
počiatočne randomizovaných na ruxolitinib v štúdiách COMFORT-I a COMFORT-II bol 33,2 mesiacov) od 457 pacientov s myelofibrózou liečených ruxolitinibom počas obdobia randomizácie a extenzie dvoch pivotných skúšaní 3 fázy. Toto hodnotenie zahŕňa údaje od pacientov, ktorí boli počiatočne randomizovaní na ruxolitinib (N=301)
a od pacientov, ktorí dostali ruxolitinib po prekrížení z kontrolných liečebných ramien (N=156). Podľa týchto aktualizovaných údajov sa ukončenie liečby z dôvodu nežiaducich účinkov pozorovalo u 17,1% pacientov liečených ruxolitinibom.
Polycytémia vera
Bezpečnosť Jakavi bola hodnotená u 110 pacientov s PV v otvorenom, randomizovanom, kontrolovanom klinickom skúšaní 3.fázy s názvom RESPONSE. Nižšie uvedené nežiaduce reakcie na
liek sa vzťahujú na počiatočnú fázu klinického skúšania (až do 32. týždňa) s rovnakou expozíciou ruxolitinibu a najlepšej dostupnej liečbe (Best Available Therapy - BAT), zodpovedajúcou mediánu trvania expozície lieku Jakavi v dĺžke 7,8 mesiaca. Priemerný vek pacientov užívajúcich Jakavi bol
približne 60 rokov.
Ukončenie liečby kvôli nežiaducim účinkom, bez ohľadu na príčiny, bolo pozorované u 3,6 %
pacientov liečených Jakavi a u 1,8 % pacientov liečených najlepšou dostupnou liečbou.
Hematologické nežiaduce reakcie (akéhokoľvek stupňa podľa CTCAE) zahrňovali anémiu (43,6 %) a trombocytopéniu (24,5 %). Anémia alebo trombocytopénia stupňa 3 a 4 podľa CTCAE bola hlásená
u 1,8 % alebo 5.5 % pacientov, v uvedenom poradí.
Tromi najčastejšími nehematologickými nežiaducimi reakciami boli závrat (15,5 %), zápcha (8,2 %) a herpes zoster (6,4 %).
Tromi najčastejšími nehematologickými laboratórnymi abnormalitami (akéhokoľvek stupňa podľa CTCAE) boli hypercholesterolémia (30,0 %), zvýšená alanínaminotransferáza (22,7 %) a zvýšená aspartátaminotransferáza (20,9 %). Všetky boli stupňa 1 a 2 podľa CTCAE okrem jedného prípadu zvýšenej alanínaminotransferázy stupňa 3 podľa CTCAE.
Dlhodobá bezpečnosť: Medián trvania expozície lieku Jakavi u pacientov bol 18,6 mesiaca (rozpätie
0,3 až 35,9 mesiaca). Pri dlhšej expozícii sa počet nežiaducich účinkov zvýšil, ale neobjavili sa žiadne nové zistenia v oblasti bezpečnosti. Po úprave na túto expozíciu bola miera výskytu nežiaducich účinkov obvykle porovnateľná s mierou ich výskytu pozorovanou v priebehu počiatočnej fázy klinického skúšania.
Tabuľkovýprehľadnežiaducichreakciízklinickýchštúdií
V programe klinických štúdií sa závažnosť nežiaducich reakcií hodnotila podľa kritérií CTCAE, kde stupeň 1 = mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4= život ohrozujúce.
Nežiaduce reakcie nahlásené z klinických štúdií (Tabuľka 1) sú zoradené podľa triedy orgánových systémov MedDRA. V každej triede orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, pričom najčastejšie sú uvedené ako prvé. Frekvencie sú definované podľa nasledovnej konvencie: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000).
Tabuľka 1 Kategória frevencie výskytu nežiaducich reakcií na liek hlásených v štúdiách fázy 3 (COMFORT-I, COMFORT-II, RESPONSE)
N
ežiaduca reakcia na liek Kategória frekvencie
výskytu u pacientov s
MF
K
ategória frekvencie výskytu u
pacientov s PV
Infekcie a nákazy
Infekcie močového traktua,d Veľmi časté Časté
Herpes zostera,d Časté Časté
Tuberkulózae Menej časté -
Poruchy krvi a lymfatického systémub,d
Anémiab - -
CTCAEc stupeň 4 (<6,5g/dl)
CTCAEc stupeň 3 (<8,0 –
6,5g/dl)
Akýkoľvek CTCAE
stupeňc
Trombocytopéniab CTCAEc stupeň 4 (<25 000/mm3)
CTCAEc stupeň 3 (50 000
– 25 000/mm3)
Akýkoľvek CTCAEc
stupeň
Neutropéniab CTCAEc stupeň 4 (<500/mm3)
CTCAEc stupeň 3 (<1 000
– 500/mm3) Akýkoľvek CTCAEc
stupeň
Krvácanie (akékoľvek krvácanie vrátane intrakraniálneho, gastrointestinálne krvácanie, podliatiny a iné krvácanie)
Veľmi časté Menej časté Veľmi časté Menej časté Veľmi časté Veľmi časté
Časté Menej časté
Časté Časté
Veľmi časté Veľmi časté
Časté - Časté - Veľmi časté -
Veľmi časté Veľmi časté
Intrakraniálne krvácanie Časté -
Gastrointestinálne krvácanie
Časté -
Podliatiny Veľmi časté Veľmi časté
Iné krvácanie (vrátane epistaxy, postprocedurálnej hemorágie a hematúrie)
Časté Veľmi časté
P
oruchy metabolizmu a
výživy
Nárast hmotnostia Veľmi časté Časté
Hypercholesterolémiab
CTCAEc stupeň 1 a 2
Hypertriglyceridémiab
CTCAEc stupeň 1
Poruchy nervového systému
Veľmi časté Veľmi časté
- Veľmi časté
Závratya Veľmi časté Veľmi časté
Bolesť hlavya Veľmi časté -
Poruchy gastrointestinálneho traktu
Plynatosťa Časté -
Zápchaa - Časté
Poruchy pečene a žlčových
ciest Zvýšená alanínaminotransferázab
CTCAEc stupeň 3 (> 5x –
20 x ULN) Akýkoľvek CTCAEc stupeň
Zvýšená aspartátaminotransferázab
Akýkoľvek CTCAEc
stupeň
Poruchy ciev
Časté Menej časté
Veľmi časté Veľmi časté
Veľmi časté Veľmi časté

Hypertenziaa - Veľmi časté
a Frekvencia vychádza z údajov o nežiaducich účinkoch.
- Subjekt, u ktorého sa vyskytlo viacero nežiaducich reakcií na liek (ADR) je v danej
ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
b Frekvencia vychádza z laboratórnych hodnôt.
- Subjekt, u ktorého sa vyskytlo viacero ADR je v danej ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
c Všeobecné terminologické kritériá pre nežiaduce účinky (CTCAE) verzia 3.0; stupeň 1 =
mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4 = život ohrozujúce
d Tieto ADR sú bližšie popísané v texte.
e Frekvencia vychádza z počtu všetkých pacientov exponovaných ruxolitinibu v klinických skúšaniach (N=4755)
Po ukončení liečby sa u pacientov s MF môžu vrátiť príznaky MF ako napr. únava, bolesť kostí,
horúčka, pruritus, nočné potenie, symptomatická splenomegália a pokles telesnej hmotnosti.
V klinických štúdiách s MF sa celkové skóre symptómov pre symptómy MF postupne vrátilo na počiatočné hodnoty do 7 dní od ukončenia liečby (pozri časť 4.4).
PopisvybranýchnežiaducichreakciínaliekAnémiaV klinických štúdiách fázy III s MF, bol medián nástupu anémie CTCAE stupňa 2 alebo vyššieho
1,5 mesiaca. Jeden pacient (0,3 %) ukončil liečbu v dôsledku anémie.
U pacientov užívajúcich Jakavi dosiahli priemerné poklesy hemoglobínu minimum približne o
10 g/liter pod počiatočnými hodnotami po 8 až 12 týždňoch liečby a potom postupne narastali, kým nedosiahli nový stabilný stav, ktorý bol približne o 5 g/liter pod počiatočnými hodnotami. Takýto
vzorec sa u pacientov pozoroval bez ohľadu na to, či počas liečby dostali transfúziu.
V randomizovanej, placebom kontrolovanej štúdii COMFORT-I 60,6 % pacientov s MF užívajúcich Jakavi a 37,7 % pacientov s MF užívajúcich placebo dostalo počas randomizovanej liečby transfúziu s červenými krvinkami. V štúdii COMFORT-II bol podiel transfúzií s červenými krvinkami 53,4 %
v ramene s Jakavi a 41,1 % v ramene s najlepšou dostupnou liečbou.
Vo fáze randomizácie v pivotných štúdiách bola anémia menej častá u pacientov s PV ako u pacientov s MF (43,6 % oproti 82,4 %). V PV populácii boli prípady CTCAE stupňa 3 a 4 hlásené u 1,8 % pacientov, kým frekvencia u pacientov s MF bola 42,56 %.
Trombocytopénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula trombocytopénia stupňa 3 alebo 4, bol medián jej nástupu približne 8 týždňov. Trombocytopénia bola vo všeobecnosti reverzibilná a ustúpila po znížení dávky alebo po prerušení liečby. Medián času potrebného na návrat počtu krvných doštičiek nad 50 000/mm3 bol 14 dní. Počas randomizovaného obdobia boli transfúzie s krvnými doštičkami podané 4,7 % pacientov užívajúcich Jakavi a 4,0 % pacientov užívajúcich
kontrolné režimy. K ukončeniu liečby v dôsledku trombocytopénie došlo u 0,7 % pacientov užívajúcich Jakavi a 0,9 % pacientov užívajúcich kontrolné režimy. U pacientov, ktorí mali pred začiatkom liečby s Jakavi počet krvných doštičiek od 100 000/mm3 do 200 000/mm3, bola vyššia frekvencia trombocytopénie stupňa 3 alebo 4 v porovnaní s pacientmi, ktorý mali počet krvných doštičiek >200 000/mm3 (64,2 % verzus 38,5 %).
Vo fáze randomizácie v pivotných štúdiách bol podiel pacientov, u ktorých sa vyskytla trombocytopénia nižší u pacientov s PV (24,5 %), ako u pacientov s MF (69,8 %). Frekvencia výskytu závažnej trombocytopénie (t.j. CTCAE stupňa 3 a 4) bola nižšia u pacientov s PV (5,5 %) ako u pacientov s MF (11,6 %).
Neutropénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula neutropénia stupňa 3 alebo 4, bol medián jej nástupu 12 týždňov. Počas randomizovaného obdobia sa pozastavenie liečby alebo zníženie dávky v dôsledku neutropénie hlásilo u 1,0 % pacientov a 0,3 % pacientov ukončilo liečbu pre neutropéniu.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV bola neutropénia hlásená u dvoch pacientov
(1,8 %), z ktorých sa u jedného pacienta vyskytla neutropénia CTCAE stupňa 4.
Krvácanie
V pivotných štúdiách fázy III s MF boli prípady krvácania (vrátane intrakraniálneho
a gastrointestinálneho krvácania, podliatin a iných prípadov krvácania) hlásené u 32,6 % pacientov liečených Jakavi a u 23,2 % pacientov liečených referenčnou liečbou (placebom alebo najlepšou dostupnou liečbou). Frekvencia udalostí stupňa 3-4 bola u pacientov liečených Jakavi alebo referenčnou liečbou podobná (4,7 % verzus 3,1 %). Väčšina pacientov s príhodou krvácania počas liečby hlásila podliatiny (65,3 %). Podliatiny boli častejšie hlásené u pacientov užívajúcich Jakavi
v porovnaní s referenčnou liečbou (21,3 % verzus 11,6 %). Intrakraniálne krvácanie bolo hlásené u
1 % pacientov exponovaných Jakavi a u 0,9 % pacientov exponovaných referenčnej liečbe. Gastrointestinálne krvácanie bolo hlásené u 5,0 % pacientov exponovaných Jakavi oproti 3,1 % pacientov exponovaných referenčnej liečbe. Iné prípady krvácania (vrátane epistaxy,
post-procedurálnej hemorágie a hematúrie) boli hlásené u 13,3 % pacientov liečených Jakavi a u
10,3 % pacientov liečených referenčnou liečbou.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV boli hlásené prípady krvácania (vrátane intrakraniálneho a gastrointestináneho krvácania, tvorby podliatin a iného krvácania) u 20 % pacientov liečených Jakavi a 15,3 % pacientov, ktorí dostávali najlepšiu dostupnú liečbu. Podliatiny boli hlásené
s rovnakou frekvenciou v liečebných skupinách s Jakavi aj BAT (10,9 % oproti 8,1 %). U pacientov užívajúcich Jakavi neboli hlásené žiadne prípady intrakraniálneho ani gastrointestinálneho krvácania.
U jedného pacienta liečeného Jakavi sa vyskytol prípad krvácania stupňa 3 (postprocedurálne krvácanie); nebolo hlásené žiadne krvácanie stupňa 4. Prípady iného krvácania (vrátane takých prípadov ako epistaxa, postprocedurálne krvácanie, gingiválne krvácanie) boli hlásené u 11,8 % pacientov liečených Jakavi a u 6,3 % pacientov liečených najlepšou dostupnou liečbou.
InfekcieV pivotných štúdiách fázy III s MF boli hlásené infekcie močových ciest stupňa 3 alebo 4 u 1,0 % pacientov, herpes zoster u 4,3 % a tuberkulóza u 1,0 %. V klinickom skúšaní fázy III bola sepsa hlásená u 3,0% pacientov. Predĺžené sledovanie pacientov liečených ruxolitinibom nepreukázalo
s postupom času žiadny zvýšený trend v rozsahu sepsy.
Vo fáze randomizácie v pivotnej štúdii bola u pacientov s PV hlásená jedna infekcia močového traktu (0,9 %) CTCAE stupňa 3 a žiadna infekcia močového traktu stupňa 4. Miera výskytu herpes zoster bola mierne vyššia u pacientov s PV (6,4 %) ako u pacientov s MF (4,0 %). U pacientov s PV sa vyskytlo jedno hlásenie postherpetickej neuralgie CTCAE stupňa 3.
Zvýšený systolický krvný tlakV pivotných štúdiách fázy III s MF bolo hlásené minimálne počas jednej vizity zvýšenie systolického
krvného tlaku oproti východiskovej hodnote o 20 mmHg alebo viac u 31,5 % pacientov v porovnaní s
19,5 % výskytom u pacientov na kontrolnej liečbe. V skúšaní COMFORT-I (pacienti s MF) bol oproti východiskovej hodnote pri Jakavi priemerne zvýšený systolický krvný tlak o 0-2 mmHg v porovnaní
s poklesom o 2-5 mmHg v ramene s placebom. V skúšaní COMFORT-II boli medzi priemernými hodnotami MF pacientov liečených ruxolitinibom alebo kontrolnou liečbou len malé rozdiely.
Vo fáze randomizácie v pivotných štúdiách bol u pacientov s PV sa priemerný systolický krvný tlak v liečebnej skupine s Jakavi zvýšil o 0,65 mmHg, kým v skupine s BAT klesol o 2 mmHg.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieNie je známe antidotum pri predávkovaní s Jakavi. Jednorazové dávky do 200 mg boli podané s prijateľnou akútnou znášanlivosťou. Vyššie ako odporúčané, opakované dávky sú spojené so zvýšenou myelosupresiou vrátane leukopénie, anémie a trombocytopénie. Je potrebné podať vhodnú podpornú liečbu.
Nepredpokladá sa, že hemodialýza zvyšuje elimináciu ruxolitinibu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE18
MechanizmusúčinkuRuxolitinib je selektívny inhibítor Janusových kináz (JAK), JAK1 a JAK2 (IC50 hodnoty 3,3 nM pre enzýmy JAK1 a 2,8 nM pre enzýmy JAK2). Tieto sprostredkujú signalizáciu mnohých cytokínov a rastových faktorov, ktoré sú dôležité pre hemopoézu a imunitné funkcie.
Myelofibróza a polycytémia vera sú myeloproliferatívne nádorové ochorenia, o ktorých je známe že súvisia s poruchou signalizácie JAK1 a JAK2. Predpokladá sa že k základom týchto porúch patrí vysoká hladina cirkulujúcich cytokínov, ktoré aktivujú JAK-STAT dráhou, mutácie pridávajúce
funkcie ako napr. JAK2V617F a utlmenie negatívnych regulačných mechanizmov. Pacienti s MF vykazujú poruchu JAK signalizácie bez ohľadu na stav JAK2V617F mutácie. U >95 % pacientov s PV sa vyskytujú aktivujúce mutácie JAK2 (V617F alebo exón 12).
Ruxolitinib inhibuje JAK-STAT signalizáciu a bunkovú proliferáciu bunkových modelov hematologických malignít závislých od cytokínov ako aj Ba/F3 buniek nezávislých od cytokínov expresiou JAK2V617F mutovaného proteínu, s hodnotou IC50 v rozsahu 80-320 nM.
Farmakodynamickéúčinky
Ruxolitinib inhibuje cytokínmi indukovanú STAT3 fosforyláciu v celej krvi od zdravých dobrovoľníkov, pacientov s MF a pacientov s PV . Ruxolitinib vyvolal maximálnu inhibíciu STAT3
fosforylácie 2 hodiny po podaní dávky, ktorá sa vrátila do normálu do 8 hodín u zdravých
dobrovoľníkov aj u pacientov s MF, čo naznačuje že nedochádza k akumulácii pôvodných ani aktívnych metabolitov.
Zvýšené východiskové zápalové markery súvisiace so základnými konštitučnými príznakmi ako TNFα, IL-6 a CRP u osôb s MF po liečbe ruxolitinibom poklesli. Počas liečby ruxolitinibom sa u pacientov s MF nevytvorila odolnosť voči jeho farmakodynamickým účinkom. Rovnako aj u pacientov s PV sa vyskytli zvýšené východiskové zápalové markery a tieto markery sa po liečbe ruxolitinibom znížili.
V podrobnej QT štúdii so zdravými dobrovoľníkmi nič nepoukazovalo na predlžujúci účinok ruxolitinibu na QT/QTc v jednotlivých dávkach až po supraterapeutickú dávku 200 mg, čo naznačuje že ruxolitinib nemá účinok na kardiálnu repolarizáciu.
Klinickáúčinnosťabezpečnosť
Myelofibróza
U pacientov s MF (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie) sa realizovali 2 randomizované štúdie fázy III (COMFORT-I a COMFORT-II). V oboch štúdiách mali pacienti hmatateľnú splenomegáliu najmenej
5 cm pod rebrovým oblúkom a rizikovú kategóriu strednú-2 alebo vysokú, podľa IWG kritérií
(International Working Group Consensus Criteria). Počiatočná dávka Jakavi sa stanovila na základe počtu krvných doštičiek.
COMFORT-I bola dvojito zaslepená, randomizovaná, placebom kontrolovaná štúdia s 309 pacientmi, ktorí nereagovali na dostupnú liečbu alebo táto pre nich nebola vhodná. Primárny cieľový ukazovateľ účinnosti bol definovaný ako podiel pacientov, u ktorých došlo k ≥35 % zmenšeniu objemu sleziny v
24. týždni v porovnaní s východiskom, merané zobrazovaním magnetickou rezonanciou (MRI) alebo počítačovou tomografiou (CT).
Sekundárne cieľové ukazovatele účinnosti zahrňovali trvanie ≥35 % zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou, podiel pacientov s ≥50 % znížením skóre celkových príznakov, zmeny v celkových skóre príznakov v porovnaní s východiskovou hodnotou v 24. týždni hodnotené pomocou modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) v2.0 denníka, a celkové prežívanie.
COMFORT-II bola otvorená, randomizovaná štúdia s 219 pacientmi. Pacienti boli randomizovaní 2:1 do skupín s Jakavi a s najlepšou dostupnou liečbou. V ramene s najlepšou dostupnou liečbou 47 % pacientov dostalo hydroxyureu a 16 % pacientov glukokortikoidy. Primárny cieľový ukazovateľ účinnosti bol podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 48. týždni v porovnaní s východiskovou hodnotou, merané MRI alebo CT.
Sekundárne cieľové ukazovatele zahrňovali podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 24. týždni v porovnaní s východiskovou hodnotou a trvanie ≥35% zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou.
Základné demografické charakteristiky a charakteristika ochorenia pacientov boli porovnateľné pre obe ramená v štúdiách COMFORT-I a COMFORT-II.
Tabuľka 2 Percento pacientov s ≥35 % zmenšením sleziny v 24. týždni v porovnaní
s východiskovou hodnotou v COMFORT-I a v 48. týždni v COMFORT-II (ITT)
COMFORT-I COMFORT-II
Jakavi
(N=155)
Placebo
(N=153)
Jakavi
(N=144)
Najlepšia dostupná liečba (N=72)
Čas Týždeň 24 Týždeň 48
Počet (%) pacientov s objemom sleziny zmenšeným o ≥35 %
95 % intervaly spoľahlivosti
65 (41,9) 1 (0,7) 41 (28,5) 0
34,1; 50,1 0; 3,6 21,3; 36,6 0,0; 5,0
p-hodnota <0,0001 <0,0001
Významne väčší podiel pacientov v skupine s Jakavi dosiahol ≥35 % zmenšenie objemu sleziny
(tabuľka 2) v porovnaní s východiskovou hodnotou bez ohľadu na prítomnosť alebo neprítomnosť
JAK2V617F mutácie alebo subtyp ochorenia (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie).
Tabuľka 3 Percento pacientov s ≥35 % zmenšením sleziny v porovnaní s východiskovou hodnotou podľa stavu mutácie JAK (súbor bezpečnosti)
COMFORT-I COMFORT-II
Jakavi Placebo Jakavi Najlepšia dostupná liečba
Stav mutácie JAK
Pozitívny (N=113) n (%)
Negatívny
(N=40)
n (%)
Pozitívny (N=121) n (%)
Negatívny
(N=27)
n (%)
Pozitívny (N=110) n (%)
Negatívny
(N=35)
n (%)
Pozitívny
(N=49)
n (%)
Negatívny
(N=20)
n (%)
Počet (%)
pacientov
s objemom sleziny zmenšeným o ≥35 %
54 (47,8)
11 (27,5)
1 (0,8)
0 36 (32,7)
5 0 0 (14,3)

Časový bod Po 24 týždňoch Po 48 týždňoch
Pravdepodobnosť udržania odpovede sleziny (≥35 % redukcia) na Jakavi v priebehu najmenej
24 týždňov bola 89 % v štúdii COMFORT I a 87 % v štúdii COMFORT II; v štúdii COMFORT II
udržalo odpoveď sleziny 52 % počas aspoň 48 týždňov.
V štúdii COMFORT I 45,9 % subjektov v skupine s Jakavi dosiahlo v 24. týždni ≥50 % zlepšenie celkového skóre príznakov v porovnaní s východiskovou hodnotou (zisťované podľa denníka MFSAF v2.0), v porovnaní s 5,3 % v skupine s placebom (p<0,0001 pomocou chi-kvadrát testu). Priemerná zmena v celkovom zdravotnom stave v 24. týždni zisťovaná podľa EORTC QLQ C30 bola +12,3 u Jakavi a -3,4 u placeba (p<0,0001).
V štúdii COMFORT-I bola úmrtnosť pacientov randomizovaných do skupiny s ruxolitinibom po priemernej dobe sledovania 34,3 mesiacov 27,1 %, v porovnaní s 35,1 % u pacientov randomizovaných na placebo; HR 0,687; 95 % IS 0,459-1,029; p=0,0668.
V štúdii COMFORT-II bola úmrtnosť pacientov randomizovaných na ruxolitinib po priemernej dobe sledovania 34,7 mesiacov 19,9 %, v porovnaní s 30,1 % u pacientov randomizovaných na najlepšiu dostupnú liečbu (BAT); HR 0,48; 95 % IS 0,28-0,85; p=0,009. V oboch štúdiách bola nižšia úmrtnosť
zaznamenaná v skupine s ruxolitinibom, čo bolo spôsobené predovšetkým výsledkami získanými v podskupinách po polycytémii vera a po esenciálnej trombocytémii.
Polycytémia vera
Randomizované, otvorené, aktívne kontrolované klinické skúšanie 3. fázy (RESPONSE) bolo vykonané u 222 pacientov s PV s rezistenciou alebo intoleranciou na hydroxyureu definované podľa
publikovaných kritérií medzinárodnej pracovnej skupiny European LeukemiaNet (ELN).
110 pacientov bolo randomizovaných do skupiny s ruxolitinibom a 112 pacientov do skupiny s BAT. Počiatočná dávka Jakavi bola 10 mg dvakrát denne. Dávky sa potom u jednotlivých pacientov upravili
podľa znášanlivosti a účinnosti na maximálnu dávku 25 mg dvakrát denne. BAT vybral skúšajúci
lekár pre každého pacienta individuálne a zahŕňala hydroxyureu (59,5 %), interferón/pegylovaný interferón (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorovanie (15,3 %).
Vstupné demografické údaje a charakteristika ochorenia boli v dvoch liečebných skupinách porovnateľné. Priemerný vek bol 60 rokov (rozpätie 33 až 90 rokov). Pacienti v skupine
s ruxolitinibom mali diagnózu PV v priemere 8,2 roka a predtým užívali v priemere približne 3 roky hydroxyureu. Väčšina pacientov (>80 %) podstúpila v priebehu posledných 24 týždňov pred skríningom najmenej dve flebotómie. Porovnávacie údaje týkajúce sa dlhodobého prežitia a výskytu
komplikácií ochorenia nie sú dostupné.
Primárnym zloženým cieľovým ukazovateľom bolo percento pacientov, ktorí dosiahli aj absenciu indikácie flebotómie (kontrola HCT) a ≥35 % zmenšenie objemu sleziny v porovnaní so vstupnými hodnotami v 32. týždni. Indikácia flebotómie bola definovaná ako potvrdené HCT s hodnotou >45 %, t.j. aspoň o 3 percentuálne body vyššie ako HCT získané pri vstupnom vyšetrení alebo potvrdené HCT s hodnotou >48 %, podľa toho, ktorá hodnota bola nižšia. Kľúčové sekundárne cieľové ukazovatele zahrňovali percento pacientov, ktorí dosiahli primárny cieľový ukazovateľ a v 48. týždni ostali bez progresie, ako aj percento pacientov, ktorí dosiahli úplnú hematologickú remisiu v 32. týždni.
Štúdia splnila svoj primárny cieľ a vyššie percento pacientov v skupine s Jakavi dosiahlo primárny zložený cieľový ukazovateľ aj každú z jeho jednotlivých zložiek. Podstatne viac pacientov liečených Jakavi (20,9 %) dosiahlo primárnu odpoveď (p<0,0001) v porovnaní s BAT (0,9 %). Kompenzácia hematokritu sa dosiahla u 60 % pacientov v skupine s Jakavi v porovnaní s 19,6 % v skupine s BAT a
≥35 % zmenšenie objemu sleziny sa dosiahlo u 38,2 % pacientov v skupine s Jakavi v porovnaní s 0,9 % v skupine s BAT (obr. č. 1). Deväťdesiatštyri (83,9 %) pacientov randomizovaných do skupiny s BAT prešlo na liečbu ruxolitinibom v 32. týždni alebo neskôr, čím bolo obmedzené porovnanie týchto dvoch skupín po 32. týždni.
Splnené boli aj oba kľúčové sekundárne cieľové ukazovatele. Percento pacientov, ktorí dosiahli úplnú hematologickú remisiu, bolo 23,6 % s Jakavi v porovnaní s 8,9 % s BAT (p=0,0028) a percento pacientov, ktorí dosiahli dlhotrvajúcu primárnu odpoveď v 48. týždni, bolo 19,1 % s Jakavi a 0,9 %
s BAT (p<0,0001).
O
brázok č. 1 Pacienti dosahujúci primárny cieľový ukazovateľ a zložky primárneho
cieľového ukazovateľa v 32. týždni
Hodnota P: < ,0001
70 Pomer š a ncí (Ruxolitinib/BAT)
a 95% IS:
60 28,64 (4,50, 12,06)

50
40 38
30
21
20
10
Jednotlivé zložky primá rnej

odpovede v 32.týždni

60

20
.RUX
.BAT
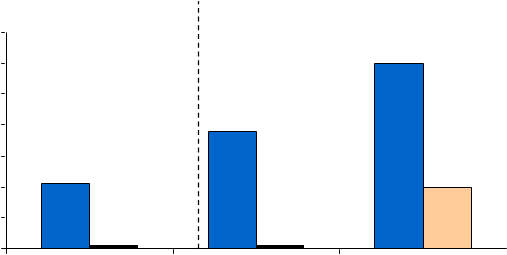
1 1
0
Pri má rny zl ožený
ci eľový uka zova teľ v
32.týždni
≥35% zníženi e obj emu
s l ezi ny
Kompenzá ci a hema tokri tu bez fl ebotómi e
Príznaková záťaž bola hodnotená pomocou MPN-SAF celkového skóre príznakov (total symptom
score - TSS) elektronického denníka pacienta, ktorý obsahoval 14 otázok. V 32. týždni dosiahlo 49 %
pacientov liečených ruxolitinibom ≥50 % zníženie TSS-14 a 64 % pacientov ≥50 % zníženie TSS-5, v porovnaní s iba 5 % a 11 % pacientov na BAT.
