
v>

Druhá
úvodnej
liečbe
Až 4 ml
(100 miliónov) PFU/ml
dávky).
· Podanie injekcie do zvyšných lézií uprednostnite podľa veľkosti lézie, kým sa nepodá maximálny objem injekcií.
N
ávšteva v rámci liečby
Všetky ďalšie
návštevy
v rámci liečby
(vrátane obnovenia
liečby)
Liečebný
interval
2 týždne po predchá- dzajúcej liečbe
Maximálny celkový objem injekcií
Až 4 ml
Koncentrácie dávky
108
(100 miliónov) PFU/ml
Prioritizácia lézií na podanie injekcie
· Najprv podajte injekciu do všetkých nových lézií (lézií, ktoré sa rozvinuli od predchádzajúcej liečebnej dávky).
· Podanie injekcie do zvyšných lézií uprednostnite podľa veľkosti lézie, kým sa nepodá maximálny objem injekcií.

 Stanovenie objemu dávky Imlygicu (na léziu)
Stanovenie objemu dávky Imlygicu (na léziu)
Objem Imlygicu injekčne podaného do každej lézie závisí od veľkosti lézie a má sa stanoviť podľa
tabuľky 2. Celkový objem injekcií pri každom podaní má byť maximálne 4 ml.
Tabuľka 2: Voľba objemu injekcie s Imlygicom na základe veľkosti lézie
Veľkosť lézie (najdlhšírozmer) Objem injekcie Imlygicu> 5 cm až 4 ml
> 2,5 cm až 5 cm až 2 ml
> 1,5 cm až 2,5 cm až 1 ml
> 0,5 cm až 1,5 cm až 0,5 ml
≤ 0,5 cm až 0,1 ml
Pred dosiahnutím odpovede sa u pacientov môže vyskytnúť zväčšenie veľkosti existujúcej lézie (lézií)
alebo sa môže objaviť nová lézia. Pokiaľ ostáva nejaká lézia (lézie) na aplikovanie lieku, v podávaní
Imlygicu sa má pokračovať najmenej 6 mesiacov, kým lekár neusúdi, že pacient nemá z liečby
Imlygicom prínos, alebo že je potrebná iná liečba.
Liečbu Imlygicom možno obnoviť vtedy, ak sa po kompletnej odpovedi objavia nové lézie a lekár usúdi, že pacient bude mať z liečby prínos.
Osobitné populáciePediatrická populáciaBezpečnosť a účinnosť Imlygicu u pediatrických pacientov nebola stanovená. K dispozícii nie sú žiadne údaje.
Staršia populáciaU pacientov vo veku ≥ 65 rokov sa nevyžaduje úprava dávky (pozri časť 5.1).
Porucha funkcie pečene a obličiekNa hodnotenie vplyvu poškodenej pečene alebo obličiek na farmakokinetiku liečiva talimogén laherparepvek sa nevykonali žiadne klinické štúdie. U pacientov s poruchou funkcie pečene alebo obličiek však úprava dávky nie je potrebná.
Spôsob podávania
Imlygic sa podáva intraléziovou injekciou do kožných, podkožných a/alebo nodálnych lézií, ktoré
možno vidieť, nahmatať alebo zistiť na základe ultrazvuku.
Ak sú zdravotnícki pracovníci náhodne vystavení Imlygicu, pozri časti 4.4 a 6.6.
Zdravotnícki pracovníci s oslabenou imunitou alebo tehotné zdravotné pracovníčky nesmú podávať Imlygic alebo prísť do priameho kontaktu s miestom (miestami) podania Imlygicu ani s telesnými tekutinami liečených pacientov (pozri časti 4.3 a 4.4).
Pri príprave a podávaní Imlygicu pacientom postupujte podľa týchto pokynov:
Pred podaním injekcie· Injekčnú liekovku (liekovky) s Imlygicom rozmrazujte pri izbovej teplote (pozri časť 6.6).
· Požadované množstvo Imlygicu natiahnite z injekčnej liekovky do injekčnej striekačky.
· Miesto podania injekcie možno ošetriť lokálnym anestetikom. Anestetikum v injekčnej forme možno aplikovať pri okraji lézie, ale nemá sa podať priamo do lézie.
· Léziu a okolitú plochu očistite liehovým tampónom a nechajte uschnúť.
Podanie injekcie· Imlygic podajte intraléziovou injekciou do kožných, podkožných a/alebo nodálnych lézií, ktoré
možno vidieť, nahmatať alebo zistiť na základe ultrazvuku.
· Stanovte objem injekcie pre každú léziu s pomocou vyššie uvedenej tabuľky 2.
· Použite jedno miesto vpichu a Imlygic podajte otáčaním ihly viacerými smermi, pokiaľ to jej radiálny dosah vnútri lézie umožní, aby sa dosiahlo rovnomerné a úplné rozptýlenie. Viaceré miesta vpichu možno použiť vtedy, ak je lézia väčšia ako radiálny dosah ihly.
Kožné lézie Podkožné lézie Nodálne lézie
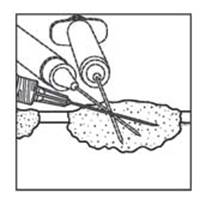
Obrázok 1.
Podanie injekcie pri kožných léziách
Obrázok 2.
Podanie injekcie pri podkožných léziách
Obrázok 3.
Podanie injekcie pri nodálnych léziách
· Imlygic rovnomerne a úplne rozptýľte v lézii ťahaním ihly nazad bez toho, aby ste ihlu z lézie
vytiahli. Počas injikovania zvyšnej dávky Imlygicu meňte smerovanie ihly toľkokrát, koľkokrát
to je potrebné. Pokračujte, kým sa celá dávka rovnomerne a úplne nerozptýli.
· Keď ihlu odstraňujete, vyťahujte ju z lézie pomaly, aby sa predišlo vytečeniu Imlygicu alebo postriekaniu Imlygicom v mieste vpichu.
· Tento postup zopakujte pri ďalších léziách, do ktorých treba podať injekciu. Keď je ihla úplne
vytiahnutá z lézie a vždy, keď sa podáva injekcia do inej lézie, použite novú ihlu.
