
br />
(>
1,5 až ≤ 3 ×
ULN)
3. stupeň
(> 3 až ≤ 10 × ULN)
|
4. stupeň
(> 10 × ULN)
|
Nepodávajte trastuzumab emtansín, kým sa celkový bilirubín neupraví na ≤ 1. stupeň (>ULN až 1,5 x ULN). Nie je potrebná žiadna úprava dávky.
|
Nepodávajte trastuzumab emtansín, kým sa celkový bilirubín neupraví na ≤ 1. stupeň (>ULN až 1,5 x ULN) a potom znížte dávku (pozri tabuľku 1).
|
Vysaďte trastuzumab emtansín.
|
ULN = horná hranica normálnej hodnoty.
Tabuľka 4 Návod na úpravu dávky z dôvodu trombocytopénie3. stupeň
| 4. stupeň
|
(Krvné doštičky: 25 000 až
< 50 000/mm3)
| (Krvné doštičky: < 25 000/mm3)
|
Nepodávajte trastuzumab emtansín, kým sa počet krvných doštičiek neupraví na
≤ 1. stupeň (t.j. krvné doštičky
≥ 75 000/mm3). Nie je potrebná žiadna úprava dávky.
|
Nepodávajte trastuzumab emtansín, kým sa počet krvných doštičiek neupraví na
≤ 1. stupeň (t.j. krvné doštičky
≥ 75 000/mm3) a potom znížte dávku
(pozri tabuľku 1).
|
Tabuľka 5 Návod na úpravu dávky z dôvodu dysfunkcie ľavej komory
LVEF < 40 %
|
LVEF > 45 %
| LVEF 40 % až
≤ 45 %
a zníženie o
< 10 % bodov od východiskovej hodnoty
| LVEF 40 % až
≤ 45 %
a zníženie o
≥ 10 % bodov od východiskovej hodnoty
|
Symptomatické
CHF
|
Nepodávajte
trastuzumab emtansín.
Opakujte vyšetrenie LVEF v priebehu
3 týždňov. Ak sa potvrdí LVEF
< 40 %, vysaďte trastuzumab emtansín.
| Pokračujte
v liečbe trastuzumab emtansínom.
| Pokračujte
v liečbe trastuzumab emtansínom.
Opakujte vyšetrenie LVEF v priebehu
3 týždňov.
| Nepodávajte
trastuzumab emtansín.
Opakujte vyšetrenie LVEF v priebehu
3 týždňov. Ak sa LVEF neupravila na 10 % bodov od východiskovej hodnoty, vysaďte trastuzumab emtansín.
| Vysaďte
trastuzumab emtansín.
|
LVEF = ejekčná frakcia ľavej komory
Periférna neuropatiaTrastuzumab emtansín sa má dočasne vysadiť u pacientov s periférnou neuropatiou 3. alebo 4. stupňa až do úpravy na ≤ 2. stupeň. Pri opakovanom nasadení liečby sa má zvážiť zníženie dávky podľa schémy pre zníženie dávky (pozri tabuľku 1).
Starší pacienti
U pacientov vo veku ≥ 65 rokov nie je potrebná žiadna úprava dávky. Na stanovenie bezpečnosti a účinnosti u pacientov vo veku ≥ 75 rokov neexistujú dostatočné údaje z dôvodu obmedzených
údajov v tejto podskupine. Populačná farmakokinetická analýza naznačuje, že vek nemá klinicky
významný vplyv na farmakokinetiku trastuzumab emtansínu (pozri časti 5.1 a 5.2).
Pacienti s poruchou funkcie obličiek
U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek nie je potrebné upraviť
úvodnú dávku (pozri časť 5.2). Potenciálnu potrebu úpravy dávky u pacientov so závažnou poruchou funkcie obličiek nie je možné stanoviť z dôvodu nedostatočných údajov a preto sa pacienti so závažnou poruchou funkcie obličiek majú starostlivo sledovať.
Pacienti s poruchou funkcie pečene
Bezpečnosť a účinnosť u pacientov s poruchou funkcie pečene neboli stanovené. Nie je možné určiť
špecifické odporúčania pre dávku (pozri časť 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť u detí a dospievajúcich vo veku do 18 rokov neboli stanovené, pretože použitie sa netýka pediatrickej populácie v indikácii metastatického karcinómu prsníka (metastatic breast
cancer - MBC).
Spôsob podávania
Trastuzumab emtansín musí rozpustiť a riediť a podávať vo forme intravenóznej infúzie zdravotnícky
pracovník. Liek sa nesmie podávať vo forme intravenóznej injekcie alebo bolusovej injekcie. Pokyny na rekonštitúciu a riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Na zlepšenie sledovateľnosti biologických liekov sa má v zdravotnom zázname pacienta jasne zaznamenať (alebo uviesť) obchodný názov podávaného lieku.
Na zabránenie chybám v medikácii je dôležité skontrolovať etikety na injekčných liekovkách, aby sa zabezpečilo, že liek, ktorý sa pripravuje a podáva, je Kadcyla (trastuzumab emtansín) a nie Herceptin (trastuzumab).
Pľúcna toxicita
V klinických štúdiách s trastuzumab emtansínom sa zaznamenali prípady intersticiálnej choroby pľúc (interstitial lung disease - ILD), vrátane pneumonitídy, ktoré niekedy vedli k syndrómu akútnej respiračnej tiesne alebo k fatálnemu následku (pozri časť 4.8). Medzi prejavy a príznaky patrí dýchavičnosť, kašeľ, vyčerpanosť a pľúcne infiltráty.
U pacientov, u ktorých bola diagnostikovaná ILD alebo pneumonitída, sa odporúča natrvalo vysadiť
trastuzumab emtansín.
U pacientov s dýchavičnosťou v pokoji v dôsledku komplikácií pokročilého nádorového ochorenia a pridružených ochorení môže byť zvýšené riziko pľúcnych príhod.
Hepatotoxicita
Hepatotoxicita, hlavne vo forme asymptomatických nárastov koncentrácií sérových transamináz (transaminitída 1.-4. stupňa), sa pozorovala počas liečby trastuzumab emtansínom v klinických štúdiách (pozri časť 4.8). Zvýšenie transamináz bolo zvyčajne prechodné s najväčším zvýšením 8. deň
po podaní liečby a následnou úpravou na 1. alebo nižší stupeň pred ďalším cyklom. Pozoroval sa aj kumulatívny účinok na transaminázy (podiel pacientov s abnormalitami ALT/AST 1.-2. stupňa sa zvyšuje v nasledujúcich cykloch).
U pacientov so zvýšenými transaminázami klesli hladiny vo väčšine prípadov na 1. stupeň alebo na normálnu hodnotu v priebehu 30 dní od poslednej dávky trastuzumab emtansínu (pozri časť 4.8).
U pacientov liečených trastuzumab emtansínom sa pozorovali závažné hepatobiliárne poruchy, vrátane nodulárnej regeneratívnej hyperplázie (NRH) pečene, pričom niektoré mali fatálny následok z dôvodu poškodenia pečene spôsobeného liekom. Diagnostika pozorovaných prípadov mohla byť skomplikovaná súbežnými ochoreniami a/alebo súbežne podávanými liekmi so známym hepatotoxickým potenciálom.
