
vaných antiemetických liekov pozri úplnú informáciu o príslušnom lieku.
Starší pacienti
U starších pacientov nie je nutná žiadna úprava dávkovania.
Poškodenie obličiek
U pacientov s renálnou insuficienciou alebo u dialyzovaných pacientov s terminálnym štádiom renálneho ochorenia nie je potrebná žiadna úprava dávkovania (pozri časť 5.2).
Poškodenie pečene
U pacientov s miernou hepatálnou insuficienciou nie je potrebná úprava dávkovania. U pacientov so stredne ťažkou hepatálnou insuficienciou sú k dispozícii iba obmedzené údaje a u pacientov
s ťažkou hepatálnou insuficienciou nie sú dostupné žiadne údaje (pozri časti 4.4 a 5.2).
Deti a adolescenti
IVEMEND sa neodporúča na použitie u detí mladších ako 18 rokov z dôvodu nedostatočných údajov o jeho bezpečnosti a účinnosti (pozri časť 5.2).
Spôsob podávania
IVEMEND sa má podať intravenózne a nesmie sa podávať intramuskulárne alebo subkutánne. Najvhodnejší spôsob intravenózneho podania je prostredníctvom kvapkajúcej intravenóznej infúzie počas 15 minút (pozri časť 6.6). IVEMEND nepodávajte vo forme bolusovej injekcie alebo nezriedeného roztoku.
4.3 Kontraindikácie
Precitlivenosť na liečivo, aprepitant alebo na polysorbát 80 alebo niektorú z ďalších pomocných látok. Súbežné podávanie s pimozidom, terfenadínom, astemizolom alebo cisapridom (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
U pacientov so stredne ťažkou hepatálnou insuficienciou sú k dispozícii iba obmedzené údaje
a u pacientov s ťažkou hepatálnou insuficienciou nie sú dostupné žiadne údaje. U týchto pacientov je pri použití lieku IVEMEND potrebná opatrnosť (pozri časť 5.2).
IVEMEND a perorálny aprepitant sa majú používať s opatrnosťou u pacientov, ktorí súbežne užívajú perorálne podávané lieky primárne metabolizované CYP3A4 (pozri časť 4.5). Ďalej je potrebné venovať zvýšenú pozornosť súbežnému podávaniu irinotekánu, pretože táto kombinácia môže vyústiť do zvýšenej toxicity.
Súbežné podanie fosaprepitantu s derivátmi ergotových alkaloidov, ktoré sú substrátmi CYP3A4, môže mať za následok zvýšené plazmatické koncentrácie týchto liekov. Preto sa v dôsledku možného rizika ergotovej toxicity odporúča zvýšená opatrnosť.
Súbežné podávanie perorálneho aprepitantu s warfarínom vedie ku skráteniu protrombínového času udávaného ako INR (International Normalised Ratio). U pacientov, ktorí sú dlhodobo liečení warfarínom, sa má pozorne monitorovať INR počas liečby perorálnym aprepitantom a počas 2 týždňov po každom trojdňovom cykle liečby chemoterapiou vyvolanej nevoľnosti a vracania fosaprepitantom a následne perorálnym aprepitantom (pozri časť 4.5).
Počas podávania a 28 dní po podávaní aprepitantu môže dôjsť k zníženiu účinnosti hormonálnych kontraceptív. Počas liečby fosaprepitantom alebo aprepitantom a počas 2 mesiacov po užití poslednej dávky aprepitantu sa majú používať alternatívne alebo zaisťovacie metódy antikoncepcie (pozri časť
4.5).
Súbežnému podávaniu fosaprepitantu s liekmi silne indukujúcimi aktivitu CYP3A4 (napr. rifampicín, fenytoín, karbamazepín, fenobarbital) je potrebné sa vyhnúť, pretože táto kombinácia spôsobuje zníženie plazmatických koncentrácií aprepitantu (pozri časť 4.5). Súbežné podávanie fosaprepitantu
a ľubovníka bodkovaného sa neodporúča.
Pri súbežnom podaní fosaprepitantu s liekmi, ktoré inhibujú aktivitu CYP3A4 (napr. ritonavir, ketokonazol, klaritromycín, telitromycín) je potrebné postupovať opatrne, pretože táto kombinácia má za následok zvýšenie plazmatickej koncentrácie aprepitantu (pozri časť 4.5).
IVEMEND sa nemá podávať ako bolusová injekcia, ale je potrebné ho vždy zriediť a podať vo forme pomalej intravenóznej infúzie (pozri časť 4.2). IVEMEND sa nemá podávať intramuskulárne alebo subkutánne. Pri vyšších dávkach sa pozorovali prípady miernej trombózy v mieste podania injekcie (pozri časť 4.9). Ak sa vyskytnú známky alebo príznaky lokálneho podráždenia, je potrebné ukončiť podávanie injekcie alebo infúzie a začať znovu do inej žily.
4.5 Liekové a iné interakcie
Fosaprepitant sa pri intravenóznom podaní rýchlo mení na aprepitant.
Liekové interakcie po podaní fosaprepitantu sa vyskytnú pravdepodobne u tých liekov, ktoré interagujú s perorálnym aprepitantom. Nasledovné informácie pochádzajú z údajov s perorálnym aprepitantom a zo štúdií uskutočnených s fosaprepitantom a midazolamom alebo diltiazemom.
Aprepitant je substrátom, stredne silným inhibítorom a induktorom CYP3A4. Aprepitant je tiež induktorom CYP2C9. Počas liečby perorálnym aprepitantom je CYP3A4 inhibovaný. Po ukončení liečby spôsobuje perorálny aprepitant prechodnú stredne silnú indukciu CYP2C9 a prechodnú miernu indukciu CYP3A4 a glukuronidácie.
