
r>
Menej časté
|
Syndróm
posteriórnej reverzibilnej
encefalopatie
|
0,3
|
0,3
|
0
|
Porychy ucha
a labyrintu
|
Časté
|
Tinnitus
|
2,2
|
0
|
0
|
Poruchy ciev
|
Veľmi časté
|
Hypertenzia
|
39,3
|
15,3
|
0,3
|
Krvácanieb, c
|
10,6
|
0,3
|
0,3
|
Časté
|
Venózne embolické
a trombotické príhodyb, c
|
1,9
|
0,8
|
0,8
|
Arteriálne
embolické
a trombotické príhodyb, c
|
1,1
|
1,1
|
0
|
Menej časté
|
Hypertenzná kríza
|
0,6
|
0,3
|
0,3
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
|
Veľmi časté
|
Dysfónia
|
28,1
|
0
|
0
|
Časté
|
Dyspnoe
|
7,0
|
0,3
|
0
|
Kašeľ
|
5,3
|
0
|
0
|
Orofaryngeálna
bolesť
|
3,3
|
0
|
0
|
Poruchy
gastrointestinálneho traktu
|
Veľmi časté
|
Hnačka
|
51,3
|
9,7
|
0,3
|
Vracanie
|
16,7
|
1,4
|
0
|
Nevoľnosť
|
28,7
|
1,4
|
0
|
Stomatitída
|
14,5
|
1,4
|
0
|
Zápcha
|
12,3
|
0
|
0
|
Časté
|
Bolesť brucha
|
8,4
|
0,6
|
0,3
|
Bolesť v epigastriu
|
6,1
|
0,3
|
0
|
Dyspepsia
|
7,8
|
0
|
0
|
Flatulencia
|
4,5
|
0
|
0
|
Hemoroidy
|
2,2
|
0
|
0
|
Menej časté
|
Gastrointestinálna perforáciab, d
|
0,3
|
0
|
0,3
|
Análna fistulab
|
0,3
|
0
|
0
|
Poruchy kože
a podkožného tkaniva
|
Veľmi časté
|
Palmárno-plantárna
erytrodyzestézia
(syndróm ruka-noha)
|
27,3
|
5,0
|
0
|
Vyrážka
|
11,7
|
0,3
|
0
|
Suchosť kože
|
10,0
|
0
|
0
|
Časté
|
Pruritus
|
5,8
|
0
|
0
|
Erytém
|
2,2
|
0
|
0
|
Alopécia
|
3,3
|
0
|
0
|
Poruchy kostrovej
a svalovej sústavy a spojivového
tkaniva
|
Časté
|
Myalgia
|
5,3
|
0,6
|
0,3
|
Artralgia
|
8,6
|
0,6
|
0
|
Bolesť končatín
|
8,9
|
0,3
|
0
|
Trieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduce reakcie
|
V
šetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
|
Poruchy obličiek
a močových ciest
|
Veľmi časté
|
Proteinúria
|
10,3
|
3,1
|
0
|
Časté
|
Zlyhanie obličiek
|
1.1
|
0.6
|
0
|
Celkové poruchy
a reakcie v mieste podania
|
Veľmi časté
|
Únava
|
34,8
|
9,5
|
0,3
|
Asténiac
|
17,5
|
3,6
|
0,3
|
Zápal sliznice
|
15,0
|
1,4
|
0
|
Laboratórne
a funkčné vyšetrenia
|
Veľmi časté
|
Úbytok hmotnosti
|
16,4
|
1,4
|
0
|
Časté
|
Zvýšenie hladiny
tyreoideu stimulujúceho hormónu
|
4,5
|
0
|
0
|
Zvýšenie hladiny
lipázy
|
2,2
|
0,6
|
0
|
Zvýšenie hladiny
alanínamino transferázy
|
1,9
|
0,3
|
0
|
Zvýšenie hladiny
aspartátamino transferázy
|
1,1
|
0,3
|
0
|
Zvýšenie hladiny
alkalickej fosfatázy
|
1,4
|
0
|
0
|
Zvýšenie hladiny
amylázy
|
1,7
|
0
|
0
|
Menej časté
|
Zvýšenie hladiny
bilirubínu v krvi
|
0,6
|
0
|
0
|
Zvýšenie hladiny
kreatinínu
|
0,6
|
0
|
0
|
|
|
a Všeobecné terminologické kritériá pre nežiaduce účinky národného inštitútu pre rakovinu verzia 3.0 (CTCAE)
b Pozri časť Popis vybraných nežiaducich účinkov
c Boli hlásené fatálne prípady (5. stupeň závažnosti)
d Výskyt nežiaducej reakcie z akejkoľvek príčiny
e Vrátane zlyhania obličiek
Opis vybraných nežiaducich reakciíDysfunkcia štítnej žľazy (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola hypotyreóza hlásená
u 18,4 % pacientov a hypertyreóza u 0,6 % pacientov. Zvýšenie hladiny thyreostimulačného hormónu
(TSH) bolo ako nežiaduci účinok hlásené u 4,5 % pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho sledovania u pacientov, ktorí mali pred liečbou TSH < 5 μU/mL, sa
objavilo zvýšenie TSH až na ≥ 10 μU/mL u 32,2 % pacientov užívajúcich axitinib.
Venózne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli venózne embolické
a trombotické nežiaduce reakcie hlásené u 1,9 % pacientov užívajúcich axitinib. Venózne embolické a trombotické nežiaduce reakcie stupňa závažnosti 3/4 boli hlásené u 1,7 % pacientov užívajúcich axitinib (vrátane pľúcnej embólie, hlbokej venóznej trombózy a oklúzie/trombózy sietnicovej vény). Fatálna pľúcna embólia bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib.
Arteriálne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli arteriálne embolické
a trombotické nežiaduce reakcie stupňa závažnosti 3/4 hlásené u 1,1 % pacientov užívajúcich axitinib.
Najčastejšou arteriálnou embolické a trombotickou príhodou bol tranzitórny ischemický atak (0,8 %). Fatálna cerebrovaskulárna príhoda bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib.
V monoterapeutických klinických štúdiách s axitinibom (N=699) boli arteriálne embolické
a trombotické nežiaduce reakcie (vrátane tranzitórneho ischemického ataku, infarktu myokardu a cerebrovaskulárnej príhody) hlásené u 1,0 % pacientov užívajúcich axitinib.
Polycytémia (pozri
Elevácia hemoglobínu alebo hematokritu v časti 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola polycytémia hlásená ako nežiaduca reakcia u 0,3 % pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho
sledovania bola zvýšená hladina hemoglobínu nad ULN zistená u 9,7 % pacientov užívajúcich
axitinib. V štyroch klinických štúdiách s axitinibom v liečbe pacientov s RCC (N=537) bola zvýšená hladina hemoglobínu nad ULN pozorovaná u 13,6 % pacientov užívajúcich axitinib.
Hemorágia (pozri časť 4,4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC, kam neboli zaradení pacienti s neliečenými metastázami v mozgu, boli hlásené hemoragické nežiaduce reakcie u 10,6 % pacientov
užívajúcich axitinib. Najčastejšími hemoragickými nežiaducimi reakciami u pacientov liečených
axitinibom boli epistaxa (5,3 %), hematúria (1,4 %), krvácanie z konečníka (1,1 %) a krvácanie z ďasien (1,1 %). Hemoragické nežiaduce reakcie stupňa závažnosti ≥ 3 boli hlásené u 0,8 %
pacientov užívajúcich axitinib (vrátane cerebrálneho krvácania, žalúdočného krvácania a krvácania
z distálneho gastrointestinálneho traktu). Fatálna hemorágia bola hlásená u jedného pacienta (0,3 %), ktorý užíval axitinib (žalúdočné krvácanie). V monoterapeutických klinických štúdiách s axitinibom (N=699) bola hemoptýza hlásená ako nežiaduca reakcia u 1,6 % pacientov, vrátane jedného prípadu (0,1 %) so stupňom závažnosti ≥ 3.
Gastrointestinálna perforácia a tvorba fistúl (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola gastrointestinálna perforácia hlásená u jedného pacienta (0,3 %, bez ohľadu na kauzalitu), ktorý užíval axitinib.
V monoterapeutických klinických štúdiách s axitinibom (N=699) boli fistuly hlásené u 0,7 %
pacientov (bez ohľadu na kauzalitu) a fatálna gastrointestinálna perforácia bola hlásená u jedného pacienta (0,1 %).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v Prílohe V.
4.9 PredávkovanieNeexistuje žiadna špecifická liečba v prípade predávkovania axitinibom.
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC jeden pacient, ktorý neúmyselne užíval dávku 20 mg dvakrát denne počas 4 dní, udával závraty (stupeň závažnosti 1).
V klinickej štúdii s axitinibom na stanovenie dávky probandi, ktorí užívali úvodnú dávku 10 mg dvakrát denne alebo 20 mg dvakrát denne mali nežiaduce reakcie, ktoré zahŕňali hypertenziu, záchvaty spojené s hypertenziou a fatálnu hemoptýzu.
V prípade podozrenia z predávkovania je potrebné vysadiť axitinib a začať podpornú starostlivosť.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
F
armakoterapeutická skupina: Cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE17
Spôsob účinku
Axitinib je účinný a selektívny inhibítor tyrozínkinázy receptorov vaskulárneho endoteliálneho rastového faktora (VEGFR-1, VEGFR-2 a VEGFR-3). Tieto receptory sa podieľajú na patologickej
angiogenéze, raste nádoru a metastatickej progresii karcinómu. Preukázalo sa, že axitinib účinne
inhibuje proliferáciu endoteliálnych buniek a ich prežívanie sprostredkované VEGF. Axitinib inhiboval fosforyláciu VEGFR-2 nádorovej vaskulatúry xenograftu, ktorá predstavovala cieľ liečby in vivo a viedol k oddialeniu rastu nádoru, regresii a inhibícii metastáz v mnohých experimentálnych modeloch rakoviny.
Vplyv na QTc interval
V randomizovanej dvojramennej štúdii s prekrížením liečebných ramien bola 35 zdravým probandom podaná jedna perorálna dávka axitinibu (5 mg) bez podávania a s podávaním 400 mg ketokonazolu
počas 7 dní. Výsledky tejto štúdie ukázali, že expozícia axitinibu v plazme až dvojnásobne vyššia ako očakávané terapeutické hladiny pri 5 mg dávkovaní neviedla ku klinicky signifikantnému predĺženiu
QT intervalu.
Klinickáúčinnosť
Bezpečnosť a účinnosť axitinibu sa hodnotili v randomizovanej, otvorenej multicentrickej klinickej štúdii fázy 3. Pacienti (N=723) s pokročilým RCC, ktorých ochorenie progredovalo počas alebo po liečbe jednou predchádzajúcou systémovou terapiou, zahrňujúcou režimy so sunitinibom, bevacizumabom, temsirolimom alebo cytokín-obsahujúce , boli randomizovaní (1:1) na užívanie axitinibu (n=361) alebo sorafenibu (n=362). Primárny cieľ, prežívanie bez progresie (PFS) sa hodnotil centrálne metódou zaslepeného nezávislého posúdenia. Medzi sekundárne ciele patrili miera objektívnej odpovede (ORR) a celkové prežívanie (OS).
Z pacientov zaradených do tejto klinickej štúdie dostalo 389 pacientov (53,8 %) jednu predchádzajúcu liečbu založenú na sunitinibe, 251 pacientov (34,7 %) dostalo jednu predchádzajúcu liečbu založenú
na cytokínoch (interleukin-2 alebo interferón-alfa), 59 pacientov (8,2%) dostalo jednu predchádzajúcu liečbu založenú na bevacizumabe a 24 pacientov (3,3 %) dostalo jednu predchádzajúcu liečbu
založenú na temsirolime. Demografické a nádorové charakteristiky na začiatku štúdie boli podobné medzi liečebnými skupinami s axitinibom a sorafenibom čo sa týka veku, pohlavia, rasy,
výkonnostného stavu podľa ECOG kritérií (Eastern Cooperative Oncology Group), geografického regiónu a predchádzajúcej liečby.
V celej populácii pacientov a dvoch hlavných podskupinách (skupina s predchádzajúcou liečbou sunitinibom a skupina s predchádzajúcou liečbou cytokínmi) pre primárny cieľ hodnotenia PFS bol preukázaný štatisticky signifikantný prínos axitinibu oproti sorafenibu (pozri Tabuľka 2 a Obrázky 1,
2 and 3). Hodnota mediánu PFS účinku bol v oboch podskupinách rozdelených podľa predchádzajúcej
liečby odlišný. Dve podskupiny boli príliš malé, aby poskytli dôveryhodné výsledky (skupina s predchádzajúcou liečbou temsirolimom a skupina s predchádzajúcou liečbou bevacizumabom).
Medzi liečebnými ramenami neboli žiadne štatisticky signifikantné rozdiely v OS v celkovej populácii ani v podskupinách rozdelených podľa predchádzajúcej liečby.
Tabuľka 2. Výsledky účinnosti
Cieľ / Populácia štúdie
|
axitinib
|
sorafenib
|
HR
(95% CI)
|
p-hodnota
|
|
Celá ITT populácia
Medián PFS v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
|
N = 361
6,8 (6,4; 8,3)
20,1 (16,7; 23,4)
|
N = 362
4,7 (4,6; 6,3)
19,2 (17,5; 22,3)
|
0,67 (0,56; 0,81)
0,97 (0,80; 1,17)
|
< 0,0001
NS
|
ORR b,e% (95% CI)
|
19,4 (15,4; 23,9)
|
9,4 (6,6; 12,9)
|
2,06f (1,41; 3,00)
|
0,0001g
|
Predchádzajúca liečba sunitinibom
|
N = 194
|
N = 195
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
4,8 (4,5; 6,5)
15,2 (12,8; 18,3)
11,3 (7,2; 16,7)
|
3,4 (2,8; 4,7)
16,5 (13,7; 19,2)
7,7 (4,4; 12,4)
|
0,74 (0,58; 0,94)
1,00 (0,78; 1,27)
1,48f (0,79; 2,75)
|
0,0063h
NS
|
Predchádzajúca liečba
cyto
k
ínmi
|
N = 126
|
N = 125
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
12,0 (10,1; 13,9)
29,4 (24,5; NE)
32,5 (24,5; 41,5)
|
6,6 (6,4; 8,3)
27,8 (23,1;, 34,5)
13,6 (8,1; 20,9)
|
0,52 (0,38;, 0,72)
0,81 (0,56; 1,19)
2,39f (1,43;3,99)
|
< 0,0001h
NS
0,0002i
|
CI =interval spoľahlivosti, HR miera rizika (axitinib/sorafenib); ITT: všetci pacienti zaradení do štúdie); NE = nemožné predpovedať; NS = štatisticky nesignifikantné; ORR Miera objektívnej
odpovede;.OS: = celkové prežívanie PFS : Prežívanie bez progresie.
a Čas od randomizácie po progresiu alebo úmrtie z akejkoľvek príčiny, podľa toho, ktorá udalosť
nastane prvá. Dátum ukončenia: 3. jún 2011.
b Stanovené podľa RECIST kritérií nezávislým rádiologickým posudkom.
c Jednostranná hodnota p podľa log-rank testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej liečby d Dátum ukončenia: 1. november 2011.
e Dátum ukončenia: 31. august 2010.
f Pre ORR je použitá veličina pomer rizík. Pomer rizík > 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene s axitinibom; pomer rizík < 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene so sorafenibom.
g Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej terapie
h Jednostranná hodnota p log-rank testu pre liečbu so stratifikáciou podľa ECOG výkonnostného stavu
i Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu
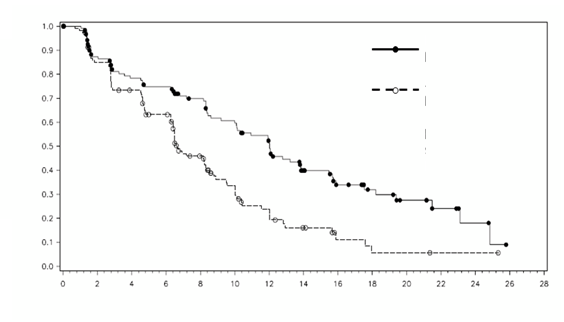
 Obrázok 1. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre celkovú populáciu
INLYTA (N=361)
m
e
d
i
á
n 6,8 mesiaca
sorafenib (N=362)
m
e
d
i
á
n 4,7 mesiaca
m
i
er
a rizika = 0,67
95
% CI [0,56, 0,81]
p hodnota <0,0001
Obrázok 1. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre celkovú populáciu
INLYTA (N=361)
m
e
d
i
á
n 6,8 mesiaca
sorafenib (N=362)
m
e
d
i
á
n 4,7 mesiaca
m
i
er
a rizika = 0,67
95
% CI [0,56, 0,81]
p hodnota <0,0001

Čas (mesiace)
Obrázok 2. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre podskupinu s predchádzajúcou liečbou sunitinibomINLYTA (N=194)medián 4,8 mesiacasorafenib (N=195)medián 3,4 mesiacamiera rizika = 0,7495% CI [0,58, 0,94]p hodnota = 0,0063
podskupinu s predchádzajúcou liečbou sunitinibomINLYTA (N=194)medián 4,8 mesiacasorafenib (N=195)medián 3,4 mesiacamiera rizika = 0,7495% CI [0,58, 0,94]p hodnota = 0,0063
Čas (mesiace)
 Obrázok 3. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre podskupinu s predchádzajúcou liečbou
c
ytokínom.
INLYTA (N=126)
m
e
d
i
á
n
12
,
0 mesiaca
sorafenib (N=125)
m
e
d
i
á
n 6,6 mesiaca
m
i
er
a rizika = 0,52
95
% CI [0,38, 0,72]
p hodnota < 0,0001
Obrázok 3. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre podskupinu s predchádzajúcou liečbou
c
ytokínom.
INLYTA (N=126)
m
e
d
i
á
n
12
,
0 mesiaca
sorafenib (N=125)
m
e
d
i
á
n 6,6 mesiaca
m
i
er
a rizika = 0,52
95
% CI [0,38, 0,72]
p hodnota < 0,0001
Čas (mesiace)
 Deti a dospievajúci
Deti a dospievajúciEurópska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií pre axitinib vo všetkých vekových podskupinách detí a dospievajúcich v liečbe karcinómu obličky a obličkovej panvičky (okrem nefroblastómu, nefroblastomatózy, sarkómu z jasných buniek, mezoblastického nefrómu, obličkového medulárneho karcinómu a rabdoidného nádoru obličky) (pozri časť 4.2 pre informácie o použití v pediatrii).
5.2 Farmakokinetické vlastnostiPo perorálnom podaní tabliet axitinibu je priemerná absolútna biologická dostupnosť 58 %
v porovnaní s intravenóznym podaním. Plazmatický polčas axitinibu kolíše od 2,5 do 6,1 hodiny. Dávkovanie axitinibu po 5 mg dvakrát denne viedlo k menej než dvojnásobnému nahromadeniu
v porovnaní s podaním v jednej dávke. Vzhľadom na krátky eliminačný polčas axitinibu sa očakáva
dosiahnutie rovnovážneho stavu v priebehu 2 až 3 dní po úvodnej dávke.
Absorpcia a distribúciaMaximálne koncentrácie axitinibu v plazme sa obvykle dosiahnu v priebehu 4 hodín po perorálnom podaní axitinibu s mediánom Tmax v rozmedzí od 2,5 do 4,1 hodiny. Podávanie axitinibu so stredne mastným jedlom viedlo k zníženiu expozície o 10 % v porovnaní s celonočným hladovaním. Veľmi mastné, vysoko kalorické jedlo viedlo k zvýšeniu expozície o 19% v porovnaní s celonočným hladovaním. Axitinib môže byť užívaný s jedlom alebo bez jedla (pozri časť 4.2).
Priemerná Cmax a AUC sa proporcionálne zvýšili v závislosti na dávkovaní axitinibu v rozsahu 5 až
10 mg.
In vitro väzba axitinibu na ľudské plazmatické proteíny je > 99 % s prednostným naviazaním
na albumín a strednou väzbou na α1-kyslý glykoproteín. Pri dávkovaní 5 mg dvakrát denne u pacientov s pokročilým RCC bola v stave nasýtenosti geometrická priemerná maximálna plazmatická koncentrácia 27,8 ng/mL a 24-hodinová AUC 265 ng.h/mL. Po perorálnom podaní bol geometrický priemerný klírens 38 l/h a distribučný objem bol 160 litrov.
Biotransformácia a elimináciaAxitinib sa metabolizuje primárne v pečeni enzýmom CYP3A4/5 a v menšej miere enzýmami
CYP1A2, CYP2C19 a UGT1A1.
Po perorálnom podaní 5 mg dávky rádioaktívne značeného axitinibu bolo 30-60 % rádioaktivity zachytenej v stolici a 23 % rádioaktivity v moči. Nemetabolizovaný axitinib, na úrovni 12 % z dávky, bol hlavnou zložkou identifikovanou v stolici. Nemetabolizovaný axitinib nebol zistený v moči; metabolity kyseliny karboxylovej a sulfoxidu predstavovali väčšinu rádioaktivity v moči. V plazme predstavoval N-glukuronidový metabolit predominantnú rádioaktívnu zložku (50 % rádioaktivity
v cirkulácii) a nemetabolizovaný axitinib a sulfoxidový metabolit každý jeden tvorili približne
20 % rádioaktivity v cirkulácii.
Sulfoxidové a N-glukuronidové metabolity vykazujú približne 400-násobne a 8000-násobne menšiu účinnosť in vitro voči VEGFR-2 v porovnaní s axitinibom.
Osobitné skupiny pacientov
Starší pacienti, pohlavie a rasa
Populačné farmakokinetické analýzy u pacientov s pokročilým karcinómom (vrátane pokročilého
RCC) a zdravých dobrovoľníkov ukazujú, že vek, pohlavie, telesná hmotnosť, rasa, obličkové funkcie, genotyp UGT1A1 alebo CYP2C19 nemajú žiadne klinicky relevantné účinky.
Deti a dospievajúci
Axitinib sa nesledoval u pacientov mladších ako 18 rokov.
Porucha funkcie pečene
In vitro a in vivo údaje ukazujú, že axitinib je primárne metabolizovaný v pečeni.
V porovnaní s pacientami s normálnou funkciou pečene je systémová expozícia po podaní jednej dávky axitinibu podobná u subjektov s ľahkou poruchou funkcie pečene (trieda A klasifikácie podľa Childa-Pugha) a vyššia (približne dvojnásobne) u pacientov so stredne závažnou poruchou funkcie pečene (trieda B klasifikácie podľa Childa-Pugha). Axitinib sa nesledoval u pacientov so závažnou poruchou funkcie pečene (trieda C klasifikácie podľa Childa-Pugha) a nemá sa používať v tejto populácii (odporúčania pre úpravu dávkovania pozri časť 4.2).