Pred začiatkom liečby a pred každou dávkou sa má sledovať funkcia pečene. Pacienti
s východiskovým zvýšením ALT (napr. z dôvodu pečeňových metastáz) môžu byť náchylní
k poškodeniu pečene s vyšším rizikom pečeňovej príhody 3.-5. stupňa alebo k zvýšeniu hodnôt testu pečeňovej funkcie. Zníženia dávky alebo vysadenie z dôvodu zvýšených sérových transamináz
a celkového bilirubínu sú uvedené v časti 4.2.U pacientov liečených trastuzumab emtansínom boli
z biopsií pečene identifikované prípady nodulárnej regeneratívnej hyperplázie (NRH) pečene. NRH je zriedkavé ochorenie pečene charakterizované rozšírenou benígnou transformáciou pečeňového parenchýmu na malé regeneratívne noduly; NRH môže viesť k ne-cirhotickej portálnej hypertenzii. Diagnózu NRH možno potvrdiť len histopatologicky. NRH sa má vziať do úvahy u všetkých pacientov s klinickými príznakmi portálnej hypertenzie a/alebo s ochorením podobným cirhóze pozorovanom na snímku počítačovej tomografie (computed tomography - CT) pečene, ale s
normálnymi hodnotami transamináz a bez iných prejavov cirhózy. Po diagnostike NRH sa musí liečba trastuzumab emtansínom natrvalo vysadiť.
Trastuzumab emtansín sa neskúmal u pacientov s vyššími sérovými transaminázami ako 2,5 × ULN alebo celkovým bilirubínom > 1,5 × ULN pred začatím liečby. Liečba pacientov so sérovými transaminázami > 3 × ULN a súčasným celkovým bilirubínom > 2 × ULN sa má natrvalo vysadiť.
Dysfunkcia ľavej komory
U pacientov liečených trastuzumab emtansínom je zvýšené riziko rozvoja dysfunkcie ľavej komory. U
pacientov liečených trastuzumab emtansínom sa pozorovala ejekčná frakcia ľavej komory (LVEF)
< 40 % a preto je potenciálnym rizikom symptomatické kongestívne zlyhanie srdca (congestive heart failure - CHF) (pozri časť 4.8). Medzi všeobecné rizikové faktory pre srdcové príhody a faktory identifikované v adjuvantných štúdiách s karcinómom prsníka liečeným trastuzumabom patrí pokročilý vek (> 50 rokov), nízke východiskové hodnoty LVEF (< 55 %), nízke hladiny LVEF pred alebo po použití paklitaxelu ako adjuvantnej liečby, predchádzajúce alebo súčasné podávanie antihypertenzív, predchádzajúca liečba antracyklínom a vysoké BMI (> 25 kg/m2).
Štandardné vyšetrenie funkcie srdca (echokardiogram alebo MUGA (multigated acquisition)) sa má vykonať pred začatím liečby a počas liečby v pravidelných intervaloch (napr. každé tri mesiace). V klinických štúdiách mali pacienti východiskovú LVEF ≥ 50 %. Pacienti s anamnézou kongestívneho zlyhania srdca (CHF), závažnej srdcovej arytmie vyžadujúcej liečbu, s anamnézou infarktu myokardu alebo nestabilnej angíny pektoris v priebehu 6 mesiacov od randomizácie alebo súčasnou pokojovou dýchavičnosťou z dôvodu pokročilej malignity boli vylúčení z klinických štúdií. Dávka sa má odložiť alebo liečba sa má podľa potreby vysadiť v prípadoch dysfunkcie ľavej komory (pozri časť 4.2).
Reakcie súvisiace s infúziou
Liečba trastuzumab emtansínom sa neskúmala u pacientov, u ktorých bola liečba trastuzumabom natrvalo vysadená z dôvodu reakcií súvisiacich s infúziou (infusion-related reactions - IRR); u týchto pacientov sa liečba neodporúča. Pacienti sa majú starostlivo sledovať kvôli reakciám súvisiacim s
infúziou, a to najmä počas prvej infúzie.
Zaznamenali sa reakcie súvisiace s infúziou (z dôvodu uvoľnenia cytokínov), ktoré sú charakterizované jedným alebo viacerými z nasledujúcich príznakov: sčervenanie, zimnica, pyrexia,
dýchavičnosť, hypotenzia, sipot, bronchospazmus a tachykardia. Vo všeobecnosti tieto príznaky nie sú závažné (pozri časť 4.8). U väčšiny pacientov tieto reakcie ustúpili v priebehu niekoľkých hodín až jedného dňa po ukončení infúzie. Liečba sa má prerušiť u pacientov so závažnými IRR až do ústupu prejavov a symptómov. Pri zvažovaní opakovanej liečby treba vychádzať z klinického posúdenia závažnosti reakcie. Liečba sa musí natrvalo vysadiť v prípade život ohrozujúcej reakcie súvisiacej s infúziou (pozri časť 4.2).
Hypersenzitívne reakcie
Liečba trastuzumab emtansínom sa neskúmala u pacientov, u ktorých bola liečba trastuzumabom natrvalo vysadená z dôvodu hypersenzitivity; liečba trastuzumab emtansínom sa u týchto pacientov
neodporúča.
Pacientov je potrebné starostlivo sledovať kvôli hypersenzitívnym/alergickým reakciám, ktoré môžu mať rovnaké klinické prejavy ako IRR. V klinických štúdiách s trastuzumab emtansínom sa pozorovali závažné anafylaktické reakcie. Lieky na liečbu takýchto reakcií, rovnako ako záchranné vybavenie, musí byť k dispozícii na okamžité použitie. V prípade skutočnej hypersenzitívnej reakcie (pri ktorej závažnosť reakcie sa zvyšuje s nasledujúcimi infúziami) sa má liečba trastuzumab emtansínom natrvalo vysadiť.
Trombocytopénia
Trombocytopénia alebo zníženie počtu krvných doštičiek bola často hlásená v súvislosti s trastuzumab emtansínom a bola to najčastejšia nežiaduca reakcia vedúca k vysadeniu liečby (pozri časť 4.8). V klinických štúdiách bola incidencia a závažnosť trombocytopénie vyššia u ázijských pacientov (pozri časť 4.8).
Pozorovali sa prípady krvácania s fatálnym následkom. V klinických štúdiách sa zaznamenali závažné prípady hemoragických príhod, vrátane hemorágie centrálneho nervového systému; tieto príhody nezáviseli na národnosti. V niektorých pozorovaných prípadoch pacienti užívali aj antikoagulačnú terapiu.
Odporúča sa sledovanie počtu krvných doštičiek a to pred každou dávkou trastuzumab emtansínu. Pacientov s trombocytopéniou (≤ 100 000/mm3) a pacientov na antikoagulačnej liečbe (napr. warfarín, heparín, nízkomolekulárne heparíny) je potrebné starostlivo sledovať počas liečby trastuzumab emtansínom. Trastuzumab emtansín sa neskúmal u pacientov s počtom krvných doštičiek
≤ 100 000/mm3 pred začatím liečby. V prípade zníženého počtu krvných doštičiek na 3. alebo vyšší stupeň (< 50 000/mm3) nepodávajte trastuzumab emtansín, kým sa počet krvných doštičiek neupraví
na 1. stupeň (≥ 75 000/mm3) (pozri časť 4.2).
Neurotoxicita
V klinických štúdiách s trastuzumab emtansínom sa zaznamenala periférna neuropatia, predovšetkým
1. stupňa a prevažne zmyslová. Pacienti s ≥ 3. stupňom periférnej neuropatie pri vstupe do štúdie boli vylúčení z klinických štúdií. Liečba trastuzumab emtansínom sa má dočasne prerušiť u pacientov
s periférnou neuropatiou 3. alebo 4. stupňa až do ústupu symptómov alebo zlepšenia na ≤ 2. stupeň. Pacienti sa majú priebežne klinicky sledovať kvôli prejavom/symptómom neurotoxicity.