Vnímanie liečebného prínosu bolo zisťované v dotazníku Celkový dojem pacienta zo zmeny (Patient Global Impression of Change - PGIC). 66 % pacientov liečených ruxolitinibom, v porovnaní s 19 % liečenými BAT, hlásilo zlepšenie už štyri týždne po začiatku liečby. Zlepšenie vo vnímaní liečebného prínosu bolo tiež vyššie u pacientov liečených ruxolitinibom v 32. týždni (78 % oproti 33 %).
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Jakavi vo všetkých podskupinách pediatrickej populácie v liečbe MF (informácie o použití v pediatrickej
populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Ruxolitinib je látka I. triedy Biofarmaceutického klasifikačného systému (BCS), s vysokou permeabilitou, vysokou rozpustnosťou a rýchlou disolúciou. V klinických štúdiách sa ruxolitinib
rýchlo absorboval po perorálnom podaní, maximálnu plazmatickú koncentráciu (Cmax) dosiahol asi po
1 hodine po podaní dávky. Na základe humánnej štúdie hmotnostnej bilancie sa zistilo, že perorálna
absorpcia ruxolitinibu, v podobe ruxolitinibu alebo metabolitov vzniknutých v priebehu presystémovej eliminácie je 95 % alebo vyššia. Priemerné hodnoty Cmax a celkovej expozície (AUC) ruxolitinibu sa priamoúmerne zvýšili po jednotlivej dávke v rozsahu 5-200 mg. Nezistili sa klinicky relevantné zmeny farmakokinetiky ruxolitinibu po podaní s jedlom s vysokým obsahom tuku. Priemerná Cmax sa mierne znížila (24 %), kým priemerná AUC zostala takmer nezmenená (4 % vzostup) po podaní s jedlom s vysokým obsahom tuku.
Distribúcia
Priemerný objem distribúcie v rovnovážnom stave je u pacientov s MF aj s PV približne 75 litrov. Pri klinicky relevantných koncentráciách ruxolitinibu je väzba na plazmatické bielkoviny približne 97 %,
prevažne na albumín. Celotelové autoradiografické štúdie na potkanoch ukázali, že ruxolinitib
nepenetruje cez krvno-mozgovú bariéru.
B
i
otransformácia
Ruxolitinib je prevažne metabolizovaný enzýmom CYP3A4 (>50 %) s čiastočnou účasťou CYP2C9. Materská zlúčenina je hlavným prvkom v ľudskej plazme, pričom predstavuje približne 60 % materiálu súvisiaceho s liekom v obehu. V plazme sú prítomné dva hlavné a aktívne metabolity, ktoré reprezentujú 25 % a 11 % materskej AUC. Tieto metabolity majú polovicu až pätinu pôvodnej farmakologickej aktivity súvisiacej s JAK. Celkové množstvo všetkých aktívnych metabolitov prispieva 18 % k celkovej farmakodynamike ruxolitinibu. Podľa in vitro štúdií, ruxolitinib v klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 alebo CYP3A4 a nie je účinný induktor CYP1A2, CYP2B6 alebo CYP3A4. In vitro údaje naznačujú, že ruxolitinib môže byť inhibítor P-gp a BCRP.
Eliminácia
Ruxolitinib sa prevažne eliminuje metabolizmom. Priemerný polčas eliminácie ruxolitinibu sú približne 3 hodiny. U zdravých dobrovoľníkov, po jednorazovej perorálnej dávke rádioaktívne značeného [14C] ruxolitinibu došlo k eliminácii prevažne metabolizáciou, pričom 74 % rádioaktivity sa vylúčilo močom a 22 % stolicou. Na nezmenenú materskú zlúčeninu pripadlo menej ako 1 % celkovej vylúčenej rádioaktivity.
Linearita/nelinearita
Priama úmera v závislosti od dávky sa dokázala v štúdiách s jednorazovými aj viacnásobnými dávkami.
Osobitépopulácie
Vplyv veku, pohlavia a rasy
Na základe štúdií so zdravými subjektmi sa nepozorovali žiadne relevantné rozdiely vo farmakokinetike ruxolitinibu v závislosti od pohlavia a rasy. Na základe analýzy farmakokinetiky u
populácie pacientov s MF sa nepotvrdil vzťah medzi perorálnym klírensom a vekom alebo rasou pacienta. Odhadovaný perorálny klírens bol u žien 17,7 l/h a u mužov 22,1 l/h, s 39 % interindividuálnou variabilitou u pacientov s MF. Klírens u pacientov s PV bol 12,7 l/h, s 42 %
interindividuálnou variabilitou a nebol zaznamenaný žiaden zjavný vzťah medzi perorálnym klírensom a pohlavím, pacientovým vekom alebo rasou, na základe farmakokinetického hodnotenia populácie pacientov s PV.
Pediatrická populácia
Bezpečnosť a účinnosť Jakavi sa u detí a dospievajúcich nesledovala (pozri časť 5.1 „Pediatrická populácia“).
Poškodenie funkcie obličiek
Funkcia obličiek sa hodnotila štúdiami úpravy diéty pri ochorení obličiek (MDRD) a kretinínu v moči. Po jednorazovej dávke ruxolitinibu 25 mg bola jeho expozícia podobná u jedincov s rôznymi stupňami
poškodenia obličiek s tými, ktorí mali normálnu funkciu obličiek. Avšak plazmatické hodnoty AUC
metabolitov ruxolitinibu mali tendenciu stúpať so závažnosťou poškodenia obličiek a najvýraznejšie zvýšenie bolo u jedincov s ťažkým poškodením obličiek. Nie je zrejmé, či zvýšená expozícia
metabolitu predstavuje bezpečnostné riziko. Odporúča sa upraviť dávku u pacientov s ťažkým
poškodením obličiek a v konečnom štádiu renálneho ochorenia (pozri časť 4.2). Podávanie iba v dňoch dialýzy znižuje expozíciu metabolitu ale zároveň aj farmakodynamický účinok, najmä počas dní medzi dialýzami.
Poškodenie funkcie pečene
U pacientov s rôznym stupňom poškodenia pečene, sa priemerná AUC ruxolitinibu po jednorazovej dávke 25 mg zvýšila u pacientov s miernym poškodením pečene o 87 %, so stredne ťažkým o 28 %
a s ťažkým o 65 %, v porovnaní s pacientmi s normálnou funkciou pečene. Nebol jasný vzťah medzi AUC a stupňom poškodenia funkcie pečene klasifikovanom podľa Childovho-Pughovho skóre. U pacientov s poškodením funkcie pečene sa konečný polčas eliminácie v porovnaní so zdravými dobrovoľníkmi predĺžil (4,1-5,0 hodín oproti 2,8 hodiny). U pacientov s poškodením funkcie pečene sa odporúča zníženie dávky približne o 50 % (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Ruxolitinib sa hodnotil na základe štúdií farmakologickej bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, reprodukčnej toxicity a karcinogenity. K cieľovým orgánom, súvisiacim s farmakologickým účinkom ruxolitinibu, v štúdiách toxicity po opakovanom podaní, patrili kostná dreň, periférna krv a lymfatické tkanivo. U psov sa pozorovali infekcie, vo všeobecnosti spojené s imunosupresiou. Nepriaznivé zníženie krvného tlaku spolu so zvýšením pulzu sa pozorovalo u psov v telemetrickej štúdii. Nepriaznivý pokles minútového objemu sa pozoroval v respiračných štúdiách u potkanov. Hranice (na základe neviazaného Cmax) na hladine bez nežiaducich účinkov v štúdiách so
psami a potkanmi boli 15,7-krát vyššie a 10,4-krát vyššie, ako maximálna dávka 25 mg dvakrát denne,
odporúčaná u ľudí. V hodnotení neurofarmakologických vplyvov ruxolitinibu sa nezaznamenali žiadne účinky.
Ruxolitinib v štúdiách u zvierat znižoval váhu plodu a zvyšoval poimpalntačné straty. U králikov a potkanov nebol zaznamenaný dôkaz o teratogénnom účinku. Rozsah expozície bol však v porovnaní s najvyššou klinickou dávkou nízky, výsledok má preto len obmedzený význam pre ľudí. Nepozorovali sa účinky na fertilitu. V štúdiách prenatálneho a postnatálneho vývoja sa pozorovala
mierne predĺžená gestačná doba, znížený počet miest implantácie a menší počet narodených šteniat. U šteniat sa pozorovala znížená priemerná počiatočná telesná hmotnosť a krátke obdobie nárastu zníženej priemernej telesnej hmotnosti. U dojčiacich potkanov sa ruxolitinib a/alebo jeho metabolity vylučovali do mlieka v koncentrácii 13-krát vyššej ako materské plazmatické koncentrácie. Ruxolitinib nebol mutagénny alebo klastogénny. Ruxolitinib nebol karcinogénny v modeloch Tg.rasH2 transgénnych myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Mikrokryštalická celulóza
Magnéziumstearát
Koloidný bezvodý oxid kremičitý
Sodná soľ karboxymetylcelulózy (typ A) Povidón
Hydroxypropylcelulóza
Monohydrát laktózy
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
6.5 Druh obalu a obsah balenia
PVC/PCTFE/hliníkové blistrové balenia obsahujúce 14 alebo 56 tabliet alebo spoločné balenia obsahujúce 168 (3 balenia po 56) tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLO(ČÍSLA)EU/1/12/773/004-006
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE23.08.2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o
bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUJakavi 10 mg tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tableta obsahuje 10 mg ruxolitinibu (ako fosfátu).
Pomocnálátkasoznámymúčinkom:
Každá tableta obsahuje 142,90 mg monohydrátu laktózy.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATableta.
Okrúhle zaoblené biele až takmer biele tablety v priemere približne 9,3 mm s pretlačeným nápisom
„NVR“ na jednej strane a „L10“ na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieMyelofibróza(MF)Jakavi je indikované na liečbu splenomegálie súvisiacej s ochorením alebo jeho príznakov u dospelých pacientov s primárnou myelofibrózou (známou aj ako chronická idiopatická myelofibróza),
myelofibrózou po polycytémii vera alebo myelofibrózou po esenciálnej trombocytémii.
Polycytémiavera(PV)Jakavi je indikované na liečbu dospelých pacientov s polycytémiou vera s rezistenciou alebo intoleranciou na hydroxyureu.
4.2 Dávkovanie a spôsob podávaniaLiečbu Jakavi má začať iba lekár, ktorý má skúsenosti s podávaním protinádorových liekov. Pred začatím liečby Jakavi sa musí vykonať kompletné vyšetrenie krvného obrazu, vrátane
diferenciálneho počtu bielych krviniek.
Pokým nie sú dávky Jakavi stabilizované, je potrebné každé 2-4 týždne monitorovať a kompletne vyšetriť krvný obraz, vrátane diferenciálneho počtu bielych krviniek, a následne podľa klinickej indikácie (pozri časť 4.4).
DávkovaniePočiatočná dávkaOdporúčaná počiatočná dávka Jakavi pri myelofibróze je 15 mg dvakrát denne u pacientov s počtom krvných doštičiek medzi 100 000/mm3 a 200 000/mm3 a 20 mg dvakrát denne u pacientov s počtom krvných doštičiek >200 000/mm3. Odporúčaná počiatočná dávka Jakavi pri polycytémii vera je 10 mg
podávaných perorálne dvakrát denne.
K dispozícii sú obmedzené informácie pre odporúčanie počiatočnej dávky u pacientov s počtom krvných doštičiek medzi 50 000/mm3 a <100 000/mm3. Maximálna odporúčaná počiatočná dávka u týchto pacientov je 5 mg dvakrát denne a dávka sa musí titrovať opatrne.
Úpravy dávky
Dávky možno titrovať na základe bezpečnosti a účinnosti. Liečba sa musí prerušiť pri počte krvných doštičiek nižšom ako 50 000/mm3 alebo pri absolútnom počte neutrofilov nižšom ako 500/mm3. Liečba pri PV sa má prerušiť aj vtedy, ak sú hodnoty hemoglobínu nižšie ako 8 g/dl. Po obnovení hodnôt krvného obrazu nad tieto hodnoty možno pokračovať v dávkovaní 5 mg dvakrát denne
a postupne zvyšovať na základe podrobného monitorovania kompletného krvného obrazu vrátane diferenciálneho počtu bielych krviniek.
Ak počet krvných doštičiek klesne pod 100 000/mm3, je potrebné zvážiť zníženie dávky, aby sa predišlo prerušeniu užívania pre trombocytopéniu. Zníženie dávky pri PV sa má zvážiť aj vtedy, ak hemoglobín klesne pod 12 g/dl a odporúča sa, ak klesne pod 10 g/dl.
Ak sa účinnosť považuje za nedostatočnú a hodnoty krvného obrazu sú adekvátne, dávky možno zvýšiť o maximálne 5 mg dvakrát denne, najviac po maximálnu dávku 25 mg dvakrát denne.
Počiatočná dávka sa nesmie zvyšovať počas prvých štyroch týždňov liečby a potom nie častejšie ako v
2-týždňových intervaloch.
Maximálna dávka Jakavi je 25 mg dvakrát denne.
Úprava dávky pri súč asnom podávaní silných CYP3A4 inhibítorov alebo flukonazolu
Ak sa Jakavi podáva so silnými CYP3A4 inhibítormi alebo s duálnymi inhibítormi enzýmov CYP2C9 a CYP3A4 (napr. flukonazol), jednotlivá dávka sa má znížiť o približne 50 % a má sa podávať dvakrát denne (pozri časť 4.5).
Počas podávania silných CYP3A4 inhibítorov alebo duálnych inhibítorov enzýmov CYP2C9 a CYP3A4 sa odporúča častejšie monitorovanie (napr. dvakrát do týždňa) hematologických parametrov a klinických prejavov a príznakov nežiaducich reakcií na liek spojených s Jakavi.
Osobité populácie
Poškodenie funkcie obličiek
U pacientov s miernym alebo stredne závažným poškodením funkcie obličiek sa nevyžaduje úprava dávky.
U pacientov so závažným poškodením funkcie obličiek (klírens kreatinínu menej ako 30 ml/min) sa má počiatočná dávka odporúčaná na základe počtu krvných doštičiek pre pacientov s MF znížiť približne o 50 % a má sa podávať dvakrát denne. Odporúčaná počiatočná dávka pre pacientov s PV so závažným poškodením funkcie obličiek je 5 mg dvakrát denne. Počas liečby Jakavi je potrebné pacientov dôsledne monitorovať so zameraním na bezpečnosť a účinnosť.
Dostupné sú iba obmedzené údaje, ktoré by pomohli určiť najvhodnejšie dávkovanie u pacientov s renálnym ochorením v konečnom štádiu (ESRD) na hemodialýze. Farmakokinetické/farmakodynamické simulácie na základe dostupných údajov u tejto populácie naznačujú, že počiatočná jednorazová dávka pre pacientov s MF s ESRD na hemodialýze je 15-20 mg alebo dve dávky po 10 mg s 12 hodinovým odstupom, podané po dialýze avšak ešte v ten istý deň. Jednorazová dávka 15 mg sa odporúča pre pacientov s MF s počtom doštičiek medzi 100 000/mm3 a
200 000/mm3. Jednorazová dávka 20 mg alebo dve dávky po 10 mg s 12 hodinovým odstupom sa odporúčajú pre pacientov s MF s počtom doštičiek >200 000/mm3. Následné dávky (jednorazové
podanie alebo dve dávky po 10 mg podané s 12 hodinovým odstupom) sa majú podať iba v dňoch hemodialýzy, po každej dialýze.
Odporúčaná počiatočná dávka pre pacientov s PV s ESRD na hemodialýze je jedna 10 mg dávka alebo dve dávky po 5 mg podané s 12 hodinovým odstupom, ktoré majú byť podané po dialýze a iba v deň hemodialýzy. Tieto dávkovacie odporúčania sú založené na simuláciach a akákoľvek úprava dávky pri ESRD sa má robiť po dôslednom sledovaní bezpečnosti a účinnosti jednotlivo u každého pacienta. Nie sú dostupné žiadne údaje o dávkovaní u pacientov, ktorí podstupujú peritoneálnu dialýzu alebo kontinuálnu venóznu hemofiltráciu (pozri časť 5.2).
Poškodenie funkcie pečene
U pacientov s akýmkoľvek poškodením funkcie pečene sa má počiatočná dávka odporúčaná na základe počtu krvných doštičiek znížiť o približne 50 % a má sa podávať dvakrát denne. Následné dávky sa majú upraviť na základe dôsledného monitorovania bezpečnosti a účinnosti. U pacientov s poškodením funkcie pečene diagnostikovaným počas užívania Jakavi sa má sledovať kompletný krvný obraz, vrátane diferenciálneho počtu bielych krviniek každý až každý druhý týždeň počas prvých 6 týždňov od začatia liečby Jakavi a podľa klinickej indikácie aj následne po stabilizácii funkcie pečene a krvného obrazu. Dávku Jakavi možno titrovať aby sa znížilo riziko cytopénie.
Starší ľudia (≥65 rokov)
U starších ľudí sa neodporúčajú žiadne dodatočné úpravy dávky.
Pediatrická populácia
Bezpečnosť a účinnosť Jakavi u detí vo veku do 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje (pozri časť 5.1).
Ukonč eni e l ieč by
V liečbe možno pokračovať dovtedy, kým podiel prínos-riziko zostáva pozitívny. Pokiaľ však do
6 mesiacov od začatia liečby nedôjde k zmenšeniu objemu sleziny alebo zlepšeniu príznakov, má byť liečba ukončená.
U pacientov, ktorí vykazujú istý stupeň klinického zlepšenia sa odporúča ukončiť liečbu ruxolitinibom v prípade, že u nich naďalej dochádza k zväčšeniu dĺžky sleziny o 40 % v porovnaní s východiskovou hodnotou (približné ekvivalentné 25 % zväčšeniu objemu sleziny) a nie je už u nich viac
preukázateľné zlepšenie príznakov súvisiacich s ochorením.
Spôsobpodávania
Jakavi sa užíva perorálne s jedlom alebo bez jedla.
Pri vynechaní dávky pacient nemá užiť dávku navyše, ale má užiť obvyklú predpísanú najbližšiu dávku.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Gravidita a laktácia.
4.4 Osobitné upozornenia a opatrenia pri používaní
Myelosupresia
Liečba s Jakavi môže spôsobiť hematologické nežiaduce reakcie na liek vrátane trombocytopénie, anémie a neutropénie. Pred začatím liečby s Jakavi je potrebné urobiť kompletný krvný obraz, vrátane diferenciálneho počtu bielych krviniek. Liečba sa má ukončiť u pacientov s počtom krvných doštičiek nižším ako 50 000/mm3 alebo s absolútnym počtom neutrofilov nižším ako 500/mm3 (pozri časť 4.2).
U pacientov s nízkym počtom krvných doštičiek (<200 000/mm3) na začiatku liečby sa pozorovala vyššia pravdepodobnosť vzniku trombocytopénie počas liečby.
Trombocytopénia je spravidla reverzibilná a obvykle sa upraví po znížení dávky alebo po dočasnom vynechaní Jakavi (pozri časti 4.2 a 4.8). Napriek tomu, môže byť potrebná transfúzia krvných doštičiek, ak je to klinicky indikované.
Pacienti, u ktorých sa vyvinie anémia, môžu potrebovať transfúziu krvi. U týchto pacientov, možno zvážiť aj úpravu dávkovania alebo prerušenie liečby.
Pacienti s hladinou hemoglobínu pod 10,0 g/dl na začiatku liečby majú vyššie riziko nábehu na pokles hladiny hemoglobínu pod 8,0 g/dl počas liečby v porovnaní s pacientmi s vyššími východiskovými hladinami hemoglobínu (79,3 % oproti 30,1 %). U pacientov s hladinou hemoglobínu pod 10,0 g/dl sa odporúča častejšie monitorovanie hematologických parametrov a klinických prejavov a príznakov nežiaducich reakcií na liek spojených s Jakavi.
Neutropénia (absolútny počet neutrofilov <500) bola spravidla reverzibilná a upravila sa po dočasnom vynechaní Jakavi (pozri časti 4.2 a 4.8).
Kompletný krvný obraz sa má sledovať podľa klinickej indikácie a dávka sa má upravovať podľa potreby (pozri časti 4.2 a 4.8).
Infekcie
U pacientov sa má vyhodnotiť riziko vzniku závažných bakteriálnych, mykobakteriálnych, hubových a vírusových infekcií. U pacientov užívajúcich Jakavi na liečbu MF bola hlásená tuberkulóza. Pred začiatkom liečby majú byť pacienti v súlade s národnými odporúčaniami vyšetrení na prítomnosť aktívnej a inaktívnej („latentnej“) tuberkulózy. Vyšetrenie môže zahŕňať anamnézu, možný kontakt
s tuberkulózou a/alebo vhodný skríning ako napr. röntgen pľúc, tuberkulínový test a/alebo imunologické vyšetrenie IGRA (interferon-gamma release assay). Predpisujúci lekár musí mať na
pamäti riziko falošne negatívnych výsledkov tuberkulínových kožných testov, najmä u ťažko chorých
alebo imunokompromitovaných pacientov. Liečba s Jakavi sa nesmie začať, pokým závažné aktívne infekcie neodoznejú. U pacientov, ktorí dostávajú Jakavi, musia lekári pozorne sledovať prejavy a príznaky infekcií a okamžite začať vhodnú liečbu (pozri časť 4.8).
U pacientov s chronickou infekciou HBV užívajúcich Jakavi bolo hlásené zvýšenie vírusovej záťaže hepatitídy B (titer HBV-DNA) s alebo bez súvisiacich zvýšení alanínaminotransferázy a aspartátaminotransferázy. Účinok Jakavi na virálnu replikáciu u pacientov s chronickou infekciou HBV nie je známy. Pacienti s chronickou infekciou HBV sa majú liečiť a monitorovať v súlade s klinickými smernicami.
Herpeszoster
Lekári musia pacientov poučiť o včasných prejavoch a príznakoch herpes zoster s odporučením, aby čo najskôr vyhľadali lekársku pomoc.
Progresívnamultifokálnaleukoencefalopatia
Pri liečbe MF s Jakavi bola hlásená progresívna multifokálna leukoencefalopatia (PML). Lekári musia byť mimoriadne ostražití na príznaky naznačujúce PML, ktoré pacienti nemusia spozorovať (napr. kognitívne, neurologické alebo psychiatrické príznaky alebo prejavy). U pacientov je potrebné
sledovať akékoľvek nové alebo zhoršujúce sa uvedené príznaky alebo prejavy a v prípade ich výskytu je potrebné zvážiť konzultáciu s neurológom a vhodné diagnostické opatrenia pre PML. Ak je
podozrenie na PML, daľšie podávanie musí byť pozastavené, až kým nie je PML vylúčená.
Nemelanómovýkožnýnádor
U pacientov liečených ruxolitinibom boli hlásené nemelanómové kožné nádory (NMSC), vrátane bazocelulárneho karcinómu, spinocelulárneho karcinómu a karcinómu Merkelových buniek. Väčšina týchto pacientov v minulosti podstúpila dlhodobú liečbu hydroxyureou a už sa u nich vyskytla NMSC alebo premalígne kožné lézie. Kauzálny vzťah k liečbe ruxolitinibom nebol stanovený. U pacientov so zvýšeným rizikom kožných nádorov sa odporúčajú pravidelné vyšetrenia kože.
O
sobité
populácie
Poškodenie funkcie obličiek
U pacientov so závažným poškodením funkcie obličiek je potrebné znížiť počiatočnú dávku Jakavi. U
pacientov s MF na hemodialýze s renálnym ochorením v konečnom štádiu sa má počiatočná dávka stanoviť na základe počtu krvných doštičiek (pozri časť 4.2). Následné dávky (jednorazová dávka
20 mg alebo dve dávky po 10 mg podané s 12 hodinovým odstupom u pacientov s MF; jednorazová dávka 10 mg alebo dve dávky po 5 mg podané s 12 hodinovým odstupom u pacientov s PV) sa majú podať iba v dňoch hemodialýzy, po každej dialýze. Ďalšia úprava dávky sa má robiť pri dôslednom
sledovaní bezpečnosti a účinnosť (pozri časti 4.2 a 5.2).
Poškodenie funkcie pečene
U pacientov s poškodením funkcie pečene sa má počiatočná dávka znížiť približne o 50 %. Následné úpravy dávky majú vychádzať z bezpečnosti a účinnosti lieku (pozri časti 4.2 a 5.2).
Interakcie
Ak sa Jakavi podáva súčasne so silnými CYP3A4 inhibítormi alebo s duálnymi inhibítormi enzýmov CYP3A4 a CYP2C9 (napr. flukonazol), jednotlivá dávka Jakavi sa má znížiť približne o 50 % a má sa podávať dvakrát denne (k frekvencii monitorovania pozri časti 4.2 a 4.5).
Súčasné podávanie cytoredukčnej liečby alebo hemopoetických rastových faktorov spolu s Jakavi nebolo skúmané. Bezpečnosť a účinnosť súčasného podávania nie je známa (pozri časť 4.5).
Príznakyzvysadenia
V dôsledku prerušenia alebo ukončenia podávania Jakavi sa približne v priebehu jedného týždňa môžu príznaky MF znovu objaviť. Boli prípady pacientov s ťažším priebehom po prerušení Jakavi, obzvlášť počas akútneho pridruženého ochorenia. Nie je jasné, či náhle prerušenie Jakavi prispelo k tomuto stavu. Pokiaľ nie je potrebné náhle prerušenie, odporúča sa zvážiť postupné znižovanie dávky Jakavi, aj keď prínos postupného znižovania nie je preukázaný.
Pomocnélátky
Jakavi obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Eliminácia ruxolitinibu je sprostredkovaná metabolizáciou katalyzovanou enzýmami CYP3A4 a
CYP2C9. Lieky inhibujúce uvedené enzými preto môžu zapríčiniť zvýšenú expozíciu ruxolitinibu.
Interakcievedúcekzníženiudávkyruxolitinibu
CYP3A4 inhibítory
Silné CYP3A4 inhibítory (ako sú, ale nie len, boceprevír, klaritromycín, indinavír, itrakonazol, ketokonazol, lopinavír/ritonavír, ritonavír, mibefradil, nefazodón, nelfinavír, posakonazol, sakvinavír,
telaprevír, telitromycín, vorikonazol)
U zdravých jedincov súčasné podávanie Jakavi (jednotlivá dávka 10 mg) so silným CYP3A4 inhibítorom ketokonazolom viedlo k zvýšeniu Cmax ruxolitinibu o 33 % a AUC o 91 %, v porovnaní so samotným ruxolitinibom. Polčas eliminácie sa pri súčasnom podávaní ketokonazolu predĺžil z 3,7 na
6,0 hodín.
Ak sa Jakavi podáva súčasne so silnými CYP3A4 inhibítormi, jednotlivá dávka sa má znížiť približne o 50 % a má sa podávať dvakrát denne. Pacienti majú byť starostlivo monitorovaní (napr. dvakrát do týždňa) so zameraním na cytopénie a dávku je potrebné titrovať na základe bezpečnosti a účinnosti (pozri časť 4.2).
D
uálne CYP2C9 a CYP3A4 inhibítory
Na základe modelových údajov in silico možno zvážiť 50 % úpravu dávkovania aj pri užívaní liekov, ktoré sú duálnymi inhibítormi enzýmov CYP2C9 a CYP3A4 (napr flukonazol).
Induktoryenzýmov
CYP3A4 induktory (ako sú, ale nie len, avasimib, karbamazepín, fenobarbital, fenytoín, rifabutín,
ri fampí n (ri f ampi cí n), ľubovní k bodkovaný (Hypericum per f oratum))
Pacienti majú byť starostlivo monitorovaní a dávku je potrebné titrovať na základe bezpečnosti a účinnosti (pozri časť 4.2).
U zdravých jedincov po podaní Jakavi (50 mg jednotlivá dávka) následne po silnom CYP3A4 induktore rifampicíne (denná dávka 600 mg počas 10 dní), bola AUC ruxolitinibu o 70 % nižšia ako po podaní samotného Jakavi. Expozícia aktívnym metabolitom ruxolitinibu nebola zmenená. Celková farmakodynamická aktivita ruxolitinibu bola podobná, naznačujúc tak minimálny vplyv indukcie CYP3A4 na farmakodynamiku. To však môže byť spojené s vysokou dávkou ruxolitinibu zapríčiňujúcou farmakodynamický účinok blízky Emax. U jednotlivých pacientov je možná potreba zvýšiť dávku ruxolitinibu v prípade začatia liečby silným enzýmovým induktorom.