Účinok aprepitantu na farmakokinetiku iných látok
Inhibícia CYP3A4
Aprepitant ako mierny inhibítor CYP3A4 môže zvýšiť plazmatické koncentrácie súbežne perorálne podávaných liekov, ktoré sa metabolizujú cez CYP3A4. Počas 3-dňovej liečby fosaprepitantom
a následne perorálne podávaným aprepitantom sa môže AUC perorálne podávaných substrátov CYP3A4 zvýšiť až približne trojnásobne. Na plazmatické koncentrácie intravenózne podávaných substrátov CYP3A4 sa očakáva nižší vplyv aprepitantu. Pri súbežnom podávaní fosaprepitantu
so substrátmi CYP3A4 sa odporúča opatrnosť.
Fosaprepitant sa nesmie používať súčasne s pimozidom, terfenadínom, astemizolom alebo cisapridom. Inhibícia CYP3A4 navodená aprepitantom by mohla viesť k zvýšeniu plazmatických koncentrácií týchto liekov a potenciálne spôsobiť závažné alebo život ohrozujúce reakcie.
Indukcia
Aprepitant ako stredne silný induktor CYP2C9 a mierny induktor CYP3A4 a glukuronidácie môže znížiť plazmatické koncentrácie substrátov eliminovaných týmito cestami v priebehu dvoch týždňov od začiatku dávkovacej schémy. Tento účinok sa môže prejaviť až po ukončení 3-dňovej schémy
s fosaprepitantom a následným aprepitantom. Pre substráty CYP2C9 a CYP3A4 je indukcia prechodná, s maximálnym účinkom dosiahnutým 3-5 dní po ukončení 3-dňovej liečby perorálnym
aprepitantom. Účinok trvá počas niekoľkých dní, potom pomaly ustupuje a klinicky nevýznamným sa
stáva do dvoch týždňov od ukončenia liečby perorálnym aprepitantom. Mierna indukcia glukuronidácie sa tiež pozoruje pri podávaní 80 mg aprepitantu perorálne počas 7 dní. Údaje
o účinkoch na CYP2C8 a CYP2C19 nie sú k dispozícii. Ak sa počas tohto časového obdobia podáva warfarín, acenokumarol, tolbutamid, fenytoín alebo iné lieky, o ktorých je známe, že sú metabolizované CYP2C9, odporúča sa opatrnosť.
Nezdá sa, že fosaprepitant alebo aprepitant interaguje s P-glykoproteínovým transportérom, čo sa preukázalo neprítomnosťou interakcie perorálneho aprepitantu s digoxínom.
Kortikosteroidy
Dexametazón:Pri súbežnom podávaní s režimom s fosaprepitantom a následným aprepitantom sa má zvyčajná dávka perorálneho dexametazónu znížiť približne o 50 %. V klinických štúdiách chemoterapiou indukovanej nevoľnosti a vracania bola dávka dexametazónu zvolená s ohľadom na liekové interakcie (pozri časť 4.2). Perorálny aprepitant podávaný v režime 125 mg súbežne
s perorálnym dexametazónom 20 mg v deň 1 a perorálny aprepitant 80 mg/deň podávaný súbežne
s perorálnym dexametazónom 8 mg počas dní 2 až 5 zvýšil AUC dexametazónu, substrátu CYP3A4,
2,2-násobne v dňoch 1 a 5.
Metylprednizolón:Pri súbežnom podávaní s režimom s fosaprepitantom a následným aprepitantom sa má zvyčajná dávka intravenózne podávaného metylprednizolónu znížiť o približne 25 % a zvyčajná dávka perorálneho metylprednizolónu o približne 50 %. Perorálny aprepitant podávaný v režime
125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 zvýšil AUC metylprednizolónu, substrátu CYP3A4, 1,3- násobne v deň 1 a 2,5-násobne v deň 3, keď sa v deň 1 metylprednizolón súbežne podával v dávke
125 mg intravenózne a v dňoch 2 a 3 v dávke 40 mg perorálne.
Počas kontinuálnej liečby metylprednizolónom sa môže AUC metylprednizolónu neskôr v priebehu 2 týždňov od začiatku podávania perorálneho aprepitantu znížiť v dôsledku indukčného účinku aprepitantu na CYP3A4. Očakáva sa, že tento účinok môže byť výraznejší pri perorálne podávanom metylprednizolóne.
Chemoterapeutiká: Keď bol perorálny aprepitant vo farmakokinetických štúdiách podávaný v režime
125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3, neovplyvnil farmakokinetiku docetaxelu podaného intravenózne v deň 1, ani vinorelbínu podaného intravenózne v deň 1 alebo deň 8. Keďže vplyv aprepitantu na farmakokinetiku perorálne podávaných substrátov CYP3A4 je väčší ako na farmakokinetiku intravenózne podávaných substrátov CYP3A4, nedá sa vylúčiť interakcia s perorálne podávanými chemoterapeutikami prevažne alebo čiastočne metabolizovanými CYP3A4 (napr.
etopozid, vinorelbín). U pacientov, ktorí užívajú takéto liečivá, sa odporúča opatrnosť a môže byť
vhodné ďalšie sledovanie (pozri časť 4.4).
Midazolam: Pri súbežnom podávaní s 3-dňovým režimom s fosaprepitantom a následným aprepitantom sa majú zohľadniť možné účinky zvýšených plazmatických koncentrácií midazolamu alebo iných benzodiazepínov, ktoré sa metabolizujú prostredníctvom CYP3A4 (alprazolam, triazolam).
Fosaprepitant v dávke 100 mg podaný v priebehu 15 minút s jednorazovou dávkou midazolamu 2 mg zvýšil AUC midazolamu 1,6-násobne. Tento účinok sa nepovažoval za klinicky významný.
Perorálny aprepitant zvýšil AUC midazolamu 2,3-násobne v deň 1 a 3,3-násobne v deň 5, keď sa jednorazová perorálna dávka midazolamu 2 mg podala v deň 1 a 5 spolu s perorálnym aprepitantom v režime 125 mg v deň 1 a 80 mg/deň v dňoch 2 až 5.
