
br />Pokyny na prípravu a podanie lieku, pozri časť 6.6.
Nesmú sa používať PVC infúzne vaky a polyuretánové infúzne súpravy.
Jevtana sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
4.3 Kontraindikácie
· Precitlivenosť na kabazitaxel, iné taxány alebo polysorbát 80 alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
· Počet neutrofilov menej ako 1500/mm3.
· Ťažká porucha funkcie pečene (celkový bilirubín >3-krát ULN).
· Súbežná vakcinácia vakcínou proti žltej zimnici (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hypersenzitívnereakcie
Pred začatím liečby infúznym roztokom kabazitaxelu musia byť všetci pacienti premedikovaní (pozri časť 4.2).
Pacienti musia byť pozorne sledovaní pre hypersenzitívne reakcie predovšetkým počas prvej a druhej infúzie. Hypersenzitívne reakcie sa môžu objaviť v priebehu niekoľkých minút po začatí infúzie kabazitaxelu, preto musia byť k dispozícii prostriedky a zariadenia na liečbu hypotenzie
a bronchospazmu. Môžu sa vyskytnúť závažné reakcie a môžu zahŕňať generalizovaný
erytém/vyrážku, hypotenziu, bronchospazmus. Závažné hypersenzitívne reakcie si vyžadujú okamžité ukončenie liečby kabazitaxelom a vhodnú liečbu. U pacientov s hypersenzitívnou reakciou, sa musí liečba Jevtanou ukončiť (pozri časť 4.3).
Útlmkostnejdrene
Môže sa vyskytnúť útlm kostnej drene prejavujúci sa ako neutropénia, anémia, trombocytopénia alebo pancytopénia (pozri nižšie časť 4.4 „Riziko neutropénie“ a „Anémia“).
Rizikoneutropénie
Podľa pokynov Americkej spoločnosti pre klinickú onkológiu (American Society of Clinical Oncology (ASCO)) a/alebo aktuálnych inštitucionálnych pokynov môžu pacienti liečení kabazitaxelom dostávať profylakticky G-CSF na zníženie rizika alebo zvládnutie komplikácií spojených s neutropéniou (prolongovaná neutropénia, febrilná neutropénia alebo neutropenická infekcia).
Primárna profylaxia G-CSF sa má zvážiť u pacientov s vysoko rizikovými klinickými faktormi (vek
vyšší ako 65 rokov, zlý zdravotný stav, predchádzajúce epizódy febrilnej neutropénie, extenzívna rádioterapia, nedostatočná výživa, alebo iné závažné komorbidity), ktoré ich predisponujú na zvýšený výskyt komplikácií z prolongovanej neutropénie. Ukázalo sa, že používanie G-CSF znižuje výskyt a závažnosť neutropénie.
Neutropénia je najčastejším nežiaducim účinkom kabazitaxelu (pozri časť 4.8). Je nevyhnutné monitorovať kompletný krvný obraz každý týždeň počas prvého cyklu liečby a pred každým cyklom liečby, aby bolo v prípade potreby možné upraviť dávkovanie.
Ak sa pri správne nastavenej liečbe vyskytne febrilná neutropénia alebo prolongovaná neutropénia,
dávku je potrebné znížiť (pozri časť 4.2).
Liečba sa môže u pacientov obnoviť, iba ak počet neutrofilov bude opäť ≥1500/mm3 (pozri časť 4.3).
Poruchygastrointestinálnehotraktu
Príznaky ako bolesť a citlivosť brucha, horúčka, pretrvávajúca zápcha, hnačka, s neutropéniou alebo bez neutropénie, môžu byť prvotnými prejavmi závažnej gastrointestinálnej toxicity a musia sa bezodkladne vyšetriť a príslušne liečiť. Môže byť potrebné liečbu kabazitaxelom odložiť alebo ukončiť.
Riziko nauzey, vracania, hnačky a dehydratácie
Ak dostane pacient po podaní kabazitaxelu hnačku, môže sa liečiť bežne používanými liekmi proti hnačke. Je potrebné urobiť príslušné opatrenia na rehydratáciu pacienta. Častejšie sa hnačka môže vyskytnúť u pacientov, ktorí absolvovali rádioterapiu v oblasti brucha a panvy. Dehydratácia sa častejšie vyskytuje u pacientov, ktorí sú starší ako 65 rokov. Je potrebné urobiť príslušné opatrenia na rehydratáciu pacientov a monitorovať a upravovať hladiny elektrolytov v sére, predovšetkým draslíka. Pri hnačke stupňa ≥3 môže byť potrebné odložiť liečbu alebo znížiť dávku (pozri časť 4.2). Ak pacienti trpia nauzeou alebo vracaním, môžu sa liečiť bežne používanými antiemetikami.
Riziko ťažkých gastrointestinálnych reakcií
U pacientov liečených kabazitaxelom bolo hlásené gastrointestinálne (GI) krvácanie a perforácia, ileus, kolitída, vrátane fatálnych prípadov (pozri časť 4.8). Opatrnosť sa odporúča najmä pri liečbe pacientov s rizikom vývoja gastrointestinálnych komplikácií: u pacientov s neutropéniou, u starších pacientov, u pacientov súčasne užívajúcich NSAID, s antiagregačnou alebo antikoagulačnou liečbou a u pacientov, ktorí majú v anamnéze rádioterapiu panvy alebo gastrointestinálne ochorenie ako je ulcerácia alebo GI krvácanie.
Periférnaneuropatia
U pacientov liečených kabazitaxelom sa pozorovali prípady periférnej neuropatie, periférnej senzorickej neuropatie (napr. parestézia, dyzestézia) a periférnej motorickej neuropatie. Pacientov liečených kabazitaxelom je potrebné poučiť, aby informovali svojho lekára o výskyte príznakov neuropatie ako je bolesť, pálenie, brnenie, necitlivosť alebo slabosť pred pokračovaním liečby. Lekár musí posúdiť prítomnosť alebo zhoršenie neuropatie pred každou liečbou. Liečba sa musí odložiť, kým sa príznaky nezlepšia. Ak pretrváva periférna neuropatia stupňa >2, musí sa dávka kabazitaxelu zredukovať z 25 mg/m2 na 20 mg/m2 (pozri časť 4.2).
Anémia
U pacientov liečených kabazitaxelom sa pozorovala anémia (pozri časť 4.8). Pred začatím liečby kabazitaxelom sa musí skontrolovať hemoglobín a hematokrit a zistiť, či pacienti vykazujú prejavy a príznaky anémie alebo straty krvi. U pacientov s hemoglobínom <10 g/dl sa odporúča opatrnosť a v prípade klinickej indikácie je potrebné urobiť vhodné opatrenia.
Rizikorenálnehozlyhania
Prípady poruchy funkcie obličiek boli hlásené v súvislosti so sepsou, ťažkou dehydratáciou spôsobenou hnačkou, vracaním a obštrukčnou uropatiou. Bolo pozorované zlyhanie obličiek, vrátane fatálnych prípadov. Je potrebné urobiť príslušné opatrenia na identifikáciu príčiny a intenzívne liečiť pacientov, ak sa to vyskytne.
Počas liečby kabazitaxelom sa má zabezpečiť adekvátna hydratácia. Pacient musí byť poučený, aby ihneď oznámil výraznú zmenu v dennom objeme moču. Koncentráciu kreatinínu v sére je nutné zmerať na začiatku liečby, pri každom krvnom obraze a vždy, keď pacient oznámi zmenu v objeme moču. Liečbu kabazitaxelom je nutné ukončiť v prípade akéhokoľvek zhoršenia funkcie až zlyhania obličiek ≥ stupeň 3 podľa CTCAE 4.0.
R
espiračné
poruchy
Boli hlásené prípady intersticiálnej pneumónie/pneumonitídy a intersticiálneho ochorenia pľúc, ktoré môžu byť spojené s fatálnymi následkami (pozri časť 4.8).
Pokiaľ sa rozvinú nové alebo sa zhoršia súčasné pľúcne príznaky, pacienti musia byť starostlivo sledovaní, ihneď vyšetrení a vhodným spôsobom liečení. Odporúča sa prerušiť liečbu kabazitaxelom do tej doby, kým je k dispozícii diagnóza. Včasné použitie podporných opatrení môže pomôcť zlepšiť stav. Prínosy obnovenia liečby kabazitaxelom sa musia starostlivo zhodnotiť.