Porucha funkcie obličiek
Nemetabolizovaný axitinib nie je detekovaný v moči.
Axitinib sa nesledoval u pacientov s poruchou funkcie obličiek. V klinických štúdiách s axitinibom
v liečbe pacientov s RCC neboli zaradení pacienti s hladinou sérového kreatinínu > 1,5 násobok ULN alebo kalkulovaným klírensom kreatinínu < 60 mL/min. Populačné farmakokinetické analýzy preukázali, že klírens axitinibu nebol zmenený u pacientov s poruchou funkcie obličiek a nevyžaduje sa žiadna úprava dávkovania.
5.3 Predklinické údaje o bezpečnosti
Toxicita pri opakovanom podávaní
Hlavné prejavy toxicity u myší a psov pri opakovanom podávaní v trvaní do 9 mesiacov boli v gastrointestinálnom, hematopoetickom, reprodukčnom, skeletálnom a dentálnom systéme,
s hladinami bez pozorovaných nežiaducich účinkov (NOAEL – No Observed Adverse Effect Levels)
približne rovnakými alebo nižšími ako je očakávaná expozícia u človeka pri odporúčanej klinickej úvodnej dávke (podľa hladín AUC).
Karcinogenicita
Nevykonali sa žiadne klinické štúdie skúmajúce karcinogenitu axitinibu.
Genotoxicita
Axitinib nebol mutagénny alebo klastogénny v konvenčných skúškach genotoxicity in vitro. Signifikantné zvýšenie polyploidity bolo pozorované in vitro pri koncentráciách > 0,22 μg/mL
a vzostup počtu mikrojadrových polychromatických erytrocytov sa pozoroval in vivo pri hladine bez
pozorovaného účinku na organizmus (NOEL - No Observed Effect Level) 69-násobne presahujúcej
očakávanú expozíciu u človeka. Nálezy genotoxicity sa nepovažujú za klinicky relevantné pri expozičných hladinách pozorovaných u ľudí.
Reprodukčná toxicita
Medzi nálezy súvisiace s axitinibom v semenníkoch a nadsemenníkoch patrili zníženie hmotnosti orgánu, atrofia alebo degenerácia, zníženie počtu germinálnych buniek, hypospermia alebo výskyt
abnormálnych foriem spermií a znížená denzita a počet spermií. Tieto nálezy sa pozorovali u myší pri
expozičných hladinách rovnajúcich sa približne 12-násobku očakávanej expozície u ľudí, u psov pri expozičných hladinách nižších ako je očakávaná expozícia u ľudí. Nepozoroval sa vplyv na párenie
alebo fertilitu u samčích myší pri expozičných hladinách rovnajúcich sa približne 57-násobku
očakávanej expozície u ľudí. K nálezom u samičiek patrili známky oneskorenia pohlavnej dospelosti, zmenšenie alebo chýbanie corpora lutea, zníženie hmotnosti maternice a atrofia maternice pri expozíciách približne rovnakých ako je očakávaná expozícia u ľudí. Znížená fertilita a viabilita embryí boli pozorované pri všetkých testovaných dávkach s expozičnými hladinami na najnižšej dávke rovnajúcimi sa približne 10-násobku očakávanej expozície u ľudí.
U gravidných myší vystavených účinku axitinibu bol preukázaný zvýšený výskyt rázštepu podnebia a skeletálnych variácií, vrátane oneskorenej osifikácie pri expozičných hladinách nižších ako očakávaná expozícia u ľudí. Štúdie perinatálnej a postnatálnej vývojovej toxicity neboli realizované.
Prejavy toxicity u nezrelých zvierat
U myší a psov, ktorí dostávali axitinib najmenej 1 mesiac pri expozičných hladinách približne
6-násobne vyšších než očakávané expozičné hladiny u ľudí bola pozorovaná reverzibilná dysplázia rastovej platničky. U myší užívajúcich axitinib viac než 1 mesiac pri expozičných hladinách
podobných očakávanej expozícii u ľudí bol pozorovaný výskyt čiastočne reverzibilných zubných
kazov. Iné toxické prejavy s možným rizikom pre pediatrických pacientov sa u juvenilných zvierat neskúmali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety mikrokryštalická celulóza monohydrát laktózy kroskarmelóza sodná stearan horečnatý
Obaľujúci film tablety hypromelóza
oxid titaničitý (E171)
monohydrát laktózy triacetín (E1518)
červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Hliníkový/hliníkový blister obsahujúci 14 filmom obalených tabliet. Každé balenie obsahuje 28 alebo
56 filmom obalených tabliet.
HDPE fľaštička s vysušovadlom obsahujúcim silikagél na absorpciu vlhkosti a polypropylénovým uzáverom obsahujúca 180 filmom obalených tabliet.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciuNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited
Ramsgate Road
Sandwich, Kent, CT13, 9NJ Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/12/777/001
EU/1/12/777/002
EU/1/12/777/003
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 3. 9. 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUInlyta 3 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá filmom obalená tableta obsahuje 3 mg axitinibu.
Pomocné látky so známym účinkom:Každá filmom obalená tableta obsahuje 35,3 mg monohydrátu laktózy. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAFilmom obalená tableta (tableta).
Červená okrúhla filmom obalená tableta s vyrytým nápisom “Pfizer“ na jednej strane a nápisom
“3 XNB“ na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieInlyta je indikovaná na liečbu dospelých pacientov s pokročilým karcinómom z obličkových buniek
(renal cell carcinoma, RCC) po zlyhaní predchádzajúcej liečby sunitinibom alebo cytokínmi.
4.2 Dávkovanie a spôsob podávaniaLiečbu Inlytou má vykonávať lekár, ktorý má skúsenosti s podávaním protinádorových liekov.
DávkovanieOdporúčaná úvodná dávka axitinibu je 5 mg dvakrát denne.
Liečba má pokračovať, pokým sa pozoruje klinický prínos alebo pokiaľ sa neprejaví neprijateľná toxicita, ktorú nie je možné zvládnuť súbežným podávaním liekov alebo úpravou dávky.
Pokiaľ pacient vracia alebo vynechá dávku, nesmie užiť dodatočnú dávku. Nasledujúca predpísaná dávka sa musí užiť v obvyklom čase.
Úpravy dávkyZvýšenie alebo zníženie dávky sa odporúča na základe individuálnej bezpečnosti a znášanlivosti.
U pacientov, ktorí tolerujú axitinib v úvodnej dávke 5 mg dvakrát denne bez nežiaducich účinkov
> ako stupeň 2 (t.j. bez závažných nežiaducich účinkov podľa Všeobecných terminologických kritérií pre nežiaduce účinky [CTCAE - Common Terminology Criteria for Adverse Events] verzia 3.0) počas dvoch po sebe idúcich týždňov, je možné dávku zvýšiť na 7 mg dvakrát denne, pokiaľ nie je krvný
tlak pacienta > 150/90 mmHg alebo pacient neužíva antihypertenzívnu liečbu. Následne podľa
rovnakých kritérií u pacientov, ktorí tolerujú axitinib v dávke 7 mg dvakrát denne, je možné zvýšiť ich dávku na maximálne 10 mg dvakrát denne.
Zvládnutie niektorých nežiaducich účinkov si môže vyžiadať prechodné alebo trvalé prerušenie liečby a/alebo zníženie dávky axitinibu (pozri časť 4.4). Ak je potrebné znížiť dávku, dávka axitinibu môže byť znížená na 3 mg dvakrát denne a ďalej na 2 mg dvakrát denne.
Úprava dávkovania nie je potrebná kvôli veku, rase, pohlaviu alebo telesnej hmotnosti.
Súčasné užívanie silných inhibítorov CYP3A4/5
Súčasné podávanie axitinibu so silnými inhibítormi CYP3A4/5 môže zvyšovať plazmatické koncentrácie axitinibu (pozri časť 4.5). Odporúča sa výber alternatívneho súčasne podávaného lieku so žiadnym alebo minimálnym potenciálom inhibície CYP3A4/5.
Hoci u pacientov užívajúcich silné inhibítory CYP3A4/5 sa neskúmala úprava dávkovania axitinibu, ak sa musí súčasne podávať silný inhibítor CYP3A4/5, odporúča sa zníženie dávky axitinibu na približne polovičnú dávku (t.j. úvodná dávka by mala byť znížená z 5 mg dvakrát denne na 2 mg dvakrát denne). Zvládnutie niektorých nežiaducich účinkov si môže vyžiadať prechodné alebo trvalé prerušenie liečby axitinibom (pozri časť 4.4). Ak sa ukončí súčasné podávanie silného inhibítora, je potrebné zvážiť návrat k dávke axitinibu užívanej pred začiatkom podávania silného inhibítora CYP3A4/5 (pozri časť 4.5).
Súčasné užívanie silných induktorov CYP3A4/5
Súčasné podávanie axitinibu so silnými induktormi CYP3A4/5 môže znižovať plazmatické koncentrácie axitinibu (pozri časť 4.5). Odporúča sa výber alternatívneho súčasne podávaného lieku so žiadnym alebo minimálnym potenciálom indukcie CYP3A4/5.
Hoci u pacientov užívajúcich silné induktory CYP3A4/5 saneskúmala úprava dávkovania axitinibu, ak sa musí súbežne podávať silný induktor CYP3A4/5, odporúča sa postupné zvyšovanie dávky
axitinibu. Maximálna indukcia pri vysokých dávkach silných induktorov CYP3A4/5 sa pozorovala
v priebehu jedného týždňa liečby induktorom. Ak sa dávka axitinibu zvýši, treba pacienta starostlivo monitorovať kvôli toxicite. Zvládnutie niektorých nežiaducich účinkov môže vyžadovať prechodné alebo trvalé prerušenie liečby a/alebo zníženie dávky axitinibu (pozri časť 4.4). Ak sa ukončí súbežné podávanie silného induktora, je treba okamžite upraviť dávku axitinibu na dávku užívanú pred začiatkom podávania silného induktora CYP3A4/5 (pozri časť 4.5).
Osobitné skupiny pacientov
Starší pacienti (≥ 65 rokov)
Nie je potrebná žiadna úprava dávkovania (pozri časti 4.4 a 5.2).
Poškodenie funkcie obličiek: Nevyžaduje sa úprava dávkovania (pozri časť 5.2). Nie sú dostupné prakticky žiadne údaje týkajúce sa liečby axitinibom u pacientov s klírensom kreatinínu < 15 ml/min.
Poškodenie funkcie pečene: Nevyžaduje sa úprava dávkovania, ak sa axitinib podáva pacientom
s ľahkým poškodením pečeňových funkcií (trieda A klasifikácie Childa-Pugha). Zníženie dávky sa odporúča pri podávaní axitinibu pacientom so stredne závažným poškodením pečeňových funkcií (trieda B klasifikácie Childa-Pugha) (t.j. úvodná dávka sa má znížiť z 5 mg dvakrát denne na 2 mg dvakrát denne). Axitinib sa neskúmal u pacientov so závažným poškodením pečeňových funkcií (trieda C klasifikácie podľa Childa-Pugha) a nemá sa preto používať v tejto skupine pacientov (pozri časti 4.4. a 5.2).
Deti a dospievajúci
Bezpečnosť a účinnosť axitinibu u detí a dospievajúcich < 18 rokov nebola stanovená. Nie sú dostupné žiadne údaje.
Spôsob podania
Axitinib sa má užívať perorálne dvakrát denne v približne 12-hodinových intervaloch s alebo bez jedla
(pozri časť 5.2). Tablety axitinibu sa majú prehltnúť celé a zapiť pohárom vody.
4.3 Kontraindikácie
Precitlivenosť na axitinib alebo na niektorú z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Pred začatím a pravidelne počas liečby axitinibom je potrebné sledovať typické príhody týkajúce sa bezpečnosti liečby ako sú uvedené ďalej.
Hypertenzia
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC sa veľmi často hlásila hypertenzia (pozri časť 4.8). Medián času do vzniku hypertenzie (systolický tlak krvi > 150 mmHg
alebo diastolický tlak krvi > 100 mmHg) sa dosiahol v priebehu prvého mesiaca od začiatku liečby
axitinibom a vzostupy krvného tlaku sa pozorovali už po 4 dňoch od začiatku užívania axitinibu.
Už pred začiatkom liečby axitinibom sa musí krvný tlak dobre kontrolovať. Pacienti sa majú sledovať kvôli hypertenzii a podľa potreby liečiť štandardnou antihypertenzívnou liečbou. V prípade pretrvávania hypertenzie napriek užívaniu antihypertenzívnych liekov sa musí znížiť dávka axitinibu. U pacientov, u ktorých sa vyvinie závažná hypertenzia, dočasne prerušte liečbu axitinibom
a pokračujte nižšou dávkou, keď je pacient normotenzný. Ak je liečba axitinibom prerušená, musia sa pacienti užívajúci antihypertenzívne lieky sledovať kvôli hypotenzii (pozri časť 4.2).
V prípade závažnej alebo pretrvávajúcej artériovej hypertenzie a príznakov pripomínajúcich syndróm posteriórnej reverzibilnej encefalopatie (viď nižšie) treba zvážiť diagnostické vyšetrenie mozgu magnetickou rezonanciou (MRI).
Dysfunkcia štítnej žľazy
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC sa hlásili prípady hypotyreózy a v menšom rozsahu prípady hypertyreózy (pozri časť 4.8).
Funkcia štítnej žľazy sa má vyšetriť ešte pred začiatkom a pravidelne kontrolovať počas liečby axitinibom. Hypotyreóza alebo hypertyreóza sa majú liečiť podľa štandardných terapeutických postupov s cieľom udržaťeutyreoidný stav.
Arteriálne embolické a trombotické príhody
V klinických štúdiách s axitinibom boli hlásené arteriálne embolické a trombotické príhody (vrátane tranzitórneho ischemického ataku, infarktu myokardu, cerebrovaskulárnej príhody a oklúzie
sietnicovej artérie) (pozri časť 4.8).
Axitinib sa má preto používať opatrne u pacientov, ktorí majú zvýšené riziko vzniku alebo sa u nich už vyskytli takéto príhody. Axitinib sa neskúmal u pacientov, ktorí mali arteriálnu embolickú
a trombotickú príhodu počas predchádzajúcich 12 mesiacov.
Venózne embolické a trombotické príhody
V klinických štúdiách s axitinibom boli hlásené venózne embolické a trombotické príhody (vrátane pľúcnej embólie, hlbokej žilovej trombózy a oklúzie/trombózy sietnicovej vény) (pozri časť 4.8).
Axitinib sa má preto používať opatrne u pacientov, ktorí majú zvýšené riziko vzniku alebo sa u nich už vyskytli takéto príhody. Axitinib sa neskúmal u pacientov, ktorí mali venóznu embolickú
a trombotickú príhodu počas predchádzajúcich 6 mesiacov.
Elevácia hemoglobínu alebo hematokritu
Počas liečby axitinibom sa môže vyskytnúť vzostup hemoglobínu alebo hematokritu ako prejav zvýšenia množstva červených krviniek (pozri časť 4.8, polycytémia). Zvýšenie množstva červených krviniek môže zvýšiť riziko vzniku embolických a trombotických príhod.
Hladina hemoglobínu alebo hodnota hematokritu sa majú vyšetriť ešte pred začiatkom a pravidelne kontrolovať počas liečby axitinibom. Ak dôjde k zvýšeniu hladiny hemoglobínu alebo hodnoty hematokritu nad normálnu úroveň, pacienti sa majú liečiť štandardnými terapeutickými postupmi
s cieľom znížiť hladinu hemoglobínu alebo hodnotu hematokritu na prijateľnú úroveň.
Hemorágia
V klinických štúdiách s axitinibom boli hlásené hemoragické príhody (pozri časť 4.8).
Axitinib sa neskúmal u pacientov s dokázanými neliečenými metastázami v mozgu alebo nedávnym aktívnym gastrointestinálnym krvácaním a nemá sa u týchto pacientov používať. Ak si akékoľvek krvácanie vyžaduje medicínsky zásah, dočasne prerušte podávanie axitinibu.
Gastrointestinálna perforácia a tvorba fistúl
V klinických štúdiách s axitinibom boli hlásené prípady gastrointestinálnej perforácie alebo vzniku fistúl (pozri časť 4.8).
Počas liečby axitinibom je potrebné sledovať príznaky svedčiace pre gastrointestinálnu perforáciu alebo vznik fistuly.
Komplikácie spojené s hojením rán
Neuskutočnili sa žiadne formálne klinické štúdie sledujúce vplyv axitinibu na hojenie rán.
Liečbu axitinibom sa má prerušiť najmenej 24 hodín pred plánovaným chirurgickým zákrokom. Rozhodnutie pokračovať v liečbe axitinibom po chirurgickom zákroku sa má opierať o klinické posúdenie adekvátneho hojenia rany.
Syndróm posteriórnej reverzibilnej encefalopatie
V klinických štúdiách s axitinibom boli hlásené prípady syndrómu posteriórnej reverzibilnej encefalopatie (PRES) (pozri časť 4.8).
PRES je neurologické ochorenie, ktoré sa môže prejaviť bolesťou hlavy, záchvatom, letargiou, zmätenosťou, slepotou a ďalšími zrakovými a neurologickými poruchami. Môže byť prítomná ľahká až závažná hypertenzia. Na potvrdenie diagnózy PRES je nevyhnutná magnetická rezonancia.
U pacientov so znakmi alebo príznakmi PRES prechodne prerušte alebo definitívne ukončite liečbu axitinibom. Bezpečnosť opätovného začatia liečby axitinibom u pacientov s anamnézou PRES nie je
známa.
Proteinúria
V klinických štúdiách s axitinibom bola hlásena proteinúria, vrátane stupňa závažnosti 3 (pozri časť
4.8).
Odporúča sa vyšetriť proteinúriu pred začiatkom liečby a kontrolovať ju pravidelne počas liečby axitinibom. Ak sa u pacientov objaví stredne závažná až závažná proteinúria, znížte dávku alebo dočasne prerušte liečbu axitinibom (pozri časť 4.2).
Nežiaduce účinky súvisiace s pečeňou
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli hlásené účinky súvisiace s pečeňou. Najčastejšie uvádzané nežiaduce účinky súvisiace s pečeňou zahŕňali zvýšenie hladiny
alanínaminotransferázy (ALT), aspartátaminotransferázy (AST) a bilirubínu v krvi (pozri časť 4.8).
Nepozorovali sa súbežné zvýšenia hladiny ALT (> 3-násobok hornej hranice normy [upper limit of normal, ULN]) a bilirubínu (> 2-násobok ULN).
V klinickej štúdii na stanovenie dávky boli súbežné zvýšenia hladín ALT (12-násobok ULN)
a bilirubínu (2,3-násobok ULN), považované za hepatotoxicitu súvisiacu s liekom, pozorované
u 1 pacienta, ktorý užíval axitinib v úvodnej dávke 20 mg dvakrát denne (4-násobok odporúčanej úvodnej dávky).
Je potrebné vyšetriť pečeňové testy pred začiatkom liečby a kontrolovať pravidelne počas liečby axitinibom.
Poškodenie pečene
V klinických štúdiách s axitinibom bola systémová expozícia axitinibu približne 2-násobne vyššia u subjektov so stredne závažným poškodením funkcie pečene (trieda B klasifikácie podľa
Childa-Pugha) oproti subjektom s normálnou funkciou pečene. U pacientov so stredne závažným
poškodením funkcie pečene (trieda B klasifikácie Childa-Pugha) sa odporúča zníženie dávky pri podávaní axitinibu (pozri časť 4.2).
Axitinib sa skúmal u pacientov so závažným poškodením funkcie pečene (trieda C klasifikácie podľa
Childa-Pugha) a nemá sa u tejto populácie používať.
Starší pacienti (≥ 65 rokov)a príslušnosťk rase
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bolo 34 % pacientov liečených axitinibom vo veku ≥ 65 rokov. Väčšina pacientov boli belosi (77 %) alebo aziati (21 %). Hoci
nemožno vylúčiť väčšiu náchylnosť na vznik nežiaducich reakcií u niektorých starších pacientov
a ázijských pacientov, celkovo neboli pozorované žiadne významné rozdiely v bezpečnosti a účinnosti axitinibu medzi pacientmi vo veku ≥ 65 rokov a mladšími a medzi príslušníkmi bielej rasy a iných rás.
Nie je potrebná úprava dávkovania na základe veku pacienta alebo príslušnosti k rase (pozri časti 4.2
a 5.2).
Laktóza
Tento liek obsahuje laktózu. Pacienti so zriedkavými vrodenými problémami intolerancie galaktózy, deficitu Lapp laktázy alebo glukózo-galaktózovej malabsorpcie nemajú užívať tento liek.
4.5 Liekové a iné interakcie
In vitro údaje ukazujú, že axitinib je metabolizovaný primárne enzýmom CYP3A4/5 a v menšej miere enzýmami CYP1A2, CYP2C19, a uridín-difosfoglukuronyltransferázou (UGT) 1A1.
Inhibítory CYP3A4/5
Ketokonazol, silný inhibítor CYP3A4/5 podávaný v dávke 400 mg raz denne počas 7 dní, zvýšil
2-násobne priemernú plochu pod krivkou (AUC – area under the curve) a 1,5-násobne Cmax
jednorazovo podanej 5 mg perorálnej dávky axitinibu u zdravých dobrovoľníkov.
Súčasné podávanie axitinibu so silnými inhibítormi CYP3A4/5 (napr. ketokonazol, itrakonazol, klaritromycín, erytromycín, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, sachinavir
a telitromycín) môže zvyšovať plazmatické koncentrácie axitinibu. Takisto grapefruit môže viesť
k zvýšeniu plazmatickej koncentrácie axitinibu. Pri súčasnom užívaní sa odporúča vyberať lieky so žiadnym alebo minimálnym inhibičným potenciálom na CYP3A4/5. Ak musí byť podávaný súbežne
silný inhibítor CYP3A4/5, odporúča sa úprava dávkovania axitinibu (pozri časť 4.2).
Inhibítory CYP1A2 a CYP2C19
CYP1A2 a CYP2C19 predstavujú minoritné (< 10%) cesty metabolizácie axitinibu. Účinok silných inhibítorov týchto izoenzýmov na farmakokinetiku axitinibu sa neskúmal. U pacientov užívajúcich
silné inhibítory týchto izoenzýmov je potrebná opatrnosť kvôli riziku zvýšenia plazmatických
koncentrácií axitinibu.