V inej štúdii s intravenózne podávaným midazolamom sa perorálny aprepitant podal v dávke 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 a midazolam 2 mg bol podaný intravenózne pred podaním
3-dňového režimu s perorálnym aprepitantom a v dňoch 4, 8 a 15. Perorálny aprepitant zvýšil AUC midazolamu o 25 % v deň 4 a znížil AUC midazolamu o 19 % v deň 8 a o 4 % v deň 15. Tieto účinky sa nepovažovali za klinicky významné.
V tretej štúdii s intravenóznym a perorálnym podaním midazolamu sa perorálny aprepitant podal v dávke 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 spolu s ondansetrónom 32 mg v deň 1, dexametazónom 12 mg v deň 1 a 8 mg v dňoch 2-4. Táto kombinácia (t.j. perorálny aprepitant, ondansetrón a dexametazón) znížila AUC perorálneho midazolamu o 16 % v deň 6, o 9 % v deň 8, o 7 % v deň 15 a o 17 % v deň 22. Tieto účinky sa nepovažovali za klinicky významné.
Bola ukončená štvrtá štúdia s intravenóznym podaním midazolamu a perorálnym aprepitantom. Midazolam v dávke 2 mg bol intravenózne podaný 1 hodinu po perorálnom podaní jednorazovej dávky perorálneho aprepitantu 125 mg. Plazmatická AUC midazolamu sa zvýšila 1,5-násobne. Tento účinok sa nepovažoval za klinicky významný.
Diltiazem: U pacientov s miernou až stredne ťažkou hypertenziou viedla infúzia 100 mg fosaprepitantu počas 15 minút s diltiazemom v dávke 120 mg 3-krát denne k 1,4-násobnému zvýšeniu AUC diltiazemu a k malému, ale klinicky významnému poklesu krvného tlaku, nedošlo však
ku klinicky významnej zmene v tepovej frekvencii alebo PR intervale.
Warfarín: U pacientov dlhodobo liečených warfarínom sa má počas liečby fosaprepitantom alebo aprepitantom a počas 2 týždňov po každom 3-dňovom režime na nevoľnosť a vracanie vyvolané chemoterapiou dôsledne monitorovať protrombínový čas (INR) (pozri časť 4.4). Keď sa podal perorálny aprepitant v jednorazovej 125-mg dávke v deň 1 a 80 mg/deň v dňoch 2 a 3 zdravým jedincom stabilizovaným na dlhodobej liečbe warfarínom, perorálny aprepitant neovplyvnil plazmatickú AUC R(+) alebo S(-) warfarínu v deň 3; 5 dní po ukončení podávania perorálneho aprepitantu sa však minimálna koncentrácia S(-) warfarínu (substrátu CYP2C9) znížila o 34 % sprevádzaná 14 % poklesom INR.
Tolbutamid: Perorálny aprepitant podávaný v dávke 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 znížil AUC tolbutamidu (substrátu CYP2C9) o 23 % v deň 4, o 28 % v deň 8 a o 15 % v deň 15, keď sa tolbutamid podal v jednorazovej dávke 500 mg perorálne pred podaním perorálneho aprepitantu
v trojdňovom režime a v deň 4, 8 a 15.
Perorálne kontraceptíva: Počas podávania a 28 dní po podávaní perorálneho aprepitantu môže dôjsť k zníženiu účinnosti hormonálnych kontraceptív. Počas liečby fosaprepitantom alebo perorálnym aprepitantom a počas 2 mesiacov po poslednej dávke aprepitantu sa majú používať alternatívne alebo zaisťovacie formy antikoncepcie. Aprepitant podávaný ako 100-mg kapsula jedenkrát denne počas
14 dní spolu s perorálnym kontraceptívom, ktoré obsahovalo 35 µg etinylestradiolu a 1 mg noretisterónu, znížil AUC etinylestradiolu o 43 % a noretisterónu o 8 %.
V inej štúdii boli v dňoch 1 až 21 podávané jednorazové dávky perorálneho kontraceptíva obsahujúceho etinylestradiol a noretisterón s perorálnym aprepitantom, ktorý sa podával v režime
125 mg v deň 8 a 80 mg/deň v dňoch 9 a 10 spolu s ondansetrónom 32 mg intravenózne v deň 8
a perorálnym dexametazónom v dávke 12 mg v deň 8 a v dávke 8 mg/deň v dňoch 9, 10 a 11. V tejto štúdii sa počas dní 9 až 21 znížili minimálne (trough) koncentrácie etinylestradiolu až o 64 %
a minimálne koncentrácie noretisterónu sa znížili až o 60 %.
Antagonisti 5-HT3: V klinických interakčných štúdiách nemal aprepitant klinicky významný účinok na farmakokinetiku ondansetrónu, granisetrónu alebo hydrodolasetrónu (aktívny metabolit dolasetrónu).
Účinok iných látok na farmakokinetiku aprepitantu
K súbežnému podávaniu fosaprepitantu alebo aprepitantu s liekmi, ktoré inhibujú aktivitu CYP3A4 (napr. ritonavir, ketokonazol, klaritromycín, telitromycín), je potrebné pristupovať s opatrnosťou, nakoľko táto kombinácia vedie k zvýšeniu plazmatických koncentrácií aprepitantu.
Súbežnému podávaniu fosaprepitantu alebo aprepitantu s liekmi, ktoré silne indukujú aktivitu CYP3A4 (napr. rifampicín, fenytoín, karbamazepín, fenobarbital), je potrebné sa vyhnúť, pretože táto kombinácia vedie k zníženým plazmatickým koncentráciám aprepitantu, čo môže spôsobiť zníženie jeho účinnosti. Súbežné podávanie fosaprepitantu a ľubovníka bodkovaného sa neodporúča.
Ketokonazol: Po podaní jednorazovej 125-mg dávky perorálneho aprepitantu v deň 5 počas
10-dňového podávania ketokonazolu 400 mg/deň, silného inhibítora CYP3A4, došlo k približne 5- násobnému zvýšeniu AUC aprepitantu a k približne 3-násobnému zvýšeniu priemerného terminálneho polčasu aprepitantu.