Obsah sodíka v pomocných látkach
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t.j. je v podstate „bez sodíka“.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne oficiálne interakčné štúdie.
In vitro štúdie metabolizmu v mikrozómoch ľudskej pečene ukazujú, že DM1, zložka trastuzumab emtansínu, sa metabolizuje predovšetkým prostredníctvom CYP3A4 a v menšej miere aj CYP3A5. Súčasnému používaniu silných inhibítorov CYP3A4 (napr. ketokonazolu, itrakonazolu, klaritromycínu, atazanaviru, indinaviru, nefazodónu, nelfinaviru, ritonaviru, sachinaviru, telitromycínu a
vorikonazolu) s trastuzumab emtansínom sa treba vyhýbať z dôvodu možnosti zvýšenia expozície a
toxicity DM1. Zvážte alternatívny liek so žiadnym alebo minimálnym potenciálom inhibície CYP3A4. Ak je súčasné používanie silných inhibítorov CYP3A4 nevyhnutné a ak je to možné, zvážte odloženie liečby trastuzumab emtansínom, kým sa silné inhibítory CYP3A4 nevylúčia z krvného obehu
(približne 3 polčasy eliminácie inhibítorov). Ak sa silný inhibítor CYP3A4 podáva súbežne a liečbu trastuzumab emtansínom nie je možné odložiť, pacienti majú byť starostlivo sledovaní z dôvodu nežiaducich reakcií.
4.6 Fertilita, gravidita a laktácia
Antikoncepcia u mužov a žien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby trastuzumab emtansínom a ešte 6 mesiacov po poslednej dávke trastuzumab emtansínu. Mužskí pacienti alebo ich partnerky majú tiež používať účinnú antikoncepciu.
Gravidita
Nie sú k dispozícii údaje o použití trastuzumab emtansínu u gravidných žien. Trastuzumab, zložka trastuzumab emtansínu, môže spôsobiť poškodenie alebo úmrtie plodu, ak sa podáva gravidnej žene. V sledovaní po uvedení lieku na trh sa u gravidných žien užívajúcich trastuzumab zaznamenali prípady oligohydramniónu, v niektorých prípadoch spojené s fatálnou hypopláziou pľúc. Štúdie na
zvieratách s maytansínom, príbuznou chemickou zlúčeninou rovnakej maytansinoidovej triedy ako
DM1, naznačujú, že DM1, cytotoxická zložka trastuzumab emtansínu inhibujúca mikrotubuly, je pravdepodobne teratogénny a potenciálne embryotoxický (pozri časť 5.3).
Podávanie trastuzumab emtansínu gravidným ženám sa neodporúča a ženy majú byť informované o možnosti poškodenia plodu predtým, ako otehotnejú. Ženy, ktoré otehotnejú, musia okamžite kontaktovať svojho lekára. Ak je liečená trastuzumab emtansínom tehotná žena, odporúča sa starostlivé sledovanie multidisciplinárnou skupinou lekárov.
Laktácia
Nie je známe, či sa trastuzumab emtansín vylučuje do ľudského mlieka. Vzhľadom k tomu, že mnoho liekov sa vylučuje do ľudského mlieka a vzhľadom na možnosť závažných nežiaducich reakcií
u dojčených dojčiat, pred začatím liečby trastuzumab emtansínom majú dojčiace ženy prerušiť
dojčenie. Ženy môžu začať dojčenie 6 mesiacov po skončení liečby.
Fertilita
Neuskutočnili sa žiadne reprodukčné a vývojové toxikologické štúdie s trastuzumab emtansínom.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Trastuzumab emtansín nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Význam hlásených nežiaducich reakcií, ako sú únava, bolesti hlavy, závrat a rozmazané videnie na schopnosť viesť vozidlá a obsluhovať stroje nie je známy. Pacientom, u ktorých sa vyskytli reakcie súvisiace s infúziou, sa má odporučiť, aby neviedli vozidlá ani obsluhovali stroje, kým príznaky neustúpia.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnosť trastuzumab emtansínu sa hodnotila u 884 pacientov s karcinómom prsníka v klinických
štúdiách. U tejto populácie pacientov:
• najčastejšie závažné nežiaduce liekové reakcie boli pyrexia, trombocytopénia, vracanie, bolesť
brucha, nauzea, zápcha, hnačka, dýchavičnosť a pneumonitída.
• najčastejšie nežiaduce liekové reakcie (adverse drug reaction - ADR) (≥ 25 %) s trastuzumab emtansínom boli hemorágia (vrátane epistaxy), zvýšenie transamináz, únava, bolesť svalov a kostí a bolesť hlavy. Väčšina ADR bola hlásená ako 1. alebo 2. stupňa závažnosti.
• najčastejšie ADR 3. alebo 4. stupňa podľa Všeobecných terminologických kritérií pre nežiaduce účinky Národného onkologického ústavu (National Cancer Institute - Common Terminology Criteria for adverse events - NCI-CTCAE) (> 2 %) boli trombocytopénia, únava, zvýšenie transamináz, anémia, hypokaliémia, bolesť svalov a kostí a neutropénia.
Zoznam nežiaducich reakcií zoradených do tabuľkyADR pozorované u 884 pacientov liečených trastuzumab emtansínom sú uvedené v tabuľke 6. ADR
sú uvedené nižšie podľa tried orgánových systémov MedDRA (TOS) a kategórií frekvencie. Kategórie frekvencie sú definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000
až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (nemožno odhadnúť z dostupných údajov). V rámci jednotlivých skupín frekvencií a TOS sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. ADR boli hlásené použitím NCI-CTCAE pre hodnotenie toxicity.