Induktory CYP3A4/5
Rifampicín, silný induktor CYP3A4/5 podávaný v dávke 600 mg raz denne počas 9 dní, znížil priemernú AUC o 79% a Cmax o 71% pri jednorazovo podanej dávke 5 mg axitinibu u zdravých dobrovoľníkov.
Súbežné podávanie axitinibu so silnými CYP3A4/5 induktormi (napr. rifampicín, dexametazón, fenytoín, karbamazepín, rifabutín, rifapentín, fenobarbital a Hypericum perforatum [Ľubovník bodkovaný]) môže znižovať plazmatické koncentrácie axitinibu. Pri súbežnom užívaní sa odporúča vyberať lieky so žiadnym alebo minimálnym indukčným potenciálom na CYP3A4/5. Ak sa musí súbežne podávať silný induktor CYP3A4/5, odporúča sa úprava dávkovania axitinibu (pozri časť 4.2).
Indukcia CYP1A2 fajčením
CYP1A2 predstavuje minoritnú (< 10%) cestu metabolizácie axitinibu. Účinok indukcie CYP1A2
súvisiaci s fajčením na farmakokinetiku axitinibu nebol úplne popísaný. Pri podávaní axitinibu fajčiarom je potrebné brať do úvahy riziko zníženia plazmatických koncentrácií axitinibu.
In vitro štúdie inhibície a indukcie CYP a UGT
In vitro štúdie ukázali, že axitinib pri terapeutických plazmatických koncentráciách neinhibuje
CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, alebo UGT1A1.
In vitro štúdie ukázali, že axitinib má potenciál inhibovať CYP1A2. Preto môže súbežné podávanie axitinibu so substrátmi CYP1A2 viesť k zvýšeniu plazmatických koncentrácií substrátov CYP1A2 (napr. teofylínu).
In vitro štúdie tiež preukázali, že axitinib má potenciál inhibovať CYP2C8. Súbežné podávanie axitinibu s paklitaxelom, známym substrátom CYP2C8 však neviedlo k zvýšeniu plazmatických koncentrácií paklitaxelu u pacientov s pokročilým karcinómom, čo dokazuje chýbanie klinicky významnej inhibície CYP2C8.
In vitro štúdie na ľudských hepatocytoch tiež ukázali, že axitinib neindukuje CYP1A1, CYP1A2 alebo CYP3A4/5. Preto sa pri súbežnom podávaní axitinibu neočakáva zníženie plazmatickej koncentrácie súbežne podávaných substrátov CYP1A1, CYP1A2 alebo CYP3A4/5 in vivo.
In vitro štúdie s P-glykoproteínom
In vitro štúdie ukázali, že axitinib inhibuje P-glykoproteín. Neočakáva sa však, že by axitinib inhiboval P-glykoproteín pri terapeutických plazmatických koncentráciách. Preto sa pri súčasnom
podávaní axitinibu neočakáva zvýšenie plazmatickej koncentrácie digoxínu alebo iných substrátov
P-glykoproteínu in vivo.
4.6 Fertilita, gravidita a laktácia
Gravidita
Neexistujú žiadne údaje týkajúce sa podávania axitinibu tehotným ženám. Na základe svojich farmakologických vlastností môže axitinib spôsobiť poškodenie plodu, ak sa podáva tehotným ženám.
Štúdie na zvieratách preukázali reprodukčnú toxicitu vrátane vzniku malformácií (pozri časť 5.3).
Axitinib sa nemá podávať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu týmto liekom.
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a až do 1 týždňa po nej.
Laktácia
Nie je známe, či sa axitinib vylučuje do ľudského mlieka. Nemožno vylúčiť riziko pre kojené dieťa. Axitinib sa nemá užívať počas laktácie.
Fertilita
Na základe neklinických zistení má axitinib potenciál poškodzovať reprodukčnú funkciu a plodnosť
u ľudí (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Axitinib má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti majú byť poučení, že sa u nich počas liečby axitinibom môžu vyskytnúť závraty a/alebo únava.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluNajdôležitejšie závažné nežiaduce účinky hlásené u pacientov užívajúcich axitinib boli arteriálne embolické a trombotické príhody, venózne embolické a trombotické príhody, krvácanie (vrátane gastrointestinálneho krvácania, cerebrálneho krvácania a hemoptýzy), gastrointestinálna perforácia a vznik fistúl, hypertenzná kríza a syndróm posteriórnej reverzibilnej encefalopatie. Tieto riziká vrátane potrebných protiopatrení sú uvedené v časti 4.4.
Najčastejšie (≥ 20 %) nežiaduce účinky pozorované po liečbe axitinibom boli hnačka, hypertenzia, únava, dysfónia, nevoľnosť, znížená chuť do jedla a syndróm palmárno-plantárnej erytrodyzestézie (syndróm ruka-noha).
Zoznam nežiaducich reakcií zostavený do tabuľkyTabuľka 1 uvádza nežiaduce reakcie hlásené u pacientov, ktorí dostávali axitinib v pivotnej klinickej štúdii na liečbu pacientov s RCC (pozri časť 5.1).
Nežiaduce reakcie sú zoradené podľa tried orgánových systémov, kategórie frekvencie výskytu
a stupňa ich závažnosti. Kategórie frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1000), veľmi zriedkavé (< 1/10 000) a neznáme (ich frekvenciu nemožno stanoviť z dostupných údajov). Súčasná databáza údajov o bezpečnosti axitinibu je príliš malá na detekovanie zriedkavých a veľmi zriedkavých nežiaducich reakcií.
Kategórie boli zoradené podľa absolútnych početností podľa údajov z klinických štúdií. V rámci každej triedy orgánového systému sú nežiaduce reakcie pri rovnakej častosti výskytu uvádzané
v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduce reakcie
| Všetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
| Poruchy krvi
a lymfatického systému
| Časté
| Anémia
| 2,8
| 0,3
| 0
| Trombocytopénia
| 1,7
| 0,3
| 0
| Menej časté
| Neutropénia
| 0,3
| 0,3
| 0
| Polycytémiab
| 0,3
| 0
| 0
| Leukopénia
| 0,3
| 0
| 0
| Poruchy
endokrinného systému
| Veľmi časté
| Hypotyreózab
| 18,4
| 0,3
| 0
| Menej časté
| Hypertyreózab
| 0,6
| 0
| 0
| Poruchy
metabolizmu a výživy
| Veľmi časté
| Znížená chuť do
jedla
| 28,4
| 3,3
| 0,3
| Časté
| Dehydratácia
| 4,7
| 2,5
| 0
| Menej časté
| Hyperkaliémia
| 0,8
| 0,6
| 0
| Hyperkalciémia
| 0,6
| 0
| 0
|
|
|
Tabuľka 1. Nežiaduce reakcie hlásené v klinickej štúdii s RCC u pacientov, ktorí dostávali axitinib (N= 359)
Trieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduce reakcie
|
V
šetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
|
Poruchy nervového
systému
|
Veľmi časté
|
Bolesť hlavy
|
10,3
|
0,6
|
0
|
Poruchy chuti
|
10,3
|
0
|
0
|
Časté
|
Závraty
|
5,6
|
0
|
0
|
Menej časté
|
Syndróm
posteriórnej reverzibilnej
encefalopatie
|
0,3
|
0,3
|
0
|
Porychy ucha
a labyrintu
|
Časté
|
Tinnitus
|
2,2
|
0
|
0
|
Poruchy ciev
|
Veľmi časté
|
Hypertenzia
|
39,3
|
15,3
|
0,3
|
Krvácanieb, c
|
10,6
|
0,3
|
0,3
|
Časté
|
Venózne embolické
a trombotické príhodyb, c
|
1,9
|
0,8
|
0,8
|
Arteriálne
embolické
a trombotické príhodyb, c
|
1,1
|
1,1
|
0
|
Menej časté
|
Hypertenzná kríza
|
0,6
|
0,3
|
0,3
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
|
Veľmi časté
|
Dysfónia
|
28,1
|
0
|
0
|
Časté
|
Dyspnoe
|
7,0
|
0,3
|
0
|
Kašeľ
|
5,3
|
0
|
0
|
Orofaryngeálna
bolesť
|
3,3
|
0
|
0
|
Poruchy
gastrointestinálneho traktu
|
Veľmi časté
|
Hnačka
|
51,3
|
9,7
|
0,3
|
Vracanie
|
16,7
|
1,4
|
0
|
Nevoľnosť
|
28,7
|
1,4
|
0
|
Stomatitída
|
14,5
|
1,4
|
0
|
Zápcha
|
12,3
|
0
|
0
|
Časté
|
Bolesť brucha
|
8,4
|
0,6
|
0,3
|
Bolesť v epigastriu
|
6,1
|
0,3
|
0
|
Dyspepsia
|
7,8
|
0
|
0
|
Flatulencia
|
4,5
|
0
|
0
|
Hemoroidy
|
2,2
|
0
|
0
|
Menej časté
|
Gastrointestinálna perforáciab, d
|
0,3
|
0
|
0,3
|
Análna fistulab
|
0,3
|
0
|
0
|
Poruchy kože
a podkožného tkaniva
|
Veľmi časté
|
Palmárno-plantárna
erytrodyzestézia
(syndróm ruka-noha)
|
27,3
|
5,0
|
0
|
Vyrážka
|
11,7
|
0,3
|
0
|
Suchosť kože
|
10,0
|
0
|
0
|
Časté
|
Pruritus
|
5,8
|
0
|
0
|
Erytém
|
2,2
|
0
|
0
|
Alopécia
|
3,3
|
0
|
0
|
Poruchy kostrovej
a svalovej sústavy a spojivového
tkaniva
|
Časté
|
Myalgia
|
5,3
|
0,6
|
0,3
|
Artralgia
|
8,6
|
0,6
|
0
|
Bolesť končatín
|
8,9
|
0,3
|
0
|
Trieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduce reakcie
|
V
šetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
|
Poruchy obličiek
a močových ciest
|
Veľmi časté
|
Proteinúria
|
10,3
|
3,1
|
0
|
Časté
|
Zlyhanie obličiek
|
1.1
|
0.6
|
0
|
Celkové poruchy
a reakcie v mieste podania
|
Veľmi časté
|
Únava
|
34,8
|
9,5
|
0,3
|
Asténiac
|
17,5
|
3,6
|
0,3
|
Zápal sliznice
|
15,0
|
1,4
|
0
|
Laboratórne
a funkčné vyšetrenia
|
Veľmi časté
|
Úbytok hmotnosti
|
16,4
|
1,4
|
0
|
Časté
|
Zvýšenie hladiny
tyreoideu stimulujúceho hormónu
|
4,5
|
0
|
0
|
Zvýšenie hladiny
lipázy
|
2,2
|
0,6
|
0
|
Zvýšenie hladiny
alanínamino transferázy
|
1,9
|
0,3
|
0
|
Zvýšenie hladiny
aspartátamino transferázy
|
1,1
|
0,3
|
0
|
Zvýšenie hladiny
alkalickej fosfatázy
|
1,4
|
0
|
0
|
Zvýšenie hladiny
amylázy
|
1,7
|
0
|
0
|
Menej časté
|
Zvýšenie hladiny
bilirubínu v krvi
|
0,6
|
0
|
0
|
Zvýšenie hladiny
kreatinínu
|
0,6
|
0
|
0
|
|
|
a Všeobecné terminologické kritériá pre nežiaduce účinky národného inštitútu pre rakovinu verzia 3.0 (CTCAE)
b Pozri časť Popis vybraných nežiaducich účinkov
c Boli hlásené fatálne prípady (5. stupeň závažnosti)
d Výskyt nežiaducej reakcie z akejkoľvek príčiny
e Vrátane zlyhania obličiek
Opis vybraných nežiaducich reakciíDysfunkcia štítnej žľazy (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola hypotyreóza hlásená
u 18,4 % pacientov a hypertyreóza u 0,6 % pacientov. Zvýšenie hladiny thyreostimulačného hormónu
(TSH) bolo ako nežiaduci účinok hlásené u 4,5 % pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho sledovania u pacientov, ktorí mali pred liečbou TSH < 5 μU/ml, sa
objavilo zvýšenie TSH až na ≥ 10 μU/ml u 32,2 % pacientov užívajúcich axitinib.
Venózne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli venózne embolické
a trombotické nežiaduce reakcie hlásené u 1,9 % pacientov užívajúcich axitinib. Venózne embolické a trombotické nežiaduce reakcie stupňa závažnosti 3/4 boli hlásené u 1,7 % pacientov užívajúcich axitinib (vrátane pľúcnej embólie, hlbokej venóznej trombózy a oklúzie/trombózy sietnicovej vény). Fatálna pľúcna embólia bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib.
Arteriálne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli arteriálne embolické
a trombotické nežiaduce reakcie stupňa závažnosti 3/4 hlásené u 1,1 % pacientov užívajúcich axitinib.
Najčastejšou arteriálnou embolické a trombotickou príhodou bol tranzitórny ischemický atak (0,8 %). Fatálna cerebrovaskulárna príhoda bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib.
V monoterapeutických klinických štúdiách s axitinibom (N=699) boli arteriálne embolické
a trombotické nežiaduce reakcie (vrátane tranzitórneho ischemického ataku, infarktu myokardu a cerebrovaskulárnej príhody) hlásené u 1,0 % pacientov užívajúcich axitinib.
Polycytémia (pozri
Elevácia hemoglobínu alebo hematokritu v časti 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola polycytémia hlásená ako nežiaduca reakcia u 0,3 % pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho
sledovania bola zvýšená hladina hemoglobínu nad ULN zistená u 9,7 % pacientov užívajúcich
axitinib. V štyroch klinických štúdiách s axitinibom v liečbe pacientov s RCC (N=537) bola zvýšená hladina hemoglobínu nad ULN pozorovaná u 13,6 % pacientov užívajúcich axitinib.
Hemorágia (pozri časť 4,4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC, kam neboli zaradení pacienti s neliečenými metastázami v mozgu, boli hlásené hemoragické nežiaduce reakcie u 10,6 % pacientov
užívajúcich axitinib. Najčastejšími hemoragickými nežiaducimi reakciami u pacientov liečených
axitinibom boli epistaxa (5,3 %), hematúria (1,4 %), krvácanie z konečníka (1,1 %) a krvácanie z ďasien (1,1 %). Hemoragické nežiaduce reakcie stupňa závažnosti ≥ 3 boli hlásené u 0,8 %
pacientov užívajúcich axitinib (vrátane cerebrálneho krvácania, žalúdočného krvácania a krvácania
z distálneho gastrointestinálneho traktu). Fatálna hemorágia bola hlásená u jedného pacienta (0,3 %), ktorý užíval axitinib (žalúdočné krvácanie). V monoterapeutických klinických štúdiách s axitinibom (N=699) bola hemoptýza hlásená ako nežiaduca reakcia u 1,6 % pacientov, vrátane jedného prípadu (0,1 %) so stupňom závažnosti ≥ 3.
Gastrointestinálna perforácia a tvorba fistúl (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola gastrointestinálna perforácia hlásená u jedného pacienta (0,3 %, bez ohľadu na kauzalitu), ktorý užíval axitinib.
V monoterapeutických klinických štúdiách s axitinibom (N=699) boli fistuly hlásené u 0,7 %
pacientov (bez ohľadu na kauzalitu) a fatálna gastrointestinálna perforácia bola hlásená u jedného pacienta (0,1 %).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v Prílohe V.
4.9 PredávkovanieNeexistuje žiadna špecifická liečba v prípade predávkovania axitinibom.
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC jeden pacient, ktorý neúmyselne užíval dávku 20 mg dvakrát denne počas 4 dní, udával závraty (stupeň závažnosti 1).
V klinickej štúdii s axitinibom na stanovenie dávky probandi, ktorí užívali úvodnú dávku 10 mg dvakrát denne alebo 20 mg dvakrát denne mali nežiaduce reakcie, ktoré zahŕňali hypertenziu, záchvaty spojené s hypertenziou a fatálnu hemoptýzu.
V prípade podozrenia z predávkovania je potrebné vysadiť axitinib a začať podpornú starostlivosť.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
F
armakoterapeutická skupina: Cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE17
Spôsob účinku
Axitinib je účinný a selektívny inhibítor tyrozínkinázy receptorov vaskulárneho endoteliálneho rastového faktora (VEGFR-1, VEGFR-2 a VEGFR-3). Tieto receptory sa podieľajú na patologickej
angiogenéze, raste nádoru a metastatickej progresii karcinómu. Preukázalo sa, že axitinib účinne
inhibuje proliferáciu endoteliálnych buniek a ich prežívanie sprostredkované VEGF. Axitinib inhiboval fosforyláciu VEGFR-2 nádorovej vaskulatúry xenograftu, ktorá predstavovala cieľ liečby in vivo a viedol k oddialeniu rastu nádoru, regresii a inhibícii metastáz v mnohých experimentálnych modeloch rakoviny.
Vplyv na QTc interval
V randomizovanej dvojramennej štúdii s prekrížením liečebných ramien bola 35 zdravým probandom podaná jedna perorálna dávka axitinibu (5 mg) bez podávania a s podávaním 400 mg ketokonazolu
počas 7 dní. Výsledky tejto štúdie ukázali, že expozícia axitinibu v plazme až dvojnásobne vyššia ako očakávané terapeutické hladiny pri 5 mg dávkovaní neviedla ku klinicky signifikantnému predĺženiu
QT intervalu.
Klinickáúčinnosť
Bezpečnosť a účinnosť axitinibu sa hodnotili v randomizovanej, otvorenej multicentrickej klinickej štúdii fázy 3. Pacienti (N=723) s pokročilým RCC, ktorých ochorenie progredovalo počas alebo po liečbe jednou predchádzajúcou systémovou terapiou, zahrňujúcou režimy so sunitinibom, bevacizumabom, temsirolimom alebo cytokín-obsahujúce , boli randomizovaní (1:1) na užívanie axitinibu (n=361) alebo sorafenibu (n=362). Primárny cieľ, prežívanie bez progresie (PFS) sa hodnotil centrálne metódou zaslepeného nezávislého posúdenia. Medzi sekundárne ciele patrili miera objektívnej odpovede (ORR) a celkové prežívanie (OS).
Z pacientov zaradených do tejto klinickej štúdie dostalo 389 pacientov (53,8 %) jednu predchádzajúcu liečbu založenú na sunitinibe, 251 pacientov (34,7 %) dostalo jednu predchádzajúcu liečbu založenú
na cytokínoch (interleukin-2 alebo interferón-alfa), 59 pacientov (8,2%) dostalo jednu predchádzajúcu liečbu založenú na bevacizumabe a 24 pacientov (3,3 %) dostalo jednu predchádzajúcu liečbu
založenú na temsirolime. Demografické a nádorové charakteristiky na začiatku štúdie boli podobné medzi liečebnými skupinami s axitinibom a sorafenibom čo sa týka veku, pohlavia, rasy,
výkonnostného stavu podľa ECOG kritérií (Eastern Cooperative Oncology Group), geografického regiónu a predchádzajúcej liečby.
V celej populácii pacientov a dvoch hlavných podskupinách (skupina s predchádzajúcou liečbou sunitinibom a skupina s predchádzajúcou liečbou cytokínmi) pre primárny cieľ hodnotenia PFS bol preukázaný štatisticky signifikantný prínos axitinibu oproti sorafenibu (pozri Tabuľka 2 a Obrázky 1,
2 and 3). Hodnota mediánu PFS účinku bol v oboch podskupinách rozdelených podľa predchádzajúcej
liečby odlišný. Dve podskupiny boli príliš malé, aby poskytli dôveryhodné výsledky (skupina
s predchádzajúcou liečbou temsirolimom a skupina s predchádzajúcou liečbou bevacizumabom). Medzi liečebnými ramenami neboli žiadne štatisticky signifikantné rozdiely v OS v celkovej populácii
ani v podskupinách rozdelených podľa predchádzajúcej liečby.
Tabuľka 2. Výsledky účinnosti
Cieľ / Populácia štúdie
|
axitinib
|
sorafenib
|
HR
(95% CI)
|
p-hodnota
|
|
Celá ITT populácia
Medián PFS v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
|
N = 361
6,8 (6,4; 8,3)
20,1 (16,7; 23,4)
|
N = 362
4,7 (4,6; 6,3)
19,2 (17,5; 22,3)
|
0,67 (0,56; 0,81)
0,97 (0,80; 1,17)
|
< 0,0001
NS
|
ORR b,e% (95% CI)
|
19,4 (15,4; 23,9)
|
9,4 (6,6; 12,9)
|
2,06f (1,41; 3,00)
|
0,0001g
|
Predchádzajúca liečba sunitinibom
|
N = 194
|
N = 195
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
4,8 (4,5; 6,5)
15,2 (12,8; 18,3)
11,3 (7,2; 16,7)
|
3,4 (2,8; 4,7)
16,5 (13,7; 19,2)
7,7 (4,4; 12,4)
|
0,74 (0,58; 0,94)
1,00 (0,78; 1,27)
1,48f (0,79; 2,75)
|
0,0063h
NS
|
Predchádzajúca liečba
cyto
k
ínmi
|
N = 126
|
N = 125
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
12,0 (10,1; 13,9)
29,4 (24,5; NE)
32,5 (24,5; 41,5)
|
6,6 (6,4; 8,3)
27,8 (23,1;, 34,5)
13,6 (8,1; 20,9)
|
0,52 (0,38;, 0,72)
0,81 (0,56; 1,19)
2,39f (1,43;3,99)
|
< 0,0001h
NS
0,0002i
|
CI =interval spoľahlivosti, HR miera rizika (axitinib/sorafenib); ITT: všetci pacienti zaradení do štúdie); NE = nemožné predpovedať; NS = štatisticky nesignifikantné; ORR Miera objektívnej
odpovede;.OS: = celkové prežívanie PFS : Prežívanie bez progresie.
a Čas od randomizácie po progresiu alebo úmrtie z akejkoľvek príčiny, podľa toho, ktorá udalosť
nastane prvá. Dátum ukončenia: 3. jún 2011.
b Stanovené podľa RECIST kritérií nezávislým rádiologickým posudkom.
c Jednostranná hodnota p podľa log-rank testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej liečby d Dátum ukončenia: 1. november 2011.
e Dátum ukončenia: 31. august 2010.
f Pre ORR je použitá veličina pomer rizík. Pomer rizík > 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene s axitinibom; pomer rizík < 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene so sorafenibom.
g Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej terapie
h Jednostranná hodnota p log-rank testu pre liečbu so stratifikáciou podľa ECOG výkonnostného stavu
i Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu
Obrázok 1. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre celkovú populáciu
INLYTA (N=361)
m
e
d
i
á
n 6,8 mesiaca
sorafenib (N=362)
m
e
d
i
á
n 4,7 mesiaca
m
i
er
a rizika = 0,67
95
% CI [0,56, 0,81]
p hodnota <0,0001
Čas (mesiace)
Obrázok 2. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia prepodskupinu s predchádzajúcou liečbou sunitinibomINLYTA (N=194)medián 4,8 mesiacasorafenib (N=195)medián 3,4 mesiacamiera rizika = 0,7495% CI [0,58, 0,94]p hodnota = 0,0063Čas (mesiace)
Obrázok 3. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre podskupinu s predchádzajúcou liečbou
c
ytokínom.