Diltiazem: Podanie infúzie fosaprepitantu v dávke 100 mg trvajúcej 15 minút spolu s diltiazemom
120 mg 3-krát denne viedlo k 1,5-násobnému zvýšeniu AUC aprepitantu. Tento účinok sa nepovažoval za klinicky významný.
Rifampicín: Po podaní jednorazovej 375-mg dávky perorálneho aprepitantu v deň 9 počas 14-dňového podávania rifampicínu 600 mg/deň, silného induktora CYP3A4, došlo k zníženiu AUC aprepitantu
o 91 % a k zníženiu priemerného terminálneho polčasu o 68 %.
4.6 Gravidita a laktácia
Nie sú k dispozícii žiadne klinické údaje o gravidných ženách vystavených účinku fosaprepitantu a aprepitantu.
Potenciálna toxicita fosaprepitantu a aprepitantu na reprodukciu nebola dostatočne skúmaná, pretože v štúdiách na zvieratách sa nedosiahli vyššie expozičné hladiny ako terapeutická expozícia u ľudí. Tieto štúdie nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3). Možné účinky zmien neurokinínovej regulácie na reprodukciu nie sú známe. IVEMEND sa nemá používať počas gravidity, pokiaľ to nie je úplne nevyhnutné.
Po intravenóznom podaní fosaprepitantu ako aj po perorálnom podaní aprepitantu sa aprepitant vylučuje do mlieka dojčiacich potkanov. Nie je známe, či sa aprepitant vylučuje do materského mlieka u ľudí. Preto sa dojčenie počas liečby liekom IVEMEND a perorálnym aprepitantom neodporúča.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch lieku IVEMEND na schopnosť viesť vozidlá a obsluhovať stroje. Pri vedení vozidla alebo obsluhovaní strojov však treba vziať do úvahy, že po použití lieku IVEMEND boli hlásené závrat a únava (pozri časť 4.8).
4.8 Nežiaduce účinky
Keďže sa fosaprepitant mení na aprepitant, očakáva sa, že sa pri fosaprepitantne vyskytnú tie nežiaduce reakcie, ktoré sú spojené s aprepitantom. Bezpečnostný profil aprepitantu sa hodnotil u približne 4 900 jedincov. Rôzne liekové formy fosaprepitantu sa podali celkovo 729 jedincom vrátane 347 zdravých jedincov a 149 pacientov s CINV.
U pacientov, ktorí dostávali vysoko emetogénnu chemoterapiu, sa nežiaduce reakcie, ktoré ošetrujúci lekár považoval za súvisiace s liekom, vyskytli u približne 17 % pacientov liečených režimom
s aprepitantom v porovnaní s približne 13 % pacientov liečených štandardnou liečbou. Liečba aprepitantom bola ukončená z dôvodu nežiaducich reakcií u 0,6 % pacientov liečených režimom s aprepitantom v porovnaní s 0,4 % pacientov liečených štandardnou liečbou. V klinickej štúdii
pacientov dostávajúcich stredne emetogénnu chemoterapiu boli klinické nežiaduce reakcie hlásené
u približne 21 % pacientov liečených režimom s aprepitantom v porovnaní s približne 20 % pacientov liečených štandardnou terapiou. Liečba aprepitantom bola ukončená z dôvodu nežiaducich reakcií
u 1,1 % pacientov liečených režimom s aprepitantom v porovnaní s 0,5 % pacientov liečených štandardnou liečbou.
U pacientov dostávajúcich vysoko emetogénnu chemoterapiu boli najčastejšími nežiaducimi
reakciami, u ktorých bola hlásená vyššia incidencia u pacientov liečených režimom s aprepitantom ako u pacientov liečených štandardnou liečbou: štikútanie (4,6 %), asténia/únava (2,9 %), zvýšenie ALT (2,8 %), zápcha (2,2 %), bolesť hlavy (2,2 %) a anorexia (2,0 %). U pacientov dostávajúcich stredne emetogénnu chemoterapiu najčastejšou nežiaducou reakciou, ktorá sa vyskytla s vyššou incidenciou
u pacientov liečených režimom s aprepitantom než u pacientov liečených štandardnou terapiou, bola únava (2,5 %).
U pacientov liečených režimom s aprepitantom sa s vyššou incidenciou ako u pacientov liečených štandardnou terapiou pozorovali tieto nežiaduce účinky:
Frekvencie sú definované ako: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až
<1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000) a neznáme (z dostupných údajov sa nedá stanoviť).
Abnormálne laboratórne a funkčné vyšetrenia:
Časté: zvýšenie ALT, zvýšenie AST.
Menej časté: zvýšenie alkalickej fosfatázy, hyperglykémia, mikroskopická hematúria, hyponatrémia, pokles hmotnosti.
Ochorenia srdca a poruchy srdcovej činnosti:
Menej časté: bradykardia.
Ochorenia krvi a lymfatického systému:
Menej časté: anémia, febrilná neutropénia.
Poruchy nervového systému:
Časté: bolesť hlavy, závrat.
Menej časté: abnormálne sny, kognitívna porucha.
Ochorenia oka:
Menej časté: konjunktivitída.
Ochorenia ucha a labyrintu:
Menej časté: tinnitus.
Ochorenia dýchacej sústavy, hrudníka a mediastína:
Časté: štikútanie.
Menej časté: faryngitída, kýchanie, kašeľ, výtok hlienu z nosnej dutiny do hrtanu, podráždenie hrdla.
Poruchy a ochorenia gastrointestinálneho traktu:
Časté: zápcha, hnačka, dyspepsia, grganie.
Menej časté: nauzea*, vracanie*, reflux žalúdočnej kyseliny, porucha chuti, diskomfort v epigastriu, zápcha, gastroezofageálna refluxová choroba, perforujúci duodenálny vred, bolesť brucha, sucho
v ústach, enterokolitída, plynatosť, stomatitída.
Poruchy obličiek a močovej sústavy:
Menej časté: polyúria, dyzúria, polakizúria.
Poruchy kože a podkožného tkaniva:
Menej časté: vyrážka, akné, fotosenzitivita, hyperhidróza, mastná pokožka, pruritus, kožná lézia.