Tabuľka 6 Zoznam ADR u pacientov liečených trastuzumab emtansínom zoradených do tabuľky
Trieda orgánových systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Infekcie a nákazy
| infekcia močových
ciest
|
|
|
Poruchy krvi a lymfatického
systému
| trombocytopénia,
anémia
| neutropénia,
leukopénia
|
|
Poruchy imunitného
systému
|
| precitlivenosť na liek
|
|
Poruchy metabolizmu
a výživy
| hypokaliémia
|
|
|
Psychické poruchy
| insomnia
|
|
|
Poruchy nervového systému
| periférna neuropatia,
bolesť hlavy, závrat
| dysgeúzia, porucha
pamäti
|
|
Poruchy oka
|
| suché oko,
konjunktivitída, rozmazané videnie, zvýšená lakrimácia
|
|
Poruchy srdca a srdcovej
činnosti
|
| dysfunkcia ľavej
komory
|
|
Poruchy ciev
| hemorágia
| hypertenzia
|
|
Poruchy dýchacej sústavy,
hrudníka a mediastína
| epistaxa, kašeľ,
dýchavičnosť
|
| pneumonitída
(ILD)
|
Poruchy
gastrointestinálneho traktu
| stomatitída, hnačka,
vracanie, nauzea, zápcha, sucho v ústach, bolesť brucha
| dyspepsia, krvácanie
z ďasien
|
|
Trieda orgánových systémov
|
Veľmi časté
|
Časté
|
Menej časté
|
Poruchy pečene a žlčových
ciest
|
|
|
hepatotoxicita,
zlyhanie pečene, nodulárna regeneratívna hyperplázia, portálna hypertenzia
|
Poruchy kože a podkožného
tkaniva
|
vyrážka
|
pruritus, alopécia,
porucha nechtov, syndróm palmárno- plantárnej erytrodyzestézie, urtikária
|
|
Poruchy kostrovej a svalovej sústavy a spojivového
tkaniva
|
muskuloskeletálna bolesť, artralgia,
myalgia
|
|
|
Celkové poruchy a reakcie v
mieste podania
|
únava, pyrexia,
asténia, zimnica
|
periférny edém
|
extravazácia
v mieste injekcie
|
Laboratórne a funkčné
vyšetrenia
|
zvýšené
transaminázy
|
zvýšená alkalická
fosfatáza v krvi
|
|
Úrazy, otravy a komplikácie
liečebného postupu
|
|
reakcie súvisiace s
infúziou
|
|
Popis vybraných nežiaducich reakcií
Zvýšené transaminázy (AST/ALT)
Zvýšenie sérových transamináz (1. -4. stupňa) sa pozorovalo počas liečby trastuzumab emtansínom v klinických štúdiách (pozri časť 4.4). Zvýšenie aminotransferáz bolo zvyčajne prechodné. Pozoroval sa
kumulatívny účinok trastuzumab emtansínu na transaminázy, ktorý zvyčajne ustúpil po skončení
liečby. Zvýšenie transamináz sa v klinických štúdiách zaznamenalo u 28 % pacientov. U 4,1 % a
2,8 % pacientov sa zaznamenali zvýšené hladiny AST a ALT 3. alebo 4. stupňa a zvyčajne sa vyskytovali počas úvodných liečebných cyklov (1-6). Vo všeobecnosti hepatálne príhody ≥ 3. stupňa nesúviseli so zlým klinickým výsledkom; následne sledované hodnoty mali tendenciu zlepšenia do rozsahu, ktoré umožňovalo pacientovi zostať v štúdii a naďalej dostávať skúmanú liečbu v rovnakej alebo nižšej dávke. Medzi expozíciou trastuzumab emtansínu (AUC), maximálnou sérovou koncentráciou trastuzumab emtansínu (Cmax), celkovou expozíciou trastuzumabu (AUC) alebo Cmax DM1 a zvýšením transamináz sa nepozoroval žiadny vzťah. Úpravy dávok v prípade zvýšenia transamináz, pozri časti 4.2 a 4.4.
Dysfunkcia ľavej komoryDysfunkcia ľavej komory sa zaznamenala u 2,0 % pacientov v klinických štúdiách s trastuzumab emtansínom. Väčšinu príhod tvorilo asymptomatické zníženie LVEF 1. alebo 2. stupňa. Príhody 3. alebo 4. stupňa sa zaznamenali u 0,3 % pacientov. Tieto menej časté príhody 3. alebo 4. stupňa sa zvyčajne vyskytovali v úvodných liečebných cykloch (1-2). Ďalšie sledovanie LVEF sa odporúča u pacientov s LVEF ≤ 45 % (pozri tabuľku 5 v časti 4.2 pre špecifické úpravy dávky).
Reakcie súvisiace s infúziouReakcie súvisiace s infúziou sú charakterizované jedným alebo viacerými z nasledujúcich symptómov: sčervenanie, zimnica, pyrexia, dýchavičnosť, hypotenzia, sipot, bronchospazmus a tachykardia. Reakcie súvisiace s infúziou boli hlásené u 4,5 % pacientov v klinických štúdiách s trastuzumab
emtansínom, pričom jedna príhoda bola 3. stupňa a neboli hlásené žiadne príhody 4. stupňa. Reakcie
súvisiace s infúziou ustúpili v priebehu niekoľkých hodín až jedného dňa po ukončení infúzie. V klinických štúdiách sa nepozoroval žiadny vzťah s dávkou. Úpravy dávok v prípade reakcií súvisiacich s infúziou, pozri časti 4.2 a 4.4.
Hypersenzitívne reakcie
Hypersenzitivita sa zaznamenala u 2,6 % pacientov v klinických štúdiách s trastuzumab emtansínom, pričom nebola hlásená žiadna príhoda 3. ani 4. stupňa. Celkovo bola väčšina hypersenzitívnych
reakcií miernej alebo strednej závažnosti a po liečbe ustúpila. Úpravy dávok v prípade hypersenzitívnych reakcií, pozri časti 4.2 a 4.4.
Trombocytopénia
Trombocytopénia alebo znížený počet krvných doštičiek sa zaznamenal u 31,4 % pacientov v klinických štúdiách s trastuzumab emtansínom a bola to najčastejšia nežiaduca reakcia vedúca k prerušeniu liečby (1,4 %). Väčšina pacientov mala príhody 1. alebo 2. stupňa (≥ 50 000/mm3), pričom najnižšia hodnota sa vyskytovala do 8. dňa a zvyčajne sa do nasledujúcej plánovanej dávky upravili na
0. alebo 1. stupeň (≥ 75 000/mm3). V klinických štúdiách bola incidencia a závažnosť
trombocytopénie vyššia u ázijských pacientov. Nezávisle od rasy bola incidencia príhod 3. alebo 4. stupňa (< 50 000/mm3) 11,3 % u pacientov liečených trastuzumab emtansínom. Závažné hemoragické príhody (≥ 3. stupeň) sa vyskytli u 1,7 % všetkých pacientov liečených trastuzumab emtansínom
a u 1 % ázijských pacientov liečených trastuzumab emtansínom. V niektorých pozorovaných prípadoch pacienti užívali aj antikoagulačnú liečbu. Pozorovali sa prípady hemoragických príhod s fatálnym následkom. Pre úpravy dávok v prípade trombocytopénie, pozri časť 4.2 a 4.4.
Imunogenicita
Rovnako ako u všetkých terapeutických proteínov, existuje možnosť imunitnej odpovede na trastuzumab emtansín. Celkovo 836 pacientov zo šiestich klinických štúdií bolo testovaných v rôznych časových bodoch na odpovede anti-terapeutických protilátok (anti-therapeutic antibody - ATA) na trastuzumab emtansín. Po podaní 5,3 % (44/836) pacientov malo pozitívne výsledky testovania
protilátok proti trastuzumab emtansínu v jednom alebo vo viacerých časových bodov po podaní dávky.
Klinický význam protilátok proti trastuzumab emtansínu nie je doposiaľ známy.
Extravazácia
V klinických štúdiách s trastuzumab emtansínom sa pozorovali sekundárne reakcie po extravazácii. Tieto reakcie boli mierne a pozostávali z erytému, citlivosti, podráždenia kože, bolesti alebo opuchu v
mieste infúzie. Tieto reakcie sa pozorovali častejšie v priebehu 24 hodín po infúzii. Špecifická liečba extravazácie trastuzumab emtansínu nie je v súčasnosti známa.
Laboratórne abnormality
V tabuľke 7 sú uvedené laboratórne anomálie pozorované u pacientov liečených trastuzumab
emtansínom v klinickej štúdii TDM4370g/BO21977.