Po podaní injekcie
· Na miesto podania injekcie pritlačte aspoň na 30 sekúnd sterilnú gázu.
· Miesto podania injekcie a okolitú plochu očistite liekovým tampónom a injikovanú léziu prekryte absorpčným tampónom a suchým oklúznym obväzom.
Likvidácia
Všetok materiál, ktorý prišiel do kontaktu s Imlygicom (napr. injekčná liekovka, injekčná striekačka, ihla, vata alebo gáza), zlikvidujte v súlade s národnými ústavnými postupmi (pozri časť 6.6).
4.3 Kontraindikácie
· Pacienti s precitlivenosťou na talimogén laherparepvek alebo na ktorúkoľvek z jeho pomocných látok v anamnéze.
· Pacienti, ktorí majú ťažko oslabený imunitný systém (napr. pacienti so závažnou vrodenou alebo získanou bunkovou a/alebo humorálnou imunodeficienciou) (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Už liečení pacienti
Údaje o účinnosti Imlygicu v súčasnej druhej alebo neskorších líniách liečby sú obmedzené.
Pacienti s oslabeným imunitným systémom
Imlygic sa neskúmal u pacientov s oslabeným imunitným systémom. Na základe údajov zo štúdií na
zvieratách u pacientov, ktorí majú ťažko oslabený imunitný systém, môže byť zvýšené riziko diseminovanej herpetickej infekcie a títo pacienti nemajú byť liečení Imlygicom (pozri časti 4.3 a 5.3). Diseminovaná herpetická infekcia sa môže vyskytnúť aj u pacientov s oslabeným imunitným systémom (napr. u pacientov s HIV/AIDS, leukémiou, lymfómom, bežnou variabilnou imunodeficienciou alebo u tých, ktorí vyžadujú dlhodobo podávané vysokodávkové steroidy alebo iné imunosupresíva). Pred podávaním Imlygicu týmto pacientom treba zvážiť riziká a prínosy liečby.
Náhodné vystavenie Imlygicu
Náhodné vystavenie môže viesť k prenosu Imlygicu a herpetickej infekcie. Zdravotnícki pracovníci
a blízke osoby (napr. členovia domácnosti, opatrovatelia, sexuálni partneri alebo osoby spiace na tej istej posteli) sa majú vyhýbať priamemu kontaktu s injikovanými léziami alebo telesnými tekutinami liečených pacientov počas celého liečebného obdobia a až 30 dní po podaní poslednej liečebnej dávky (pozri časť 6.6). Počas prípravy a podávania Imlygicu boli zdravotníckymi pracovníkmi hlásené náhodné pichnutie ihlou a postriekanie.
Blízke osoby, ktoré sú tehotné alebo majú oslabenú imunitu, nemajú vymieňať obväz pacienta ani čistiť jeho miesto podania injekcie. Tehotné ženy, novorodenci a jednotlivci s oslabenou imunitou nesmú byť vystavení potenciálne kontaminovaným materiálom.
Zdravotnícki pracovníci majú zabezpečiť, aby pacienti mohli splniť požiadavku zakryť miesta podania injekcie oklúznymi obväzmi (pozri časť 6.6). Pacientov tiež treba upozorniť, aby sa vyhýbali
dotýkaniu alebo škriabaniu miesta podania injekcie, pretože by to mohlo mať za následok neúmyselný prenos Imlygicu na iné oblasti ich tela alebo na ich blízke osoby.
Hoci nie je známe, či sa Imlygic môže preniesť pohlavným stykom, je doložené, že HSV-1 divokého typu sa pohlavným stykom môže preniesť. Pacientov treba upozorniť, aby počas pohlavného styku používali latexový kondóm, a zabránili tak možnému prenosu Imlygicu. Ženy schopné otehotnieť treba upozorniť, aby na zabránenie otehotneniu počas liečby Imlygicom používali účinnú antikoncepčnú metódu (pozri časť 4.6).
Opatrovateľov treba upozorniť, aby pri pomáhaní pacientom aplikovať alebo vymieňať oklúzne obväzy nosili ochranné rukavice a dodržiavali bezpečnostné opatrenia na likvidáciu použitých obväzov a čistiacich materiálov (pozri časti 4.2 a 6.6).
V prípade náhodného vystavenia Imlygicu treba zasiahnutých jednotlivcov poučiť, aby si postihnuté miesto dôkladne umyli mydlom a vodou a/alebo dezinfekčným prostriedkom. Ak sa rozvinú prejavy
alebo príznaky herpetickej infekcie, majú sa obrátiť na svojho lekára. Talimogén laherparepvek je citlivý na acyklovir.
Herpetická infekcia u pacientov liečených Imlygicom
V klinických štúdiách boli u pacientov liečených Imlygicom hlásené herpetické infekcie (vrátane
oparu pery a herpetickej keratitídy). Predpokladá sa, že príznaky lokálnej alebo systémovej infekcie v možnej súvislosti s Imlygicom sa budú podobať príznakom, ktoré vyvolávajú infekcie spôsobené
HSV-1 divokého typu.
Je známe, že jednotlivci s infekciou HSV-1 divokého typu sú v dôsledku reaktivácie latentného HSV-
1 divokého typu celoživotne ohrození symptomatickou herpetickou infekciou. Treba vziať do úvahy
symptomatickú herpetickú infekciu v dôsledku možnej reaktivácie Imlygicu.
Pacientov, u ktorých sa rozvinú herpetické infekcie, treba poučiť, aby na zabránenie prenosu vírusu dodržiavali štandardné hygienické postupy.
Talimogén laherparepvek je citlivý na acyklovir. Pred podaním acykloviru alebo iných antivirotík indikovaných na liečbu herpetickej infekcie treba zvážiť riziká a prínosy liečby Imlygicom. Ak sa tieto lieky podajú systémovo alebo lokálne priamo na miesto podania injekcie, môžu interferovať s účinnosťou Imlygicu.
Celulitída v mieste podania injekcie
Po liečbe Imlygicom môže dôjsť k nekróze alebo ulcerácii nádorového tkaniva. Boli hlásené celulitída
a systémová bakteriálna infekcia. Odporúča sa dôkladná starostlivosť o ranu a preventívne opatrenia proti infekcii, najmä ak nekróza tkaniva prejde do otvorených rán.