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva:
Menej časté: svalový kŕč, myalgia.
Poruchy metabolizmu a výživy:
Časté: anorexia.
Menej časté: prírastok hmotnosti, polydipsia.
Infekcie a nákazy:
Menej časté: kandidóza, stafylokoková infekcia.
Cievne poruchy:
Menej časté: sčervenanie/nával tepla.
Celkové ochorenia a reakcie v mieste podania:
Časté: asténia/únava.
Menej časté: edém, diskomfort hrudníka, letargia, smäd.
Psychiatrické poruchy a ochorenia:
Menej časté: dezorientácia, eufória, úzkosť.
* Nauzea a vracanie boli v prvých 5 dňoch po chemoterapeutickej liečbe parametrami účinnosti a až potom sa hlásili ako nežiaduce reakcie.
Profily nežiaducich reakcií v predĺžení s viacnásobnými cyklami až na 5 ďalších cyklov chemoterapie boli vo všeobecnosti podobné tým, ktoré sa pozorovali v 1. cykle.
Jeden prípad Stevens-Johnsonovho syndrómu bol hlásený ako závažný nežiaduci účinok u pacienta, ktorý dostával aprepitant s protirakovinovou chemoterapiou.
Okrem toho, v bioekvivalenčnej štúdii, v ktorej sa 66 jedincom intravenózne podalo 115 mg fosaprepitantu, boli časté nežiaduce reakcie indurácia v mieste podania infúzie a bolesť v mieste podania infúzie.
Ďalšie nežiaduce reakcie boli pozorované u pacientov liečených aprepitantom (40 mg) na pooperačnú nevoľnosť a vracanie a s incidenciou vyššou ako pri ondansetróne: bolesť hornej časti brucha, abnormálne zvuky čriev, dyzartria, dyspnoe, znížená citlivosť, insomnia, mióza, nauzea, senzorická porucha, žalúdočné ťažkosti, zníženie zrakovej ostrosti, sipot.
V klinických štúdiách pooperačnej nevoľnosti a vracania (PONV) boli navyše u pacientov užívajúcich vyššiu dávku aprepitantu hlásené dve závažné nežiaduce reakcie: jeden prípad zápchy a jeden prípad subilea.
Jeden prípad angioedému a urtikárie bol hlásený ako závažný nežiaduci účinok u pacienta, ktorý dostával aprepitant v non-CINV/non-PONV štúdii.
4.9 Predávkovanie
Nie sú k dispozícii žiadne špecifické informácie o liečbe predávkovania.
U jedného pacienta, ktorý požil 1 440 mg aprepitantu, bola hlásená ospalosť a bolesť hlavy. Jednorazové dávky fosaprepitantu až do 200 mg boli u zdravých osôb vo všeobecnosti dobre
tolerované.
U troch z 33 osôb, ktoré dostali 200 mg fosaprepitantu, sa vyskytla mierna trombóza v mieste podania injekcie.
V prípade predávkovania je potrebné prerušiť liečbu fosaprepitantom a prejsť na celkovú podpornú liečbu a monitorovanie pacienta. Pretože aprepitant má antiemetický účinok, nemusí byť farmakologicky navodené vracanie efektívne.
Aprepitant sa nedá odstrániť hemodialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiemetiká a antinauzeiká, ATC kód:[navrhnutý: podaná žiadosť]. Fosaprepitant je proliečivo aprepitantu a po intravenóznom podaní sa rýchlo mení na aprepitant (pozri
časť 5.2). Podiel fosaprepitantu na celkovom antiemetickom účinku nebol plne charakterizovaný, ale nedá sa vylúčiť prechodné prispenie počas počiatočnej fázy. Aprepitant je selektívny vysokoafinitný antagonista na neurokinín 1 (NK1) receptoroch pre ľudskú substanciu P. Farmakologický účinok fosaprepitantu sa prisudzuje aprepitantu.
Dve randomizované dvojito zaslepené štúdie, ktoré celkovo zahŕňali 1 094 pacientov dostávajúcich chemoterapiu zahŕňajúcu cisplatinu ≥70 mg/m2, porovnávali schému s aprepitantom v kombinácii
s ondansetrónom/dexametazónom (pozri časť 4.2) a štandardnú schému (placebo plus ondansetrón
32 mg intravenózne v deň 1 plus dexametazón 20 mg perorálne v deň 1 a 8 mg perorálne dvakrát denne počas dní 2 až 4).
Účinnosť vychádzala z hodnotenia nasledujúceho kompozitného ukazovateľa: úplná odpoveď (definovaná ako stav bez epizód vracania a bez použitia záchrannej liečby) hlavne počas 1. cyklu. Výsledky sa hodnotili pre každú individuálnu štúdiu a pre 2 štúdie v kombinácii.
V tabuľke 1 je súhrn hlavných výsledkov štúdií z kombinovanej analýzy.
Tabuľka 1
 Percento odpovedajúcich pacientov, ktorí dostávali vysoko emetogénnu chemoterapiu podľa liečebnej skupiny a fázy - 1. cyklus
Percento odpovedajúcich pacientov, ktorí dostávali vysoko emetogénnu chemoterapiu podľa liečebnej skupiny a fázy - 1. cyklus
KOMPOZITNÉ UKAZOVATELE
Schéma
s aprepitantom
(N=521)†
%
Štandardná
liečba
(N=524)†
%
Rozdiely*
% (95 % IS)
 Úplná odpoveď (žiadne vracanie a žiadna záchranná terapia)
Úplná odpoveď (žiadne vracanie a žiadna záchranná terapia)
Celkovo (0-120 hodín)
|
67,7
|
47,8
|
19,9
|
(14,0; 25,8)
|
0-24 hodín
|
86,0
|
73,2
|
12,7
|
(7,9; 17,6)
|
25-120 hodín
|
71,5
|
51,2
|
20,3
|
(14,5; 26,1)
|
JEDNOTLIVÉ UKAZOVATELE
|
|
|
|
|
Bez vracania (žiadne epizódy vracania, bez ohľadu na použitie záchrannej terapie)
Celkovo (0-120 hodín)
|
71,9
|
49,7
|
22,2
|
(16,4; 28,0)
|
0-24 hodín
|
86,8
|
74,0
|
12,7
|
(8,0; 17,5)
|
25-120 hodín
|
76,2
|
53,5
|
22,6
|
(17,0; 28,2)
|
Bez signifikantnej nauzey (maximálny VAS <25 mm na stupnici 0-100 mm)
Celkovo (0-120 hodín)
|
72,1
|
64,9
|
7,2
|
(1,6; 12,8)
|
25-120 hodín
|
74,0
|
66,9
|
7,1
|
(1,5; 12,6)
|
* Intervaly spoľahlivosti boli vypočítané bez úpravy podľa pohlavia a súbežnej chemoterapiu, ktoré
boli zahrnuté v primárnej analýze miery pravdepodobnosti a logistických modeloch.
