
v priestore medzistavcovej platničky je limitovaná 8 mg
(5,3 cm3 namočeného matrixu). InductOs musí byť umiestnený v prístroji (prístrojoch) lumbálnej fúzie
tiel stavcov alebo v prednej časti priestoru medzistavcovej platničky.
Chirurgia akútnej fraktúry tíbie
Objem InductOsu pre implantáciu určuje anatómia zlomeniny a možnosť uzavrieť ranu bez nadmerného stlačenia prípravku. Všeobecne je možné každú zlomeninu liečiť obsahom jedného 12 mg balenia. Maximálna dávka je limitovaná na 24 mg (dve celé 12 mg balenia matrixu).
Deti a dospievajúci
Bezpečnosťa účinnosť InductOs u detí vo veku menej ako 18 rokov neboli doteraz stanovené.
K dispozícii nie sú žiadne údaje.
Spôsob podania
Liek je podávaný implantáciou.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6. Nedodržanie spôsobu podania InductOsu
môže ohroziť jeho bezpečnosť a účinnosť.
Na manipuláciu s InductOsom by sa mali používať kliešte. Pri manipulácii a implantácii lieku minimalizujte straty tekutín z matrixu. Nestláčajte.
Chirurgia lumbálnej medzitelovej fúzie
Pri tejto indikácii sa InductOs nesmie použiť samostatne, ale má sa použiť spolu so schváleným
(označenie CE) prístrojom (prístrojmi) lumbálnej medzitelovej fúzie. Kompatibilita bola preukázaná
u titánu, polyetereterketónu (PEEK) a alogénneho štepu kosti. Je potrebná opatrnosť a pozornosť, aby
sa predišlo preplneniu zariadenia lumbálnej medzitelovej fúzie a/alebo prednej časti priestoru
medzistavcovej platničky (pozri časť 4.4).
Pred implantáciou
4 mg balenie
Matrix je predrezaný na 2 diely – každý po 2,5 cm x 5 cm.
12 mg balenie:
Jeden diel matrixu veľkosti 7,5 cm x 10 cm.Namočený matrix by mal byť rozdelený na 6 rovnakých dielov (približne 2,5 cm x 5 cm) ako pomôcka pre výber dávky. Vybrané diely môžu byť ďalej rozdelené ako je požadované.
Dutá geometria zariadenia lumbálnej medzitelovej fúzie musí byť starostlivo a voľne naplnená
objemom InductOsu zodpovedajúcim vnútornému objemu prístroja.
Implantácia
Podľa štandardných postupov hmota disku a chrupavkovitých častí vertebrálnych zakončení sa má odstrániť tak, aby sa zachovali kortikálne časti zakončení a má byť dosiahnutá hemostáza (pozri
časť 4.5).
Pokyny na implantáciu zariadenia lumbálnej medzitelovej fúzie, nájdete v návode na použitie od výrobcu.
InductOs nesmie byť implantovaný posteriórne k zariadeniu lumbálnej fúzie tiel stavcov, kde je možný priamy prístup k spinálnemu kanálu a/alebo nervovému koreňu (koreňom). Ak je možné presakovanie do spinálneho kanála a nervového koreňa, musí byť fyzikálna bariéra medzi matrix a akýmkoľvek nervovým tkanivom znovu vytvorená použitím napríklad lokálnej kosti alebo alogénneho kostného štepu (pozri časť 4.5).
Po implantácii
Ak boli už InductOs a prístroj (prístroje) lumbálnej medzitelovej fúzie implantované, vnútro priestoru medzistavcovej platničky nesmie byť vymývané. Mimo priestoru medzistavcovej platničky by malo byť chirurgické pole vymývané podľa potreby a akékoľvek straty tekutín z namočeného matrixu by mali byť odplavené.
Ak sa vyžaduje chirurgická drenáž, drenáž by mala byť umiestnená ďalej od miesta implantácie alebo
prednostne jednu vrstvu superficiálne nad implantačným miestom.
Chirurgia akútnej fraktúry tíbie
Pred implantáciou
Pred implantáciou InductOsu by mala byť vykonaná konečná redukcia zlomeniny, fixácia a malo by
byť zastavené krvácanie.
Pred implantáciou by sa mal InductOs zahnúť alebo orezať podľa potreby.
Implantácia
InductOs je implantovaný po štandardnom ošetrení rany a fraktúry ( t.j. pri uzavretí mäkkých tkanív).
Pokiaľ je to možné, InductOs by mal pokryť dosiahnuteľný povrch zlomeniny (lomné línie a defekty). InductOs by mal byť umiestnený tak, aby premostil oblasť zlomeniny a zabezpečil dobrý kontakt hlavných proximálnych a distálnych fragmentov.
InductOs môže byť vložený do dutiny (voľne), zahnutý, zrolovaný alebo zbalený, tak ako to dovoľuje geometria zlomeniny. InductOs neposkytuje mechanickú stabilitu a nemá sa používať na vyplnenie prázdneho miesta v prítomnosti kompresných síl.
Po implantácii
Ak bol už InductOs implantovaný, ranu nevymývajte.