Rizikosrdcovejarytmie
Zaznamenali sa srdcové arytmie, najčastejšie tachykardia a atriálna fibrilácia (pozri časť 4.8).
Staršípacienti
Starší pacienti (≥65 rokov) majú zvýšenú pravdepodobnosť výskytu nežiaducich reakcií vrátane neutropénie a febrilnej neutropénie (pozri časť 4.8).
Pacientispoškodenímpečene
Liečba Jevtanou je kontraindikovaná u pacientov s ťažkou poruchou funkcie pečene (celkový bilirubín
>3-krát ULN) (pozri časti 4.3 a 5.2).
Dávka sa musí znížiť pacientom s miernou poruchou funkcie pečene (celkový bilirubín >1 až ≤1,5- krát ULN alebo AST >1,5-krát ULN) (pozri časti 4.2 a 5.2).
Interakcie
Je potrebné vyhnúť sa súbežnému podávaniu silných CYP3A inhibítorov, pretože môžu zvýšiť plazmatické koncentrácie kabazitaxelu (pozri časti 4.2 a 4.5). Ak nie je možné vyhnúť sa súbežnému
podávaniu so silným CYP3A inhibítorom, musí sa zvážiť dôsledné sledovanie toxicity a zníženie
dávky kabazitaxelu (pozri časti 4.2 a 4.5).
Je potrebné vyhnúť sa súbežnému podávaniu silných CYP3A induktorov, pretože môžu znížiť plazmatické koncentrácie kabazitaxelu (pozri časti 4.2 a 4.5).
Pomocnélátky
Rozpúšťadlo obsahuje 573,3 mg etanolu 96 % (15 % v/v), čo zodpovedá 14 ml piva alebo 6 ml vína. Škodlivé pre tých, ktorí trpia alkoholizmom.
Potrebné vziať do úvahy u vysoko rizikových skupín, ako sú pacienti s ochorením pečene alebo s epilepsiou.
4.5 Liekové a iné interakcie
In vitro štúdie ukázali, že kabazitaxel je metabolizovaný predovšetkým prostredníctvom CYP3A (80 % až 90 %) (pozri časť 5.2).
InhibítoryCYP3A
Opakované podávanie ketokonazolu (400 mg raz denne), silného CYP3A inhibítora, viedlo k 20 % zníženiu klírensu kabazitaxelu zodpovedajúcemu 25 % nárastu AUC. Preto je nutné vyhnúť sa súbežnému podávaniu silných CYP3A inhibítorov (napr. ketokonazol, itrakonazol, klaritromycín, indinavir, nefazodón, nelfinavir, ritonavir, sachinavir, telitromycín, vorikonazol), keďže sa môže vyskytnúť zvýšenie plazmatických koncentrácií kabazitaxelu (pozri časti 4.2 a 4.4).
Súbežné podávanie aprepitantu, stredne silného CYP3A inhibítora, neovplyvnilo klírens kabazitaxelu. InduktoryCYP3A
Opakované podávanie rifampicínu (600 mg raz denne), silného CYP3A induktora, viedlo k 21 %
zvýšeniu klírensu kabazitaxelu zodpovedajúcemu 17 % zníženiu AUC. Preto je nutné vyhnúť sa súbežnému podávaniu silných CYP3A induktorov (napr. fenytoín, karbamazepín, rifampicín, rifabutín, rifapentín, fenobarbital), keďže sa môže vyskytnúť zníženie plazmatických koncentrácií kabazitaxelu (pozri časti 4.2 a 4.4). Pacienti taktiež nemajú užívať ľubovník bodkovaný.
OA
T
P1B1
Kabazitaxel preukázal in vitro inhibíciu transportných proteínov organických aniónových transportných polypeptidov OATP1B1. Riziko interakcie so substrátmi OATP1B1 (napr. statíny, valsartan, repaglinid) existuje najmä v priebehu trvania infúzie (1 hodina) a až do 20 minút po ukončení infúzie. Pred podaním substrátov OATP1B1 sa odporúča dodržať časový odstup 12 hodín pred podaním infúzie a najmenej 3 hodiny po ukončení infúzie.
Vakcinácie
Podávanie živých alebo atenuovaných vakcín u pacientov, ktorí sú imunokompromitovaní chemoterapiou, môže mať za následok ťažké alebo fatálne infekcie. U pacientov dostávajúcich kabazitaxel je potrebné zabrániť očkovaniu živými atenuovanými vakcínami. Môžu sa podávať mŕtve alebo inaktivované vakcíny, avšak odpoveď na očkovanie môže byť v tomto prípade znížená.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití kabazitaxelu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu pri maternotoxických dávkach (pozri časť 5.3) a že kabazitaxel prechádza cez placentárnu bariéru (pozri časť 5.3). Rovnako ako aj iné cytostatiká, kabazitaxel môže poškodiť plod u exponovaných gravidných žien.
Gravidným ženám a ženám vo fertilnom veku, ktoré nepoužívajú účinnú antikoncepciu, sa užívanie kabazitaxelu neodporúča.
Dojčenie
Dostupné farmakokinetické údaje u zvierat ukázali, že kabazitaxel a jeho metabolity sa vylučujú do mlieka (pozri časť 5.3). Nie je možné vylúčiť riziko u dojčených detí.
Kabazitaxel sa nesmie užívať počas dojčenia.
Fertilita
Štúdie na zvieratách ukázali, že kabazitaxel postihuje reprodukčný systém samcov potkanov a psov bez akéhokoľvek funkčného účinku na fertilitu (pozri časť 5.3). Avšak, ak sa vezme do úvahy
farmakologická aktivita taxánov, ich genotoxický potenciál a účinok niektorých zlúčenín z tejto triedy na fertilitu v štúdiách na zvieratách, nie je možné vylúčiť účinok na fertilitu mužov u ľudí.
Kvôli potenciálnym účinkom na mužské gaméty a potenciálnej expozícii spermy musia muži liečení kabazitaxelom počas liečby a ešte 6 mesiacov po podaní poslednej dávky kabazitaxelu používať účinnú antikoncepciu. Kvôli potenciálnej expozícii spermy musia muži liečení kabazitaxelom počas liečby zabrániť kontaktu druhých osôb s ejakulátom. Mužov treba poučiť, aby pred liečbou vyhľadali poradenstvo o konzervácii spermií.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kabazitaxel môže ovplyvniť schopnosť viesť vozidlá a obsluhovať stroje, pretože môže spôsobovať únavu a závraty. Pacientov treba poučiť, aby neviedli vozidlá a neobsluhovali stroje, ak pocítia počas liečby tieto nežiaduce účinky.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Bezpečnosť Jevtany v kombinácii s prednizónom alebo prednizolónom bola hodnotená u 371
pacientov s metastatickým kastračne rezistentným karcinómom prostaty, ktorí boli liečení dávkou
25 mg/m2 kabazitaxelu raz za tri týždne v randomizovanej, otvorenej kontrolovanej štúdii fázy III. Pacienti dostávali kabazitaxel v priemere 6 cyklov.
Najčastejšie (≥ 10 %) nežiaduce účinky vo všetkých stupňoch boli anémia (97,3 %), leukopénia
(95,7 %), neutropénia (93,5 %), trombocytopénia (47,4 %) a hnačka (46,6 %). Najčastejšie nežiaduce
účinky (≥5 %) stupňa ≥3 v skupine pacientov s kabazitaxelom boli neutropénia (81,7 %), leukopénia
(68,2 %), anémia (10,5 %), febrilná neutropénia (7,5 %), hnačka (6,2 %).
Liečba bola kvôli nežiaducim účinkom ukončená u 68 pacientov (18,3 %) dostávajúcich kabazitaxel. Najčastejším nežiaducim účinkom, ktorý viedol k ukončeniu liečby kabazitaxelom, bola neutropénia.
Zoznamnežiaducichúčinkovvtabuľkovomformáte
Nežiaduce účinky rozdelené do tried podľa orgánových systémov MedDRA s uvedením frekvencie ich výskytu sú uvedené v tabuľke 2. V rámci každej skupiny s rovnakou frekvenciou sú nežiaduce účinky
zoradené podľa klesajúcej závažnosti. Intenzita nežiaducich účinkov je odstupňovaná
podľa CTCAE 4.0 (stupeň ≥3 = G≥3). Frekvencie výskytu sú určené podľa všetkých stupňov
a definované ako: veľmi časté (≥1/10), časté (≥1/100 až <1/10); menej časté (≥1/1000 až <1/100);
zriedkavé (≥1/10 000 až <1/1000); veľmi zriedkavé (<1/10 000); neznáme (z dostupných údajov).