Inéinterakcievyžadujúceobozretnosťsvplyvomnaruxolitinib
Mierne alebo stredne silné CYP3A4 inhibítory (ako sú, ale nie len, ciprofloxacín, erytromycín,amprenavír, atazanavír, diltiazém, cimetidín)
U zdravých jedincov súčasné podávanie ruxolitinibu (jednotlivá dávka 10 mg) s erytromycínom
500 mg dvakrát denne počas štyroch dní, viedlo k zvýšeniu Cmax ruxolitinibu o 8 % a AUC o 27 %, v porovnaní so samotným ruxolitinibom.
Keď sa ruxolitinib podáva súčasne s miernymi alebo stredne silnými CYP3A4 inhibítormi
(napr. erytromycín), nie je potrebná úprava dávky. Pri začatí liečby so stredne silnými CYP3A4
inhibítormi je však potrebné pacientov dôsledne monitorovať so zameraním na cytopénie.
Vplyvruxolitinibunainélieky
Lie či vá pre nášané P-glykoproteínom a inými transportérmi
Ruxolitinib môže inhibovať P-glykoproteín a proteín rezistencie rakoviny prsníka (BCRP) v čreve. To môže mať za následok zvýšenú expozíciu substrátom týchto transportérov, ako napr. dabigatran
etexilát, cyklosporín, rosuvastatín a možno digoxín. Odporúča sa terapeutické monitorovanie lieku
alebo klinické monitorovanie ovplyvneného liečiva.
Je možné, že inhibícia P-glykoproteínu a proteínu rezistencie rakoviny prsníka môže byť minimalizovaná, ak sa oddelí čas spoločného podania na čo najdlhší časový interval.
Hematopoetické rastové faktory
Súčasné užívanie hematopoetických rastových faktorov a Jakavi sa nesledovalo. Nie je známe, či inhibícia Janusových kináz (JAK) prostredníctvom Jakavi znižuje účinnosť hematopoetických rastových faktorov alebo či hematopoetické rastové faktory ovplyvňujú účinnosť Jakavi (pozri časť 4.4).
Cyt oredukt ívne l i ečby
Súčasné používanie cytoreduktívnej liečby a Jakavi sa nesledovalo. Bezpečnosť a účinnosť súčasného podávania nie je známa (pozri časť 4.4).
Skúšanie na zdravých jedincoch naznačilo, že ruxolitinib nemal inhibičný účinok na metabolizmus perorálne podaného midazolamu, substrátu CYP3A4. Pri súčasnom podaní substrátov CYP3A4 s Jakavi sa preto nepredpokladá zvýšenie ich expozície. Ďalšie skúšanie na zdravých jedincoch nazančilo, že Jakavi nemá vplyv na farmakokinetiku perorálnych kontraceptív obsahujúcich etinylestradiol a levonorgestrel. Nie je preto predpoklad, že by bol účinok uvedenej kombinácie kontraceptív oslabený spoločným podávaním s ruxolitinibom.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
a
k
ontraceptíva
u
ž
ien
Nie sú k dispozícii žiadne údaje o použití Jakavi u gravidných žien.
Štúdie na zvieratách preukázali že ruxolitinib je embryotoxický a fetotoxický. Teratogenicita sa nezaznamenala u potkanov a králikov. Rozsah expozície bol však v porovnaní s najvyššou klinickou dávkou nízky, výsledok má preto len obmedzený význam pre ľudí (pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe. Ako preventívne opatrenie, je použitie Jakavi počas gravidity kontraindikované (pozri časť 4.3). Ženy vo fertilnom veku musia počas liečby s Jakavi používať účinnú antikoncepciu. V prípade, že by došlo ku gravidite počas liečby s Jakavi, je potrebné individuálne prehodnotiť prínos/riziko s dôkladnou konzultáciou ohľadom možných rizík pre plod (pozri časť 5.3).
Laktácia
Jakavi sa nemá užívať počas laktácie (pozri časť 4.3), preto musí byť pri začatí liečby dojčenie ukončené. Nie je známe, či sa ruxolitinib a/alebo jeho metabolity vylučujú do ľudského mlieka. Riziko
u dojčiat nemôže byť vylúčené. Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali
vylučovanie ruxolitinibu a jeho metabolitov do mlieka (pozri časť 5.3).
Fertilita
Nie sú dostupné údaje o účinkoch ruxolitinibu na fertilitu u ľudí. V štúdiách na zvieratách sa nepozorovali účinky na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Jakavi nemá žiaden alebo má zanedbateľný sedatívny účinok. Pacienti, u ktorých sa po užití Jakavi vyskytnú závraty, nemajú viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
Celkovo bolo hodnotenie bezpečnosti založené na 855 pacientoch (s MF alebo PV) užívajúcich Jakavi v klinických skúšaniach 2. a 3. fázy.
Myelofibróza
V dvoch pivotných štúdiách, COMFORT-I a COMFORT-II, bol vo fáze randomizácie medián trvania expozície lieku Jakavi 10,8 mesiaca (rozpätie 0,3 až 23,5 mesiaca). Väčšina pacientov (68,4 %) sa liečila minimálne 9 mesiacov. U 111 pacientov z 301 (36,9 %) bol počet krvných doštičiek na začiatku liečenia medzi 100 000/mm3 a 200 000/mm3 a u 190 (63,1 %) bol počet krvných doštičiek
>200 000/mm3.
V týchto klinických skúšaniach bolo u 11,3 % pacientov pozorované ukončenie liečby kvôli nežiaducim účinkom, bez ohľadu na kauzalitu.
Najčastejšie hlásenými nežiaducimi reakciami boli trombocytopénia a anémia.
K hematologickým nežiaducim reakciám na liek (všetkých stupňov škály spoločných kritérií pre názvoslovie nežiaducich udalostí [CTCAE]) patrila anémia (82,4 %), trombocytopénia (69,8 %) a neutropénia (16,6 %).
Anémia, trombocytopénia a neutropénia sú reakcie úmerné dávke.
Tri najčastejšie iné ako hematologické nežiaduce reakcie na liek boli tvorenie podliatin (21,3 %), závraty (15,3 %) a bolesť hlavy (14,0 %).
Tri najčastejšie iné ako hematologické abnormálne laboratórne nálezy boli zvýšená
alanín aminotrasferáza (27,2 %), zvýšená aspartát aminotransferáza (19,9 %) a hypercholesterolémia (16,9 %).V 3.fáze klinických skúšaní u MF nebola pozorovaná ani cholesterolémia stupňa 3 alebo 4 podľa CTCAE, zvýšená aspartátaminotransferáza ani zvýšená alanínaminotransferáza stupňa 4 podľa CTCAE.
Dlhodobá bezpečnosť: Podľa očakávania sa pri predĺžení doby ďalšieho sledovania zvýšila kumulatívna frekvencia niektorých nežiaducich udalostí pri hodnotení bezpečnostných údajov z 3- ročného obdobia (medián trvania expozície u pacientov počiatočne randomizovaných na ruxolitinib v štúdiách COMFORT-I a COMFORT-II bol 33,2 mesiacov) od 457 pacientov s myelofibrózou liečených ruxolitinibom počas obdobia randomizácie a extenzie dvoch pivotných skúšaní 3 fázy. Toto hodnotenie zahŕňa údaje od pacientov, ktorí boli počiatočne randomizovaní na ruxolitinib (N=301)
a od pacientov, ktorí dostali ruxolitinib po prekrížení z kontrolných liečebných ramien (N=156). Podľa týchto aktualizovaných údajov sa ukončenie liečby z dôvodu nežiaducich účinkov pozorovalo u 17,1% pacientov liečených ruxolitinibom.
Polycytémia vera
Bezpečnosť Jakavi bola hodnotená u 110 pacientov s PV v otvorenom, randomizovanom, kontrolovanom klinickom skúšaní 3.fázy s názvom RESPONSE. Nižšie uvedené nežiaduce reakcie na
liek sa vzťahujú na počiatočnú fázu klinického skúšania (až do 32. týždňa) s rovnakou expozíciou ruxolitinibu a najlepšej dostupnej liečbe (Best Available Therapy - BAT), zodpovedajúcou mediánu trvania expozície lieku Jakavi v dĺžke 7,8 mesiaca. Priemerný vek pacientov užívajúcich Jakavi bol
približne 60 rokov.
Ukončenie liečby kvôli nežiaducim účinkom, bez ohľadu na príčiny, bolo pozorované u 3,6 %
pacientov liečených Jakavi a u 1,8 % pacientov liečených najlepšou dostupnou liečbou.
Hematologické nežiaduce reakcie (akéhokoľvek stupňa podľa CTCAE) zahrňovali anémiu (43,6 %) a trombocytopéniu (24,5 %). Anémia alebo trombocytopénia stupňa 3 a 4 podľa CTCAE bola hlásená
u 1,8 % alebo 5.5 % pacientov, v uvedenom poradí.
Tromi najčastejšími nehematologickými nežiaducimi reakciami boli závrat (15,5 %), zápcha (8,2 %) a herpes zoster (6,4 %).
Tromi najčastejšími nehematologickými laboratórnymi abnormalitami (akéhokoľvek stupňa podľa CTCAE) boli hypercholesterolémia (30,0 %), zvýšená alanínaminotransferáza (22,7 %) a zvýšená aspartátaminotransferáza (20,9 %). Všetky boli stupňa 1 a 2 podľa CTCAE okrem jedného prípadu zvýšenej alanínaminotransferázy stupňa 3 podľa CTCAE.
Dlhodobá bezpečnosť: Medián trvania expozície lieku Jakavi u pacientov bol 18,6 mesiaca (rozpätie
0,3 až 35,9 mesiaca). Pri dlhšej expozícii sa počet nežiaducich účinkov zvýšil, ale neobjavili sa žiadne nové zistenia v oblasti bezpečnosti. Po úprave na túto expozíciu bola miera výskytu nežiaducich účinkov obvykle porovnateľná s mierou ich výskytu pozorovanou v priebehu počiatočnej fázy klinického skúšania.
Tabuľkovýprehľadnežiaducichreakciízklinickýchštúdií
V programe klinických štúdií sa závažnosť nežiaducich reakcií hodnotila podľa kritérií CTCAE, kde stupeň 1 = mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4= život ohrozujúce.
Nežiaduce reakcie nahlásené z klinických štúdií (Tabuľka 1) sú zoradené podľa triedy orgánových systémov MedDRA. V každej triede orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, pričom najčastejšie sú uvedené ako prvé. Frekvencie sú definované podľa nasledovnej konvencie: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000).
Tabuľka 1 Kategória frevencie výskytu nežiaducich reakcií na liek hlásených v štúdiách fázy 3 (COMFORT-I, COMFORT-II, RESPONSE)
N
ežiaduca reakcia na liek Kategória frekvencie
výskytu u pacientov s
MF
K
ategória frekvencie výskytu u
pacientov s PV
Infekcie a nákazy
Infekcie močového traktua,d Veľmi časté Časté
Herpes zostera,d Časté Časté
Tuberkulózae Menej časté -
Poruchy krvi a lymfatického systémub,d
Anémiab - -
CTCAEc stupeň 4 (<6,5g/dl)
CTCAEc stupeň 3 (<8,0 –
6,5g/dl)
Akýkoľvek CTCAE
stupeňc
Trombocytopéniab CTCAEc stupeň 4 (<25 000/mm3)
CTCAEc stupeň 3 (50 000
– 25 000/mm3)
Akýkoľvek CTCAEc
stupeň
Neutropéniab CTCAEc stupeň 4 (<500/mm3)
CTCAEc stupeň 3 (<1 000
– 500/mm3) Akýkoľvek CTCAEc
stupeň
Krvácanie (akékoľvek krvácanie vrátane intrakraniálneho, gastrointestinálne krvácanie, podliatiny a iné krvácanie)
Veľmi časté Menej časté Veľmi časté Menej časté Veľmi časté Veľmi časté
Časté Menej časté
Časté Časté
Veľmi časté Veľmi časté
Časté - Časté - Veľmi časté -
Veľmi časté Veľmi časté
Intrakraniálne krvácanie Časté -
Gastrointestinálne krvácanie
Časté -
Podliatiny Veľmi časté Veľmi časté
Iné krvácanie (vrátane epistaxy, postprocedurálnej hemorágie a hematúrie)
Časté Veľmi časté
P
oruchy metabolizmu a
výživy
Nárast hmotnostia Veľmi časté Časté
Hypercholesterolémiab
CTCAEc stupeň 1 a 2
Hypertriglyceridémiab
CTCAEc stupeň 1
Poruchy nervového systému
Veľmi časté Veľmi časté
- Veľmi časté
Závratya Veľmi časté Veľmi časté
Bolesť hlavya Veľmi časté -
Poruchy gastrointestinálneho traktu
Plynatosťa Časté -
Zápchaa - Časté
Poruchy pečene a žlčových
ciest Zvýšená alanínaminotransferázab
CTCAEc stupeň 3 (> 5x –
20 x ULN) Akýkoľvek CTCAEc stupeň
Zvýšená aspartátaminotransferázab
Akýkoľvek CTCAEc
stupeň
Poruchy ciev
Časté Menej časté
Veľmi časté Veľmi časté
Veľmi časté Veľmi časté
Hypertenziaa - Veľmi časté
a Frekvencia vychádza z údajov o nežiaducich účinkoch.
- Subjekt, u ktorého sa vyskytlo viacero nežiaducich reakcií na liek (ADR) je v danej
ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
b Frekvencia vychádza z laboratórnych hodnôt.
- Subjekt, u ktorého sa vyskytlo viacero ADR je v danej ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
c Všeobecné terminologické kritériá pre nežiaduce účinky (CTCAE) verzia 3.0; stupeň 1 =
mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4 = život ohrozujúce
d Tieto ADR sú bližšie popísané v texte.
e Frekvencia vychádza z počtu všetkých pacientov exponovaných ruxolitinibu v klinických skúšaniach (N=4755)
Po ukončení liečby sa u pacientov s MF môžu vrátiť príznaky MF ako napr. únava, bolesť kostí,
horúčka, pruritus, nočné potenie, symptomatická splenomegália a pokles telesnej hmotnosti.
V klinických štúdiách s MF sa celkové skóre symptómov pre symptómy MF postupne vrátilo na počiatočné hodnoty do 7 dní od ukončenia liečby (pozri časť 4.4).
PopisvybranýchnežiaducichreakciínaliekAnémiaV klinických štúdiách fázy III s MF, bol medián nástupu anémie CTCAE stupňa 2 alebo vyššieho
1,5 mesiaca. Jeden pacient (0,3 %) ukončil liečbu v dôsledku anémie.
U pacientov užívajúcich Jakavi dosiahli priemerné poklesy hemoglobínu minimum približne o
10 g/liter pod počiatočnými hodnotami po 8 až 12 týždňoch liečby a potom postupne narastali, kým nedosiahli nový stabilný stav, ktorý bol približne o 5 g/liter pod počiatočnými hodnotami. Takýto
vzorec sa u pacientov pozoroval bez ohľadu na to, či počas liečby dostali transfúziu.
V randomizovanej, placebom kontrolovanej štúdii COMFORT-I 60,6 % pacientov s MF užívajúcich Jakavi a 37,7 % pacientov s MF užívajúcich placebo dostalo počas randomizovanej liečby transfúziu s červenými krvinkami. V štúdii COMFORT-II bol podiel transfúzií s červenými krvinkami 53,4 %
v ramene s Jakavi a 41,1 % v ramene s najlepšou dostupnou liečbou.
Vo fáze randomizácie v pivotných štúdiách bola anémia menej častá u pacientov s PV ako u pacientov s MF (43,6 % oproti 82,4 %). V PV populácii boli prípady CTCAE stupňa 3 a 4 hlásené u 1,8 % pacientov, kým frekvencia u pacientov s MF bola 42,56 %.
Trombocytopénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula trombocytopénia stupňa 3 alebo 4, bol medián jej nástupu približne 8 týždňov. Trombocytopénia bola vo všeobecnosti reverzibilná a ustúpila po znížení dávky alebo po prerušení liečby. Medián času potrebného na návrat počtu krvných doštičiek nad 50 000/mm3 bol 14 dní. Počas randomizovaného obdobia boli transfúzie s krvnými doštičkami podané 4,7 % pacientov užívajúcich Jakavi a 4,0 % pacientov užívajúcich
kontrolné režimy. K ukončeniu liečby v dôsledku trombocytopénie došlo u 0,7 % pacientov užívajúcich Jakavi a 0,9 % pacientov užívajúcich kontrolné režimy. U pacientov, ktorí mali pred začiatkom liečby s Jakavi počet krvných doštičiek od 100 000/mm3 do 200 000/mm3, bola vyššia frekvencia trombocytopénie stupňa 3 alebo 4 v porovnaní s pacientmi, ktorý mali počet krvných doštičiek >200 000/mm3 (64,2 % verzus 38,5 %).
Vo fáze randomizácie v pivotných štúdiách bol podiel pacientov, u ktorých sa vyskytla trombocytopénia nižší u pacientov s PV (24,5 %), ako u pacientov s MF (69,8 %). Frekvencia výskytu závažnej trombocytopénie (t.j. CTCAE stupňa 3 a 4) bola nižšia u pacientov s PV (5,5 %) ako u pacientov s MF (11,6 %).
Neutropénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula neutropénia stupňa 3 alebo 4, bol medián jej nástupu 12 týždňov. Počas randomizovaného obdobia sa pozastavenie liečby alebo zníženie dávky v dôsledku neutropénie hlásilo u 1,0 % pacientov a 0,3 % pacientov ukončilo liečbu pre neutropéniu.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV bola neutropénia hlásená u dvoch pacientov
(1,8 %), z ktorých sa u jedného pacienta vyskytla neutropénia CTCAE stupňa 4.
Krvácanie
V pivotných štúdiách fázy III s MF boli prípady krvácania (vrátane intrakraniálneho
a gastrointestinálneho krvácania, podliatin a iných prípadov krvácania) hlásené u 32,6 % pacientov liečených Jakavi a u 23,2 % pacientov liečených referenčnou liečbou (placebom alebo najlepšou dostupnou liečbou). Frekvencia udalostí stupňa 3-4 bola u pacientov liečených Jakavi alebo referenčnou liečbou podobná (4,7 % verzus 3,1 %). Väčšina pacientov s príhodou krvácania počas liečby hlásila podliatiny (65,3 %). Podliatiny boli častejšie hlásené u pacientov užívajúcich Jakavi
v porovnaní s referenčnou liečbou (21,3 % verzus 11,6 %). Intrakraniálne krvácanie bolo hlásené u
1 % pacientov exponovaných Jakavi a u 0,9 % pacientov exponovaných referenčnej liečbe. Gastrointestinálne krvácanie bolo hlásené u 5,0 % pacientov exponovaných Jakavi oproti 3,1 % pacientov exponovaných referenčnej liečbe. Iné prípady krvácania (vrátane epistaxy,
post-procedurálnej hemorágie a hematúrie) boli hlásené u 13,3 % pacientov liečených Jakavi a u
10,3 % pacientov liečených referenčnou liečbou.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV boli hlásené prípady krvácania (vrátane intrakraniálneho a gastrointestináneho krvácania, tvorby podliatin a iného krvácania) u 20 % pacientov liečených Jakavi a 15,3 % pacientov, ktorí dostávali najlepšiu dostupnú liečbu. Podliatiny boli hlásené
s rovnakou frekvenciou v liečebných skupinách s Jakavi aj BAT (10,9 % oproti 8,1 %). U pacientov užívajúcich Jakavi neboli hlásené žiadne prípady intrakraniálneho ani gastrointestinálneho krvácania.
U jedného pacienta liečeného Jakavi sa vyskytol prípad krvácania stupňa 3 (postprocedurálne krvácanie); nebolo hlásené žiadne krvácanie stupňa 4. Prípady iného krvácania (vrátane takých prípadov ako epistaxa, postprocedurálne krvácanie, gingiválne krvácanie) boli hlásené u 11,8 % pacientov liečených Jakavi a u 6,3 % pacientov liečených najlepšou dostupnou liečbou.
InfekcieV pivotných štúdiách fázy III s MF boli hlásené infekcie močových ciest stupňa 3 alebo 4 u 1,0 % pacientov, herpes zoster u 4,3 % a tuberkulóza u 1,0 %. V klinickom skúšaní fázy III bola sepsa hlásená u 3,0% pacientov. Predĺžené sledovanie pacientov liečených ruxolitinibom nepreukázalo
s postupom času žiadny zvýšený trend v rozsahu sepsy.
Vo fáze randomizácie v pivotnej štúdii bola u pacientov s PV hlásená jedna infekcia močového traktu (0,9 %) CTCAE stupňa 3 a žiadna infekcia močového traktu stupňa 4. Miera výskytu herpes zoster bola mierne vyššia u pacientov s PV (6,4 %) ako u pacientov s MF (4,0 %). U pacientov s PV sa vyskytlo jedno hlásenie postherpetickej neuralgie CTCAE stupňa 3.
Zvýšený systolický krvný tlakV pivotných štúdiách fázy III s MF bolo hlásené minimálne počas jednej vizity zvýšenie systolického
krvného tlaku oproti východiskovej hodnote o 20 mmHg alebo viac u 31,5 % pacientov v porovnaní s
19,5 % výskytom u pacientov na kontrolnej liečbe. V skúšaní COMFORT-I (pacienti s MF) bol oproti východiskovej hodnote pri Jakavi priemerne zvýšený systolický krvný tlak o 0-2 mmHg v porovnaní
s poklesom o 2-5 mmHg v ramene s placebom. V skúšaní COMFORT-II boli medzi priemernými hodnotami MF pacientov liečených ruxolitinibom alebo kontrolnou liečbou len malé rozdiely.
Vo fáze randomizácie v pivotných štúdiách bol u pacientov s PV sa priemerný systolický krvný tlak v liečebnej skupine s Jakavi zvýšil o 0,65 mmHg, kým v skupine s BAT klesol o 2 mmHg.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieNie je známe antidotum pri predávkovaní s Jakavi. Jednorazové dávky do 200 mg boli podané s prijateľnou akútnou znášanlivosťou. Vyššie ako odporúčané, opakované dávky sú spojené so zvýšenou myelosupresiou vrátane leukopénie, anémie a trombocytopénie. Je potrebné podať vhodnú podpornú liečbu.
Nepredpokladá sa, že hemodialýza zvyšuje elimináciu ruxolitinibu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE18
MechanizmusúčinkuRuxolitinib je selektívny inhibítor Janusových kináz (JAK), JAK1 a JAK2 (IC50 hodnoty 3,3 nM pre enzýmy JAK1 a 2,8 nM pre enzýmy JAK2). Tieto sprostredkujú signalizáciu mnohých cytokínov a rastových faktorov, ktoré sú dôležité pre hemopoézu a imunitné funkcie.
Myelofibróza a polycytémia vera sú myeloproliferatívne nádorové ochorenia, o ktorých je známe že súvisia s poruchou signalizácie JAK1 a JAK2. Predpokladá sa že k základom týchto porúch patrí vysoká hladina cirkulujúcich cytokínov, ktoré aktivujú JAK-STAT dráhou, mutácie pridávajúce
funkcie ako napr. JAK2V617F a utlmenie negatívnych regulačných mechanizmov. Pacienti s MF vykazujú poruchu JAK signalizácie bez ohľadu na stav JAK2V617F mutácie. U >95 % pacientov s PV sa vyskytujú aktivujúce mutácie JAK2 (V617F alebo exón 12).
Ruxolitinib inhibuje JAK-STAT signalizáciu a bunkovú proliferáciu bunkových modelov hematologických malignít závislých od cytokínov ako aj Ba/F3 buniek nezávislých od cytokínov expresiou JAK2V617F mutovaného proteínu, s hodnotou IC50 v rozsahu 80-320 nM.
Farmakodynamickéúčinky
Ruxolitinib inhibuje cytokínmi indukovanú STAT3 fosforyláciu v celej krvi od zdravých dobrovoľníkov, pacientov s MF a pacientov s PV . Ruxolitinib vyvolal maximálnu inhibíciu STAT3
fosforylácie 2 hodiny po podaní dávky, ktorá sa vrátila do normálu do 8 hodín u zdravých
dobrovoľníkov aj u pacientov s MF, čo naznačuje že nedochádza k akumulácii pôvodných ani aktívnych metabolitov.
Zvýšené východiskové zápalové markery súvisiace so základnými konštitučnými príznakmi ako TNFα, IL-6 a CRP u osôb s MF po liečbe ruxolitinibom poklesli. Počas liečby ruxolitinibom sa u pacientov s MF nevytvorila odolnosť voči jeho farmakodynamickým účinkom. Rovnako aj u pacientov s PV sa vyskytli zvýšené východiskové zápalové markery a tieto markery sa po liečbe ruxolitinibom znížili.
V podrobnej QT štúdii so zdravými dobrovoľníkmi nič nepoukazovalo na predlžujúci účinok ruxolitinibu na QT/QTc v jednotlivých dávkach až po supraterapeutickú dávku 200 mg, čo naznačuje že ruxolitinib nemá účinok na kardiálnu repolarizáciu.
Klinickáúčinnosťabezpečnosť
Myelofibróza
U pacientov s MF (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie) sa realizovali 2 randomizované štúdie fázy III (COMFORT-I a COMFORT-II). V oboch štúdiách mali pacienti hmatateľnú splenomegáliu najmenej
5 cm pod rebrovým oblúkom a rizikovú kategóriu strednú-2 alebo vysokú, podľa IWG kritérií
(International Working Group Consensus Criteria). Počiatočná dávka Jakavi sa stanovila na základe počtu krvných doštičiek.
COMFORT-I bola dvojito zaslepená, randomizovaná, placebom kontrolovaná štúdia s 309 pacientmi, ktorí nereagovali na dostupnú liečbu alebo táto pre nich nebola vhodná. Primárny cieľový ukazovateľ účinnosti bol definovaný ako podiel pacientov, u ktorých došlo k ≥35 % zmenšeniu objemu sleziny v
24. týždni v porovnaní s východiskom, merané zobrazovaním magnetickou rezonanciou (MRI) alebo počítačovou tomografiou (CT).
Sekundárne cieľové ukazovatele účinnosti zahrňovali trvanie ≥35 % zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou, podiel pacientov s ≥50 % znížením skóre celkových príznakov, zmeny v celkových skóre príznakov v porovnaní s východiskovou hodnotou v 24. týždni hodnotené pomocou modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) v2.0 denníka, a celkové prežívanie.
COMFORT-II bola otvorená, randomizovaná štúdia s 219 pacientmi. Pacienti boli randomizovaní 2:1 do skupín s Jakavi a s najlepšou dostupnou liečbou. V ramene s najlepšou dostupnou liečbou 47 % pacientov dostalo hydroxyureu a 16 % pacientov glukokortikoidy. Primárny cieľový ukazovateľ účinnosti bol podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 48. týždni v porovnaní s východiskovou hodnotou, merané MRI alebo CT.
Sekundárne cieľové ukazovatele zahrňovali podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 24. týždni v porovnaní s východiskovou hodnotou a trvanie ≥35% zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou.
Základné demografické charakteristiky a charakteristika ochorenia pacientov boli porovnateľné pre obe ramená v štúdiách COMFORT-I a COMFORT-II.
Tabuľka 2 Percento pacientov s ≥35 % zmenšením sleziny v 24. týždni v porovnaní
s východiskovou hodnotou v COMFORT-I a v 48. týždni v COMFORT-II (ITT)
COMFORT-I COMFORT-II
Jakavi
(N=155)
Placebo
(N=153)
Jakavi
(N=144)
Najlepšia dostupná liečba (N=72)
Čas Týždeň 24 Týždeň 48
Počet (%) pacientov s objemom sleziny zmenšeným o ≥35 %
95 % intervaly spoľahlivosti
65 (41,9) 1 (0,7) 41 (28,5) 0
34,1; 50,1 0; 3,6 21,3; 36,6 0,0; 5,0
p-hodnota <0,0001 <0,0001
Významne väčší podiel pacientov v skupine s Jakavi dosiahol ≥35 % zmenšenie objemu sleziny
(tabuľka 2) v porovnaní s východiskovou hodnotou bez ohľadu na prítomnosť alebo neprítomnosť
JAK2V617F mutácie alebo subtyp ochorenia (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie).
Tabuľka 3 Percento pacientov s ≥35 % zmenšením sleziny v porovnaní s východiskovou
hodnotou podľa stavu mutácie JAK (súbor bezpečnosti)
COMFORT-I COMFORT-II
Jakavi Placebo Jakavi Najlepšia dostupná liečba
Stav mutácie JAK
Pozitívny (N=113) n (%)
Negatívny
(N=40)
n (%)
Pozitívny (N=121) n (%)
Negatívny
(N=27)
n (%)
Pozitívny (N=110) n (%)
Negatívny
(N=35)
n (%)
Pozitívny
(N=49)
n (%)
Negatívny
(N=20)
n (%)
Počet (%)
pacientov
s objemom sleziny zmenšeným o ≥35 %
54 (47,8)
11 (27,5)
1 (0,8)
0 36 (32,7)
5 0 0 (14,3)
Časový bod Po 24 týždňoch Po 48 týždňoch
Pravdepodobnosť udržania odpovede sleziny (≥35 % redukcia) na Jakavi v priebehu najmenej
24 týždňov bola 89 % v štúdii COMFORT I a 87 % v štúdii COMFORT II; v štúdii COMFORT II
udržalo odpoveď sleziny 52 % počas aspoň 48 týždňov.