Ak sa vyžaduje chirurgická drenáž, drenáž by mala byť umiestnená ďalej od miesta implantácie alebo prednostne jednu vrstvu superficiálne nad implantačným miestom.
Na dosiahnutie maximálnej potenciálnej účinnosti je potrebné po implantácii dosiahnuť úplné prekrytie InductOsu mäkkými tkanivami.
4.3 Kontraindikácie
InductOs je kontraindikovaný u pacientov so:
• Známou precitlivenosťou na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti
6.1.
• Skeletálnou nezrelosťou.
• S akýmkoľvek aktívnym zhubným nádorom alebo u pacientov podstupujúcich liečbu zhubného
nádoru.
• Aktívnou infekciou v blízkosti operačnej rany.
• Perzistentým kompartmentovým syndrómom alebo neurovaskulárnymi zvyškami kompartmentového syndrómu.
• Patologickými zlomeninami ako tie, ktoré sú pozorované pri (ale nielen pri) Pagetovej chorobe alebo pri metastatickej kosti.
4.4 Osobitné upozornenia a opatrenia pri používaní
Nedodržanie pokynov na prípravu prípravku v časti 6.6 a spôsobu podania v časti 4.2 može negatívne
ovplyvniť bezpečnosť a účinnosť InductOsu.
Chirurgiakrčnejchrbtice
Bezpečnosť a účinnosť InductOsu v chirurgii krčnej chrbtice neboli stanovené a InductOs sa nemá pri tomto stave používať. Lokalizovaný edém sprevádzajúci užívanie InductOsu bol hlásený u pacientov, ktorí sa podrobili chirurgickému zákroku na krčnej chrbtici. Vznik edému bol oneskorený a zvyčajne sa objavil počas prvého týždňa po zákroku. V niektorých prípadoch bol edém natoľko závažný, že obmedzoval dýchacie cesty.
Malignita
InductOs sa nemá používať u pacientov, ktorí mali v anamnéze výskyt malignity alebo je u nich podozrenie na výskyt malignity v mieste podania (pozri časť 4.3).
Heterotopická osifikácia
Použitie IndukOsu môže spôsobiť heterotopickú osifikáciu v mieste implantácie a/alebo v okolitých
tkanivách, čo môže viesť ku komplikáciám.
Zvýšená resorpcia kostného tkaniva
InductOs môže spôsobiť počiatočnú resorpciu okolitého trabekulárneho kostného tkaniva čo dokazuje
rádiolucencia. Preto pri absencii klinických údajov sa liek nemá používať k priamej aplikácii na
trabekulárne kostné tkanivo, kde by mohla prechodná resorpcia kostného tkaniva spôsobiť riziko
krehkosti kostí (pozri časť 4.8).
Hromadenie tekutiny
V súvislosti s použitím InductOsu bolo hlásené nahromadenie tekutiny (pseudocysty, lokalizovaný edém, tekutina v implantovanej súprave), niekedy enkapsulované, a v niektorých prípadoch vedúce ku
kompresii nervu a bolesti. Ak symptómy pretrvávajú, môže byť potrebná klinická intervencia
(aspirácia a/alebo chirurgické odstránenie) (pozri časť 4.8).
I
m
unitná
odpoveď
U dibotermínu alfa ako i u hovädzieho kolagénu typu I sa zistilo, že u pacientov vyvolávajú imunitnú
odpoveď.
Protilátky anti-dibotermín alfa: V štúdiach so spinálnou fúziou pri použití InductOsu vznikli protilátky proti dibotermínu alfa u 1,3 % pacientov oproti 0,8 % pacientov pri použití autológneho kostného štepu. Pri zlomeninách dlhých kostí vznikli protilátky proti dibotermínu alfa u 6,3 % pacientov liečených dibotermínom alfa s hovädzím kolagénovým matrixom typu I voči 1,3 % v kontrolnej skupine. Všetci pacienti, ktorí boli testovaní na neutralizujúce protilátky proti kostnému morfogenetickému proteínu- 2 boli negatívni.
Protilátky proti hovädziemu kolagénu typu I: V štúdiách so spinálnou fúziou vznikli protilátky proti hovädziemu kolagénu typu I u 13,5 % pacientov po aplikácii InductOsu oproti 14,3 % pacientov po aplikácii autológneho kostného štepu. V štúdiách zlomenín dlhých kostí sa protilátky proti hovädziemu kolagénu typu I vyvinuli u 13,0% pacientov liečených dibotermínom alfa s hovädzím kolagénovým matrixom typu I oproti 5,3% kontrolných pacientov. Žiadny z pacientov s pozitívnymi titrami na hovädzí kolagén typu I nemal krížovú reakciu protilátok na humánny kolagén typu I.
Hoci v klinických štúdiách sa nepozorovalo spojenie s klinickým dôsledkom alebo nežiaducimi účinkami, nemožno vylúčiť možnosť tvorby neutralizačných protilátok alebo reakcií z precitlivenosti. Možnosť vzniku imunitnej odpovede na liek má byť zvážená v prípadoch, kde existuje podozrenie na nežiaduce účinky imunologického pôvodu. Zvlášť sa má zvážiť riziko a prínos u pacientov, ktorým bol pred tým podaný kolagén injekčne (pozri tiež časť 4.3). Pri absencii akýchkoľvek skúseností sa opakované použitie InductOsu neodporúča.