Tabuľka 2: Hlásené nežiaduce účinky a hematologické abnormality pri kabazitaxeli v kombinácii s prednizónom alebo prednizolónom v štúdii TROPIC (n=371)
T
rieda orgánového
systému
N
ežiaduci účinok Všetky stupne
n (%) Veľmi časté Časté
Stupeň > 3
n (%)
Septický šok 4 (1,1) 4 (1,1)
Sepsa 4 (1,1) 4 (1,1) Celulitída 6 (1,6) 2 (0,5)
Infekcie močových ciest
27 (7,3) 4 (1,1)
Infekcie a nákazy
Chrípka 11 (3) 0
Cystitída 10 (2,7) 1 (0,3)
Infekcie horných dýchacích ciest
10 (2,7) 0
Pásový opar 5 (1,3) 0
Kandidóza 4 (1,1) 0
Neutropéniaa* 347 (93,5) 303 (81,7)

a
Poruchy krvi a lymfatického systému
Anémia
Leukopéniaa
361 (97,3) 39 (10,5)
355 (95,7) 253 (68,2)
Poruchy imunitného systému
Poruchy metabolizmu a výživy
Trombocytopéniaa 176 (47,4) 15 (4)
Febrilná neutropénia 28 (7,5) 28 (7,5) Hypersenzitivita 5 (1,3) 0
Anorexia 59 (15,9) 3 (0,8) Dehydratácia 18 (4,9) 8 (2,2) Hyperglykémia 4 (1,1) 3 (0,8) Hypokaliémia 4 (1,1) 2 (0,5)
Psychické poruchy Úzkosť 11 (3) 0
Stav zmätenosti 5 (1,3) 0
Dysgeúzia 41 (11.1) 0
Periférna neuropatia 30 (8,1) 2 (0,5)
Periférna senzorická neuropatia
20 (5,4) 1 (0,3)
Poruchy nervového systému
Závrat 30 (8,1) 0
Bolesť hlavy 28 (7,5) 0
Parestézia 17 (4,6) 0
Letargia 5 (1,3) 1 (0,3) Hypoestézia 5 (1,3) 0
Ischias 4 (1,1) 1 (0,3)
T
rieda orgánového systému
N
ežiaduci účinok Všetky stupne n (%)
V
eľmi časté Časté
Stupeň > 3
n (%)
Poruchy oka Konjunktivitída 5 (1,3) 0
Zvýšená lakrimácia 5 (1,3) 0
Poruchy ucha a labyrintu Poruchy srdca a
srdcovej činnosti*
Tinitus 5 (1,3) 0
Vertigo 5 (1,3) 0
Atriálna fibrilácia 4 (1,1) 2 (0,5) Tachykardia 6 (1,6) 0
Hypotenzia 20 (5,4) 2 (0,5)
Hlboká žilová trombóza
8 (2,2) 7 (1,9)
Hypertenzia 6 (1,6) 1 (0,3)
Poruchy ciev
Ortostatická hypotenzia
5 (1,3) 1 (0,3)
Návaly tepla 5 (1,3) 0
Sčervenanie pokožky
4 (1,1) 0
Poruchy dýchacej
Dyspnoe 44 (11,9) 5 (1,3) Kašeľ 40 (10,8) 0
sústavy, hrudníka a
mediastína
Orofaryngálna bolesť
13 (3,5) 0
Pneumónia 9 (2,4) 6 (1,6) Hnačka 173 (46,6) 23 (6,2) Nauzea 127 (34,2) 7 (1,9) Vracanie 84 (22,6) 7 (1,9) Zápcha 76 (20,5) 4 (1,1) Bolesť brucha 43 (11,6) 7 (1,9) Dyspepsia 25 (6,7) 0
Poruchy gastrointestinálneho
Bolesť brucha v
hornej časti
20 (5,4) 0
traktu
Hemoroidy 14 (3,8) 0
Gastroezofageálny
reflux Krvácanie z konečníka
12 (3,2) 0
8 (2,2) 2 (0,5)
Sucho v ústach 8 (2,2) 1 (0,3)
Abdominálna distenzia
5 (1,3) 1 (0,3)
Poruchy kože a podkožného tkaniva
Alopécia 37 (10) 0
Suchá koža 9 (2,4) 0
Erytém 5 (1,3) 0
Bolesť chrbta 60 (16,2) 14 (3,8) Artralgia 39 (10,5) 4 (1,1)
Poruchy kostrovej a
Bolesť v
končatinách
30 (8,1) 6 (1,6)
svalovej sústavy a spojivového tkaniva
Svalové kŕče 27 (7,3) 0
Myalgia 14 (3,8) 1 (0,3)
Muskuloskeletálna bolesť na hrudníku
11 (3) 1 (0,3)
Bolesť v trieslach 7 (1,9) 3 (0,8)
Poruchy obličiek a močových ciest
Akútne zlyhanie obličiek
8 (2,2) 6 (1,6)
Zlyhanie obličiek 7 (1,9) 6 (1,6)
T
rieda orgánového systému
N
ežiaduci účinok Všetky stupne n (%)
V
eľmi časté Časté
Stupeň > 3
n (%)
Dysúria 25 (6,7) 0
Obličková kolika 5 (1,3) 1 (0,3) Hematúria 62 (16.7) 7 (1,9) Polakizúria 13 (3,5) 1 (0,3) Hydronefróza 9 (2,4) 3 (0,8) Retencia moču 9 (2,4) 3 (0,8) Inkontinencia moču 9 (2,4) 0
Obštrukcia
močovodov
7 (1,9) 5 (1,3)
Poruchy reprodukčného systému a prsníkov
Celkové poruchy a reakcie v mieste podania
Bolesť panvy 7 (1,9) 1 (0,3)
Únava 136 (36,7) 18 (4,9) Asténia 76 (20,5) 17 (4,6) Pyrexia 45 (12,1) 4 (1,1) Periférny edém 34 (9,2) 2 (0,5) Zápal slizníc 22 (5,9) 1 (0,3) Bolesť 20 (5,4) 4 (1,1) Bolesť na hrudníku 9 (2,4) 2 (0,5) Edém 7 (1,9) 1 (0,3) Zimnica 6 (1,6) 0
Malátnosť 5 (1,3) 0
Úbytok hmotnosti 32 (8,6) 0
Laboratórne a funkčné vyšetrenia
Zvýšená hladina aspartátaminotransfe rázy
Zvýšená hladina transamináz
4 (1,1) 0
4 (1,1) 0
a na základe laboratórnych hodnôt
* podrobnejšie informácie v časti dolu
Opis vybranýchnežiaducichúčinkovNeutropénia a pridružené klinické udalostiVýskyt neutropénie stupňa ≥ 3 bol 81,7 % na základe laboratórnych údajov. Výskyt nežiaducich účinkov klinickej neutropénie stupňa ≥ 3 bol 21,3 % a febrilnej neutropénie 7,5 %. Neutropénia bola najčastejším nežiaducim účinkom, ktorý viedol k ukončeniu liečby (2,4 %).
Komplikácie neutropénie zahŕňali neutropenické infekcie (0,5 %), neutropenickú sepsu (0,8 %)
a septický šok (1,1 %), v niektorých prípadoch s fatálnym koncom.
Ukázalo sa, že užívanie G-CSF limituje výskyt a závažnosť neutropénie (pozri časti 4.2 a 4.4).
Poruchy srdca a arytmiePri poruchách srdca boli všetky udalosti stupňa častejšie s kabazitaxelom, z nich 6 pacientov (1,6 %) malo srdcové arytmie stupňa ≥ 3. Výskyt tachykardie s kabazitaxelom bol 1,6 %, žiadny z nich nebol stupňa ≥ 3. Výskyt atriálnej fibrilácie v skupine s kabazitaxelom bol 1,1 %. Udalosti srdcového zlyhania boli častejšie u kabazitaxelu, udalosť bola takto hlásená u 2 pacientov (0,5 %). Jeden pacient v skupine s kabazitaxelom zomrel na zlyhanie srdca. Fatálna ventrikulárna fibrilácia bola zaznamenaná u 1 pacienta (0,3 %) a zástava srdca u 2 pacientov (0,5 %). Žiadna nebola investigátorom posúdená ako súvisiaca s liekom.