INLYTA (N=126)
m
e
d
i
á
n
12
,
0 mesiaca
sorafenib (N=125)
m
e
d
i
á
n 6,6 mesiaca
m
i
er
a rizika = 0,52
95
% CI [0,38, 0,72]
p hodnota < 0,0001
Čas (mesiace)
Deti a dospievajúciEurópska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií pre axitinib vo všetkých vekových podskupinách detí a dospievajúcich v liečbe karcinómu obličky a obličkovej panvičky (okrem nefroblastómu, nefroblastomatózy, sarkómu z jasných buniek, mezoblastického nefrómu, obličkového medulárneho karcinómu a rabdoidného nádoru obličky) (pozri časť 4.2 pre informácie o použití v pediatrii).
5.2 Farmakokinetické vlastnostiPo perorálnom podaní tabliet axitinibu je priemerná absolútna biologická dostupnosť 58 %
v porovnaní s intravenóznym podaním. Plazmatický polčas axitinibu kolíše od 2,5 do 6,1 hodiny. Dávkovanie axitinibu po 5 mg dvakrát denne viedlo k menej než dvojnásobnému nahromadeniu
v porovnaní s podaním v jednej dávke. Vzhľadom na krátky eliminačný polčas axitinibu sa očakáva
dosiahnutie rovnovážneho stavu v priebehu 2 až 3 dní po úvodnej dávke.
Absorpcia a distribúciaMaximálne koncentrácie axitinibu v plazme sa obvykle dosiahnu v priebehu 4 hodín po perorálnom podaní axitinibu s mediánom Tmax v rozmedzí od 2,5 do 4,1 hodiny. Podávanie axitinibu so stredne mastným jedlom viedlo k zníženiu expozície o 10 % v porovnaní s celonočným hladovaním. Veľmi mastné, vysoko kalorické jedlo viedlo k zvýšeniu expozície o 19% v porovnaní s celonočným hladovaním. Axitinib môže byť užívaný s jedlom alebo bez jedla (pozri časť 4.2).
Priemerná Cmax a AUC sa proporcionálne zvýšili v závislosti na dávkovaní axitinibu v rozsahu 5 až
10 mg.
In vitro väzba axitinibu na ľudské plazmatické proteíny je > 99 % s prednostným naviazaním
na albumín a strednou väzbou na α1-kyslý glykoproteín. Pri dávkovaní 5 mg dvakrát denne u pacientov s pokročilým RCC bola v stave nasýtenosti geometrická priemerná maximálna plazmatická koncentrácia 27,8 ng/ml a 24-hodinová AUC 265 ng.h/ml. Po perorálnom podaní bol geometrický priemerný klírens 38 l/h a distribučný objem bol 160 litrov.
Biotransformácia a elimináciaAxitinib sa metabolizuje primárne v pečeni enzýmom CYP3A4/5 a v menšej miere enzýmami
CYP1A2, CYP2C19 a UGT1A1.
Po perorálnom podaní 5 mg dávky rádioaktívne značeného axitinibu bolo 30-60 % rádioaktivity zachytenej v stolici a 23 % rádioaktivity v moči. Nemetabolizovaný axitinib, na úrovni 12 % z dávky, bol hlavnou zložkou identifikovanou v stolici. Nemetabolizovaný axitinib nebol zistený v moči; metabolity kyseliny karboxylovej a sulfoxidu predstavovali väčšinu rádioaktivity v moči. V plazme predstavoval N-glukuronidový metabolit predominantnú rádioaktívnu zložku (50 % rádioaktivity
v cirkulácii) a nemetabolizovaný axitinib a sulfoxidový metabolit každý jeden tvorili približne
20 % rádioaktivity v cirkulácii.
Sulfoxidové a N-glukuronidové metabolity vykazujú približne 400-násobne a 8000-násobne menšiu účinnosť in vitro voči VEGFR-2 v porovnaní s axitinibom.
Osobitné skupiny pacientov
Starší pacienti, pohlavie a rasa
Populačné farmakokinetické analýzy u pacientov s pokročilým karcinómom (vrátane pokročilého
RCC) a zdravých dobrovoľníkov ukazujú, že vek, pohlavie, telesná hmotnosť, rasa, obličkové funkcie, genotyp UGT1A1 alebo CYP2C19 nemajú žiadne klinicky relevantné účinky.
Deti a dospievajúci
Axitinib sa nesledoval u pacientov mladších ako 18 rokov.
Porucha funkcie pečene
In vitro a in vivo údaje ukazujú, že axitinib je primárne metabolizovaný v pečeni.
V porovnaní s pacientami s normálnou funkciou pečene je systémová expozícia po podaní jednej dávky axitinibu podobná u subjektov s ľahkou poruchou funkcie pečene (trieda A klasifikácie podľa Childa-Pugha) a vyššia (približne dvojnásobne) u pacientov so stredne závažnou poruchou funkcie pečene (trieda B klasifikácie podľa Childa-Pugha). Axitinib sa nesledoval u pacientov so závažnou poruchou funkcie pečene (trieda C klasifikácie podľa Childa-Pugha) a nemá sa používať v tejto populácii (odporúčania pre úpravu dávkovania pozri časť 4.2).
Porucha funkcie obličiek
Nemetabolizovaný axitinib nie je detekovaný v moči.
Axitinib sa nesledoval u pacientov s poruchou funkcie obličiek. V klinických štúdiách s axitinibom
v liečbe pacientov s RCC neboli zaradení pacienti s hladinou sérového kreatinínu > 1,5 násobok ULN alebo kalkulovaným klírensom kreatinínu < 60 ml/min. Populačné farmakokinetické analýzy preukázali, že klírens axitinibu nebol zmenený u pacientov s poruchou funkcie obličiek a nevyžaduje sa žiadna úprava dávkovania.
5.3 Predklinické údaje o bezpečnosti
Toxicita pri opakovanom podávaní
Hlavné prejavy toxicity u myší a psov pri opakovanom podávaní v trvaní do 9 mesiacov boli v gastrointestinálnom, hematopoetickom, reprodukčnom, skeletálnom a dentálnom systéme,
s hladinami bez pozorovaných nežiaducich účinkov (NOAEL – No Observed Adverse Effect Levels)
približne rovnakými alebo nižšími ako je očakávaná expozícia u človeka pri odporúčanej klinickej úvodnej dávke (podľa hladín AUC).
Karcinogenicita
Nevykonali sa žiadne klinické štúdie skúmajúce karcinogenitu axitinibu.
Genotoxicita
Axitinib nebol mutagénny alebo klastogénny v konvenčných skúškach genotoxicity in vitro. Signifikantné zvýšenie polyploidity bolo pozorované in vitro pri koncentráciách > 0,22 μg/ml
a vzostup počtu mikrojadrových polychromatických erytrocytov sa pozoroval in vivo pri hladine bez
pozorovaného účinku na organizmus (NOEL - No Observed Effect Level) 69-násobne presahujúcej
očakávanú expozíciu u človeka. Nálezy genotoxicity sa nepovažujú za klinicky relevantné pri expozičných hladinách pozorovaných u ľudí.
Reprodukčná toxicita
Medzi nálezy súvisiace s axitinibom v semenníkoch a nadsemenníkoch patrili zníženie hmotnosti orgánu, atrofia alebo degenerácia, zníženie počtu germinálnych buniek, hypospermia alebo výskyt
abnormálnych foriem spermií a znížená denzita a počet spermií. Tieto nálezy sa pozorovali u myší pri
expozičných hladinách rovnajúcich sa približne 12-násobku očakávanej expozície u ľudí, u psov pri expozičných hladinách nižších ako je očakávaná expozícia u ľudí. Nepozoroval sa vplyv na párenie
alebo fertilitu u samčích myší pri expozičných hladinách rovnajúcich sa približne 57-násobku
očakávanej expozície u ľudí. K nálezom u samičiek patrili známky oneskorenia pohlavnej dospelosti, zmenšenie alebo chýbanie corpora lutea, zníženie hmotnosti maternice a atrofia maternice pri expozíciách približne rovnakých ako je očakávaná expozícia u ľudí. Znížená fertilita a viabilita embryí boli pozorované pri všetkých testovaných dávkach s expozičnými hladinami na najnižšej dávke rovnajúcimi sa približne 10-násobku očakávanej expozície u ľudí.
U gravidných myší vystavených účinku axitinibu bol preukázaný zvýšený výskyt rázštepu podnebia a skeletálnych variácií, vrátane oneskorenej osifikácie pri expozičných hladinách nižších ako očakávaná expozícia u ľudí. Štúdie perinatálnej a postnatálnej vývojovej toxicity neboli realizované.
Prejavy toxicity u nezrelých zvierat
U myší a psov, ktorí dostávali axitinib najmenej 1 mesiac pri expozičných hladinách približne
6-násobne vyšších než očakávané expozičné hladiny u ľudí bola pozorovaná reverzibilná dysplázia rastovej platničky. U myší užívajúcich axitinib viac než 1 mesiac pri expozičných hladinách
podobných očakávanej expozícii u ľudí bol pozorovaný výskyt čiastočne reverzibilných zubných
kazov. Iné toxické prejavy s možným rizikom pre pediatrických pacientov sa u juvenilných zvierat neskúmali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety mikrokryštalická celulóza monohydrát laktózy kroskarmelóza sodná stearan horečnatý
Obaľujúci film tablety hypromelóza
oxid titaničitý (E171)
monohydrát laktózy triacetín (E1518)
červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Hliníkový/hliníkový blister obsahujúci 14 filmom obalených tabliet. Každé balenie obsahuje 28 alebo
56 filmom obalených tabliet.
HDPE fľaštička s vysušovadlom obsahujúcim silikagél na absorpciu vlhkosti a polypropylénovým uzáverom obsahujúca 60 filmom obalených tabliet.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciuNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited
Ramsgate Road
Sandwich, Kent, CT13, 9NJ Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/12/777/007
EU/1/12/777/008
EU/1/12/777/009
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 3. 9. 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUInlyta 5 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá filmom obalená tableta obsahuje 5 mg axitinibu.
Každá filmom obalená tableta obsahuje 58,8 mg monohydrátu laktózy. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAFilmom obalená tableta (tableta).
Červená trojuholníková filmom obalená tableta s vyrytým nápisom “Pfizer“ na jednej strane a nápisom “5 XNB“ na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieInlyta je indikovaná na liečbu dospelých pacientov s pokročilým karcinómom z obličkových buniek
(renal cell carcinoma, RCC) po zlyhaní predchádzajúcej liečby sunitinibom alebo cytokínmi.
4.2 Dávkovanie a spôsob podávaniaLiečbu Inlytou má vykonávať lekár, ktorý má skúsenosti s podávaním protinádorových liekov.
DávkovanieOdporúčaná úvodná dávka axitinibu je 5 mg dvakrát denne.
Liečba má pokračovať, pokým sa pozoruje klinický prínos alebo pokiaľ sa neprejaví neprijateľná toxicita, ktorú nie je možné zvládnuť súbežným podávaním liekov alebo úpravou dávky.
Pokiaľ pacient vracia alebo vynechá dávku, nesmie užiť dodatočnú dávku. Nasledujúca predpísaná dávka sa musí užiť v obvyklom čase.
Úpravy dávkyZvýšenie alebo zníženie dávky sa odporúča na základe individuálnej bezpečnosti a znášanlivosti.
U pacientov, ktorí tolerujú axitinib v úvodnej dávke 5 mg dvakrát denne bez nežiaducich účinkov
> ako stupeň 2 (t.j. bez závažných nežiaducich účinkov podľa Všeobecných terminologických kritérií pre nežiaduce účinky [CTCAE - Common Terminology Criteria for Adverse Events] verzia 3.0) počas
dvoch po sebe idúcich týždňov, je možné dávku zvýšiť na 7 mg dvakrát denne, pokiaľ nie je krvný
tlak pacienta > 150/90 mmHg alebo pacient neužíva antihypertenzívnu liečbu. Následne podľa rovnakých kritérií u pacientov, ktorí tolerujú axitinib v dávke 7 mg dvakrát denne, je možné zvýšiť ich dávku na maximálne 10 mg dvakrát denne.
Zvládnutie niektorých nežiaducich účinkov si môže vyžiadať prechodné alebo trvalé prerušenie liečby a/alebo zníženie dávky axitinibu (pozri časť 4.4). Ak je potrebné znížiť dávku, dávka axitinibu môže byť znížená na 3 mg dvakrát denne a ďalej na 2 mg dvakrát denne.
Úprava dávkovania nie je potrebná kvôli veku, rase, pohlaviu alebo telesnej hmotnosti.
Súčasné užívanie silných inhibítorov CYP3A4/5
Súčasné podávanie axitinibu so silnými inhibítormi CYP3A4/5 môže zvyšovať plazmatické koncentrácie axitinibu (pozri časť 4.5). Odporúča sa výber alternatívneho súčasne podávaného lieku so žiadnym alebo minimálnym potenciálom inhibície CYP3A4/5.
Hoci u pacientov užívajúcich silné inhibítory CYP3A4/5 sa neskúmala úprava dávkovania axitinibu, ak sa musí súčasne podávať silný inhibítor CYP3A4/5, odporúča sa zníženie dávky axitinibu na približne polovičnú dávku (t.j. úvodná dávka by mala byť znížená z 5 mg dvakrát denne na 2 mg dvakrát denne). Zvládnutie niektorých nežiaducich účinkov si môže vyžiadať prechodné alebo trvalé prerušenie liečby axitinibom (pozri časť 4.4). Ak sa ukončí súčasné podávanie silného inhibítora, je potrebné zvážiť návrat k dávke axitinibu užívanej pred začiatkom podávania silného inhibítora CYP3A4/5 (pozri časť 4.5).
Súčasné užívanie silných induktorov CYP3A4/5
Súčasné podávanie axitinibu so silnými induktormi CYP3A4/5 môže znižovať plazmatické koncentrácie axitinibu (pozri časť 4.5). Odporúča sa výber alternatívneho súčasne podávaného lieku so
žiadnym alebo minimálnym potenciálom indukcie CYP3A4/5.
Hoci u pacientov užívajúcich silné induktory CYP3A4/5 saneskúmala úprava dávkovania axitinibu, ak sa musí súbežne podávať silný induktor CYP3A4/5, odporúča sa postupné zvyšovanie dávky
axitinibu. Maximálna indukcia pri vysokých dávkach silných induktorov CYP3A4/5 sa pozorovala
v priebehu jedného týždňa liečby induktorom. Ak sa dávka axitinibu zvýši, treba pacienta starostlivo monitorovať kvôli toxicite. Zvládnutie niektorých nežiaducich účinkov môže vyžadovať prechodné
alebo trvalé prerušenie liečby a/alebo zníženie dávky axitinibu (pozri časť 4.4). Ak sa ukončí súbežné podávanie silného induktora, je treba okamžite upraviť dávku axitinibu na dávku užívanú pred
začiatkom podávania silného induktora CYP3A4/5 (pozri časť 4.5).
Osobitné skupiny pacientov
Starší pacienti (≥ 65 rokov)
Nie je potrebná žiadna úprava dávkovania (pozri časti 4.4 a 5.2).
Poškodenie funkcie obličiek: Nevyžaduje sa úprava dávkovania (pozri časť 5.2). Nie sú dostupné prakticky žiadne údaje týkajúce sa liečby axitinibom u pacientov s klírensom kreatinínu < 15 mL/min.
Poškodenie funkcie pečene: Nevyžaduje sa úprava dávkovania, ak sa axitinib podáva pacientom
s ľahkým poškodením pečeňových funkcií (trieda A klasifikácie podľa Childa-Pugha). Zníženie dávky sa odporúča pri podávaní axitinibu pacientom so stredne závažným poškodením pečeňových funkcií
(trieda B klasifikácie podľa Childa-Pugha) (t.j. úvodná dávka sa má znížiť z 5 mg dvakrát denne na
2 mg dvakrát denne). Axitinib sa neskúmal u pacientov so závažným poškodením pečeňových funkcií
(trieda C klasifikácie podľa Childa-Pugha) a nemá sa preto používať v tejto skupine pacientov (pozri
časti 4.4. a 5.2).
Deti a dospievajúci
Bezpečnosť a účinnosť axitinibu u detí a dospievajúcich < 18 rokov nebola stanovená. Nie sú dostupné žiadne údaje.
Spôsob podania
Axitinib sa má užívať perorálne dvakrát denne v približne 12-hodinových intervaloch s alebo bez jedla
(pozri časť 5.2). Tablety axitinibu sa majú prehltnúť celé a zapiť pohárom vody.
4.3 Kontraindikácie
Precitlivenosť na axitinib alebo na niektorú z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Pred začatím a pravidelne počas liečby axitinibom je potrebné sledovať typické príhody týkajúce sa bezpečnosti liečby ako sú uvedené ďalej.
Hypertenzia
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC sa veľmi často hlásila hypertenzia (pozri časť 4.8). Medián času do vzniku hypertenzie (systolický tlak krvi > 150 mmHg
alebo diastolický tlak krvi > 100 mmHg) sa dosiahol v priebehu prvého mesiaca od začiatku liečby
axitinibom a vzostupy krvného tlaku sa pozorovali už po 4 dňoch od začiatku užívania axitinibu.
Už pred začiatkom liečby axitinibom sa musí krvný tlak dobre kontrolovať. Pacienti sa majú sledovať kvôli hypertenzii a podľa potreby liečiť štandardnou antihypertenzívnou liečbou. V prípade pretrvávania hypertenzie napriek užívaniu antihypertenzívnych liekov sa musí znížiť dávka axitinibu. U pacientov, u ktorých sa vyvinie závažná hypertenzia, dočasne prerušte liečbu axitinibom
a pokračujte nižšou dávkou, keď je pacient normotenzný. Ak je liečba axitinibom prerušená, musia sa pacienti užívajúci antihypertenzívne lieky sledovať kvôli hypotenzii (pozri časť 4.2).
V prípade závažnej alebo pretrvávajúcej artériovej hypertenzie a príznakov pripomínajúcich syndróm posteriórnej reverzibilnej encefalopatie (viď nižšie) treba zvážiť diagnostické vyšetrenie mozgu magnetickou rezonanciou (MRI).
Dysfunkcia štítnej žľazy
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC sa hlásili prípady hypotyreózy a v menšom rozsahu prípady hypertyreózy (pozri časť 4.8).
Funkcia štítnej žľazy sa má vyšetriť ešte pred začiatkom a pravidelne kontrolovať počas liečby axitinibom. Hypotyreóza alebo hypertyreóza sa majú liečiť podľa štandardných terapeutických postupov s cieľom udržaťeutyreoidný stav.
Arteriálne embolické a trombotické príhody
V klinických štúdiách s axitinibom boli hlásené arteriálne embolické a trombotické príhody (vrátane tranzitórneho ischemického ataku, infarktu myokardu, cerebrovaskulárnej príhody a oklúzie
sietnicovej artérie) (pozri časť 4.8).
Axitinib sa má preto používať opatrne u pacientov, ktorí majú zvýšené riziko vzniku alebo sa u nich už vyskytli takéto príhody. Axitinib sa neskúmal u pacientov, ktorí mali arteriálnu embolickú a trombotickú príhodu počas predchádzajúcich 12 mesiacov.
Venózne embolické a trombotické príhody
V klinických štúdiách s axitinibom boli hlásené venózne embolické a trombotické príhody (vrátane pľúcnej embólie, hlbokej žilovej trombózy a oklúzie/trombózy sietnicovej vény) (pozri časť 4.8).
Axitinib sa má preto používať opatrne u pacientov, ktorí majú zvýšené riziko vzniku alebo sa u nich už vyskytli takéto príhody. Axitinib sa neskúmal u pacientov, ktorí mali venóznu embolickú
a trombotickú príhodu počas predchádzajúcich 6 mesiacov.
Elevácia hemoglobínu alebo hematokritu
Počas liečby axitinibom sa môže vyskytnúť vzostup hemoglobínu alebo hematokritu ako prejav zvýšenia množstva červených krviniek (pozri časť 4.8, polycytémia). Zvýšenie množstva červených krviniek môže zvýšiť riziko vzniku embolických a trombotických príhod.
Hladina hemoglobínu alebo hodnota hematokritu sa majú vyšetriť ešte pred začiatkom a pravidelne kontrolovať počas liečby axitinibom. Ak dôjde k zvýšeniu hladiny hemoglobínu alebo hodnoty hematokritu nad normálnu úroveň, pacienti sa majú liečiť štandardnými terapeutickými postupmi
s cieľom znížiť hladinu hemoglobínu alebo hodnotu hematokritu na prijateľnú úroveň.
Hemorágia
V klinických štúdiách s axitinibom boli hlásené hemoragické príhody (pozri časť 4.8).
Axitinib sa neskúmal u pacientov s dokázanými neliečenými metastázami v mozgu alebo nedávnym aktívnym gastrointestinálnym krvácaním a nemá sa u týchto pacientov používať. Ak si akékoľvek krvácanie vyžaduje medicínsky zásah, dočasne prerušte podávanie axitinibu.
Gastrointestinálna perforácia a tvorba fistúl
V klinických štúdiách s axitinibom boli hlásené prípady gastrointestinálnej perforácie alebo vzniku fistúl (pozri časť 4.8).
Počas liečby axitinibom je potrebné sledovať príznaky svedčiace pre gastrointestinálnu perforáciu alebo vznik fistuly.
Komplikácie spojené s hojením rán
Neuskutočnili sa žiadne formálne klinické štúdie sledujúce vplyv axitinibu na hojenie rán.
Liečbu axitinibom sa má prerušiť najmenej 24 hodín pred plánovaným chirurgickým zákrokom. Rozhodnutie pokračovať v liečbe axitinibom po chirurgickom zákroku sa má opierať o klinické posúdenie adekvátneho hojenia rany.