Osobitné populácie
Bezpečnosť a účinnosť použitia InductOsu u pacientov so známym autoimúnym ochorením neboli stanovené. Tieto autoimúnne ochorenia zahŕňajú reumatoidnú artritídu, systemový lupus erythematosus, sklerodermiu, Sjögrenov syndróm a dermatomyozitídu/polymyozitídu.
Bezpečnosť a účinnosť InductOsu nebola preukázaná u pacientov s metabolickými kostnými ochoreniami.
Žiadne štúdie sa nevykonali u pacientov s poškodením pečene, obličiek alebo srdca.
Pri liečbe týchto osobitných populácií sa odporúča, aby lekár pozorne zvážil prospech a riziká pre daného pacienta predtým, ako sa použije InductOs. Odporúča sa starostlivé monitorovanie pacienta kvôli nežiaducim reakciám a úspechu liečby.
Pomocné látky
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v maximálnej dávke (dve 12 mg balenia), t.j. je v podstate „bez sodíka“.
Osobitné upozornenia a opatrenia pri používaní u prednej lumbálnej medzitelovej fúzie
Bezpečnosť a účinnosť InductOsu neboli stanovené v nasledujúcich podmienkach:
• s použitím prístrojov medzitelovej fúzie vyrobených z iného materiálu ako titán, PEEK alebo
kosť
• implantovaného na mieste inom ako lumbálna chrbtica
• s použitím chirurgických techník iných ako lumbálna medzitelová fúzia
Aby nedošlo k zvýšenému farmakologickým účinkom InductOsu, je potrebná opatrnosť a pozornosť, aby sa predišlo preplneniu prístroja lumbálnej medzitelovej fúzie a/alebo prednej časti priestoru medzistavcovej platničky.
Heterotopická osifikácia
Tvorba kosti mimo priestoru medzistavcovej platničky nie je žiaduca, keďže môže mať nepriaznivý
vplyv na lokálne neurovaskulárne štruktúry.
V klinických štúdiách, v ktorých bolo degeneratívne ochorenie disku liečené postupom zadnej lumbálnej medzitelovej fúzie s dibotermínom alfa, bola na CT skenoch pozorovaná zadná tvorba kostí. V niektorých prípadoch to môže viesť k nervovej kompresii potenciálne vyžadujúcej chirurgický zákrok (pozri časť 4.8). Ako preventívne opatrenie musí byť znova vytvorená fyzikálna bariéra medzi matrixom a akýmkoľvek neurologickým tkanivom (pozri časť 4.2).
Dislokácia zariadenia
K dislokácii zariadenia môže dôjsť po použití InductOsu v chirurgii spinálnej fúzie, čo si môže vynútiť operačnú revíziu (pozri časť 4.8).
Osobitné upozornenia a opatrenia pri používaní u tibiálnych fraktúr
InductOs je určený pre použitie u pacientov s:
• adekvátnou repozíciou a stabilizáciou zlomeniny zaisťujúcou mechanickú stabilitu kostí
• adekvátnym neurovaskulárnym statusom (t.j. neprítomnosť kompartment syndrómu, nízke
riziko amputácie)
• adekvátnou hemostázou (t.j. poskytujúcou relatívne suché prostredie pre implantáciu)
• absenciou hojenia veľkých segmentálnych defektov dlhých kostí, pri ktorých môže vzniknúť
významná kompresia mäkkých tkanív
Implantát sa môže použiť len v adekvátne prehľadnom teréne zlomeniny a s najväčšou opatrnosťou
(pozri časť 4.2).
Informácie o účinnosti liečby zlomenín tíbie sa získali len v kontrolovaných klinických štúdiách, v ktorých boli otvorené fraktúry tíbie liečené fixáciou intramedulárnym klincom (pozri časť 5.1). V klinickej štúdii, v ktorej bol intramedulárny kanál rozšírený ku kortikálnej hranici, sa pozoroval zvýšený podiel infekcií v skupine liečenej InductOsom, v porovnaní s kontrolnou skupinou so
štandardnou starostlivosťou (pozri časť 4.8). Použitie InductOsu s rozšírenými klincami sa pri náprave
otvorenej tibiálnej fraktúry neodporúča.
InductOs neposkytuje mechanickú stabilitu a nemá sa používať na vyplnenie prázdneho miesta v prítomnosti kompresných síl. Liečba zlomenín dlhých kostí a starostlivosť o mäkké tkanivá nemá byť založená na štandardných terapeutických postupoch vrátane kontroly infekcií.
4.5 Liekové a iné interakcie
Nevykonali sa žiadne interakčné štúdie.
Pretože dibotermín alfa je proteín a nebol identifikovaný v krvnom obehu, nepredpokladajú sa interakcie s inými liekmi.