Hematúria
Hematúria všetkých frekvenčných stupňov bola pozorovaná v 20,8 % pri 25 mg/m2 v štúdii EFC11785 (pozri časť 5.1). Takmer v dvoch tretinách prípadov boli identifikované mätúce príčiny, ako napr. progresia ochorenia, inštrumentácia, infekcia alebo antikoagulačná/NSAID/aspirínová liečba.
Ďalšie laboratórne abnormalityNa základe laboratórnych abnormalít bol výskyt anémie stupňa ≥ 3 10,5 %, zvýšené hladiny AST
0,7 %, ALT 0,9 % a bilirubínu 0,6 %.
Poruchy gastrointestinálneho traktuBola hlásená kolitída, enterokolitída, gastritída, neutropenická enterokolitída. Ďalej bolo zaznamenané gastrointestinálne krvácanie a perforácia, ileus a intestinálna obštrukcia (pozri časť 4.4).
Respiračné poruchyPrípady intersticiálnej pneumónie/pneumonitídy a intersticiálneho ochorenia pľúc, niekedy fatálne, boli hlásené s neznámou frekvenciou výskytu (nie je možné odhadnúť z dostupných údajov) (pozri časť 4.4).
Poruchy obličiek a močových ciestMenej často bola hlásená cystitída, vrátane hemoragickej cystitídy, spôsobená recall fenoménom po liečbe ožarovaním.
PediatrickápopuláciaPozri časť 4.2
InéšpeciálneskupinypacientovStarší pacientiMedzi 371 pacientmi, ktorí boli v štúdii karcinómu prostaty liečení kabazitaxelom, bolo 240 pacientov starších ako 65 rokov a z nich 70 starších ako 75 rokov.
Nasledujúce nežiaduce účinky hlásené v pomere ³ 5 % vyššom u pacientov vo veku 65 rokov
a starších v porovnaní s mladšími pacientmi boli únava (40,4 % oproti 29,8 %), klinická neutropénia (24,2 % oproti 17,6 %), asténia (23,8 % oproti 14,5 %), pyrexia (14,6 % oproti 7,6 %), závrat (10,0 % oproti 4,6 %), infekcie močových ciest (9,6 % oproti 3,1 %) a dehydratácia (6,7 % oproti 1,5 %),
v uvedenom poradí.
Výskyt nasledujúcich nežiaducich účinkov stupňa ≥ 3 bol u pacientov ³ 65 rokov vyšší v porovnaní
s mladšími pacientmi: neutropénia založená na laboratórnych abnormalitách (86,3 % oproti 73,3 %), klinická neutropénia (23,8 % oproti 16,8 %) a febrilná neutropénia (8,3 % oproti 6,1 %) (pozri
časti 4.2 a 4.4).
Z 595 pacientov liečených kabazitaxelom 25 mg/m2 v štúdii karcinómu prostaty EFC 11785, malo 420
pacientov 65 rokov alebo viac. Nežiaducimi reakciami hlásenými u pacientov vo veku 65 rokov
a starších, s výskytom aspoň o 5 % vyšším v porovnaní s mladšími pacientmi boli hnačka (42,9 %
oproti 32,6 %), únava (30,2 % oproti 19,4 %), asténia (22,4 % oproti 13,1 %), zápcha (20,2 % oproti
12,6 %), klinická neutropénia (12,9 % oproti 6,3 %), febrilná neutropénia (11,2 % oproti 4,6 %) a dyspnoe (9,5 % oproti 3,4 %).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNie je známe žiadne antidotum pre kabazitaxel. Predpokladané komplikácie z predávkovania sú exacerbácia nežiaducich účinkov ako útlm kostnej drene a gastrointestinálne poruchy.
V prípade predávkovania musí pacient zostať na špecializovanom pracovisku a byť pozorne monitorovaný. Ihneď po zistení, že došlo k predávkovaniu, musí pacient dostávať terapeutické dávky G-CSF. Podľa okolností treba začať ďalšie vhodné symptomatické opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antineoplastiká, taxány, ATC kód: L01CD04
Mechanizmusúčinku
Kabazitaxel je antineoplastická látka, ktorá narušuje v bunkách mikrotubulárnu sieť. Kabazitaxel sa viaže na tubulín a podporuje zabudovávanie tubulínu do mikrotubulov a zároveň inhibuje ich depolymerizáciu. Toto vedie k stabilizácii mikrotubulov a výsledkom je inhibícia mitotických a medzifázových bunkových funkcií.
Farmakodynamickéúčinky
Kabazitaxel preukázal široké spektrum experimentálnej protinádorovej aktivity voči pokročilým ľudským nádorovým transplantátom u myší. Kabazitaxel je aktívny v nádoroch senzitívnych na
docetaxel. Navyše kabazitaxel preukázal aktivitu v tumorových modeloch nesenzitívnych na
chemoterapiu vrátane docetaxelu.
Klinickáúčinnosťabezpečnosť
Účinnosť a bezpečnosť Jevtany v kombinácii s prednizónom alebo prednizolónom boli vyhodnotené v randomizovanej, otvorenej, medzinárodnej, multicentrickej štúdii fázy III (štúdia EFC6193) na pacientoch s metastatickým kastračne rezistentným karcinómom prostaty, ktorí boli v predchádzajúcom období liečení režimom obsahujúcim docetaxel.
Primárnym koncovým ukazovateľom účinnosti v štúdii bolo celkové prežívanie (Overall Survival - OS).
Sekundárne koncové ukazovatele zahŕňali čas prežívania bez progresie [PFS, (definovaný ako čas od
randomizácie do progresie tumoru, progresie prostatického špecifického antigénu (PSA), progresie bolesti alebo do úmrtia z rôznych príčin, podľa toho, čo sa objavilo skôr], mieru odpovede nádoru (Tumour Response Rate) podľa kritérií hodnotenia odpovede solídnych tumorov (Response Evaluation Criteria in Solid Tumours – RECIST), progresiu PSA (definovaná ako ≥ 25 % nárast u PSA bez odpovede alebo > 50 % u PSA s odpoveďou), PSA odpoveď (pokles hladiny PSA v sére aspoň
o 50 %), progresiu bolesti [vyhodnotené podľa škály intenzity prítomnej bolesti (Present Pain Intensity
- PPI) z McGill-Melzackovho dotazníka bolesti a podľa analgetického skóre (Analgesic Score - AS) ]
a odpoveď bolesti (definovaná ako viac ako 2 bodový pokles zo vstupného mediánu PPI
bez súčasného zvýšenia v AS, alebo pokles ≥ 50 % v užívaní analgetík z priemernej počiatočnej hodnoty AS bez súčasného zvýšenia bolesti).
Celkovo bolo randomizovaných 755 pacientov dostávajúcich buď intravenózne Jevtanu 25 mg/m2 každé 3 týždne v maximálne 10 cykloch s prednizónom alebo s prednizolónom 10 mg denne perorálne (n=378), alebo dostávajúcich intravenózne mitoxantrón 12 mg/m2 každé 3 týždne v maximálne
10 cykloch s prednizónom alebo s prednizolónom 10 mg denne perorálne (n=377).
Do štúdie boli zaradení pacienti starší ako 18 rokov s metastatickým kastračne rezistentným karcinómom prostaty buď merateľným prostredníctvom kritérií RECIST alebo nemerateľným typom so zvýšenými hladinami PSA alebo vznikom nových lézií a výkonnostným stavom 0 až 2 podľa Eastern Cooperative Oncology Group (ECOG). Pacienti museli mať hladinu neutrofilov > 1500/mm3, krvných doštičiek > 100000/mm3, hemoglobínu > 10 g/dl, kreatinínu < 1,5-krát ULN, celkového bilirubínu < 1-krát ULN, AST a ALT < 1,5-krát ULN.
Pacienti s osobnou anamnézou kongestívneho zlyhania srdca alebo infarktu myokardu počas posledných 6 mesiacov alebo pacienti s nekontrolovanou srdcovou arytmiou, anginou pectoris, a/alebo hypertenziou neboli zaradení do štúdie.
