
2.png" />zlej tolerancii vysokých dávok v prípade príliš rýchlej titrácie nahor. V závislosti od tolerancie sa dávky pacientov môžu titrovať aj nadol na nižšie dávky. Pacienti sa musia udržiavať na najvyššej tolerovanej dávke tolvaptánu.
Cieľom titrácie dávky je čo najúplnejšie a najtrvalejšie zablokovať pôsobenie vazopresínu na obličkový receptor V2 pri zachovaní prijateľnej rovnováhy tekutín (pozri časť 4.4).
Odporúča sa merať osmolalitu moču, pomocou ktorej sa monitoruje primeranosť inhibície
vazopresínu. Má sa zvážiť pravidelné meranie osmolality plazmy alebo hladiny sodíka v sére (na výpočet osmolarity plazmy) a/alebo telesnej hmotnosti s cieľom monitorovať riziko dehydratácie v dôsledku účinkov tolvaptánu na zvýšenie vylučovania vody v prípade, že pacient neprijíma dostatočnémnožstvo vody.Bezpečnosť a účinnosť Jinarcu v štádiu 5 CKD nie sú dostatočne
preskúmané, preto sa má liečba tolvaptánom ukončiť, ak obličková nedostatočnosť pokročí do štádia 5
CKD.Ranná dávka Jinarcu sa má užiť aspoň 30 minút pred ranným jedlom. Druhú dennú dávku možno užiť s jedlom alebo bez jedla.V prípade obmedzenia schopnosti piť alebo dostupnosti vody sa liečba musí prerušiť (pozri časť 4.4).
Tolvaptán sa nesmie užívať s grapefruitovou šťavou (pozri časť 4.5). Pacienti musia byť poučení, aby pili dostatočné množstvo vody alebo iných vodných tekutín (pozri časť 4.4).
Úprava dávkovania u pacientov užívajúcich silné inhibítory CYP3A
U pacientov užívajúcich silné inhibítory CYP3A (pozri časť 4.5) sa dávky tolvaptánu musia znížiť
takto:
Denná delená dávka tolvaptánu Znížená dávka (raz denne)
90 + 30 mg 30 mg(ďalšie zníženie na 15 mg, ak dávka 30 mg nie je dobre tolerovaná)
60 + 30 mg 30 mg(ďalšie zníženie na 15 mg, ak dávka 30 mg nie je dobre tolerovaná)
45 + 15 mg 15 mg
Úprava dávkovania u pacientov užívajúcich stredne silné inhibítory CYP3A
U pacientov užívajúcich stredné silné inhibítory CYP3A sa dávky tolvaptánu musia znížiť takto:
Denná delená dávka tolvaptánu Znížená delená dávka
90 + 30 mg 45 + 15 mg
60 + 30 mg 30 + 15 mg
45 + 15 mg 15 + 15 mg
Ďalšie zníženie treba zvážiť v prípade, že pacienti netolerujú znížené dávky tolvaptánu.
Staršia populácia
Vyšší vek nemá žiadny vplyv na koncentráciu tolvaptánu v plazme. Bezpečnosť a účinnosť tolvaptánu u pacientov s ADPKD starších ako 50 rokov nebola doteraz stanovená.
Porucha funkcie obličiek
Tolvaptán je kontraindikovaný u anurických pacientov (pozri časť 4.3).
U pacientov s poruchou funkcie obličiek sa úprava dávky nevyžaduje. Neuskutočnili sa žiadne klinické skúšania u pacientov s klírensom kreatinínu < 10 ml/min., ani u pacientov podstupujúcich
dialýzu. Riziko poškodenia pečene u pacientov so závažne zníženou funkciou obličiek (t. j. hodnota
eGFR < 20) môže byť zvýšené. Títo pacienti sa majú pozorne monitorovať na pečeňovú toxicitu. Údaje o pacientoch v štádiu 3 CKD sú obmedzenejšie ako údaje o pacientoch v štádiu 1 alebo 2 (pozri časť 5.1).
Porucha funkcie pečene
U pacientov so závažnou poruchou funkcie pečene sa musia starostlivo zvážiť prínosy a riziká liečby Jinarcom. Liečba pacientov sa musí starostlivo riadiť a musia sa pravidelne monitorovať pečeňové enzýmy (pozri časť 4.4).Jinarc je kontraindikovaný u pacientov, ktorí majú pred začatím liečby zvýšenú hladinu pečeňových enzýmov a/alebo prejavy alebo príznaky poškodenia pečene spĺňajúce požiadavky na trvalé ukončenie liečby tolvaptánom (pozri časti 4.3 a 4.4).U pacientov s miernou až stredne závažnou poruchou funkcie pečene (triedy A a B podľa Child-Pughovej klasifikácie) nie je potrebná žiadna úprava dávky.
Pediatrická populácia
Bezpečnosť a účinnosť tolvaptánu u detí a dospievajúcich nebola doteraz stanovená. K dispozícii nie
sú žiadne údaje. Použitie tolvaptánu v pediatrickej vekovej skupine sa neodporúča.
Spôsob podávania
Perorálne použitie.
Tablety sa musia prehltnúť bez žuvania a zapiť pohárom vody.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1alebo
na benzazepín alebo deriváty benzazepínu (pozri časť 4.4).
• Zvýšená hladina pečeňových enzýmov a/alebo prejavy alebo príznaky poškodenia pečene pred začatím liečby, ktoré spĺňajú požiadavky na trvalé ukončenie liečby tolvaptánom (pozri
časť 4.4).
• Anúria.
• Deplécia objemu.
• Hypernatrémia.
• Pacienti, ktorí nevnímajú smäd alebo naň nereagujú.
• Gravidita (pozri časť 4.6).
• Laktácia (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Idiosynkratická pečeňovátoxicita
Tolvaptán bol spájaný s idiosynkratickým zvýšením hladiny alanínaminotransferázy (ALT)
a aspartátaminotransferázy (AST) v krvi s občasnými prípadmi súčasného zvýšenia hladiny celkového
bilirubínu (BT).
Po uvedení lieku na trh bolo pri používaní tolvaptánu u ADPKD hlásené akútne zlyhanie pečene vyžadujúce transplantáciu pečene.
V dvojito zaslepenom placebom kontrolovanom skúšaní u pacientov s ADPKD sa pozorovalo
zvýšenie (> 3 × horná hranica normálu [upper limit of normal, ULN]) hladiny ALT u 4,4 % (42/958)
pacientov liečených tolvaptánom a u 1,0 % (5/484) pacientov dostávajúcich placebo, kým zvýšenie
(> 3 × ULN) hladiny AST sa pozorovalo u 3,1 % (30/958) pacientov liečených tolvaptánom a u 0,8 % (4/484) pacientov dostávajúcich placebo. U dvoch (2/957, 0,2 %) z týchto pacientov liečených
tolvaptánom a u tretieho pacienta z rozšíreného otvoreného skúšania došlo k zvýšeniu hladiny pečeňových enzýmov (> 3 × ULN) za súčasného zvýšenia hladiny BT (> 2 × ULN). Obdobie nástupu
hepatocelulárneho poškodenia (na základe zvýšenia hladiny ALT > 3 × ULN) bolo do 3 až
14 mesiacov od začiatku liečby a toto zvýšenie bolo vratné s návratom hladiny ALT na
úroveň < 3 × ULN v priebehu 1 až 4 mesiacov. Hoci tieto súčasné zvýšenia boli v prípade rýchleho
ukončenia liečby tolvaptánom vratné, predstavujú potenciál závažného poškodenia pečene. Podobné zmeny v prípade iných liekov boli spojené s potenciálom spôsobiť nevratné a potenciálne život ohrozujúce poškodenie pečene.