† U jedného pacienta v režime s aprepitantom boli dostupné iba údaje z akútnej fázy a bol vylúčený z celkovej analýzy a analýzy oneskorenej fázy; u jedného pacienta so štandardnou liečbou boli dostupné iba údaje z oneskorenej fázy a bol vylúčený z celkovej analýzy a analýzy akútnej fázy.
Odhadovaný čas do prvého vracania v kombinovanej analýze je zobrazený na Kaplan-Meierovom grafe na obrázku 1.
Obrázok 1Percento pacientov dostávajúcich vysoko emetogénnu chemoterapiu, ktorí boli v časovom priebehu bez vracania - 1. cyklus
Aprepitant Regimen (N=520) Standard Therapy (N=523)
|
|
 ▬▬
▬▬ schéma s aprepitantom (N=520)
100%
------- štandardná liečba (N=523)

90%

80%
70%
60%
50%
40%
0
0 12 24 36 48 60 72 84 96 108 120
čas (hodiny)
Štatisticky významné rozdiely v účinnosti sa pozorovali aj v každej z 2 individuálnych štúdií.
V tých istých 2 klinických štúdiách pokračovalo 851 pacientov v predĺžení s opakovanými cyklami, až do 5 ďalších cyklov chemoterapie. Režim s aprepitantom si zachoval účinnosť počas všetkých cyklov.
V randomizovanej dvojito zaslepenej štúdii u celkovo 866 pacientov (864 žien, 2 muži) dostávajúcich chemoterapiu, ktorá zahŕňala cyklofosfamid 750-1 500 mg/m2 alebo cyklofosfamid 500-1 500 mg/m2 a doxorubicín (≤60 mg/m2) alebo epirubicín (≤100 mg/m2), sa porovnávala schéma s aprepitantom
v kombinácii s ondansetrónom/dexametazónom (pozri časť 4.2) so štandardnou terapiou (placebo plus
ondansetrón 8 mg perorálne (dvakrát v deň 1 a každých 12 hodín v dňoch 2 a 3) plus dexametazón
20 mg perorálne v deň 1).
Účinnosť vychádzala z hodnotenia kompozitného ukazovateľa: úplná odpoveď (definovaná ako stav bez epizód vracania a bez použitia záchrannej liečby) hlavne počas 1. cyklu.
Súhrn najdôležitejších výsledkov štúdie je uvedený v tabuľke 2.
Tabuľka 2Percento odpovedajúcich pacientov podľa liečebnej skupiny a fázy - 1. cyklus stredne emetogénna chemoterapia
KOMPOZITNÉ UKAZOVATELE
Schéma
s aprepitantom
(N=433)†
%
Štandardná
liečba
(N=424)
%
Rozdiely*
% (95 % IS)
'
Úplná odpoveď (žiadne vracanie a žiadna záchranná terapia)
Celkovo (0-120 hodín)
|
50,8
|
42,5
|
8,3
|
(1,6; 15,0)
|
0-24 hodín
|
75,7
|
69,0
|
6,7
|
(0,7; 12,7)
|
25-120 hodín
|
55,4
|
49,1
|
6,3
|
(-0,4; 13,0)
|
JEDNOTLIVÉ UKAZOVATELE
|
|
|
|
|
Bez vracania (žiadne epizódy vracania, bez ohľadu na použitie záchrannej terapie)
Celkovo (0-120 hodín)
|
75,7
|
58,7
|
17,0
|
(10,8; 23,2)
|
0-24 hodín
|
87,5
|
77,3
|
10,2
|
(5,1;15,3)
|
25-120 hodín
|
80,8
|
69,1
|
11,7
|
(5,9; 17,5)
|
Bez signifikantnej nauzey (maximálny VAS <25 mm na stupnici 0-100 mm)
Celkovo (0-120 hodín)
|
60,9
|
55,7
|
5,3
|
(-1,3; 11,9)
|
0-24 hodín
|
79,5
|
78,3
|
1,3
|
(-4,2; 6,8)
|
25-120 ho hodín
|
65,3
|
61,5
|
3,9
|
(-2,6; 10,3)
|
* Intervaly spoľahlivosti boli vypočítané bez úpravy podľa vekovej kategórie (<55 rokov, ≥55 rokov)
a vyšetrovanej skupiny, ktoré boli zahrnuté v primárnej analýze miery pravdepodobnosti a logistických modeloch.
† U jedného pacienta v režime s aprepitantom boli dostupné iba údaje z akútnej fázy a bol vylúčený z celkovej analýzy a analýzy oneskorenej fázy.
Odhadovaný čas do prvého vracania v štúdii je zobrazený na Kaplan-Meierovom grafe na obrázku 2.
Obrázok 2
Percento pacientov dostávajúcich stredne emetogénnu chemoterapiu, ktorí boli v časovom priebehu bez vracania - 1. cyklus
100% schéma s aprepitanton (N=432)






štandardná liečba (N=424)
90%
80%

70%
60%
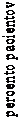
50%
40%
0 12 24 36 48 60 72 84 96 108 120
čas (hodiny)
V tej istej klinickej štúdii pokračovalo 744 pacientov v predĺžení s opakovanými cyklami až do 3
ďalších cyklov chemoterapie. Režim s aprepitantom si zachovala účinnosť počas všetkých cyklov.