Demografické charakteristiky vrátane veku, rasy a výkonnostného stavu ECOG (0 až 2), boli vyvážené medzi obidvomi liečebnými ramenami štúdie. V skupine s Jevtanou bol priemerný vek
68 rokov (od 42 do 92) a rasové zastúpenie bolo 83,9 % belochov, 6,9 % aziatov, 5,3 % černochov a
4 % iných.
V skupine s Jevtanou bol medián počtu cyklov 6 a v skupine s mitoxantrónom 4. Počet pacientov, ktorí dokončili liečbu v rámci štúdie (10 cyklov) bol 29,4 % v skupine s Jevtanou a 13,5 %
v porovnávacej skupine.
Celkové prežívanie bolo štatisticky významne dlhšie pri Jevtane (15,1 mesiacov) v porovnaní
s mitoxantrónom (12,7 mesiaca), s 30 % znížením rizika smrti v porovnaní s mitoxantrónom (pozri tabuľku 3 a graf 1).
Podskupina 59 pacientov dostávala predtým kumulatívnu dávku docetaxelu < 225 mg/m²
(29 pacientov v ramene s Jevtanou, 30 pacientov v ramene s mitoxantrónom). V tejto skupine pacientov nebol žiadny významný rozdiel v celkovom prežívaní pacientov (HR (95 %CI) 0,96 (0,49-
1,86)).

Tabuľka 3 - Účinnosť Jevtany v štúdii EFC6193 v liečbe pacientov s metastatickým kastračne rezistentným karcinómom prostaty
Jevtana + prednizón
m
i
t
oxantrón + prednizón
n=378 n=377
Celkový čas prežívania
Počet úmrtí (%) 234 (61,9 %) 279 (74 %)
Medián prežívania (mesiace) (95 % CI)
15,1 (14,1-16,3) 12,7 (11,6-13,7)
Hazard Ratio (HR)1 (95 % CI) 0,70 (0,59-0,83)
p-hodnota <0,0001

1HR hodnota vypočítaná podľa Coxovho modelu; hodnota HR menšia ako 1 je v prospech Jevtany
Graf 1: Kaplan-Meierove krivky celkového prežívania (EFC6193)

100
mitoxantrón + prednizón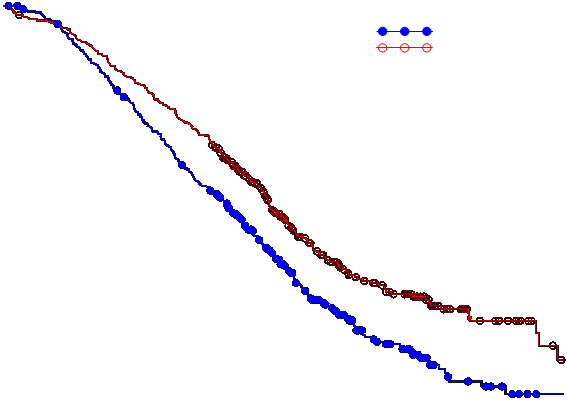

90
kabazitaxel + prednizón
80
70
60
50
40
30
20
10
0
0 6 12 18 24 30
P
očet rizikových pacientov
Č
as (mesiace)
m
i
t
o
x
a
nt
rón
+ prednizón kabazitaxel
+ prednizón
377 300 188 67 11 1
378 321 231 90 28 4
V skupine s Jevtanou bolo zlepšenie prežívania bez progresie v porovnaní so skupinou
s mitoxantrónom, 2,8 (2,4–3,0) mesiaca oproti 1,4 (1,4–1,7) mesiaca v uvedenom poradí, HR (95 % CI) 0,74 (0,64-0,86), p<0,0001.
U pacientov v ramene s Jevtanou bola štatisticky významne vyššia miera odpovede tumoru, a to
14,4 % (95 % CI: 9,6-19,3) v porovnaní so 4,4 % (95 % CI: 1,6-7,2) u pacientov v ramene s mitoxantrónom, p=0,0005.
Sekundárne koncové ukazovatele PSA boli pozitívne v ramene s Jevtanou. Medián progresie PSA bol
6,4 mesiacov (95 % CI: 5,1-7,3) u pacientov v ramene s Jevtanou, v porovnaní s 3,1 mesiacov (95 % CI: 2,2-4,4) v ramene s mitoxantrónom, HR 0,75 mesiaca (95 % CI 0,63-0,90), p=0,0010. Odpoveď
PSA bola 39,2 % u pacientov v ramene s Jevtanou (95 % CI: 33,9-44,5) oproti 17,8 % pacientov,
ktorým bol podávaný mitoxantrón (95 % CI: 13,7-22,0), p=0,0002.
V progresii bolesti a odpovedi bolesti nebol medzi ramenami štatisticky významný rozdiel.
V neinferiórnej, multicentrickej, viacnárodnej, randomizovanej otvorenej štúdii (EFC11785 štúdia) fázy III bolo randomizovaných 1200 pacientov s metastatickým kastračne rezistentným karcinómom prostaty, v minulosti liečených režimom obsahujúcim docetaxel, ktorí dostávali buď dávku kabazitaxelu 25 mg/m2 (n = 602) alebo 20 mg/m2 (n = 598). Primárnym koncovým ukazovateľom účinnosti bolo celkové prežívanie (overall survival, OS).
Štúdia splnila svoj primárny cieľ preukázania neinferiority kabazitaxelu 20 mg/m2 v porovnaní s
25 mg/m2 (pozri tabuľku 4). Štatisticky významne vyššie percento (p < 0,001) pacientov preukázalo
PSA odpoveď v skupine s 25 mg/m2 (42,9 %) oproti skupine s 20 mg/m2 (29,5 %). Bolo pozorované
štatisticky významne vyššie riziko progresie PSA u pacientov s dávkou 20 mg/m2, voči dávke
25 mg/m2 (HR 1,195; 95 % CI: 1,025 až 1,393). Neboli zistené žiadne štatistické rozdiely v súvislosti s ďalšími sekundárnymi koncovými ukazovateľmi (PFS, tumor a reakcia na bolesť, tumor a progresia bolesti a štyri podkategórie FACT-P).
Tabuľka 4 - Celkové prežívanie v EFC11785 štúdii, v ramene s kabazitaxelom 25 mg/m2 oproti ramenu s kabazitaxelom 20 mg/m2 (analýza pri zámere liečiť, Intent-to-treat analysis) – primárny koncový ukazovateľ účinnosti
C
elkové prežívanie
C
B
Z
20 + PRED
n = 598
C
B
Z
25 + PRED
n = 602
Počet úmrtí, n (%) 497 (83,1 %) 501 (83,2 %)
Stredná doba prežívania (95 % CI) (mesiace)
Miera rizikaa
13,4 (12,19 až 14,88) 14,5 (13,47 až 15,28)
oproti CBZ25 + PRED 1,024 - jednostranná 98,89 % UCI 1,184 - jednostranná 95 % LCI 0,922 -
CBZ20 = kabazitaxel 20 mg/m2, CBZ25 = kabazitaxel 25 mg/m2, PRED = prednizón/prednizolón CI = interval spoľahlivosti (confidence interval), LCI = dolná hranica intervalu spoľahlivosti (lower bound of the confidence interval), UCI = horná hranica intervalu spoľahlivosti (upper bound of the
confidence interval)
a Miera rizika sa odhaduje použitím Coxového regresného modelu pomerného rizika. Miera rizika
< 1 indikuje nižšie riziko kabazitaxelu 20 mg/m2 oproti 25 mg/m2.
Bezpečnostný profil kabazitaxelu 25 mg/m2 pozorovaný v štúdii EFC11785 bol kvalitatívne a kvantitatívne podobný tomu, ktorý bol pozorovaný v štúdii EFC6193. Štúdia EFC11785 preukázala lepší bezpečnostný profil pre dávku kabazitaxelu 20 mg/m2.