Syndróm posteriórnej reverzibilnej encefalopatie
V klinických štúdiách s axitinibom boli hlásené prípady syndrómu posteriórnej reverzibilnej encefalopatie (PRES) (pozri časť 4.8).
PRES je neurologické ochorenie, ktoré sa môže prejaviť bolesťou hlavy, záchvatom, letargiou, zmätenosťou, slepotou a ďalšími zrakovými a neurologickými poruchami. Môže byť prítomná ľahká až závažná hypertenzia. Na potvrdenie diagnózy PRES je nevyhnutná magnetická rezonancia.
U pacientov so znakmi alebo príznakmi PRES prechodne prerušte alebo definitívne ukončite liečbu axitinibom. Bezpečnosť opätovného začatia liečby axitinibom u pacientov s anamnézou PRES nie je
známa.
Proteinúria
V klinických štúdiách s axitinibom bola hlásena proteinúria, vrátane stupňa závažnosti 3 (pozri časť
4.8).
Odporúča sa vyšetriť proteinúriu pred začiatkom liečby a kontrolovať ju pravidelne počas liečby axitinibom. Ak sa u pacientov objaví stredne závažná až závažná proteinúria, znížte dávku alebo dočasne prerušte liečbu axitinibom (pozri časť 4.2).
Nežiaduce účinky súvisiace s pečeňou
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli hlásené nežiaduce účinky súvisiace s pečeňou. Najčastejšie uvádzané nežiaduce účinky súvisiace s pečeňou zahŕňali zvýšenie
hladiny alanínaminotransferázy (ALT), aspartátaminotransferázy (AST) a bilirubínu v krvi (pozri časť
4.8). Nepozorovali sa súbežné zvýšenia hladiny ALT (> 3-násobok hornej hranice normy [upper limit of normal, ULN]) a bilirubínu (> 2-násobok ULN).
V klinickej štúdii na stanovenie dávky boli súbežné zvýšenia hladín ALT (12-násobok ULN)
a bilirubínu (2,3-násobok ULN), považované za hepatotoxicitu súvisiacu s liekom, pozorované
u 1 pacienta, ktorý užíval axitinib v úvodnej dávke 20 mg dvakrát denne (4-násobok odporúčanej úvodnej dávky).
Je potrebné vyšetriť pečeňové testy pred začiatkom liečby a kontrolovať pravidelne počas liečby axitinibom.
Poškodenie pečene
V klinických štúdiách s axitinibom bola systémová expozícia axitinibu približne 2-násobne vyššia u subjektov so stredne závažným poškodením funkcie pečene (trieda B klasifikácie podľa
Childa-Pugha) oproti subjektom s normálnou funkciou pečene. U pacientov so stredne závažným
poškodením funkcie pečene (trieda B klasifikácie podľa Childa-Pugha) sa odporúča zníženie dávky pri podávaní axitinibu (pozri časť 4.2).
Axitinib saskúmal u pacientov so závažným poškodením funkcie pečene (trieda C klasifikácie podľa
Childa-Pugha) a nemá sa u tejto populácie používať.
Starší pacienti (≥ 65 rokov)a príslušnosťk rase
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bolo 34 % pacientov liečených axitinibom vo veku ≥ 65 rokov. Väčšina pacientov boli belosi (77 %) alebo aziati (21 %). Hoci
nemožno vylúčiť väčšiu náchylnosť na vznik nežiaducich reakcií u niektorých starších pacientov
a ázijských pacientov, celkovo neboli pozorované žiadne významné rozdiely v bezpečnosti a účinnosti axitinibu medzi pacientmi vo veku ≥ 65 rokov a mladšími a medzi príslušníkmi bielej rasy a iných rás.
Nie je potrebná úprava dávkovania na základe veku pacienta alebo príslušnosti k rase (pozri časti 4.2
a 5.2).
Laktóza
Tento liek obsahuje laktózu. Pacienti so zriedkavými vrodenými problémami intolerancie galaktózy, deficitu Lapp laktázy alebo glukózo-galaktózovej malabsorpcie nemajú užívať tento liek.
4.5 Liekové a iné interakcie
In vitro údaje ukazujú, že axitinib je metabolizovaný primárne enzýmom CYP3A4/5 a v menšej miere enzýmami CYP1A2, CYP2C19 a uridín-difosfoglukuronyltransferázou (UGT) 1A1.
Inhibítory CYP3A4/5
Ketokonazol, silný inhibítor CYP3A4/5 podávaný v dávke 400 mg raz denne počas 7 dní, zvýšil
2-násobne priemernú plochu pod krivkou (AUC – area under the curve) a 1,5-násobne Cmax
jednorazovo podanej 5 mg perorálnej dávky axitinibu u zdravých dobrovoľníkov.
Súčasné podávanie axitinibu so silnými inhibítormi CYP3A4/5 (napr. ketokonazol, itrakonazol, klaritromycín, erytromycín, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, sachinavir
a telitromycín) môže zvyšovať plazmatické koncentrácie axitinibu. Takisto grapefruit môže viesť
k zvýšeniu plazmatickej koncentrácie axitinibu. Pri súčasnom užívaní sa odporúča vyberať lieky so žiadnym alebo minimálnym inhibičným potenciálom na CYP3A4/5. Ak musí byť podávaný súbežne
silný inhibítor CYP3A4/5, odporúča sa úprava dávkovania axitinibu (pozri časť 4.2).
Inhibítory CYP1A2 a CYP2C19
CYP1A2 a CYP2C19 predstavujú minoritné (< 10%) cesty metabolizácie axitinibu. Účinok silných inhibítorov týchto izoenzýmov na farmakokinetiku axitinibu sa neskúmal. U pacientov užívajúcich
silné inhibítory týchto izoenzýmov je potrebná opatrnosť kvôli riziku zvýšenia plazmatických
koncentrácií axitinibu.
Induktory CYP3A4/5
Rifampicín, silný induktor CYP3A4/5 podávaný v dávke 600 mg raz denne počas 9 dní, znížil priemernú AUC o 79% a Cmax o 71% pri jednorazovo podanej dávke 5 mg axitinibu u zdravých dobrovoľníkov.
Súbežné podávanie axitinibu so silnými CYP3A4/5 induktormi (napr. rifampicín, dexametazón, fenytoín, karbamazepín, rifabutín, rifapentín, fenobarbital a Hypericum perforatum [Ľubovník bodkovaný]) môže znižovať plazmatické koncentrácie axitinibu. Pri súbežnom užívaní sa odporúča vyberať lieky so žiadnym alebo minimálnym indukčným potenciálom na CYP3A4/5. Ak sa musí súbežne podávať silný induktor CYP3A4/5, odporúča sa úprava dávkovania axitinibu (pozri časť 4.2).
Indukcia CYP1A2 fajčením
CYP1A2 predstavuje minoritnú (< 10 %) cestu metabolizácie axitinibu. Účinok indukcie CYP1A2
súvisiaci s fajčením na farmakokinetiku axitinibu nebol úplne popísaný. Pri podávaní axitinibu fajčiarom je potrebné brať do úvahy riziko zníženia plazmatických koncentrácií axitinibu.
In vitro štúdie inhibície a indukcie CYP a UGT
In vitro štúdie ukázali, že axitinib pri terapeutických plazmatických koncentráciách neinhibuje
CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, alebo UGT1A1.
In vitro štúdie ukázali, že axitinib má potenciál inhibovať CYP1A2. Preto môže súbežné podávanie axitinibu so substrátmi CYP1A2 viesť k zvýšeniu plazmatických koncentrácií substrátov CYP1A2 (napr. teofylínu).
In vitro štúdie tiež preukázali, že axitinib má potenciál inhibovať CYP2C8. Súbežné podávanie axitinibu s paklitaxelom, známym substrátom CYP2C8 však neviedlo k zvýšeniu plazmatických koncentrácií paklitaxelu u pacientov s pokročilým karcinómom, čo dokazuje chýbanie klinicky významnej inhibície CYP2C8.
In vitro štúdie na ľudských hepatocytoch tiež ukázali, že axitinib neindukuje CYP1A1, CYP1A2 alebo CYP3A4/5. Preto sa pri súbežnom podávaní axitinibu neočakáva zníženie plazmatickej koncentrácie súbežne podávaných substrátov CYP1A1, CYP1A2 alebo CYP3A4/5 in vivo.
In vitro štúdie s P-glykoproteínom
In vitro štúdie ukázali, že axitinib inhibuje P-glykoproteín. Neočakáva sa však, že by axitinib inhiboval P-glykoproteín pri terapeutických plazmatických koncentráciách. Preto sa pri súčasnom
podávaní axitinibu neočakáva zvýšenie plazmatickej koncentrácie digoxínu alebo iných substrátov
P-glykoproteínu in vivo.
4.6 Fertilita, gravidita a laktácia
Gravidita
Neexistujú žiadne údaje týkajúce sa podávania axitinibu tehotným ženám. Na základe svojich farmakologických vlastností môže axitinib spôsobiť poškodenie plodu, ak sa podáva tehotným ženám.
Štúdie na zvieratách preukázali reprodukčnú toxicitu vrátane vzniku malformácií (pozri časť 5.3).
Axitinib sa nemá podávať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu týmto liekom.
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a až do 1 týždňa po nej.
Laktácia
Nie je známe, či sa axitinib vylučuje do ľudského mlieka. Nemožno vylúčiť riziko pre kojené dieťa. Axitinib sa nemá užívať počas laktácie.
Fertilita
Na základe neklinických zistení má axitinib potenciál poškodzovať reprodukčnú funkciu a plodnosť
u ľudí (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Axitinib má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti majú byť poučení, že sa u nich počas liečby axitinibom môžu vyskytnúť závraty a/alebo únava.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluNajdôležitejšie závažné nežiaduce účinky hlásené u pacientov užívajúcich axitinib boli arteriálne embolické a trombotické príhody, venózne embolické a trombotické príhody, krvácanie (vrátane gastrointestinálneho krvácania, cerebrálneho krvácania a hemoptýzy), gastrointestinálna perforácia a vznik fistúl, hypertenzná kríza a syndróm posteriórnej reverzibilnej encefalopatie. Tieto riziká vrátane potrebných protiopatrení sú uvedené v časti 4.4.
Najčastejšie (≥ 20 %) nežiaduce účinky pozorované po liečbe axitinibom boli hnačka, hypertenzia, únava, dysfónia, nevoľnosť, znížená chuť do jedla a syndróm palmárno-plantárnej erytrodyzestézie (syndróm ruka-a noha).
Zoznam nežiaducich reakcií zostavený do tabuľkyTabuľka 1 uvádza nežiaduce reakcie hlásené u pacientov, ktorí dostávali axitinib v pivotnej klinickej štúdii na liečbu pacientov s RCC (pozri časť 5.1).
Nežiaduce reakcie sú zoradené podľa tried orgánových systémov, kategórie frekvencie výskytu
a stupňa ich závažnosti. Kategórie frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1000), veľmi zriedkavé (< 1/10 000) a neznáme (ich frekvenciu nemožno stanoviť z dostupných údajov). Súčasná databáza údajov o bezpečnosti axitinibu je príliš malá na detekovanie zriedkavých a veľmi zriedkavých nežiaducich reakcií.
Kategórie boli zoradené podľa absolútnych početností podľa údajov z klinických štúdií. V rámci každej triedy orgánového systému sú nežiaduce reakcie pri rovnakej častosti výskytu uvádzané
v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduce reakcie
| Všetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
| Poruchy krvi
a lymfatického systému
| Časté
| Anémia
| 2,8
| 0,3
| 0
| Trombocytopénia
| 1,7
| 0,3
| 0
|
| Neutropénia
| 0,3
| 0,3
| 0
| Menej časté
| Polycytémiab
| 0,3
| 0
| 0
| Leukopénia
| 0,3
| 0
| 0
| Poruchy
endokrinného systému
| Veľmi časté
| Hypotyreózab
| 18,4
| 0,3
| 0
| Menej časté
| Hypertyreózab
| 0,6
| 0
| 0
| Poruchy
metabolizmu a výživy
| Veľmi časté
| Znížená chuť do
jedla
| 28,4
| 3,3
| 0,3
| Časté
| Dehydratácia
| 4,7
| 2,5
| 0
| Menej časté
| Hyperkaliémia
| 0,8
| 0,6
| 0
| Hyperkalciémia
| 0,6
| 0
| 0
|
|
|
Tabuľka 1. Nežiaduce reakcie hlásené v klinickej štúdii s RCC u pacientov, ktorí dostávali axitinib (N= 359)
Trieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduce reakcie
|
V
šetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
|
Poruchy nervového
systému
|
Veľmi časté
|
Bolesť hlavy
|
10,3
|
0,6
|
0
|
Poruchy chuti
|
10,3
|
0
|
0
|
Časté
|
Závraty
|
5,6
|
0
|
0
|
Menej časté
|
Syndróm
posteriórnej reverzibilnej
encefalopatie
|
0,3
|
0,3
|
0
|
Porychy ucha
a labyrintu
|
Časté
|
Tinnitus
|
2,2
|
0
|
0
|
Poruchy ciev
|
Veľmi časté
|
Hypertenzia
|
39,3
|
15,3
|
0,3
|
Krvácanieb, c
|
10,6
|
0,3
|
0,3
|
Časté
|
Venózne embolické
a trombotické príhodyb, c
|
1,9
|
0,8
|
0,8
|
Arteriálne
embolické
a trombotické príhodyb, c
|
1,1
|
1,1
|
0
|
Menej časté
|
Hypertenzná kríza
|
0,6
|
0,3
|
0,3
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
|
Veľmi časté
|
Dysfónia
|
28,1
|
0
|
0
|
Časté
|
Dyspnoe
|
7,0
|
0,3
|
0
|
Kašeľ
|
5,3
|
0
|
0
|
Orofaryngeálna
bolesť
|
3,3
|
0
|
0
|
Poruchy
gastrointestinálneho traktu
|
Veľmi časté
|
Hnačka
|
51,3
|
9,7
|
0,3
|
Vracanie
|
16,7
|
1,4
|
0
|
Nevoľnosť
|
28,7
|
1,4
|
0
|
Stomatitída
|
14,5
|
1,4
|
0
|
Zápcha
|
12,3
|
0
|
0
|
Časté
|
Bolesť brucha
|
8,4
|
0,6
|
0,3
|
Bolesť v epigastriu
|
6,1
|
0,3
|
0
|
Dyspepsia
|
7,8
|
0
|
0
|
Flatulencia
|
4,5
|
0
|
0
|
Hemoroidy
|
2,2
|
0
|
0
|
Menej časté
|
Gastrointestinálna perforáciab, d
|
0,3
|
0
|
0,3
|
Análna fistulab
|
0,3
|
0
|
0
|
Poruchy kože
a podkožného tkaniva
|
Veľmi časté
|
Palmárno-plantárna
erytrodyzestézia
(syndróm ruka- noha)
|
27,3
|
5,0
|
0
|
Vyrážka
|
11,7
|
0,3
|
0
|
Suchosť kože
|
10,0
|
0
|
0
|
Časté
|
Pruritus
|
5,8
|
0
|
0
|
Erytém
|
2,2
|
0
|
0
|
Alopécia
|
3,3
|
0
|
0
|
Poruchy kostrovej
a svalovej sústavy a spojivového
tkaniva
|
Časté
|
Myalgia
|
5,3
|
0,6
|
0,3
|
Artralgia
|
8,6
|
0,6
|
0
|
Bolesť končatín
|
8,9
|
0,3
|
0
|
Trieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduce reakcie
|
V
šetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
|
Poruchy obličiek
a močových ciest
|
Veľmi časté
|
Proteinúria
|
10,3
|
3,1
|
0
|
Časté
|
Zlyhanie obličiek
|
1.1
|
0.6
|
0
|
Celkové poruchy
a reakcie v mieste podania
|
Veľmi časté
|
Únava
|
34,8
|
9,5
|
0,3
|
Asténiac
|
17,5
|
3,6
|
0,3
|
Zápal sliznice
|
15,0
|
1,4
|
0
|
Laboratórne
a funkčné vyšetrenia
|
Veľmi časté
|
Úbytok hmotnosti
|
16,4
|
1,4
|
0
|
Časté
|
Zvýšenie hladiny
tyreoideu stimulujúceho hormónu
|
4,5
|
0
|
0
|
Zvýšenie hladiny
lipázy
|
2,2
|
0,6
|
0
|
Zvýšenie hladiny
alanínamino transferázy
|
1,9
|
0,3
|
0
|
Zvýšenie hladiny
aspartátamino transferázy
|
1,1
|
0,3
|
0
|
Zvýšenie hladiny
alkalickej fosfatázy
|
1,4
|
0
|
0
|
Zvýšenie hladiny
amylázy
|
1,7
|
0
|
0
|
Menej časté
|
Zvýšenie hladiny
bilirubínu v krvi
|
0,6
|
0
|
0
|
Zvýšenie hladiny
kreatinínu
|
0,6
|
0
|
0
|
|
|
a Všeobecné terminologické kritériá pre nežiaduce účinky národného inštitútu pre rakovinu verzia 3.0 (CTCAE)
b Pozri časť Popis vybraných nežiaducich účinkov
c Boli hlásené fatálne prípady (5. stupeň závažnosti)
d Výskyt nežiaducej reakcie z akejkoľvek príčiny
e Vrátane zlyhania obličiek
Opis vybraných nežiaducich reakciíDysfunkcia štítnej žľazy (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola hypotyreóza hlásená
u 18,4% pacientov a hypertyreóza u 0,6 % pacientov. Zvýšenie hladiny thyreostimulačného hormónu (TSH) bolo ako nežiaduci účinok hlásené u 4,5% pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho sledovania u pacientov, ktorí mali pred liečbou TSH < 5μU/ml,L sa objavilo zvýšenie TSH až na ≥ 10 μU/mL u 32,2 % pacientov užívajúcich axitinib.
Venózne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli venózne embolické
a trombotické nežiaduce reakcie hlásené u 1,9 % pacientov užívajúcich axitinib. Venózne embolické a trombotické nežiaduce reakcie stupňa závažnosti 3/4 boli hlásené u 1,7 % pacientov užívajúcich
axitinib (vrátane pľúcnej embólie, hlbokej venóznej trombózy a oklúzie/trombózy sietnicovej vény).
Fatálna pľúcna embólia bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib.
Arteriálne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli arteriálne embolické
a trombotické nežiaduce reakcie stupňa závažnosti 3/4 hlásené u 1,1 % pacientov užívajúcich axitinib. Najčastejšou arteriálnou embolické a trombotickou príhodou bol tranzitórny ischemický atak (0,8 %).
Fatálna cerebrovaskulárna príhoda bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib. V monoterapeutických klinických štúdiách s axitinibom (N=699) boli arteriálne embolické
a trombotické nežiaduce reakcie (vrátane tranzitórneho ischemického ataku, infarktu myokardu
a cerebrovaskulárnej príhody) hlásené u 1,0 % pacientov užívajúcich axitinib.
Polycytémia (pozri
Elevácia hemoglobínu alebo hematokritu v časti 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola polycytémia hlásená ako nežiaduca reakcia u 0,3 % pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho
sledovania bola zvýšená hladina hemoglobínu nad ULN zistená u 9,7% pacientov užívajúcich axitinib. V štyroch klinických štúdiách s axitinibom v liečbe pacientov s RCC (N=537) bola zvýšená hladina
hemoglobínu nad ULN pozorovaná u 13,6 % pacientov užívajúcich axitinib.
Hemorágia (pozri časť 4,4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC, kam neboli zaradení pacienti s neliečenými metastázami v mozgu, boli hlásené hemoragické nežiaduce reakcie u 10,6% pacientov užívajúcich axitinib. Najčastejšími hemoragickými nežiaducimi reakciami u pacientov liečených axitinibom boli epistaxa (5,3 %), hematúria (1,4 %), krvácanie z konečníka (1,1 %) a krvácanie
z ďasien (1,1 %). Hemoragické nežiaduce reakcie stupňa závažnosti ≥ 3 boli hlásené u 0,8 %
pacientov užívajúcich axitinib (vrátane cerebrálneho krvácania, žalúdočného krvácania a krvácania
z distálneho gastrointestinálneho traktu). Fatálna hemorágia bola hlásená u jedného pacienta (0,3 %), ktorý užíval axitinib (žalúdočné krvácanie). V monoterapeutických klinických štúdiách s axitinibom
(N=699) bola hemoptýza hlásená ako nežiaduca reakcia u 1,6 % pacientov, vrátane jedného prípadu
(0,1 %) so stupňom závažnosti ≥ 3.
Gastrointestinálna perforácia a tvorba fistúl (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola gastrointestinálna perforácia hlásená u jedného pacienta (0,3 %, bez ohľadu na kauzalitu), ktorý užíval axitinib. V monoterapeutických klinických štúdiách s axitinibom (N=699) boli fistuly hlásené u 0,7 %
pacientov (bez ohľadu na kauzalitu) a fatálna gastrointestinálna perforácia bola hlásená u jedného pacienta (0,1%).
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieNeexistuje žiadna špecifická liečba v prípade predávkovania axitinibom.
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC jeden pacient, ktorý neúmyselne užíval dávku 20 mg dvakrát denne počas 4 dní, udával závraty (stupeň závažnosti 1).
V klinickej štúdii s axitinibom na stanovenie dávky probandi, ktorí užívali úvodnú dávku 10 mg dvakrát denne alebo 20 mg dvakrát denne mali nežiaduce reakcie, ktoré zahŕňali hypertenziu, záchvaty spojené s hypertenziou a fatálnu hemoptýzu.
V prípade podozrenia z predávkovania je potrebné vysadiť axitinib a začať podpornú starostlivosť.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
F
armakoterapeutická skupina: Cytostatiká, inhibítory proteínovej kinázy, ATC kód: L01XE17
Spôsob účinku
Axitinib je účinný a selektívny inhibítor tyrozínkinázy receptorov vaskulárneho endoteliálneho rastového faktora (VEGFR- 1, VEGFR-2 a VEGFR-3). Tieto receptory sa podieľajú na patologickej
angiogenéze, raste nádoru a metastatickej progresii karcinómu. Preukázalo sa, že axitinib účinne
inhibuje proliferáciu endoteliálnych buniek a ich prežívanie sprostredkované VEGF. Axitinib inhiboval fosforyláciu VEGFR-2 nádorovej vaskulatúry xenograftu, ktorá predstavovala cieľ liečby in vivo a viedol k oddialeniu rastu nádoru, regresii a inhibícii metastáz v mnohých experimentálnych modeloch rakoviny.