5.2 Farmakokinetické vlastnostiFosaprepitant, proliečivo aprepitantu, sa po intravenóznom podaní rýchlo mení na aprepitant. Plazmatické koncentrácie fosaprepitantu sú do 30 minút po ukončení podávania infúzie pod hranicou merateľných hladín.
Aprepitant po podaní fosaprepitantuAUC aprepitantu po podaní 115 mg fosaprepitantu bola ekvivalentná s AUC perorálneho aprepitantu v dávke 125 mg, zatiaľ čo Cmax bola 2,5-násobne vyššia.
Po podaní jednorazovej intravenóznej dávky fosaprepitantu vo forme 15-minútovej infúzie zdravým dobrovoľníkom bola priemerná AUC0-24hod aprepitantu 19,8 µg•hod/ml a priemerná maximálna koncentrácia aprepitantu bola 3,26 µg/ml. Priemerné plazmatické koncentrácie aprepitantu od 4 hodín po podaní dávky (vrátane koncentrácie 24 hodín po podaní) boli u perorálneho aprepitantu v dávke
125 mg a intravenózneho fosaprepitantu v dávke 115 mg podobné.
DistribúciaAprepitant sa silne viaže na proteíny, v priemere 97 %. Geometrický priemer zdanlivého distribučného objemu v rovnovážnom stave (Vdss) je u ľudí približne 66 l.
MetabolizmusV
in vitro inkubáciách s ľudskými pečeňovými preparátmi bol fosaprepitant rýchlo konvertovaný na aprepitant. Okrem toho, fosaprepitant prešiel rýchlou a takmer úplnou konverziou na aprepitant v S9 preparátoch z iných ľudských tkanív vrátane obličiek, pľúc a ilea. Preto je pravdepodobné, že
ku konverzii fosaprepitantu na aprepitant môže dôjsť v mnohých tkanivách. U ľudí sa intravenózne podaný fosaprepitant rýchlo zmenil na aprepitant v priebehu 30 minút po ukončení podávania infúzie.
Aprepitant je značne metabolizovaný. U zdravých mladých dospelých po podaní jednorazovej intravenóznej 100-mg dávky proliečiva [14C]-aprepitantu tvorí aprepitant počas 72 hodín približne
19 % rádioaktivity v plazme, čo ukazuje na významnú prítomnosť metabolitov v plazme. V ľudskej plazme sa identifikovalo dvanásť metabolitov aprepitantu. Aprepitant sa do značnej miery
metabolizuje oxidáciou na morfolínovom kruhu a jeho vedľajších reťazcoch, a výsledné metabolity boli iba slabo aktívne. In vitro štúdie s použitím ľudských hepatálnych mikrozómov naznačujú, že aprepitant je primárne metabolizovaný CYP3A4 a potenciálne s malým prispením CYP1A2
a CYP2C19.
Všetky metabolity, ktoré sa po intravenóznom podaní [14C]-fosaprepitantu v dávke 100 mg vyskytli v moči, stolici a v plazme, boli pozorované aj po perorálnej dávke [14C]-aprepitantu. Po konverzii
115 mg fosaprepitantu na aprepitant sa z fosaprepitantu uvoľní 18,3 mg fosfátu.
Eliminácia
Aprepitant sa nevylučuje močom nezmenený. Metabolity sú vylučované v moči a prostredníctvom biliárnej exkrécie v stolici. Po podaní jednorazovej intravenóznej 100-mg dávky [14C]-fosaprepitantu zdravým jedincom sa 57 % rádioaktivity vylúčilo v moči a 45 % v stolici.
Farmakokinetika aprepitantu je v rozsahu klinického dávkovania nelineárna. Terminálny polčas aprepitantu po perorálnom podaní sa pohyboval v rozmedzí približne od 9 do 13 hodín.
Farmakokinetika u osobitných skupín pacientov
Farmakokinetika fosaprepitantu sa u osobitných skupín pacientov nehodnotila. Nie je predpoklad, že by v súvislosti s vekom alebo pohlavím došlo ku klinicky významným rozdielom vo farmakokinetike aprepitantu.
Pediatrickí pacienti: Farmakokinetika fosaprepitantu sa u pacientov mladších ako 18 rokov nehodnotila.
Hepatálna insuficiencia: Fosaprepitant sa metabolizuje v rôznych mimopečeňových tkanivách; preto sa neočakáva, že hepatálna insuficiencia zmení konverziu fosaprepitantu na aprepitant. Mierna hepatálna insuficiencia (Child-Pughovo skóre 5 až 6) nemá klinicky významný vplyv na farmakokinetiku aprepitantu. U pacientov s miernou hepatálnou insuficienciou nie je potrebná úprava dávkovania. Z dostupných údajov nie je možné vyvodiť závery týkajúce sa vplyvu stredne ťažkej hepatálnej insuficiencie (Child-Pughovo skóre 7 až 8) na farmakokinetiku aprepitantu. U pacientov
s ťažkou hepatálnou insuficienciou (Child-Pughovo skóre > 9) nie sú dostupné žiadne klinické alebo farmakokinetické údaje.
Renálna insuficiencia: Jednorazová 240-mg dávka perorálneho aprepitantu sa podala pacientom
s ťažkou renálnou insuficienciou (klírens kreatinínu <30 ml/min) a pacientom s terminálnym štádiom renálneho ochorenia (ESRD) vyžadujúcim hemodialýzu.
U pacientov s ťažkou renálnou insuficienciou poklesla AUC0-∞ celkového aprepitantu (viazaného aj neviazaného na proteíny) v porovnaní so zdravými jedincami o 21 % a Cmax poklesla o 32 %.