Tabuľka 5 - Súhrn bezpečnostných údajov v ramene s kabazitaxelom 25 mg/m2 oproti ramenu s kabazitaxelom 20 mg/m2 v štúdii EFC11785
CBZ20 + PRED
n = 580
CBZ25 + PRED
n = 595
Priemerný počet cyklov/ priemerná doba trvania liečby
Počet pacientov s redukciou
6/18 týždňov 7/21 týždňov
Od 25 do 20 mg/m2: 128 (21,5 %)
2
dávky n (%) Od 20 do 15 mg/m2: 58 (10,0 %)
Od 15 do 12 mg/m2: 9 (1,6 %)
Nežiaduce reakcie všetkých stupňova (%)
Od 20 do 15 mg/m : 19 (3,2 %)
Od 15 do 12 mg/m2: 1 (0,2 %)


Hnačka 30,7 39,8
Nauzea 24,5 32,1
Únava 24,7 27,1
Hematúria 14,1 20,8
Asténia 15,3 19,7
Znížená chuť do jedla 13,1 18,5
Vracanie 14,5 18,2
Zápcha 17,6 18,0
Bolesť chrbta 11,0 13,9
Klinická neutropénia 3,1 10,9
Infekcie močového traktu 6,9 10,8
Periférna senzorická neuropatia
6,6 10,6
Dysgeúzia 7,1 10,6
Nežiaduce reakcie stupňa ≥ 3b (%)
Klinická neutropénia 2,4 9,6
Febrilná neutropénia 2,1 9,2
Hematologické abnormalityc (%)
Neutropénia stupňa ≥ 3 41,8 73,3
Anémia stupňa ≥ 3 9,9 13,7
Trombocytopénia stupňa ≥ 3 2,6 4,2
CBZ20 = kabazitaxel 20 mg/m2, CBZ25 = kabazitaxel 25 mg/m2, PRED=Prednizón/Prednizolón
a Nežiaduce reakcie všetkých stupňov s výskytom vyšším ako 10 %
b Nežiaduce reakcie stupňa ≥ 3 s výskytom vyšším ako 5 %
c Založené na laboratórnych hodnotách
PediatrickápopuláciaEurópska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií s Jevtanou vo všetkých podskupinách pediatrickej populácie pre schválenú indikáciu karcinómu prostaty (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
Jevtana bola hodnotená vo fázach 1 a 2 otvorenej multicentrickej štúdii vykonanej celkovo u 39 pediatrických pacientov (vo veku od 4 do 18 rokov pre časť štúdie fázy 1 a vo veku od 3 do 16 rokov pre časť štúdie fázy 2). Štúdia fázy 2 nepreukázala účinnosť kabazitaxelu v monoterapii v pediatrickej populácii s rekurentným alebo refraktérnym difúznym vnútornym gliómom mozgového kmeňa a high- grade gliómom (HGG) liečeným 30 mg/m².
5.2 Farmakokinetické vlastnostiPopulačná farmakokinetická analýza sa uskutočnila u 170 pacientov vrátane pacientov s pokročilým solídnym tumorom (n=69), metastatickým karcinómom prsníka (n=34) a metastatickým karcinómom prostaty (n=67). Títo pacienti dostávali kabazitaxel v dávkach od 10 do 30 mg/m2 každý týždeň alebo každé 3 týždne.
AbsorpciaU pacientov s metastatickým karcinómom prostaty bola po 1-hodinovej intravenóznej infúzii
25 mg/m2 kabazitaxelu Cmax 226 ng/ml (variačný koeficient - Coefficient of Variation (CV): 107 %)
a bola dosiahnutá na konci 1-hodinovej infúzie (Tmax). Priemerná hodnota AUC bola 991 ng.h/ml
(CV: 34 %).
U pacientov s pokročilým solídnym tumorom (n=126) nebola pozorovaná veľká odchýlka vo vzťahu k proporcionalite dávky od 10 do 30 mg/m².
DistribúciaDistribučný objem (Vss) bol 4870 l (2640 l/m² u pacientov s mediánom BSA 1,84 m²) v rovnovážnom stave.
I
n vitro, viazanie kabazitaxelu na ľudské sérové proteíny bolo 89-92 % a nebolo saturovateľné'
do 50 000 ng/ml, čo pokrýva maximálnu koncentráciu v klinických štúdiách. Kabazitaxel sa viaže predovšetkým na ľudský sérový albumín (82,0 %) a lipoproteíny (87,9 % pre HDL, 69,8 % pre LDL, a 55,8 % pre VLDL).
V ľudskej krvi bol in vitro pomer koncentrácie v krvi a plazme od 0,90 do 0,99, čo naznačuje, že kabazitaxel sa distribuoval do krvi a plazmy rovnomerne.
Biotransformácia
Kabazitaxel sa extenzívne metabolizuje v pečeni (> 95 %), predovšetkým izoenzýmom CYP3A (80 % až 90 %). V ľudskej plazme cirkuluje ako hlavná zlúčenina kabazitaxel. V plazme bolo zistených sedem metabolitov (vrátane 3 aktívnych metabolitov, ktoré vznikli z O-demetylácií), pričom hlavný bol zodpovedný za 5 % expozície. Okolo 20 metabolitov kabazitaxelu sa vylučuje do ľudského moču
a stolice.
Na základe in vitro štúdií je v klinicky relevantných koncentráciách možné riziko inhibície kabazitaxelom vo vzťahu k tým liekom, ktoré sú predovšetkým substrátmi CYP3A.
Avšak v klinickej štúdii sa preukázalo, že kabazitaxel (25 mg/m2 podávaný vo forme samostatnej
1 hodinovej infúzie) nemenil plazmatické hladiny midazolamu, skúšobného substrátu CYP3A. Pri súčasnom podávaní substrátov CYP3A a kabazitaxelu v terapeutických dávkach sa preto neočakáva žiadny klinický dopad na pacienta.
Nejestvuje potenciálne riziko inhibície liekov, ktoré sú substrátmi iných CYP enzýmov (1A2, 2B6,
2C9, 2C8, 2C19, 2E1 a2D6), rovnako ako neexistuje potenciálne riziko indukcie kabazitaxelom tých liekov, ktoré sú substrátmi CYP1A, CYP2C9 a CYP3A. Kabazitaxel in vitro neinhiboval hlavnú cestu biotransformácie warfarínu na 7-hydroxywarfarín sprostredkovaný CYP2C9. Preto sa in vivo neočakáva žiadna farmakokinetická interakcia kabazitaxelu na warfarín. In vitro kabazitaxel neinhiboval proteíny mnohopočetnej liekovej rezistencie (Multidrug-Resistant Proteins, MRP): MRP1 a MRP2 ani transportér organických katiónov (Organic Cation Transporter, OCT1). Kabazitaxel inhiboval transport P-glykoproteínov (PgP) (digoxín, vinblastín), proteínov rezistencie rakoviny prsníka (Breast-Cancer-Resistant-Proteins, BCRP) (metotrexát) a organického aniónového transportného polypeptidu (Organic Anion Transporting Polypeptide) OATP1B3 (CCK8) pri koncentráciách minimálne 15-násobne vyšších ako v klinických podmienkach, zatiaľ čo inhiboval transport OATP1B1 (estradiol-17β-glukuronid) pri koncentráciách len 5-násobne vyšších ako
v klinických podmienkach. Preto je pri dávke 25 mg/m2 riziko interakcie so substrátmi MRP, OCT1, PgP a OATP1B3 in vivo nepravdepodobné. Existuje riziko interakcie s transportérom OATP1B1, predovšetkým v priebehu trvania infúzie (1 hodina) a až do 20 minút po ukončení infúzie (pozri
časť 4.5).
Vylučovanie
Po 1-hodinovej intravenóznej infúzii [14C]-kabazitaxelu v dávke 25 mg/m2 sa u pacientov približne
80 % podanej dávky eliminovalo v priebehu 2 týždňov. Kabazitaxel je vylučovaný predovšetkým v stolici, ako mnohé metabolity, (76 % dávky); zatiaľ čo renálne vylučovanie kabazitaxelu
a metabolitov predstavujú menej ako 4 % dávky (2,3 % sa v nezmenenej podobe vylúči v moči).
Kabazitaxel mal vysoký klírens plazmy: 48,5 l/h (26,4 l/h/m² u pacientov s mediánom BSA 1,84 m²)
a dlhý terminálny polčas: 95 hodín.
Špeciálneskupinypacientov
Starší pacienti
V populačnej farmakokinetickej analýze u 70 pacientov, ktorí boli starší ako 65 rokov (57 pacientov
vo veku od 65 do 75 rokov a 13 pacientov starších ako 75 rokov) nebol pozorovaný žiadny vplyv veku na farmakokinetiku kabazitaxelu.