V klinických štúdiách zlomenín tíbie boli zaznamenané mierne alebo stredne závažné nežiaduce udalosti súvisiace s hojením rán (napr. drenáž rany) u viacerých pacientov liečených InductOsom, súčasne s užívaním nesteroidných protizápalových liekov (NSAIDs) nepretržite počas 14 dní
v porovnaní s pacientmi liečenými InductOsom, ktorí neužívali NSAIDs. Hoci dôsledok u pacienta
nebol ovplyvnený, nemožno vylúčiť interakcie medzi NSAIDs a InductOsom.
Informácie z klinických štúdií akútnych fraktúr tíbie poukazujú na to, že použitie InductOsu
u pacientov užívajúcich glukokortikoidy nebolo spojené so žiadnymi zjavnými nežiaducimi reakciami. V neklinických štúdiách súbežné podanie glukokortikoidov znížilo hojenie kostí (merané ako
% zmeny oproti kontrole), avšak účinky InductOsu neboli ovplyvnené.
V štúdii in vitro bola dokázaná väzba dibotermínu alfa na hemostatické faktory na báze fibrínu alebo tesniace hmoty. Použitie týchto prípravkov v tesnej blízkosti na InductOs sa neodporúča, keďže to môže viesť k tvorbe kosti v mieste implantácie hemostatických faktorov na báze fibrínu alebo tesniacich hmôt (pozri časť 4.2).
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití InductOsu u gravidných žien.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
Vzhľadom na neznáme riziká na plod spojené s možným rozvojom neutralizačných protilátok proti dibotermínu alfa, IndukOs sa neodporúča počas gravidity alebo u žien vo fertilnom veku, ktoré neužívajú antikoncepciu (pozri časť 4.4).
Laktácia
Neexistujú žiadne informácie o vylučovaní dibotermínu alfa/metabolitov do ľudského mlieka.
S ohľadom na typ prípravku, systémová expozícia u dojčaťa sa neočakáva, avšak riziko
u novorodenca/dojčiaťa nemôže byť vylúčené.
S ohľadom na prínos dojčenia pre dieťa a prínos liečby pre ženu sa musí urobiť rozhodnutie, či prerušiť dojčenie alebo odstúpiť od liečby InductOsom.
Fertilita
V neklinických štúdiách sa nezaznamenal vplyv na fertilitu. Nie sú k dispozícii klinické údaje,
potenciálne riziko pre ľudí nie je známe.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
InductOs nemá žiadny alebo len zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Najčastejšie nežiaduce reakcie InductOsu v chirurgii lumbálnej medzitelovej fúzie boli radikulopatie
a pri chirurgii akútnej fraktury tibie to bola lokalizovaná infekcia. Najzávažnejšia nežiaduca reakcia je
lokalizovaný edém pri chirurgickom zákroku krčnej chrbtice. Pohlavie, vek ani rasa nemali vplyv na
výskyt nežiaducich reakcií s InductOsom.
Tabuľkový prehľad nežiaducichreakcií
Viac ako 1700 pacientov dostali InductOs v klinických štúdiách. V štúdiách so zlomeninami dlhých
kostí dostalo InductOs viac ako 500 pacientov. V štúdiách lumbálnej medzitelovej fúzie dostalo
InductOs viac ako 600 pacientov. Zvyšní pacienti sa zúčastnili štúdií s použitím InductOsu
v indikáciách, ktoré v súčasnosti nie sú v EÚ schválené. Tieto údaje sú doplnené informáciami z použitia InductOsu vo všeobecnej populácii.
Frekvencia výskytu nežiaducich reakcií u pacientov liečených InductOsom je uvedená v tabuľke
nižšie. Frekvencie sú definované ako veľmi časté (≥1/10) alebo časté (≥1/100 do <1/10). Nepozorovali sa reakcie s frekvenciou menej časté (≥1/1 000 do <1/100), zriedkavé (≥1/10 000 do <1/1 000) alebo
veľmi zriedkavé (<1/10 000).
Frekvencie výskytu nežiaducich reakcií identifikovaných počas postmarketingového použitia
InductOsu nie sú známe, keďže tieto reakcie boli hlásené v populácii s neurčitou veľkosťou.
T
rieda orgánového systému
F
r
ekvencie
V
eľmi časté Časté Neznáme
Celkové poruchy a reakcie v mieste podania
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Poruchy nervového systému
Infekcie a nákazy Lokalizovaná infekcia5*
Dislokácia prístroja1*
Hromadenie tekutiny2*
Heterotopická osifikácia1,
3*
Radikulopatické udalosti1, 4
Osteolýza* Zvýšená resorpcia kostí*

1 Pozorovali sa počas použitia pri lumbálnej medzitelovej fúzii.
2 Nahromadenie tekutiny vrátane lokalizovaného edému, pseudocysty a výpotku v mieste implantátu.
3 Heterotopická osifikácia vrátane exostózy, extraskeletálnej osifikácie, pooperačnej heterotopickej kalcifikácie, zvýšenej tvorby kosti a kalcifikácie v mieste implantátu.
4 Radikulopatické udalosti vrátane radikulitídy, lumbálnej radikulopatie, radikulárnej bolesti, lumbosakrálnej radikulitídy, radikulopatie a ischias.