 P
r
edpisujúci lekári musia v plnej miere dodržiavať nevyhnutné bezpečnostné opatrenia uvedené ďalej.
P
r
edpisujúci lekári musia v plnej miere dodržiavať nevyhnutné bezpečnostné opatrenia uvedené ďalej.
Na zmiernenie rizika závažného a/alebo nevratného poškodenia pečene sa pred začatím liečby Jinarcom vyžadujú krvné testy na pečeňové transaminázy a bilirubín, v ktorých sa má pokračovať v mesačných intervaloch počas 18 mesiacov a potom v pravidelných 3-mesačných intervaloch.
Odporúča sa súbežné monitorovanie príznakov, ktoré môžu nasvedčovať poškodeniu pečene (ako sú
únava, anorexia, nauzea, nepohodlie v pravej hornej časti brucha, vracanie, horúčka, vyrážka, pruritus, tmavý moč alebo žltačka).
Ak má pacient pred začatím liečby abnormálnu hladinu ALT, AST alebo BT, ktorá spĺňa kritériá na trvalé ukončenie (pozri ďalej), použitie tolvaptánu je kontraindikované (pozri časť 4.3). V prípade abnormálnej východiskovej úrovne nižšej ako hranica pre trvalé ukončenie možno liečbu začať iba vtedy, keď potenciálny prínos liečby prevažuje nad možnými rizikami, a testy funkcie pečene sa musia vykonávať vo zvýšenej frekvencii. Odporúča sa získať vyjadrenie hepatológa.
Počas prvých 18 mesiacov liečby možno Jinarc podávať iba pacientom, u ktorých lekár zistil, že
funkcia pečene umožňuje nepretržitú liečbu.
V prípade nástupu príznakov alebo prejavov zodpovedajúcich poškodeniu pečene, alebo ak sa počas liečby zistí klinicky významný abnormálny nárast hladiny ALT alebo AST, sa podávanie Jinarcu musí okamžite prerušiť a čo najskôr sa musia znova vykonať testy vrátane ALT, AST, BT a alkalickej fosfatázy (AP) (ideálne do 48-72 hodín). Testovanie musí pokračovať v zvýšenej frekvencii, kým sa príznaky/prejavy/laboratórne abnormality neustália alebo kým nezmiznú. Potom možno znova začať liečbu Jinarcom.
Podľa súčasnej klinickej praxe sa liečba Jinarcom má prerušiť, ak sa potvrdia trvalo zvýšené alebo zvyšujúce sa hladiny transamináz, a natrvalo ukončiť, ak pretrváva výrazné zvýšenie týchto hladín a/alebo klinické príznaky poškodenia pečene.
Hodnoty, pri ktorých sa odporúča trvalé ukončenie liečby:
• ALT alebo AST > 8-násobok ULN,
• ALT alebo AST > 5-násobok ULN počas viac ako 2 týždňov,
• ALT alebo AST > 3-násobok ULN a (BT > 2-násobok ULN alebo medzinárodný normalizovaný pomer [INR] > 1,5),
• ALT alebo AST > 3-násobok ULN s pretrvávajúcimi príznakmi poškodenia pečene uvedenými
vyššie.
Ak hladiny ALT a AST zostanú na úrovni nižšej ako 3-násobok hornej hranice normálu (ULN), s liečbou Jinarcom sa môže s opatrnosťou znova začať pri rovnakej alebo nižšej dávke s častým monitorovaním, pretože sa zdá, že hladiny transamináz sa počas pokračujúcej liečby u niektorých pacientov ustália.
Prístup k vodeTolvaptán môže spôsobiť nežiaduce reakcie súvisiace so stratou vody, ako je smäd, polyúria, noktúria
a polakizúria (pozri časť 4.8). Pacienti preto musia mať prístup k vode (alebo iným vodným tekutinám) a musia byť schopní vypiť dostatočné množstvo týchto tekutín (pozri časť 4.2). Pacienti
musia byť poučení, aby pili vodu alebo iné vodné tekutiny pri prvom náznaku smädu, aby predišli
nadmernému smädu alebo dehydratácii.
Pacienti navyše musia vypiť 1-2 poháre tekutiny pred spaním bez ohľadu na vnímaný smäd a dopĺňať tekutiny cez noc pri každej epizóde noktúrie.
Dehydratácia
U pacientov užívajúcich tolvaptán sa musí monitorovať stav objemu, lebo liečba tolvaptánom môže viesť k závažnej dehydratácii, ktorá predstavuje rizikový faktor dysfunkcie obličiek. Ak je dehydratácia zjavná, prijmite vhodné opatrenia, ktoré môžu zahŕňať prerušenie alebo zníženie dávky tolvaptánu a zvýšenie príjmu tekutín. Osobitná pozornosť sa musí venovať pacientom s ochoreniami, ktoré narušujú primeraný príjem tekutín, alebo pacientom, ktorým hrozí zvýšené riziko straty vody, napr. v prípade vracania alebo hnačky.
Upchatie močovéhovýstupu
Musí sa zabezpečiť priechodnosť močového výstupu. Pacientom s čiastočným upchatím močového
výstupu, napríklad pacientom s hypertrofiou prostaty alebo poruchou močenia, hrozí zvýšené riziko
rozvoja akútnej retencie.
Rovnováha tekutín a elektrolytov
U všetkých pacientov sa musí monitorovať stav tekutín a elektrolytov. Podávanie tolvaptánu vyvoláva
výdatné vylučovanie vody a môže spôsobiť dehydratáciu a zvýšenie hladiny sodíka v sére (pozri
časť 4.8) a je kontraindikované u pacientov s hypernatrémiou (pozri časť 4.3). Pred začatím a po začatí užívania tolvaptánu je preto nutné vyhodnotiť hladinu kreatinínu v sére, hladinu elektrolytov
a príznaky nerovnováhy elektrolytov (napr. závraty, mdloby, palpitácie, zmätenosť, slabosť, nestabilita
chôdze, hyperreflexia, záchvaty, kóma) na účely monitorovania dehydratácie. Počas dlhodobej liečby sa elektrolyty musia monitorovať aspoň každé tri mesiace.
Abnormality hladiny sodíka v sére
Pred začatím liečby tolvaptánom sa musia odstrániť prípadné existujúce abnormality hladiny sodíka
(hyponatrémia alebo hypernatrémia).
Anafylaxia
V rámci skúseností po uvedení na trh sa po podaní tolvaptánu veľmi zriedkavo hlásila anafylaxia
(vrátane anafylaktického šoku a generalizovanej vyrážky). Tento druh reakcie sa vyskytol po prvom
podaní tolvaptánu. Pacientov je potrebné počas liečby dôkladne sledovať. U pacientov so známymi reakciami z precitlivenosti na benzazepín alebo deriváty benzazepínu (napr. benazepril, konivaptán, fenoldopam mesylát alebo mirtazapín) môže existovať riziko reakcie z precitlivenosti na tolvaptán (pozri časť 4.3).
Ak sa vyskytne anafylaktická reakcia alebo iné závažné alergické reakcie, podávanie tolvaptánu sa musí ihneď ukončiť a musí sa začať vhodná liečba. Keďže precitlivelosť je kontraindikovaná (pozri časť 4.3), po anafylaktickej reakcii alebo iných závažných alergických reakciách sa už liečba nikdy nesmie znova začať.
Laktóza
Jinarc obsahuje laktózu ako pomocnú látku. Pacienti so zriedkavými dedičnými problémami
galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie
nesmú užívať tento liek.
Diabetes mellitus
U diabetických pacientov so zvýšenou koncentráciou glukózy (napr. viac ako 300 mg/dl) sa môže
vyskytnúť pseudohyponatrémia. Tento stav sa musí vylúčiť pred začiatkom liečby tolvaptánom i počas
nej.
Tolvaptán môže spôsobiť hyperglykémiu (pozri časť 4.8). Diabetickí pacienti sa preto musia liečiť tolvaptánom so zvýšenou opatrnosťou. Platí to najmä pre pacientov s nedostatočne kontrolovaným diabetom typu II.
Zvýšenie hladinykyselinymočovej
Zníženie klírensu kyseliny močovej obličkami je známym účinkom tolvaptánu. V dvojito zaslepenom
placebom kontrolovanom skúšaní u pacientov s ADPKD bolo hlásené potenciálne klinicky významné
zvýšenie hladiny kyseliny močovej (viac ako 10 mg/dl) s vyšším výskytom u pacientov liečených tolvaptánom (6,2 %) v porovnaní s pacientmi liečenými placebom (1,7 %). Dna bola ako nežiaduca reakcia hlásená častejšie u pacientov liečených tolvaptánom (28/961, 2,9 %) ako u pacientov dostávajúcich placebo (7/483, 1,4 %). V dvojito zaslepenom placebom kontrolovanom skúšaní sa navyše pozorovalo zvýšené používanie alopurinolu a iných liekov slúžiacich na liečbu dny. Účinky na hladinu kyseliny močovej v sére možno pripísať vratným zmenám hemodynamiky obličiek, ktoré sa vyskytujú v reakcii na účinky tolvaptánu na osmolalitu moču a môžu byť klinicky významné. Epizódy zvýšenej hladiny kyseliny močovej a/alebo dny v dvojito zaslepenom placebom kontrolovanom skúšaní však neboli závažné a neviedli k ukončeniu liečby. Koncentrácia kyseliny močovej sa má vyhodnotiť pred začatím liečby Jinarcom a podľa potreby počas liečby na základe príznakov.
Vplyv tolvaptánu narýchlosťglomerulárnejfiltrácie
V skúšaniach s pacientmi s ADPKD sa na začiatku liečby tolvaptánom pozorovalo vratné zníženie
rýchlosti glomerulárnej filtrácie.
4.5 Liekové a iné interakcie
Účinok iných liekovnafarmakokinetikutolvaptánu
Inhibítory CYP3A
Súčasné užívanie liekov, ktoré sú stredne silné inhibítory CYP3A (napr. amprenavir, aprepitant, atazanavir, ciprofloxacín, krizotinib, darunavir/ritonavir, diltiazem, erytromycín, flukonazol,
fosamprenavir, imatinib, verapamil) alebo silné inhibítory CYP3A (napr. itrakonazol, ketokonazol, ritonavir, klaritromycín) zvyšuje expozíciu tolvaptánu.
Spoločné podávanie tolvaptánu a ketokonazolu viedlo k zvýšeniu plochy pod krivkou koncentrácie
tolvaptánu v čase (AUC) o 440 % a k zvýšeniu maximálnej pozorovanej koncentrácie tolvaptánu
v plazme (Cmax) o 248 %.
Súbežné podávanie tolvaptánu a flukonazolu, stredne silného inhibítora CYP3A, spôsobilo nárast
AUC tolvaptánu o 200 % a nárast Cmaxtolvaptánu o 80 %.
Spoločné podávanie tolvaptánu a grapefruitovej šťavy, ktorá je stredne silný až silný inhibítor CYP3A,
viedlo k zdvojnásobeniu vrcholovej koncentrácie tolvaptánu (Cmax).
U pacientov užívajúcich stredne silné alebo silné inhibítory CYP3A sa odporúča znížiť dávku tolvaptánu (pozri časť 4.2). Liečba pacientov užívajúcich stredne silné alebo silné inhibítory CYP3A
sa musí riadiť so zvýšenou opatrnosťou, najmä ak sa inhibítory užívajú častejšie ako raz denne.
Induktory CYP3A
Súčasné užívanie liekov, ktoré sú silné induktory CYP3A (napr. rifampicín), zníži expozíciu
a účinnosť tolvaptánu. Spoločné podávanie tolvaptánu a rifampicínu znižuje hodnoty Cmax a AUC
tolvaptánu o približne 85 %. Preto je potrebné vyhnúť sa súčasnému podávaniu tolvaptánu so silnými induktormi CYP3A (ako sú rifampicín, rifabutín, rifapentín, fenytoín, karbamazepín a ľubovník bodkovaný).
Spoločné podávanie s liekmi, ktoré zvyšujú koncentráciu sodíka v sére
Nie sú žiadne skúsenosti z kontrolovaných klinických skúšaní so súčasným užívaním tolvaptánu a hypertonického roztokuchloridu sodného, perorálnych liekov s obsahom sodíka a liekov, ktoré zvyšujú koncentráciu sodíka v sére. Lieky s vysokým obsahom sodíka, napríklad šumivé analgetické
prípravky a niektoré lieky obsahujúce sodík na liečbu dyspepsie, môžu takisto zvýšiť koncentráciu sodíka v sére. Súčasné užívanie tolvaptánu s liekmi, ktoré zvyšujú koncentráciu sodíka v sére, môže mať za následok vyššie riziko rozvoja hypernatrémie (pozri časť 4.4), a preto sa neodporúča.
Diuretiká
Tolvaptán sa podrobne neskúmal u pacientov s ADPKD pri podávaní v kombinácii s diuretikami. Aj
keď sa nezdá, že súčasné užívanie tolvaptánu so slučkovými a tiazidovými diuretikami má synergický alebo aditívny účinok, každá zo skupín týchto látok má potenciál spôsobiť závažnú dehydratáciu, ktorá je rizikovým faktorom dysfunkcie obličiek. V prípade zjavnej dehydratácie alebo dysfunkcie obličiek sa musia prijať vhodné opatrenia, ktoré môžu zahŕňať prerušenie liečby alebo zníženie dávky tolvaptánu a/alebo diuretík a zvýšenie príjmu tekutín. Musia sa vyhodnotiť a odstrániť aj ďalšie možné príčiny dysfunkcie obličiek alebo dehydratácie.
Vplyv tolvaptánu na farmakokinetiku iných liekov
Substráty CYP3A
U zdravých ľudí nemal tolvaptán, ktorý je substrátom CYP3A, žiadny vplyv na koncentrácie niektorých ďalších substrátov CYP3A v plazme (napr. warfarínu alebo amiodarónu). Tolvaptán zvýšil
hladinu lovastatínu v plazme 1,3-násobne až 1,5-násobne. Hoci toto zvýšenie nemá klinický význam, nasvedčuje tomu, že tolvaptán môže potenciálne zvýšiť expozíciu substrátom CYP3A4.
Substráty transportérov
V štúdiách in vitro sa ukázalo, že tolvaptán je substrát a kompetitívny inhibítor P-glykoproteínu(P-gp). V štúdiách in vitro sa ukázalo, že tolvaptán alebo jeho oxymaslový metabolit môže mať potenciál
inhibovať transportéry OATP1B1, OATP1B3, OAT3, BCRP a OCT1.Koncentrácia digoxínu
v rovnovážnom stave sa zvýšila (1,3-násobne pre maximálnu pozorovanú koncentráciu v plazme
[Cmax] a 1,2-násobne pre plochu pod krivkou koncentrácie v čase v priebehu intervalu dávkovania
[AUCτ]), keď sa digoxín podával súčasne s opakovanými dávkami tolvaptánu 60 mg raz denne. U pacientov užívajúcich digoxín alebo iné úzko špecifické terapeutické substráty P-gp (napr.
dabigatran) sa preto liečbatolvaptánom musí riadiť so zvýšenou opatrnosťou a musia sa vyhodnotiť nadmerné účinky.Statíny bežne používané v hlavnom skúšaní fázy 3 s tolvaptánom (napr.
rosuvastatín, pitavastatín) sú substráty OATP1B1 alebo OATP1B3, v tomto skúšaní sa však nepozoroval žiadny rozdiel v profile nežiaducich účinkov u pacientov s ADPKD.Ak sa spoločne
s tolvaptánom podávajú substráty OATP1B1 a OATP1B3 (napr. statíny ako rosuvastatín
a pitavastatín), substráty OAT3 (napr. metotrexát, ciprofloxacín), substráty BCRP (napr. sulfasalazín)
alebo substráty OCT1 (napr. metformín), liečba pacientov sa musí riadiť so zvýšenou opatrnosťou
a musia sa vyhodnotiť nadmerné účinky týchto liekov.
Diuretiká alebo nediuretické lieky proti vysokému krvnému tlaku
V skúšaniach s pacientmi s ADPKD sa bežne nemeral krvný tlak v stoji, takže nemožno vylúčiť riziko
ortostatickej/posturálnej hypotenzie v dôsledku farmakodynamickej interakcie s tolvaptánom.
Spoločné podávanie s analógmi vazopresínu
Okrem toho, že tolvaptán spôsobuje vylučovanie vody obličkami, je schopný blokovať receptory
vazopresínu V2 v cievach, ktoré sa podieľajú na uvoľňovaní koagulačných faktorov (napr. von
Willebrandovho faktora) z endotelových buniek. Preto u pacientov, ktorým sa podávajú analógy vazopresínu, napr. dezmopresín, na prevenciu alebo kontrolu krvácania, môže byť účinok týchto analógov oslabený, ak sa podávajú spoločne s tolvaptánom. Neodporúča sa podávať Jinarc s analógmi vazopresínu.
Fajčenie a alkohol
Údaje zo skúšaní s pacientmi s ADPKD týkajúce sa fajčenia alebo požívania alkoholu v anamnéze sú
príliš obmedzené na to, aby sa dali určiť možné interakcie fajčenia alebo alkoholu s účinnosťou
a bezpečnosťou liečby ADPKD tolvaptánom.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Nie sú k dispozícii dostatočné údaje o použití tolvaptánu u gravidných žien. Štúdie na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3). Potenciálne riziko u ľudí nie je známe.
Ženy vo fertilnom veku musia počas užívania Jinarcu používať primerané metódy antikoncepcie. Jinarc sa nesmie užívať počas gravidity (pozri časť 4.3).
Dojčenie
Nie je známe, či sa tolvaptán vylučuje do ľudského materského mlieka. Štúdie na potkanoch
preukázali vylučovanie tolvaptánu do mlieka.
Potenciálne riziko u ľudí nie je známe. Jinarc je kontraindikovaný počas laktácie (pozri časť 4.3).
Fertilita
Štúdie na zvieratách preukázali účinky na fertilitu (pozri časť 5.3). Potenciálne riziko pre ľudí nie je
známe.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Jinarc má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri vedení vozidiel alebo obsluhe strojov je potrebné vziať do úvahy, že občas sa môžu vyskytnúť závraty, asténia alebo únava.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Farmakodynamicky predvídateľné a najčastejšie hlásené nežiaduce reakcie sú smäd, polyúria,
noktúria, a polakizúria, ktoré sa vyskytujú približne u 55 %, 38 %, 29 %, resp. 23 % pacientov v uvedenom poradí. Tolvaptán bol navyše spájaný s idiosynkratickým zvýšením hladiny alanínaminotransferázy (ALT) a aspartátaminotransferázy (AST) v krvi s občasnými prípadmi súčasného zvýšenia hladiny celkového bilirubínu (BT).
Tabuľkový zoznamnežiaducichreakcií
Výskyt nežiaducich účinkov lieku (ADR, z angl. adverse drug reactions) spojených s liečbou
tolvaptánom je uvedený v tabuľke nižšie. Tabuľka vychádza z nežiaducich udalostí hlásených počas klinických skúšaní a/alebo po uvedení na trh.
Všetky nežiaduce reakcie lieku sú zoradené podľa triedy orgánových systémov a frekvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov). Pri každej frekvencii výskytu sú nežiaduce reakcie zoradené podľa klesajúcej závažnosti.
Frekvenciu výskytu nežiaducich reakcií hlásených po uvedení na trh nie je možné určiť, pretože pochádzajú zo spontánnych hlásení. Frekvencia výskytu týchto nežiaducich udalostí je následne klasifikovaná ako „neznáma“.
 P
oruchy
im
unitného systému
V
eľmi časté Časté Menej časté Neznáme*
P
oruchy
im
unitného systému
V
eľmi časté Časté Menej časté Neznáme*
Anafylaktický šok, Generalizovaná vyrážka
P
oruchy metabolizmu a výživy
V
eľmi časté Časté Menej časté Neznáme*
Polydipsia Dehydratácia, Hypernatrémia, Znížená chuť do jedla, Hyperurikémia, Hyperglykémia,
Dna
P
sychické poruchy Nespavosť
P
oruchy nervového systému
P
oruchy srdca
a srdcovej činnosti Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy
gastrointestinálneho
t
raktu
P
oruchy pečene
a žlčových ciest
P
oruchy kože a podkožného tkaniva
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
P
oruchy obličiek
a močových ciest
C
elkové poruchy a reakcie v mieste podania Laboratórne
a funkčné
vyšetrenia
Bolesť hlavy,
Závraty
Hnačka,
Sucho
v ústach
Noktúria, Polakizúria, Polyúria Únava, Smäd
Palpitácie
Dyspnoe
Bolesť brucha, Abdominálna distenzia, Zápcha,
Dyspepsia, Gastroezofageálna
refluxná choroba
Abnormálna funkcia
pečene
Vyrážka,
Pruritus
Svalové kŕče
Asténia
Zvýšená hladina alanínaminotransferázy, Zvýšená hladina aspartátaminotransferázy, Pokles hmotnosti
Zvýšená hladina bilirubínu
Akútne zlyhanie pečene1
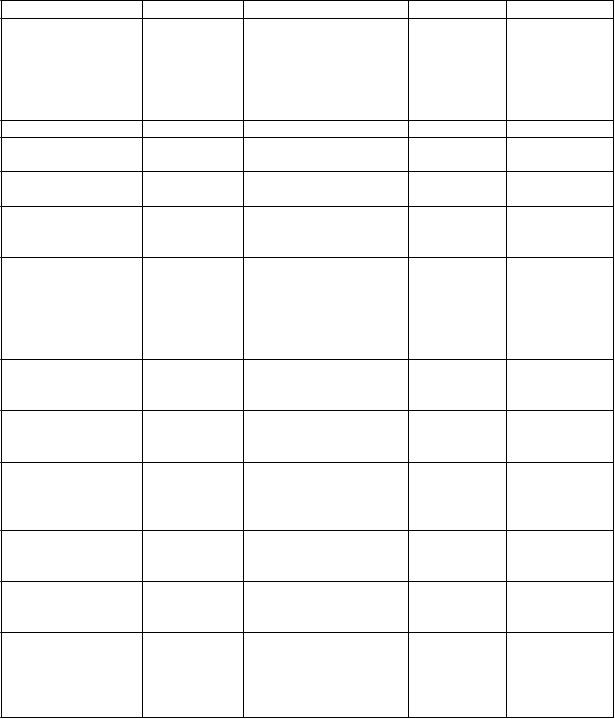
* hlásené počas sledovania po uvedení tolvaptánu na trh schváleného na iné indikácie
1 pozorované po uvedení tolvaptánu na trh pri ADPKD. Bola potrebná transplantácia pečene.
Opis vybranýchnežiaducichreakciíNa zmiernenie rizika závažného alebo nevratného poškodenia pečene sa pred začatím liečby Jinarcom
vyžadujú krvné testy na pečeňové transaminázy, v ktorých sa má pokračovať v mesačných intervaloch
počas 18 mesiacov a potom v pravidelných 3-mesačných intervaloch (pozri časť 4.4).
Najčastejšie nežiaduce reakcie súvisia so stratou vody. Je preto nanajvýš dôležité, aby pacienti mali prístup k vode a dokázali vypiť dostatočné množstvo tekutín. Stav objemu pacientov užívajúcich tolvaptán sa musí monitorovať, aby nedošlo k dehydratácii (pozri časť 4.4).
H
l
ásenie
podozrení
na
nežiaduce
r
eakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 Predávkovanie
Jednorazové perorálne dávky do 480 mg (4-násobok maximálnej odporúčanej dennej dávky)
a opakované dávky do 300 mg raz denne počas 5 dní boli v skúšaniach so zdravými účastníkmi dobre
tolerované. Neexistuje žiadne špecifické antidotum proti intoxikácii tolvaptánom. Možno očakávať, že
prejavy a príznaky akútneho predávkovania budú zodpovedať nadmernému farmakologickému účinku: zvýšenie koncentrácie sodíka v sére, polyúria, smäd a dehydratácia/hypovolémia.
U potkanov a psov sa po jednorazovej perorálnej dávke 2 000 mg/kg (maximálna možná dávka)
nepozorovala žiadna úmrtnosť. U myší bola jednorazová perorálna dávka 2 000 mg/kg smrteľná, pričom medzi príznaky toxicity u postihnutých myší patrilo zníženie pohybovej aktivity, potácanie,
tras a hypotermia.
U pacientov s podozrením na predávkovanie tolvaptánom sa odporúča vyhodnotiť vitálne funkcie,
koncentrácie elektrolytov, EKG a stav tekutín. Vhodná náhrada vody a/alebo elektrolytov musí pokračovať, až kým vylučovanie vody nepoľaví. Dialýza nemusí byť účinná na odstránenie tolvaptánu vzhľadom na jeho vysokú väzbovú afinitu k ľudskému plazmatickému proteínu (> 98 %).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Diuretiká, antagonisty vazopresínu, ATC kód: C03XA01.
Mechanizmus účinku
Tolvaptán je antagonista vazopresínu, ktorý špecificky blokuje väzbu arginínvazopresínu (AVP) na
receptory V2 v distálnych častiach nefrónu. Afinita tolvaptánu k ľudskému receptoru V2 je 1,8-krát
vyššia ako afinita natívneho AVP.
Farmakodynamické účinky
Farmakodynamické účinky tolvaptánu sa stanovili u zdravých ľudí a pacientov s ADPKD v štádiu
CKD 1 až 4.Účinky na klírens voľnej vody a objem moču sú zjavné vo všetkých štádiách CKD,
pričom v neskorších štádiách sa pozorovali slabšie celkové účinky, čo zodpovedá klesajúcemu počtu plne funkčných nefrónov.Pozorovalo sa aj akútne zníženie priemerného celkového objemu obličiek po
3 týždňoch liečby vo všetkých štádiách CKD v rozmedzí od –4,6 % v štádiu 1 do –1,9 % v štádiu 4.
Klinická účinnosťabezpečnosť
Hlavným prvkom klinického programu vývoja tabliet tolvaptánu na liečbu ADPKD je jedno hlavné
nadnárodné randomizované placebom kontrolované skúšanie 3. fázy, v ktorom sa dlhodobá
bezpečnosť a účinnosť režimov perorálnej delenej dávky tolvaptánu (titrovanej medzi 60 mg/deň
a 120 mg/deň) porovnávala s placebom u 1 445 dospelých pacientov s ADPKD. Celkovo sa na celom
svete uskutočnilo 14 klinických skúšaní s tolvaptánom na podporu indikácie ADPKD vrátane 8
skúšaní v USA, 1 v Holandsku, 3 v Japonsku, 1 v Južnej Kórei a nadnárodného hlavného skúšania 3.
fázy.
Hlavné skúšanie 3. fázy (TEMPO 3:4, 156-04-251) zahŕňalo pacientov zo 129 centier v Severnej a Južnej Amerike, Japonsku, Európe a ďalších krajinách.Hlavným cieľom tohto skúšania bolo
vyhodnotiť dlhodobú účinnosť tolvaptánu u pacientov s ADPKD liečených tolvaptánom v porovnaní s pacientmi dostávajúcimi placebo na základe miery zmeny (%) celkového objemu obličiek (total kidney volume, TKV).V tomto skúšaní sa celkovo 1 445 dospelých pacientov (vo veku 18 až
50 rokov) s preukázaným rýchlo postupujúcim skorým ADPKD (ktorí spĺňali upravené Ravineho kritériá, celkový objem obličiek (TKV) ≥ 750 ml, odhadovaný klírens kreatinínu ≥ 60 ml/min.) randomizovalo v pomere 2:1 na liečbu tolvaptánom alebo placebom. Dĺžka liečby pacientov bola až
3 roky.
Skupiny s tolvaptánom (n = 961) a placebom (n = 484) si zodpovedali z hľadiska pohlavia
a priemerného veku 39 rokov.Podľa kritérií pre zaradenie sa identifikovali pacienti, u ktorých sa na začiatku skúšania preukázalo skoré štádium ochorenia.Na začiatku skúšania mali pacienti priemernú odhadovanú rýchlosť glomerulárnej filtrácie (estimated glomerular filtration rate, eGFR)
82 ml/min./1,73 m2 (CKD-EPI), pričom 79 % pacientov malo hypertenziu a priemerná hodnota TKV
bola 1 692 ml (972 ml/m po úprave podľa výšky). Približne 35 % pacientov malo chronické ochorenie
obličiek (CKD) v štádiu 1, 48 % CKD v štádiu 2 a 17 % CKD v štádiu 3 (eGFRCKD-EPI). Kým tieto kritériá boli užitočné na obohatenie skúmanej populácie o pacientov s rýchlym postupom ochorenia, analýzy podskupín na základe kritérií stratifikácie (vek, TKV, GFR, albuminúria, hypertenzia) ukázali, že prítomnosť týchto rizikových faktorov v mladšom veku predpovedá rýchlejší postup ochorenia.
Výsledky primárneho sledovaného parametra, teda pomeru miery zmeny TKV u pacientov randomizovaných do skupiny s tolvaptánom (normalizovanej ako percento) voči rýchlosti zmeny
u pacientov užívajúcich placebo, boli vysoko štatisticky významné. Miera nárastu TKV počas 3 rokov
bola výrazne nižšia u pacientov liečených tolvaptánom ako u pacientov, ktorí dostávali placebo:
2,80 % za rok, resp. 5,51 % za rok (pomer geometrického priemeru 0,974, 95 % IS 0,969-0,980, p < 0,0001).'
Postupne sa testovali aj vopred stanovené sekundárne sledované parametre. Kľúčovým sekundárnym zloženým sledovaným parametrom (postup ADPKD) bol čas do výskytu rôznych udalostí poukazujúcich na klinický postup ochorenia:
1) zhoršenia funkcie obličiek (definovaného ako pretrvávajúce [opakujúce sa aspoň 2 týždne]
zníženie hladiny recipročného kreatinínu v sére o 25 % v priebehu liečby [od konca titrácie do poslednej návštevy počas užívania lieku]);
2) medicínsky významnej bolesti obličiek (definovanejakobolesťvyžadujúcapráceneschopnosť,
analgetiká poslednej záchrany, narkotiká a antinociceptíva, rádiologické alebo chirurgickézákroky);
3) zhoršenia hypertenzie;
4) zhoršenia albuminúrie.
Relatívna miera udalostí súvisiacich s ADPKD sa znížila o 13,5 % u pacientov liečených tolvaptánom
(pomer nebezpečenstva 0,87, 95 % IS 0,78až0,97, p = 0,0095).
Výsledok kľúčového sekundárneho zloženého sledovaného parametra sa primárne pripisuje účinkom na zhoršenie funkcie obličiek a medicínsky významnú bolesť obličiek. Udalosti súvisiace s funkciou
obličiek sa v prípade tolvaptánu vyskytovali s pravdepodobnosťou o 61,4 % menšou ako v prípade
placeba (pomer nebezpečenstva 0,39, 95 % IS 0,26až0,57, nominálna p < 0,0001), kým udalosti súvisiace s bolesťou obličiek sa v prípade tolvaptánu vyskytovali s pravdepodobnosťou menšou
o 35,8 % (pomer nebezpečenstva 0,64, 95 % IS 0,47až0,89, nominálna p = 0,007). Naopak, tolvaptán
nemal žiadny vplyv na postup hypertenzie ani albuminúrie.
TEMPO 4:4 je otvorená rozšírená štúdia zahŕňajúca 871 účastníkov, ktorí dokončili štúdiu TEMPO 3:4 na 106 pracoviských v 13 krajinách. Toto skúšanie hodnotilo účinky tolvaptánu na bezpečnosť, TKV a eGFR u účastníkov dostávajúcich aktívnu liečbu po dobu 5 rokov (včasné liečenie), v porovnaní s účastníkmi, ktorí boli liečení placebom po dobu 3 rokov a potom prešli na aktívnu liečbu po dobu 2 rokov (oneskorené liečenie).
Primárny výsledok pre TKV nepreukázal rozdiel (–1,7 %) pri 5-ročnej liečbe medzi účastníkmi so včasnou a oneskoreou liečbou pri vopred definovanom prahu štatistického významu (p = 0,3580).
Rastová trajektória TKV u oboch skupín sa spomalila v relatívnom pomere k placebu počas prvých
3 rokov, čo naznačujte, že účastníci so včasnou aj oneskorenou liečbou tolvaptánom mali podobný stupeň prínosu.
Sekundárny výsledok testoval pretrvávanie pozitívnych účinkov na funkciu obličiek a preukázal, že
pretrvávanie eGFR pozorované na konci TEMPO 3:4 pivotného skúšania (3,01 až
3,34 ml/min/1,73 m2 pri kontrolnej návšteve 1 a 2) je možné zachovať počas otvorenej liečby. Tento rozdiel sa udržal pri vopred definovanej analýze MMRM (3,15 ml/min/1,73 m2, 95 %CI 1,462 až
4,836, p = 0,0003) a aj pri analýzach citlivosti, pri ktorých boli údaje eGFR prenesené vpred
(2,64 ml/min/1,73 m2, 95 %CI 0,672 až 4,603, p = 0,0086). Tieto údaje naznačujú, že Jinarc môže spomaľovať pokles funkcie obličiek a tieto prínosy pretrvávajú počas trvania liečby.
Dlhodobejšie údaje nie sú momentálne dostupné, aby preukázali, či dlhodobá liečba s Jinarc pokračuje v spomaľovaní rýchlosti poklesu funkcie obličiek a ovplyvňujte klinické výsledky ADPKD, vrátane oneskoreného nástupu koncového štádia ochorenia obličiek.
Genotypizácia génov PKD1 a PKD2 bola vykonaná u väčšiny pacientov, ktorí vstúpili do otvorenej rozšírenej štúdie (TEMPO 4:4), výsledky však ešte nie sú známe.
Po ďalších 2 rokoch liečby tolvaptánom, čiže celkovej liečbe tolvaptánom v trvaní 5 rokov, neboli
identifikované žiadne nové bezpečnostné signály.
Multicentrické, medzinárodné, placebom kontrolované, dvojito zaslepené skúšanie fázy 3 s randomizovane ukončenou účasťou 156-13-210 porovnávalo účinnosť a bezpečnosť tolvaptánu (45 až 120 mg/deň) s placebom u pacientov, ktorí znášali tolvaptán počas 5-týždňového obdobia
titrácie a úvodného podávania tolvaptánu. Skúšanie malo dizajn randomizovaného ukončenia účasti pre obohatenie o pacientov, ktorí dokázali tolerovať tolvaptán počas 5-týždňového jednoducho
zaslepeného obdobia pred randomizáciou, ktoré zahŕňalo 2-týždňové obdobie titrácie a 3-týždňové
úvodné obdobie. Dizajn slúžil na minimalizáciu vplyvu predčasného ukončenia a nedostatku údajov o výsledkoch skúšania.
Celkovo 1370 pacientov (vo veku 18-65 rokov) s chronickým ochorením obličiek (CKD) s eGFR
v rozsahu 25 až 65 ml/min/1,73 m2, akboli mladší ako 56 rokov alebo s eGFR v rozsahu 25 až
44 ml/min/1,73 m2 a s poklesom eGFR > 2,0 ml/min/1,73 m2/rok, ak boli vo veku 56-65 rokov, bolo randomizovaných na tolvaptán (n = 683) alebo placebo (n = 687) a boli liečení po dobu 12 mesiacov.
U randomizovaných účastníkov bola východisková priemerná odhadovaná rýchlosť glomerulárnej filtrácie (eGFR) 41 ml/min/1,73 m2 (epidemiologická rovnica pre CKD) a celkový objem obličiek v minulosti, dostupný u 318 (23 %) účastníkov, bol v priemere 2026 ml. Približne 5 % malo eGFR
60 ml/min/1,73 m2alebo vyššiu (CKD štádium 2), 75% malo eGFR nižšiu ako 60 a vyššiu ako
30 ml/min/1,73 m2 (CKD štádium 3) a 20 % malo eGFR nižšiu ako 30, ale vyššiu ako
15 ml/min/1,73 m2 (CKD štádium4). CKD v štádiu 3 je možné ďalej rozdeliť na štádium 3a 30 %, (eGFR 45 ml/min/1,73 m2až menej ako 60 ml/min/1,73 m2) a štádium 3b 45 %, (eGFR v rozsahu 30-
45 ml/min/1,73 m2).
Primárnym výsledkom štúdie bola zmena odhadovanej rýchlosti glomerulárnej filtrácie (eGFR)
z východiskovej úrovne pred liečou na hodnotenie po liečbe. U pacientov liečených tolvaptánom bolo zníženie eGFR menej výraznejšie ako u pacientov liečených placebom (p < 0,0001). Liečebný rozdiel v zmene eGFR pozorovanej v tomto skúšaní je 1,27 ml/min/1,73 m2, čo zodpovedá 35 % poklesu priemerov najmenších štvorcov zmeny eGFR -2,34 ml/min/1,73 m2v skupine s tolvaptánom v porovnaní s -3,61 ml/min/1,73 m2v skupine s placebom pozorované v priebehu jedného roka. Hlavným sekundárnym výsledkom bolo porovnanie účinnosti liečby tolvaptánom a placebom znížením poklesu ročnej krivky eGFR vo všetkých meraných časových bodoch počas štúdie. Tieto údaje preukázali aj výrazný prínos liečby tolvaptánom v porovnaní s placebom (p < 0,0001).
Analýza podskupín primárnych a sekundárnych výsledkov podľa rýchlosti CKD preukázala podobné konzistentné účinky liečby vo vzťahu k placebu u účastníkov, ktorí boli na začiatku v štádiu 2, 3a, 3b a 4.
Vopred špecifikovaná analýza podskupiny naznačila, že tolvaptán má nižší účinok u pacientov starších ako 55 rokov, u malej podskupiny s evidentne pomalšou rýchlosťou poklesu eGFR.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s tolvaptánom
v jednej alebo vo viacerých podskupinách pediatrickej populácie s polycystickým ochorením obličiek
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po perorálnom podaní sa tolvaptán rýchlo absorbuje, pričom vrcholové koncentrácie v plazme sa
vyskytujú asi 2 hodiny po podaní. Absolútna biologická dostupnosť tolvaptánu je približne 56 %.Po podaní tolvaptánu spolu s jedlom s vysokým obsahom tuku sa zvýšila vrcholová koncentrácia tolvaptánu až 2-násobne, hodnota AUC však zostala nezmenená. Hoci klinický význam tohto zistenia nie je známy, na minimalizovanie zbytočného rizika zvýšenia maximálnej expozície sa ranná dávka má užívať na lačno (pozri časť 4.2).
Distribúcia
Po jednorazových perorálnych dávkach ≥ 300 mg dosiahli vrcholové koncentrácie v plazme plató,
pravdepodobne z dôvodu nasýtenia absorpcie. Tolvaptán sa vratne (98 %) viaže na plazmatické
proteíny.
Biotransformácia
Tolvaptán sa v značnej miere metabolizuje v pečeni, a to takmer výlučne prostredníctvom CYP3A.
Tolvaptán je slabý substrát CYP3A4 a nezdá sa, že by mal nejaký inhibičný účinok.V štúdiách in vitro sa ukázalo, že tolvaptán nemá inhibičný účinok na CYP3A. V plazme, moči a stolici sa zistilo štrnásť metabolitov, z ktorých všetky okrem jedného sa metabolizovali aj prostredníctvom CYP3A. Len metabolit kyselina oxomaslová je prítomný vo väčšom množstve ako 10 % celkovej rádioaktivity plazmy. Všetky ostatné sú prítomné v koncentráciách nižších ako tolvaptán.Metabolity tolvaptánu len málo prispievajú alebo nijak neprispievajú k farmakologickému účinku tolvaptánu. Všetky metabolity majú len slabý alebo nemajú žiadny antagonistický účinok na ľudské receptory V2 v porovnaní
s tolvaptánom. Polčas konečnej eliminácie je približne 8 hodín a rovnovážna koncentrácia tolvaptánu
sa dosiahne po prvej dávke.
Eliminácia
Menej ako 1 % nevyužitého liečiva sa vylúči v nezmenenej forme močom. V pokusoch s rádioaktívne
označeným tolvaptánom sa zistilo, že 40 % rádioaktivity sa vylúčilo v moči a 59 % v stolici, kde nezmenený tolvaptán predstavoval 32 % rádioaktivity. Tolvaptán je len menšinová zložka plazmy
(3 %).
Linearita
Po jednotlivých perorálnych dávkach sa hodnoty Cmax zvyšovali menej ako proporcionálne k dávke pri dávkach od 30 do 240 mg a potom dosiahli plató pri dávkach od 240 do 480 mg. Hodnota AUC sa
zvyšovala lineárne.
Po opakovanom dávkovaní 300 mg raz denne sa expozícia tolvaptánu zvýšila len 6,4-násobne
v porovnaní s dávkou 30 mg. V režimoch delenej dávky 30, 60 a 120 mg/deň sa expozícia tolvaptánu
(AUC) u pacientov s ADPKD zvyšuje lineárne.
Farmakokinetika v osobitných populáciách
Vek
Vek nemá významný vplyv na klírens tolvaptánu.
Poruchy funkcie pečene
Účinok miernej alebo stredne závažnej poruchy funkcie pečene (triedy A a B podľa Child-Pughovej klasifikácie) na farmakokinetiku tolvaptánu sa skúmal u 87 pacientov s ochorením pečene rôzneho
pôvodu. Nepozorovali sa žiadne klinicky významné zmeny v klírense pri dávkach v rozmedzí od 5 do
60 mg. Informácie o pacientoch so závažnou poruchou funkcie pečene (trieda C podľa Child-Pughovej
klasifikácie) sú veľmi obmedzené.
V populačnej farmakokinetickej analýze pacientov s pečeňovým edémom bola hodnota AUC tolvaptánu u pacientov so závažnou poruchou funkcie pečene (trieda C podľa Child-Pughovej klasifikácie) 3,1-krát vyššia a u pacientov s mierne alebo stredne závažnou poruchou funkcie pečene (triedy A a B podľa Child-Pughovej klasifikácie) 2,3-vyššia ako u zdravých účastníkov.
Poruchy funkcie obličiek
V populačnej farmakokinetickej analýze pacientov s ADPKD bola koncentrácia tolvaptánu zvýšená
v porovnaní so zdravými účastníkmi, pričom funkcia obličiek klesla pod úroveň eGFR
60 ml/min./1,73 m2. Pokles hodnoty eGFRCKD-EPI zo 72,2 na 9,79 (ml/min./1,73 m2) bol spojený so
znížením klírensu celého tela 32 %.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity alebo karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.U králikov, ktoré dostávali dávku 1 000 mg/kg/deň (7,5-násobok expozície pri ľudskej dávke 120 mg/deň na základe AUC), sa pozorovala teratogenicita. Pri dávke 300 mg/kg/deň (asi
1,25-násobok až 2,65-násobok expozície u ľudí v dávke 120 mg/deň na základe AUC) sa u králikov
nepozorovali žiadne teratogénne účinky.V perinatálnej a postnatálnej štúdii na potkanoch sa
pozorovala oneskorená osifikácia a znížená telesná hmotnosť mláďat pri vysokej dávke
1 000 mg/kg/deň.
V dvoch štúdiách fertility na potkanoch sa preukázali účinky na rodičovskú generáciu (znížená
konzumácia potravy a prírastok telesnej hmotnosti, slinenie), ale tolvaptán neovplyvnil rozmnožovaciu výkonnosť samcov a nezistili sa žiadne účinky na plody. U samíc sa v oboch štúdiách pozorovali abnormálne cykly ruje.
Úroveň bez pozorovaného nežiaduceho účinku (no observed adverse effect level, NOAEL) pre účinky na rozmnožovanie samíc (100 mg/kg/deň) bola asi 8-krát vyššia ako maximálna odporúčaná dávka
u človeka 120 mg/deň stanovovaná v mg/m2.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
kukuričný škrob hydroxypropylcelulóza monohydrát laktózy stearan horečnatý mikrokryštalická celulóza hlinitý lak indigokarmínu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v pôvodnom obale na ochranu pred svetlom a vlhkosťou.
6.5 Druh obalu a obsah balenia
Jinarc 15 mg tablety
7 alebo 28 tabliet v blistri z PVC/hliníkovej fólie
Jinarc 30 mg tablety
7 alebo 28 tabliet v blistri z PVC/hliníkovej fólie
Jinarc 15 mg tablety + Jinarc 45 mg tablety
14 tabliet v 1 blistri z PVC/hliníkovej fólie so 7 × 15 mg a 7 × 45 mg tabletami
28 tabliet v 2 blistroch z PVC/hliníkovej fólie so 7 × 15 mg a 7 × 45 mg tabletami
56 tabliet v 4 blistroch z PVC/hliníkovej fólie so 7 × 15 mg a 7 × 45 mg tabletami
14 tabliet v 1 blistri z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 15 mg a 7 × 45 mg tabletami
28 tabliet v 2 blistroch z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 15 mg a 7 × 45 mg tabletami
56 tabliet v 4 blistroch z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 15 mg a 7 × 45 mg
tabletami
Jinarc 30 mg tablety + Jinarc 60 mg tablety
14 tabliet v 1 blistri z PVC/hliníkovej fólie so 7 × 30 mg a 7 × 60 mg tabletami
28 tabliet v 2 blistroch z PVC/hliníkovej fólie so 7 × 30 mg a 7 × 60 mg tabletami
56 tabliet v 4 blistroch z PVC/hliníkovej fólie so 7 × 30 mg a 7 × 60 mg tabletami
14 tabliet v 1 blistri z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 30 mg a 7 × 60 mg tabletami
28 tabliet v 2 blistroch z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 30 mg a 7 × 60 mg tabletami
56 tabliet v 4 blistroch z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 30 mg a 7 × 60 mg tabletami
Jinarc 30 mg tablety + Jinarc 90 mg tablety
14 tabliet v 1 blistri z PVC/hliníkovej fólie so 7 × 30 mg a 7 × 90 mg tabletami
28 tabliet v 2 blistroch z PVC/hliníkovej fólie so 7 × 30 mg a 7 × 90 mg tabletami
56 tabliet v 4 blistroch z PVC/hliníkovej fólie so 7 × 30 mg a 7 × 90 mg tabletami
14 tabliet v 1 blistri z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 30 mg a 7 × 90 mg tabletami
28 tabliet v 2 blistroch z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 30 mg a 7 × 90 mg tabletami
56 tabliet v 4 blistroch z PVC/hliníkovej fólie na pohotovostnej karte so 7 × 30 mg a 7 × 90 mg
tabletami
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIOtsuka Pharmaceutical Netherlands B.V. Herikerbergweg 292
1101 CT, Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLOJinarc 15 mg tabletyEU/1/15/1000/001-002 (blister)
Jinarc 30 mg tabletyEU/1/15/1000/003-004 (blister)
Jinarc 15 mg tablety + Jinarc 45 mg tabletyEU/1/15/1000/005-007 (blister)
EU/1/15/1000/014-016 (blister na pohotovostnej karte)
Jinarc 30 mg tablety + Jinarc 60 mg tabletyEU/1/15/1000/008-010 (blister)
EU/1/15/1000/017-019 (blister na pohotovostnej karte)
Jinarc 30 mg tablety + Jinarc 90 mg tabletyEU/1/15/1000/011-013 (blistri)
EU/1/15/1000/020-022 (blister na pohotovostnej karte)
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:27. máj2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.