Pediatrickí pacienti
Bezpečnosť a účinnosť Jevtany neboli u detí a dospievajúcich vo veku do 18 rokov stanovené.
Poškodenie pečene
Kabazitaxel je eliminovaný predovšetkým metabolizáciou v pečeni.
Špecializovaná štúdia so 43 pacientami s rakovinou s poškodením pečene nepreukázala žiadny vplyv mierneho (celkový bilirubín >1 až ≤1,5-krát ULN alebo AST >1,5-krát ULN) alebo stredne ťažkého (celkový bilirubín >1,5 až ≤3-krát ULN) poškodenia pečene na farmakokinetiku kabazitaxelu. Maximálna tolerovaná dávka (MTD) kabazitaxelu bola 20 a 15 mg/m2 v danom poradí.
U 3 pacientov s ťažkým poškodením pečene (celkový bilirubín >3 ULN) bolo pozorované zníženie klírens o 39 % v porovnaní s pacientmi s miernym poškodením pečene, čo naznačuje určitý vplyv ťažkého poškodenia pečene na farmakokinetiku kabazitaxelu. MTD kabazitaxelu u pacientov
s ťažkým poškodením pečene nebola stanovená.
Na základe údajov o bezpečnosti a znášanlivosti by mala byť u pacientov s miernym poškodením pečene dávka kabazitaxelu znížená (pozri časti 4.2, 4.4).U pacientov s ťažkým poškodením pečene je Jevtana kontraindikovaná (pozri časť 4.3).
Poškodenie obličiek
Kabazitaxel sa vylučuje obličkami iba minimálne (2,3 % dávky). V populačnej farmakokinetickej analýze uskutočnenej na 170 pacientoch, medzi ktorými bolo 14 pacientov so stredne ťažkým poškodením obličiek (klírens kreatinínu v rozmedzí 30 až 50 ml/min) a 59 pacientov s miernym poškodením obličiek (klírens kreatinínu v rozmedzí 50 až 80 ml/min) bolo preukázané, že mierne až stredne ťažké poškodenie obličiek nemá významný účinok na farmakokinetiku kabazitaxelu. Toto bolo potvrdené aj v špecializovanej porovnávacej farmakokinetickej štúdii u pacientov so zhubnými solídnymi nádormi s normálnou funkciou obličiek (8 pacientov), so stredne ťažkou (8 pacientov)
a ťažkou (9 pacientov) poruchou funkcie obličiek, ktorí sa liečili niekoľkými cyklami kabazitaxelu v samostatnej intravenóznej infúzii až do 25 mg/m2.
5.3 Predklinické údaje o bezpečnosti
Nežiaduce reakcie, ktoré neboli pozorované v klinických štúdiách, ale boli pozorované u psov po podaní jedinej dávky pri 5-dňovom a týždennom podávaní pri expozíciách nižších ako sú klinické a s možným významom pre klinické použitie boli arteriolárna/periarteriolárna nekróza pečene, hyperplázia žlčovodu a/alebo hepatocelulárna nekróza (pozri časť 4.2).
Nežiaduce reakcie, ktoré neboli pozorované v klinických štúdiách, ale boli pozorované u potkanov v štúdiách toxicity po opakovanom podávaní dávky pri expozíciách vyšších ako sú klinické a s možným významom pre klinické použitie, boli poruchy oka charakterizované opuchom/degeneráciou subkapsulárnych vlákien šošovky. Po 8 týždňoch boli tieto účinky čiastočne reverzibilné.
Štúdie karcinogénneho potenciálu kabazitaxelu neboli vykonané.
V teste bakteriálnej reverznej mutácie (Ames) kabazitaxel neindukoval mutácie. Nebol klastogenický v ľudských lymfocytoch in vitro (neindukoval štrukturálne chromozomálne aberácie, ale zvýšil počet polyploidných buniek) a indukoval zvýšenie počtu mikrojadier u potkanov v in vivo testoch. Avšak tieto výsledky genotoxicity sú inherentné s farmakologickou aktivitou zlúčeniny (inhibícia tubulínovej depolymerizácie) a boli pozorované u liekov, ktoré vykazovali rovnakú farmakologickú aktivitu.
Kabazitaxel nemal žiadny účinok na aktivitu pri párení alebo fertilitu liečených samcov potkanov. Avšak v štúdiách toxicity po opakovanom podávaní dávky bola u potkanov pozorovaná degenerácia semenných vačkov a atrofia semenotvorných kanálikov v semenníkoch a u psov bola pozorovaná testikulárna degenerácia (minimálna nekróza jednotlivých epitelových buniek v epididymis). Expozície u zvierat boli podobné alebo nižšie než u ľudí, ktorým boli podávané klinicky relevantné dávky kabazitaxelu.
Kabazitaxel indukoval embryofetálnu toxicitu u samíc potkanov, ktorým bol podávaný intravenózne raz denne v 6. až 17. gestačnom dni spojenú s materskou toxicitou, došlo k úmrtiu plodu a zníženiu priemernej hmotnosti plodu, ktoré súviselo s oneskorením skeletálnej osifikácie. Expozície u zvierat boli nižšie než u ľudí, ktorým boli podávané klinicky relevantné dávky kabazitaxelu. Kabazitaxel
u myší prechádzal placentárnou bariérou.
U myší sa kabazitaxel a jeho metabolity vylučovali do materského mlieka v množstve až 1,5 %
podanej dávky viac ako 24 hodín.
Hodnotenieenviromentálnehorizika(ERA)
Výsledky štúdií hodnotenia environmentálneho rizika naznačujú, že používanie Jevtany nepredstavuje významné riziko pre vodné životné prostredie (pozri časť 6.6 o likvidácii nepoužitého lieku).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Koncentrát
Polysorbát 80
Kyselina citrónová
Rozpúšťadlo Etanol 96 % Voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
PVC infúzne vaky alebo polyuretánové infúzne súpravy sa nesmú používať na prípravu a podávanie infúzneho roztoku.
6.3 Čas použiteľnosti
Neotvorenéinjekčnéliekovky
3 roky.
Pootvorení
Injekčné liekovky s koncentrátom a rozpúšťadlom sa musia okamžite použiť. Ak sa nepoužijú okamžite, za čas a podmienky uchovávania pri používaní preberá zodpovednosť používateľ.
Popočiatočnomzriedeníkoncentrátusrozpúšťadlom
Chemická a fyzikálna stabilita pri používaní bola preukázaná po dobu 1 hodiny pri izbovej teplote
(15°C-30°C). Z mikrobiologického hľadiska je nutné, aby sa použila zmes koncentrátu
s rozpúšťadlom okamžite. Ak sa nepoužije okamžite, za čas a podmienky uchovávania pri používaní preberá zodpovednosť používateľ a normálne by čas nemal byť dlhší ako 24 hodín pri teplote 2°C –
8°C, ak riedenie neprebehlo v kontrolovaných a validovaných aseptických podmienkach.
Pokonečnomzriedenívinfúznomvaku/fľaši
Chemická a fyzikálna stabilita infúzneho roztoku bola preukázaná po dobu 8 hodín pri izbovej teplote
(vrátane 1 hodiny podávania infúzie) a 48 hodín pri uchovávaní v chladničke (vrátane 1 hodiny podávania infúzie).
Z mikrobiologického hľadiska je nutné, aby sa použila zmes koncentrátu s rozpúšťadlom okamžite.
Ak sa nepoužije okamžite, za čas a podmienky uchovávania pri používaní preberá zodpovednosť používateľ a normálne by čas nemal byť dlhší ako 24 hodín pri teplote 2°C – 8°C, ak riedenie neprebehlo v kontrolovaných a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30 °C. Neuchovávajte v chladničke.