Vplyv na QTc interval
V randomizovanej dvojramennej štúdii s prekrížením liečebných ramien bola 35 zdravým probandom podaná jedna perorálna dávka axitinibu (5 mg) bez podávania a s podávaním 400 mg ketokonazolu
počas 7 dní. Výsledky tejto štúdie ukázali, že expozícia axitinibu v plazme až dvojnásobne vyššia ako očakávané terapeutické hladiny pri 5 mg dávkovaní neviedla ku klinicky signifikantnému predĺženiu
QT intervalu.
Klinickáúčinnosť
Bezpečnosť a účinnosť axitinibu sa hodnotili v randomizovanej, otvorenej multicentrickej klinickej štúdii fázy 3. Pacienti (N=723) s pokročilým RCC, ktorých ochorenie progredovalo počas alebo po liečbe jednou predchádzajúcou systémovou terapiou, zahrňujúcou režimy so sunitinibom, bevacizumabom, temsirolimom alebo cytokín-obsahujúce , boli randomizovaní (1:1) na užívanie axitinibu (n=361) alebo sorafenibu (n=362). Primárny cieľ, prežívanie bez progresie (PFS) sa hodnotil centrálne metódou zaslepeného nezávislého posúdenia. Medzi sekundárne ciele patrili miera objektívnej odpovede (ORR) a celkové prežívanie (OS).
Z pacientov zaradených do tejto klinickej štúdie dostalo 389 pacientov (53,8 %) jednu predchádzajúcu liečbu založenú na sunitinibe, 251 pacientov (34,7 %) dostalo jednu predchádzajúcu liečbu založenú
na cytokínoch (interleukin-2 alebo interferón-alfa), 59 pacientov (8,2%) dostalo jednu predchádzajúcu liečbu založenú na bevacizumabe a 24 pacientov (3,3 %) dostalo jednu predchádzajúcu liečbu
založenú na temsirolime. Demografické a nádorové charakteristiky na začiatku štúdie boli podobné medzi liečebnými skupinami s axitinibom a sorafenibom čo sa týka veku, pohlavia, rasy,
výkonnostného stavu podľa ECOG kritérií (Eastern Cooperative Oncology Group), geografického regiónu a predchádzajúcej liečby.
V celej populácii pacientov a dvoch hlavných podskupinách (skupina s predchádzajúcou liečbou sunitinibom a skupina s predchádzajúcou liečbou cytokínmi) pre primárny cieľ hodnotenia PFS bol preukázaný štatisticky signifikantný prínos axitinibu oproti sorafenibu (pozri Tabuľka 2 a Obrázky 1,
2 and 3). Hodnota mediánu PFS účinku bol v oboch podskupinách rozdelených podľa predchádzajúcej
liečby odlišný. Dve podskupiny boli príliš malé, aby poskytli dôveryhodné výsledky (skupina
s predchádzajúcou liečbou temsirolimom a skupina s predchádzajúcou liečbou bevacizumabom). Medzi liečebnými ramenami neboli žiadne štatisticky signifikantné rozdiely v OS v celkovej populácii
ani v podskupinách rozdelených podľa predchádzajúcej liečby.
Tabuľka 2. Výsledky účinnosti
Cieľ / Populácia štúdie
|
axitinib
|
sorafenib
|
HR
(95% CI)
|
p-hodnota
|
|
Celá ITT populácia
Medián PFS v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
|
N = 361
6,8 (6,4; 8,3)
20,1 (16,7; 23,4)
|
N = 362
4,7 (4,6; 6,3)
19,2 (17,5; 22,3)
|
0,67 (0,56; 0,81)
0,97 (0,80; 1,17)
|
< 0,0001
NS
|
ORR b,e% (95% CI)
|
19,4 (15,4; 23,9)
|
9,4 (6,6; 12,9)
|
2,06f (1,41; 3,00)
|
0,0001g
|
Predchádzajúca liečba sunitinibom
|
N = 194
|
N = 195
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
4,8 (4,5; 6,5)
15,2 (12,8; 18,3)
11,3 (7,2; 16,7)
|
3,4 (2,8; 4,7)
16,5 (13,7; 19,2)
7,7 (4,4; 12,4)
|
0,74 (0,58; 0,94)
1,00 (0,78; 1,27)
1,48f (0,79; 2,75)
|
0,0063h
NS
|
Predchádzajúca liečba
cyto
k
ínmi
|
N = 126
|
N = 125
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
12,0 (10,1; 13,9)
29,4 (24,5; NE)
32,5 (24,5; 41,5)
|
6,6 (6,4; 8,3)
27,8 (23,1;, 34,5)
13,6 (8,1; 20,9)
|
0,52 (0,38;, 0,72)
0,81 (0,56; 1,19)
2,39f (1,43;3,99)
|
< 0,0001h
NS
0,0002i
|
CI =interval spoľahlivosti, HR miera rizika (axitinib/sorafenib); ITT: všetci pacienti zaradení do štúdie); NE = nemožné predpovedať; NS = štatisticky nesignifikantné; ORR Miera objektívnej'
odpovede;.OS: = celkové prežívanie PFS : Prežívanie bez progresie.,
a Čas od randomizácie po progresiu alebo úmrtie z akejkoľvek príčiny, podľa toho, ktorá udalosť
nastane prvá. Dátum ukončenia: 3. jún 2011.
b Stanovené podľa RECIST kritérií nezávislým rádiologickým posudkom.
c Jednostranná hodnota p podľa log-rank testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej liečby d Dátum ukončenia: 1. november 2011.
e Dátum ukončenia: 31. august 2010.
f Pre ORR je použitá veličina pomer rizík. Pomer rizík > 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene s axitinibom; pomer rizík < 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene so sorafenibom.
g Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej terapie
h Jednostranná hodnota p log-rank testu pre liečbu so stratifikáciou podľa ECOG výkonnostného stavu
i Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu
Obrázok 1. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre celkovú populáciu
INLYTA (N=361)
m
e
d
i
á
n 6,8 mesiaca
sorafenib (N=362)
m
e
d
i
á
n 4,7 mesiaca
m
i
er
a rizika = 0,67
95
% CI [0,56, 0,81]
p hodnota <0,0001
Čas (mesiace)
Obrázok 2. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia prepodskupinu s predchádzajúcou liečbou sunitinibomINLYTA (N=194)medián 4,8 mesiacasorafenib (N=195)medián 3,4 mesiacamiera rizika = 0,7495% CI [0,58, 0,94]p hodnota = 0,0063Čas (mesiace)
Obrázok 3. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre podskupinu s predchádzajúcou liečbou
c
ytokínom.
INLYTA (N=126)
m
e
d
i
á
n
12
,
0 mesiaca
sorafenib (N=125)
m
e
d
i
á
n 6,6 mesiaca
m
i
er
a rizika = 0,52
95
% CI [0,38, 0,72]
p hodnota < 0,0001
Čas (mesiace)
 Deti a dospievajúci
Deti a dospievajúciEurópska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií pre axitinib vo všetkých vekových podskupinách detí a dospievajúcich v liečbe karcinómu obličky a obličkovej panvičky (okrem nefroblastómu, nefroblastomatózy, sarkómu z jasných buniek, mezoblastického nefrómu, obličkového medulárneho karcinómu a rabdoidného nádoru obličky) (pozri časť 4.2 pre informácie o použití v pediatrii).
5.2 Farmakokinetické vlastnostiPo perorálnom podaní tabliet axitinibu je priemerná absolútna biologická dostupnosť 58 %
v porovnaní s intravenóznym podaním. Plazmatický polčas axitinibu kolíše od 2,5 do 6,1 hodiny. Dávkovanie axitinibu po 5 mg dvakrát denne viedlo k menej než dvojnásobnému nahromadeniu
v porovnaní s podaním v jednej dávke. Vzhľadom na krátky eliminačný polčas axitinibu sa očakáva
dosiahnutie rovnovážneho stavu v priebehu 2 až 3 dní po úvodnej dávke.
Absorpcia a distribúciaMaximálne koncentrácie axitinibu v plazme sa obvykle dosiahnu v priebehu 4 hodín po perorálnom podaní axitinibu s mediánom Tmax v rozmedzí od 2,5 do 4,1 hodiny. Podávanie axitinibu so stredne mastným jedlom viedlo k zníženiu expozície o 10% v porovnaní s celonočným hladovaním. Veľmi mastné, vysoko kalorické jedlo viedlo k zvýšeniu expozície o 19% v porovnaní s celonočným hladovaním. Axitinib môže byť užívaný s jedlom alebo bez jedla (pozri časť 4.2).
Priemerná Cmax a AUC sa proporcionálne zvýšili v závislosti na dávkovaní axitinibu v rozsahu 5 až
10 mg.
In vitro väzba axitinibu na ľudské plazmatické proteíny je > 99 % s prednostným naviazaním
na albumín a strednou väzbou na α1-kyslý glykoproteín. Pri dávkovaní 5 mg dvakrát denne u pacientov s pokročilým RCC bola v stave nasýtenosti geometrická priemerná maximálna plazmatická koncentrácia 27,8 ng/mL a 24-hodinová AUC 265 ng.h/mL. Po perorálnom podaní bol geometrický priemerný klírens 38 l/h a distribučný objem bol 160 litrov.
Biotransformácia a elimináciaAxitinib sa metabolizuje primárne v pečeni enzýmom CYP3A4/5 a v menšej miere enzýmami
CYP1A2, CYP2C19 a UGT1A1.
Po perorálnom podaní 5 mg dávky rádioaktívne značeného axitinibu bolo 30-60 % rádioaktivity zachytenej v stolici a 23 % rádioaktivity v moči. Nemetabolizovaný axitinib, na úrovni 12% z dávky, bol hlavnou zložkou identifikovanou v stolici. Nemetabolizovaný axitinib nebol zistený v moči; metabolity kyseliny karboxylovej a sulfoxidu predstavovali väčšinu rádioaktivity v moči. V plazme predstavoval N-glukuronidový metabolit predominantnú rádioaktívnu zložku (50 % rádioaktivity
v cirkulácii) a nemetabolizovaný axitinib a sulfoxidový metabolit každý jeden tvorili približne 20 %
rádioaktivity v cirkulácii.
Sulfoxidové a N-glukuronidové metabolity vykazujú približne 400-násobne a 8000-násobne menšiu účinnosť in vitro voči VEGFR-2 v porovnaní s axitinibom.
Osobitné skupiny pacientov
Starší pacienti, pohlavie a rasa
Populačné farmakokinetické analýzy u pacientov s pokročilým karcinómom (vrátane pokročilého
RCC) a zdravých dobrovoľníkov ukazujú, že vek, pohlavie, telesná hmotnosť, rasa, obličkové funkcie, genotyp UGT1A1 alebo CYP2C19 nemajú žiadne klinicky relevantné účinky.
Deti a dospievajúci
Axitinib sa nesledoval u pacientov mladších ako 18 rokov.
Porucha funkcie pečene
In vitro a in vivo údaje ukazujú, že axitinib je primárne metabolizovaný v pečeni.
V porovnaní s pacientami s normálnou funkciou pečene je systémová expozícia po podaní jednej dávky axitinibu podobná u subjektov s ľahkou poruchou funkcie pečene (trieda A podľa Childa- Pugha) a vyššia (približne dvojnásobne) u pacientov so stredne závažnou poruchou funkcie pečene (trieda B klasifikácie podľa Childa-Pugha). Axitinib sa nesledoval u pacientov so závažnou poruchou funkcie pečene (trieda C klasifikácie podľa Childa-Pugha) a nemá sa používať v tejto populácii (odporúčania pre úpravu dávkovania pozri časť 4.2).
Porucha funkcie obličiek
Nemetabolizovaný axitinib nie je detekovaný v moči.
Axitinib sa nesledoval u pacientov s poruchou funkcie obličiek. V klinických štúdiách s axitinibom
v liečbe pacientov s RCC neboli zaradení pacienti s hladinou sérového kreatinínu > 1,5 násobok ULN alebo kalkulovaným klírensom kreatinínu < 60 mL/min. Populačné farmakokinetické analýzy preukázali, že klírens axitinibu nebol zmenený u pacientov s poruchou funkcie obličiek a nevyžaduje sa žiadna úprava dávkovania.
5.3 Predklinické údaje o bezpečnosti
Toxicita pri opakovanom podávaní
Hlavné prejavy toxicity u myší a psov pri opakovanom podávaní v trvaní do 9 mesiacov boli v gastrointestinálnom, hematopoetickom, reprodukčnom, skeletálnom a dentálnom systéme,
s hladinami bez pozorovaných nežiaducich účinkov (NOAEL - No Observed Adverse Effect Levels)
približne rovnakými alebo nižšími ako je očakávaná expozícia u človeka pri odporúčanej klinickej úvodnej dávke (podľa hladín AUC).
Karcinogenicita
Nevykonali sa žiadne klinické štúdie skúmajúce karcinogenitu axitinibu.
Genotoxicita
Axitinib nebol mutagénny alebo klastogénny v konvenčných skúškach genotoxicity in vitro. Signifikantné zvýšenie polyploidity bolo pozorované in vitro pri koncentráciách > 0,22 μg/mL
a vzostup počtu mikrojadrových polychromatických erytrocytov sa pozoroval in vivo pri hladine bez
pozorovaného účinku na organizmus (NOEL - No Observed Effect Level) 69-násobne presahujúcej
očakávanú expozíciu u človeka. Nálezy genotoxicity sa nepovažujú za klinicky relevantné pri expozičných hladinách pozorovaných u ľudí.
Reprodukčná toxicita
Medzi nálezy súvisiace s axitinibom v semenníkoch a nadsemenníkoch patrili zníženie hmotnosti orgánu, atrofia alebo degenerácia, zníženie počtu germinálnych buniek, hypospermia alebo výskyt
abnormálnych foriem spermií a znížená denzita a počet spermií. Tieto nálezy sa pozorovali u myší pri
expozičných hladinách rovnajúcich sa približne 12-násobku očakávanej expozície u ľudí, u psov pri expozičných hladinách nižších ako je očakávaná expozícia u ľudí. Nepozoroval sa vplyv na párenie
alebo fertilitu u samčích myší pri expozičných hladinách rovnajúcich sa približne 57-násobku
očakávanej expozície u ľudí. K nálezom u samičiek patrili známky oneskorenia pohlavnej dospelosti, zmenšenie alebo chýbanie corpora lutea, zníženie hmotnosti maternice a atrofia maternice pri expozíciách približne rovnakých ako je očakávaná expozícia u ľudí. Znížená fertilita a viabilita embryí boli pozorované pri všetkých testovaných dávkach s expozičnými hladinami na najnižšej dávke rovnajúcimi sa približne 10-násobku očakávanej expozície u ľudí.
U gravidných myší vystavených účinku axitinibu bol preukázaný zvýšený výskyt rázštepu podnebia a skeletálnych variácií, vrátane oneskorenej osifikácie pri expozičných hladinách nižších ako očakávaná expozícia u ľudí. Štúdie perinatálnej a postnatálnej vývojovej toxicity neboli realizované.
Prejavy toxicity u nezrelých zvierat
U myší a psov, ktorí dostávali axitinib najmenej 1 mesiac pri expozičných hladinách približne
6-násobne vyšších než očakávané expozičné hladiny u ľudí bola pozorovaná reverzibilná dysplázia rastovej platničky. U myší užívajúcich axitinib viac než 1 mesiac pri expozičných hladinách
podobných očakávanej expozícii u ľudí bol pozorovaný výskyt čiastočne reverzibilných zubných
kazov. Iné toxické prejavy s možným rizikom pre pediatrických pacientov sa u juvenilných zvierat neskúmali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety mikrokryštalická celulóza monohydrát laktózy kroskarmelóza sodná stearan horečnatý
Obaľujúci film tablety hypromelóza
oxid titaničitý (E171)
monohydrát laktózy triacetín (E1518)
červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Hliníkový/hliníkový blister obsahujúci 14 filmom obalených tabliet. Každé balenie obsahuje 28 alebo
56 filmom obalených tabliet.
HDPE fľaštička s vysušovadlom obsahujúcim silikagél na absorpciu vlhkosti a polypropylénovým uzáverom obsahujúca 60 filmom obalených tabliet.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciuNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited
Ramsgate Road
Sandwich, Kent, CT13, 9NJ Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/12/777/004
EU/1/12/777/005
EU/1/12/777/006
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 3. 9. 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUInlyta 7 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá filmom obalená tableta obsahuje 7 mg axitinibu.
Pomocné látky so známym účinkom:Každá filmom obalená tableta obsahuje 82,3 mg monohydrátu laktózy. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAFilmom obalená tableta.
Červená filmom obalená tableta v tvare kosoštvorca s vyrytým nápisom “Pfizer“ na jednej strane a nápisom “7 XNB“ na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieInlyta je indikovaná na liečbu dospelých pacientov s pokročilým karcinómom z obličkových buniek
(renal cell carcinoma, RCC) po zlyhaní predchádzajúcej liečby sunitinibom alebo cytokínmi.
4.2 Dávkovanie a spôsob podávaniaLiečbu Inlytou má vykonávať lekár, ktorý má skúsenosti s podávaním protinádorových liekov.
DávkovanieOdporúčaná úvodná dávka axitinibu je 5 mg dvakrát denne.
Liečba má pokračovať, pokým sa pozoruje klinický prínos alebo pokiaľ sa neprejaví neprijateľná toxicita, ktorú nie je možné zvládnuť súbežným podávaním liekov alebo úpravou dávky.
Pokiaľ pacient vracia alebo vynechá dávku, nesmie užiť dodatočnú dávku. Nasledujúca predpísaná dávka sa musí užiť v obvyklom čase.
Úpravy dávkyZvýšenie alebo zníženie dávky sa odporúča na základe individuálnej bezpečnosti a znášanlivosti.
U pacientov, ktorí tolerujú axitinib v úvodnej dávke 5 mg dvakrát denne bez nežiaducich účinkov
> ako stupeň 2 (t.j. bez závažných nežiaducich účinkov podľa Všeobecných terminologických kritérií pre nežiaduce účinky [CTCAE - Common Terminology Criteria for Adverse Events] verzia 3.0) počas dvoch po sebe idúcich týždňov, je možné dávku zvýšiť na 7 mg dvakrát denne, pokiaľ nie je krvný
tlak pacienta > 150/90 mmHg alebo pacient neužíva antihypertenzívnu liečbu. Následne podľa
rovnakých kritérií u pacientov, ktorí tolerujú axitinib v dávke 7 mg dvakrát denne, je možné zvýšiť ich dávku na maximálne 10 mg dvakrát denne.
Zvládnutie niektorých nežiaducich účinkov si môže vyžiadať prechodné alebo trvalé prerušenie liečby a/alebo zníženie dávky axitinibu (pozri časť 4.4). Ak je potrebné znížiť dávku, dávka axitinibu môže byť znížená na 3 mg dvakrát denne a ďalej na 2 mg dvakrát denne.
Úprava dávkovania nie je potrebná kvôli veku, rase, pohlaviu alebo telesnej hmotnosti.
Súčasné užívanie silných inhibítorov CYP3A4/5
Súčasné podávanie axitinibu so silnými inhibítormi CYP3A4/5 môže zvyšovať plazmatické koncentrácie axitinibu (pozri časť 4.5). Odporúča sa výber alternatívneho súčasne podávaného lieku so žiadnym alebo minimálnym potenciálom inhibície CYP3A4/5.
Hoci u pacientov užívajúcich silné inhibítory CYP3A4/5 sa neskúmala úprava dávkovania axitinibu, ak sa musí súčasne podávať silný inhibítor CYP3A4/5, odporúča sa zníženie dávky axitinibu na približne polovičnú dávku (t.j. úvodná dávka by mala byť znížená z 5 mg dvakrát denne na 2 mg dvakrát denne). Zvládnutie niektorých nežiaducich účinkov si môže vyžiadať prechodné alebo trvalé prerušenie liečby axitinibom (pozri časť 4.4). Ak sa ukončí súčasné podávanie silného inhibítora, je potrebné zvážiť návrat k dávke axitinibu užívanej pred začiatkom podávania silného inhibítora CYP3A4/5 (pozri časť 4.5).
Súčasné užívanie silných induktorov CYP3A4/5
Súčasné podávanie axitinibu so silnými induktormi CYP3A4/5 môže znižovať plazmatické koncentrácie axitinibu (pozri časť 4.5). Odporúča sa výber alternatívneho súčasne podávaného lieku so žiadnym alebo minimálnym potenciálom indukcie CYP3A4/5.
Hoci u pacientov užívajúcich silné induktory CYP3A4/5 saneskúmala úprava dávkovania axitinibu, ak sa musí súbežne podávať silný induktor CYP3A4/5, odporúča sa postupné zvyšovanie dávky
axitinibu. Maximálna indukcia pri vysokých dávkach silných induktorov CYP3A4/5 sa pozorovala
v priebehu jedného týždňa liečby induktorom. Ak sa dávka axitinibu zvýši, treba pacienta starostlivo monitorovať kvôli toxicite. Zvládnutie niektorých nežiaducich účinkov môže vyžadovať prechodné alebo trvalé prerušenie liečby a/alebo zníženie dávky axitinibu (pozri časť 4.4). Ak sa ukončí súbežné podávanie silného induktora, je treba okamžite upraviť dávku axitinibu na dávku užívanú pred začiatkom podávania silného induktora CYP3A4/5 (pozri časť 4.5).
Osobitné skupiny pacientov
Starší pacienti (≥ 65 rokov)
Nie je potrebná žiadna úprava dávkovania (pozri časti 4.4 a 5.2).
Poškodenie funkcie obličiek: Nevyžaduje sa úprava dávkovania (pozri časť 5.2). Nie sú dostupné prakticky žiadne údaje týkajúce sa liečby axitinibom u pacientov s klírensom kreatinínu < 15 ml/min.
Poškodenie funkcie pečene: Nevyžaduje sa úprava dávkovania, ak sa axitinib podáva pacientom
s ľahkým poškodením pečeňových funkcií (trieda A klasifikácie Childa-Pugha). Zníženie dávky sa odporúča pri podávaní axitinibu pacientom so stredne závažným poškodením pečeňových funkcií (trieda B klasifikácie Childa-Pugha) (t.j. úvodná dávka sa má znížiť z 5 mg dvakrát denne na 2 mg dvakrát denne). Axitinib sa neskúmal u pacientov so závažným poškodením pečeňových funkcií (trieda C klasifikácie podľa Childa-Pugha) a nemá sa preto používať v tejto skupine pacientov (pozri časti 4.4. a 5.2).
Deti a dospievajúci
Bezpečnosť a účinnosť axitinibu u detí a dospievajúcich < 18 rokov nebola stanovená. Nie sú dostupné žiadne údaje.