Zhoršené hojenie v mieste podania injekcie
V klinických štúdiách bolo hlásené zhoršené hojenie v mieste podania injekcie. Imlygic môže zvýšiť
riziko zhoršeného hojenia u pacientov so základnými rizikovými faktormi (napr. predchádzajúce ožarovanie v mieste podania injekcie alebo lézie v nedostatočne vaskularizovaných oblastiach).
Ak sa vyvinie pretrvávajúca infekcia alebo zdĺhavé hojenie, pred pokračovaním v liečbe treba zvážiť
riziká a prínosy Imlygicu.
Imunitne sprostredkované udalosti
V klinických štúdiách boli u pacientov liečených Imlygicom hlásené imunitne sprostredkované
udalosti vrátane glomerulonefritídy, vaskulitídy, pneumonitídy, zhoršenej psoriázy a vitiliga.
Pred začatím liečby u pacientov, ktorí majú autoimúnne ochorenie, alebo pred pokračovaním v liečbe u pacientov, u ktorých sa vyskytnú imunitne sprostredkované udalosti, treba zvážiť riziká a prínosy Imlygicu.
Plazmocytóm v mieste podania injekcie
Po podaní Imlygicu v blízkosti miesta vpichu injekcie bol hlásený plazmocytóm. U pacientov
s mnohopočetným myelómom alebo u pacientov, u ktorých sa počas liečby vyvinie plazmocytóm, treba zvážiť riziká a prínosy Imlygicu.
Obštrukčné ochorenie dýchacích ciest
Po liečbe Imlygicom bolo hlásené obštrukčné ochorenie dýchacích ciest. Pri podávaní injekcie do lézií
v tesnej blízkosti hlavných dýchacích ciest sa má postupovať opatrne.
H
SV-1 séronegatívni pacienti
Bolo hlásené, že u pacientov, ktorí boli na začiatku liečby HSV-1 séronegatívni, sa častejšie vyskytla
horúčka, triaška a ochorenie podobného chrípke v porovnaní s pacientmi, ktorí boli na začiatku liečby
HSV-1 séropozitívni, predovšetkým v období prvých 6 liečebných dávok (pozri časť 4.8).
Všetci pacienti
Imlygic obsahuje sorbitol (E420). Pacienti so zriedkavými dedičnými problémami fruktózovej
intolerancie nesmú užívať tento liek.
Každá 4 ml dávka Imlygicu obsahuje približne 30 mg (1,3 mmol) sodíka. Má sa to vziať do úvahy
u pacientov na diéte s kontrolovaným obsahom sodíka.
Sledovanie Imlygicu
Na zlepšenie sledovania biologických liekov sa má názov a číslo šarže podávaného lieku jasne uviesť
(alebo zaznamenať) v zdravotnej dokumentácii pacienta.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie s Imlygicom. Ak sa acyklovir a iné antivirotiká podávajú systémovo alebo lokálne priamo do miesta podania injekcie, môžu interferovať s účinnosťou Imlygicu. Pred podávaním acykloviru alebo iných antivirotík indikovaných na liečbu herpetickej infekcie zvážte riziká a prínosy liečby Imlygicom.
4.6 Fertilita, gravidita a laktácia
Ženy schopné otehotnieť/antikoncepcia
Ženy schopné otehotnieť treba upozorniť, aby na zabránenie otehotneniu počas liečby Imlygicom
používali účinnú antikoncepčnú metódu.
Všetkých pacientov treba upozorniť, aby počas pohlavného styku používali latexový kondóm, a tým predišli možnému prenosu Imlygicu (pozri časť 4.4).
Gravidita
Vhodné a dostatočne kontrolované štúdie s liečivom talimogén laherparepvek u tehotných žien sa
neuskutočnili.
Ak má tehotná žena infekciu HSV-1 divokého typu (primárnu alebo reaktiváciu), existuje možnosť, že vírus prejde cez placentárnu bariéru, a aj riziko prenosu počas pôrodu v dôsledku uvoľňovania vírusu. Infekcie HSV-1 divokého typu sa spájajú so závažnými nežiaducimi účinkami vrátane multiorgánového zlyhania a úmrtia, ak plod alebo novorodenec dostane herpetickú infekciu vyvolanú vírusom divokého typu. Hoci o infekciách liečivom talimogén laherparepvek u gravidných žien nie sú klinické údaje, ak by talimogén laherparepvek pôsobil rovnako, môže existovať riziko pre plod alebo novorodenca. V štúdiách na zvieratách sa nepozorovali žiadne účinky na embryonálny/fetálny vývoj (pozri časť 5.3). V rámci bezpečnostných opatrení je najlepšie vyhnúť sa užívaniu liečiva talimogén laherparepvek počas gravidity.
Môžu sa vyskytnúť transplacentárne metastázy malígneho melanómu. Pretože talimogén laherparepvek je navrhnutý tak, aby prenikol do nádorového tkaniva a replikoval sa v ňom, môže existovať riziko vystavenia plodu liečivu talimogén laherparepvek z nádorového tkaniva, ktoré prenikne cez placentu.

Ak sa Imlygic užíva počas gravidity alebo ak pri užívaní Imlygicu pacientka otehotnie, treba ju
informovať o možnom riziku pre plod a/alebo novorodenca.
DojčenieNie je známe, či sa talimogén laherparepvek vylučuje do materského mlieka. Rozhodnutie o tom, či
ukončiť dojčenie, alebo ukončiť/prerušiť liečbu Imlygicom, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
FertilitaNevykonali sa klinické štúdie na vyhodnotenie účinku liečiva talimogén laherparepvek na fertilitu
(pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeTalimogén laherparepvek môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
V dôsledku možných nežiaducich reakcií, ako sú závrat a stavy zmätenosti (pozri časť 4.8), pacientov
treba upozorniť, aby boli obozretní pri vedení vozidla alebo obsluhovaní strojov, pokým si nie sú istí, že talimogén laherparepvek na nich nepôsobí nepriaznivo.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluBezpečnosť Imlygicu sa hodnotila v pivotnej štúdii, kde 292 pacientov dostalo najmenej jednu dávku
Imlygicu (pozri časť 5.1). Medián trvania expozície Imlygicu bol 23 týždňov (5,3 mesiaca).