5 Pozorovali sa počas použitia pri akútnych frakciách tíbie
* Dodatočné informácie sú uvedené nižšie
Opis vybraných nežiaducich reakciíNovotvorba kostí a remodelácia kostíV rámci farmakologického mechanizmu účinku dibotermínu alfa sa objavuje remodelácia kostí (pozri
časť 5.1). Počas tohto procesu sa uskutočňuje resorpcia a aj tvorba kosti. Za niektorých okolností môže vystupňovanie týchto procesov viesť ku komplikáciám, ako sú kompresia nervov (spôsobená heterotopickou osifikáciou) alebo dislokácia prístroja (súvisiaca s resorpciou kosti alebo osteolýzou).
Počas dvoch rokov sledovania v klinických štúdiách lumbálnej medzitelovej fúzie použitím zadného prístupu, sa heterotopická osifikácia zobrazená na röntgenologických snímkach objavila častejšie u pacientov liečených InductOsom v porovnaní s autogénnym štepom (pozri časť 4.4). Tieto röntgenologické nálezy môžu byť asymptomatické alebo symptomatické.
Nahromadenie tekutinyVzhľadom na angiogénnu aktivitu InductOsu sa môže objaviť nahromadenia tekutiny (pseudocysta, lokalizovaný edém, výpotok v mieste implantátu), niekedy opuzdrené, niekedy vedúce ku kompresii nervu a/alebo bolesti.
Lokalizovaný edém bol častý, keď bol InductOs použitý pre spinálnu fúziu krčnej chrbtice. Nástup edému sa oneskoril a v niektorých prípadoch bol taký závažný, že viedol k oslabeniu funkcie dýchacích ciest (pozri časť 4.4).
Lokalizovaná infekciaLokalizovaná infekcia špecifická pre zlomenú končatinu sa objavila veľmi často (>1/10) u pacientov v klinickej štúdii, kde bol intramedulárny kanál rozšírený ku kortikálnej hranici. Zvýšený výskyt infekcií sa pozoroval v skupine liečenej InductOsom v porovnaní s kontrolnou skupinou liečenou štandardnou liečbou (19 % v porovnaní s 9 %, pozri časť 4.4). Pri použití InductOsu s nerozšírenými klincami bol odhadovaný výskyt infekcií podobný medzi liečenými skupinami štúdie (21 % v porovnaní s 23 %).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieV prípade predávkovania (t.j. pacient užíva väčšiu koncentráciu alebo množstvo dibotermínu alfa ako je odporúčané), by mala byť liečba podporná.
Používanie InductOsu u pacientov podstupujúcich operáciu krčnej chrbtice v množstvách nižších alebo podobných ako pri lumbálnej medzitelovej fúzii bolo spojené s hláseniami lokalizovaného edému natoľko závažného, že viedlo k ohrozeniu dýchacích ciest (pozri časť 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Lieky na liečbu kostných ochorení: ATC kód: M05BC01
Dibotermín alfa je osteoinduktívny proteín, ktorého následkom dochádza k indukcii nového kostného tkaniva v mieste jeho implantácie. Dibotermín alfa sa viaže na receptory na povrchu mezenchymálnych buniek a spôsobuje diferenciáciu buniek na bunky chrupky a bunky kostného tkaniva. Diferencované bunky tvoria trabekulárnu kosť, ako je degradovaný matrix, súčasne
s evidentnou cievnou inváziou. Proces tvorby kostného tkaniva sa vyvíja zvonku od implantátu
smerom k stredu, až kým je celý implantát InductOsu nahradený trabekulárnou kosťou.
Uloženie InductOsu do trabekulárnej kosti vyústilo do prechodnej resorpcie kosti v okolí implantátu, čo následne viedlo k jeho nahradeniu novou kosťou s vyššou hustotou. Remodelácia okolitej kosti nastáva spôsobom, ktorý je konzistentný s biomechanickými silami, ktoré na ňu pôsobia. Schopnosť InductOsu podporiť remodeláciu kosti môže byť zodpovedná za biologickú a iomechanickú integráciu nového kostného tkaniva indukovaného InductOsom s tkanivom okolitej kosti. Rádiografické, biomechanické a histologické hodnotenie indukovaného kostného tkaniva naznačuje,
že je biologicky a biomechanicky rovnako funkčné ako prirodzená kosť. Okrem toho neklinické štúdie naznačujú, že ak dôjde k zlomenine kosti, ktorej rast bol indukovaný InductOsom, hojí sa spôsobom nerozoznateľným od prirodzenej kosti.
Neklinické štúdie naznačili, že tvorba kostného tkaniva iniciovaná InductOsom je samolimitujúcim procesom, kedy sa tvorí dobre definovaný objem kostného tkaniva. Tento samolimitujúci proces nastáva pre pokles dibotermínu alfa v mieste implantátu ako aj prítomnosťou BMP inhibítorov
v okolitých tkanivách. Navyše viaceré nedklinické štúdie naznačujú, že na molekulárnej úrovni existuje negatívna spätná väzba, ktorá limituje kostnú indukciu spôsobenú BMP.