Tabuľka 7 Laboratórne abnormality pozorované u pacientov liečených trastuzumab emtansínom v štúdii TDM4370g/BO21977
|
Trastuzumab emtansín
|
|
|
|
Paramete
r
|
stupne (%)
|
3.stupeň (%)
|
4.stupeň (%)
|
Hepatálne
|
Zvýšený bilirubín
|
20
|
< 1
|
0
|
Zvýšené AST
|
98
|
7
|
< 1
|
Zvýšené ALT
|
82
|
5
|
< 1
|
Hematologické
|
Znížený počet trombocytov
|
84
|
14
|
3
|
Znížený hemoglobín
|
62
|
4
|
1
|
Znížený počet neutrofilov
|
39
|
4
|
< 1
|
Draslík
|
Znížená hladina draslíka
|
34
|
3
|
<1
|
|
|
Všetky
Hlásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieNeexistuje žiadne známe antidotum pri predávkovaní trastuzumab emtansínom. V prípade predávkovania sa má pacient starostlivo sledovať kvôli prejavom alebo príznakom nežiaducich reakcií a zahájeniu vhodnej symptomatickej liečby. V súvislosti s liečbou trastuzumab emtansínom sa zaznamenali prípady predávkovania, pričom väčšina súvisela s trombocytopéniou a zaznamenalo sa jedno úmrtie. Vo fatálnom prípade bol pacientovi podaný trastuzumab emtansín 6 mg/kg nesprávne
a pacient zomrel približne tri týždne po predávkovaní; kauzálny vzťah s trastuzumab emtansínom sa nestanovil.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Cytostatikum; iné cytostatiká; monoklonálne protilátky
ATC kód: L01XC14
Mechanizmus účinkuKadcyla, trastuzumab emtansín, je konjugát HER2-cielenej protilátky a liečiva, ktorý obsahuje humanizovaný anti-HER2 IgG1, trastuzumab, kovalentne naviazaný na inhibítora mikrotubulov DM1 (derivát maytansínu) prostredníctvom stabilného tioéterového linkera MCC (4-[N-maleimidometyl] cyklohexán-1-karboxylát). Emtansín sa vzťahuje na MCC-DM1 komplex. Na každú molekulu trastuzumabu je konjugovaných priemerne 3,5 molekúl DM1.
Konjugácia DM1 na trastuzumab spôsobuje selektivitu cytostatika pre nádorové bunky s nadmernou expresiou HER2, a tým zvyšuje intracelulárne dodanie DM1 priamo do malígnych buniek. Po naviazaní na HER2 trastuzumab emtansín podstupuje receptorom sprostredkovanú internalizáciu a
a následnú lyzozomálnu degradáciu, čo má za následok uvoľňovanie cytotoxických katabolitov obsahujúcich DM1 (predovšetkým lyzín-MCC-DM1).
Trastuzumab emtansín má mechanizmus účinku oboch trastuzumabu aj DM1:
• Trastuzumab emtansín, tak ako aj trastuzumab, sa viaže na doménu IV HER2 extracelulárnej domény (extracellular domain - ECD), ako aj na Fcγ receptory a komplement C1q. Okrem toho trastuzumab emtansín, tak ako trastuzumab, inhibuje odštiepenie HER2 ECD, inhibuje signalizáciu prostredníctvom fosfatidylinozitol 3-kinázovej (PI3-K) dráhy a sprostredkováva na protilátke závislú bunkami sprostredkovanú cytotoxicitu (antibody-dependent cell-mediated cytotoxicity - ADCC) v ľudských bunkách karcinómu prsníka s nadmernou expresiou HER2.
• DM1, cytotoxická zložka trastuzumab emtansínu, sa viaže na tubulín. Inhibíciou polymerizácie tubulínu, DM1 aj trastuzumab emtansín spôsobuje zastavenie buniek v G2/M fáze bunkového cyklu, čo nakoniec vedie k apoptotickej bunkovej smrti. Výsledky cytotoxických testov in vitro ukazujú, že DM1 je 20- 200-krát silnejší ako taxány a vinka alkaloidy.
• MCC linker je určený na obmedzenie systémového uvoľňovania a zvýšenie cielenej dodávky
DM1, o čom svedčí detekcia veľmi nízkych hladín voľného DM1 v plazme.
Klinická účinnosť
TDM4370g/BO21977
Randomizovaná, multicentrická, medzinárodná, nezaslepená klinická štúdia fázy III sa uskutočnila u pacientov s HER2-pozitívnym neoperovateľným lokálne pokročilým karcinómom prsníka (locally
advanced breast cancer - LABC) alebo MBC, ktorí boli predtým liečení taxánmi a trastuzumabom, vrátane pacientov, ktorí boli predtým liečení trastuzumabom a taxánom v adjuvantnej liečbe
a u ktorých sa vyskytol relaps v priebehu adjuvantnej liečby alebo do šiestich mesiacov po ukončení adjuvantnej liečby. Vhodní boli iba pacienti s výkonnostným stavom Východnej spolupracujúcej onkologickej skupiny (Eastern Cooperative Oncology Group - ECOG) 0 alebo 1. Pred zaradením do
štúdie museli mať vzorky nádoru centrálne potvrdený pozitívny HER2 stav definovaný ako skóre 3 +
pomocou IHC alebo amplifikácie génu pomocou ISH. Východiskové charakteristiky pacientov a nádoru boli dobre vyvážené medzi liečebnými skupinami. Pacienti s liečenými mozgovými metastázami boli spôsobilí na zaradenie do štúdie, pokiaľ nepotrebovali liečbu na kontrolu príznakov. U pacientov randomizovaných na trastuzumab emtansín bol medián veku 53 rokov, pričom väčšina pacientov boli ženy (99,8 %), väčšina boli belosi (72 %) a 57 % pacientov malo ochorenie pozitivitou estrogénových receptorov a/alebo progesterónových receptorov. Štúdia porovnávala bezpečnosť a účinnosť trastuzumabu emtansínu s bezpečnosťou a účinnosťou lapatinibu v kombinácii s kapecitabínom. Celkovo bolo randomizovaných 991 pacientov na trastuzumab emtansín alebo lapatinib v kombinácii s kapecitabínom a to nasledovne:
• Skupina s trastuzumab emtansínom: trastuzumab emtansín 3,6 mg/kg intravenózne po dobu
30-90 minút v 1. deň 21-dňového cyklu
• Kontrolná skupina (lapatinib v kombinácii s kapecitabínom): lapatinib 1 250 mg/denne perorálne jedenkrát denne v priebehu 21-dňového cyklu v kombinácii s kapecitabínom
1 000 mg/m2 perorálne dvakrát denne 1.-14. deň 21-dňového cyklu
Ko-primárne ciele účinnosti štúdie boli prežívanie bez progresie (progression-free survival - PFS) podľa posúdenia nezávislou hodnotiacou komisiou (IRC) a celkové prežívanie (overall survival - OS) (pozri tabuľku 8 a obrázky 1 až 2).
Počas klinickej štúdie bol tiež hodnotený čas do progresie symptómov, definovaný poklesom skóre o 5 bodov odvodeným z podškály indexu výsledkov klinických štúdií s karcinómom prsníka (Trials Outcome Index-Breast – TOI-B) funkčného hodnotenia dotazníka kvality života pri liečbe karcinómu prsníka (Functional Assessment of Cancer Therapy-Breast Quality of Life - FACT-B QoL) . Zmena TOI-B o 5 bodov sa považuje za klinicky významnú. Kadcyla predĺžila čas do progresie symptómov hlásených pacientom na 7,1 mesiaca v porovnaní s 4,6 mesiacov u kontrolnej skupiny (Hazard Ratio
0,796 (0,667, 0,951); p-hodnota 0,0121). Údaje pochádzajú z nezaslepenej štúdie a nie je možné vyvodiť žiadne jednoznačné závery.