V štúdii COMFORT I 45,9 % subjektov v skupine s Jakavi dosiahlo v 24. týždni ≥50 % zlepšenie celkového skóre príznakov v porovnaní s východiskovou hodnotou (zisťované podľa denníka MFSAF v2.0), v porovnaní s 5,3 % v skupine s placebom (p<0,0001 pomocou chi-kvadrát testu). Priemerná zmena v celkovom zdravotnom stave v 24. týždni zisťovaná podľa EORTC QLQ C30 bola +12,3 u Jakavi a -3,4 u placeba (p<0,0001).
V štúdii COMFORT-I bola úmrtnosť pacientov randomizovaných do skupiny s ruxolitinibom po priemernej dobe sledovania 34,3 mesiacov 27,1 %, v porovnaní s 35,1 % u pacientov randomizovaných na placebo; HR 0,687; 95 % IS 0,459-1,029; p=0,0668.
V štúdii COMFORT-II bola úmrtnosť pacientov randomizovaných na ruxolitinib po priemernej dobe sledovania 34,7 mesiacov 19,9 %, v porovnaní s 30,1 % u pacientov randomizovaných na najlepšiu dostupnú liečbu (BAT); HR 0,48; 95 % IS 0,28-0,85; p=0,009. V oboch štúdiách bola nižšia úmrtnosť
zaznamenaná v skupine s ruxolitinibom, čo bolo spôsobené predovšetkým výsledkami získanými v podskupinách po polycytémii vera a po esenciálnej trombocytémii.
Polycytémia vera
Randomizované, otvorené, aktívne kontrolované klinické skúšanie 3. fázy (RESPONSE) bolo vykonané u 222 pacientov s PV s rezistenciou alebo intoleranciou na hydroxyureu definované podľa
publikovaných kritérií medzinárodnej pracovnej skupiny European LeukemiaNet (ELN).
110 pacientov bolo randomizovaných do skupiny s ruxolitinibom a 112 pacientov do skupiny s BAT. Počiatočná dávka Jakavi bola 10 mg dvakrát denne. Dávky sa potom u jednotlivých pacientov upravili
podľa znášanlivosti a účinnosti na maximálnu dávku 25 mg dvakrát denne. BAT vybral skúšajúci
lekár pre každého pacienta individuálne a zahŕňala hydroxyureu (59,5 %), interferón/pegylovaný interferón (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorovanie (15,3 %).
Vstupné demografické údaje a charakteristika ochorenia boli v dvoch liečebných skupinách porovnateľné. Priemerný vek bol 60 rokov (rozpätie 33 až 90 rokov). Pacienti v skupine
s ruxolitinibom mali diagnózu PV v priemere 8,2 roka a predtým užívali v priemere približne 3 roky hydroxyureu. Väčšina pacientov (>80 %) podstúpila v priebehu posledných 24 týždňov pred skríningom najmenej dve flebotómie. Porovnávacie údaje týkajúce sa dlhodobého prežitia a výskytu
komplikácií ochorenia nie sú dostupné.
Primárnym zloženým cieľovým ukazovateľom bolo percento pacientov, ktorí dosiahli aj absenciu indikácie flebotómie (kontrola HCT) a ≥35 % zmenšenie objemu sleziny v porovnaní so vstupnými hodnotami v 32. týždni. Indikácia flebotómie bola definovaná ako potvrdené HCT s hodnotou >45 %, t.j. aspoň o 3 percentuálne body vyššie ako HCT získané pri vstupnom vyšetrení alebo potvrdené HCT s hodnotou >48 %, podľa toho, ktorá hodnota bola nižšia. Kľúčové sekundárne cieľové ukazovatele zahrňovali percento pacientov, ktorí dosiahli primárny cieľový ukazovateľ a v 48. týždni ostali bez progresie, ako aj percento pacientov, ktorí dosiahli úplnú hematologickú remisiu v 32. týždni.
Štúdia splnila svoj primárny cieľ a vyššie percento pacientov v skupine s Jakavi dosiahlo primárny zložený cieľový ukazovateľ aj každú z jeho jednotlivých zložiek. Podstatne viac pacientov liečených Jakavi (20,9 %) dosiahlo primárnu odpoveď (p<0,0001) v porovnaní s BAT (0,9 %). Kompenzácia hematokritu sa dosiahla u 60 % pacientov v skupine s Jakavi v porovnaní s 19,6 % v skupine s BAT a
≥35 % zmenšenie objemu sleziny sa dosiahlo u 38,2 % pacientov v skupine s Jakavi v porovnaní s 0,9 % v skupine s BAT (obr. č. 1). Deväťdesiatštyri (83,9 %) pacientov randomizovaných do skupiny s BAT prešlo na liečbu ruxolitinibom v 32. týždni alebo neskôr, čím bolo obmedzené porovnanie týchto dvoch skupín po 32. týždni.
Splnené boli aj oba kľúčové sekundárne cieľové ukazovatele. Percento pacientov, ktorí dosiahli úplnú hematologickú remisiu, bolo 23,6 % s Jakavi v porovnaní s 8,9 % s BAT (p=0,0028) a percento pacientov, ktorí dosiahli dlhotrvajúcu primárnu odpoveď v 48. týždni, bolo 19,1 % s Jakavi a 0,9 %
s BAT (p<0,0001).
O
brázok č. 1 Pacienti dosahujúci primárny cieľový ukazovateľ a zložky primárneho cieľového ukazovateľa v 32. týždni
Hodnota P: < ,0001
70 Pomer š a ncí (Ruxolitinib/BAT)
a 95% IS:
60 28,64 (4,50, 12,06)
50
40 38
30
21
20
10
Jednotlivé zložky primá rnej
odpovede v 32.týždni
60
20
.RUX
.BAT
1 1
0
Pri má rny zl ožený
ci eľový uka zova teľ v
32.týždni
≥35% zníženi e obj emu
s l ezi ny
Kompenzá ci a hema tokri tu bez fl ebotómi e
Príznaková záťaž bola hodnotená pomocou MPN-SAF celkového skóre príznakov (total symptom
score - TSS) elektronického denníka pacienta, ktorý obsahoval 14 otázok. V 32. týždni dosiahlo 49 %
pacientov liečených ruxolitinibom ≥50 % zníženie TSS-14 a 64 % pacientov ≥50 % zníženie TSS-5, v porovnaní s iba 5 % a 11 % pacientov na BAT.
Vnímanie liečebného prínosu bolo zisťované v dotazníku Celkový dojem pacienta zo zmeny (Patient Global Impression of Change - PGIC). 66 % pacientov liečených ruxolitinibom, v porovnaní s 19 % liečenými BAT, hlásilo zlepšenie už štyri týždne po začiatku liečby. Zlepšenie vo vnímaní liečebného prínosu bolo tiež vyššie u pacientov liečených ruxolitinibom v 32. týždni (78 % oproti 33 %).
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Jakavi vo všetkých podskupinách pediatrickej populácie v liečbe MF (informácie o použití v pediatrickej
populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Ruxolitinib je látka I. triedy Biofarmaceutického klasifikačného systému (BCS), s vysokou permeabilitou, vysokou rozpustnosťou a rýchlou disolúciou. V klinických štúdiách sa ruxolitinib
rýchlo absorboval po perorálnom podaní, maximálnu plazmatickú koncentráciu (Cmax) dosiahol asi po
1 hodine po podaní dávky. Na základe humánnej štúdie hmotnostnej bilancie sa zistilo, že perorálna
absorpcia ruxolitinibu, v podobe ruxolitinibu alebo metabolitov vzniknutých v priebehu presystémovej eliminácie je 95 % alebo vyššia. Priemerné hodnoty Cmax a celkovej expozície (AUC) ruxolitinibu sa priamoúmerne zvýšili po jednotlivej dávke v rozsahu 5-200 mg. Nezistili sa klinicky relevantné zmeny farmakokinetiky ruxolitinibu po podaní s jedlom s vysokým obsahom tuku. Priemerná Cmax sa mierne znížila (24 %), kým priemerná AUC zostala takmer nezmenená (4 % vzostup) po podaní s jedlom s vysokým obsahom tuku.
Distribúcia
Priemerný objem distribúcie v rovnovážnom stave je u pacientov s MF aj s PV približne 75 litrov. Pri klinicky relevantných koncentráciách ruxolitinibu je väzba na plazmatické bielkoviny približne 97 %,
prevažne na albumín. Celotelové autoradiografické štúdie na potkanoch ukázali, že ruxolinitib
nepenetruje cez krvno-mozgovú bariéru.
B
i
otransformácia
Ruxolitinib je prevažne metabolizovaný enzýmom CYP3A4 (>50 %) s čiastočnou účasťou CYP2C9. Materská zlúčenina je hlavným prvkom v ľudskej plazme, pričom predstavuje približne 60 % materiálu súvisiaceho s liekom v obehu. V plazme sú prítomné dva hlavné a aktívne metabolity, ktoré reprezentujú 25 % a 11 % materskej AUC. Tieto metabolity majú polovicu až pätinu pôvodnej farmakologickej aktivity súvisiacej s JAK. Celkové množstvo všetkých aktívnych metabolitov prispieva 18 % k celkovej farmakodynamike ruxolitinibu. Podľa in vitro štúdií, ruxolitinib v klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 alebo CYP3A4 a nie je účinný induktor CYP1A2, CYP2B6 alebo CYP3A4. In vitro údaje naznačujú, že ruxolitinib môže byť inhibítor P-gp a BCRP.
Eliminácia
Ruxolitinib sa prevažne eliminuje metabolizmom. Priemerný polčas eliminácie ruxolitinibu sú približne 3 hodiny. U zdravých dobrovoľníkov, po jednorazovej perorálnej dávke rádioaktívne značeného [14C] ruxolitinibu došlo k eliminácii prevažne metabolizáciou, pričom 74 % rádioaktivity sa vylúčilo močom a 22 % stolicou. Na nezmenenú materskú zlúčeninu pripadlo menej ako 1 % celkovej vylúčenej rádioaktivity.
Linearita/nelinearita
Priama úmera v závislosti od dávky sa dokázala v štúdiách s jednorazovými aj viacnásobnými dávkami.
Osobitépopulácie
Vplyv veku, pohlavia a rasy
Na základe štúdií so zdravými subjektmi sa nepozorovali žiadne relevantné rozdiely vo farmakokinetike ruxolitinibu v závislosti od pohlavia a rasy. Na základe analýzy farmakokinetiky u
populácie pacientov s MF sa nepotvrdil vzťah medzi perorálnym klírensom a vekom alebo rasou pacienta. Odhadovaný perorálny klírens bol u žien 17,7 l/h a u mužov 22,1 l/h, s 39 % interindividuálnou variabilitou u pacientov s MF. Klírens u pacientov s PV bol 12,7 l/h, s 42 %
interindividuálnou variabilitou a nebol zaznamenaný žiaden zjavný vzťah medzi perorálnym klírensom a pohlavím, pacientovým vekom alebo rasou, na základe farmakokinetického hodnotenia populácie pacientov s PV.
Pediatrická populácia
Bezpečnosť a účinnosť Jakavi sa u detí a dospievajúcich nesledovala (pozri časť 5.1 „Pediatrická populácia“).
Poškodenie funkcie obličiek
Funkcia obličiek sa hodnotila štúdiami úpravy diéty pri ochorení obličiek (MDRD) a kretinínu v moči. Po jednorazovej dávke ruxolitinibu 25 mg bola jeho expozícia podobná u jedincov s rôznymi stupňami
poškodenia obličiek s tými, ktorí mali normálnu funkciu obličiek. Avšak plazmatické hodnoty AUC
metabolitov ruxolitinibu mali tendenciu stúpať so závažnosťou poškodenia obličiek a najvýraznejšie zvýšenie bolo u jedincov s ťažkým poškodením obličiek. Nie je zrejmé, či zvýšená expozícia
metabolitu predstavuje bezpečnostné riziko. Odporúča sa upraviť dávku u pacientov s ťažkým
poškodením obličiek a v konečnom štádiu renálneho ochorenia (pozri časť 4.2). Podávanie iba v dňoch dialýzy znižuje expozíciu metabolitu ale zároveň aj farmakodynamický účinok, najmä počas dní medzi dialýzami.
Poškodenie funkcie pečene
U pacientov s rôznym stupňom poškodenia pečene, sa priemerná AUC ruxolitinibu po jednorazovej dávke 25 mg zvýšila u pacientov s miernym poškodením pečene o 87 %, so stredne ťažkým o 28 %
a s ťažkým o 65 %, v porovnaní s pacientmi s normálnou funkciou pečene. Nebol jasný vzťah medzi AUC a stupňom poškodenia funkcie pečene klasifikovanom podľa Childovho-Pughovho skóre. U pacientov s poškodením funkcie pečene sa konečný polčas eliminácie v porovnaní so zdravými dobrovoľníkmi predĺžil (4,1-5,0 hodín oproti 2,8 hodiny). U pacientov s poškodením funkcie pečene sa odporúča zníženie dávky približne o 50 % (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Ruxolitinib sa hodnotil na základe štúdií farmakologickej bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, reprodukčnej toxicity a karcinogenity. K cieľovým orgánom, súvisiacim s farmakologickým účinkom ruxolitinibu, v štúdiách toxicity po opakovanom podaní, patrili kostná dreň, periférna krv a lymfatické tkanivo. U psov sa pozorovali infekcie, vo všeobecnosti spojené s imunosupresiou. Nepriaznivé zníženie krvného tlaku spolu so zvýšením pulzu sa pozorovalo u psov v telemetrickej štúdii. Nepriaznivý pokles minútového objemu sa pozoroval v respiračných štúdiách u potkanov. Hranice (na základe neviazaného Cmax) na hladine bez nežiaducich účinkov v štúdiách so
psami a potkanmi boli 15,7-krát vyššie a 10,4-krát vyššie, ako maximálna dávka 25 mg dvakrát denne,
odporúčaná u ľudí. V hodnotení neurofarmakologických vplyvov ruxolitinibu sa nezaznamenali žiadne účinky.
Ruxolitinib v štúdiách u zvierat znižoval váhu plodu a zvyšoval poimpalntačné straty. U králikov a potkanov nebol zaznamenaný dôkaz o teratogénnom účinku. Rozsah expozície bol však v porovnaní s najvyššou klinickou dávkou nízky, výsledok má preto len obmedzený význam pre ľudí. Nepozorovali sa účinky na fertilitu. V štúdiách prenatálneho a postnatálneho vývoja sa pozorovala
mierne predĺžená gestačná doba, znížený počet miest implantácie a menší počet narodených šteniat. U šteniat sa pozorovala znížená priemerná počiatočná telesná hmotnosť a krátke obdobie nárastu zníženej priemernej telesnej hmotnosti. U dojčiacich potkanov sa ruxolitinib a/alebo jeho metabolity vylučovali do mlieka v koncentrácii 13-krát vyššej ako materské plazmatické koncentrácie. Ruxolitinib nebol mutagénny alebo klastogénny. Ruxolitinib nebol karcinogénny v modeloch Tg.rasH2 transgénnych myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Mikrokryštalická celulóza
Magnéziumstearát
Koloidný bezvodý oxid kremičitý
Sodná soľ karboxymetylcelulózy (typ A) Povidón
Hydroxypropylcelulóza
Monohydrát laktózy
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
6.5 Druh obalu a obsah balenia
PVC/PCTFE/hliníkové blistrové balenia obsahujúce 14 alebo 56 tabliet alebo spoločné balenia obsahujúce 168 (3 balenia po 56) tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLO(ČÍSLA)EU/1/12/773/014-016
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE23.08.2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o
bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUJakavi 15 mg tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tableta obsahuje 15 mg ruxolitinibu (ako fosfátu).
Pomocnálátkasoznámymúčinkom:
Každá tableta obsahuje 214,35 mg monohydrátu laktózy.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATableta.
Oválne zaoblené biele až takmer biele tablety približne 15,0 x 7,0 mm s pretlačeným nápisom „NVR“
na jednej strane a „L15“ na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieMyelofibróza(MF)Jakavi je indikované na liečbu splenomegálie súvisiacej s ochorením alebo jeho príznakov u dospelých pacientov s primárnou myelofibrózou (známou aj ako chronická idiopatická myelofibróza), myelofibrózou po polycytémii vera alebo myelofibrózou po esenciálnej trombocytémii.
Polycytémiavera(PV)Jakavi je indikované na liečbu dospelých pacientov s polycytémiou vera s rezistenciou alebo intoleranciou na hydroxyureu.
4.2 Dávkovanie a spôsob podávaniaLiečbu Jakavi má začať iba lekár, ktorý má skúsenosti s podávaním protinádorových liekov. Pred začatím liečby Jakavi sa musí vykonať kompletné vyšetrenie krvného obrazu, vrátane
diferenciálneho počtu bielych krviniek.
Pokým nie sú dávky Jakavi stabilizované, je potrebné každé 2-4 týždne monitorovať a kompletne vyšetriť krvný obraz, vrátane diferenciálneho počtu bielych krviniek, a následne podľa klinickej indikácie (pozri časť 4.4).
DávkovaniePočiatočná dávkaOdporúčaná počiatočná dávka Jakavi pri myelofibróze je 15 mg dvakrát denne u pacientov s počtom krvných doštičiek medzi 100 000/mm3 a 200 000/mm3 a 20 mg dvakrát denne u pacientov s počtom krvných doštičiek >200 000/mm3. Odporúčaná počiatočná dávka Jakavi pri polycytémii vera je 10 mg
podávaných perorálne dvakrát denne.
K dispozícii sú obmedzené informácie pre odporúčanie počiatočnej dávky u pacientov s počtom krvných doštičiek medzi 50 000/mm3 a <100 000/mm3. Maximálna odporúčaná počiatočná dávka u týchto pacientov je 5 mg dvakrát denne a dávka sa musí titrovať opatrne.
Úpravy dávky
Dávky možno titrovať na základe bezpečnosti a účinnosti. Liečba sa musí prerušiť pri počte krvných doštičiek nižšom ako 50 000/mm3 alebo pri absolútnom počte neutrofilov nižšom ako 500/mm3. Liečba pri PV sa má prerušiť aj vtedy, ak sú hodnoty hemoglobínu nižšie ako 8 g/dl. Po obnovení hodnôt krvného obrazu nad tieto hodnoty možno pokračovať v dávkovaní 5 mg dvakrát denne
a postupne zvyšovať na základe podrobného monitorovania kompletného krvného obrazu vrátane diferenciálneho počtu bielych krviniek.
Ak počet krvných doštičiek klesne pod 100 000/mm3, je potrebné zvážiť zníženie dávky, aby sa predišlo prerušeniu užívania pre trombocytopéniu. Zníženie dávky pri PV sa má zvážiť aj vtedy, ak hemoglobín klesne pod 12 g/dl a odporúča sa, ak klesne pod 10 g/dl.
Ak sa účinnosť považuje za nedostatočnú a hodnoty krvného obrazu sú adekvátne, dávky možno zvýšiť o maximálne 5 mg dvakrát denne, najviac po maximálnu dávku 25 mg dvakrát denne.
Počiatočná dávka sa nesmie zvyšovať počas prvých štyroch týždňov liečby a potom nie častejšie ako v
2-týždňových intervaloch.
Maximálna dávka Jakavi je 25 mg dvakrát denne.
Úprava dávky pri súč asnom podávaní silných CYP3A4 inhibítorov alebo flukonazolu
Ak sa Jakavi podáva so silnými CYP3A4 inhibítormi alebo s duálnymi inhibítormi enzýmov CYP2C9 a CYP3A4 (napr. flukonazol), jednotlivá dávka sa má znížiť o približne 50 % a má sa podávať dvakrát denne (pozri časť 4.5).
Počas podávania silných CYP3A4 inhibítorov alebo duálnych inhibítorov enzýmov CYP2C9 a CYP3A4 sa odporúča častejšie monitorovanie (napr. dvakrát do týždňa) hematologických parametrov a klinických prejavov a príznakov nežiaducich reakcií na liek spojených s Jakavi.
Osobité populácie
Poškodenie funkcie obličiek
U pacientov s miernym alebo stredne závažným poškodením funkcie obličiek sa nevyžaduje úprava dávky.
U pacientov so závažným poškodením funkcie obličiek (klírens kreatinínu menej ako 30 ml/min) sa má počiatočná dávka odporúčaná na základe počtu krvných doštičiek pre pacientov s MF znížiť približne o 50 % a má sa podávať dvakrát denne. Odporúčaná počiatočná dávka pre pacientov s PV so závažným poškodením funkcie obličiek je 5 mg dvakrát denne. Počas liečby Jakavi je potrebné pacientov dôsledne monitorovať so zameraním na bezpečnosť a účinnosť.
Dostupné sú iba obmedzené údaje, ktoré by pomohli určiť najvhodnejšie dávkovanie u pacientov s renálnym ochorením v konečnom štádiu (ESRD) na hemodialýze. Farmakokinetické/farmakodynamické simulácie na základe dostupných údajov u tejto populácie naznačujú, že počiatočná jednorazová dávka pre pacientov s MF s ESRD na hemodialýze je 15-20 mg alebo dve dávky po 10 mg s 12 hodinovým odstupom, podané po dialýze avšak ešte v ten istý deň. Jednorazová dávka 15 mg sa odporúča pre pacientov s MF s počtom doštičiek medzi 100 000/mm3 a
200 000/mm3. Jednorazová dávka 20 mg alebo dve dávky po 10 mg s 12 hodinovým odstupom sa odporúčajú pre pacientov s MF s počtom doštičiek >200 000/mm3. Následné dávky (jednorazové
podanie alebo dve dávky po 10 mg podané s 12 hodinovým odstupom) sa majú podať iba v dňoch hemodialýzy, po každej dialýze.
Odporúčaná počiatočná dávka pre pacientov s PV s ESRD na hemodialýze je jedna 10 mg dávka alebo dve dávky po 5 mg podané s 12 hodinovým odstupom, ktoré majú byť podané po dialýze a iba v deň hemodialýzy. Tieto dávkovacie odporúčania sú založené na simuláciach a akákoľvek úprava dávky pri ESRD sa má robiť po dôslednom sledovaní bezpečnosti a účinnosti jednotlivo u každého pacienta. Nie sú dostupné žiadne údaje o dávkovaní u pacientov, ktorí podstupujú peritoneálnu dialýzu alebo kontinuálnu venóznu hemofiltráciu (pozri časť 5.2).
Poškodenie funkcie pečene
U pacientov s akýmkoľvek poškodením funkcie pečene sa má počiatočná dávka odporúčaná na základe počtu krvných doštičiek znížiť o približne 50 % a má sa podávať dvakrát denne. Následné dávky sa majú upraviť na základe dôsledného monitorovania bezpečnosti a účinnosti. U pacientov s poškodením funkcie pečene diagnostikovaným počas užívania Jakavi sa má sledovať kompletný krvný obraz, vrátane diferenciálneho počtu bielych krviniek každý až každý druhý týždeň počas prvých 6 týždňov od začatia liečby Jakavi a podľa klinickej indikácie aj následne po stabilizácii funkcie pečene a krvného obrazu. Dávku Jakavi možno titrovať aby sa znížilo riziko cytopénie.
Starší ľudia (≥65 rokov)
U starších ľudí sa neodporúčajú žiadne dodatočné úpravy dávky.
Pediatrická populácia
Bezpečnosť a účinnosť Jakavi u detí vo veku do 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje (pozri časť 5.1).
Ukonč eni e l ieč by
V liečbe možno pokračovať dovtedy, kým podiel prínos-riziko zostáva pozitívny. Pokiaľ však do
6 mesiacov od začatia liečby nedôjde k zmenšeniu objemu sleziny alebo zlepšeniu príznakov, má byť liečba ukončená.
U pacientov, ktorí vykazujú istý stupeň klinického zlepšenia sa odporúča ukončiť liečbu ruxolitinibom v prípade, že u nich naďalej dochádza k zväčšeniu dĺžky sleziny o 40 % v porovnaní s východiskovou hodnotou (približné ekvivalentné 25 % zväčšeniu objemu sleziny) a nie je už u nich viac
preukázateľné zlepšenie príznakov súvisiacich s ochorením.
Spôsobpodávania
Jakavi sa užíva perorálne s jedlom alebo bez jedla.
Pri vynechaní dávky pacient nemá užiť dávku navyše, ale má užiť obvyklú predpísanú najbližšiu dávku.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Gravidita a laktácia.
4.4 Osobitné upozornenia a opatrenia pri používaní
Myelosupresia
Liečba s Jakavi môže spôsobiť hematologické nežiaduce reakcie na liek vrátane trombocytopénie, anémie a neutropénie. Pred začatím liečby s Jakavi je potrebné urobiť kompletný krvný obraz, vrátane diferenciálneho počtu bielych krviniek. Liečba sa má ukončiť u pacientov s počtom krvných doštičiek nižším ako 50 000/mm3 alebo s absolútnym počtom neutrofilov nižším ako 500/mm3 (pozri časť 4.2).
U pacientov s nízkym počtom krvných doštičiek (<200 000/mm3) na začiatku liečby sa pozorovala vyššia pravdepodobnosť vzniku trombocytopénie počas liečby.
Trombocytopénia je spravidla reverzibilná a obvykle sa upraví po znížení dávky alebo po dočasnom vynechaní Jakavi (pozri časti 4.2 a 4.8). Napriek tomu, môže byť potrebná transfúzia krvných doštičiek, ak je to klinicky indikované.
Pacienti, u ktorých sa vyvinie anémia, môžu potrebovať transfúziu krvi. U týchto pacientov, možno zvážiť aj úpravu dávkovania alebo prerušenie liečby.
Pacienti s hladinou hemoglobínu pod 10,0 g/dl na začiatku liečby majú vyššie riziko nábehu na pokles hladiny hemoglobínu pod 8,0 g/dl počas liečby v porovnaní s pacientmi s vyššími východiskovými hladinami hemoglobínu (79,3 % oproti 30,1 %). U pacientov s hladinou hemoglobínu pod 10,0 g/dl sa odporúča častejšie monitorovanie hematologických parametrov a klinických prejavov a príznakov nežiaducich reakcií na liek spojených s Jakavi.
Neutropénia (absolútny počet neutrofilov <500) bola spravidla reverzibilná a upravila sa po dočasnom vynechaní Jakavi (pozri časti 4.2 a 4.8).
Kompletný krvný obraz sa má sledovať podľa klinickej indikácie a dávka sa má upravovať podľa potreby (pozri časti 4.2 a 4.8).
Infekcie
U pacientov sa má vyhodnotiť riziko vzniku závažných bakteriálnych, mykobakteriálnych, hubových a vírusových infekcií. U pacientov užívajúcich Jakavi na liečbu MF bola hlásená tuberkulóza. Pred začiatkom liečby majú byť pacienti v súlade s národnými odporúčaniami vyšetrení na prítomnosť aktívnej a inaktívnej („latentnej“) tuberkulózy. Vyšetrenie môže zahŕňať anamnézu, možný kontakt
s tuberkulózou a/alebo vhodný skríning ako napr. röntgen pľúc, tuberkulínový test a/alebo imunologické vyšetrenie IGRA (interferon-gamma release assay). Predpisujúci lekár musí mať na
pamäti riziko falošne negatívnych výsledkov tuberkulínových kožných testov, najmä u ťažko chorých
alebo imunokompromitovaných pacientov. Liečba s Jakavi sa nesmie začať, pokým závažné aktívne infekcie neodoznejú. U pacientov, ktorí dostávajú Jakavi, musia lekári pozorne sledovať prejavy a príznaky infekcií a okamžite začať vhodnú liečbu (pozri časť 4.8).
U pacientov s chronickou infekciou HBV užívajúcich Jakavi bolo hlásené zvýšenie vírusovej záťaže hepatitídy B (titer HBV-DNA) s alebo bez súvisiacich zvýšení alanínaminotransferázy a aspartátaminotransferázy. Účinok Jakavi na virálnu replikáciu u pacientov s chronickou infekciou HBV nie je známy. Pacienti s chronickou infekciou HBV sa majú liečiť a monitorovať v súlade s klinickými smernicami.
Herpeszoster
Lekári musia pacientov poučiť o včasných prejavoch a príznakoch herpes zoster s odporučením, aby čo najskôr vyhľadali lekársku pomoc.
Progresívnamultifokálnaleukoencefalopatia
Pri liečbe MF s Jakavi bola hlásená progresívna multifokálna leukoencefalopatia (PML). Lekári musia byť mimoriadne ostražití na príznaky naznačujúce PML, ktoré pacienti nemusia spozorovať (napr. kognitívne, neurologické alebo psychiatrické príznaky alebo prejavy). U pacientov je potrebné
sledovať akékoľvek nové alebo zhoršujúce sa uvedené príznaky alebo prejavy a v prípade ich výskytu je potrebné zvážiť konzultáciu s neurológom a vhodné diagnostické opatrenia pre PML. Ak je
podozrenie na PML, daľšie podávanie musí byť pozastavené, až kým nie je PML vylúčená.