U pacientov s ESRD na hemodialýze poklesla AUC0-∞ celkového aprepitantu o 42 % a Cmax poklesla o 32 %. Vzhľadom na mierne zníženie väzby aprepitantu na proteíny u pacientov s renálnym ochorením nebola AUC farmakologicky aktívneho neviazaného aprepitantu u pacientov s renálnou insuficienciou v porovnaní so zdravými jedincami signifikantne ovplyvnená. Hemodialýza, ktorá prebehla 4 alebo 48 hodín po podaní dávky, nemala na farmakokinetiku aprepitantu žiaden signifikantný vplyv, v dialyzáte sa vylúčilo menej ako 0,2 % dávky.
U pacientov s renálnou insuficienciou alebo u pacientov s ESRD na hemodialýze nie je potrebná úprava dávkovania.
Vzťah medzi koncentráciou a účinkom
Fosaprepitant je proliečivo aprepitantu. PET zobrazovacie štúdie s použitím vysoko špecifického značkovača NK1-receptora u zdravých mladých mužov preukázali, že po podaní perorálneho aprepitantu, aprepitant preniká do mozgu a obsadzuje NK1 receptory v závislosti od dávky
a plazmatickej koncentrácie. Predpokladá sa, že plazmatické koncentrácie aprepitantu, ktoré sa dosahujú po 3-dňovom režime podávania perorálneho aprepitantu, zabezpečujú obsadenie viac ako
95 % mozgových NK1 receptorov. Vzťah medzi koncentráciou a účinkom sa po podaní fosaprepitantu nehodnotil.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané pri intravenóznom podávaní fosaprepitantu a perorálnom podávaní aprepitantu na základe obvyklých štúdií toxicity po jednorazovom a opakovanom podávaní, genotoxicity (vrátane in vitro testov) a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí. U laboratórnych zvierat spôsobil fosaprepitant v nekomerčných liekových formách vaskulárnu toxicitu a hemolýzu pri koncentráciách pod 1 mg/ml a vyšších, v závislosti od liekovej formy. Dôkaz
hemolýzy sa zistil tiež na ľudských premytých krvinkách s nekomerčnou liekovou formou fosaprepitantu v koncentráciách 2,3 mg/ml a vyšších, hoci testy s plnou ľudskou krvou boli negatívne. S komerčnou liekovou formou fosaprepitantu v koncentráciách do 1 mg/ml sa v plnej ľudskej krvi, ani na premytých ľudských krvinkách hemolýza nezistila.
Karcinogénny potenciál u hlodavcov sa skúmal len u perorálne podávaného aprepitantu. Treba však podotknúť, že význam štúdií toxicity vykonaných na hlodavcoch, králikoch a opiciach vrátane štúdií reprodukčnej toxicity je obmedzený, pretože systémové expozície fosaprepitantu a aprepitantu boli iba podobné alebo dokonca nižšie ako terapeutická expozícia u ľudí. Vo farmakologických štúdiách bezpečnosti a v štúdiách toxicity po opakovanej dávke vykonaných na psoch boli hodnoty Cmax fosaprepitantu 4-7-krát vyššie a AUC aprepitantu 60-70-krát vyššie ako klinické hodnoty.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Edetan disodný (E386) Polysorbát 80 (E433) Bezvodá laktóza
Hydroxid sodný (E524) (na úpravu pH) a/alebo
Zriedená kyselina chlorovodíková (E507) (na úpravu pH)
6.2 Inkompatibility
IVEMEND je inkompatibilný s akýmikoľvek roztokmi obsahujúcimi dvojmocné katióny (napr. Ca2+, Mg2+) vrátane Hartmanovho a Ringerovho laktátového roztoku. Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sa uvádzajú v časti 6.6.
6.3 Čas použiteľnosti
2 roky.
Po rekonštitúcii a zriedení bola chemická a fyzikálna stabilita preukázaná počas 24 hodín pri teplote
25°C.
Z mikrobiologického hľadiska sa má liek použiť ihneď. Ak sa nepoužije okamžite, zodpovednosť za čas a podmienky skladovania po príprave má používateľ a normálne by nemali byť dlhšie ako 24 hodín pri teplote 2 až 8ºC.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (pri teplote 2°C - 8°C).
Podmienky na uchovávanie rekonštituovaného a zriedeného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
10 ml injekčná liekovka z číreho skla typu I s chlórbutylovou alebo brómbutylovou gumovou zátkou a hliníkovou fóliou s modrým plastovým odklápacím viečkom.
IVEMEND je dostupný ako balenie po 1 injekčnej liekovke so 115 mg fosaprepitantu alebo
10 injekčných liekovkách so 115 mg fosaprepitantu.
Nie všetky veľkosti balenia musia byť uvedené do obehu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomŽiadne zvláštne požiadavky na likvidáciu.
IVEMEND sa pred podaním musí rekonštituovať a následne zriediť. Príprava na intravenózne podanie:
1. Do injekčnej liekovky preneste 5 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %).
Zabezpečte, aby bol injekčný roztok chloridu sodného 9 mg/ml (0,9 %) pridaný do injekčnej liekovky aplikáciou na stenu liekovky, aby sa predišlo peneniu. Injekčnou liekovkou jemne zakrúžte. Vyhnite sa traseniu a vstrekovaniu injekčného roztoku chloridu sodného 9 mg/ml (0,9 %) do injekčnej liekovky.
2. Pripravte infúzny vak naplnený 110 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %) (napríklad pridaním 10 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %) do infúzneho vaku so 100 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %)).
3. Natiahnite celý objem injekčnej liekovky a preneste ho do infúzneho vaku obsahujúceho 110 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), čím sa dosiahne celkový objem 115 ml. Vak 2-3 razy jemne prevráťte.
Tento liek sa nesmie rekonštituovať alebo miešať s roztokmi, pre ktoré nebola stanovená fyzikálna a chemická kompatibilita (pozri časť 6.2).
Vzhľad rekonštituovaného roztoku je rovnaký ako vzhľad rozpúšťadla.
Tento liek sa má pred podaním skontrolovať zrakom na prítomnosť cudzorodých častíc a zmenu farby.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Veľká Británia
8. REGISTRAČNÉ ČÍSLA9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.emea.europa.eu/.