukazujú fyzikálno-chemické,
in-vitro a údaje u zvierat.
Eliminácia
U zdravých osôb je primárny spôsob eliminácie difelikefalínu obličkami – približne 81 % dávky sa
vylučuje močom v porovnaní s 11 %, ktoré sa vylučujú stolicou. U zdravých dobrovoľníkov aj osôb na hemodialýze väčšinu dávky vylúčenej do moču a stolice tvoril nezmenený difelikefalín s malými množstvami predpokladaných metabolitov, z čoho žiaden neprekračoval 2,5 %. Priemerný celkový klírens bol v rozsahu 54 až 71 ml/h/kg a priemerný polčas rozpadu bol v rozsahu 2 až 3 hodiny. Naopak, u pacientov na hemodialýze bola eliminácia najmä stolicou podieľajúca sa priemerne na eliminácii 59 % dávky, približne 19 % sa získalo z dialyzátu a približne 11 % sa našlo v moči.
V porovnaní s účastníkmi s normálnou funkciou obličiek sa priemerný celkový klírens (5,3 až
7,5 ml/h/kg) znížil a polčas rozpadu (23 až 31 hodín) zvýšil približne 10-násobne.
Interakcie s inými liekmi
Difelikefalín nie je substrátom CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ani CYP3A4. Nie
je ani inhibítorom CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 alebo CYP3A4/5
a má minimálny alebo žiadny potenciál na indukciu ľudského CYP1A2, CYP2B6 alebo CYP3A. Nie je ani inhibítorom glukuronidačných enzýmov (UGT1A3, UGT1A9 alebo UGT2B7).
Okrem toho, difelikefalín nie je inhibítorom BCRP, BSEP, LAT1, MATE1, MATE2-K, MRP2, OAT1, OAT3, OATP1A2, OATP1B1, OATP1B3, OCT1, OCT2, OCT3, P-glykoproteínu, PEPT1 ani PEPT2 a nie je substrátom pre ASBT, BCRP, BSEP, LAT1, MATE1, MATE2-K, MRP2, OAT1, OAT2, OAT3, OATP1A2, OATP1B1, OATP1B3, OATP2B1, OCT1, OCT2, OCT3, OCTN1, OCTN2, OSTαβ, P-glykoproteín, PEPT1 alebo PEPT2.
Linearita/nelinearita
Farmakokinetika difelikefalínu sa u zdravých osôb preukázala ako lineárna a priamo úmerná oproti
dávke (testované v rozsahoch dávky 1 až 40 v štúdiách s jednou dávkou a 1 až 20 mikrogramov/kg v štúdiách s opakovaným podávaním dávok). U pacientov s chronickým ochorením obličiek na hemodialýze dostávajúcich opakované dávky od 0,5 do 2,5 mikrogramu/kg 3-krát týždenne počas
1 týždňa bola tiež stanovená proporcionalita dávok v rovnovážnom stave. V inej štúdii sa však proporcionalita pozorovala pri dávkach 0,5 a 1 mikrogram/kg, ale nie pri dávke 1,5 mikrogramu/kg. Hodnoty maximálnej koncentrácie v plazme dosiahli ustálený stav do druhej dávky a pre dávku
0,5 mikrogramu/kg bol v jednej štúdii priemerný pomer akumulácie 1,144 na základe AUC0 – 48 h
a v druhej štúdii 1,33 na základe AUC0 – 44 h, čo poukazuje na to, že variabilita parametrov kumulácie
môže presiahnuť 30 %.
Charakteristiky v špecifických skupinách osôb alebo pacientov
Na základe dostupných dôkazov nič nenaznačuje tomu, že by faktory, ako napríklad vek, pohlavie,
etnická príslušnosť alebo mierna až stredne závažná porucha funkcie pečene mali nejaký vplyv na farmakokinetiku difelikefalínu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanej dávke, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
Reprodukčná toxicita
U samcov a samíc potkanov nebola ovplyvnená fertilita, raný embryonálny a prenatálny ani
postnatálny vývin až do 2000-násobku hodnoty AUC u ľudí. U králikov nedošlo k žiadnemu zhoršeniu prenatálneho vývinu napriek výraznej toxicite pre matku pri 30-násobku hodnoty AUC u ľudí.
Difelikefalín u potkanov prechádza cez placentárnu bariéru.
Potenciál zneužívania a závislosti
Štúdie potenciálu zneužívania a závislosti u potkanov naznačujú, že je nepravdepodobné, že by
difelikefalín predstavoval riziko fyzickej závislosti alebo zneužívania.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Kyselina octová (na úpravu pH)
Trihydrát octanu sodného (na úpravu pH) Chlorid sodný
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5. Druh obalu a obsah balenia
Kapruvia sa dodáva v jednorazovej 2 ml sklenenej injekčnej liekovke (typ I) s gumovou zátkou z bromobutylu, hliníkovým tesnením a modrým plastovým odklápacím viečkom.
Veľkosti balení s 3 a 12 injekčnými liekovkami obsahujúcimi 1 ml injekčného roztoku. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Len na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Vifor Fresenius Medical Care Renal Pharma France
100–101 Terrasse Boieldieu
Tour Franklin La Défense 8
92042 Paris la Défense Cedex
Francúzsko
8
. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/22/1643/001
EU/1/22/1643/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 25. apríl 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.