Nemelanómovýkožnýnádor
U pacientov liečených ruxolitinibom boli hlásené nemelanómové kožné nádory (NMSC), vrátane bazocelulárneho karcinómu, spinocelulárneho karcinómu a karcinómu Merkelových buniek. Väčšina týchto pacientov v minulosti podstúpila dlhodobú liečbu hydroxyureou a už sa u nich vyskytla NMSC alebo premalígne kožné lézie. Kauzálny vzťah k liečbe ruxolitinibom nebol stanovený. U pacientov so zvýšeným rizikom kožných nádorov sa odporúčajú pravidelné vyšetrenia kože.
O
sobité
populácie
Poškodenie funkcie obličiek
U pacientov so závažným poškodením funkcie obličiek je potrebné znížiť počiatočnú dávku Jakavi. U
pacientov s MF na hemodialýze s renálnym ochorením v konečnom štádiu sa má počiatočná dávka stanoviť na základe počtu krvných doštičiek (pozri časť 4.2). Následné dávky (jednorazová dávka
20 mg alebo dve dávky po 10 mg podané s 12 hodinovým odstupom u pacientov s MF; jednorazová dávka 10 mg alebo dve dávky po 5 mg podané s 12 hodinovým odstupom u pacientov s PV) sa majú podať iba v dňoch hemodialýzy, po každej dialýze. Ďalšia úprava dávky sa má robiť pri dôslednom
sledovaní bezpečnosti a účinnosť (pozri časti 4.2 a 5.2).
Poškodenie funkcie pečene
U pacientov s poškodením funkcie pečene sa má počiatočná dávka znížiť približne o 50 %. Následné úpravy dávky majú vychádzať z bezpečnosti a účinnosti lieku (pozri časti 4.2 a 5.2).
Interakcie
Ak sa Jakavi podáva súčasne so silnými CYP3A4 inhibítormi alebo s duálnymi inhibítormi enzýmov CYP3A4 a CYP2C9 (napr. flukonazol), jednotlivá dávka Jakavi sa má znížiť približne o 50 % a má sa podávať dvakrát denne (k frekvencii monitorovania pozri časti 4.2 a 4.5).
Súčasné podávanie cytoredukčnej liečby alebo hemopoetických rastových faktorov spolu s Jakavi nebolo skúmané. Bezpečnosť a účinnosť súčasného podávania nie je známa (pozri časť 4.5).
Príznakyzvysadenia
V dôsledku prerušenia alebo ukončenia podávania Jakavi sa približne v priebehu jedného týždňa môžu príznaky MF znovu objaviť. Boli prípady pacientov s ťažším priebehom po prerušení Jakavi, obzvlášť počas akútneho pridruženého ochorenia. Nie je jasné, či náhle prerušenie Jakavi prispelo k tomuto stavu. Pokiaľ nie je potrebné náhle prerušenie, odporúča sa zvážiť postupné znižovanie dávky Jakavi, aj keď prínos postupného znižovania nie je preukázaný.
Pomocnélátky
Jakavi obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Eliminácia ruxolitinibu je sprostredkovaná metabolizáciou katalyzovanou enzýmami CYP3A4 a
CYP2C9. Lieky inhibujúce uvedené enzými preto môžu zapríčiniť zvýšenú expozíciu ruxolitinibu.
Interakcievedúcekzníženiudávkyruxolitinibu
CYP3A4 inhibítory
Silné CYP3A4 inhibítory (ako sú, ale nie len, boceprevír, klaritromycín, indinavír, itrakonazol, ketokonazol, lopinavír/ritonavír, ritonavír, mibefradil, nefazodón, nelfinavír, posakonazol, sakvinavír,
telaprevír, telitromycín, vorikonazol)
U zdravých jedincov súčasné podávanie Jakavi (jednotlivá dávka 10 mg) so silným CYP3A4 inhibítorom ketokonazolom viedlo k zvýšeniu Cmax ruxolitinibu o 33 % a AUC o 91 %, v porovnaní so samotným ruxolitinibom. Polčas eliminácie sa pri súčasnom podávaní ketokonazolu predĺžil z 3,7 na
6,0 hodín.
Ak sa Jakavi podáva súčasne so silnými CYP3A4 inhibítormi, jednotlivá dávka sa má znížiť približne o 50 % a má sa podávať dvakrát denne. Pacienti majú byť starostlivo monitorovaní (napr. dvakrát do týždňa) so zameraním na cytopénie a dávku je potrebné titrovať na základe bezpečnosti a účinnosti (pozri časť 4.2).
D
uálne CYP2C9 a CYP3A4 inhibítory
Na základe modelových údajov in silico možno zvážiť 50 % úpravu dávkovania aj pri užívaní liekov, ktoré sú duálnymi inhibítormi enzýmov CYP2C9 a CYP3A4 (napr flukonazol).
Induktoryenzýmov
CYP3A4 induktory (ako sú, ale nie len, avasimib, karbamazepín, fenobarbital, fenytoín, rifabutín,
ri fampí n (ri f ampi cí n), ľubovní k bodkovaný (Hypericum per f oratum))
Pacienti majú byť starostlivo monitorovaní a dávku je potrebné titrovať na základe bezpečnosti a účinnosti (pozri časť 4.2).
U zdravých jedincov po podaní Jakavi (50 mg jednotlivá dávka) následne po silnom CYP3A4 induktore rifampicíne (denná dávka 600 mg počas 10 dní), bola AUC ruxolitinibu o 70 % nižšia ako po podaní samotného Jakavi. Expozícia aktívnym metabolitom ruxolitinibu nebola zmenená. Celková farmakodynamická aktivita ruxolitinibu bola podobná, naznačujúc tak minimálny vplyv indukcie CYP3A4 na farmakodynamiku. To však môže byť spojené s vysokou dávkou ruxolitinibu zapríčiňujúcou farmakodynamický účinok blízky Emax. U jednotlivých pacientov je možná potreba zvýšiť dávku ruxolitinibu v prípade začatia liečby silným enzýmovým induktorom.
Inéinterakcievyžadujúceobozretnosťsvplyvomnaruxolitinib
Mierne alebo stredne silné CYP3A4 inhibítory (ako sú, ale nie len, ciprofloxacín, erytromycín,amprenavír, atazanavír, diltiazém, cimetidín)
U zdravých jedincov súčasné podávanie ruxolitinibu (jednotlivá dávka 10 mg) s erytromycínom
500 mg dvakrát denne počas štyroch dní, viedlo k zvýšeniu Cmax ruxolitinibu o 8 % a AUC o 27 %, v porovnaní so samotným ruxolitinibom.
Keď sa ruxolitinib podáva súčasne s miernymi alebo stredne silnými CYP3A4 inhibítormi
(napr. erytromycín), nie je potrebná úprava dávky. Pri začatí liečby so stredne silnými CYP3A4
inhibítormi je však potrebné pacientov dôsledne monitorovať so zameraním na cytopénie.
Vplyvruxolitinibunainélieky
Lie či vá pre nášané P-glykoproteínom a inými transportérmi
Ruxolitinib môže inhibovať P-glykoproteín a proteín rezistencie rakoviny prsníka (BCRP) v čreve. To môže mať za následok zvýšenú expozíciu substrátom týchto transportérov, ako napr. dabigatran
etexilát, cyklosporín, rosuvastatín a možno digoxín. Odporúča sa terapeutické monitorovanie lieku
alebo klinické monitorovanie ovplyvneného liečiva.
Je možné, že inhibícia P-glykoproteínu a proteínu rezistencie rakoviny prsníka môže byť minimalizovaná, ak sa oddelí čas spoločného podania na čo najdlhší časový interval.
Hematopoetické rastové faktory
Súčasné užívanie hematopoetických rastových faktorov a Jakavi sa nesledovalo. Nie je známe, či inhibícia Janusových kináz (JAK) prostredníctvom Jakavi znižuje účinnosť hematopoetických rastových faktorov alebo či hematopoetické rastové faktory ovplyvňujú účinnosť Jakavi (pozri časť 4.4).
Cyt oredukt ívne l i ečby
Súčasné používanie cytoreduktívnej liečby a Jakavi sa nesledovalo. Bezpečnosť a účinnosť súčasného podávania nie je známa (pozri časť 4.4).
Skúšanie na zdravých jedincoch naznačilo, že ruxolitinib nemal inhibičný účinok na metabolizmus perorálne podaného midazolamu, substrátu CYP3A4. Pri súčasnom podaní substrátov CYP3A4 s Jakavi sa preto nepredpokladá zvýšenie ich expozície. Ďalšie skúšanie na zdravých jedincoch nazančilo, že Jakavi nemá vplyv na farmakokinetiku perorálnych kontraceptív obsahujúcich etinylestradiol a levonorgestrel. Nie je preto predpoklad, že by bol účinok uvedenej kombinácie kontraceptív oslabený spoločným podávaním s ruxolitinibom.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
a
k
ontraceptíva
u
ž
ien
Nie sú k dispozícii žiadne údaje o použití Jakavi u gravidných žien.
Štúdie na zvieratách preukázali že ruxolitinib je embryotoxický a fetotoxický. Teratogenicita sa nezaznamenala u potkanov a králikov. Rozsah expozície bol však v porovnaní s najvyššou klinickou dávkou nízky, výsledok má preto len obmedzený význam pre ľudí (pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe. Ako preventívne opatrenie, je použitie Jakavi počas gravidity kontraindikované (pozri časť 4.3). Ženy vo fertilnom veku musia počas liečby s Jakavi používať účinnú antikoncepciu. V prípade, že by došlo ku gravidite počas liečby s Jakavi, je potrebné individuálne prehodnotiť prínos/riziko s dôkladnou konzultáciou ohľadom možných rizík pre plod (pozri časť 5.3).
Laktácia
Jakavi sa nemá užívať počas laktácie (pozri časť 4.3), preto musí byť pri začatí liečby dojčenie ukončené. Nie je známe, či sa ruxolitinib a/alebo jeho metabolity vylučujú do ľudského mlieka. Riziko
u dojčiat nemôže byť vylúčené. Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali
vylučovanie ruxolitinibu a jeho metabolitov do mlieka (pozri časť 5.3).
Fertilita
Nie sú dostupné údaje o účinkoch ruxolitinibu na fertilitu u ľudí. V štúdiách na zvieratách sa nepozorovali účinky na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Jakavi nemá žiaden alebo má zanedbateľný sedatívny účinok. Pacienti, u ktorých sa po užití Jakavi vyskytnú závraty, nemajú viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
Celkovo bolo hodnotenie bezpečnosti založené na 855 pacientoch (s MF alebo PV) užívajúcich Jakavi v klinických skúšaniach 2. a 3. fázy.
Myelofibróza
V dvoch pivotných štúdiách, COMFORT-I a COMFORT-II, bol vo fáze randomizácie medián trvania expozície lieku Jakavi 10,8 mesiaca (rozpätie 0,3 až 23,5 mesiaca). Väčšina pacientov (68,4 %) sa liečila minimálne 9 mesiacov. U 111 pacientov z 301 (36,9 %) bol počet krvných doštičiek na začiatku liečenia medzi 100 000/mm3 a 200 000/mm3 a u 190 (63,1 %) bol počet krvných doštičiek
>200 000/mm3.
V týchto klinických skúšaniach bolo u 11,3 % pacientov pozorované ukončenie liečby kvôli nežiaducim účinkom, bez ohľadu na kauzalitu.
Najčastejšie hlásenými nežiaducimi reakciami boli trombocytopénia a anémia.
K hematologickým nežiaducim reakciám na liek (všetkých stupňov škály spoločných kritérií pre názvoslovie nežiaducich udalostí [CTCAE]) patrila anémia (82,4 %), trombocytopénia (69,8 %) a neutropénia (16,6 %).
Anémia, trombocytopénia a neutropénia sú reakcie úmerné dávke.
Tri najčastejšie iné ako hematologické nežiaduce reakcie na liek boli tvorenie podliatin (21,3 %), závraty (15,3 %) a bolesť hlavy (14,0 %).
Tri najčastejšie iné ako hematologické abnormálne laboratórne nálezy boli zvýšená
alanín aminotrasferáza (27,2 %), zvýšená aspartát aminotransferáza (19,9 %) a hypercholesterolémia (16,9 %).V 3.fáze klinických skúšaní u MF nebola pozorovaná ani cholesterolémia stupňa 3 alebo 4 podľa CTCAE, zvýšená aspartátaminotransferáza ani zvýšená alanínaminotransferáza stupňa 4 podľa CTCAE.
Dlhodobá bezpečnosť: Podľa očakávania sa pri predĺžení doby ďalšieho sledovania zvýšila kumulatívna frekvencia niektorých nežiaducich udalostí pri hodnotení bezpečnostných údajov z 3- ročného obdobia (medián trvania expozície u pacientov počiatočne randomizovaných na ruxolitinib v štúdiách COMFORT-I a COMFORT-II bol 33,2 mesiacov) od 457 pacientov s myelofibrózou liečených ruxolitinibom počas obdobia randomizácie a extenzie dvoch pivotných skúšaní 3 fázy. Toto hodnotenie zahŕňa údaje od pacientov, ktorí boli počiatočne randomizovaní na ruxolitinib (N=301)
a od pacientov, ktorí dostali ruxolitinib po prekrížení z kontrolných liečebných ramien (N=156). Podľa týchto aktualizovaných údajov sa ukončenie liečby z dôvodu nežiaducich účinkov pozorovalo u 17,1% pacientov liečených ruxolitinibom.
Polycytémia vera
Bezpečnosť Jakavi bola hodnotená u 110 pacientov s PV v otvorenom, randomizovanom, kontrolovanom klinickom skúšaní 3.fázy s názvom RESPONSE. Nižšie uvedené nežiaduce reakcie na
liek sa vzťahujú na počiatočnú fázu klinického skúšania (až do 32. týždňa) s rovnakou expozíciou ruxolitinibu a najlepšej dostupnej liečbe (Best Available Therapy - BAT), zodpovedajúcou mediánu trvania expozície lieku Jakavi v dĺžke 7,8 mesiaca. Priemerný vek pacientov užívajúcich Jakavi bol
približne 60 rokov.
Ukončenie liečby kvôli nežiaducim účinkom, bez ohľadu na príčiny, bolo pozorované u 3,6 %
pacientov liečených Jakavi a u 1,8 % pacientov liečených najlepšou dostupnou liečbou.
Hematologické nežiaduce reakcie (akéhokoľvek stupňa podľa CTCAE) zahrňovali anémiu (43,6 %) a trombocytopéniu (24,5 %). Anémia alebo trombocytopénia stupňa 3 a 4 podľa CTCAE bola hlásená
u 1,8 % alebo 5.5 % pacientov, v uvedenom poradí.
Tromi najčastejšími nehematologickými nežiaducimi reakciami boli závrat (15,5 %), zápcha (8,2 %) a herpes zoster (6,4 %).
Tromi najčastejšími nehematologickými laboratórnymi abnormalitami (akéhokoľvek stupňa podľa CTCAE) boli hypercholesterolémia (30,0 %), zvýšená alanínaminotransferáza (22,7 %) a zvýšená aspartátaminotransferáza (20,9 %). Všetky boli stupňa 1 a 2 podľa CTCAE okrem jedného prípadu zvýšenej alanínaminotransferázy stupňa 3 podľa CTCAE.
Dlhodobá bezpečnosť: Medián trvania expozície lieku Jakavi u pacientov bol 18,6 mesiaca (rozpätie
0,3 až 35,9 mesiaca). Pri dlhšej expozícii sa počet nežiaducich účinkov zvýšil, ale neobjavili sa žiadne nové zistenia v oblasti bezpečnosti. Po úprave na túto expozíciu bola miera výskytu nežiaducich účinkov obvykle porovnateľná s mierou ich výskytu pozorovanou v priebehu počiatočnej fázy klinického skúšania.
Tabuľkovýprehľadnežiaducichreakciízklinickýchštúdií
V programe klinických štúdií sa závažnosť nežiaducich reakcií hodnotila podľa kritérií CTCAE, kde stupeň 1 = mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4= život ohrozujúce.
Nežiaduce reakcie nahlásené z klinických štúdií (Tabuľka 1) sú zoradené podľa triedy orgánových systémov MedDRA. V každej triede orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, pričom najčastejšie sú uvedené ako prvé. Frekvencie sú definované podľa nasledovnej konvencie: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000).
Tabuľka 1 Kategória frevencie výskytu nežiaducich reakcií na liek hlásených v štúdiách fázy 3 (COMFORT-I, COMFORT-II, RESPONSE)
N
ežiaduca reakcia na liek Kategória frekvencie
výskytu u pacientov s
MF
K
ategória frekvencie výskytu u
pacientov s PV
Infekcie a nákazy
Infekcie močového traktua,d Veľmi časté Časté
Herpes zostera,d Časté Časté
Tuberkulózae Menej časté -
Poruchy krvi a lymfatického systémub,d
Anémiab - -
CTCAEc stupeň 4 (<6,5g/dl)
CTCAEc stupeň 3 (<8,0 –
6,5g/dl)
Akýkoľvek CTCAE
stupeňc
Trombocytopéniab CTCAEc stupeň 4 (<25 000/mm3)
CTCAEc stupeň 3 (50 000
– 25 000/mm3)
Akýkoľvek CTCAEc
stupeň
Neutropéniab CTCAEc stupeň 4 (<500/mm3)
CTCAEc stupeň 3 (<1 000
– 500/mm3) Akýkoľvek CTCAEc
stupeň
Krvácanie (akékoľvek krvácanie vrátane intrakraniálneho, gastrointestinálne krvácanie, podliatiny a iné krvácanie)
Veľmi časté Menej časté Veľmi časté Menej časté Veľmi časté Veľmi časté
Časté Menej časté
Časté Časté
Veľmi časté Veľmi časté
Časté - Časté - Veľmi časté -
Veľmi časté Veľmi časté
Intrakraniálne krvácanie Časté -
Gastrointestinálne krvácanie
Časté -
Podliatiny Veľmi časté Veľmi časté
Iné krvácanie (vrátane epistaxy, postprocedurálnej hemorágie a hematúrie)
Časté Veľmi časté
P
oruchy metabolizmu a
výživy
Nárast hmotnostia Veľmi časté Časté
Hypercholesterolémiab
CTCAEc stupeň 1 a 2
Hypertriglyceridémiab
CTCAEc stupeň 1
Poruchy nervového systému
Veľmi časté Veľmi časté
- Veľmi časté
Závratya Veľmi časté Veľmi časté
Bolesť hlavya Veľmi časté -
Poruchy gastrointestinálneho traktu
Plynatosťa Časté -
Zápchaa - Časté
Poruchy pečene a žlčových
ciest Zvýšená alanínaminotransferázab
CTCAEc stupeň 3 (> 5x –
20 x ULN) Akýkoľvek CTCAEc stupeň
Zvýšená aspartátaminotransferázab
Akýkoľvek CTCAEc
stupeň
Poruchy ciev
Časté Menej časté
Veľmi časté Veľmi časté
Veľmi časté Veľmi časté
Hypertenziaa - Veľmi časté
a Frekvencia vychádza z údajov o nežiaducich účinkoch.
- Subjekt, u ktorého sa vyskytlo viacero nežiaducich reakcií na liek (ADR) je v danej
ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
b Frekvencia vychádza z laboratórnych hodnôt.
- Subjekt, u ktorého sa vyskytlo viacero ADR je v danej ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
c Všeobecné terminologické kritériá pre nežiaduce účinky (CTCAE) verzia 3.0; stupeň 1 =
mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4 = život ohrozujúce
d Tieto ADR sú bližšie popísané v texte.
e Frekvencia vychádza z počtu všetkých pacientov exponovaných ruxolitinibu v klinických skúšaniach (N=4755)
Po ukončení liečby sa u pacientov s MF môžu vrátiť príznaky MF ako napr. únava, bolesť kostí,
horúčka, pruritus, nočné potenie, symptomatická splenomegália a pokles telesnej hmotnosti.
V klinických štúdiách s MF sa celkové skóre symptómov pre symptómy MF postupne vrátilo na počiatočné hodnoty do 7 dní od ukončenia liečby (pozri časť 4.4).
PopisvybranýchnežiaducichreakciínaliekAnémiaV klinických štúdiách fázy III s MF, bol medián nástupu anémie CTCAE stupňa 2 alebo vyššieho
1,5 mesiaca. Jeden pacient (0,3 %) ukončil liečbu v dôsledku anémie.
U pacientov užívajúcich Jakavi dosiahli priemerné poklesy hemoglobínu minimum približne o
10 g/liter pod počiatočnými hodnotami po 8 až 12 týždňoch liečby a potom postupne narastali, kým nedosiahli nový stabilný stav, ktorý bol približne o 5 g/liter pod počiatočnými hodnotami. Takýto
vzorec sa u pacientov pozoroval bez ohľadu na to, či počas liečby dostali transfúziu.
V randomizovanej, placebom kontrolovanej štúdii COMFORT-I 60,6 % pacientov s MF užívajúcich Jakavi a 37,7 % pacientov s MF užívajúcich placebo dostalo počas randomizovanej liečby transfúziu s červenými krvinkami. V štúdii COMFORT-II bol podiel transfúzií s červenými krvinkami 53,4 %
v ramene s Jakavi a 41,1 % v ramene s najlepšou dostupnou liečbou.
Vo fáze randomizácie v pivotných štúdiách bola anémia menej častá u pacientov s PV ako u pacientov s MF (43,6 % oproti 82,4 %). V PV populácii boli prípady CTCAE stupňa 3 a 4 hlásené u 1,8 % pacientov, kým frekvencia u pacientov s MF bola 42,56 %.
Trombocytopénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula trombocytopénia stupňa 3 alebo 4, bol medián jej nástupu približne 8 týždňov. Trombocytopénia bola vo všeobecnosti reverzibilná a ustúpila po znížení dávky alebo po prerušení liečby. Medián času potrebného na návrat počtu krvných doštičiek nad 50 000/mm3 bol 14 dní. Počas randomizovaného obdobia boli transfúzie s krvnými doštičkami podané 4,7 % pacientov užívajúcich Jakavi a 4,0 % pacientov užívajúcich
kontrolné režimy. K ukončeniu liečby v dôsledku trombocytopénie došlo u 0,7 % pacientov užívajúcich Jakavi a 0,9 % pacientov užívajúcich kontrolné režimy. U pacientov, ktorí mali pred začiatkom liečby s Jakavi počet krvných doštičiek od 100 000/mm3 do 200 000/mm3, bola vyššia frekvencia trombocytopénie stupňa 3 alebo 4 v porovnaní s pacientmi, ktorý mali počet krvných doštičiek >200 000/mm3 (64,2 % verzus 38,5 %).
Vo fáze randomizácie v pivotných štúdiách bol podiel pacientov, u ktorých sa vyskytla trombocytopénia nižší u pacientov s PV (24,5 %), ako u pacientov s MF (69,8 %). Frekvencia výskytu závažnej trombocytopénie (t.j. CTCAE stupňa 3 a 4) bola nižšia u pacientov s PV (5,5 %) ako u pacientov s MF (11,6 %).
Neutropénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula neutropénia stupňa 3 alebo 4, bol medián jej nástupu 12 týždňov. Počas randomizovaného obdobia sa pozastavenie liečby alebo zníženie dávky v dôsledku neutropénie hlásilo u 1,0 % pacientov a 0,3 % pacientov ukončilo liečbu pre neutropéniu.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV bola neutropénia hlásená u dvoch pacientov
(1,8 %), z ktorých sa u jedného pacienta vyskytla neutropénia CTCAE stupňa 4.
Krvácanie
V pivotných štúdiách fázy III s MF boli prípady krvácania (vrátane intrakraniálneho
a gastrointestinálneho krvácania, podliatin a iných prípadov krvácania) hlásené u 32,6 % pacientov liečených Jakavi a u 23,2 % pacientov liečených referenčnou liečbou (placebom alebo najlepšou dostupnou liečbou). Frekvencia udalostí stupňa 3-4 bola u pacientov liečených Jakavi alebo referenčnou liečbou podobná (4,7 % verzus 3,1 %). Väčšina pacientov s príhodou krvácania počas liečby hlásila podliatiny (65,3 %). Podliatiny boli častejšie hlásené u pacientov užívajúcich Jakavi
v porovnaní s referenčnou liečbou (21,3 % verzus 11,6 %). Intrakraniálne krvácanie bolo hlásené u
1 % pacientov exponovaných Jakavi a u 0,9 % pacientov exponovaných referenčnej liečbe. Gastrointestinálne krvácanie bolo hlásené u 5,0 % pacientov exponovaných Jakavi oproti 3,1 % pacientov exponovaných referenčnej liečbe. Iné prípady krvácania (vrátane epistaxy,
post-procedurálnej hemorágie a hematúrie) boli hlásené u 13,3 % pacientov liečených Jakavi a u
10,3 % pacientov liečených referenčnou liečbou.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV boli hlásené prípady krvácania (vrátane intrakraniálneho a gastrointestináneho krvácania, tvorby podliatin a iného krvácania) u 20 % pacientov liečených Jakavi a 15,3 % pacientov, ktorí dostávali najlepšiu dostupnú liečbu. Podliatiny boli hlásené
s rovnakou frekvenciou v liečebných skupinách s Jakavi aj BAT (10,9 % oproti 8,1 %). U pacientov užívajúcich Jakavi neboli hlásené žiadne prípady intrakraniálneho ani gastrointestinálneho krvácania.
U jedného pacienta liečeného Jakavi sa vyskytol prípad krvácania stupňa 3 (postprocedurálne krvácanie); nebolo hlásené žiadne krvácanie stupňa 4. Prípady iného krvácania (vrátane takých prípadov ako epistaxa, postprocedurálne krvácanie, gingiválne krvácanie) boli hlásené u 11,8 % pacientov liečených Jakavi a u 6,3 % pacientov liečených najlepšou dostupnou liečbou.
InfekcieV pivotných štúdiách fázy III s MF boli hlásené infekcie močových ciest stupňa 3 alebo 4 u 1,0 % pacientov, herpes zoster u 4,3 % a tuberkulóza u 1,0 %. V klinickom skúšaní fázy III bola sepsa hlásená u 3,0% pacientov. Predĺžené sledovanie pacientov liečených ruxolitinibom nepreukázalo
s postupom času žiadny zvýšený trend v rozsahu sepsy.
Vo fáze randomizácie v pivotnej štúdii bola u pacientov s PV hlásená jedna infekcia močového traktu (0,9 %) CTCAE stupňa 3 a žiadna infekcia močového traktu stupňa 4. Miera výskytu herpes zoster bola mierne vyššia u pacientov s PV (6,4 %) ako u pacientov s MF (4,0 %). U pacientov s PV sa vyskytlo jedno hlásenie postherpetickej neuralgie CTCAE stupňa 3.
Zvýšený systolický krvný tlakV pivotných štúdiách fázy III s MF bolo hlásené minimálne počas jednej vizity zvýšenie systolického
krvného tlaku oproti východiskovej hodnote o 20 mmHg alebo viac u 31,5 % pacientov v porovnaní s
19,5 % výskytom u pacientov na kontrolnej liečbe. V skúšaní COMFORT-I (pacienti s MF) bol oproti východiskovej hodnote pri Jakavi priemerne zvýšený systolický krvný tlak o 0-2 mmHg v porovnaní
s poklesom o 2-5 mmHg v ramene s placebom. V skúšaní COMFORT-II boli medzi priemernými hodnotami MF pacientov liečených ruxolitinibom alebo kontrolnou liečbou len malé rozdiely.
Vo fáze randomizácie v pivotných štúdiách bol u pacientov s PV sa priemerný systolický krvný tlak v liečebnej skupine s Jakavi zvýšil o 0,65 mmHg, kým v skupine s BAT klesol o 2 mmHg.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieNie je známe antidotum pri predávkovaní s Jakavi. Jednorazové dávky do 200 mg boli podané s prijateľnou akútnou znášanlivosťou. Vyššie ako odporúčané, opakované dávky sú spojené so zvýšenou myelosupresiou vrátane leukopénie, anémie a trombocytopénie. Je potrebné podať vhodnú podpornú liečbu.
Nepredpokladá sa, že hemodialýza zvyšuje elimináciu ruxolitinibu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE18
MechanizmusúčinkuRuxolitinib je selektívny inhibítor Janusových kináz (JAK), JAK1 a JAK2 (IC50 hodnoty 3,3 nM pre enzýmy JAK1 a 2,8 nM pre enzýmy JAK2). Tieto sprostredkujú signalizáciu mnohých cytokínov a rastových faktorov, ktoré sú dôležité pre hemopoézu a imunitné funkcie.