Spôsob podania
Axitinib sa má užívať perorálne dvakrát denne v približne 12-hodinových intervaloch s alebo bez jedla
(pozri časť 5.2). Tablety axitinibu sa majú prehltnúť celé a zapiť pohárom vody.
4.3 Kontraindikácie
Precitlivenosť na axitinib alebo na niektorú z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Pred začatím a pravidelne počas liečby axitinibom je potrebné sledovať typické príhody týkajúce sa bezpečnosti liečby ako sú uvedené ďalej.
Hypertenzia
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC sa veľmi často hlásila hypertenzia (pozri časť 4.8). Medián času do vzniku hypertenzie (systolický tlak krvi > 150 mmHg
alebo diastolický tlak krvi > 100 mmHg) sa dosiahol v priebehu prvého mesiaca od začiatku liečby
axitinibom a vzostupy krvného tlaku sa pozorovali už po 4 dňoch od začiatku užívania axitinibu.
Už pred začiatkom liečby axitinibom sa musí krvný tlak dobre kontrolovať. Pacienti sa majú sledovať kvôli hypertenzii a podľa potreby liečiť štandardnou antihypertenzívnou liečbou. V prípade pretrvávania hypertenzie napriek užívaniu antihypertenzívnych liekov sa musí znížiť dávka axitinibu. U pacientov, u ktorých sa vyvinie závažná hypertenzia, dočasne prerušte liečbu axitinibom
a pokračujte nižšou dávkou, keď je pacient normotenzný. Ak je liečba axitinibom prerušená, musia sa pacienti užívajúci antihypertenzívne lieky sledovať kvôli hypotenzii (pozri časť 4.2).
V prípade závažnej alebo pretrvávajúcej artériovej hypertenzie a príznakov pripomínajúcich syndróm posteriórnej reverzibilnej encefalopatie (viď nižšie) treba zvážiť diagnostické vyšetrenie mozgu magnetickou rezonanciou (MRI).
Dysfunkcia štítnej žľazy
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC sa hlásili prípady hypotyreózy a v menšom rozsahu prípady hypertyreózy (pozri časť 4.8).
Funkcia štítnej žľazy sa má vyšetriť ešte pred začiatkom a pravidelne kontrolovať počas liečby axitinibom. Hypotyreóza alebo hypertyreóza sa majú liečiť podľa štandardných terapeutických postupov s cieľom udržaťeutyreoidný stav.
Arteriálne embolické a trombotické príhody
V klinických štúdiách s axitinibom boli hlásené arteriálne embolické a trombotické príhody (vrátane tranzitórneho ischemického ataku, infarktu myokardu, cerebrovaskulárnej príhody a oklúzie
sietnicovej artérie) (pozri časť 4.8).
Axitinib sa má preto používať opatrne u pacientov, ktorí majú zvýšené riziko vzniku alebo sa u nich už vyskytli takéto príhody. Axitinib sa neskúmal u pacientov, ktorí mali arteriálnu embolickú
a trombotickú príhodu počas predchádzajúcich 12 mesiacov.
Venózne embolické a trombotické príhody
V klinických štúdiách s axitinibom boli hlásené venózne embolické a trombotické príhody (vrátane pľúcnej embólie, hlbokej žilovej trombózy a oklúzie/trombózy sietnicovej vény) (pozri časť 4.8).
Axitinib sa má preto používať opatrne u pacientov, ktorí majú zvýšené riziko vzniku alebo sa u nich už vyskytli takéto príhody. Axitinib sa neskúmal u pacientov, ktorí mali venóznu embolickú
a trombotickú príhodu počas predchádzajúcich 6 mesiacov.
Elevácia hemoglobínu alebo hematokritu
Počas liečby axitinibom sa môže vyskytnúť vzostup hemoglobínu alebo hematokritu ako prejav zvýšenia množstva červených krviniek (pozri časť 4.8, polycytémia). Zvýšenie množstva červených krviniek môže zvýšiť riziko vzniku embolických a trombotických príhod.
Hladina hemoglobínu alebo hodnota hematokritu sa majú vyšetriť ešte pred začiatkom a pravidelne kontrolovať počas liečby axitinibom. Ak dôjde k zvýšeniu hladiny hemoglobínu alebo hodnoty hematokritu nad normálnu úroveň, pacienti sa majú liečiť štandardnými terapeutickými postupmi
s cieľom znížiť hladinu hemoglobínu alebo hodnotu hematokritu na prijateľnú úroveň.
Hemorágia
V klinických štúdiách s axitinibom boli hlásené hemoragické príhody (pozri časť 4.8).
Axitinib sa neskúmal u pacientov s dokázanými neliečenými metastázami v mozgu alebo nedávnym aktívnym gastrointestinálnym krvácaním a nemá sa u týchto pacientov používať. Ak si akékoľvek krvácanie vyžaduje medicínsky zásah, dočasne prerušte podávanie axitinibu.
Gastrointestinálna perforácia a tvorba fistúl
V klinických štúdiách s axitinibom boli hlásené prípady gastrointestinálnej perforácie alebo vzniku fistúl (pozri časť 4.8).
Počas liečby axitinibom je potrebné sledovať príznaky svedčiace pre gastrointestinálnu perforáciu alebo vznik fistuly.
Komplikácie spojené s hojením rán
Neuskutočnili sa žiadne formálne klinické štúdie sledujúce vplyv axitinibu na hojenie rán.
Liečbu axitinibom sa má prerušiť najmenej 24 hodín pred plánovaným chirurgickým zákrokom. Rozhodnutie pokračovať v liečbe axitinibom po chirurgickom zákroku sa má opierať o klinické posúdenie adekvátneho hojenia rany.
Syndróm posteriórnej reverzibilnej encefalopatie
V klinických štúdiách s axitinibom boli hlásené prípady syndrómu posteriórnej reverzibilnej encefalopatie (PRES) (pozri časť 4.8).
PRES je neurologické ochorenie, ktoré sa môže prejaviť bolesťou hlavy, záchvatom, letargiou, zmätenosťou, slepotou a ďalšími zrakovými a neurologickými poruchami. Môže byť prítomná ľahká až závažná hypertenzia. Na potvrdenie diagnózy PRES je nevyhnutná magnetická rezonancia.
U pacientov so znakmi alebo príznakmi PRES prechodne prerušte alebo definitívne ukončite liečbu axitinibom. Bezpečnosť opätovného začatia liečby axitinibom u pacientov s anamnézou PRES nie je
známa.
Proteinúria
V klinických štúdiách s axitinibom bola hlásena proteinúria, vrátane stupňa závažnosti 3 (pozri časť
4.8).
Odporúča sa vyšetriť proteinúriu pred začiatkom liečby a kontrolovať ju pravidelne počas liečby axitinibom. Ak sa u pacientov objaví stredne závažná až závažná proteinúria, znížte dávku alebo dočasne prerušte liečbu axitinibom (pozri časť 4.2).
Nežiaduce účinky súvisiace s pečeňou
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli hlásené účinky súvisiace s pečeňou. Najčastejšie uvádzané nežiaduce účinky súvisiace s pečeňou zahŕňali zvýšenie hladiny
alanínaminotransferázy (ALT), aspartátaminotransferázy (AST) a bilirubínu v krvi (pozri časť 4.8).
Nepozorovali sa súbežné zvýšenia hladiny ALT (> 3-násobok hornej hranice normy [upper limit of normal, ULN]) a bilirubínu (> 2-násobok ULN).
V klinickej štúdii na stanovenie dávky boli súbežné zvýšenia hladín ALT (12-násobok ULN)
a bilirubínu (2,3-násobok ULN), považované za hepatotoxicitu súvisiacu s liekom, pozorované
u 1 pacienta, ktorý užíval axitinib v úvodnej dávke 20 mg dvakrát denne (4-násobok odporúčanej úvodnej dávky).
Je potrebné vyšetriť pečeňové testy pred začiatkom liečby a kontrolovať pravidelne počas liečby axitinibom.
Poškodenie pečene
V klinických štúdiách s axitinibom bola systémová expozícia axitinibu približne 2-násobne vyššia u subjektov so stredne závažným poškodením funkcie pečene (trieda B klasifikácie podľa
Childa-Pugha) oproti subjektom s normálnou funkciou pečene. U pacientov so stredne závažným
poškodením funkcie pečene (trieda B klasifikácie Childa-Pugha) sa odporúča zníženie dávky pri podávaní axitinibu (pozri časť 4.2).
Axitinib sa skúmal u pacientov so závažným poškodením funkcie pečene (trieda C klasifikácie podľa
Childa-Pugha) a nemá sa u tejto populácie používať.
Starší pacienti (≥ 65 rokov)a príslušnosťk rase
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bolo 34 % pacientov liečených axitinibom vo veku ≥ 65 rokov. Väčšina pacientov boli belosi (77 %) alebo aziati (21 %). Hoci
nemožno vylúčiť väčšiu náchylnosť na vznik nežiaducich reakcií u niektorých starších pacientov
a ázijských pacientov, celkovo neboli pozorované žiadne významné rozdiely v bezpečnosti a účinnosti axitinibu medzi pacientmi vo veku ≥ 65 rokov a mladšími a medzi príslušníkmi bielej rasy a iných rás.
Nie je potrebná úprava dávkovania na základe veku pacienta alebo príslušnosti k rase (pozri časti 4.2
a 5.2).
Laktóza
Tento liek obsahuje laktózu. Pacienti so zriedkavými vrodenými problémami intolerancie galaktózy, deficitu Lapp laktázy alebo glukózo-galaktózovej malabsorpcie nemajú užívať tento liek.
4.5 Liekové a iné interakcie
In vitro údaje ukazujú, že axitinib je metabolizovaný primárne enzýmom CYP3A4/5 a v menšej miere enzýmami CYP1A2, CYP2C19, a uridín-difosfoglukuronyltransferázou (UGT) 1A1.
Inhibítory CYP3A4/5
Ketokonazol, silný inhibítor CYP3A4/5 podávaný v dávke 400 mg raz denne počas 7 dní, zvýšil
2-násobne priemernú plochu pod krivkou (AUC – area under the curve) a 1,5-násobne Cmax
jednorazovo podanej 5 mg perorálnej dávky axitinibu u zdravých dobrovoľníkov.
Súčasné podávanie axitinibu so silnými inhibítormi CYP3A4/5 (napr. ketokonazol, itrakonazol, klaritromycín, erytromycín, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, sachinavir
a telitromycín) môže zvyšovať plazmatické koncentrácie axitinibu. Takisto grapefruit môže viesť
k zvýšeniu plazmatickej koncentrácie axitinibu. Pri súčasnom užívaní sa odporúča vyberať lieky so žiadnym alebo minimálnym inhibičným potenciálom na CYP3A4/5. Ak musí byť podávaný súbežne
silný inhibítor CYP3A4/5, odporúča sa úprava dávkovania axitinibu (pozri časť 4.2).
Inhibítory CYP1A2 a CYP2C19
CYP1A2 a CYP2C19 predstavujú minoritné (< 10%) cesty metabolizácie axitinibu. Účinok silných inhibítorov týchto izoenzýmov na farmakokinetiku axitinibu sa neskúmal. U pacientov užívajúcich
silné inhibítory týchto izoenzýmov je potrebná opatrnosť kvôli riziku zvýšenia plazmatických
koncentrácií axitinibu.
Induktory CYP3A4/5
Rifampicín, silný induktor CYP3A4/5 podávaný v dávke 600 mg raz denne počas 9 dní, znížil priemernú AUC o 79% a Cmax o 71% pri jednorazovo podanej dávke 5 mg axitinibu u zdravých dobrovoľníkov.
Súbežné podávanie axitinibu so silnými CYP3A4/5 induktormi (napr. rifampicín, dexametazón, fenytoín, karbamazepín, rifabutín, rifapentín, fenobarbital a Hypericum perforatum [Ľubovník bodkovaný]) môže znižovať plazmatické koncentrácie axitinibu. Pri súbežnom užívaní sa odporúča vyberať lieky so žiadnym alebo minimálnym indukčným potenciálom na CYP3A4/5. Ak sa musí súbežne podávať silný induktor CYP3A4/5, odporúča sa úprava dávkovania axitinibu (pozri časť 4.2).
Indukcia CYP1A2 fajčením
CYP1A2 predstavuje minoritnú (< 10%) cestu metabolizácie axitinibu. Účinok indukcie CYP1A2
súvisiaci s fajčením na farmakokinetiku axitinibu nebol úplne popísaný. Pri podávaní axitinibu fajčiarom je potrebné brať do úvahy riziko zníženia plazmatických koncentrácií axitinibu.
In vitro štúdie inhibície a indukcie CYP a UGT
In vitro štúdie ukázali, že axitinib pri terapeutických plazmatických koncentráciách neinhibuje
CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, alebo UGT1A1.
In vitro štúdie ukázali, že axitinib má potenciál inhibovať CYP1A2. Preto môže súbežné podávanie axitinibu so substrátmi CYP1A2 viesť k zvýšeniu plazmatických koncentrácií substrátov CYP1A2 (napr. teofylínu).
In vitro štúdie tiež preukázali, že axitinib má potenciál inhibovať CYP2C8. Súbežné podávanie axitinibu s paklitaxelom, známym substrátom CYP2C8 však neviedlo k zvýšeniu plazmatických koncentrácií paklitaxelu u pacientov s pokročilým karcinómom, čo dokazuje chýbanie klinicky významnej inhibície CYP2C8.
In vitro štúdie na ľudských hepatocytoch tiež ukázali, že axitinib neindukuje CYP1A1, CYP1A2 alebo CYP3A4/5. Preto sa pri súbežnom podávaní axitinibu neočakáva zníženie plazmatickej koncentrácie súbežne podávaných substrátov CYP1A1, CYP1A2 alebo CYP3A4/5 in vivo.
In vitro štúdie s P-glykoproteínom
In vitro štúdie ukázali, že axitinib inhibuje P-glykoproteín. Neočakáva sa však, že by axitinib inhiboval P-glykoproteín pri terapeutických plazmatických koncentráciách. Preto sa pri súčasnom
podávaní axitinibu neočakáva zvýšenie plazmatickej koncentrácie digoxínu alebo iných substrátov
P-glykoproteínu in vivo.
4.6 Fertilita, gravidita a laktácia
Gravidita
Neexistujú žiadne údaje týkajúce sa podávania axitinibu tehotným ženám. Na základe svojich farmakologických vlastností môže axitinib spôsobiť poškodenie plodu, ak sa podáva tehotným ženám.
Štúdie na zvieratách preukázali reprodukčnú toxicitu vrátane vzniku malformácií (pozri časť 5.3).
Axitinib sa nemá podávať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu týmto liekom.
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a až do 1 týždňa po nej.
Laktácia
Nie je známe, či sa axitinib vylučuje do ľudského mlieka. Nemožno vylúčiť riziko pre kojené dieťa. Axitinib sa nemá užívať počas laktácie.
Fertilita
Na základe neklinických zistení má axitinib potenciál poškodzovať reprodukčnú funkciu a plodnosť
u ľudí (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Axitinib má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti majú byť poučení, že sa u nich počas liečby axitinibom môžu vyskytnúť závraty a/alebo únava.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluNajdôležitejšie závažné nežiaduce účinky hlásené u pacientov užívajúcich axitinib boli arteriálne embolické a trombotické príhody, venózne embolické a trombotické príhody, krvácanie (vrátane gastrointestinálneho krvácania, cerebrálneho krvácania a hemoptýzy), gastrointestinálna perforácia a vznik fistúl, hypertenzná kríza a syndróm posteriórnej reverzibilnej encefalopatie. Tieto riziká vrátane potrebných protiopatrení sú uvedené v časti 4.4.
Najčastejšie (≥ 20 %) nežiaduce účinky pozorované po liečbe axitinibom boli hnačka, hypertenzia, únava, dysfónia, nevoľnosť, znížená chuť do jedla a syndróm palmárno-plantárnej erytrodyzestézie (syndróm ruka-noha).
Zoznam nežiaducich reakcií zostavený do tabuľkyTabuľka 1 uvádza nežiaduce reakcie hlásené u pacientov, ktorí dostávali axitinib v pivotnej klinickej štúdii na liečbu pacientov s RCC (pozri časť 5.1).
Nežiaduce reakcie sú zoradené podľa tried orgánových systémov, kategórie frekvencie výskytu
a stupňa ich závažnosti. Kategórie frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1000), veľmi zriedkavé (< 1/10 000) a neznáme (ich frekvenciu nemožno stanoviť z dostupných údajov). Súčasná databáza údajov o bezpečnosti axitinibu je príliš malá na detekovanie zriedkavých a veľmi zriedkavých nežiaducich reakcií.
Kategórie boli zoradené podľa absolútnych početností podľa údajov z klinických štúdií. V rámci každej triedy orgánového systému sú nežiaduce reakcie pri rovnakej častosti výskytu uvádzané
v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduce reakcie
| Všetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
| Poruchy krvi
a lymfatického systému
| Časté
| Anémia
| 2,8
| 0,3
| 0
| Trombocytopénia
| 1,7
| 0,3
| 0
| Menej časté
| Neutropénia
| 0,3
| 0,3
| 0
| Polycytémiab
| 0,3
| 0
| 0
| Leukopénia
| 0,3
| 0
| 0
| Poruchy
endokrinného systému
| Veľmi časté
| Hypotyreózab
| 18,4
| 0,3
| 0
| Menej časté
| Hypertyreózab
| 0,6
| 0
| 0
| Poruchy
metabolizmu a výživy
| Veľmi časté
| Znížená chuť do
jedla
| 28,4
| 3,3
| 0,3
| Časté
| Dehydratácia
| 4,7
| 2,5
| 0
| Menej časté
| Hyperkaliémia
| 0,8
| 0,6
| 0
| Hyperkalciémia
| 0,6
| 0
| 0
|
|
|
Tabuľka 1. Nežiaduce reakcie hlásené v klinickej štúdii s RCC u pacientov, ktorí dostávali axitinib (N= 359)
Trieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduce reakcie
|
V
šetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
|
Poruchy nervového
systému
|
Veľmi časté
|
Bolesť hlavy
|
10,3
|
0,6
|
0
|
Poruchy chuti
|
10,3
|
0
|
0
|
Časté
|
Závraty
|
5,6
|
0
|
0
|
Menej časté
|
Syndróm
posteriórnej reverzibilnej
encefalopatie
|
0,3
|
0,3
|
0
|
Porychy ucha
a labyrintu
|
Časté
|
Tinnitus
|
2,2
|
0
|
0
|
Poruchy ciev
|
Veľmi časté
|
Hypertenzia
|
39,3
|
15,3
|
0,3
|
Krvácanieb, c
|
10,6
|
0,3
|
0,3
|
Časté
|
Venózne embolické
a trombotické príhodyb, c
|
1,9
|
0,8
|
0,8
|
Arteriálne
embolické
a trombotické príhodyb, c
|
1,1
|
1,1
|
0
|
Menej časté
|
Hypertenzná kríza
|
0,6
|
0,3
|
0,3
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
|
Veľmi časté
|
Dysfónia
|
28,1
|
0
|
0
|
Časté
|
Dyspnoe
|
7,0
|
0,3
|
0
|
Kašeľ
|
5,3
|
0
|
0
|
Orofaryngeálna
bolesť
|
3,3
|
0
|
0
|
Poruchy
gastrointestinálneho traktu
|
Veľmi časté
|
Hnačka
|
51,3
|
9,7
|
0,3
|
Vracanie
|
16,7
|
1,4
|
0
|
Nevoľnosť
|
28,7
|
1,4
|
0
|
Stomatitída
|
14,5
|
1,4
|
0
|
Zápcha
|
12,3
|
0
|
0
|
Časté
|
Bolesť brucha
|
8,4
|
0,6
|
0,3
|
Bolesť v epigastriu
|
6,1
|
0,3
|
0
|
Dyspepsia
|
7,8
|
0
|
0
|
Flatulencia
|
4,5
|
0
|
0
|
Hemoroidy
|
2,2
|
0
|
0
|
Menej časté
|
Gastrointestinálna perforáciab, d
|
0,3
|
0
|
0,3
|
Análna fistulab
|
0,3
|
0
|
0
|
Poruchy kože
a podkožného tkaniva
|
Veľmi časté
|
Palmárno-plantárna
erytrodyzestézia
(syndróm ruka-noha)
|
27,3
|
5,0
|
0
|
Vyrážka
|
11,7
|
0,3
|
0
|
Suchosť kože
|
10,0
|
0
|
0
|
Časté
|
Pruritus
|
5,8
|
0
|
0
|
Erytém
|
2,2
|
0
|
0
|
Alopécia
|
3,3
|
0
|
0
|
Poruchy kostrovej
a svalovej sústavy a spojivového
tkaniva
|
Časté
|
Myalgia
|
5,3
|
0,6
|
0,3
|
Artralgia
|
8,6
|
0,6
|
0
|
Bolesť končatín
|
8,9
|
0,3
|
0
|
Trieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduce reakcie
|
V
šetky stupne závažnosti n (%)
|
3. stupeň závažnosti n (%)
|
4. stupeň závažnosti n (%)
|
Poruchy obličiek
a močových ciest
|
Veľmi časté
|
Proteinúria
|
10,3
|
3,1
|
0
|
Časté
|
Zlyhanie obličiek
|
1.1
|
0.6
|
0
|
Celkové poruchy
a reakcie v mieste podania
|
Veľmi časté
|
Únava
|
34,8
|
9,5
|
0,3
|
Asténiac
|
17,5
|
3,6
|
0,3
|
Zápal sliznice
|
15,0
|
1,4
|
0
|
Laboratórne
a funkčné vyšetrenia
|
Veľmi časté
|
Úbytok hmotnosti
|
16,4
|
1,4
|
0
|
Časté
|
Zvýšenie hladiny
tyreoideu stimulujúceho hormónu
|
4,5
|
0
|
0
|
Zvýšenie hladiny
lipázy
|
2,2
|
0,6
|
0
|
Zvýšenie hladiny
alanínamino transferázy
|
1,9
|
0,3
|
0
|
Zvýšenie hladiny
aspartátamino transferázy
|
1,1
|
0,3
|
0
|
Zvýšenie hladiny
alkalickej fosfatázy
|
1,4
|
0
|
0
|
Zvýšenie hladiny
amylázy
|
1,7
|
0
|
0
|
Menej časté
|
Zvýšenie hladiny
bilirubínu v krvi
|
0,6
|
0
|
0
|
Zvýšenie hladiny
kreatinínu
|
0,6
|
0
|
0
|
|
|
a Všeobecné terminologické kritériá pre nežiaduce účinky národného inštitútu pre rakovinu verzia 3.0 (CTCAE)
b Pozri časť Popis vybraných nežiaducich účinkov
c Boli hlásené fatálne prípady (5. stupeň závažnosti)
d Výskyt nežiaducej reakcie z akejkoľvek príčiny
e Vrátane zlyhania obličiek
Opis vybraných nežiaducich reakciíDysfunkcia štítnej žľazy (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola hypotyreóza hlásená
u 18,4 % pacientov a hypertyreóza u 0,6 % pacientov. Zvýšenie hladiny thyreostimulačného hormónu
(TSH) bolo ako nežiaduci účinok hlásené u 4,5 % pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho sledovania u pacientov, ktorí mali pred liečbou TSH < 5 μU/ml, sa
objavilo zvýšenie TSH až na ≥ 10 μU/ml u 32,2 % pacientov užívajúcich axitinib.