Dvadsaťšesť (26) pacientov bolo vystavených Imlygicu najmenej jeden rok.
U pacientov liečených Imlygicom boli najčastejšie hlásenými nežiaducimi reakciami (≥ 25 %) únava
(50,3 %), triaška (48,6 %), horúčka (42,8 %), nevoľnosť (35,6 %), ochorenie podobné chrípke
(30,5 %) a bolesť v mieste podania injekcie (27,7 %). Celkovo deväťdesiatosem percent (98 %) týchto hlásených nežiaducich reakcií bolo miernych alebo stredne závažných. Najčastejšou nežiaducou
reakciou 3. alebo vyššieho stupňa bola celulitída (2,1 %) (pozri časť 4.4).
Zoznam nežiaducich reakcií zoradených dotabuľkyTabuľka 3 uvádza nežiaduce reakcie pozorované v klinických skúšaniach u pacientov s melanómom
liečených Imlygicom v porovnaní s GM-CSF. Reakcie sú zoradené podľa triedy systémových orgánov
a podľa frekvencie. Frekvencie sú definované ako: veľmi časté (≥ 1/10), časté (> 1/100 až < 1/10) a menej časté (≥ 1/1 000 až < 1/100
). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 3. Nežiaduce reakcie pozorované v klinických skúšaniach u pacientov s melanómom
liečených Imlygicom
Infekcie a nákazyČasté Celulitída, orálny herpes
Menej časté Infekcia v mieste incízie
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)Časté Nádorová bolesť, infikovaný novotvar Menej časté Plazmocytóm v mieste podania injekcie
Poruchy krvi a lymfatického systémuVeľmi časté Periférny edém
Časté Anémia
Poruchy imunitného systémuČasté Imunitne sprostredkované udalosti†
 P
oruchy metabolizmu a výživy
P
oruchy metabolizmu a výživy Časté Dehydratácia
Poruchy nervového systému Veľmi časté Bolesť hlavy
Časté Stavy zmätenosti, úzkosť, depresia, závrat, insomnia
Poruchy okaMenej časté Herpetická keratitída
Poruchy ucha a labyrintuČasté Bolesť ucha
Poruchy srdca a srdcovej činnosti Časté Tachykardia
Poruchy cievČasté Hlboká žilová trombóza, hypertenzia, návaly tepla
Poruchy dýchacej sústavy, hrudníka a mediastínaVeľmi časté Kašeľ
Časté Dyspnoe pri námahe, orofaryngeálna bolesť, infekcia horných dýchacích ciest
Menej časté Obštrukčné ochorenie dýchacích ciest
Poruchy gastrointestinálneho traktuVeľmi časté Vracanie, diarea, konstipácia, nauzea Časté Bolesť brucha, abdominálny diskomfort
Poruchy kože a podkožného tkanivaČasté Vitiligo, vyrážka, dermatitída
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté Myalgia, artralgia, bolesť končatín Časté Bolesť chrbta, bolesť v slabinách
Celkové poruchy a reakcie v mieste podaniaVeľmi časté Ochorenie podobné chrípke, horúčka, triaška, únava, bolesť, reakcie v mieste podania injekcie§
Časté Celkový pocit choroby, axilárna bolesť
Laboratórne a funkčné vyšetreniaČasté Úbytok na hmotnosti
Úrazy, otravy a komplikácie liečebného postupuČasté Komplikácia rany, výtok z rany, pomliaždenina, procedurálna bolesť
§ Medzi reakcie v mieste podania injekcie patrí: veľmi častý výskyt bolesti v mieste podania injekcie, časté výskyty erytému, krvácania, opuchu, reakcie, zápalu, sekrécie, výtoku v mieste podania injekcie, menej častý výskyt tepla v mieste podania injekcie.
† Medzi imunitne sprostredkované udalosti patria: menej častý výskyt vaskulitídy, pneumonitídy, zhoršenej psoriázy a glomerulonefritídy.
Opis vybraných nežiaducich reakciíImunitne sprostredkované udalostiImunitne sprostredkované udalosti hlásené v pivotnej klinickej štúdii zahŕňali prípad zhoršenej psoriázy u pacienta s predchádzajúcou anamnézou psoriázy, jeden prípad pneumonitídy u pacienta s predchádzajúcou anamnézou autoimúnneho ochorenia, jeden prípad vaskulitídy a dva prípady glomerulonefritídy, z ktorých jeden sa prejavil akútnym zlyhaním obličiek.
PlazmocytómV klinických skúšaniach bol pozorovaný jeden prípad plazmocytómu v mieste podania injekcie u pacienta, u ktorého sa zistil mnohopočetný myelóm.
C
elulitída
V pivotnom klinickom skúšaní (štúdia 005/05) boli zaznamenané prípady celulitídy, pričom niektoré sa považovali za závažné nežiaduce udalosti. Žiadne však neviedli k trvalému ukončeniu liečby Imlygicom. Odporúča sa dôkladná starostlivosť o ranu a preventívne opatrenia proti infekcii, najmä ak nekróza tkaniva prejde do otvorených rán.
Príznaky podobné chrípkeU 90 % pacientov liečených Imlygicom sa vyskytli príznaky podobné chrípke. Horúčka, triaška
a ochorenie podobné chrípke, ktoré sa môžu objaviť kedykoľvek počas liečby Imlygicom, zvyčajne dozneli v priebehu 72 hodín. Tieto udalosti boli hlásené častejšie v období prvých 6 liečebných dávok, predovšetkým u pacientov, ktorí boli na začiatku liečby HSV-1 negatívni.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieNeexistujú žiadne klinické skúsenosti s predávkovaním Imlygicom. V klinických skúšaniach boli podávané dávky až do objemu 4 ml v koncentrácii 108 PFU/ml každé 2 týždne bez preukázania toxicity limitujúcej dávku. Maximálna dávka Imlygicu, ktorú možno bezpečne podať, nebola
stanovená. V prípade podozrenia na predávkovanie alebo náhodné intravenózne podanie treba pacienta liečiť symptomaticky, napr. acyklovirom alebo inými antivirotikami (pozri časť 4.4), a v prípade potreby začať s podpornou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká a imunomodulátory, ATC kód: zatiaľ nepridelený.