Myelofibróza a polycytémia vera sú myeloproliferatívne nádorové ochorenia, o ktorých je známe že súvisia s poruchou signalizácie JAK1 a JAK2. Predpokladá sa že k základom týchto porúch patrí vysoká hladina cirkulujúcich cytokínov, ktoré aktivujú JAK-STAT dráhou, mutácie pridávajúce
funkcie ako napr. JAK2V617F a utlmenie negatívnych regulačných mechanizmov. Pacienti s MF vykazujú poruchu JAK signalizácie bez ohľadu na stav JAK2V617F mutácie. U >95 % pacientov s PV sa vyskytujú aktivujúce mutácie JAK2 (V617F alebo exón 12).
Ruxolitinib inhibuje JAK-STAT signalizáciu a bunkovú proliferáciu bunkových modelov hematologických malignít závislých od cytokínov ako aj Ba/F3 buniek nezávislých od cytokínov expresiou JAK2V617F mutovaného proteínu, s hodnotou IC50 v rozsahu 80-320 nM.
Farmakodynamickéúčinky
Ruxolitinib inhibuje cytokínmi indukovanú STAT3 fosforyláciu v celej krvi od zdravých dobrovoľníkov, pacientov s MF a pacientov s PV . Ruxolitinib vyvolal maximálnu inhibíciu STAT3
fosforylácie 2 hodiny po podaní dávky, ktorá sa vrátila do normálu do 8 hodín u zdravých
dobrovoľníkov aj u pacientov s MF, čo naznačuje že nedochádza k akumulácii pôvodných ani aktívnych metabolitov.
Zvýšené východiskové zápalové markery súvisiace so základnými konštitučnými príznakmi ako TNFα, IL-6 a CRP u osôb s MF po liečbe ruxolitinibom poklesli. Počas liečby ruxolitinibom sa u pacientov s MF nevytvorila odolnosť voči jeho farmakodynamickým účinkom. Rovnako aj u pacientov s PV sa vyskytli zvýšené východiskové zápalové markery a tieto markery sa po liečbe ruxolitinibom znížili.
V podrobnej QT štúdii so zdravými dobrovoľníkmi nič nepoukazovalo na predlžujúci účinok ruxolitinibu na QT/QTc v jednotlivých dávkach až po supraterapeutickú dávku 200 mg, čo naznačuje že ruxolitinib nemá účinok na kardiálnu repolarizáciu.
Klinickáúčinnosťabezpečnosť
Myelofibróza
U pacientov s MF (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie) sa realizovali 2 randomizované štúdie fázy III (COMFORT-I a COMFORT-II). V oboch štúdiách mali pacienti hmatateľnú splenomegáliu najmenej
5 cm pod rebrovým oblúkom a rizikovú kategóriu strednú-2 alebo vysokú, podľa IWG kritérií
(International Working Group Consensus Criteria). Počiatočná dávka Jakavi sa stanovila na základe počtu krvných doštičiek.
COMFORT-I bola dvojito zaslepená, randomizovaná, placebom kontrolovaná štúdia s 309 pacientmi, ktorí nereagovali na dostupnú liečbu alebo táto pre nich nebola vhodná. Primárny cieľový ukazovateľ účinnosti bol definovaný ako podiel pacientov, u ktorých došlo k ≥35 % zmenšeniu objemu sleziny v
24. týždni v porovnaní s východiskom, merané zobrazovaním magnetickou rezonanciou (MRI) alebo počítačovou tomografiou (CT).
Sekundárne cieľové ukazovatele účinnosti zahrňovali trvanie ≥35 % zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou, podiel pacientov s ≥50 % znížením skóre celkových príznakov, zmeny v celkových skóre príznakov v porovnaní s východiskovou hodnotou v 24. týždni hodnotené pomocou modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) v2.0 denníka, a celkové prežívanie.
COMFORT-II bola otvorená, randomizovaná štúdia s 219 pacientmi. Pacienti boli randomizovaní 2:1 do skupín s Jakavi a s najlepšou dostupnou liečbou. V ramene s najlepšou dostupnou liečbou 47 % pacientov dostalo hydroxyureu a 16 % pacientov glukokortikoidy. Primárny cieľový ukazovateľ účinnosti bol podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 48. týždni v porovnaní s východiskovou hodnotou, merané MRI alebo CT.
Sekundárne cieľové ukazovatele zahrňovali podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 24. týždni v porovnaní s východiskovou hodnotou a trvanie ≥35% zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou.
Základné demografické charakteristiky a charakteristika ochorenia pacientov boli porovnateľné pre obe ramená v štúdiách COMFORT-I a COMFORT-II.
Tabuľka 2 Percento pacientov s ≥35 % zmenšením sleziny v 24. týždni v porovnaní
s východiskovou hodnotou v COMFORT-I a v 48. týždni v COMFORT-II (ITT)
COMFORT-I COMFORT-II
Jakavi
(N=155)
Placebo
(N=153)
Jakavi
(N=144)
Najlepšia dostupná liečba (N=72)
Čas Týždeň 24 Týždeň 48
Počet (%) pacientov s objemom sleziny zmenšeným o ≥35 %
95 % intervaly spoľahlivosti
65 (41,9) 1 (0,7) 41 (28,5) 0
34,1; 50,1 0; 3,6 21,3; 36,6 0,0; 5,0
p-hodnota <0,0001 <0,0001
Významne väčší podiel pacientov v skupine s Jakavi dosiahol ≥35 % zmenšenie objemu sleziny
(tabuľka 2) v porovnaní s východiskovou hodnotou bez ohľadu na prítomnosť alebo neprítomnosť
JAK2V617F mutácie alebo subtyp ochorenia (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie).
Tabuľka 3 Percento pacientov s ≥35 % zmenšením sleziny v porovnaní s východiskovou
hodnotou podľa stavu mutácie JAK (súbor bezpečnosti)
COMFORT-I COMFORT-II
Jakavi Placebo Jakavi Najlepšia dostupná liečba
Stav mutácie JAK
Pozitívny (N=113) n (%)
Negatívny
(N=40)
n (%)
Pozitívny (N=121) n (%)
Negatívny
(N=27)
n (%)
Pozitívny (N=110) n (%)
Negatívny
(N=35)
n (%)
Pozitívny
(N=49)
n (%)
Negatívny
(N=20)
n (%)
Počet (%)
pacientov
s objemom sleziny zmenšeným o ≥35 %
54 (47,8)
11 (27,5)
1 (0,8)
0 36 (32,7)
5 0 0 (14,3)
Časový bod Po 24 týždňoch Po 48 týždňoch
Pravdepodobnosť udržania odpovede sleziny (≥35 % redukcia) na Jakavi v priebehu najmenej
24 týždňov bola 89 % v štúdii COMFORT I a 87 % v štúdii COMFORT II; v štúdii COMFORT II
udržalo odpoveď sleziny 52 % počas aspoň 48 týždňov.
V štúdii COMFORT I 45,9 % subjektov v skupine s Jakavi dosiahlo v 24. týždni ≥50 % zlepšenie celkového skóre príznakov v porovnaní s východiskovou hodnotou (zisťované podľa denníka MFSAF v2.0), v porovnaní s 5,3 % v skupine s placebom (p<0,0001 pomocou chi-kvadrát testu). Priemerná zmena v celkovom zdravotnom stave v 24. týždni zisťovaná podľa EORTC QLQ C30 bola +12,3 u Jakavi a -3,4 u placeba (p<0,0001).
V štúdii COMFORT-I bola úmrtnosť pacientov randomizovaných do skupiny s ruxolitinibom po priemernej dobe sledovania 34,3 mesiacov 27,1 %, v porovnaní s 35,1 % u pacientov randomizovaných na placebo; HR 0,687; 95 % IS 0,459-1,029; p=0,0668.
V štúdii COMFORT-II bola úmrtnosť pacientov randomizovaných na ruxolitinib po priemernej dobe sledovania 34,7 mesiacov 19,9 %, v porovnaní s 30,1 % u pacientov randomizovaných na najlepšiu dostupnú liečbu (BAT); HR 0,48; 95 % IS 0,28-0,85; p=0,009. V oboch štúdiách bola nižšia úmrtnosť zaznamenaná v skupine s ruxolitinibom, čo bolo spôsobené predovšetkým výsledkami získanými
v podskupinách po polycytémii vera a po esenciálnej trombocytémii.
Polycytémia vera
Randomizované, otvorené, aktívne kontrolované klinické skúšanie 3. fázy (RESPONSE) bolo vykonané u 222 pacientov s PV s rezistenciou alebo intoleranciou na hydroxyureu definované podľa
publikovaných kritérií medzinárodnej pracovnej skupiny European LeukemiaNet (ELN).
110 pacientov bolo randomizovaných do skupiny s ruxolitinibom a 112 pacientov do skupiny s BAT. Počiatočná dávka Jakavi bola 10 mg dvakrát denne. Dávky sa potom u jednotlivých pacientov upravili podľa znášanlivosti a účinnosti na maximálnu dávku 25 mg dvakrát denne. BAT vybral skúšajúci
lekár pre každého pacienta individuálne a zahŕňala hydroxyureu (59,5 %), interferón/pegylovaný interferón (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorovanie (15,3 %).
Vstupné demografické údaje a charakteristika ochorenia boli v dvoch liečebných skupinách porovnateľné. Priemerný vek bol 60 rokov (rozpätie 33 až 90 rokov). Pacienti v skupine
s ruxolitinibom mali diagnózu PV v priemere 8,2 roka a predtým užívali v priemere približne 3 roky hydroxyureu. Väčšina pacientov (>80 %) podstúpila v priebehu posledných 24 týždňov pred
skríningom najmenej dve flebotómie. Porovnávacie údaje týkajúce sa dlhodobého prežitia a výskytu komplikácií ochorenia nie sú dostupné.
Primárnym zloženým cieľovým ukazovateľom bolo percento pacientov, ktorí dosiahli aj absenciu indikácie flebotómie (kontrola HCT) a ≥35 % zmenšenie objemu sleziny v porovnaní so vstupnými hodnotami v 32. týždni. Indikácia flebotómie bola definovaná ako potvrdené HCT s hodnotou >45 %, t.j. aspoň o 3 percentuálne body vyššie ako HCT získané pri vstupnom vyšetrení alebo potvrdené HCT s hodnotou >48 %, podľa toho, ktorá hodnota bola nižšia. Kľúčové sekundárne cieľové ukazovatele zahrňovali percento pacientov, ktorí dosiahli primárny cieľový ukazovateľ a v 48. týždni ostali bez progresie, ako aj percento pacientov, ktorí dosiahli úplnú hematologickú remisiu v 32. týždni.'
Štúdia splnila svoj primárny cieľ a vyššie percento pacientov v skupine s Jakavi dosiahlo primárny zložený cieľový ukazovateľ aj každú z jeho jednotlivých zložiek. Podstatne viac pacientov liečených Jakavi (20,9 %) dosiahlo primárnu odpoveď (p<0,0001) v porovnaní s BAT (0,9 %). Kompenzácia hematokritu sa dosiahla u 60 % pacientov v skupine s Jakavi v porovnaní s 19,6 % v skupine s BAT a
≥35 % zmenšenie objemu sleziny sa dosiahlo u 38,2 % pacientov v skupine s Jakavi v porovnaní s 0,9 % v skupine s BAT (obr. č. 1). Deväťdesiatštyri (83,9 %) pacientov randomizovaných do skupiny s BAT prešlo na liečbu ruxolitinibom v 32. týždni alebo neskôr, čím bolo obmedzené porovnanie týchto dvoch skupín po 32. týždni.
Splnené boli aj oba kľúčové sekundárne cieľové ukazovatele. Percento pacientov, ktorí dosiahli úplnú hematologickú remisiu, bolo 23,6 % s Jakavi v porovnaní s 8,9 % s BAT (p=0,0028) a percento pacientov, ktorí dosiahli dlhotrvajúcu primárnu odpoveď v 48. týždni, bolo 19,1 % s Jakavi a 0,9 %
s BAT (p<0,0001).
O
brázok č. 1 Pacienti dosahujúci primárny cieľový ukazovateľ a zložky primárneho cieľového ukazovateľa v 32. týždni
Hodnota P: < ,0001
70 Pomer š a ncí (Ruxolitinib/BAT)
a 95% IS:
60 28,64 (4,50, 12,06)
50
40 38
30
21
20
10
Jednotlivé zložky primá rnej
odpovede v 32.týždni
60
20
.RUX
.BAT
1 1
0
Pri má rny zl ožený
ci eľový uka zova teľ v
32.týždni
≥35% zníženi e obj emu
s l ezi ny
Kompenzá ci a hema tokri tu bez fl ebotómi e
Príznaková záťaž bola hodnotená pomocou MPN-SAF celkového skóre príznakov (total symptom
score - TSS) elektronického denníka pacienta, ktorý obsahoval 14 otázok. V 32. týždni dosiahlo 49 %
pacientov liečených ruxolitinibom ≥50 % zníženie TSS-14 a 64 % pacientov ≥50 % zníženie TSS-5, v porovnaní s iba 5 % a 11 % pacientov na BAT.
Vnímanie liečebného prínosu bolo zisťované v dotazníku Celkový dojem pacienta zo zmeny (Patient Global Impression of Change - PGIC). 66 % pacientov liečených ruxolitinibom, v porovnaní s 19 % liečenými BAT, hlásilo zlepšenie už štyri týždne po začiatku liečby. Zlepšenie vo vnímaní liečebného prínosu bolo tiež vyššie u pacientov liečených ruxolitinibom v 32. týždni (78 % oproti 33 %).
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Jakavi vo všetkých podskupinách pediatrickej populácie v liečbe MF (informácie o použití v pediatrickej
populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Ruxolitinib je látka I. triedy Biofarmaceutického klasifikačného systému (BCS), s vysokou permeabilitou, vysokou rozpustnosťou a rýchlou disolúciou. V klinických štúdiách sa ruxolitinib
rýchlo absorboval po perorálnom podaní, maximálnu plazmatickú koncentráciu (Cmax) dosiahol asi po
1 hodine po podaní dávky. Na základe humánnej štúdie hmotnostnej bilancie sa zistilo, že perorálna
absorpcia ruxolitinibu, v podobe ruxolitinibu alebo metabolitov vzniknutých v priebehu presystémovej eliminácie je 95 % alebo vyššia. Priemerné hodnoty Cmax a celkovej expozície (AUC) ruxolitinibu sa priamoúmerne zvýšili po jednotlivej dávke v rozsahu 5-200 mg. Nezistili sa klinicky relevantné zmeny farmakokinetiky ruxolitinibu po podaní s jedlom s vysokým obsahom tuku. Priemerná Cmax sa mierne znížila (24 %), kým priemerná AUC zostala takmer nezmenená (4 % vzostup) po podaní s jedlom s vysokým obsahom tuku.
Distribúcia
Priemerný objem distribúcie v rovnovážnom stave je u pacientov s MF aj s PV približne 75 litrov. Pri klinicky relevantných koncentráciách ruxolitinibu je väzba na plazmatické bielkoviny približne 97 %,
prevažne na albumín. Celotelové autoradiografické štúdie na potkanoch ukázali, že ruxolinitib
nepenetruje cez krvno-mozgovú bariéru.
B
i
otransformácia
Ruxolitinib je prevažne metabolizovaný enzýmom CYP3A4 (>50 %) s čiastočnou účasťou CYP2C9. Materská zlúčenina je hlavným prvkom v ľudskej plazme, pričom predstavuje približne 60 % materiálu súvisiaceho s liekom v obehu. V plazme sú prítomné dva hlavné a aktívne metabolity, ktoré reprezentujú 25 % a 11 % materskej AUC. Tieto metabolity majú polovicu až pätinu pôvodnej farmakologickej aktivity súvisiacej s JAK. Celkové množstvo všetkých aktívnych metabolitov prispieva 18 % k celkovej farmakodynamike ruxolitinibu. Podľa in vitro štúdií, ruxolitinib v klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 alebo CYP3A4 a nie je účinný induktor CYP1A2, CYP2B6 alebo CYP3A4. In vitro údaje naznačujú, že ruxolitinib môže byť inhibítor P-gp a BCRP.
Eliminácia
Ruxolitinib sa prevažne eliminuje metabolizmom. Priemerný polčas eliminácie ruxolitinibu sú približne 3 hodiny. U zdravých dobrovoľníkov, po jednorazovej perorálnej dávke rádioaktívne značeného [14C] ruxolitinibu došlo k eliminácii prevažne metabolizáciou, pričom 74 % rádioaktivity sa vylúčilo močom a 22 % stolicou. Na nezmenenú materskú zlúčeninu pripadlo menej ako 1 % celkovej vylúčenej rádioaktivity.
Linearita/nelinearita
Priama úmera v závislosti od dávky sa dokázala v štúdiách s jednorazovými aj viacnásobnými dávkami.
Osobitépopulácie
Vplyv veku, pohlavia a rasy
Na základe štúdií so zdravými subjektmi sa nepozorovali žiadne relevantné rozdiely vo farmakokinetike ruxolitinibu v závislosti od pohlavia a rasy. Na základe analýzy farmakokinetiky u
populácie pacientov s MF sa nepotvrdil vzťah medzi perorálnym klírensom a vekom alebo rasou pacienta. Odhadovaný perorálny klírens bol u žien 17,7 l/h a u mužov 22,1 l/h, s 39 % interindividuálnou variabilitou u pacientov s MF. Klírens u pacientov s PV bol 12,7 l/h, s 42 %
interindividuálnou variabilitou a nebol zaznamenaný žiaden zjavný vzťah medzi perorálnym klírensom a pohlavím, pacientovým vekom alebo rasou, na základe farmakokinetického hodnotenia populácie pacientov s PV.
Pediatrická populácia
Bezpečnosť a účinnosť Jakavi sa u detí a dospievajúcich nesledovala (pozri časť 5.1 „Pediatrická populácia“).
Poškodenie funkcie obličiek
Funkcia obličiek sa hodnotila štúdiami úpravy diéty pri ochorení obličiek (MDRD) a kretinínu v moči. Po jednorazovej dávke ruxolitinibu 25 mg bola jeho expozícia podobná u jedincov s rôznymi stupňami
poškodenia obličiek s tými, ktorí mali normálnu funkciu obličiek. Avšak plazmatické hodnoty AUC
metabolitov ruxolitinibu mali tendenciu stúpať so závažnosťou poškodenia obličiek a najvýraznejšie zvýšenie bolo u jedincov s ťažkým poškodením obličiek. Nie je zrejmé, či zvýšená expozícia
metabolitu predstavuje bezpečnostné riziko. Odporúča sa upraviť dávku u pacientov s ťažkým
poškodením obličiek a v konečnom štádiu renálneho ochorenia (pozri časť 4.2). Podávanie iba v dňoch dialýzy znižuje expozíciu metabolitu ale zároveň aj farmakodynamický účinok, najmä počas dní medzi dialýzami.
Poškodenie funkcie pečene
U pacientov s rôznym stupňom poškodenia pečene, sa priemerná AUC ruxolitinibu po jednorazovej dávke 25 mg zvýšila u pacientov s miernym poškodením pečene o 87 %, so stredne ťažkým o 28 %
a s ťažkým o 65 %, v porovnaní s pacientmi s normálnou funkciou pečene. Nebol jasný vzťah medzi AUC a stupňom poškodenia funkcie pečene klasifikovanom podľa Childovho-Pughovho skóre. U pacientov s poškodením funkcie pečene sa konečný polčas eliminácie v porovnaní so zdravými dobrovoľníkmi predĺžil (4,1-5,0 hodín oproti 2,8 hodiny). U pacientov s poškodením funkcie pečene sa odporúča zníženie dávky približne o 50 % (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Ruxolitinib sa hodnotil na základe štúdií farmakologickej bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, reprodukčnej toxicity a karcinogenity. K cieľovým orgánom, súvisiacim s farmakologickým účinkom ruxolitinibu, v štúdiách toxicity po opakovanom podaní, patrili kostná dreň, periférna krv a lymfatické tkanivo. U psov sa pozorovali infekcie, vo všeobecnosti spojené s imunosupresiou. Nepriaznivé zníženie krvného tlaku spolu so zvýšením pulzu sa pozorovalo u psov v telemetrickej štúdii. Nepriaznivý pokles minútového objemu sa pozoroval v respiračných štúdiách u potkanov. Hranice (na základe neviazaného Cmax) na hladine bez nežiaducich účinkov v štúdiách so
psami a potkanmi boli 15,7-krát vyššie a 10,4-krát vyššie, ako maximálna dávka 25 mg dvakrát denne,
odporúčaná u ľudí. V hodnotení neurofarmakologických vplyvov ruxolitinibu sa nezaznamenali žiadne účinky.
Ruxolitinib v štúdiách u zvierat znižoval váhu plodu a zvyšoval poimpalntačné straty. U králikov a potkanov nebol zaznamenaný dôkaz o teratogénnom účinku. Rozsah expozície bol však v porovnaní s najvyššou klinickou dávkou nízky, výsledok má preto len obmedzený význam pre ľudí. Nepozorovali sa účinky na fertilitu. V štúdiách prenatálneho a postnatálneho vývoja sa pozorovala
mierne predĺžená gestačná doba, znížený počet miest implantácie a menší počet narodených šteniat. U šteniat sa pozorovala znížená priemerná počiatočná telesná hmotnosť a krátke obdobie nárastu zníženej priemernej telesnej hmotnosti. U dojčiacich potkanov sa ruxolitinib a/alebo jeho metabolity vylučovali do mlieka v koncentrácii 13-krát vyššej ako materské plazmatické koncentrácie. Ruxolitinib nebol mutagénny alebo klastogénny. Ruxolitinib nebol karcinogénny v modeloch Tg.rasH2 transgénnych myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Mikrokryštalická celulóza
Magnéziumstearát
Koloidný bezvodý oxid kremičitý
Sodná soľ karboxymetylcelulózy (typ A) Povidón
Hydroxypropylcelulóza
Monohydrát laktózy
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
6.5 Druh obalu a obsah balenia
PVC/PCTFE/hliníkové blistrové balenia obsahujúce 14 alebo 56 tabliet alebo spoločné balenia obsahujúce 168 (3 balenia po 56) tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLO(ČÍSLA)EU/1/12/773/007-009
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE23.08.2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o
bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUJakavi 20 mg tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tableta obsahuje 20 mg ruxolitinibu (ako fosfátu).
Pomocnálátkasoznámymúčinkom:
Každá mg tableta obsahuje 285,80 mg monohydrátu laktózy.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMATableta.
Podlhovasté zaoblené biele až takmer biele tablety približne 16,5 x 7,4 mm s pretlačeným nápisom
„NVR“ na jednej strane a „L20“ na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieMyelofibróza(MF)Jakavi je indikované na liečbu splenomegálie súvisiacej s ochorením alebo jeho príznakov u dospelých pacientov s primárnou myelofibrózou (známou aj ako chronická idiopatická myelofibróza), myelofibrózou po polycytémii vera alebo myelofibrózou po esenciálnej trombocytémii.
Polycytémiavera(PV)Jakavi je indikované na liečbu dospelých pacientov s polycytémiou vera s rezistenciou alebo intoleranciou na hydroxyureu.
4.2 Dávkovanie a spôsob podávaniaLiečbu Jakavi má začať iba lekár, ktorý má skúsenosti s podávaním protinádorových liekov. Pred začatím liečby Jakavi sa musí vykonať kompletné vyšetrenie krvného obrazu, vrátane
diferenciálneho počtu bielych krviniek.
Pokým nie sú dávky Jakavi stabilizované, je potrebné každé 2-4 týždne monitorovať a kompletne vyšetriť krvný obraz, vrátane diferenciálneho počtu bielych krviniek, a následne podľa klinickej indikácie (pozri časť 4.4).
DávkovaniePočiatočná dávkaOdporúčaná počiatočná dávka Jakavi pri myelofibróze je 15 mg dvakrát denne u pacientov s počtom krvných doštičiek medzi 100 000/mm3 a 200 000/mm3 a 20 mg dvakrát denne u pacientov s počtom krvných doštičiek >200 000/mm3. Odporúčaná počiatočná dávka Jakavi pri polycytémii vera je 10 mg
podávaných perorálne dvakrát denne.
K dispozícii sú obmedzené informácie pre odporúčanie počiatočnej dávky u pacientov s počtom krvných doštičiek medzi 50 000/mm3 a <100 000/mm3. Maximálna odporúčaná počiatočná dávka u týchto pacientov je 5 mg dvakrát denne a dávka sa musí titrovať opatrne.
Úpravy dávky
Dávky možno titrovať na základe bezpečnosti a účinnosti. Liečba sa musí prerušiť pri počte krvných doštičiek nižšom ako 50 000/mm3 alebo pri absolútnom počte neutrofilov nižšom ako 500/mm3. Liečba pri PV sa má prerušiť aj vtedy, ak sú hodnoty hemoglobínu nižšie ako 8 g/dl. Po obnovení hodnôt krvného obrazu nad tieto hodnoty možno pokračovať v dávkovaní 5 mg dvakrát denne
a postupne zvyšovať na základe podrobného monitorovania kompletného krvného obrazu vrátane diferenciálneho počtu bielych krviniek.
Ak počet krvných doštičiek klesne pod 100 000/mm3, je potrebné zvážiť zníženie dávky, aby sa predišlo prerušeniu užívania pre trombocytopéniu. Zníženie dávky pri PV sa má zvážiť aj vtedy, ak hemoglobín klesne pod 12 g/dl a odporúča sa, ak klesne pod 10 g/dl.
Ak sa účinnosť považuje za nedostatočnú a hodnoty krvného obrazu sú adekvátne, dávky možno zvýšiť o maximálne 5 mg dvakrát denne, najviac po maximálnu dávku 25 mg dvakrát denne.
Počiatočná dávka sa nesmie zvyšovať počas prvých štyroch týždňov liečby a potom nie častejšie ako v
2-týždňových intervaloch.
Maximálna dávka Jakavi je 25 mg dvakrát denne.
Úprava dávky pri súč asnom podávaní silných CYP3A4 inhibítorov alebo flukonazolu
Ak sa Jakavi podáva so silnými CYP3A4 inhibítormi alebo s duálnymi inhibítormi enzýmov CYP2C9 a CYP3A4 (napr. flukonazol), jednotlivá dávka sa má znížiť o približne 50 % a má sa podávať dvakrát denne (pozri časť 4.5).
Počas podávania silných CYP3A4 inhibítorov alebo duálnych inhibítorov enzýmov CYP2C9 a CYP3A4 sa odporúča častejšie monitorovanie (napr. dvakrát do týždňa) hematologických parametrov a klinických prejavov a príznakov nežiaducich reakcií na liek spojených s Jakavi.
Osobité populácie
Poškodenie funkcie obličiek
U pacientov s miernym alebo stredne závažným poškodením funkcie obličiek sa nevyžaduje úprava dávky.
U pacientov so závažným poškodením funkcie obličiek (klírens kreatinínu menej ako 30 ml/min) sa má počiatočná dávka odporúčaná na základe počtu krvných doštičiek pre pacientov s MF znížiť približne o 50 % a má sa podávať dvakrát denne. Odporúčaná počiatočná dávka pre pacientov s PV so závažným poškodením funkcie obličiek je 5 mg dvakrát denne. Počas liečby Jakavi je potrebné pacientov dôsledne monitorovať so zameraním na bezpečnosť a účinnosť.
Dostupné sú iba obmedzené údaje, ktoré by pomohli určiť najvhodnejšie dávkovanie u pacientov s renálnym ochorením v konečnom štádiu (ESRD) na hemodialýze. Farmakokinetické/farmakodynamické simulácie na základe dostupných údajov u tejto populácie naznačujú, že počiatočná jednorazová dávka pre pacientov s MF s ESRD na hemodialýze je 15-20 mg alebo dve dávky po 10 mg s 12 hodinovým odstupom, podané po dialýze avšak ešte v ten istý deň. Jednorazová dávka 15 mg sa odporúča pre pacientov s MF s počtom doštičiek medzi 100 000/mm3 a
200 000/mm3. Jednorazová dávka 20 mg alebo dve dávky po 10 mg s 12 hodinovým odstupom sa odporúčajú pre pacientov s MF s počtom doštičiek >200 000/mm3. Následné dávky (jednorazové
podanie alebo dve dávky po 10 mg podané s 12 hodinovým odstupom) sa majú podať iba v dňoch hemodialýzy, po každej dialýze.
Odporúčaná počiatočná dávka pre pacientov s PV s ESRD na hemodialýze je jedna 10 mg dávka alebo dve dávky po 5 mg podané s 12 hodinovým odstupom, ktoré majú byť podané po dialýze a iba v deň hemodialýzy. Tieto dávkovacie odporúčania sú založené na simuláciach a akákoľvek úprava dávky pri ESRD sa má robiť po dôslednom sledovaní bezpečnosti a účinnosti jednotlivo u každého pacienta. Nie sú dostupné žiadne údaje o dávkovaní u pacientov, ktorí podstupujú peritoneálnu dialýzu alebo kontinuálnu venóznu hemofiltráciu (pozri časť 5.2).