Venózne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli venózne embolické
a trombotické nežiaduce reakcie hlásené u 1,9 % pacientov užívajúcich axitinib. Venózne embolické a trombotické nežiaduce reakcie stupňa závažnosti 3/4 boli hlásené u 1,7 % pacientov užívajúcich axitinib (vrátane pľúcnej embólie, hlbokej venóznej trombózy a oklúzie/trombózy sietnicovej vény). Fatálna pľúcna embólia bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib.
Arteriálne embolické a trombotické príhody (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC boli arteriálne embolické
a trombotické nežiaduce reakcie stupňa závažnosti 3/4 hlásené u 1,1 % pacientov užívajúcich axitinib.
Najčastejšou arteriálnou embolické a trombotickou príhodou bol tranzitórny ischemický atak (0,8 %). Fatálna cerebrovaskulárna príhoda bola hlásená u jedného pacienta (0,3 %) užívajúceho axitinib.
V monoterapeutických klinických štúdiách s axitinibom (N=699) boli arteriálne embolické
a trombotické nežiaduce reakcie (vrátane tranzitórneho ischemického ataku, infarktu myokardu a cerebrovaskulárnej príhody) hlásené u 1,0 % pacientov užívajúcich axitinib.
Polycytémia (pozri
Elevácia hemoglobínu alebo hematokritu v časti 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola polycytémia hlásená ako nežiaduca reakcia u 0,3 % pacientov užívajúcich axitinib. V rámci pravidelného laboratórneho
sledovania bola zvýšená hladina hemoglobínu nad ULN zistená u 9,7 % pacientov užívajúcich
axitinib. V štyroch klinických štúdiách s axitinibom v liečbe pacientov s RCC (N=537) bola zvýšená hladina hemoglobínu nad ULN pozorovaná u 13,6 % pacientov užívajúcich axitinib.
Hemorágia (pozri časť 4,4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC, kam neboli zaradení pacienti s neliečenými metastázami v mozgu, boli hlásené hemoragické nežiaduce reakcie u 10,6 % pacientov
užívajúcich axitinib. Najčastejšími hemoragickými nežiaducimi reakciami u pacientov liečených
axitinibom boli epistaxa (5,3 %), hematúria (1,4 %), krvácanie z konečníka (1,1 %) a krvácanie z ďasien (1,1 %). Hemoragické nežiaduce reakcie stupňa závažnosti ≥ 3 boli hlásené u 0,8 %
pacientov užívajúcich axitinib (vrátane cerebrálneho krvácania, žalúdočného krvácania a krvácania
z distálneho gastrointestinálneho traktu). Fatálna hemorágia bola hlásená u jedného pacienta (0,3 %), ktorý užíval axitinib (žalúdočné krvácanie). V monoterapeutických klinických štúdiách s axitinibom (N=699) bola hemoptýza hlásená ako nežiaduca reakcia u 1,6 % pacientov, vrátane jedného prípadu (0,1 %) so stupňom závažnosti ≥ 3.
Gastrointestinálna perforácia a tvorba fistúl (pozri časť 4.4)
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC bola gastrointestinálna perforácia hlásená u jedného pacienta (0,3 %, bez ohľadu na kauzalitu), ktorý užíval axitinib.
V monoterapeutických klinických štúdiách s axitinibom (N=699) boli fistuly hlásené u 0,7 %
pacientov (bez ohľadu na kauzalitu) a fatálna gastrointestinálna perforácia bola hlásená u jedného pacienta (0,1 %).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v Prílohe V.
4.9 PredávkovanieNeexistuje žiadna špecifická liečba v prípade predávkovania axitinibom.
V kontrolovanej klinickej štúdii s axitinibom v liečbe pacientov s RCC jeden pacient, ktorý neúmyselne užíval dávku 20 mg dvakrát denne počas 4 dní, udával závraty (stupeň závažnosti 1).
V klinickej štúdii s axitinibom na stanovenie dávky probandi, ktorí užívali úvodnú dávku 10 mg dvakrát denne alebo 20 mg dvakrát denne mali nežiaduce reakcie, ktoré zahŕňali hypertenziu, záchvaty spojené s hypertenziou a fatálnu hemoptýzu.
V prípade podozrenia z predávkovania je potrebné vysadiť axitinib a začať podpornú starostlivosť.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
F
armakoterapeutická skupina: Cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE17
Spôsob účinku
Axitinib je účinný a selektívny inhibítor tyrozínkinázy receptorov vaskulárneho endoteliálneho rastového faktora (VEGFR-1, VEGFR-2 a VEGFR-3). Tieto receptory sa podieľajú na patologickej
angiogenéze, raste nádoru a metastatickej progresii karcinómu. Preukázalo sa, že axitinib účinne
inhibuje proliferáciu endoteliálnych buniek a ich prežívanie sprostredkované VEGF. Axitinib inhiboval fosforyláciu VEGFR-2 nádorovej vaskulatúry xenograftu, ktorá predstavovala cieľ liečby in vivo a viedol k oddialeniu rastu nádoru, regresii a inhibícii metastáz v mnohých experimentálnych modeloch rakoviny.
Vplyv na QTc interval
V randomizovanej dvojramennej štúdii s prekrížením liečebných ramien bola 35 zdravým probandom podaná jedna perorálna dávka axitinibu (5 mg) bez podávania a s podávaním 400 mg ketokonazolu
počas 7 dní. Výsledky tejto štúdie ukázali, že expozícia axitinibu v plazme až dvojnásobne vyššia ako očakávané terapeutické hladiny pri 5 mg dávkovaní neviedla ku klinicky signifikantnému predĺženiu
QT intervalu.
Klinickáúčinnosť
Bezpečnosť a účinnosť axitinibu sa hodnotili v randomizovanej, otvorenej multicentrickej klinickej štúdii fázy 3. Pacienti (N=723) s pokročilým RCC, ktorých ochorenie progredovalo počas alebo po liečbe jednou predchádzajúcou systémovou terapiou, zahrňujúcou režimy so sunitinibom, bevacizumabom, temsirolimom alebo cytokín-obsahujúce , boli randomizovaní (1:1) na užívanie axitinibu (n=361) alebo sorafenibu (n=362). Primárny cieľ, prežívanie bez progresie (PFS) sa hodnotil centrálne metódou zaslepeného nezávislého posúdenia. Medzi sekundárne ciele patrili miera objektívnej odpovede (ORR) a celkové prežívanie (OS).
Z pacientov zaradených do tejto klinickej štúdie dostalo 389 pacientov (53,8 %) jednu predchádzajúcu liečbu založenú na sunitinibe, 251 pacientov (34,7 %) dostalo jednu predchádzajúcu liečbu založenú
na cytokínoch (interleukin-2 alebo interferón-alfa), 59 pacientov (8,2%) dostalo jednu predchádzajúcu liečbu založenú na bevacizumabe a 24 pacientov (3,3 %) dostalo jednu predchádzajúcu liečbu
založenú na temsirolime. Demografické a nádorové charakteristiky na začiatku štúdie boli podobné medzi liečebnými skupinami s axitinibom a sorafenibom čo sa týka veku, pohlavia, rasy,
výkonnostného stavu podľa ECOG kritérií (Eastern Cooperative Oncology Group), geografického regiónu a predchádzajúcej liečby.
V celej populácii pacientov a dvoch hlavných podskupinách (skupina s predchádzajúcou liečbou sunitinibom a skupina s predchádzajúcou liečbou cytokínmi) pre primárny cieľ hodnotenia PFS bol preukázaný štatisticky signifikantný prínos axitinibu oproti sorafenibu (pozri Tabuľka 2 a Obrázky 1,
2 and 3). Hodnota mediánu PFS účinku bol v oboch podskupinách rozdelených podľa predchádzajúcej
liečby odlišný. Dve podskupiny boli príliš malé, aby poskytli dôveryhodné výsledky (skupina
s predchádzajúcou liečbou temsirolimom a skupina s predchádzajúcou liečbou bevacizumabom). Medzi liečebnými ramenami neboli žiadne štatisticky signifikantné rozdiely v OS v celkovej populácii
ani v podskupinách rozdelených podľa predchádzajúcej liečby.
Tabuľka 2. Výsledky účinnosti
Cieľ / Populácia štúdie
|
axitinib
|
sorafenib
|
HR
(95% CI)
|
p-hodnota
|
|
Celá ITT populácia
Medián PFS v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
|
N = 361
6,8 (6,4; 8,3)
20,1 (16,7; 23,4)
|
N = 362
4,7 (4,6; 6,3)
19,2 (17,5; 22,3)
|
0,67 (0,56; 0,81)
0,97 (0,80; 1,17)
|
< 0,0001
NS
|
ORR b,e% (95% CI)
|
19,4 (15,4; 23,9)
|
9,4 (6,6; 12,9)
|
2,06f (1,41; 3,00)
|
0,0001g
|
Predchádzajúca liečba sunitinibom
|
N = 194
|
N = 195
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
4,8 (4,5; 6,5)
15,2 (12,8; 18,3)
11,3 (7,2; 16,7)
|
3,4 (2,8; 4,7)
16,5 (13,7; 19,2)
7,7 (4,4; 12,4)
|
0,74 (0,58; 0,94)
1,00 (0,78; 1,27)
1,48f (0,79; 2,75)
|
0,0063h
NS
|
Predchádzajúca liečba
cyto
k
ínmi
|
N = 126
|
N = 125
|
|
|
Medián PFS a,b v mesiacoch
(95% CI)
Medián OS d v mesiacoch
(95% CI)
ORR b,e % (95% CI)
|
12,0 (10,1; 13,9)
29,4 (24,5; NE)
32,5 (24,5; 41,5)
|
6,6 (6,4; 8,3)
27,8 (23,1;, 34,5)
13,6 (8,1; 20,9)
|
0,52 (0,38;, 0,72)
0,81 (0,56; 1,19)
2,39f (1,43;3,99)
|
< 0,0001h
NS
0,0002i
|
CI =interval spoľahlivosti, HR miera rizika (axitinib/sorafenib); ITT: všetci pacienti zaradení do štúdie); NE = nemožné predpovedať; NS = štatisticky nesignifikantné; ORR Miera objektívnej
odpovede;.OS: = celkové prežívanie PFS : Prežívanie bez progresie.
a Čas od randomizácie po progresiu alebo úmrtie z akejkoľvek príčiny, podľa toho, ktorá udalosť
nastane prvá. Dátum ukončenia: 3. jún 2011.
b Stanovené podľa RECIST kritérií nezávislým rádiologickým posudkom.
c Jednostranná hodnota p podľa log-rank testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej liečby d Dátum ukončenia: 1. november 2011.
e Dátum ukončenia: 31. august 2010.
f Pre ORR je použitá veličina pomer rizík. Pomer rizík > 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene s axitinibom; pomer rizík < 1 znamenal väčšiu pravdepodobnosť
liečebnej odpovede v ramene so sorafenibom.
g Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu a predchádzajúcej terapie
h Jednostranná hodnota p log-rank testu pre liečbu so stratifikáciou podľa ECOG výkonnostného stavu
i Jednostranná hodnota p Cochran-Mantel-Haenszel testu pre liečbu so stratifikáciou podľa ECOG
výkonnostného stavu
Obrázok 1. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre celkovú populáciu
INLYTA (N=361)
m
e
d
i
á
n 6,8 mesiaca
sorafenib (N=362)
m
e
d
i
á
n 4,7 mesiaca
m
i
er
a rizika = 0,67
95
% CI [0,56, 0,81]
p hodnota <0,0001
Čas (mesiace)
Obrázok 2. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia prepodskupinu s predchádzajúcou liečbou sunitinibomINLYTA (N=194)medián 4,8 mesiacasorafenib (N=195)medián 3,4 mesiacamiera rizika = 0,7495% CI [0,58, 0,94]p hodnota = 0,0063Čas (mesiace)
Obrázok 3. Kaplan-Meierova krivka prežívania bez progresie podľa nezávislého hodnotenia pre podskupinu s predchádzajúcou liečbou
c
ytokínom.
INLYTA (N=126)
m
e
d
i
á
n
12
,
0 mesiaca
sorafenib (N=125)
m
e
d
i
á
n 6,6 mesiaca
m
i
er
a rizika = 0,52
95
% CI [0,38, 0,72]
p hodnota < 0,0001
Čas (mesiace)
Deti a dospievajúciEurópska lieková agentúra udelila výnimku z povinnosti predložiť výsledky štúdií pre axitinib vo všetkých vekových podskupinách detí a dospievajúcich v liečbe karcinómu obličky a obličkovej panvičky (okrem nefroblastómu, nefroblastomatózy, sarkómu z jasných buniek, mezoblastického nefrómu, obličkového medulárneho karcinómu a rabdoidného nádoru obličky) (pozri časť 4.2 pre informácie o použití v pediatrii).
5.2 Farmakokinetické vlastnostiPo perorálnom podaní tabliet axitinibu je priemerná absolútna biologická dostupnosť 58 %
v porovnaní s intravenóznym podaním. Plazmatický polčas axitinibu kolíše od 2,5 do 6,1 hodiny. Dávkovanie axitinibu po 5 mg dvakrát denne viedlo k menej než dvojnásobnému nahromadeniu
v porovnaní s podaním v jednej dávke. Vzhľadom na krátky eliminačný polčas axitinibu sa očakáva
dosiahnutie rovnovážneho stavu v priebehu 2 až 3 dní po úvodnej dávke.
Absorpcia a distribúciaMaximálne koncentrácie axitinibu v plazme sa obvykle dosiahnu v priebehu 4 hodín po perorálnom podaní axitinibu s mediánom Tmax v rozmedzí od 2,5 do 4,1 hodiny. Podávanie axitinibu so stredne mastným jedlom viedlo k zníženiu expozície o 10 % v porovnaní s celonočným hladovaním. Veľmi mastné, vysoko kalorické jedlo viedlo k zvýšeniu expozície o 19% v porovnaní s celonočným hladovaním. Axitinib môže byť užívaný s jedlom alebo bez jedla (pozri časť 4.2).
Priemerná Cmax a AUC sa proporcionálne zvýšili v závislosti na dávkovaní axitinibu v rozsahu 5 až
10 mg.
In vitro väzba axitinibu na ľudské plazmatické proteíny je > 99 % s prednostným naviazaním
na albumín a strednou väzbou na α1-kyslý glykoproteín. Pri dávkovaní 5 mg dvakrát denne u pacientov s pokročilým RCC bola v stave nasýtenosti geometrická priemerná maximálna plazmatická koncentrácia 27,8 ng/ml a 24-hodinová AUC 265 ng.h/ml. Po perorálnom podaní bol geometrický priemerný klírens 38 l/h a distribučný objem bol 160 litrov.
Biotransformácia a elimináciaAxitinib sa metabolizuje primárne v pečeni enzýmom CYP3A4/5 a v menšej miere enzýmami
CYP1A2, CYP2C19 a UGT1A1.
Po perorálnom podaní 5 mg dávky rádioaktívne značeného axitinibu bolo 30-60 % rádioaktivity zachytenej v stolici a 23 % rádioaktivity v moči. Nemetabolizovaný axitinib, na úrovni 12 % z dávky, bol hlavnou zložkou identifikovanou v stolici. Nemetabolizovaný axitinib nebol zistený v moči; metabolity kyseliny karboxylovej a sulfoxidu predstavovali väčšinu rádioaktivity v moči. V plazme predstavoval N-glukuronidový metabolit predominantnú rádioaktívnu zložku (50 % rádioaktivity
v cirkulácii) a nemetabolizovaný axitinib a sulfoxidový metabolit každý jeden tvorili približne
20 % rádioaktivity v cirkulácii.
Sulfoxidové a N-glukuronidové metabolity vykazujú približne 400-násobne a 8000-násobne menšiu účinnosť in vitro voči VEGFR-2 v porovnaní s axitinibom.
Osobitné skupiny pacientov
Starší pacienti, pohlavie a rasa
Populačné farmakokinetické analýzy u pacientov s pokročilým karcinómom (vrátane pokročilého
RCC) a zdravých dobrovoľníkov ukazujú, že vek, pohlavie, telesná hmotnosť, rasa, obličkové funkcie, genotyp UGT1A1 alebo CYP2C19 nemajú žiadne klinicky relevantné účinky.
Deti a dospievajúci
Axitinib sa nesledoval u pacientov mladších ako 18 rokov.
Porucha funkcie pečene
In vitro a in vivo údaje ukazujú, že axitinib je primárne metabolizovaný v pečeni.
V porovnaní s pacientami s normálnou funkciou pečene je systémová expozícia po podaní jednej dávky axitinibu podobná u subjektov s ľahkou poruchou funkcie pečene (trieda A klasifikácie podľa Childa-Pugha) a vyššia (približne dvojnásobne) u pacientov so stredne závažnou poruchou funkcie pečene (trieda B klasifikácie podľa Childa-Pugha). Axitinib sa nesledoval u pacientov so závažnou poruchou funkcie pečene (trieda C klasifikácie podľa Childa-Pugha) a nemá sa používať v tejto populácii (odporúčania pre úpravu dávkovania pozri časť 4.2).
Porucha funkcie obličiek
Nemetabolizovaný axitinib nie je detekovaný v moči.
Axitinib sa nesledoval u pacientov s poruchou funkcie obličiek. V klinických štúdiách s axitinibom
v liečbe pacientov s RCC neboli zaradení pacienti s hladinou sérového kreatinínu > 1,5 násobok ULN alebo kalkulovaným klírensom kreatinínu < 60 ml/min. Populačné farmakokinetické analýzy preukázali, že klírens axitinibu nebol zmenený u pacientov s poruchou funkcie obličiek a nevyžaduje sa žiadna úprava dávkovania.
5.3 Predklinické údaje o bezpečnosti
Toxicita pri opakovanom podávaní
Hlavné prejavy toxicity u myší a psov pri opakovanom podávaní v trvaní do 9 mesiacov boli v gastrointestinálnom, hematopoetickom, reprodukčnom, skeletálnom a dentálnom systéme,
s hladinami bez pozorovaných nežiaducich účinkov (NOAEL – No Observed Adverse Effect Levels)
približne rovnakými alebo nižšími ako je očakávaná expozícia u človeka pri odporúčanej klinickej úvodnej dávke (podľa hladín AUC).
Karcinogenicita
Nevykonali sa žiadne klinické štúdie skúmajúce karcinogenitu axitinibu.
Genotoxicita
Axitinib nebol mutagénny alebo klastogénny v konvenčných skúškach genotoxicity in vitro. Signifikantné zvýšenie polyploidity bolo pozorované in vitro pri koncentráciách > 0,22 μg/ml
a vzostup počtu mikrojadrových polychromatických erytrocytov sa pozoroval in vivo pri hladine bez
pozorovaného účinku na organizmus (NOEL - No Observed Effect Level) 69-násobne presahujúcej
očakávanú expozíciu u človeka. Nálezy genotoxicity sa nepovažujú za klinicky relevantné pri expozičných hladinách pozorovaných u ľudí.
Reprodukčná toxicita
Medzi nálezy súvisiace s axitinibom v semenníkoch a nadsemenníkoch patrili zníženie hmotnosti orgánu, atrofia alebo degenerácia, zníženie počtu germinálnych buniek, hypospermia alebo výskyt
abnormálnych foriem spermií a znížená denzita a počet spermií. Tieto nálezy sa pozorovali u myší pri
expozičných hladinách rovnajúcich sa približne 12-násobku očakávanej expozície u ľudí, u psov pri expozičných hladinách nižších ako je očakávaná expozícia u ľudí. Nepozoroval sa vplyv na párenie
alebo fertilitu u samčích myší pri expozičných hladinách rovnajúcich sa približne 57-násobku
očakávanej expozície u ľudí. K nálezom u samičiek patrili známky oneskorenia pohlavnej dospelosti, zmenšenie alebo chýbanie corpora lutea, zníženie hmotnosti maternice a atrofia maternice pri expozíciách približne rovnakých ako je očakávaná expozícia u ľudí. Znížená fertilita a viabilita embryí boli pozorované pri všetkých testovaných dávkach s expozičnými hladinami na najnižšej dávke rovnajúcimi sa približne 10-násobku očakávanej expozície u ľudí.
U gravidných myší vystavených účinku axitinibu bol preukázaný zvýšený výskyt rázštepu podnebia a skeletálnych variácií, vrátane oneskorenej osifikácie pri expozičných hladinách nižších ako očakávaná expozícia u ľudí. Štúdie perinatálnej a postnatálnej vývojovej toxicity neboli realizované.
Prejavy toxicity u nezrelých zvierat
U myší a psov, ktorí dostávali axitinib najmenej 1 mesiac pri expozičných hladinách približne
6-násobne vyšších než očakávané expozičné hladiny u ľudí bola pozorovaná reverzibilná dysplázia rastovej platničky. U myší užívajúcich axitinib viac než 1 mesiac pri expozičných hladinách
podobných očakávanej expozícii u ľudí bol pozorovaný výskyt čiastočne reverzibilných zubných
kazov. Iné toxické prejavy s možným rizikom pre pediatrických pacientov sa u juvenilných zvierat neskúmali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety mikrokryštalická celulóza monohydrát laktózy kroskarmelóza sodná stearan horečnatý
Obaľujúci film tablety hypromelóza
oxid titaničitý (E171)
monohydrát laktózy triacetín (E1518)
červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Hliníkový/hliníkový blister obsahujúci 14 filmom obalených tabliet. Každé balenie obsahuje 28 alebo
56 filmom obalených tabliet.
HDPE fľaštička s vysušovadlom obsahujúcim silikagél na absorpciu vlhkosti a polypropylénovým uzáverom obsahujúca 60 filmom obalených tabliet.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciuNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited
Ramsgate Road
Sandwich, Kent, CT13, 9NJ Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/12/777/010
EU/1/12/777/011
EU/1/12/777/012
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 3. 9. 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.