Mechanizmus účinkuTalimogén laherparepvek je liek na onkolytickú imunoterapiu, ktorý je získaný z HSV-1. Talimogén

laherparepvek bol modifikovaný tak, aby sa replikoval vnútri nádorov a produkoval imunostimulačný proteín ľudský GM-CSF (faktor stimulujúci kolónie granulocytov a makrofágov). Talimogén laherparepvek spôsobuje odumretie nádorových buniek a uvoľnenie antigénov pochádzajúcich z nádoru. Dá sa predpokladať, že spolu s GM-CSF bude podporovať systémovú protinádorovú imunitnú odpoveď a odpoveď efektorových T-lymfocytov. Myši, ktoré mali kompletnú regresiu svojich primárnych nádorov po liečbe, boli rezistentné na následné obnovenie liečby nádoru.
Medzi modifikácie liečiva talimogén laherparepvek z HSV-1 patrí delécia ICP34.5 a ICP47. Hoci protivírusové imunitné odpovede bránia normálne bunky po infekcii liečivom talimogén laherparepvek, ukázalo sa, že nádory sú vnímavé na poškodenie a odumretie buniek ICP34.5- deficientnými HSV-1 vírusmi vrátane liečiva talimogén laherparepvek. Delécia ICP47 zabraňuje negatívnej (tlmiacej) regulácii antigén prezentujúcich molekúl a zvyšuje expresiu génu HSV US11, čím stupňuje vírusovú replikáciu v nádorových bunkách.
K
l
i
n
i
cká účinnosť abezpečnosť
Štúdia 005/05
Účinnosť a bezpečnosť monoterapie Imlygicom v porovnaní so subkutánne podávaným GM-CSF sa hodnotili v medzinárodnej, otvorenej a randomizovanej klinickej štúdii fázy 3 s pacientmi
s melanómom štádia IIIB, IIIC a IV, ktorý sa považoval za chirurgicky neresekovateľný.
Predchádzajúca systémová liečba melanómu bola povolená, ale nevyžadovala sa. Pacienti s aktívnymi metastázami v mozgu, kostnými metastázami, rozsiahlym viscerálnym ochorením, primárnym očným
alebo slizničným melanómom, preukázanou imunosupresiou alebo podstupujúci liečbu systémovým
antiherpetikom boli zo štúdie vylúčení.
Pacienti boli randomizovaní v pomere 2 : 1 na užívanie buď Imlygicu, alebo GM-CSF (N = 436; 295
s Imlygicom, 141 s GM-CSF). Imlygic bol 1. deň podaný vo forme intraléziovej injekcie v začiatočnej
koncentrácii 106 (1 milión) PFU/ml, za ktorou nasledovala koncentrácia 108 (100 miliónov) PFU/ml
21. deň a potom každé 2 týždne v dávke až 4 ml. GM-CSF bol podávaný subkutánne v dávke
125 µg/m2 každý deň po dobu 14 dní, potom nasledovala 14-dňová prestávka v opakovaných intervaloch.
S cieľom umožniť výskyt oneskorených imunitne sprostredkovaných protinádorových účinkov boli pacienti liečení najmenej 6 mesiacov alebo kým už nemali žiadne lézie na podanie injekcie. Počas tohto obdobia liečba pokračovala napriek zväčšeniu existujúcej lézie (lézií) a/alebo vzniku novej lézie (lézií), kým sa u pacienta nevyvinula neprijateľná toxicita alebo sa skúšajúci lekár nedomnieval, že je v najlepšom záujme pacienta ukončiť liečbu melanómu alebo dostávať inú. Po 6 mesiacoch liečby
mali pacienti v terapii pokračovať do klinicky významnej progresie ochorenia (t. j. progresie ochorenia
spojenej so zhoršením výkonnostného stavu a/alebo potrebou alternatívnej liečby podľa názoru skúšajúceho lekára). Pacienti, u ktorých nastala odpoveď po 12 mesiacoch liečby, mohli pokračovať v liečbe až ďalších 6 mesiacov. Priemerné (SD) trvanie liečby celej populácie s liečebným zámerom (ITT, intent-to-treat) bolo 15,76 týždňa (15,79) v ramene s GM-CSF a 26,83 týždňa (18,39) v ramene
s Imlygicom. Primárnym koncovým ukazovateľom bola miera trvalej odpovede (DRR) [definovaná ako percentuálny podiel pacientov s úplnou odpoveďou (CR) alebo čiastočnou odpoveďou (PR)
udržaná nepretržite najmenej 6 mesiacov] na základe zaslepeného centrálneho posudku. Medzi
sekundárne koncové ukazovatele patrili celkové prežívanie (OS), celková miera odpovede (ORR)
[PR+CR], čas do odpovede, trvanie odpovede a čas do zlyhania liečby (čas od randomizácie po prvú epizódu klinicky relevantnej progresie ochorenia, keď sa po progresii nedosiahne odpoveď, alebo po
úmrtie).
Priemerný vek bol 63 (rozpätie: 22 až 94) rokov; 26,5 % účastníkov bolo vo veku vyše 65 rokov
a 23,3 % účastníkov vo veku vyše 74 rokov. Väčšina pacientov, 98 %, boli belosi. Pacienti mužského pohlavia tvorili 57 % populácie klinickej štúdie a 70 % pacientov malo východiskový výkonnostný
stav 0 podľa ECOG. Zo zaradených pacientov malo 22 % ochorenie štádia IV M1c a 53 % pacientov okrem chirurgického zákroku, adjuvantnej terapie alebo ožarovania absolvovalo predchádzajúcu
liečbu melanómu, ako je chemoterapia a imunoterapia cytokínmi. Celkovo 58 % všetkých pacientov zaradených do štúdie bolo na začiatku liečby séropozitívnych na HSV-1 divokého typu a 32,6 % bolo séronegatívnych; sérologický stav HSV-1 zvyšných 9,4 % nie je známy.
Rozdiel v DDR medzi Imlygicom a GM-CSF bol v populácii ITT štatisticky významný (pozri
tabuľku 4) v prospech Imlygicu.