Histologický dôkaz zo štúdií lumbálnej medzitelovej fúzie na zvieratách použitím predného alebo zadného chirurgického prístupu ukázal, že dibotermín alfa podávaný medzitelovými prístrojmi
z titánu, PEEK alebo štepu bol biokompatibilný a produkoval trvalo vysokú mieru fúzie nezávislej od
chirurgického prístupu alebo materiálu prístroja s menej evidentným fibróznym tkanivom v porovnaní s autológnym štepom.
Klinické farmakologické štúdie preukazujú, že samotný matrix nie je osteoinduktívnym a nie je viac
prítomný v biopsiách odobratých 16 týždňov po implantácii.
Farmakodynamické informácie špecifické pre štúdie lumbálnej medzitelovej fúzieÚčinnosť a bezpečnosť InductOsu bola preukázaná randomizovanou, kontrolovanou, multicentrickou
štúdiou non-inferiority u 279 pacientov vo veku od 19 do 78 rokov, ktorí podstúpili otvorenú prednú lumbálnu fúziu tiel stavcov. Pacienti boli liečení minimálne 6 mesiacov neoperačnými postupmi pred liečbou s použitím InductOsu pri prednej lumbálnej spinálnej fúzii. Pacienti boli randomizovaní do
skupín s použitím titánového prístroja medzitelovej fúzie s náplňou InductOsu alebo s náplňou
autológneho kostného štepu z lopaty bedrovej kosti.
24 mesiacov po operácii nebol účinok InductOsu štatisticky nižší ako pri použití autológneho kostného štepu s úspešnosťou rádiologicky znázornenej fúzie 94,4 % pre InductOs oproti 88,9 % pre autológny kostný štep (95% dvojstranný interval spoľahlivosti: -1,53; 12,46). Čo sa týka bolesti a zneschopnenia (Oswestry skóre), podiel úspešnej terapie bol 72,9 % v skupine používajúcej InductOs oproti 72,5 %
v skupine používajúcej autológny kostný štep (95% dvojstranný interval spoľahlivosti: -11,2; 12,0).
Následná meta analýza 6 kontrolovaných klinických štúdií s dátami pacientov liečených InductOsom alebo autogénnym kostným štepom podávaným použitím prístrojov lumbálnej medzitelovej fúzie označených CE alebo kostným štepom a rôznymi chirurgickými prístupmi ukázala, že 24 mesiacov po operácii bol InductOs spojený s vyššou mierou úspešnosti (95 %, 241 z 255 pacientov) v porovnaní s autogénnym kostným štepom (85 %, 177 z 209 pacientov), s pomerom pravdepodobnosti (odds ratio)'
3,26 (95% CI: 1,172, 9,075; P=0,024). Odhadovaný absolútny rozdiel miery úspešnosti fúzie medzi
InductOsom a autogénnym kostným štepom bol 11,7 % (95 % CI: 0,8 %, 22,5 %; P=0,035).
V súhrnnej bezpečnostnej analýze 8 klinických štúdií 24 mesiacov po operácii, bola frekvencia pacientov so pseudoartrózou približne dvojnásobne nižšia po liečbe InductOsom (4,8 %, 22 zo 456 pacientov) v porovnaní s autogénnym kostným štepom (12,7 %, 31 z 244 pacientov).
Farmakodynamické vlastnosti špecifické pre štúdie zlomeniny tíbie
Účinnosť InductOsu bola preukázaná multinacionálnou, randomizovanou, kontrolovanou, jednoducho
zaslepenou štúdiou u 450 pacientov (vo veku od 18 do 87 rokov; 81% mužov) s otvorenou zlomeninou
tíbie, ktorá si vyžadovala chirurgický zákrok. Pacienti boli liečení (v pomere 1:1:1) štandardným postupom (kontrolná skupina) pozostávajúcim z intramedulárnej (IM) klincovej fixácie a bežného ošetrenia mäkkých tkanív, štandardným postupom a navyše InductOsom 0,75 mg/ml alebo štandardným postupom a navyše InductOsom 1,5 mg/ml. Pacienti boli sledovaní počas 12 mesiacov od zacelenia mäkkého tkaniva.
V rámci pivotnej štúdie zlomenín tíbie InductOs zvýšil pravdepodobnosť vyliečenia zlomeniny;
u pacientov liečených InductOsom 1,5 mg/ml bolo riziko zlyhania liečby o 44% nižšie (sekundárny
zásah na podporu liečenia zlomeniny) v porovnaní s pacientmi liečenými bežnou starostlivosťou
(RR = 0,56; 95% CI = 0,40 do 0,78). Tieto výsledky boli nezávisle potvrdené rádiologickou skupinou
odborníkov, ktorí nepoznali spôsob liečby. Počet sekundárnych a následných zásahov bol signifikantne nižší u pacientov s InductOsom, predovšetkým s ohľadom na invazívnejšie zásahy ako sú kostné štepy a výmena klinov (P=0,0326).
Podiel pacientov vyliečených po liečbe InductOsom 1,5 mg/ml bol signifikantne vyšší pri všetkých kontrolných vyšetreniach od 10 týždňov do 12 mesiacov po operácii, čo svedčí o zrýchlenom liečení zlomeniny.
InductOs 1,5 mg/ml bol signifikantne účinnejší (v porovnaní so štandardnou starostlivosťou) ako
u fajčiarov tak i nefajčiarov.
Závažnosť zlomenín: Liečba InductOsom 1,5 mg/ml bola signifikantne účinnejšia u všetkých tried zlomeniny vrátane závažných zlomenín triedy Gustilo IIIB (52% zníženie rizika sekundárnych intervencií v porovnaní s pacientmi so štandardnou starostlivosťou).
Podiel pacientov s vyliečenými poraneniami mäkkých tkanív bol pri kontrole 6 týždňov po liečbe signifikantne vyšší u skupiny liečenej InductOsom 1,5 mg/ml v porovnaní so skupinou pacientov liečených štandardnou starostlivosťou (83% oproti 65%; P=0,0010). Podiel pacientov so zlyhaním pevných súčastí (fixujúce skrutky ohnuté alebo zlomené) bol signifikantne nižší u skupiny liečenej InductOsom 1,5 mg/ml v porovnaní so skupinou liečenou štandardnou starostlivosťou (11% oproti
22%; P=0,0174).
5.2 Farmakokinetické vlastnosti
InductOs účinkuje v mieste implantácie. V dvoch výskumných štúdiách boli zozbierané vzorky séra pred a po chirurgickom zákroku od niekoľkých pacientov so zlomeninami dlhých kostí. V sére nebol zistený dibotermín alfa.
V štúdiách na zvieratách (potkany) pri použití InductOsu obsahujúceho dibotermín alfa označený rádionuklidom, bol stredný čas prítomnosti v mieste implantácie 4-8 dní. Najvyššie hladiny cirkulujúceho dibotermínu alfa (0,1% implantovanej dávky) boli zaznamenané v limite 6 hodín po implantácii. Pri intravenóznej injekčnej aplikácii bol terminálny polčas premeny dibotermínu alfa
16 minút u potkanov a 6,7 minúty u opíc makak cynomolgus. Preto sa usudzuje, že v mieste
implantácie sa dibotermín alfa pomaly uvoľňuje z matrixu a rýchlo opúšťa systémovú cirkuláciu.
5.3 Predklinické údaje o bezpečnosti
Neklinické údaje na základe obvyklých bezpečnostne farmakologických štúdií, štúdií akútnej toxicity, toxicity po opakovanom podaní a genotoxicity neodhalili žiadne špeciálne riziká pre ľudí.
V štúdiách reprodukčnej toxicity u potkanov, kedy bol dibotermin alfa aplikovaný intravenózne na
dosiahnutie maximálnych systémových účinkov, bola pozorovaná zvýšená fetálna hmotnosť
a nemožno vylúčiť efekt v spojitosti s liečbou. Klinický význam týchto pozorovaní nie je známy.
Protilátky proti dibotermínu boli skúmané u gravidných potkanov následne po hyperimunizácii dibotermínom alfa pre experimentálnu indukciu protilátok proti dibotermínu alfa. U niektorých plodov so zníženou hmotnosťou bol pozorované pokles osifikácie frontálnych a parietálnych kostí (4 z 151 plodov), ktorý sa všeobecne považuje za reverzibilný, pričom účinok v súvislosti s protilátkami nemožno vylúčiť. Neboli pozorované iné odlišnosti fetálnej vonkajšej, viscerálnej alebo kostnej morfológie. Dibotermín alfa preukázal rôzne účinky na rad ľudských tumorových buniek in vitro.
Dostupné in vivo údaje o rade ľudských tumorových buniek nenaznačujú možnosť pre podporu rastu tumoru alebo metastázy. Ako u jednorazového produktu, u InductOsu nebola skúmaná karcinogenita in vivo (pozri tiež časť 4.3).
InductOs bol skúmaný na modeli spinálnej implantácie u psov. InductOs bol aplikovaný priamo na exponovanú duru po laminektómii. I keď bolo pozorované zúženie a stenóza otvorov pre miechové nervy, mineralizácia dury, stenóza miechy a neurologické deficity neboli zaznamenané ako následok aplikácie InductOsu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok sacharóza glycín
kyselina glutámová chlorid sodný
polysorbát 80
hydroxid sodný
Rozpúšťadlo
voda na injekciu
Matrix
hovädzí kolagén typ I.
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi, okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C. Nezmrazujte. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
4 mg balenie InductOsu obsahuje:
• Prášok v injekčnej liekovke (10 ml; zo skla typu I) s uzáverom (bromobutylová zátka).
• Rozpúšťadlo v injekčnej liekovke (10 ml; zo skla typu I) s uzáverom (bromobutylová zátka).
• Dva matrixy (2,5 cm x 5 cm) v blistri (polyvinylchlorid - PVC).
• Dve injekčné striekačky (5 ml; polypropylénové).
• Dve ihly (nehrdzavejúca oceľ).
12 mg balenie InductOsu obsahuje :
• Prášok v injekčnej liekovke (20 ml; zo skla typu I) s uzáverom (bromobutylová zátka).
• Rozpúšťadlo v injekčnej liekovke (10 ml; zo skla typu I) s uzáverom (bromobutylová zátka).
• Jeden matrix v blistri (7,5 cm x 10 cm) polyvinylchlorid - PVC).
• Dve injekčné striekačky (10 ml; polypropylénové).
• Dve ihly (nehrdzavejúca oceľ).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
InductOs sa pripravuje tesne pred použitím. Dibotermín alfa musí byť používaný len po rekonštitúcii s
rozpúšťadlom a matrixom, ktoré sú súčasťou balenia InductOsu.
Po príprave InductOs obsahuje dibotermín alfa v koncentrácii 1,5 mg/ml.InductOs sa nesmie používať
v koncentráciách vyšších ako 1,5 mg/ml (pozri časť 4.9).
Príprava lieku
Aby sa zabránilo presýteniu matrixu, je dôležité dibotermín alfa rozpustiť a zvlhčiť celý nosič ako je
popísané nižšie.
4 mg balenie:
V nesterilnom poli
1. Použitím sterilnej techniky položte jednu striekačku, jednu ihlu a vnútorný obal s matrixami do
sterilného poľa.
2. Vydezinfikujte zátky injekčných liekoviek dibotermínu alfa a rozpúšťadla.
3. Použitím zvyšnej striekačky a ihly z balenia rozpusťte obsah injekčnej liekovky dibotermínu
alfa s 3,2 ml rozpúšťadla. Pomaly vstreknite rozpúšťadlo do injekčnej liekovky obsahujúcej
lyofilizovaný dibotermín alfa. Jemne premiešajte obsah injekčnej liekovky otáčavým pohybom, čím sa napomôže rekonštitúcii. Netrepať. Po použití zlikvidujte striekačku a ihlu.
 3,2 ml
3,2 mlrozpúšťadla
4. Vydezinfikujte zátku injekčnej liekovky rozpusteného dibotermínu alfa.
V sterilnom poli5. Otvorte vnútorný obal matrixov a nechajte matrixy na ich podnosoch.
6. Použite aseptickú prenosovú techniku, striekačku a ihlu z kroku 1, vytiahnite 2,8 ml pripraveného roztoku dibotermínu alfa z injekčnej liekovky v nesterilnom poli. Injekčnú liekovku držte hore dnom na uľahčenie vytiahnutia.
7. Ponechajúc matrix na podnose, ROVNOMERNE rozdeľte 1,4 ml roztoku dibotermínu alfa na každý z dvoch 2,5 cm x 5 cm matrixov podľa schémy na obrázku nižšie.
8. Počkajte MINIMÁLNE 15 minút pred tým, ako použijete pripravený InductOs. Prípravok sa
musí použiť do 2 hodín po jeho príprave.
12 mg balenie:V nesterilnom poli1. Použitím sterilnej techniky položte jednu striekačku, jednu ihlu a vnútorný obal s matrixom do
sterilného poľa.
2. Vydezinfikujte zátky injekčných liekoviek dibotermínu alfa a rozpúšťadla.
3. Použitím zvyšnej striekačky a ihly z balenia rozpusťte obsah injekčnej liekovky dibotermínu alfa s 8,4 ml rozpúšťadla. Pomaly vstreknite rozpúšťadlo do injekčnej liekovky obsahujúcej lyofilizovaný dibotermín alfa. Jemne premiešajte obsah injekčnej liekovky otáčavým pohybom, čím sa napomôže rekonštitúcii. Netrepať. Po použití zlikvidujte striekačku a ihlu.
 8,4 ml
rozpúšťadla
8,4 ml
rozpúšťadla
4. Vydezinfikujte zátku injekčnej liekovky rozpusteného dibotermínu alfa.
V sterilnom poli5. Otvorte vnútorný obal matrixu a nechajte matrix na jeho podnose.
6. Použite aseptickú prenosovú techniku, striekačku a ihlu z kroku 1, vytiahnite 8,0 ml pripraveného roztoku dibotermínu alfa z injekčnej liekovky v nesterilnom poli. Injekčnú liekovku držte hore dnom na uľahčenie vytiahnutia.
7. Ponechajúc matrix na podnose, ROVNOMERNE rozdeľte roztok dibotermínu alfa na matrix podľa schémy na obrázku nižšie.
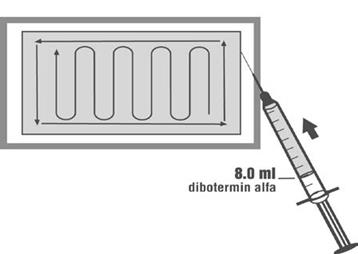
8. Počkajte MINIMÁLNE 15 minút pred tým, ako použijete pripravený InductOs. Prípravok sa
musí použiť do 2 hodín po jeho príprave.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMedtronic BioPharma B.V. Earl Bakkenstraat 10
6422 PJ Heerlen
Holandsko
tel +31 (0) 45 566 8000
fax +31 (0) 45 566 8012
8. REGISTRAČNÉ ČÍSLO
EU/1/02/226/001
EU/1/02/226/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 9. september 2002
Dátum posledného predĺženia: 20. júla 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry http:
/www.ema.europa.euDoplnkové vzdelávacie materiály pre zdravotníckych pracovníkov sú k dispozícii na nasledujúcej
adrese URL: [doplniť URL] <a na internetovej stránke <členský štát> >.