Podmienky na uchovávanie lieku po nariedení, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Jedno balenie obsahuje jednu injekčnú liekovku s koncentrátom a jednu injekčnú liekovku s rozpúšťadlom:
· Koncentrát: 1,5 ml koncentrátu v 15 ml čírej sklenenej injekčnej liekovke (typ I), s uzáverom zo šedej chlórbutylovej gumy, ktorý je zapečatený hliníkovým vrchnákom a zakrytý bledozeleným plastovým odlamovacím viečkom. Jedna injekčná liekovka obsahuje 60 mg kabazitaxelu na
1,5 ml nominálneho objemu (plniaci objem: 73,2 mg kabazitaxelu/1,83 ml). Tento plniaci objem sa stanovil v priebehu vývoja Jevtany na kompenzáciu straty roztoku pri príprave premixu. Preplnenie zaručuje, že po zriedení celým obsahom priloženého rozpúšťadla pre Jevtanu, je minimálny využiteľný objem 6 ml premixu Jevtany s koncentráciou 10 mg/ml, čo zodpovedá uvádzanému množstvu 60 mg v jednej injekčnej liekovke.
· Rozpúšťadlo: 4,5 ml rozpúšťadla v 15 ml čírej sklenenej injekčnej liekovke (typ I), s uzáverom zo šedej chlórbutylovej gumy, ktorý je zapečatený zlatým hliníkovým vrchnákom a zakrytý
bezfarebným plastovým odlamovacím viečkom. Nominálny objem jednej injekčnej liekovky je
4,5 ml (plniaci objem: 5,67 ml). Tento plniaci objem sa stanovil v priebehu vývoja Jevtany a po pridaní celého objemu rozpúšťadla k obsahu koncentrátu Jevtany 60 mg zaručuje preplnenie koncentráciu roztoku premixu Jevtany 10 mg/ml.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Jevtanu má pripravovať a podávať len personál vyškolený na prácu s cytostatikami. Tehotné ženy nesmú narábať s liekom. Tak ako aj pri iných antineoplastikách, je potrebné byť opatrný pri zaobchádzaní a príprave roztoku Jevtany a mať na pamäti používanie bezpečnostných prostriedkov, výbavu osobnej ochrany (napr. rukavice) a dodržiavanie postupu na prípravu. Ak sa Jevtana
v ktoromkoľvek kroku počas narábania s ňou dostane do kontaktu s pokožkou, okamžite ju dôkladne umyte mydlom a vodou. Ak sa dostane do kontatu so sliznicou, okamžite ju dôkladne umyte vodou.
Koncentrát na infúzny roztok sa má vždy pred pridaním do infúzneho roztoku riediť s celým
dodávaným rozpúšťadlom.
Pred miešaním a riedením si pozorne prečítajte túto CELÚ časť. Jevtana vyžaduje DVE riedenia pred podaním. Postupujte podľa pokynov na prípravu uvedených nižšie.
Poznámka: Obe injekčné liekovky Jevtany 60 mg/1,5 ml, injekčná liekovka koncentrátu (plniaci objem: 73,2 mg kabazitaxelu/1,83 ml) a injekčná liekovka rozpúšťadla (plniaci objem: 5,67 ml), obsahujú preplnenie na kompenzáciu straty roztoku počas prípravy. Toto preplnenie zaručuje, že po zriedení CELÝM obsahom priloženého rozpúšťadla vznikne roztok obsahujúci 10 mg/ml kabazitaxelu.
Na prípravu infúzneho roztoku je nutné dodržať postup v nasledujúcich 2 krokoch a aseptické podmienky.
K
rok
1:Počiatočnézriedeniekoncentrátunainfúznyroztoksdodávanýmrozpúšťadlom.
K
rok 1.1
Skontrolujte injekčnú liekovku koncentrátu a dodávaného rozpúšťadla. Roztok koncentrátu a rozpúšťadla musia byť číre.
K
rok 1.2
Asepticky natiahnite
celý obsah dodaného rozpúšťadla pomocou striekačky s upevnenou ihlou tak, že čiastočne natočíte injekčnú liekovku.
Injekčná liekovka
koncentrátu
(60 mg – 1,5 ml)
Injekčná
liekovka rozpúšťadla
K
rok 1.3
Aplikujte
celý obsah do príslušnej injekčnej liekovky s koncentrátom.
Aby ste pri aplikovaní rozpúšťadla minimalizovali spenenie, namierte ihlu na vnútornú stenu injekčnej liekovky
s koncentrátom a pomaly aplikujte.
Injekčná liekovka
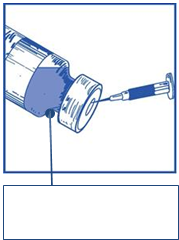
rozpúšťadla
Po rekonštitúcii obsahuje výsledný roztok
10 mg/ml kabazitaxelu.
Krok 1.4Odstráňte injekčnú striekačku a ihlu a jemne manuálne premiešajte opakovaným prevracaním, pokiaľ nedosiahnete číry
a homogénny roztok. Malo by to trvať približne
45 sekúnd.
Zmes koncentrátu a rozpúšťadla

10 mg/ml
Injekčná liekovka rozpúšťadla

Zmes koncentrátu a
rozpúšťadla 10 mg/ml
K
rok 1.5
Nechajte tento roztok približne 5 minút odstáť
a potom skontrolujte, či je roztok homogénny a číry.
Spenenie je normálne aj po uplynutí tohto času.

Zmes koncentrátu a
rozpúšťadla 10 mg/ml
Výsledná zmes koncentrátu a rozpúšťadla obsahuje 10 mg/ml kabazitaxelu (minimálne 6 ml
aplikovateľného objemu). Druhé riedenie sa musí urobiť okamžite (do 1 hodiny) podľa podrobného návodu uvedeného v kroku 2.
Na podanie predpísanej dávky môže byť potrebná viac ako jedna injekčná liekovka zmesi koncentrátu a rozpúšťadla.
Krok 2:Druhé(konečné)riedenienainfúziuKrok 2.1Asepticky natiahnite požadované množstvo zmesi koncentrátu a rozpúšťadla (10 mg/ml kabazitaxelu) kalibrovanou injekčnou striekačkou s upevnenou ihlou. Napríklad dávka
45 mg Jevtany by si vyžadovala 4,5 ml zmesi koncentrátu a rozpúšťadla pripravenej podľa postupu uvedeného v kroku 1.
Keďže na stenách injekčnej liekovky tohto
roztoku môže zostať pena, tak ako je to uvedené v postupe prípravy v kroku 1, odporúča sa
umiestniť ihlu striekačky pri naťahovaní
doprostred.
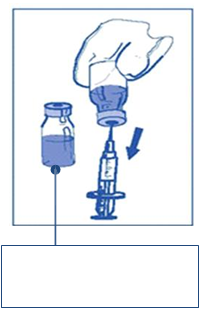
Zmes koncentrátu a rozpúšťadla 10 mg/ml
K
rok
2.2
Aplikujte do sterilnej infúznej nádoby bez
obsahu PVC buď do 5 % roztoku glukózy alebo do roztoku chloridu sodného 9 mg/ml (0,9 %).
Koncentrácia infúzneho roztoku musí byť medzi
0,10 mg/ml a 0,26 mg/ml.
Krok 2.3Odstráňte injekčnú striekačku a ručne premiešajte obsah infúzneho vaku alebo fľaše kývavým pohybom.
Krok 2.4 Tak ako pri všetkých parenterálnych liekoch, výsledný infúzny roztok sa musí pred použitím vizuálne skontrolovať. Infúzny roztok je supersaturovaný a preto môže po čase skryštalizovať. V takomto prípade sa roztok nesmie použiť a musí sa znehodnotiť.
Požadované množstvo zmesi koncentrátu a rozpúšťadla
5 % roztok glukózy alebo roztok chloridu sodného
9 mg/ml (0,9 %)

Infúzny roztok sa má podať ihneď. Avšak čas použitia pri správnom uchovávaní môže byť dlhší, ak sú
dodržané špeciálne podmienky popísané v časti 6.3.
Na podávanie sa odporúča použiť filter, ktorého nominálna veľkosť pórov je 0,22 mikrometrov
(označovaný tiež 0,2 mikrometrov).
Nepoužívajte infúzne vaky alebo fľaše obsahujúce PVC alebo polyuretánové infúzne súpravy na prípravu a podávanie Jevtany.
Jevtana sa nesmie miešať so žiadnymi inými liekmi okrem tých, ktoré sú uvedené. Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIsanofi-aventis groupe
54, rue La Boétie
F - 75008 Paris
Francúzsko
8. REGISTRAČNÉ ČÍSLO
EU/1/11/676/001
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 17. marec 2011
Dátum posledného predĺženia registrácie: 19. november 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.