Tabuľka 8 Súhrn účinnosti zo štúdie TDM4370g/BO21977 (EMILIA)
|
Lapatinib + Kapecitabín n = 496
|
Trastu
z
u
mab emtansín
n = 495
|
Primárne ciele
|
Prežívanie bez progresie (PFS)
hodnotené IRC
|
|
Počet (%) pacientov s udalosťou
|
304 (61,3 %)
|
265 (53,5 %)
|
Medián trvania PFS (mesiace)
|
6,4
|
9,6
|
Hazard Ratio (stratifikovaný*)
|
0,650
|
95 % IS pre Hazard Ratio
|
(0,549, 0,771)
|
p-hodnota (Log-rank test, stratifikovaný*)'
|
< 0,0001
|
Celkové prežívanie (OS)**
|
|
Počet (%) pacientov, ktorí zomreli
|
182 (36,7 %)
|
149 (30,1 %)
|
Medián trvania prežívania (mesiace)
|
25,1
|
30,9
|
Hazard Ratio (stratifikovaný*)
|
0,682
|
95 % IS pre pomer rizika
|
(0,548, 0,849)
|
p-hodnota (Log-rank test*)
|
0,0006
|
Kľúčové sekundárne ciele
|
PFS hodnotené skúšajúcim
|
|
Počet (%) pacientov s udalosťou
|
335 (67,5 %)
|
287 (58,0 %)
|
Medián trvania PFS (mesiace)
|
5,8
|
9,4
|
Hazard Ratio (95 % IS)
|
0,658 (0,560, 0,774)
|
p-hodnota (Log-rank test*)
|
< 0,0001
|
Miera objektívnej odpovede (ORR)
|
|
Pacienti s merateľným ochorením
|
389
|
397
|
Počet pacientov s OR (%)
|
120 (30,8 %)
|
173 (43,6 %)
|
Rozdiel (95 % IS)
|
12,7 % (6,0, 19,4)
|
p-hodnota (Mantel-Haenszelov chí- kvadrát test*)
|
0,0002
|
Trvanie objektívnej odpovede
(mesiace)
|
|
Počet pacientov s OR
|
120
|
173
|
Medián 95 % IS
|
6,5 (5,5, 7,2)
|
12,6 (8,4, 20,8)
|
OS (overall survival): celkové prežívanie; PFS (progression-free survival): prežívanie bez
progresie; ORR (objective response rate): miera objektívnej odpovede; OR (objective response): objektívna odpoveď; IRC (independent review committee): nezávislá hodnotiaca komisia; HR (hazard ratios): pomery rizika; IS: interval spoľahlivosti
* Stratifikované: celosvetovo (USA, západná Európa, iné), počet predchádzajúcich chemoterapeutických režimov pre lokálne pokročilé alebo metastatické ochorenie (0-1
oproti > 1) a
viscerálne
oproti neviscerálnemu ochoreniu.
** Predbežná analýza OS sa uskutočnila, keď sa pozorovalo 331 udalostí. Vzhľadom k tomu, že hranica účinnosti bola prekročená v tejto analýze, táto analýza sa považuje za konečnú.
Liečebný prínos sa pozoroval v podskupine pacientov, u ktorých došlo k relapsu v priebehu 6 mesiacov od ukončenia adjuvantnej liečby a ktorí neboli predtým liečení systémovou protinádorovou liečbou metastatického ochorenia (n=118); Hazard Ratio pre PFS a OS bol 0,51 (95 % IS: 0,30, 0,85) a 0,61 (95 % IS: 0,32, 1,16), v uvedenom poradí. Medián PFS pre skupinu s trastuzumab emtansínom bol 10,8 mesiacov a medián OS sa nedosiahol, v porovnaní s 5,7 mesiacov a 27,9 mesiacov,
v uvedenom poradí, pre skupinu s lapatinibom v kombinácii s kapecitabínom.
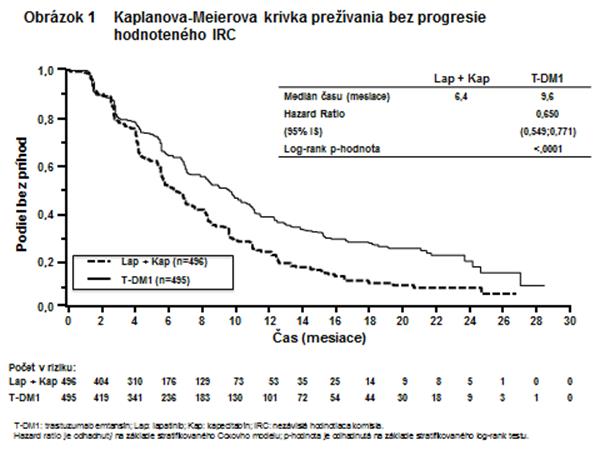

V štúdii TDM4370g/BO21977 sa pozoroval pretrvávajúci prínos liečby trastuzumab emtansínom vo väčšine vopred špecifikovaných hodnotených podskupín, čo podporuje robustnosť celkového výsledku. V podskupine pacientov s ochorením s negatívnymi hormonálnymi receptormi (n=426) bol Hazard Ratio pre PFS 0,56 (95 % IS: 0,44, 0,72) a pre OS 0,75 (95 % IS: 0,54, 1,03). V podskupine pacientov
s ochorením s pozitívnymi hormonálnymi receptormi (n=545) bol Hazard Ratio pre PFS 0,72 (95 % IS: 0,58, 0,91) a pre OS 0,62 (95 % IS: 0,46, 0,85).
V podskupine pacientov s nemerateľným ochorením (n=205) bol Hazard Ratio, na základe hodnotenia
IRC, pre PFS 0,91 (95 % IS: 0,59, 1,42) a pre OS 0,96 (95 % IS: 0,54, 1,68). U pacientov vo veku
≥ 65 rokov (n=138 v oboch liečebných skupinách) bol Hazard Ratio pre prežívanie bez progresie (PFS) 1,06 (95 % IS: 0,68, 1,66) a pre celkové prežívanie (OS) 1,05 (95% IS: 0,58, 1,91). U pacientov vo veku 65 až 74 rokov (n=113) bol Hazard Ratio, na základe hodnotenia IRC, pre PFS 0,88 (95 % IS:
0,53, 1,45) a pre OS 0,74 (95 % IS: 0,37, 1,47). U pacientov vo veku 75 rokov alebo starších bol Hazard Ratio, na základe hodnotenia IRC, pre PFS 3,51 (95 % IS: 1,22, 10,13) a pre OS 3,45 (95 % IS: 0,94, 12,65). Podskupina pacientov vo veku 75 rokov alebo starších nepreukázala prínos pre PFS ani OS, bola však príliš malá (n = 25) na vyvodenie definitívnych záverov.
TDM4450g
Randomizovaná, multicentrická, nezaslepená štúdia fázy II hodnotila účinok trastuzumab emtansínu oproti trastuzumabu v kombinácii s docetaxelom u pacientov s HER2-pozitívnym MBC, ktorí neboli predtým liečení chemoterapiou pre metastatické ochorenie. Pacienti boli randomizovaní na liečbu
3,6 mg/kg trastuzumab emtansínu intravenózne každé 3 týždne (n = 67) alebo trastuzumabom v nárazovej dávke 8 mg/kg, po ktorej nasleduje dávka 6 mg/kg intravenózne každé 3 týždne
v kombinácii s docetaxelom v dávke 75-100 mg/m2 intravenózne každé 3 týždne (n = 70).
Primárnym cieľom bolo prežívanie bez progresie ochorenia (PFS) hodnotené skúšajúcim. Medián PFS bol 9,2 mesiaca v skupine s trastuzumabom v kombinácii s docetaxelom a 14,2 mesiaca v skupine s trastuzumab emtansínom (Hazard Ratio 0,59; p = 0,035) s mediánom sledovania približne 14 mesiacov v oboch skupinách. Miera objektívnej odpovede (ORR) bol 58,0 % pre trastuzumab
v kombinácii s docetaxelom a 64,2 % pre trastuzumab emtansín. Medián trvania odpovede sa nedosiahol pre trastuzumab emtansín oproti 9,5 mesiaca v kontrolnej skupine.
TDM4374g
Nezaslepená štúdia fázy II s jedným ramenom hodnotila účinok trastuzumab emtansínu u pacientov s
HER2-pozitívnym, nevyliečiteľným LABC alebo MBC. Všetci pacienti boli predtým liečení HER2 cielenými liečbami (trastuzumab a lapatinib) a chemoterapiou (antracyklín, taxán a kapecitabín) v neoadjuvantnej, adjuvantnej liečbe lokálne pokročilého alebo metastatického ochorenia. Medián počtu protirakovinových liekov, ktoré pacienti dostávali v ktoromkoľvek štádiu bol 8,5 (rozmedzie 5-19) a pre metastatické ochorenie bol 7,0 (rozmedzie 3-17), vrátane všetkých liekov určených na liečbu karcinómu prsníka.
Pacienti (n = 110) dostávali 3,6 mg/kg trastuzumab emtansínu intravenózne každé 3 týždne až do progresie ochorenia alebo do neakceptovateľnej toxicity.
Kľúčové analýzy účinnosti boli ORR založená na nezávislej rádiologickej kontrole a trvanie objektívnej odpovede. ORR bola 32,7 % (95 % IS: 24,1, 42,1), n = 36 pacientov odpovedalo na liečbu pri posudzovaní IRC aj skúšajúcim. Medián trvania odpovede hodnotený IRC sa nedosiahol (95 % IS,
4,6 mesiaca až neodhadnuteľný).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s trastuzumab emtansínom vo všetkých podskupinách pediatrickej populácie s karcinómom prsníka (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Trastuzumab emtansín sa podáva intravenózne. Neuskutočnili sa žiadne štúdie s inými spôsobmi podávania.
Distribúcia
Pacienti v štúdii TDM4370g/BO21977, ktorí dostávali 3,6 mg/kg trastuzumab emtansínu intravenózne každé 3 týždne, mali priemernú maximálnu sérovú koncentráciu (Cmax) trastuzumab emtansínu
83,4 (± 16,5) µg/ml. Na základe populačnej FK analýzy po intravenóznom podaní bol centrálny
distribučný objem trastuzumab emtansínu (3,13 l) a približoval sa plazmatickému objemu.
Biotransformácia (trastuzumab emtansín a DM1)
Predpokladá sa, že trastuzumab emtansín podstupuje dekonjugáciu a katabolizmus prostredníctvom proteolýzy v bunkových lyzozómoch.
In vitro štúdie metabolizmu s ľudskými pečeňovými mikrozómami naznačujú, že DM1, zložka trastuzumab emtansínu s malou molekulou, je metabolizovaný predovšetkým prostredníctvom CYP3A4 a v menšej miere CYP3A5. DM1 neinhibuje hlavné enzýmy CYP450 in vitro. V ľudskej plazme boli katabolity trastuzumab emtansínu MCC-DM1, Lys-MCC-DM1 a DM1 zistené v nízkych koncentráciách. In vitro bol DM1 substrátom P-glykoproteínu (P-gp).
Eliminácia
Na základe populačnej farmakokinetickej (FK) analýzy po intravenóznom podaní trastuzumab emtansínu u pacientov s HER2-pozitívnym metastatickým karcinómom prsníka bol klírens trastuzumab emtansínu 0,68 l/deň a eliminačný polčas (t1/2) približne 4 dni. Po opakovanom podávaní intravenóznej infúzie každé 3 týždne sa nepozorovala kumulácia trastuzumab emtansínu.
Na základe populačnej FK analýzy boli telesná hmotnosť, albumín, súčet najdlhšieho priemeru cieľových lézií podľa kritérií pre hodnotenie odpovede u solídnych nádorov (Response Evaluation Criteria In Solid Tumors - RECIST), HER2 extracelulárna doména (ECD), východiskové koncentrácie trastuzumabu a aspartátaminotransferáza (AST) boli identifikované ako štatisticky významné
kovariáty pre FK parametre trastuzumab emtansínu. Rozsah účinku týchto kovariátov na expozíciu trastuzumab emtansínu však naznačuje, že je nepravdepodobné, že tieto kovariáty majú klinicky významný vplyv na expozície trastuzumab emtansínu. Okrem toho exploračná analýza ukázala, že vplyv kovariátov (t.j. renálna funkcia, rasa a vek) na farmakokinetiku celkového trastuzumabu a DM1 bol obmedzený a nebol klinicky významný. V predklinických štúdiách katabolity trastuzumab emtansínu, vrátane DM1, Lys-MCC-DM1 a MCC-DM1, sa vylučujú hlavne žlčou s minimálnym vylučovaním do moču.
Linearita/nelinearita
Trastuzumab emtansín, ak je podávaný intravenózne každé 3 týždne, vykazuje lineárnu FK v rozmedzí dávok 2,4 až 4,8 mg/kg; pacienti, ktorí dostávali dávky menšie alebo rovnajúce sa 1,2 mg/kg, mali rýchlejší klírens.
Starší pacienti
Populačná FK analýza ukázala, že vek neovplyvňuje FK trastuzumab emtansínu. Žiadny významný rozdiel sa nepozoroval v FK trastuzumab emtansínu u pacientov vo veku < 65 rokov (n = 577), u pacientov vo veku 65-75 rokov (n = 78) a u pacientov vo veku > 75 rokov (n = 16).
Pacienti s poruchou funkcie obličiek
Žiadne oficiálne FK štúdie u pacientov s poruchou funkcie obličiek sa neuskutočnili. Populačná FK analýza preukázala, že klírens kreatinínu neovplyvňuje FK trastuzumab emtansínu. Farmakokinetika trastuzumab emtansínu u pacientov s miernou (klírens kreatinínu CLcr 60 až 89 ml/min, n = 254)
alebo stredne závažnou (CLcr 30 až 59 ml/min, n = 53) poruchou funkcie obličiek bola podobná ako u pacientov s normálnou funkciou obličiek (CLcr ≥ 90 ml/min, n = 361). Farmakokinetické údaje u pacientov so závažnou poruchou funkcie obličiek (CLcr 15 až 29 ml/min) sú obmedzené (n = 1), preto nie je možné stanoviť odporúčania pre dávkovanie.
Pacienti s poruchou funkcie pečene
Žiadne oficiálne FK štúdie u pacientov s poruchou funkcie pečene sa neuskutočnili.
Ďalšie osobitné populácie
Populačná FK analýza preukázala, že rasa pravdepodobne neovplyvňuje FK trastuzumab emtansínu. Pretože väčšina pacientov v klinických štúdiách s trastuzumab emtansínom boli ženy, vplyv pohlavia na FK trastuzumab emtansínu nebol oficiálne hodnotený.
5.3 Predklinické údaje o bezpečnosti
Toxikológia a/alebo farmakológia u zvierat
Podávanie trastuzumab emtansínu bolo dobre tolerované u potkanov a opíc pri dávkach až do 20
a 10 mg/kg, čo zodpovedá 2 040 µg DM1/m2 u obidvoch druhov, čo je približne ekvivalentné klinickej dávke trastuzumab emtansínu u pacientov. V SLP toxikologických štúdiách, s výnimkou ireverzibilnej
periférnej axonálnej toxicity (pozorovanej len u opíc pri dávke ≥ 10 mg/kg) a reprodukčnej orgánovej toxicity (pozorovanej len u potkanov pri dávke 60 mg/kg), boli identifikované čiastočne alebo úplne reverzibilné na dávke závislé toxicity u oboch zvieracích modelov. Hlavné toxicity zahŕňali pečeň (zvýšenie pečeňových enzýmov) pri dávke ≥ 20 mg/kg a ≥ 10 mg/kg, kostnú dreň (zníženie počtu krvných doštičiek a bielych krviniek)/hematologické toxicity pri dávke ≥ 20 mg/kg a ≥ 10 mg/kg a lymfoidné orgány pri dávke ≥ 20 mg/kg a ≥ 3 mg/kg u potkanov a opíc, v uvedenom poradí.
Mutagenicita
DM1 bol aneugénny alebo klastogénny v in vivo mikronukleárnom teste kostnej drene u potkanov s jednorazovou dávkou pri expozíciách, ktoré boli porovnateľné s priemernými maximálnymi
koncentráciami DM1 nameranými u človeka, ktorý dostával trastuzumab emtansín. DM1 nebol mutagénny v in vitro (Amesovom) teste bakteriálnej reverznej mutácie.
Porucha fertility a teratogenita
Špecializované štúdie fertility s trastuzumab emtansínom sa neuskutočnili. Avšak, na základe výsledkov zo všeobecných toxikologických štúdií na zvieratách je možné očakávať negatívne účinky
na fertilitu.
Špecializované štúdie embryo-fetálneho vývoja u zvierat s trastuzumab emtansínom sa neuskutočnili. Vývojová toxicita trastuzumabu bola identifikovaná v klinických štúdiách, hoci v predklinickom štúdiách sa nepredpokladala. Okrem toho bola identifikovaná vývojová toxicita maytansínu v predklinických štúdiách, čo naznačuje, že DM1, cytotoxická maytansinoidová zložka trastuzumab emtansínu inhibujúca mikrotubuly, bude podobne teratogénny a potenciálne embryotoxický.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Kyselina sukcínová Hydroxid sodný Sacharóza Polysorbát 20
6.2 Inkompatibility
Tento liek sa nesmie miešať ani riediť s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6. Roztok glukózy (5 %) sa nemá používať na rekonštitúciu ani riedenie, pretože to môže spôsobiť
agregáciu proteínu.
6.3 Čas použiteľnosti
3 roky.
Čas použiteľnosti rekonštituovaného roztoku
Chemická a fyzikálna stabilita pri používaní rekonštituovaného roztoku sa stanovila na 24 hodín pri
2 °C až 8 °C. Z mikrobiologického hľadiska sa liek má použiť ihneď. Ak sa nepoužije ihneď, rekonštituované injekčné liekovky sa majú uchovávať 24 hodín pri 2 °C až 8 °C za podmienky, že rekonštitúcia sa uskutočnila za kontrolovaných a validovaných aseptických podmienok a potom sa musia zlikvidovať.
Čas použiteľnosti zriedeného roztoku
Rekonštituovaný roztok Kadcyly obsahujúci infúzny roztok chloridu sodného 9 mg/ml (0,9 %) alebo
infúzny roztok chloridu sodného 4,5 mg/ml (0,45 %) je stabilný 24 hodín pri 2 °C až 8 °C za podmienky, že bol pripravený za kontrolovaných a validovaných aseptických podmienok. Počas uchovávania sa môžu pozorovať častice pri nariedení v roztoku chloridu sodného 0,9 % (pozri časť 6.6).
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Podmienky na uchovávanie po rekonštitúcii a riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Kadcyla je dostupná v 15 ml (100 mg) alebo 20 ml (160 mg) injekčných liekovkách zo skla typu 1
uzatvorených sivou butylovou gumovou zátkou potiahnutou fluoro-živicovým laminátom
a zapečatených hliníkovým tesnením s bielym alebo červeným odklápacím viečkom z plastickej hmoty.
Balenie s 1 injekčnou liekovkou.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Je potrebné používať vhodný aseptický postup. Je potrebné používať vhodné postupy pre prípravu cytostatík.
Rekonštituovaný roztok Kadcyly sa má riediť v polyvinylchloridových (PVC) alebo polyolefínových infúznych vakoch bez latexu a bez PVC.
Ak je infúzny koncentrát zriedený infúznym roztokom chloridu sodného 9 mg/ml (0,9 %), na infúziu sa má použiť in-line polyétersulfónový (PES) filter 0,22 mikrónu.
Na zabránenie chybám v medikácii je dôležité skontrolovať etikety na injekčných liekovkách, aby sa zabezpečilo, že liek, ktorý sa pripravuje a podáva, je Kadcyla (trastuzumab emtansín) a nie Herceptin (trastuzumab).
Pokyny na rekonštitúciu
• injekčná liekovka so 100 mg trastuzumab emtansínu: Pomocou sterilnej injekčnej striekačky pomaly vstreknite 5 ml sterilnej vody na injekciu do injekčnej liekovky.
• injekčná liekovka so 160 mg trastuzumab emtansínu: Pomocou sterilnej injekčnej striekačky pomaly vstreknite 8 ml sterilnej vody na injekciu do injekčnej liekovky.
• Jemne premiešajte, kým sa prášok úplne nerozpustí. Netrepte.
Rekonštituovaný roztok sa má pred podaním vizuálne skontrolovať, či neobsahuje častice alebo či nezmenil farbu. Rekonštituovaný roztok nemá obsahovať viditeľné častice, má byť číry až slabo
opalizujúci. Farba rekonštituovaného roztoku má byť bezfarebná až svetlo hnedá. Nepoužívajte ho, ak rekonštituovaný roztok obsahuje viditeľné častice alebo ak je zakalený alebo inak sfarbený.
Pokyny na riedenieUrčite objem potrebného rekonštituovaného roztoku na základe dávky 3,6 mg trastuzumab emtansínu/kg telesnej hmotnosti (pozri časť 4.2):
Objem (ml) =
Celková dávka, ktorá sa má podať (telesná hmotnosť(kg) x dávka (mg/kg))20 (mg/ml, koncentrácia rekonštituovaného roztoku)
Príslušné množstvo roztoku sa má odobrať z injekčnej liekovky a pridať do infúzneho vaku obsahujúceho 250 ml infúzneho roztoku chloridu sodného 4,5 mg/ml (0,45 %) alebo infúzneho roztoku chloridu sodného 9 mg/ml (0,9 %). Roztok glukózy (5 %) sa nemá používať (pozri časť 6.2). Infúzny roztok chloridu sodného 4,5 mg/ml (0,45 %) sa môže použiť bez polyétersulfónového (PES)
0,22 µm in-line filtra. Ak sa na infúziu používa infúzny roztok chloridu sodného 9 mg/ml (0,9 %), je potrebný polyétersulfónový (PES) 0,22 µm in-line filter. Po príprave infúzie sa má roztok podať okamžite. Počas uchovávania infúziu neuchovávajte v mrazničke ani netrepte.
LikvidáciaRekonštituovaný liek neobsahuje žiadne konzervačné látky a je určený len na jednorazové použitie. Nespotrebovaný roztok zlikvidujte.
Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.