Poškodenie funkcie pečene
U pacientov s akýmkoľvek poškodením funkcie pečene sa má počiatočná dávka odporúčaná na základe počtu krvných doštičiek znížiť o približne 50 % a má sa podávať dvakrát denne. Následné dávky sa majú upraviť na základe dôsledného monitorovania bezpečnosti a účinnosti. U pacientov s poškodením funkcie pečene diagnostikovaným počas užívania Jakavi sa má sledovať kompletný krvný obraz, vrátane diferenciálneho počtu bielych krviniek každý až každý druhý týždeň počas prvých 6 týždňov od začatia liečby Jakavi a podľa klinickej indikácie aj následne po stabilizácii funkcie pečene a krvného obrazu. Dávku Jakavi možno titrovať aby sa znížilo riziko cytopénie.
Starší ľudia (≥65 rokov)
U starších ľudí sa neodporúčajú žiadne dodatočné úpravy dávky.
Pediatrická populácia
Bezpečnosť a účinnosť Jakavi u detí vo veku do 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje (pozri časť 5.1).
Ukonč eni e l ieč by
V liečbe možno pokračovať dovtedy, kým podiel prínos-riziko zostáva pozitívny. Pokiaľ však do
6 mesiacov od začatia liečby nedôjde k zmenšeniu objemu sleziny alebo zlepšeniu príznakov, má byť liečba ukončená.
U pacientov, ktorí vykazujú istý stupeň klinického zlepšenia sa odporúča ukončiť liečbu ruxolitinibom v prípade, že u nich naďalej dochádza k zväčšeniu dĺžky sleziny o 40 % v porovnaní s východiskovou hodnotou (približné ekvivalentné 25 % zväčšeniu objemu sleziny) a nie je už u nich viac
preukázateľné zlepšenie príznakov súvisiacich s ochorením.
Spôsobpodávania
Jakavi sa užíva perorálne s jedlom alebo bez jedla.
Pri vynechaní dávky pacient nemá užiť dávku navyše, ale má užiť obvyklú predpísanú najbližšiu dávku.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Gravidita a laktácia.
4.4 Osobitné upozornenia a opatrenia pri používaní
Myelosupresia
Liečba s Jakavi môže spôsobiť hematologické nežiaduce reakcie na liek vrátane trombocytopénie, anémie a neutropénie. Pred začatím liečby s Jakavi je potrebné urobiť kompletný krvný obraz, vrátane diferenciálneho počtu bielych krviniek. Liečba sa má ukončiť u pacientov s počtom krvných doštičiek nižším ako 50 000/mm3 alebo s absolútnym počtom neutrofilov nižším ako 500/mm3 (pozri časť 4.2).
U pacientov s nízkym počtom krvných doštičiek (<200 000/mm3) na začiatku liečby sa pozorovala vyššia pravdepodobnosť vzniku trombocytopénie počas liečby.
Trombocytopénia je spravidla reverzibilná a obvykle sa upraví po znížení dávky alebo po dočasnom vynechaní Jakavi (pozri časti 4.2 a 4.8). Napriek tomu, môže byť potrebná transfúzia krvných doštičiek, ak je to klinicky indikované.
Pacienti, u ktorých sa vyvinie anémia, môžu potrebovať transfúziu krvi. U týchto pacientov, možno zvážiť aj úpravu dávkovania alebo prerušenie liečby.
Pacienti s hladinou hemoglobínu pod 10,0 g/dl na začiatku liečby majú vyššie riziko nábehu na pokles hladiny hemoglobínu pod 8,0 g/dl počas liečby v porovnaní s pacientmi s vyššími východiskovými hladinami hemoglobínu (79,3 % oproti 30,1 %). U pacientov s hladinou hemoglobínu pod 10,0 g/dl sa odporúča častejšie monitorovanie hematologických parametrov a klinických prejavov a príznakov nežiaducich reakcií na liek spojených s Jakavi.
Neutropénia (absolútny počet neutrofilov <500) bola spravidla reverzibilná a upravila sa po dočasnom vynechaní Jakavi (pozri časti 4.2 a 4.8).
Kompletný krvný obraz sa má sledovať podľa klinickej indikácie a dávka sa má upravovať podľa potreby (pozri časti 4.2 a 4.8).
Infekcie
U pacientov sa má vyhodnotiť riziko vzniku závažných bakteriálnych, mykobakteriálnych, hubových a vírusových infekcií. U pacientov užívajúcich Jakavi na liečbu MF bola hlásená tuberkulóza. Pred začiatkom liečby majú byť pacienti v súlade s národnými odporúčaniami vyšetrení na prítomnosť aktívnej a inaktívnej („latentnej“) tuberkulózy. Vyšetrenie môže zahŕňať anamnézu, možný kontakt
s tuberkulózou a/alebo vhodný skríning ako napr. röntgen pľúc, tuberkulínový test a/alebo imunologické vyšetrenie IGRA (interferon-gamma release assay). Predpisujúci lekár musí mať na
pamäti riziko falošne negatívnych výsledkov tuberkulínových kožných testov, najmä u ťažko chorých
alebo imunokompromitovaných pacientov. Liečba s Jakavi sa nesmie začať, pokým závažné aktívne infekcie neodoznejú. U pacientov, ktorí dostávajú Jakavi, musia lekári pozorne sledovať prejavy a príznaky infekcií a okamžite začať vhodnú liečbu (pozri časť 4.8).
U pacientov s chronickou infekciou HBV užívajúcich Jakavi bolo hlásené zvýšenie vírusovej záťaže hepatitídy B (titer HBV-DNA) s alebo bez súvisiacich zvýšení alanínaminotransferázy a aspartátaminotransferázy. Účinok Jakavi na virálnu replikáciu u pacientov s chronickou infekciou HBV nie je známy. Pacienti s chronickou infekciou HBV sa majú liečiť a monitorovať v súlade s klinickými smernicami.
Herpeszoster
Lekári musia pacientov poučiť o včasných prejavoch a príznakoch herpes zoster s odporučením, aby čo najskôr vyhľadali lekársku pomoc.
Progresívnamultifokálnaleukoencefalopatia
Pri liečbe MF s Jakavi bola hlásená progresívna multifokálna leukoencefalopatia (PML). Lekári musia byť mimoriadne ostražití na príznaky naznačujúce PML, ktoré pacienti nemusia spozorovať (napr. kognitívne, neurologické alebo psychiatrické príznaky alebo prejavy). U pacientov je potrebné
sledovať akékoľvek nové alebo zhoršujúce sa uvedené príznaky alebo prejavy a v prípade ich výskytu je potrebné zvážiť konzultáciu s neurológom a vhodné diagnostické opatrenia pre PML. Ak je
podozrenie na PML, daľšie podávanie musí byť pozastavené, až kým nie je PML vylúčená.
Nemelanómovýkožnýnádor
U pacientov liečených ruxolitinibom boli hlásené nemelanómové kožné nádory (NMSC), vrátane bazocelulárneho karcinómu, spinocelulárneho karcinómu a karcinómu Merkelových buniek. Väčšina týchto pacientov v minulosti podstúpila dlhodobú liečbu hydroxyureou a už sa u nich vyskytla NMSC alebo premalígne kožné lézie. Kauzálny vzťah k liečbe ruxolitinibom nebol stanovený. U pacientov so zvýšeným rizikom kožných nádorov sa odporúčajú pravidelné vyšetrenia kože.
O
sobité
populácie
Poškodenie funkcie obličiek
U pacientov so závažným poškodením funkcie obličiek je potrebné znížiť počiatočnú dávku Jakavi. U
pacientov s MF na hemodialýze s renálnym ochorením v konečnom štádiu sa má počiatočná dávka stanoviť na základe počtu krvných doštičiek (pozri časť 4.2). Následné dávky (jednorazová dávka
20 mg alebo dve dávky po 10 mg podané s 12 hodinovým odstupom u pacientov s MF; jednorazová dávka 10 mg alebo dve dávky po 5 mg podané s 12 hodinovým odstupom u pacientov s PV) sa majú podať iba v dňoch hemodialýzy, po každej dialýze. Ďalšia úprava dávky sa má robiť pri dôslednom
sledovaní bezpečnosti a účinnosť (pozri časti 4.2 a 5.2).
Poškodenie funkcie pečene
U pacientov s poškodením funkcie pečene sa má počiatočná dávka znížiť približne o 50 %. Následné úpravy dávky majú vychádzať z bezpečnosti a účinnosti lieku (pozri časti 4.2 a 5.2).
Interakcie
Ak sa Jakavi podáva súčasne so silnými CYP3A4 inhibítormi alebo s duálnymi inhibítormi enzýmov CYP3A4 a CYP2C9 (napr. flukonazol), jednotlivá dávka Jakavi sa má znížiť približne o 50 % a má sa podávať dvakrát denne (k frekvencii monitorovania pozri časti 4.2 a 4.5).
Súčasné podávanie cytoredukčnej liečby alebo hemopoetických rastových faktorov spolu s Jakavi nebolo skúmané. Bezpečnosť a účinnosť súčasného podávania nie je známa (pozri časť 4.5).
Príznakyzvysadenia
V dôsledku prerušenia alebo ukončenia podávania Jakavi sa približne v priebehu jedného týždňa môžu príznaky MF znovu objaviť. Boli prípady pacientov s ťažším priebehom po prerušení Jakavi, obzvlášť počas akútneho pridruženého ochorenia. Nie je jasné, či náhle prerušenie Jakavi prispelo k tomuto stavu. Pokiaľ nie je potrebné náhle prerušenie, odporúča sa zvážiť postupné znižovanie dávky Jakavi, aj keď prínos postupného znižovania nie je preukázaný.
Pomocnélátky
Jakavi obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Eliminácia ruxolitinibu je sprostredkovaná metabolizáciou katalyzovanou enzýmami CYP3A4 a
CYP2C9. Lieky inhibujúce uvedené enzými preto môžu zapríčiniť zvýšenú expozíciu ruxolitinibu.
Interakcievedúcekzníženiudávkyruxolitinibu
CYP3A4 inhibítory
Silné CYP3A4 inhibítory (ako sú, ale nie len, boceprevír, klaritromycín, indinavír, itrakonazol, ketokonazol, lopinavír/ritonavír, ritonavír, mibefradil, nefazodón, nelfinavír, posakonazol, sakvinavír,
telaprevír, telitromycín, vorikonazol)
U zdravých jedincov súčasné podávanie Jakavi (jednotlivá dávka 10 mg) so silným CYP3A4 inhibítorom ketokonazolom viedlo k zvýšeniu Cmax ruxolitinibu o 33 % a AUC o 91 %, v porovnaní so samotným ruxolitinibom. Polčas eliminácie sa pri súčasnom podávaní ketokonazolu predĺžil z 3,7 na
6,0 hodín.
Ak sa Jakavi podáva súčasne so silnými CYP3A4 inhibítormi, jednotlivá dávka sa má znížiť približne o 50 % a má sa podávať dvakrát denne. Pacienti majú byť starostlivo monitorovaní (napr. dvakrát do týždňa) so zameraním na cytopénie a dávku je potrebné titrovať na základe bezpečnosti a účinnosti (pozri časť 4.2).
D
uálne CYP2C9 a CYP3A4 inhibítory
Na základe modelových údajov in silico možno zvážiť 50 % úpravu dávkovania aj pri užívaní liekov, ktoré sú duálnymi inhibítormi enzýmov CYP2C9 a CYP3A4 (napr flukonazol).
Induktoryenzýmov
CYP3A4 induktory (ako sú, ale nie len, avasimib, karbamazepín, fenobarbital, fenytoín, rifabutín,
ri fampí n (ri f ampi cí n), ľubovní k bodkovaný (Hypericum per f oratum))
Pacienti majú byť starostlivo monitorovaní a dávku je potrebné titrovať na základe bezpečnosti a účinnosti (pozri časť 4.2).
U zdravých jedincov po podaní Jakavi (50 mg jednotlivá dávka) následne po silnom CYP3A4 induktore rifampicíne (denná dávka 600 mg počas 10 dní), bola AUC ruxolitinibu o 70 % nižšia ako po podaní samotného Jakavi. Expozícia aktívnym metabolitom ruxolitinibu nebola zmenená. Celková farmakodynamická aktivita ruxolitinibu bola podobná, naznačujúc tak minimálny vplyv indukcie CYP3A4 na farmakodynamiku. To však môže byť spojené s vysokou dávkou ruxolitinibu zapríčiňujúcou farmakodynamický účinok blízky Emax. U jednotlivých pacientov je možná potreba zvýšiť dávku ruxolitinibu v prípade začatia liečby silným enzýmovým induktorom.
Inéinterakcievyžadujúceobozretnosťsvplyvomnaruxolitinib
Mierne alebo stredne silné CYP3A4 inhibítory (ako sú, ale nie len, ciprofloxacín, erytromycín,amprenavír, atazanavír, diltiazém, cimetidín)
U zdravých jedincov súčasné podávanie ruxolitinibu (jednotlivá dávka 10 mg) s erytromycínom
500 mg dvakrát denne počas štyroch dní, viedlo k zvýšeniu Cmax ruxolitinibu o 8 % a AUC o 27 %, v porovnaní so samotným ruxolitinibom.
Keď sa ruxolitinib podáva súčasne s miernymi alebo stredne silnými CYP3A4 inhibítormi
(napr. erytromycín), nie je potrebná úprava dávky. Pri začatí liečby so stredne silnými CYP3A4
inhibítormi je však potrebné pacientov dôsledne monitorovať so zameraním na cytopénie.
Vplyvruxolitinibunainélieky
Lie či vá pre nášané P-glykoproteínom a inými transportérmi
Ruxolitinib môže inhibovať P-glykoproteín a proteín rezistencie rakoviny prsníka (BCRP) v čreve. To môže mať za následok zvýšenú expozíciu substrátom týchto transportérov, ako napr. dabigatran
etexilát, cyklosporín, rosuvastatín a možno digoxín. Odporúča sa terapeutické monitorovanie lieku
alebo klinické monitorovanie ovplyvneného liečiva.
Je možné, že inhibícia P-glykoproteínu a proteínu rezistencie rakoviny prsníka môže byť minimalizovaná, ak sa oddelí čas spoločného podania na čo najdlhší časový interval.
Hematopoetické rastové faktory
Súčasné užívanie hematopoetických rastových faktorov a Jakavi sa nesledovalo. Nie je známe, či inhibícia Janusových kináz (JAK) prostredníctvom Jakavi znižuje účinnosť hematopoetických rastových faktorov alebo či hematopoetické rastové faktory ovplyvňujú účinnosť Jakavi (pozri časť 4.4).
Cyt oredukt ívne l i ečby
Súčasné používanie cytoreduktívnej liečby a Jakavi sa nesledovalo. Bezpečnosť a účinnosť súčasného podávania nie je známa (pozri časť 4.4).
Skúšanie na zdravých jedincoch naznačilo, že ruxolitinib nemal inhibičný účinok na metabolizmus perorálne podaného midazolamu, substrátu CYP3A4. Pri súčasnom podaní substrátov CYP3A4 s Jakavi sa preto nepredpokladá zvýšenie ich expozície. Ďalšie skúšanie na zdravých jedincoch nazančilo, že Jakavi nemá vplyv na farmakokinetiku perorálnych kontraceptív obsahujúcich etinylestradiol a levonorgestrel. Nie je preto predpoklad, že by bol účinok uvedenej kombinácie kontraceptív oslabený spoločným podávaním s ruxolitinibom.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
a
k
ontraceptíva
u
ž
ien
Nie sú k dispozícii žiadne údaje o použití Jakavi u gravidných žien.
Štúdie na zvieratách preukázali že ruxolitinib je embryotoxický a fetotoxický. Teratogenicita sa nezaznamenala u potkanov a králikov. Rozsah expozície bol však v porovnaní s najvyššou klinickou dávkou nízky, výsledok má preto len obmedzený význam pre ľudí (pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe. Ako preventívne opatrenie, je použitie Jakavi počas gravidity kontraindikované (pozri časť 4.3). Ženy vo fertilnom veku musia počas liečby s Jakavi používať účinnú antikoncepciu. V prípade, že by došlo ku gravidite počas liečby s Jakavi, je potrebné individuálne prehodnotiť prínos/riziko s dôkladnou konzultáciou ohľadom možných rizík pre plod (pozri časť 5.3).
Laktácia
Jakavi sa nemá užívať počas laktácie (pozri časť 4.3), preto musí byť pri začatí liečby dojčenie ukončené. Nie je známe, či sa ruxolitinib a/alebo jeho metabolity vylučujú do ľudského mlieka. Riziko
u dojčiat nemôže byť vylúčené. Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali
vylučovanie ruxolitinibu a jeho metabolitov do mlieka (pozri časť 5.3).
Fertilita
Nie sú dostupné údaje o účinkoch ruxolitinibu na fertilitu u ľudí. V štúdiách na zvieratách sa nepozorovali účinky na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Jakavi nemá žiaden alebo má zanedbateľný sedatívny účinok. Pacienti, u ktorých sa po užití Jakavi vyskytnú závraty, nemajú viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
Celkovo bolo hodnotenie bezpečnosti založené na 855 pacientoch (s MF alebo PV) užívajúcich Jakavi v klinických skúšaniach 2. a 3. fázy.
Myelofibróza
V dvoch pivotných štúdiách, COMFORT-I a COMFORT-II, bol vo fáze randomizácie medián trvania expozície lieku Jakavi 10,8 mesiaca (rozpätie 0,3 až 23,5 mesiaca). Väčšina pacientov (68,4 %) sa liečila minimálne 9 mesiacov. U 111 pacientov z 301 (36,9 %) bol počet krvných doštičiek na začiatku liečenia medzi 100 000/mm3 a 200 000/mm3 a u 190 (63,1 %) bol počet krvných doštičiek
>200 000/mm3.
V týchto klinických skúšaniach bolo u 11,3 % pacientov pozorované ukončenie liečby kvôli nežiaducim účinkom, bez ohľadu na kauzalitu.
Najčastejšie hlásenými nežiaducimi reakciami boli trombocytopénia a anémia.
K hematologickým nežiaducim reakciám na liek (všetkých stupňov škály spoločných kritérií pre názvoslovie nežiaducich udalostí [CTCAE]) patrila anémia (82,4 %), trombocytopénia (69,8 %) a neutropénia (16,6 %).
Anémia, trombocytopénia a neutropénia sú reakcie úmerné dávke.
Tri najčastejšie iné ako hematologické nežiaduce reakcie na liek boli tvorenie podliatin (21,3 %), závraty (15,3 %) a bolesť hlavy (14,0 %).
Tri najčastejšie iné ako hematologické abnormálne laboratórne nálezy boli zvýšená
alanín aminotrasferáza (27,2 %), zvýšená aspartát aminotransferáza (19,9 %) a hypercholesterolémia
(16,9 %).V 3.fáze klinických skúšaní u MF nebola pozorovaná ani cholesterolémia stupňa 3 alebo 4 podľa CTCAE, zvýšená aspartátaminotransferáza ani zvýšená alanínaminotransferáza stupňa 4 podľa CTCAE.
Dlhodobá bezpečnosť: Podľa očakávania sa pri predĺžení doby ďalšieho sledovania zvýšila kumulatívna frekvencia niektorých nežiaducich udalostí pri hodnotení bezpečnostných údajov z 3- ročného obdobia (medián trvania expozície u pacientov počiatočne randomizovaných na ruxolitinib v štúdiách COMFORT-I a COMFORT-II bol 33,2 mesiacov) od 457 pacientov s myelofibrózou liečených ruxolitinibom počas obdobia randomizácie a extenzie dvoch pivotných skúšaní 3 fázy. Toto hodnotenie zahŕňa údaje od pacientov, ktorí boli počiatočne randomizovaní na ruxolitinib (N=301)
a od pacientov, ktorí dostali ruxolitinib po prekrížení z kontrolných liečebných ramien (N=156). Podľa týchto aktualizovaných údajov sa ukončenie liečby z dôvodu nežiaducich účinkov pozorovalo u 17,1%
pacientov liečených ruxolitinibom.
Polycytémia vera
Bezpečnosť Jakavi bola hodnotená u 110 pacientov s PV v otvorenom, randomizovanom, kontrolovanom klinickom skúšaní 3.fázy s názvom RESPONSE. Nižšie uvedené nežiaduce reakcie na
liek sa vzťahujú na počiatočnú fázu klinického skúšania (až do 32. týždňa) s rovnakou expozíciou ruxolitinibu a najlepšej dostupnej liečbe (Best Available Therapy - BAT), zodpovedajúcou mediánu trvania expozície lieku Jakavi v dĺžke 7,8 mesiaca. Priemerný vek pacientov užívajúcich Jakavi bol
približne 60 rokov.
Ukončenie liečby kvôli nežiaducim účinkom, bez ohľadu na príčiny, bolo pozorované u 3,6 %
pacientov liečených Jakavi a u 1,8 % pacientov liečených najlepšou dostupnou liečbou.
Hematologické nežiaduce reakcie (akéhokoľvek stupňa podľa CTCAE) zahrňovali anémiu (43,6 %) a trombocytopéniu (24,5 %). Anémia alebo trombocytopénia stupňa 3 a 4 podľa CTCAE bola hlásená
u 1,8 % alebo 5.5 % pacientov, v uvedenom poradí.
Tromi najčastejšími nehematologickými nežiaducimi reakciami boli závrat (15,5 %), zápcha (8,2 %) a herpes zoster (6,4 %).
Tromi najčastejšími nehematologickými laboratórnymi abnormalitami (akéhokoľvek stupňa podľa CTCAE) boli hypercholesterolémia (30,0 %), zvýšená alanínaminotransferáza (22,7 %) a zvýšená aspartátaminotransferáza (20,9 %). Všetky boli stupňa 1 a 2 podľa CTCAE okrem jedného prípadu zvýšenej alanínaminotransferázy stupňa 3 podľa CTCAE.
Dlhodobá bezpečnosť: Medián trvania expozície lieku Jakavi u pacientov bol 18,6 mesiaca (rozpätie
0,3 až 35,9 mesiaca). Pri dlhšej expozícii sa počet nežiaducich účinkov zvýšil, ale neobjavili sa žiadne nové zistenia v oblasti bezpečnosti. Po úprave na túto expozíciu bola miera výskytu nežiaducich
účinkov obvykle porovnateľná s mierou ich výskytu pozorovanou v priebehu počiatočnej fázy
klinického skúšania.
Tabuľkovýprehľadnežiaducichreakciízklinickýchštúdií
V programe klinických štúdií sa závažnosť nežiaducich reakcií hodnotila podľa kritérií CTCAE, kde stupeň 1 = mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4= život ohrozujúce.
Nežiaduce reakcie nahlásené z klinických štúdií (Tabuľka 1) sú zoradené podľa triedy orgánových systémov MedDRA. V každej triede orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, pričom najčastejšie sú uvedené ako prvé. Frekvencie sú definované podľa nasledovnej konvencie: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000).
Tabuľka 1 Kategória frevencie výskytu nežiaducich reakcií na liek hlásených v štúdiách fázy 3 (COMFORT-I, COMFORT-II, RESPONSE)
N
ežiaduca reakcia na liek Kategória frekvencie
výskytu u pacientov s
MF
K
ategória frekvencie výskytu u
pacientov s PV
Infekcie a nákazy
Infekcie močového traktua,d Veľmi časté Časté
Herpes zostera,d Časté Časté
Tuberkulózae Menej časté -
Poruchy krvi a lymfatického systémub,d
Anémiab - -
CTCAEc stupeň 4 (<6,5g/dl)
CTCAEc stupeň 3 (<8,0 –
6,5g/dl)
Akýkoľvek CTCAE
stupeňc
Trombocytopéniab CTCAEc stupeň 4 (<25 000/mm3)
CTCAEc stupeň 3 (50 000
– 25 000/mm3)
Akýkoľvek CTCAEc
stupeň
Neutropéniab CTCAEc stupeň 4 (<500/mm3)
CTCAEc stupeň 3 (<1 000
– 500/mm3) Akýkoľvek CTCAEc
stupeň
Krvácanie (akékoľvek krvácanie vrátane intrakraniálneho, gastrointestinálne krvácanie, podliatiny a iné krvácanie)
Veľmi časté Menej časté Veľmi časté Menej časté Veľmi časté Veľmi časté
Časté Menej časté
Časté Časté
Veľmi časté Veľmi časté
Časté - Časté - Veľmi časté -
Veľmi časté Veľmi časté
Intrakraniálne krvácanie Časté -
Gastrointestinálne krvácanie
Časté -
Podliatiny Veľmi časté Veľmi časté
Iné krvácanie (vrátane epistaxy, postprocedurálnej hemorágie a hematúrie)
Časté Veľmi časté
P
oruchy metabolizmu a
výživy
Nárast hmotnostia Veľmi časté Časté
Hypercholesterolémiab
CTCAEc stupeň 1 a 2
Hypertriglyceridémiab
CTCAEc stupeň 1
Poruchy nervového systému
Veľmi časté Veľmi časté
- Veľmi časté
Závratya Veľmi časté Veľmi časté
Bolesť hlavya Veľmi časté -
Poruchy gastrointestinálneho traktu
Plynatosťa Časté -
Zápchaa - Časté
Poruchy pečene a žlčových
ciest Zvýšená alanínaminotransferázab
CTCAEc stupeň 3 (> 5x –
20 x ULN) Akýkoľvek CTCAEc stupeň
Zvýšená aspartátaminotransferázab
Akýkoľvek CTCAEc
stupeň
Poruchy ciev
Časté Menej časté
Veľmi časté Veľmi časté
Veľmi časté Veľmi časté
Hypertenziaa - Veľmi časté
a Frekvencia vychádza z údajov o nežiaducich účinkoch.
- Subjekt, u ktorého sa vyskytlo viacero nežiaducich reakcií na liek (ADR) je v danej
ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
b Frekvencia vychádza z laboratórnych hodnôt.
- Subjekt, u ktorého sa vyskytlo viacero ADR je v danej ADR kategórii započítaný iba raz.
- Nahlásené ADR sa vyskytli počas liečby alebo do 28 dní od ukončenia liečby.
c Všeobecné terminologické kritériá pre nežiaduce účinky (CTCAE) verzia 3.0; stupeň 1 =
mierne, stupeň 2 = stredné, stupeň 3 = závažné, stupeň 4 = život ohrozujúce
d Tieto ADR sú bližšie popísané v texte.
e Frekvencia vychádza z počtu všetkých pacientov exponovaných ruxolitinibu v klinických skúšaniach (N=4755)
Po ukončení liečby sa u pacientov s MF môžu vrátiť príznaky MF ako napr. únava, bolesť kostí,
horúčka, pruritus, nočné potenie, symptomatická splenomegália a pokles telesnej hmotnosti.
V klinických štúdiách s MF sa celkové skóre symptómov pre symptómy MF postupne vrátilo na počiatočné hodnoty do 7 dní od ukončenia liečby (pozri časť 4.4).
PopisvybranýchnežiaducichreakciínaliekAnémiaV klinických štúdiách fázy III s MF, bol medián nástupu anémie CTCAE stupňa 2 alebo vyššieho
1,5 mesiaca. Jeden pacient (0,3 %) ukončil liečbu v dôsledku anémie.
U pacientov užívajúcich Jakavi dosiahli priemerné poklesy hemoglobínu minimum približne o
10 g/liter pod počiatočnými hodnotami po 8 až 12 týždňoch liečby a potom postupne narastali, kým nedosiahli nový stabilný stav, ktorý bol približne o 5 g/liter pod počiatočnými hodnotami. Takýto
vzorec sa u pacientov pozoroval bez ohľadu na to, či počas liečby dostali transfúziu.
V randomizovanej, placebom kontrolovanej štúdii COMFORT-I 60,6 % pacientov s MF užívajúcich Jakavi a 37,7 % pacientov s MF užívajúcich placebo dostalo počas randomizovanej liečby transfúziu s červenými krvinkami. V štúdii COMFORT-II bol podiel transfúzií s červenými krvinkami 53,4 %
v ramene s Jakavi a 41,1 % v ramene s najlepšou dostupnou liečbou.
Vo fáze randomizácie v pivotných štúdiách bola anémia menej častá u pacientov s PV ako u pacientov s MF (43,6 % oproti 82,4 %). V PV populácii boli prípady CTCAE stupňa 3 a 4 hlásené u 1,8 % pacientov, kým frekvencia u pacientov s MF bola 42,56 %.
Trombocytopénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula trombocytopénia stupňa 3 alebo 4, bol medián jej nástupu približne 8 týždňov. Trombocytopénia bola vo všeobecnosti reverzibilná a ustúpila po znížení dávky alebo po prerušení liečby. Medián času potrebného na návrat počtu krvných doštičiek nad 50 000/mm3 bol 14 dní. Počas randomizovaného obdobia boli transfúzie s krvnými doštičkami podané 4,7 % pacientov užívajúcich Jakavi a 4,0 % pacientov užívajúcich
kontrolné režimy. K ukončeniu liečby v dôsledku trombocytopénie došlo u 0,7 % pacientov užívajúcich Jakavi a 0,9 % pacientov užívajúcich kontrolné režimy. U pacientov, ktorí mali pred začiatkom liečby s Jakavi počet krvných doštičiek od 100 000/mm3 do 200 000/mm3, bola vyššia frekvencia trombocytopénie stupňa 3 alebo 4 v porovnaní s pacientmi, ktorý mali počet krvných doštičiek >200 000/mm3 (64,2 % verzus 38,5 %).