Tabuľka 4 Súhrn výsledkov za populáciu ITT z Imlygic štúdie 005/05
K
oncový
ukazovateľ
štúdie
Imlygic N = 295 GM-CSF N = 141
Miera trvalej odpovede
Primárny 16,3 % (n = 48)
(95 % CI: 12,1; 20,5)
2,1 % (n = 3) (95 % CI:
0,0; 4,5)
Pomer pravdepodobností 8,9; (95 % CI: 2,7; 29,2) P < 0,0001
Celková miera odpovede
(% CR, % PR)'
Sekundárny 26,4 % (n = 78)
(95 % CI: 21,4 %, 31,5 %) (10,8 % CR, 15,6 % PR)
5,7 % (n = 8)
(95 % CI: 1,9 %, 9,5 %) (0,7 % CR, 5 % PR)
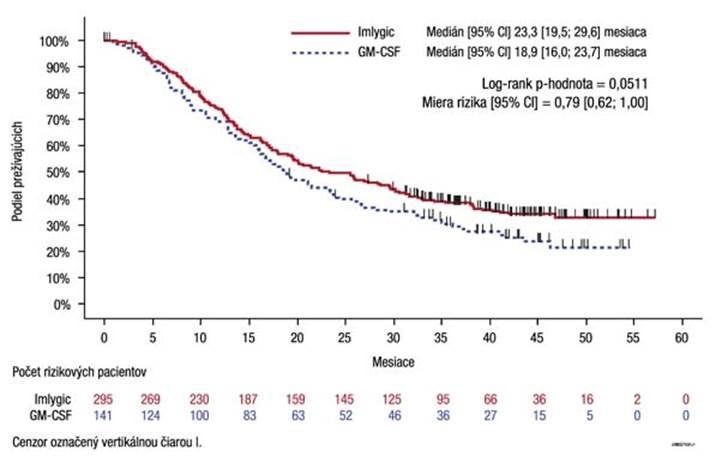
Celkové prežívanie Sekundárny Medián 23,3 (95% CI:
19,5; 29,6) mesiaca
Medián 18,9
(95 % CI: 16,0; 23,7)
mesiaca
HR: 0,79; (95 % CI: 0,62; 1,00) p = 0,051
Trvanie odpovede (prebiehajúca odpoveď pri
Sekundárny Nedosiahnutý
(Rozsah: > 0,0 až > 16,8
mesiaca)
Medián 2,8 mesiaca (Rozsah: 1,2 až > 14,9 mesiaca)
poslednom hodnotení nádoru)
Čas do odpovede
(medián)
HR: 0,46; (95 % CI: 0,35; 0,60) Sekundárny 4,1 mesiaca 3,7 mesiaca
Čas do zlyhania liečby
(medián)
Sekundárny 8,2 mesiaca
(95 % CI: 6,5; 9,9)
2,9 mesiaca
(95 % CI: 2,8; 4,0)

HR: 0,42; (95 % CI: 0,32; 0,54)
Medzi respondérmi liečenými Imlygicom 56 (72 %) odpovedí stále prebiehalo v čase primárnej
analýzy. Z týchto respondérov u 42 (54 %) nastalo ≥ 25 % zväčšenie celkovej veľkosti existujúcej lézie (lézií) a/alebo sa vyvinula nová lézia (lézie) predtým, ako napokon dosiahli odpoveď.
V analýze na vyhodnotenie systémového účinku Imlygicu 27 zo 79 pacientov (34,2 %) malo ≥ 50 % celkové zmenšenie neviscerálnych lézií, do ktorých nebola podaná injekcia Imlygicu, a 8 zo 71 pacientov (11,3 %) malo ≥ 50 % celkové zmenšenie viscerálnych lézií, do ktorých nebola podaná injekcia Imlygicu.
O
brázok 4: Kaplanov-Meierov graf – celkové prežívanie (ITT populácia)
Medzi staršími (vo veku ≥ 65 rokov) a mladšími dospelými pacientmi sa nepozorovali žiadne celkové rozdiely v bezpečnosti alebo účinnosti.
Výskumné podskupinyVykonali sa aj exploratórne analýzy podskupín pre DRR a celkové prežívanie podľa štádia ochorenia (pozri obrázok 5 a tabuľku 5). Hoci cieľom pivotnej štúdie nebolo hodnotiť účinnosť v týchto jednotlivých podskupinách, väčší prínos z liečby Imlygicom mali pacienti bez viscerálneho ochorenia ako pacienti s pokročilejším ochorením.
Tabuľka 5 Súhrn výsledkov exploratórnej analýzy podskupín z Imlygic štúdie 005/05
DRR (%) ORR (%) OS (pomer rizík)Imlygic GM-CSF Imlygic GM-CSF Imlygic vs GM-CSF
Štádium§ IIIB/IIIC/
IVM1a
(Imlygic, n = 163; GM-CSF, n = 86)
25,2 1,2 40,5 2,3 0,57; (95% CI: 0,40;
0,80);
Štádium§
IVM1B/IVM1C (Imlygic, n = 131; GM-CSF, n = 55)
5,3 3,6 9,2 10,9 1,07; (95% CI: 0,75;
1,52);
§Kategorizácia podľa Amerického spoločného výboru pre rakovinu (American Joint Committee on Cancer, AJCC), 6. vydanie
Obrázok 5: Kaplanov-Meierov odhad celkového prežívania podľa ramena randomizovanej liečby pre
ochorenie štádia IIIB/IIIC/IVM1a (exploratórna analýza podskupín)
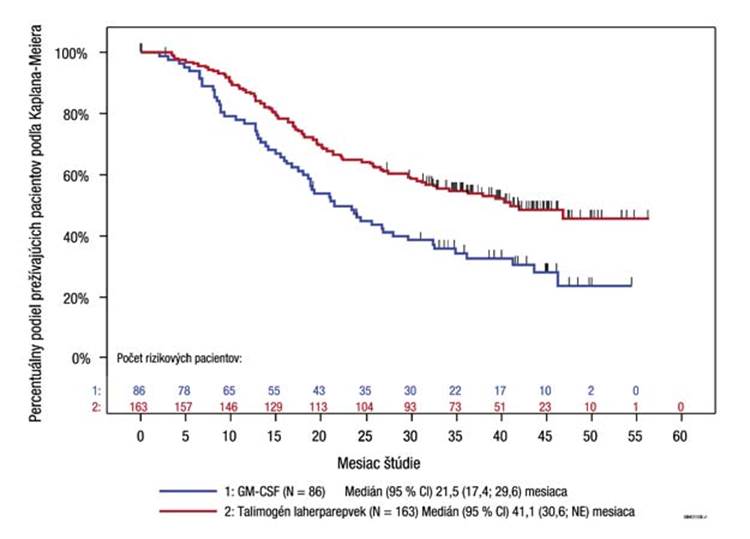
Cenzor označený vertikálnou čiarou
ǀNE = nehodnotiteľné
Vzhľadom na exploratórnu povahu analýzy a na základe súčasných dôkazov sa nepotvrdilo, že sa
Imlygic spája s účinkom na celkové prežívanie.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad povinnosti predložiť výsledky štúdií s Imlygicom v jednej
alebo vo viacerých podskupinách pediatrickej populácie s melanómom (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiTalimogén laherparepvek je geneticky modifikovaný a replikačno kompetentný HSV-1 vírus. V dôsledku toho sa jeho farmakokinetika a biodistribúcia riadia miestom podania intraléziovej injekcie, nádorovo selektívnou replikáciou a uvoľnením z nádorového tkaniva.
AbsorpciaBunkové vychytávanie liečiva talimogén laherparepvek nastáva prostredníctvom receptorov HSV-1 na
nádorových a nenádorových bunkách po podaní lokálnej injekcie do nádorov. Keďže talimogén laherparepvek sa podáva injekčne a replikuje sa intratumorálne, biologická dostupnosť a systémová koncentrácia liečiva talimogén laherparepvek nepredikujú účinnosť liečiva, a preto neboli hodnotené.
Metabolizmus/eliminácia
Talimogén laherparepvek sa odstraňuje pomocou zvyčajných obranných mechanizmov hostiteľa (ako
je napr. autofágia, adaptívne imunitné odpovede). Talimogén laherparepvek sa odbúrava katabolickými cestami pre endogénne proteíny a DNA. Tak ako pri iných infekciách HSV-1 divokého typu, aj latentný rezervoár DNA liečiva talimogén laherparepvek môže pretrvávať v tele nervových buniek inervujúcich miesta podania injekcie; v dôsledku toho výskyt latentnej infekcie liečivom talimogén laherparepvek nemožno vylúčiť.
Biodistribúcia (v organizme) a vylučovanie vírusu (exkrécia/sekrécia)
DNA liečiva talimogén laherparepvek bola kvantifikovaná na základe vysoko citlivej a špecifickej
analýzy kvantitatívna polymerázová reťazová reakcia (quantitative Polymerase Chain Reaction, qPCR), čo nemusí korelovať s rizikom infekčnosti vírusu. Talimogén laherparepvek bol
kvantifikovaný aj vo vzorkách získaných od vybraných pacientov v klinických štúdiách s použitím
analýz infekčnosti vírusu v miestach podania injekcie a v niektorých prípadoch možných herpetických lézií.
Klinická biodistribúcia, eliminácia a vylučovanie
V štúdii s melanómom sa skúma biodistribúcia a vylučovanie intraléziovo podaného liečiva talimogén laherparepvek. Predbežné výsledky od 30 pacientov ukazujú, že DNA liečiva talimogén laherparepvek bola v štúdii zistená v premenlivých a nízkych koncentráciách v krvi u 90 % pacientov a v moči
u 20 % pacientov. Podiel pacientov s preukázateľnou DNA liečiva talimogén laherparepvek v krvi a moči bol najvyšší počas druhého cyklu. DNA liečiva talimogén laherparepvek bola zistená vo
vzorkách z injikovaných lézií približne u 90 % pacientov. Iba 14 % pacientov však malo pozitívny test
na infekčný vírus podľa analýzy 50 % infekčnej dávky pre tkanivové kultúry (50% Tissue Culture
Infectious Dose, TCID50), všetci v priebehu 8 dní od podania lieku. Sedemnásť percent vzoriek z povrchu oklúzneho obväzu malo pozitívny výsledok testu na DNA liečiva talimogén laherparepvek,
ale ani jedna nebola pozitívna na prítomnosť infekčného vírusu. Spomedzi vzoriek ústnej sliznice iba 1
vzorka mala zistiteľnú DNA liečiva talimogén laherparepvek počas štúdie, ale táto vzorka nebola pozitívna na prítomnosť infekčného vírusu.
Farmakokinetika v osobitných populáciách
V osobitných populáciách sa neuskutočnili žiadne farmakokinetické štúdie s liečivom talimogén
laherparepvek.
5.3 Predklinické údaje o bezpečnosti
V dávkach až 4 x 108 PFU/kg alebo 107 PFU/dávka (60-násobne vyšších, ako je najvyššia navrhovaná klinická dávka) boli jednorazové alebo opakované dávky liečiva talimogén laherparepvek podané s.c., i.v. alebo intratumorálnou injekciou imunokompetentným myšiam, potkanom a psom dobre tolerované. Nepozorovala sa neuropatológia ani nežiaduce neurologické účinky. V in vivo štúdii
s intracerebrálnym injekčným podaním bol talimogén laherparepvek 10 000-násobne menej virulentný v porovnaní s dávkou HSV-1 divokého typu, ktorá viedla k 50 % úhynu myší v čase.
Talimogén laherparepvek bol injekčne podaný do rôznych nádorových xenoštepov v dávkach až
2 x 108 PFU/kg (30-násobne vyšších, ako je navrhovaná klinická dávka) imunodeficientným myšiam
(nahým (nude mice) a SCID). Letálna systémová vírusová infekcia sa pozorovala až pri 20 % nahých myší (primárne s funkčným deficitom T-lymfocytov) a pri 100 % myší SCID (bez T aj B lymfocytov).
V štúdiách sa fatálna diseminovaná vírusová infekcia pozorovala v 14 % nahých myší po liečbe
s liečivom talimogén laherparepvek v dávkach 10- až 100-násobne vyšších ako dávky, ktoré viedli k 100 % letalite s HSV-1 divokého typu.
Mutagenita
V dlhodobých štúdiách na zvieratách ani v štúdiách u ľudí sa genotoxický potenciál liečiva talimogén
laherparepvek nehodnotil. Pretože HSV-1 divokého typu sa neintegruje do genómu hostiteľa, riziko
inzerčnej mutagenézy liečivom talimogén laherparepvek je zanedbateľné.
Karcinogenita
V dlhodobých štúdiách na zvieratách ani v štúdiách u ľudí sa karcinogénny potenciál liečiva talimogén
laherparepvek nehodnotil. Dostupné údaje pre talimogén laherparepvek a HSV-1 divokého typu však
nesvedčia o karcinogénnom riziku u ľudí. Reprodukčná a vývojová toxicita
Po liečbe dospelých myší v dávkach až 4 x 108 PFU/kg (na základe PFU/kg 60-násobne vyšších v porovnaní s maximálnou klinickou dávkou) sa nepozoroval vplyv na samčie alebo samičie
reprodukčné tkanivá. Pri podávaní liečiva talimogén laherparepvek gravidným myšiam počas
organogenézy v dávkach až 4 x 108 (400 miliónov) PFU/kg (na základe PFU/kg 60-násobne vyšších v porovnaní s maximálnou klinickou dávkou) sa nepozorovali žiadne účinky na embryonálny/fetálny
vývoj. Vo fetálnej krvi sa zistili zanedbateľné množstvá (< 0,001 % hladín v materskej krvi) DNA
liečiva talimogén laherparepvek.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dihydrát hydrogenfosforečnanu sodného dihydrát dihydrogenfosforečnanu sodného chlorid sodný
myoinozitol sorbitol (E420) voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie inkompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
4 roky.
Rozmrazovanie injekčných liekoviek s Imlygicom
· Pred použitím rozmrazujte injekčné liekovky s Imlygicom pri izbovej teplote (20 °C až 25 °C),
kým nie je Imlygic tekutý (približne 30 minút). Jemne krúžte. NETRASTE.
· Injekčné liekovky sa majú rozmrazovať a uchovávať v pôvodnom obale na ochranu pred svetlom.
Po rozmrazení
· Po rozmrazení sa má Imlygic uchovávať pri teplote 2 °C až 8 °C do podania.
· Imlygic 106 plakotvorných jednotiek (PFU)/ml injekčný roztok: rozmrazený Imlygic
106 (1 milión) PFU/ml, injekčné liekovky, možno uchovávať pri teplote 2 °C až 8 °C najviac
12 hodín.
Imlygic 108 plakotvorných jednotiek (PFU)/ml injekčný roztok: rozmrazený Imlygic
108 (100 miliónov) PFU/ml, injekčné liekovky, možno uchovávať pri teplote 2 °C až 8 °C
najviac 48 hodín.
· Imlygic sa po rozmrazení nesmie znovu zmrazovať. Zlikvidujte každú rozmrazenú injekčnú
liekovku s Imlygicom uchovávanú dlhšie, ako je presne stanovený čas.
· Imlygic natiahnite do injekčnej striekačky až tesne pred podaním.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte a prepravujte zmrazené (−90 °C až −70 °C). Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Počas rozmrazovania a podávania má byť Imlygic vystavený iba izbovej teplote (20 °C až 25 °C)
(pozri časť 6.3).
6.5 Druh obalu a obsah balenia
Jeden ml roztoku bez konzervačných látok v jednorazovej injekčnej liekovke (cykloolefínový
polymér) so zátkou (chlórbutylkaučuk) a tesnením (hliník) s odklápacím viečkom (polypropylén).
Viečko injekčnej liekovky je farebne označené: 106 (1 milión) PFU/ml je svetlozelené a
108 (100 miliónov) PFU/ml je tmavomodrej farby.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Dodržiavajte národné ústavné smernice týkajúce sa zaobchádzania a podávania, osobných ochranných prostriedkov, náhodného rozliatia a likvidácie odpadu.
· Pri príprave a podávaní Imlygicu používajte ochranný alebo laboratórny plášť, bezpečnostné okuliare alebo ochranný štít na tvár a rukavice. Pred podávaním zakryte všetky odkryté rany. Vyhýbajte sa kontaktu s kožou, očami alebo sliznicami.
· Po podaní si vymeňte rukavice pred aplikovaním oklúznych obväzov na injikované lézie.
Povrch oklúzneho obväzu utrite liehovým tampónom. Odporúča sa mať miesta podania injekcie podľa možnosti stále obviazané vzduchotesnými a vodotesnými obväzmi. Na minimalizovanie rizika prenosu vírusu majú mať pacienti svoje miesto podania injekcie zakryté aspoň 8 dní od poslednej liečby alebo dlhšie, ak miesto podania injekcie mokvá alebo presakuje. Pacientov poučte, aby si obväz dávali podľa pokynov zdravotníckeho pracovníka a aby si spadnutý obväz vymenili za nový.
· Všetok materiál, ktorý prišiel do kontaktu s Imlygicom (napr. injekčná liekovka, injekčná striekačka, ihla, tampón alebo gáza), zlikvidujte v súlade s národnými ústavnými postupmi.
Náhodné vystavenie
· V prípade náhodného vystavenia Imlygicu pri práci (napr. vniknutie do očí alebo na sliznicu)
počas prípravy a podávania, oplachujte miesto čistou vodou najmenej 15 minút. V prípade vystavenia poškodenej koži alebo pichnutia ihlou postihnutú oblasť dôkladne umyte mydlom a vodou a/alebo dezinfekčným prostriedkom.
· Rozliaty Imlygic očistite virucídnym prípravkom a absorpčnými materiálmi.
· Pacientov treba poučiť, aby použité obväzy a čistiace materiály vložili do plastového vreca
a dobre ho uzavreli, pretože môžu byť kontaminované, a vrece vyhodili do domového odpadu.
Tento liek obsahuje geneticky modifikované organizmy. Nepoužitý liek sa musí zlikvidovať v súlade s ústavnými smernicami pre geneticky modifikované organizmy, prípadne pre biologicky nebezpečný odpad.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAmgen Europe B.V. Minervum 7061
NL-4817 ZK Breda
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/15/1064/001
EU/1/15/1064/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.