Vo fáze randomizácie v pivotných štúdiách bol podiel pacientov, u ktorých sa vyskytla trombocytopénia nižší u pacientov s PV (24,5 %), ako u pacientov s MF (69,8 %). Frekvencia výskytu závažnej trombocytopénie (t.j. CTCAE stupňa 3 a 4) bola nižšia u pacientov s PV (5,5 %) ako u pacientov s MF (11,6 %).
Neutropénia
V klinických štúdiách fázy III s MF, u pacientov, u ktorých sa vyvinula neutropénia stupňa 3 alebo 4, bol medián jej nástupu 12 týždňov. Počas randomizovaného obdobia sa pozastavenie liečby alebo zníženie dávky v dôsledku neutropénie hlásilo u 1,0 % pacientov a 0,3 % pacientov ukončilo liečbu pre neutropéniu.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV bola neutropénia hlásená u dvoch pacientov
(1,8 %), z ktorých sa u jedného pacienta vyskytla neutropénia CTCAE stupňa 4.
Krvácanie
V pivotných štúdiách fázy III s MF boli prípady krvácania (vrátane intrakraniálneho
a gastrointestinálneho krvácania, podliatin a iných prípadov krvácania) hlásené u 32,6 % pacientov liečených Jakavi a u 23,2 % pacientov liečených referenčnou liečbou (placebom alebo najlepšou dostupnou liečbou). Frekvencia udalostí stupňa 3-4 bola u pacientov liečených Jakavi alebo referenčnou liečbou podobná (4,7 % verzus 3,1 %). Väčšina pacientov s príhodou krvácania počas liečby hlásila podliatiny (65,3 %). Podliatiny boli častejšie hlásené u pacientov užívajúcich Jakavi
v porovnaní s referenčnou liečbou (21,3 % verzus 11,6 %). Intrakraniálne krvácanie bolo hlásené u
1 % pacientov exponovaných Jakavi a u 0,9 % pacientov exponovaných referenčnej liečbe. Gastrointestinálne krvácanie bolo hlásené u 5,0 % pacientov exponovaných Jakavi oproti 3,1 % pacientov exponovaných referenčnej liečbe. Iné prípady krvácania (vrátane epistaxy,
post-procedurálnej hemorágie a hematúrie) boli hlásené u 13,3 % pacientov liečených Jakavi a u
10,3 % pacientov liečených referenčnou liečbou.
Vo fáze randomizácie v pivotnej štúdii u pacientov s PV boli hlásené prípady krvácania (vrátane intrakraniálneho a gastrointestináneho krvácania, tvorby podliatin a iného krvácania) u 20 % pacientov liečených Jakavi a 15,3 % pacientov, ktorí dostávali najlepšiu dostupnú liečbu. Podliatiny boli hlásené
s rovnakou frekvenciou v liečebných skupinách s Jakavi aj BAT (10,9 % oproti 8,1 %). U pacientov užívajúcich Jakavi neboli hlásené žiadne prípady intrakraniálneho ani gastrointestinálneho krvácania.
U jedného pacienta liečeného Jakavi sa vyskytol prípad krvácania stupňa 3 (postprocedurálne krvácanie); nebolo hlásené žiadne krvácanie stupňa 4. Prípady iného krvácania (vrátane takých prípadov ako epistaxa, postprocedurálne krvácanie, gingiválne krvácanie) boli hlásené u 11,8 % pacientov liečených Jakavi a u 6,3 % pacientov liečených najlepšou dostupnou liečbou.
InfekcieV pivotných štúdiách fázy III s MF boli hlásené infekcie močových ciest stupňa 3 alebo 4 u 1,0 % pacientov, herpes zoster u 4,3 % a tuberkulóza u 1,0 %. V klinickom skúšaní fázy III bola sepsa hlásená u 3,0% pacientov. Predĺžené sledovanie pacientov liečených ruxolitinibom nepreukázalo
s postupom času žiadny zvýšený trend v rozsahu sepsy.
Vo fáze randomizácie v pivotnej štúdii bola u pacientov s PV hlásená jedna infekcia močového traktu (0,9 %) CTCAE stupňa 3 a žiadna infekcia močového traktu stupňa 4. Miera výskytu herpes zoster bola mierne vyššia u pacientov s PV (6,4 %) ako u pacientov s MF (4,0 %). U pacientov s PV sa vyskytlo jedno hlásenie postherpetickej neuralgie CTCAE stupňa 3.
Zvýšený systolický krvný tlakV pivotných štúdiách fázy III s MF bolo hlásené minimálne počas jednej vizity zvýšenie systolického
krvného tlaku oproti východiskovej hodnote o 20 mmHg alebo viac u 31,5 % pacientov v porovnaní s
19,5 % výskytom u pacientov na kontrolnej liečbe. V skúšaní COMFORT-I (pacienti s MF) bol oproti východiskovej hodnote pri Jakavi priemerne zvýšený systolický krvný tlak o 0-2 mmHg v porovnaní
s poklesom o 2-5 mmHg v ramene s placebom. V skúšaní COMFORT-II boli medzi priemernými hodnotami MF pacientov liečených ruxolitinibom alebo kontrolnou liečbou len malé rozdiely.
Vo fáze randomizácie v pivotných štúdiách bol u pacientov s PV sa priemerný systolický krvný tlak v liečebnej skupine s Jakavi zvýšil o 0,65 mmHg, kým v skupine s BAT klesol o 2 mmHg.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieNie je známe antidotum pri predávkovaní s Jakavi. Jednorazové dávky do 200 mg boli podané s prijateľnou akútnou znášanlivosťou. Vyššie ako odporúčané, opakované dávky sú spojené so zvýšenou myelosupresiou vrátane leukopénie, anémie a trombocytopénie. Je potrebné podať vhodnú podpornú liečbu.
Nepredpokladá sa, že hemodialýza zvyšuje elimináciu ruxolitinibu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE18
MechanizmusúčinkuRuxolitinib je selektívny inhibítor Janusových kináz (JAK), JAK1 a JAK2 (IC50 hodnoty 3,3 nM pre enzýmy JAK1 a 2,8 nM pre enzýmy JAK2). Tieto sprostredkujú signalizáciu mnohých cytokínov a rastových faktorov, ktoré sú dôležité pre hemopoézu a imunitné funkcie.
Myelofibróza a polycytémia vera sú myeloproliferatívne nádorové ochorenia, o ktorých je známe že súvisia s poruchou signalizácie JAK1 a JAK2. Predpokladá sa že k základom týchto porúch patrí vysoká hladina cirkulujúcich cytokínov, ktoré aktivujú JAK-STAT dráhou, mutácie pridávajúce
funkcie ako napr. JAK2V617F a utlmenie negatívnych regulačných mechanizmov. Pacienti s MF vykazujú poruchu JAK signalizácie bez ohľadu na stav JAK2V617F mutácie. U >95 % pacientov s PV sa vyskytujú aktivujúce mutácie JAK2 (V617F alebo exón 12).
Ruxolitinib inhibuje JAK-STAT signalizáciu a bunkovú proliferáciu bunkových modelov hematologických malignít závislých od cytokínov ako aj Ba/F3 buniek nezávislých od cytokínov expresiou JAK2V617F mutovaného proteínu, s hodnotou IC50 v rozsahu 80-320 nM.
Farmakodynamickéúčinky
Ruxolitinib inhibuje cytokínmi indukovanú STAT3 fosforyláciu v celej krvi od zdravých dobrovoľníkov, pacientov s MF a pacientov s PV . Ruxolitinib vyvolal maximálnu inhibíciu STAT3
fosforylácie 2 hodiny po podaní dávky, ktorá sa vrátila do normálu do 8 hodín u zdravých
dobrovoľníkov aj u pacientov s MF, čo naznačuje že nedochádza k akumulácii pôvodných ani aktívnych metabolitov.
Zvýšené východiskové zápalové markery súvisiace so základnými konštitučnými príznakmi ako TNFα, IL-6 a CRP u osôb s MF po liečbe ruxolitinibom poklesli. Počas liečby ruxolitinibom sa u pacientov s MF nevytvorila odolnosť voči jeho farmakodynamickým účinkom. Rovnako aj u pacientov s PV sa vyskytli zvýšené východiskové zápalové markery a tieto markery sa po liečbe ruxolitinibom znížili.
V podrobnej QT štúdii so zdravými dobrovoľníkmi nič nepoukazovalo na predlžujúci účinok ruxolitinibu na QT/QTc v jednotlivých dávkach až po supraterapeutickú dávku 200 mg, čo naznačuje že ruxolitinib nemá účinok na kardiálnu repolarizáciu.
Klinickáúčinnosťabezpečnosť
Myelofibróza
U pacientov s MF (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie) sa realizovali 2 randomizované štúdie fázy III (COMFORT-I a COMFORT-II). V oboch štúdiách mali pacienti hmatateľnú splenomegáliu najmenej
5 cm pod rebrovým oblúkom a rizikovú kategóriu strednú-2 alebo vysokú, podľa IWG kritérií
(International Working Group Consensus Criteria). Počiatočná dávka Jakavi sa stanovila na základe počtu krvných doštičiek.
COMFORT-I bola dvojito zaslepená, randomizovaná, placebom kontrolovaná štúdia s 309 pacientmi, ktorí nereagovali na dostupnú liečbu alebo táto pre nich nebola vhodná. Primárny cieľový ukazovateľ účinnosti bol definovaný ako podiel pacientov, u ktorých došlo k ≥35 % zmenšeniu objemu sleziny v
24. týždni v porovnaní s východiskom, merané zobrazovaním magnetickou rezonanciou (MRI) alebo počítačovou tomografiou (CT).
Sekundárne cieľové ukazovatele účinnosti zahrňovali trvanie ≥35 % zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou, podiel pacientov s ≥50 % znížením skóre celkových príznakov, zmeny v celkových skóre príznakov v porovnaní s východiskovou hodnotou v 24. týždni hodnotené pomocou modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) v2.0 denníka, a celkové prežívanie.
COMFORT-II bola otvorená, randomizovaná štúdia s 219 pacientmi. Pacienti boli randomizovaní 2:1 do skupín s Jakavi a s najlepšou dostupnou liečbou. V ramene s najlepšou dostupnou liečbou 47 % pacientov dostalo hydroxyureu a 16 % pacientov glukokortikoidy. Primárny cieľový ukazovateľ účinnosti bol podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 48. týždni v porovnaní s východiskovou hodnotou, merané MRI alebo CT.
Sekundárne cieľové ukazovatele zahrňovali podiel pacientov, ktorí dosiahli ≥35 % zmenšenie objemu sleziny v 24. týždni v porovnaní s východiskovou hodnotou a trvanie ≥35% zmenšenia objemu sleziny v porovnaní s východiskovou hodnotou.
Základné demografické charakteristiky a charakteristika ochorenia pacientov boli porovnateľné pre obe ramená v štúdiách COMFORT-I a COMFORT-II.
Tabuľka 2 Percento pacientov s ≥35 % zmenšením sleziny v 24. týždni v porovnaní
s východiskovou hodnotou v COMFORT-I a v 48. týždni v COMFORT-II (ITT)
COMFORT-I COMFORT-II
Jakavi
(N=155)
Placebo
(N=153)
Jakavi
(N=144)
Najlepšia dostupná liečba (N=72)
Čas Týždeň 24 Týždeň 48
Počet (%) pacientov s objemom sleziny zmenšeným o ≥35 %
95 % intervaly spoľahlivosti
65 (41,9) 1 (0,7) 41 (28,5) 0
34,1; 50,1 0; 3,6 21,3; 36,6 0,0; 5,0
p-hodnota <0,0001 <0,0001
Významne väčší podiel pacientov v skupine s Jakavi dosiahol ≥35 % zmenšenie objemu sleziny
(tabuľka 2) v porovnaní s východiskovou hodnotou bez ohľadu na prítomnosť alebo neprítomnosť
JAK2V617F mutácie alebo subtyp ochorenia (primárna myelofibróza, myelofibróza v dôsledku polycytémia vera, alebo myelofibróza v dôsledku esenciálnej trombocytémie).
Tabuľka 3 Percento pacientov s ≥35 % zmenšením sleziny v porovnaní s východiskovou
hodnotou podľa stavu mutácie JAK (súbor bezpečnosti)
COMFORT-I COMFORT-II
Jakavi Placebo Jakavi Najlepšia dostupná liečba
Stav mutácie JAK
Pozitívny (N=113) n (%)
Negatívny
(N=40)
n (%)
Pozitívny (N=121) n (%)
Negatívny
(N=27)
n (%)
Pozitívny (N=110) n (%)
Negatívny
(N=35)
n (%)
Pozitívny
(N=49)
n (%)
Negatívny
(N=20)
n (%)
Počet (%)
pacientov
s objemom sleziny zmenšeným o ≥35 %
54 (47,8)
11 (27,5)
1 (0,8)
0 36 (32,7)
5 0 0 (14,3)
Časový bod Po 24 týždňoch Po 48 týždňoch
Pravdepodobnosť udržania odpovede sleziny (≥35 % redukcia) na Jakavi v priebehu najmenej
24 týždňov bola 89 % v štúdii COMFORT I a 87 % v štúdii COMFORT II; v štúdii COMFORT II
udržalo odpoveď sleziny 52 % počas aspoň 48 týždňov.
V štúdii COMFORT I 45,9 % subjektov v skupine s Jakavi dosiahlo v 24. týždni ≥50 % zlepšenie celkového skóre príznakov v porovnaní s východiskovou hodnotou (zisťované podľa denníka MFSAF v2.0), v porovnaní s 5,3 % v skupine s placebom (p<0,0001 pomocou chi-kvadrát testu). Priemerná zmena v celkovom zdravotnom stave v 24. týždni zisťovaná podľa EORTC QLQ C30 bola +12,3 u Jakavi a -3,4 u placeba (p<0,0001).
V štúdii COMFORT-I bola úmrtnosť pacientov randomizovaných do skupiny s ruxolitinibom po priemernej dobe sledovania 34,3 mesiacov 27,1 %, v porovnaní s 35,1 % u pacientov randomizovaných na placebo; HR 0,687; 95 % IS 0,459-1,029; p=0,0668.
V štúdii COMFORT-II bola úmrtnosť pacientov randomizovaných na ruxolitinib po priemernej dobe sledovania 34,7 mesiacov 19,9 %, v porovnaní s 30,1 % u pacientov randomizovaných na najlepšiu dostupnú liečbu (BAT); HR 0,48; 95 % IS 0,28-0,85; p=0,009. V oboch štúdiách bola nižšia úmrtnosť
zaznamenaná v skupine s ruxolitinibom, čo bolo spôsobené predovšetkým výsledkami získanými v podskupinách po polycytémii vera a po esenciálnej trombocytémii.
Polycytémia vera
Randomizované, otvorené, aktívne kontrolované klinické skúšanie 3. fázy (RESPONSE) bolo vykonané u 222 pacientov s PV s rezistenciou alebo intoleranciou na hydroxyureu definované podľa
publikovaných kritérií medzinárodnej pracovnej skupiny European LeukemiaNet (ELN).
110 pacientov bolo randomizovaných do skupiny s ruxolitinibom a 112 pacientov do skupiny s BAT. Počiatočná dávka Jakavi bola 10 mg dvakrát denne. Dávky sa potom u jednotlivých pacientov upravili
podľa znášanlivosti a účinnosti na maximálnu dávku 25 mg dvakrát denne. BAT vybral skúšajúci
lekár pre každého pacienta individuálne a zahŕňala hydroxyureu (59,5 %), interferón/pegylovaný interferón (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorovanie (15,3 %).
Vstupné demografické údaje a charakteristika ochorenia boli v dvoch liečebných skupinách porovnateľné. Priemerný vek bol 60 rokov (rozpätie 33 až 90 rokov). Pacienti v skupine
s ruxolitinibom mali diagnózu PV v priemere 8,2 roka a predtým užívali v priemere približne 3 roky hydroxyureu. Väčšina pacientov (>80 %) podstúpila v priebehu posledných 24 týždňov pred skríningom najmenej dve flebotómie. Porovnávacie údaje týkajúce sa dlhodobého prežitia a výskytu
komplikácií ochorenia nie sú dostupné.
Primárnym zloženým cieľovým ukazovateľom bolo percento pacientov, ktorí dosiahli aj absenciu indikácie flebotómie (kontrola HCT) a ≥35 % zmenšenie objemu sleziny v porovnaní so vstupnými hodnotami v 32. týždni. Indikácia flebotómie bola definovaná ako potvrdené HCT s hodnotou >45 %, t.j. aspoň o 3 percentuálne body vyššie ako HCT získané pri vstupnom vyšetrení alebo potvrdené HCT s hodnotou >48 %, podľa toho, ktorá hodnota bola nižšia. Kľúčové sekundárne cieľové ukazovatele zahrňovali percento pacientov, ktorí dosiahli primárny cieľový ukazovateľ a v 48. týždni ostali bez progresie, ako aj percento pacientov, ktorí dosiahli úplnú hematologickú remisiu v 32. týždni.
Štúdia splnila svoj primárny cieľ a vyššie percento pacientov v skupine s Jakavi dosiahlo primárny zložený cieľový ukazovateľ aj každú z jeho jednotlivých zložiek. Podstatne viac pacientov liečených Jakavi (20,9 %) dosiahlo primárnu odpoveď (p<0,0001) v porovnaní s BAT (0,9 %). Kompenzácia hematokritu sa dosiahla u 60 % pacientov v skupine s Jakavi v porovnaní s 19,6 % v skupine s BAT a
≥35 % zmenšenie objemu sleziny sa dosiahlo u 38,2 % pacientov v skupine s Jakavi v porovnaní s 0,9 % v skupine s BAT (obr. č. 1). Deväťdesiatštyri (83,9 %) pacientov randomizovaných do skupiny s BAT prešlo na liečbu ruxolitinibom v 32. týždni alebo neskôr, čím bolo obmedzené porovnanie týchto dvoch skupín po 32. týždni.
Splnené boli aj oba kľúčové sekundárne cieľové ukazovatele. Percento pacientov, ktorí dosiahli úplnú hematologickú remisiu, bolo 23,6 % s Jakavi v porovnaní s 8,9 % s BAT (p=0,0028) a percento pacientov, ktorí dosiahli dlhotrvajúcu primárnu odpoveď v 48. týždni, bolo 19,1 % s Jakavi a 0,9 %
s BAT (p<0,0001).
O
brázok č. 1 Pacienti dosahujúci primárny cieľový ukazovateľ a zložky primárneho cieľového ukazovateľa v 32. týždni
Hodnota P: < ,0001
70 Pomer š a ncí (Ruxolitinib/BAT)
a 95% IS:
60 28,64 (4,50, 12,06)
50
40 38
30
21
20
10
Jednotlivé zložky primá rnej
odpovede v 32.týždni
60
20
.RUX
.BAT
1 1
0
Pri má rny zl ožený
ci eľový uka zova teľ v
32.týždni
≥35% zníženi e obj emu
s l ezi ny
Kompenzá ci a hema tokri tu bez fl ebotómi e
Príznaková záťaž bola hodnotená pomocou MPN-SAF celkového skóre príznakov (total symptom
score - TSS) elektronického denníka pacienta, ktorý obsahoval 14 otázok. V 32. týždni dosiahlo 49 %
pacientov liečených ruxolitinibom ≥50 % zníženie TSS-14 a 64 % pacientov ≥50 % zníženie TSS-5, v porovnaní s iba 5 % a 11 % pacientov na BAT.
Vnímanie liečebného prínosu bolo zisťované v dotazníku Celkový dojem pacienta zo zmeny (Patient Global Impression of Change - PGIC). 66 % pacientov liečených ruxolitinibom, v porovnaní s 19 % liečenými BAT, hlásilo zlepšenie už štyri týždne po začiatku liečby. Zlepšenie vo vnímaní liečebného prínosu bolo tiež vyššie u pacientov liečených ruxolitinibom v 32. týždni (78 % oproti 33 %).
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Jakavi vo všetkých podskupinách pediatrickej populácie v liečbe MF (informácie o použití v pediatrickej
populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Ruxolitinib je látka I. triedy Biofarmaceutického klasifikačného systému (BCS), s vysokou permeabilitou, vysokou rozpustnosťou a rýchlou disolúciou. V klinických štúdiách sa ruxolitinib
rýchlo absorboval po perorálnom podaní, maximálnu plazmatickú koncentráciu (Cmax) dosiahol asi po
1 hodine po podaní dávky. Na základe humánnej štúdie hmotnostnej bilancie sa zistilo, že perorálna
absorpcia ruxolitinibu, v podobe ruxolitinibu alebo metabolitov vzniknutých v priebehu presystémovej eliminácie je 95 % alebo vyššia. Priemerné hodnoty Cmax a celkovej expozície (AUC) ruxolitinibu sa priamoúmerne zvýšili po jednotlivej dávke v rozsahu 5-200 mg. Nezistili sa klinicky relevantné zmeny farmakokinetiky ruxolitinibu po podaní s jedlom s vysokým obsahom tuku. Priemerná Cmax sa mierne znížila (24 %), kým priemerná AUC zostala takmer nezmenená (4 % vzostup) po podaní s jedlom s vysokým obsahom tuku.
Distribúcia
Priemerný objem distribúcie v rovnovážnom stave je u pacientov s MF aj s PV približne 75 litrov. Pri klinicky relevantných koncentráciách ruxolitinibu je väzba na plazmatické bielkoviny približne 97 %,
prevažne na albumín. Celotelové autoradiografické štúdie na potkanoch ukázali, že ruxolinitib
nepenetruje cez krvno-mozgovú bariéru.
B
i
otransformácia
Ruxolitinib je prevažne metabolizovaný enzýmom CYP3A4 (>50 %) s čiastočnou účasťou CYP2C9. Materská zlúčenina je hlavným prvkom v ľudskej plazme, pričom predstavuje približne 60 % materiálu súvisiaceho s liekom v obehu. V plazme sú prítomné dva hlavné a aktívne metabolity, ktoré reprezentujú 25 % a 11 % materskej AUC. Tieto metabolity majú polovicu až pätinu pôvodnej farmakologickej aktivity súvisiacej s JAK. Celkové množstvo všetkých aktívnych metabolitov prispieva 18 % k celkovej farmakodynamike ruxolitinibu. Podľa in vitro štúdií, ruxolitinib v klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 alebo CYP3A4 a nie je účinný induktor CYP1A2, CYP2B6 alebo CYP3A4. In vitro údaje naznačujú, že ruxolitinib môže byť inhibítor P-gp a BCRP.
Eliminácia
Ruxolitinib sa prevažne eliminuje metabolizmom. Priemerný polčas eliminácie ruxolitinibu sú približne 3 hodiny. U zdravých dobrovoľníkov, po jednorazovej perorálnej dávke rádioaktívne značeného [14C] ruxolitinibu došlo k eliminácii prevažne metabolizáciou, pričom 74 % rádioaktivity sa vylúčilo močom a 22 % stolicou. Na nezmenenú materskú zlúčeninu pripadlo menej ako 1 % celkovej vylúčenej rádioaktivity.
Linearita/nelinearita
Priama úmera v závislosti od dávky sa dokázala v štúdiách s jednorazovými aj viacnásobnými dávkami.
Osobitépopulácie
Vplyv veku, pohlavia a rasy
Na základe štúdií so zdravými subjektmi sa nepozorovali žiadne relevantné rozdiely vo farmakokinetike ruxolitinibu v závislosti od pohlavia a rasy. Na základe analýzy farmakokinetiky u
populácie pacientov s MF sa nepotvrdil vzťah medzi perorálnym klírensom a vekom alebo rasou pacienta. Odhadovaný perorálny klírens bol u žien 17,7 l/h a u mužov 22,1 l/h, s 39 % interindividuálnou variabilitou u pacientov s MF. Klírens u pacientov s PV bol 12,7 l/h, s 42 %
interindividuálnou variabilitou a nebol zaznamenaný žiaden zjavný vzťah medzi perorálnym klírensom a pohlavím, pacientovým vekom alebo rasou, na základe farmakokinetického hodnotenia populácie pacientov s PV.
Pediatrická populácia
Bezpečnosť a účinnosť Jakavi sa u detí a dospievajúcich nesledovala (pozri časť 5.1 „Pediatrická populácia“).
Poškodenie funkcie obličiek
Funkcia obličiek sa hodnotila štúdiami úpravy diéty pri ochorení obličiek (MDRD) a kretinínu v moči. Po jednorazovej dávke ruxolitinibu 25 mg bola jeho expozícia podobná u jedincov s rôznymi stupňami
poškodenia obličiek s tými, ktorí mali normálnu funkciu obličiek. Avšak plazmatické hodnoty AUC
metabolitov ruxolitinibu mali tendenciu stúpať so závažnosťou poškodenia obličiek a najvýraznejšie zvýšenie bolo u jedincov s ťažkým poškodením obličiek. Nie je zrejmé, či zvýšená expozícia
metabolitu predstavuje bezpečnostné riziko. Odporúča sa upraviť dávku u pacientov s ťažkým
poškodením obličiek a v konečnom štádiu renálneho ochorenia (pozri časť 4.2). Podávanie iba v dňoch dialýzy znižuje expozíciu metabolitu ale zároveň aj farmakodynamický účinok, najmä počas dní medzi dialýzami.
Poškodenie funkcie pečene
U pacientov s rôznym stupňom poškodenia pečene, sa priemerná AUC ruxolitinibu po jednorazovej dávke 25 mg zvýšila u pacientov s miernym poškodením pečene o 87 %, so stredne ťažkým o 28 %
a s ťažkým o 65 %, v porovnaní s pacientmi s normálnou funkciou pečene. Nebol jasný vzťah medzi AUC a stupňom poškodenia funkcie pečene klasifikovanom podľa Childovho-Pughovho skóre. U pacientov s poškodením funkcie pečene sa konečný polčas eliminácie v porovnaní so zdravými dobrovoľníkmi predĺžil (4,1-5,0 hodín oproti 2,8 hodiny). U pacientov s poškodením funkcie pečene sa odporúča zníženie dávky približne o 50 % (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Ruxolitinib sa hodnotil na základe štúdií farmakologickej bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, reprodukčnej toxicity a karcinogenity. K cieľovým orgánom, súvisiacim s farmakologickým účinkom ruxolitinibu, v štúdiách toxicity po opakovanom podaní, patrili kostná dreň, periférna krv a lymfatické tkanivo. U psov sa pozorovali infekcie, vo všeobecnosti spojené s imunosupresiou. Nepriaznivé zníženie krvného tlaku spolu so zvýšením pulzu sa pozorovalo u psov v telemetrickej štúdii. Nepriaznivý pokles minútového objemu sa pozoroval v respiračných štúdiách u potkanov. Hranice (na základe neviazaného Cmax) na hladine bez nežiaducich účinkov v štúdiách so
psami a potkanmi boli 15,7-krát vyššie a 10,4-krát vyššie, ako maximálna dávka 25 mg dvakrát denne,
odporúčaná u ľudí. V hodnotení neurofarmakologických vplyvov ruxolitinibu sa nezaznamenali žiadne účinky.
Ruxolitinib v štúdiách u zvierat znižoval váhu plodu a zvyšoval poimpalntačné straty. U králikov a potkanov nebol zaznamenaný dôkaz o teratogénnom účinku. Rozsah expozície bol však v porovnaní s najvyššou klinickou dávkou nízky, výsledok má preto len obmedzený význam pre ľudí. Nepozorovali sa účinky na fertilitu. V štúdiách prenatálneho a postnatálneho vývoja sa pozorovala
mierne predĺžená gestačná doba, znížený počet miest implantácie a menší počet narodených šteniat. U šteniat sa pozorovala znížená priemerná počiatočná telesná hmotnosť a krátke obdobie nárastu zníženej priemernej telesnej hmotnosti. U dojčiacich potkanov sa ruxolitinib a/alebo jeho metabolity vylučovali do mlieka v koncentrácii 13-krát vyššej ako materské plazmatické koncentrácie. Ruxolitinib nebol mutagénny alebo klastogénny. Ruxolitinib nebol karcinogénny v modeloch Tg.rasH2 transgénnych myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Mikrokryštalická celulóza
Magnéziumstearát
Koloidný bezvodý oxid kremičitý
Sodná soľ karboxymetylcelulózy (typ A) Povidón
Hydroxypropylcelulóza
Monohydrát laktózy
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
6.5 Druh obalu a obsah balenia
PVC/PCTFE/hliníkové blistrové balenia obsahujúce 14 alebo 56 tabliet alebo spoločné balenia obsahujúce 168 (3 balenia po 56) tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLO(ČÍSLA)EU/1/12/773/010-012
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE23.08.2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu