
lobulín triedy G (IgG1), predpokladá sa transplacentárny prenos u žien. Klinický dopad tohto zistenia nie je známy. Podávanie živých vakcín novorodencom, vystaveným kanakinumabu v maternici sa však neodporúča 16 týždňov od podania poslednej dávky Ilarisu matke pred pôrodom. Ženy, ktoré dostali kanakinumab počas gravidity, je potrebné poučiť, aby o tom informovali detského lekára pred akoukoľvek vakcináciou ich novorodenca.
Dojčenie
Nie je známe, či sa kanakinumab vylučuje do ľudského mlieka. Rozhodnutie, či dojčiť počas liečby
Ilarisom, sa preto má urobiť len po dôkladnom zhodnotení prínosu a rizika liečby.
Štúdie na zvieratách ukázali, že myšacia protilátka proti myšaciemu IL-1 beta nemala nežiaduce účinky na vývoj dojčených myšacích mláďat a že táto protilátka sa na ne preniesla (pozri časť 5.3).
Fertilita
Formálne štúdie možného účinku Ilarisu na ľudskú plodnosť sa nevykonali.
Kanakinumab nemal účinok na parametre plodnosti u samcov opíc kozmáčov (C. jacchus). Myšacia protilátka proti myšaciemu IL-1 beta nemala nežiaduce účinky na plodnosť samcov ani samíc myši (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ilaris má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Liečba Ilarisom môže vyvolať závraty/vertigo alebo asténiu (pozri časť 4.8). Pacienti, u ktorých sa počas liečby Ilarisom vyskytnú takéto symptómy, majú počkať do ich úplného vymiznutia, kým budú viesť vozidlo alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Viac ako 2 600 osôb, vrátane približne 480 detí (vo veku 2 až 17 rokov), bolo liečených Ilarisom v intervenčných klinických skúšaniach s pacientmi s CAPS, TRAPS, HIDS/MKD, FMF, SJIA, dnavou artritídou alebo s inými ochoreniami sprostredkovanými IL-1 beta, ako aj zdravých dobrovoľníkov. Pozorovali sa závažné infekcie. Najčastejšími nežiaducimi reakciami na liek boli infekcie predovšetkým horného dýchacieho traktu. Pri dlhodobejšej liečbe sa nezaznamenal žiadny vplyv na typ ani frekvenciu nežiaducich reakcií na liek.
U pacientov liečených Ilarisom boli hlásené reakcie z precitlivenosti (pozri časti 4.3 a 4.4). U pacientov liečených Ilarisom boli hlásené oportúnne infekcie (pozri časť 4.4).
C
APS
V klinických skúšaniach dostávalo Ilaris celkovo 211 dospelých a pediatrických pacientov s CAPS (vrátane FCAS/FCU, MWS a NOMID/CINCA). Bezpečnosť Ilarisu v porovnaní s placebom sa skúmala v pivotnom skúšaní fázy III, ktoré pozostávalo z 8-týždňového obdobia otvoreného podávania (časť I), 24-týždňového randomizovaného, dvojito zaslepeného a placebom kontrolovaného obdobia vysadenia lieku (časť II) a 16-týždňového obdobia otvoreného podávania Ilarisu (časť III). Všetci pacienti dostávali subkutánne Ilaris v dávke 150 mg alebo 2 mg/kg, ak ich telesná hmotnosť bola
≥ 15 kg a ≤ 40 kg.
TRAPS, HIDS/MKD, FMF
V jednom pivotnom klinickom skúšaní fázy III dostávalo Ilaris celkovo 169 dospelých a pediatrických pacientov s TRAPS, HIDS/MKD a FMF vo veku 2 roky a starších. Bezpečnosť Ilarisu v porovnaní
s placebom sa skúmala v tomto skúšaní, ktoré pozostávalo z 12-týždňového obdobia skríningu (časť I) a 16-týždňového randomizovaného, dvojito zaslepeného, placebom kontrolovaného obdobia liečby (časť II). Pacientom, ktorí dostávali Ilaris, sa podávalo subkutánne 150 mg, alebo 2 mg/kg, ak ich telesná hmotnosť bola ≤ 40 kg (pozri časť 5.1).
Stillova choroba
Celkovo 324 pacientov so SJIA vo veku 2 až < 20 rokov dostávalo Ilaris v klinických skúšaniach, vrátane 293 pacientov vo veku 2 až < 16 rokov, 21 pacientov vo veku 16 až < 18 rokov a 10 pacientov vo veku 18 až < 20 rokov. Bezpečnosť Ilarisu sa skúmala v porovnaní s placebom v dvoch pivotných skúšaniach fázy III (pozri časť 5.1).
Dnavá artritída
Viac ako 700 pacientov s dnavou artritídou bolo liečených Ilarisom v dávkach od 10 mg do 300 mg v randomizovaných, dvojito zaslepených a účinným liekom kontrolovaných klinických skúšaniach trvajúcich do 24 týždňov. Viac ako 250 pacientov dostávalo odporúčanú dávku 150 mg v klinických skúšaniach fázy II a III (pozri časť 5.1).
T
abuľkový
z
oznam
nežiaducich
reakcií
Nežiaduce reakcie sú usporiadané podľa tried orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa kategórie frekvencie, najčastejšie ako prvé. Kategórie frekvencie sú definované pomocou nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až< 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov).V rámci každej skupiny frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 1 Tabuľkový zoznam nežiaducich reakcií pri CAPS, TRAPS, HIDS/MKD, FMF, SJIA a dnavej artritíde
T
rieda
orgánových systémov MedDRA
Infekcie a nákazy
V
šetky indikácie:
CA
P
S, TRAPS, HIDS/MKD, FMF, SJIA, dnavá artritída
Veľmi časté Infekcie dýchacích ciest (vrátane pneumónie, bronchitídy, chrípky, vírusovej infekcie, sínusitídy, rinitídy, faryngitídy, tonzilitídy, nazofaryngitídy, infekcie horných dýchacích ciest)
Infekcia ucha Celulitída Gastroenteritída
Infekcia močových ciest Časté Vulvovaginálna kandidóza
Poruchy nervového systémuČasté Závraty/vertigo
Poruchy gastrointestinálneho traktuVeľmi časté Bolesť hornej časti brucha 1
Menej časté Gastroezofágová refluxná choroba 2
Poruchy kože a podkožného tkanivaVeľmi časté Reakcia v mieste vpichu
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté Artralgia 1
Časté Bolesť svalov a kostí 1
Bolesť chrbta 2
Celkové poruchy a reakcie v mieste podaniaČasté Únava/asténia 2
Laboratórne a funkčné vyšetreniaVeľmi časté Zníženie obličkového klírensu kreatinínu 1,3
Proteinúria 1,4
Leukopénia 1,5
Časté Neutropénia 5
Menej časté Pokles počtu trombocytov 5
1 Pri SJIA
2 Pri dnavej artritíde
3 Založené na odhadovanom klírense kreatinínu, väčšinou prechodné
4 Väčšinu predstavovali prechodné stopy až 1+ pozitivita bielkovín v moči, stanovené indikátorovým papierikom
5 Pozri ďalšie informácie nižšie
V podskupine mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov (N=31) bol profil
bezpečnosti Ilarisu v súlade s tým, ktorý sa pozoroval u pacientov so SJIA mladších ako 16 rokov. Na základe literárnych údajov sa očakáva podobný profil bezpečnosti u AOSD pacientov ako u pacientov so SJIA.
Popis
vy
braných
nežiaducich
reakcií
D
l
hodobé údaje a laboratórne abnormality u pacientov s CAPS
Počas klinických skúšaní Ilarisu u pacientov s CAPS sa priemerné hodnoty hemoglobínu zvýšili
a hodnoty leukocytov, neutrofilov a trombocytov znížili.
Zvýšenie hladín aminotransferáz sa u pacientov s CAPS pozorovalo zriedkavo.
U pacientov s CAPS liečených Ilarisom sa pozorovali asymptomatické a mierne zvýšenia bilirubínu
v sére, bez súčasného zvýšenia aminotransferáz.
V dlhodobých, otvorených klinických skúšaniach so zvyšovaním dávky boli ako udalosti častejšie hlásené infekcie (gastroenteritída, infekcia dýchacieho traktu, infekcia horného dýchacieho traktu), vracanie a závraty v skupine s dávkou 600 mg alebo 8 mg/kg ako v skupinách s inými dávkami.
Laboratórne abnormality u pacientov s TRAPS, HIDS/MKD a FMF Neutrofily
Hoci sa zníženie počtu neutrofilov ≥ stupňa 2 vyskytlo u 6,5 % pacientov (časté) a zníženie stupňa 1 sa vyskytlo u 9,5 % pacientov, zníženie je všeobecne prechodné a infekcia spojená s neutropéniou nebola identifikovaná ako nežiaduca reakcia.
Trombocyty
Hoci sa zníženie počtu trombocytov (≥ stupňa 2) vyskytlo u 0,6 % pacientov, krvácanie nebolo identifikované ako nežiaduca reakcia. Mierne a prechodné zníženie trombocytov stupňa 1 sa vyskytlo u 15,9 % pacientov bez akýchkoľvek súvisiacich krvácavých nežiaducich príhod.
Laboratórne abnormality u pacientov so SJIA Hematológia
V celkovom programe SJIA bol prechodný pokles počtu bielych krviniek (WBC) ≤ 0,8 x LLN hlásený u 33 pacientov (16,5 %).
V celkovom programe SJIA bol prechodný pokles absolútneho počtu neutrofilov (ANC) na menej ako
1 x 109/l hlásený u 12 pacientov (6,0 %).
V celkovom programe SJIA sa prechodný pokles trombocytov (< LLN) pozoroval u 19 pacientov
(9,5 %).
ALT/AST
V celkovom programe SJIA boli vysoké hodnoty ALT a/alebo AST > 3 x hornej hranice normálneho rozmedzia (ULN) hlásené u 19 pacientov (9,5 %).
Laboratórne abnormality u pacientov s dnavou artritídou
Hematológia
Znížený počet leukocytov (white blood cell counts, WBC) ≤ 0,8 x dolná hranica normálu (lower limit of normal, LLN) bol hlásený u 6,7 % pacientov liečených Ilarisom v porovnaní s 1,4 % pacientov liečených triamcinolónacetonidom. V porovnávacích klinických skúšaniach bol u 2 % pacientov hlásený pokles absolútneho počtu neutrofilov (ANC) na menej ako 1 x 109/l. Pozorovali sa aj ojedinelé prípady ANC < 0,5 x 109/l (pozri časť 4.4).
V klinických skúšaniach kontrolovaných účinným liekom sa u pacientov s dnavou artritídou pozorovalo mierne (< LLN a > 75 x 109/l) a prechodné zníženie počtu trombocytov s vyššou incidenciou (12,7 %) pri Ilarise oproti komparátoru (7,7 %).
K
yselina močová
V porovnávacích klinických skúšaniach pri dnavej artritíde sa po liečbe Ilarisom pozorovalo zvýšenie koncentrácie kyseliny močovej (0,7 mg/dl po 12 týždňoch a 0,5 mg/dl po 24 týždňoch). V inom klinickom skúšaní sa nepozorovalo zvýšenie kyseliny močovej u pacientov, ktorí začínali ULT. Zvýšenie kyseliny močovej sa nepozorovalo v klinických skúšaniach u populácií bez dnavej artritídy (pozri časť 5.1).
ALT/ASTZvýšenie priemeru o 3,0 U/l a mediánu o 2,0 U/l pri alanínaminotransferáze (ALT) a o 2,7 U/l
a o 2,0 U/l pri aspartátaminotransferáze (AST) sa pozorovalo oproti východiskovým hodnotám do konca klinického skúšania v skupinách liečených Ilarisom oproti skupinám liečeným triamcinolónacetonidom, avšak incidencia klinicky významných zmien (≥ 3 x horná hranica normálu) bola vyššia u pacientov liečených triamcinolónacetonidom (2,5 % pri AST aj ALT) v porovnaní s pacientmi liečenými Ilarisom (1,6 % pri ALT a 0,8 % pri AST).
TriacylglycerolyV klinických skúšaniach kontrolovaných účinným liekom pri dnavej artritíde sa triacylglyceroly zvýšili v priemere o 33,5 mg/dl u pacientov liečených Ilarisom v porovnaní s miernym poklesom o -3,1 mg/dl pri triamcinolónacetonide. Incidencia pacientov so zvýšením triacylglycerolov > 5 x horná hranica normálu (ULN) bola 2,4 % pri Ilarise a 0,7 % pri triamcinolónacetonide. Klinická významnosť tohto pozorovania nie je známa.
Pediatrická populáciaV štúdiách bolo 80 pediatrických pacientov s CAPS (vo veku 2–17 rokov), ktorí dostávali kanakinumab. Celkovo sa nevyskytli žiadne klinicky významné rozdiely v profiloch bezpečnosti a znášanlivosti Ilarisu u pediatrických pacientov v porovnaní s celkovou populáciou s CAPS
(tvorenou dospelými aj pediatrickými pacientmi, N=211), vrátane celkovej frekvencie a závažnosti epizód infekcií. Infekcie horného dýchacieho traktu boli najčastejšie hlásenými udalosťami infekcií.
Okrem toho sa 6 pediatrických pacientov mladších ako 2 roky vyhodnotilo v malej, otvorenej klinickej štúdii. Ukázalo sa, že profil bezpečnosti Ilarisu je podobný ako u pacientov vo veku 2 rokov a starších.
V 16-týždňovej štúdii bolo 102 pacientov s TRAPS, HIDS/MKD a FMF (vo veku 2–17 rokov), ktorí dostávali kanakinumab. Celkovo sa nevyskytli žiadne klinicky významné rozdiely v profiloch bezpečnosti a znášanlivosti kanakinumabu u pediatrických pacientov v porovnaní s celkovou populáciou.
StaršiapopuláciaV profile bezpečnosti pozorovanom u pacientov vo veku ≥ 65 rokov nie je významný rozdiel.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieZaznamenané skúsenosti s predávkovaním sú obmedzené. V počiatočných klinických skúšaniach dostali pacienti a zdraví dobrovoľníci intravenózne alebo subkutánne podané dávky až 10 mg/kg bez preukázanej akútnej toxicity.
V prípade predávkovania sa odporúča u pacienta sledovať akékoľvek príznaky a prejavy nežiaducich
reakcií a okamžite začať náležitú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu, ATC kód: L04AC08
Mechanizmus účinku
Kanakinumab je ľudská monoklonálna protilátka proti ľudskému interleukínu-1 beta (IL-1 beta) izotypu IgG1/κ. Kanakinumab sa s vysokou afinitou viaže špecificky na ľudský IL-1 beta a neutralizuje biologickú aktivitu ľudského IL-1 beta blokovaním jeho interakcie s receptormi IL-1, čím bráni aktivácii génu vyvolanej IL-1 beta a tvorbe zápalových mediátorov.
Farmakodynamickéúčinky
CAPS, TRAPS, HIDS/MKD a FMF
V klinických štúdiách pacienti s CAPS, TRAPS, HIDS/MKD a FMF, ktorí majú nekontrolovanú nadmernú tvorbu IL-1 beta, vykazujú rýchlu a pretrvávajúcu odpoveď na liečbu kanakinumabom, t.j. laboratórne parametre, ako vysoké hladiny C-reaktívneho proteínu (CRP) a sérového amyloidu A (SAA), vysoký počet neutrofilov a trombocytov a leukocytóza, sa rýchlo vrátili na normálne hodnoty.
Stillova choroba (AOSD a SJIA)
Stillova choroba s nástupom ochorenia v dospelosti (AOSD) a systémová juvenilná idiopatická artritída sú závažné autoinflamačné choroby spôsobené vrodenou imunitou a sprostredkovaná prozápalovými cytokínmi, z ktorých kľúčový je IL-1-beta.
Časté príznaky AOSD a SJIA sú horúčka, exantém, hepatosplenomegália, lymfadenopatia, polyserozitída a artritída. Liečba kanakinumabom spôsobila rýchle a pretrvávajúce zmiernenie kĺbových aj systémových príznakov SJIA s významným poklesom počtu zapálených kĺbov, rýchlym odznením horúčky a znížením reaktantov akútnej fázy u väčšiny pacientov (pozri časť Klinická účinnosť a bezpečnosť).
Dnavá artritída
Záchvat dnavej artritídy je spôsobený kryštálmi urátu (monohydrát mononátriumurátu) v kĺbe a v okolitých tkanivách, čo spúšťa tvorbu IL-1 beta rezidentnými makrofágmi prostredníctvom
„inflamazómového komplexu NALP3“. Aktivácia makrofágov a súčasná nadmerná tvorba IL-1 beta vyvolávajú akútnu bolestivú zápalovú reakciu. Iné aktivátory vrodeného imunitného systému, ako sú endogénne agonisty „toll-like“ receptorov, môžu prispieť k transkripčnej aktivácii génu IL-1 beta a vyvolať záchvat dnavej artritídy. Po liečbe kanakinumabom rýchlo poklesli zápalové markery CRP alebo SAA a ustúpili prejavy akútneho zápalu (napr. bolesť, opuch, sčervenanie) v postihnutom kĺbe.
Klinická účinnosťabezpečnosť
CAPS
Účinnosť a bezpečnosť Ilarisu sa preukázali u pacientov s rôznymi stupňami závažnosti ochorenia a rôznymi fenotypmi CAPS (vrátane FCAS/FCU, MWS a NOMID/CINCA). Do hlavnej štúdie boli zaradení len pacienti s potvrdenou mutáciou NLRP3.
V štúdii fázy I/II mala liečba Ilarisom rýchly nástup účinku, s vymiznutím alebo klinicky významným zlepšením symptómov do jedného dňa po podaní. Laboratórne parametre, ako vysoké hladiny CRP a SAA, vysoký počet neutrofilov a trombocytov, sa normalizovali rýchlo, počas niekoľkých dní po injekcii Ilarisu.
Hlavná štúdia pozostávala zo 48-týždňovej multicentrickej štúdie zloženej z troch častí, t.j. 8- týždňového otvoreného obdobia (časť I), 24-týždňového randomizovaného, dvojito zaslepeného a placebom kontrolovaného obdobia vysadenia lieku (časť II), po ktorom nasledovalo 16-týždňové otvorené obdobie (časť III). Cieľom tejto štúdie bolo hodnotenie účinnosti, bezpečnosti a znášanlivosti Ilarisu (150 mg alebo 2 mg/kg každých 8 týždňov) u pacientov s CAPS.
- Časť I: Úplná klinická odpoveď a odpoveď biomarkerov na Ilaris (definovaná ako súhrn celkového hodnotenia autoinflamačného a kožného ochorenia lekárom ≤ minimálne a hodnôt CRP alebo SAA < 10 mg/l) sa pozorovala u 97 % pacientov a objavila sa do 7 dní od začatia liečby. V klinickom hodnotení aktivity autoinflamačného ochorenia lekárom sa pozorovali významné zlepšenia: pri celkovom hodnotení aktivity autoinflamačného ochorenia, hodnotení kožného ochorenia (kožný exantém typu urtikárie), artralgie, myalgie, bolesti hlavy/migrény, konjunktivitídy, únavy/pocitu nepohody, hodnotení iných súvisiacich symptómov a hodnotení symptómov pacientom.
- Časť II: V období vysadenia lieku počas hlavnej štúdie bol primárny koncový parameter definovaný ako podiel pacientov s relapsom/vzplanutím ochorenia: žiadny (0 %) z pacientov randomizovaných do skupiny Ilarisu nemal vzplanutie ochorenia, v porovnaní s 81 % pacientov randomizovaných do skupiny placeba.
- Časť III: Pacienti, ktorí v časti II dostávali placebo a u ktorých došlo k vzplanutiu ochorenia, po zaradení do otvorenej extenzie štúdie Ilarisu opäť dosiahli a udržali si klinickú a sérologickú odpoveď.
Tabuľka 2 Tabuľkové zhrnutie účinnosti v klinickom skúšaní fázy III, hlavné obdobie vysadenia lieku kontrolované placebom (časť II)
Klinické skúšanie fázy III, hlavné obdobie vysadenia lieku kontrolované placebom (časť II)
Ilaris N=15 n(%)
Placebo N=16 n(%)
Hodnota p
P
rimárny parameter (vzplanutie ochorenia)
Podiel pacientov so vzplanutím ochorenia v
časti II
0 (0 %) 13 (81 %) < 0,001
 Z
ápalové markery*
Z
ápalové markery*
C-reaktívny proteín, mg/l 1,10 (0,40) 19,93 (10,50) < 0,001
Sérový amyloid A, mg/l 2,27 (-0,20) 71,09 (14,35) 0,002
* priemerná (medián) zmena od začiatku časti II
Vykonali sa dve otvorené, nekontrolované dlhodobé klinické skúšania fázy III. Jedno z nich bolo
skúšanie bezpečnosti, znášanlivosti a účinnosti kanakinumabu u pacientov s CAPS. Celkové trvanie liečby bolo v rozmedzí od 6 mesiacov do 2 rokov. Druhé bolo otvorené skúšanie kanakinumabu, ktoré hodnotilo účinnosť a bezpečnosť u japonských pacientov s CAPS počas 24 týždňov, s fázou extenzie až do 48 týždňov. Primárnym cieľom bolo stanovenie podielu pacientov, ktorí boli bez relapsov po
24. týždni, vrátane tých pacientov, ktorým sa dávka zvýšila.
Podľa združenej analýzy účinnosti týchto dvoch skúšaní dosiahlo 65,6 % pacientov, ktorí sa predtým neliečili kanakinumabom, úplnú odpoveď pri dávke 150 mg alebo 2 mg/kg, zatiaľ čo 85,2 % pacientov dosiahlo úplnú odpoveď pri akejkoľvek dávke. Z pacientov, ktorí dostávali 600 mg alebo
8 mg/kg (alebo ešte vyššiu dávku), 43,8 % dosiahlo úplnú odpoveď. Menej pacientov vo veku 2 až
< 4 roky dosiahlo úplnú odpoveď (57,1 %) v porovnaní so staršími pediatrickými a dospelými pacientmi. Z pacientov, ktorí dosiahli úplnú odpoveď, si 89,3 % odpoveď udržalo bez relapsov.
Skúsenosti u jednotlivých pacientov, ktorí dosiahli úplnú odpoveď po zvýšení dávky na 600 mg
(8 mg/kg) každých 8 týždňov, naznačujú, že vyššia dávka môže byť prínosom pre pacientov, ktorí nedosiahnu úplnú odpoveď alebo si neudržia úplnú odpoveď pri odporúčaných dávkach (150 mg alebo 2 mg/kg u pacientov ≥ 15 kg a ≤ 40 kg). Zvýšená dávka sa častejšie podávala pacientom vo veku 2 až < 4 roky a pacientom so symptómami NOMID/CINCA v porovnaní s pacientmi s FCAS alebo MWS.
Pediatrická populácia
Do klinických skúšaní Ilarisu pri CAPS bolo zaradených celkovo 80 pediatrických pacientov vo vekovom rozmedzí od 2 do 17 rokov (približne polovica z nich bola liečená na základe mg/kg). Celkovo sa nezistili žiadne klinicky významné rozdiely v účinnosti, bezpečnosti a znášanlivosti Ilarisu u detí a dospievajúcich v porovnaní s celkovou populáciou s CAPS. Väčšina pediatrických pacientov dosiahla zlepšenie v klinických príznakoch a v objektívnych zápalových markeroch (napr. SAA
a CRP).
Na vyhodnotenie účinnosti, bezpečnosti a znášanlivosti Ilarisu u pediatrických pacientov s CAPS vo
veku ≤ 4 roky sa vykonala otvorená štúdia trvajúca 56 týždňov. Vyhodnotilo sa 17 pacientov (vrátane
6 pacientov vo veku menej ako 2 roky), ktorí v závislosti od telesnej hmotnosti dostali začiatočné dávky 2-8 mg/kg. Štúdia tiež vyhodnotila účinok kanakinumabu na vznik protilátok proti vakcínam štandardne podávaným deťom. Nepozorovali sa rozdiely v bezpečnosti alebo účinnosti u pacientov mladších ako 2 roky v porovnaní s pacientmi vo veku 2 roky a staršími. U všetkých pacientov, ktorí dostali neživú vakcínu štandardne používanú u detí (N=7), sa vyvinuli ochranné hladiny protilátok.
TRAPS, HIDS/MKD a FMF
Účinnosť a bezpečnosť Ilarisu v liečbe TRAPS, HIDS/MKD a FMF sa preukázali v jednej pivotnej
štúdii fázy III so štyrmi časťami (N2301), ktorá pozostávala z troch samostatných skupín chorôb.
- Časť I: Pacienti v každej skupine chorôb vo veku 2 roky a starší sa zúčastnili 12-týždňového obdobia skríningu, počas ktorého sa u nich hodnotil nástup vzplanutia ochorenia.
- Časť II: Pacienti boli pri nástupe vzplanutia randomizovaní do 16-týždňového, dvojito zaslepeného, placebom kontrolovaného obdobia liečby, počas ktorého dostávali buď 150 mg Ilarisu (2 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) subkutánne (s.c.), alebo placebo každé 4 týždne. Pacientom vo veku > 28 dní, ale < 2 roky bolo povolené vstúpiť do štúdie priamo do skupiny otvorenej liečby v časti II ako nerandomizovaní pacienti (a boli vylúčení
z primárnej analýzy účinnosti).
- Časť III: Pacienti, ktorí ukončili 16 týždňov liečby a boli klasifikovaní ako pacienti odpovedajúci na liečbu, boli opäť randomizovaní do 24-týždňového, dvojito zaslepeného obdobia vysadenia lieku, počas ktorého dostávali 150 mg Ilarisu (2 mg/kg u pacientov
s telesnou hmotnosťou ≤ 40 kg) s.c., alebo placebo každých 8 týždňov.
- Časť IV: Všetci pacienti z časti III liečení Ilarisom boli vhodní na účasť v 72-týždňovom období predĺženia otvorenej liečby.
Celkovo 185 pacientov vo veku 28 dní a starších bolo zaradených a celkovo 181 pacientov vo veku
2 roky a starších bolo randomizovaných do časti II tejto štúdie.
Primárnym parametrom účinnosti randomizovaného obdobia liečby (časť II) bol podiel pacientov
s odpoveďou na liečbu v rámci každej skupiny, u ktorých došlo k poklesu indexu vzplanutia ochorenia na 15. deň a u ktorých sa nevyskytlo nové vzplanutie počas zostávajúceho 16-týždňového obdobia liečby (definované ako úplná odpoveď). Pokles indexu vzplanutia ochorenia bolo definovaný ako
skóre aktivity ochorenia < 2 („minimálne alebo žiadne ochorenie“) podľa globálneho hodnotenia
lekárom (Physician´s Global Assessment, PGA) a CRP v rozmedzí normy (≤ 10 mg/l) alebo zníženie
≥ 70 % oproti východiskovej hodnote. Nové vzplanutie bolo definované ako skóre ≥ 2 („mierne, stredne ťažké alebo ťažké ochorenie“) podľa PGA a CRP ≥ 30 mg/ml. Sekundárne parametre, všetky založené na výsledkoch v 16. týždni (koniec časti II), zahŕňali podiel pacientov, ktorí dosiahli skóre
< 2 podľa PGA, podiel pacientov so sérologickou remisiou (definovaná ako CRP ≤ 10 mg/ml)
a podiel pacientov s normalizovanou hladinou SAA (definovaná ako SAA ≤ 10 mg/ml).
Z hľadiska primárneho parametra účinnosti bol Ilaris účinnejší ako placebo vo všetkých troch skupinách chorôb. Ilaris tiež preukázal vyššiu účinnosť v porovnaní s placebom pre sekundárne parametre PGA < 2 a CRP ≤ 10 mg/ml vo všetkých troch skupinách. Vyššie podiely pacientov mali normalizovanú SAA (≤ 10 mg/ml) v 16. týždni liečby Ilarisom v porovnaní s placebom vo všetkých troch skupinách, so štatisticky významným rozdielom pozorovaným u pacientov s TRAPS (pozri nižšie Tabuľku 3 s výsledkami štúdie).
Tabuľka 3 Tabuľkové zhrnutie účinnosti v klinickom skúšaní fázy III, pivotné,
randomizované, placebom kontrolované obdobie liečby (časť II)
Klinické skúšanie fázy III, pivotné, randomizované placebom kontrolované obdobie liečby
(časť II)
Ilaris n/N (%)
Placebo
n/N (%) Hodnota p
 P
rimárny parameter (vzplanutie ochorenia) –
P
rimárny parameter (vzplanutie ochorenia) – podiel pacientov, u ktorých došlo k poklesu indexu vzplanutia ochorenia na 15. deň a u ktorých sa nevyskytlo nové vzplanutie počas zostávajúceho 16- týždňového obdobia liečby
FMF 19/31 (61,29) 2/32 (6,25) < 0,0001* HIDS/MKD 13/37 (35,14) 2/35 (5,71) 0,0020* TRAPS 10/22 (45,45) 2/24 (8,33) 0,0050*
Sekundárne parametre (ochorenie a zápalové markery)Globálne hodnotenie lekárom < 2
FMF 20/31 (64,52) 3/32 (9,38) < 0,0001** HIDS/MKD 17/37 (45,95) 2/35 (5,71) 0,0006** TRAPS 10/22 (45,45) 1/24 (4,17) 0,0028**
C-reaktívny proteín ≤ 10 mg/l
FMF 21/31 (67,74) 2/32 (6,25) < 0,0001** HIDS/MKD 15/37 (40,54) 2/35 (5,71) 0,0010** TRAPS 8/22 (36,36) 2/24 (8,33) 0,0149**
Sérový amyloid A ≤ 10 mg/l
FMF 8/31 (25,81) 0/32 (0,00) 0,0286
HIDS/MKD 5/37 (13,51) 1/35 (2,86) 0,0778
TRAPS 6/22 (27,27) 0/24 (0,00) 0,0235**
n=počet pacientov s odpoveďou na liečbu; N=počet vyhodnotiteľných pacientov
* indikuje štatistickú významnosť (jednostranná) s hladinou 0,025 na základe Fisherovho presného testu
** indikuje štatistickú významnosť (jednostranná) s hladinou 0,025 na základe modelu logistickej regresie so skupinou liečby a východiskovými hodnotami PGA, CRP alebo SAA ako vysvetľujúce premenné pre každú skupinu
Titrácia nahorV časti II štúdie dostali pacienti liečení Ilarisom, ktorí mali pretrvávajúcu aktivitu ochorenia, ďalšiu dávku 150 mg (alebo 2 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) počas prvého mesiaca. Táto ďalšia dávka sa mohla podať už 7 dní po prvej liečebnej dávke. Všetci pacienti s dávkou
titrovanou nahor zostali pri zvýšenej dávke 300 mg (alebo 4 mg/kg u pacientov s telesnou hmotnosťou
≤ 40 kg) každé 4 týždne.
V exploračnej analýze primárneho parametra sa pozorovalo, že u pacientov s nedostatočnou odpoveďou na liečbu po prvej dávke titrácia nahor počas prvého mesiaca na dávku 300 mg (alebo
4 mg/kg) každé 4 týždne ďalej zlepšila kontrolu vzplanutia, znížila aktivitu ochorenia a normalizovala hladiny CRP a SAA.
Pediatrickí pacienti:
Dvaja nerandomizovaní pacienti s HIDS/MKD vo veku > 28 dní, ale < 2 roky, boli zaradení do štúdie a dostávali kanakinumab. Jeden pacient mal pokles indexu vzplanutia na 15. deň po podaní jednej jednorazovej dávky kanakinumabu 2 mg/kg, ale liečbu prerušil po tejto prvej dávke pre závažné nežiaduce udalosti (pancytopénia a zlyhanie pečene). Tento pacient mal pri vstupe do štúdie imunitnú trombocytopenickú purpuru v anamnéze a aktívne ochorenie s abnormálnou funkciou pečene. Druhý pacient dostal začiatočnú dávku kanakinumabu 2 mg/kg a doplnkovú dávku 2 mg/kg v 3. týždni,
s titráciou dávky nahor v 5. týždni na podávanie 4 mg/kg každé 4 týždne až do konca časti II štúdie. Vymiznutie vzplanutia ochorenia sa dosiahlo v 5. týždni a u pacienta sa nevyskytlo žiadne nové vzplanutie na konci časti II štúdie (16. týždeň).
Stillova choroba
SJIA
Účinnosť Ilarisu v liečbe aktívnej SJIA sa vyhodnotila v dvoch pivotných klinických skúšaniach (G2305 a G2301). Pacienti zaradení do skúšaní boli vo veku 2 až < 20 rokov (priemerný vek 8,5 roka a priemerné trvanie choroby v čase zaradenia do skúšania 3,5 roka) a mali aktívnu chorobu definovanú ako ≥ 2 kĺby s aktívnou artritídou, horúčkou a zvýšeným CRP.
Klinické skúšanie G2305
G2305 bolo randomizované, dvojito zaslepené, placebom kontrolované, 4 týždne trvajúce skúšanie na
stanovenie krátkodobej účinnosti Ilarisu u 84 pacientov randomizovaných na jednorazovú dávku
4 mg/kg (do 300 mg) Ilarisu alebo na placebo. Primárnym ukazovateľom bol podiel pacientov, u ktorých sa na 15. deň dosiahlo zlepšenie minimálne o 30 % v zmysle kritérií odpovede podľa pediatrickej American College of Rheumatology (ACR) upravených tak, aby zahŕňali neprítomnosť horúčky. Liečba Ilarisom zlepšila všetky skóre odpovede podľa pediatrickej ACR v porovnaní
s placebom na 15. aj 29. deň (Tabuľka 4).
Tabuľka 4 Odpoveď podľa pediatrickej ACR a stav choroby na 15. a 29. deň
15. deň 29. deň
Ilaris
N=43
Placebo
N=41
Ilaris
N=43
Placebo
N=41

ACR30 84 % 10 % 81 % 10 % ACR50 67 % 5 % 79 % 5 % ACR70 61 % 2 % 67 % 2 % ACR90 42 % 0 % 47 % 2 % ACR100 33 % 0 % 33 % 2 % Neaktívna choroba 33 % 0 % 30 % 0 % Rozdiel v liečbe pri všetkých skóre ACR bol významný (p ≤ 0,0001)
Výsledky zložiek upravenej odpovede podľa pediatrickej ACR, ktoré zahŕňali systémové a artritické
zložky, boli v súlade s celkovými výsledkami odpovede podľa ACR. Na 15. deň bol medián zmien oproti východiskovej hodnote v počte kĺbov s aktívnou artritítou -67 % a s obmedzeným rozsahom pohybu -73 % pri Ilarise (N=43) v porovnaní s mediánom zmien pri placebe 0 % a 0 % (N=41). Priemerná zmena v skóre bolesti podľa pacienta (vizuálna analogická škála 0-100 mm) na 15. deň bola pri Ilarise -50,0 mm (N=43) v porovnaní s placebom +4,5 mm (N=25). Priemerná zmena v skóre bolesti u pacientov liečených Ilarisom pretrvávala na 29. deň.
K
li
nické skúšanie G2301
G2301 bolo randomizované, dvojito zaslepené, placebom kontrolované skúšanie Ilarisu na prevenciu vzplanutí pri vysadení liečby. Skúšanie pozostávalo z dvoch častí s dvoma nezávislými primárnymi ukazovateľmi (úspešné zníženie dávky steroidov a čas do vzplanutia). Do časti I (otvorená liečba) bolo zaradených 177 pacientov, ktorí dostávali 4 mg/kg (do 300 mg) Ilarisu každé 4 týždne počas najviac 32 týždňov. Pacienti v časti II (dvojito zaslepená liečba) dostávali buď 4 mg/kg Ilarisu, alebo placebo každé 4 týždne, až do výskytu 37 udalostí vzplanutia.
Znižovanie dávky kortikosteroidov:
Z celkového počtu 128 pacientov, ktorí pri vstupe do časti I užívali kortikosteroidy, sa 92 pokúsilo o zníženie dávky kortikosteroidov. Päťdesiatsedem (62 %) z 92 pacientov, ktorí sa pokúsili znížiť svoju dávku, bolo pri znížení dávky kortikosteroidov úspešných a 42 (46 %) kortikosteroidy vysadilo.
Čas do vzplanutia:
Pacienti, ktorí používali Ilaris v časti II, mali riziko udalosti vzplanutia znížené o 64 % v porovnaní so skupinou placeba (pomer rizika 0,36; 95 % IS: 0,17 až 0,75; p=0,0032). U 63 zo 100 pacientov, ktorí vstúpili do časti II, bez ohľadu na to, či boli pridelení na placebo alebo kanakinumab, sa nevyskytlo vzplanutie počas sledovaného obdobia (najviac 80 týždňov).
Výsledky klinických skúšaní G2305 a G2301 týkajúce sa zdravia a kvality života
Liečba Ilarisom vyvolala klinicky významné zlepšenie telesných funkcií a kvality života pacientov. V skúšaní G2305 bolo zlepšenie podľa dotazníka na hodnotenie zdravia v detskom veku (Childhood Health Assessment Questionnaire) metódou najmenších štvorcov pri Ilarise 0,69 oproti placebu, čo predstavuje 3,6-násobok minimálneho klinicky významného rozdielu 0,19 (p=0,0002). Medián zlepšenia oproti východiskovej hodnote do konca časti I skúšania G2301 bol 0,88 (79 %). Štatisticky významné zlepšenie skóre v dotazníku zdravia dieťaťa (Child Health Questionnaire-PF50) pri Ilarise v porovnaní s placebom bolo hlásené v skúšaní G2305 (telesné p=0,0012; psychosociálne zdravie p=0,0017).
Združená analýza účinnosti
Údaje z prvých 12 týždňov liečby Ilarisom v skúšaniach G2305, G2301 a v extenzii klinického skúšania sa združili na vyhodnotenie zachovania účinnosti. Tieto údaje preukázali do 12. týždňa oproti východiskovým hodnotám podobné zlepšenie podľa upravených odpovedí pediatrickej ACR a ich zložiek, aké sa pozorovalo v skúšaní kontrolovanom placebom (G2305). Po 12. týždni boli upravené odpovede podľa pediatrickej ACR30, 50, 70, 90 a 100: 70 %, 69 %, 61 %, 49 % a 30 %, choroba bola neaktívna u 28 % pacientov (N=178).
Účinnosť pozorovaná v skúšaniach G2305 a G2301 sa udržala počas pokračujúcej otvorenej, dlhodobej extenzie (údaje sú dostupné pre medián 49 týždňov následného sledovania). V tomto skúšaní sa u 25 pacientov, ktorí mali silnú odpoveď na liečbu podľa ACR počas najmenej 5 mesiacov, znížila dávka Ilarisu na 2 mg/kg každé 4 týždne, pričom odpoveď si títo pacienti udržali podľa pediatrickej ACR100 počas celého obdobia, keď sa im podávala znížená dávka (medián 32 týždňov,
8-124 týždňov).
Hoci sú údaje obmedzené, dôkazy z klinických skúšaní naznačujú, že pacienti, ktorí nereagujú na tocilizumab alebo anakinru, môžu reagovať na kanakinumab.
SJIA u mladých dospelých a AOSD
Účinnosť Ilarisu v podskupine mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov bola v súlade s účinnosťou, pozorovanou u pacientov mladších ako 16 rokov. Na základe literárnych údajov sa očakáva podobný profil účinnosti u AOSD pacientov ako u pacientov so SJIA.
D
navá artritída
Účinnosť Ilarisu v liečbe akútnych záchvatov dnavej artritídy sa preukázala v dvoch multicentrických, randomizovaných, dvojito zaslepených klinických skúšaniach kontrolovaných účinným liekom u pacientov s častou dnavou artritídou (≥ 3 záchvaty počas predošlých 12 mesiacov), ktorí nemohli používať NSAID alebo kolchicín (pre kontraindikáciu, neznášanlivosť alebo nedostatočnú účinnosť). Klinické skúšania trvali 12 týždňov, po ktorých nasledovala 12-týždňová dvojito zaslepená extenzia. Celkovo 225 pacientov bolo liečených subkutánne podaným Ilarisom 150 mg a 229 pacientov bolo liečených intramuskulárne podaným triamcinolónacetonidom (TA) 40 mg pri zaradení do klinického skúšania a neskôr pri výskyte nového záchvatu. Priemerný počet záchvatov dnavej artritídy počas predošlých 12 mesiacov bol 6,5. Viac ako 85 % pacientov malo sprievodné choroby vrátane hypertenzie (60 %), diabetu (15 %), ischemickej choroby srdca (12 %) a chronickej choroby obličiek
v štádiu ≥ 3 (25 %). Približne u jednej tretiny zaradených pacientov (76 [33,8 %] v skupine Ilarisu a
84 [36,7 %] v skupine triamcinolónacetonidu) bolo zdokumentované, že nemôžu (pre intoleranciu, kontraindikáciu alebo nedostatočnú odpoveď) používať NSAID ani kolchicín. Súbežnú liečbu ULT hlásilo 42 % pacientov pri zaradení do klinického skúšania.
Súbežné primárne parametre boli: (i) intenzita bolesti pri dnavej artritíde (vizuálna analógová škála,
VAS) 72 hodín po podaní, a (ii) čas do prvého nového záchvatu dnavej artritídy.
V celkovej populácii v klinickom skúšaní bola intenzita bolesti po 72 hodinách štatisticky významne nižšia pri Ilarise 150 mg v porovnaní s triamcinolónacetonidom. Ilaris tiež znížil riziko ďalších záchvatov (pozri Tabuľku 5).
Výsledky účinnosti u pacientov, ktorí nemohli užívať NSAID ani kolchicín a ktorí dostávali ULT,
u ktorých ULT nebola účinná alebo u ktorých bola ULT kontraindikovaná (N=101), sa zhodovali s výsledkami celkovej populácie v klinickom skúšaní, so štatisticky významným rozdielom oproti triamcinolónacetonidu pri intenzite bolesti po 72 hodinách (-10,2 mm, p=0,0208) a pri znížení rizika ďalších záchvatov (pomer rizika 0,39, p=0,0047 po 24 týždňoch).
Výsledky účinnosti v užšej podskupine obmedzenej na súčasných používateľov ULT (N=62) sú uvedené v Tabuľke 5. Liečba Ilarisom vyvolala zmiernenie bolesti a znížila riziko ďalších záchvatov u pacientov, ktorí používali ULT a nemohli používať NSAID ani kolchicín, hoci rozdiel pozorovaný pri liečbe v porovnaní s triamcinolónacetonidom bol menej výrazný ako v celkovej populácii v klinickom skúšaní.
 Tabuľka 5 Účinnosť u celkovej populácie v klinickom skúšaní a u podskupiny pacientov v súčasnosti používajúcich ULT, ktorí nemôžu používať NSAID ani kolchicín
Tabuľka 5 Účinnosť u celkovej populácie v klinickom skúšaní a u podskupiny pacientov v súčasnosti používajúcich ULT, ktorí nemôžu používať NSAID ani kolchicín
P
arameter účinnosti Celková populácia
v klinickom skúšaní; N=454
N
emožnosť používať NSAID
ani kolchicín; používanie
ULT
N=62
Liečbazáchvatovdnavej
artritídy
m
eraná ako intenzita bolesti (VAS) po 72 hodinách
Priemerný rozdiel oproti triamcinolónacetonidu odhadovaný metódou najmenších štvorcov
IS
Hodnota p, 1-stranná
-10,7
(-15,4, -6,0)
p < 0,0001*
-3,8
(-16,7, 9,1)
p=0,2798
Z
níženie rizika ďalších záchvatov dnavej artritídy merané ako čas do prvého nového vzplanutia
(
24 týždňov)
Pomer rizika oproti triamcinolón- acetonidu
IS
Hodnota p, 1-stranná

* Označuje významnú hodnotu p < 0,025
0,44
(0,32, 0,60)
p < 0,0001*
0,71
(0,29, 1,77)
p=0,2337
Výsledky bezpečnosti ukázali zvýšenú incidenciu nežiaducich udalostí pri kanakinumabe v porovnaní s triamcinolónacetonidom, keď počas 24 týždňov akúkoľvek nežiaducu udalosť hlásilo 66 % oproti
53 % pacientov a infekciu ako nežiaducu udalosť hlásilo 20 % oproti 10 % pacientov.
Starší pacienti
Celkovo bol profil účinnosti, bezpečnosti a znášanlivosti Ilarisu u starších pacientov vo veku
≥ 65 rokov a vo veku < 65 rokov porovnateľný.
Pacienti, ktorí dostávajú liečbu znižujúcu urát (ULT)
V klinických skúšaniach sa Ilaris bezpečne podával s ULT. V celkovej populácii v klinickom skúšaní bol u pacientov, ktorí dostávali ULT, pri liečbe menej výrazný rozdiel v zmiernení bolesti aj znížení rizika ďalších záchvatov dnavej artritídy v porovnaní s pacientmi, ktorí nedostávali ULT.
Imunogenita
Protilátky proti Ilarisu sa pozorovali u približne 1,5 % pacientov liečených Ilarisom pre CAPS, u 3% pacientov liečených pre SJIA a u 2 % pacientov liečených pre dnavú artritídu. Nezistili sa žiadne neutralizujúce protilátky. Nepozorovala sa žiadna zjavná korelácia medzi tvorbou protilátok
a klinickou odpoveďou alebo nežiaducimi udalosťami.
Nepozorovali sa žiadne protilátky proti Ilarisu u pacientov s TRAPS, HIDS/MKD a FMF, ktorí dostávali dávky 150 mg a 300 mg počas 16 týždňov liečby.
Tento liek bol registrovaný pre CAPS za tzv. mimoriadnych okolností. To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku. Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
Pediatrická populácia
Držiteľ rozhodnutia o registrácii uskutočnil štyri výskumné pediatrické plány pre Ilaris (pre CAPS, SJIA, FMF - HIDS/MKD a TRAPS). Táto informácia o lieku bola aktualizovaná, aby zahrnula výsledky štúdií s Ilarisom v pediatrickej populácii.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Ilarisom vo všetkých podskupinách pediatrickej populácie pre dnavú artritídu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
CAPS
Absorpcia
Maximálna koncentrácia kanakinumabu v sére (Cmax) sa dosiahla asi 7 dní po jednorazovom subkutánnom podaní 150 mg dospelým pacientom s CAPS. Priemerný terminálny polčas bol 26 dní. Priemerné hodnoty Cmax a AUCinf po jednorazovej subkutánnej dávke 150 mg u typického dospelého pacienta s CAPS (70 kg) boli 15,9 µg/ml a 708 µg*deň/ml. Odhad absolútnej biologickej dostupnosti subkutánne podaného kanakinumabu bol 66 %. Parametre expozície (napr. AUC a Cmax) sa zvyšovali úmerne dávke v rozmedzí dávok 0,30 až 10,0 mg/kg podávaných intravenóznou infúziou alebo od 150 do 600 mg subkutánnou injekciou. Predpokladané hodnoty expozície v rovnovážnom stave (Cmin,ss, Cmax,ss, AUC,ss,8w) po subkutánnom podávaní 150 mg (alebo 2 mg/kg) každých 8 týždňov boli o niečo vyššie v hmotnostnej kategórii 40-70 kg (6,6 µg/ml, 24,3 µg/ml, 767 µg*d/ml) v porovnaní
s hmotnostnými kategóriami < 40 kg (4,0 µg/ml, 19,9 µg/ml, 566 µg*d/ml) a > 70 kg (4,6 µg/ml,
17,8 µg/ml, 545 µg*d/ml). Očakávaný pomer akumulácie bol 1,3-násobný po 6 mesiacoch subkutánneho podávania 150 mg kanakinumabu každých 8 týždňov.
D
i
stribúcia
Kanakinumab sa viaže na sérový IL-1 beta. Distribučný objem (Vss) kanakinumabu sa líšil podľa
telesnej hmotnosti. U pacienta s CAPS a telesnou hmotnosťou 70 kg sa odhadol na 6,2 l.
Eliminácia
Zdanlivý klírens (CL/F) kanakinumabu sa zvyšuje s telesnou hmotnosťou. U pacienta s CAPS
s telesnou hmotnosťou 70 kg sa odhadol na 0,17 l/d a u pacienta so SJIA s telesnou hmotnosťou 33 kg na 0,11 l/d. Po zohľadnení rozdielov v telesnej hmotnosti sa nepozorovali žiadne klinicky významné rozdiely vo farmakokinetických vlastnostiach kanakinumabu medzi pacientmi s CAPS a SJIA.
Po opakovanom podávaní kanakinumabu sa nezistil žiadny náznak zrýchleného klírensu alebo zmeny farmakokinetických vlastností v závislosti od času. Po korekcii na telesnú hmotnosť sa nepozorovali žiadne farmakokinetické rozdiely v súvislosti s pohlavím alebo vekom.
TRAPS, HIDS/MKD a FMF
Biologická dostupnosť sa nestanovila nezávisle u pacientov s TRAPS, HIDS/MKD a FMF. Zdanlivý klírens (CL/F) bol porovnateľný medzi populáciami s TRAPS, HIDS/MKD a FMF s telesnou hmotnosťou 55 kg (0,14 l/d) a s CAPS s telesnou hmotnosťou 70 kg (0,17 l/d). Zdanlivý distribučný objem (V/F) bol 4,96 l pri telesnej hmotnosti 55 kg.
Po opakovanom subkutánnom podávaní 150 mg každé 4 týždne bola minimálna koncentrácia kanakinumabu v 16. týždni (Cmin) odhadovaná na 15,4 ± 6,6 µg/ml. Odhadovaný rovnovážny stav AUCtau bol 636,7 ± 260,2 µg*d/ml.
Stillova choroba (AOSD a SJIA)
Biologická dostupnosť sa nestanovila nezávisle u pacientov so SJIA. Zdanlivý klírens na kg telesnej hmotnosti (CL/F na kg) bol porovnateľný medzi populáciami so SJIA a s CAPS (0,004 l/d na kg). Zdanlivý distribučný objem na kg (V/F na kg) bol 0,14 l/kg.
Po opakovanom podávaní 4 mg/kg každé 4 týždne pacientom so SJIA bol pomer akumulácie kanakinumabu 1,6-násobný. Rovnovážny stav sa dosiahol po 110 dňoch. Celkové predpokladané priemerné (±SD) hodnoty boli pre Cmin,ss 14,7±8,8 μg/ml, Cmax,ss 36,5 ± 14,9 μg/ml a AUC,ss4w 696,1 ±
326,5 μg*d/ml.
AUCss4w bola vo vekovej skupine 2-3 roky 692 µg*d/ml, 4-5 rokov 615 µg*d/ml, 6-11 rokov
707 µg*d/ml a 12-19 rokov 742 µg*d/ml. Po stratifikácii podľa telesnej hmotnosti sa pozoroval nižší
(30-40%) medián expozície pre Cmin,ss (11,4 oproti 19 µg/ml) a AUCss (594 oproti 880 µg*d/ml) pre
kategóriu nižšej telesnej hmotnosti (≤ 40 kg) oproti kategórii vyššej telesnej hmotnosti (> 40 kg).
Na základe analýzy populačného farmakokinetického modelovania bola farmakokinetika kanakinumabu u mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov podobná ako
u pacientov mladších ako 16 rokov. Predpokladané expozície kanakinumabu v rovnovážnom stave pri
veľkosti dávky 4 mg/kg (maximálne 300 mg) boli porovnateľné u pacientov nad 20 rokov a u pacientov so SJIA mladších ako 20 rokov.
Populácia s dnavou artritídou
Biologická dostupnosť u pacientov s dnavou artritídou sa nezávisle nestanovila. Zdanlivý klírens na
kg telesnej hmotnosti (CL/F na kg) bol porovnateľný medzi populáciami s dnavou artritídou a s CAPS (0,004 l/deň/kg). Priemerná expozícia u typického pacienta s dnavou artritídou (93 kg) po
jednorazovej subkutánnej dávke 150 mg (Cmax: 10,8 µg/ml a AUCinf: 495 µg*deň/ml) bola nižšia ako u typického pacienta s CAPS s telesnou hmotnosťou 70 kg (15,9 µg/ml a 708 µg*deň/ml). Zhoduje sa to s pozorovaným zvyšovaním CL/F s telesnou hmotnosťou.
Očakávaný pomer akumulácie bol 1,1-násobný po subkutánnom podávaní 150 mg kanakinumabu
každých 12 týždňov.
Pediatrická populácia
Maximálne koncentrácie kanakinumabu u pediatrických pacientov vo veku 4 rokov a starších sa dosiahli medzi 2. a 7. dňom (Tmax) po jednorazovom subkutánnom podaní 150 mg alebo 2 mg/kg kanakinumabu. Terminálny polčas bol v rozmedzí 22,9 až 25,7 dní, čo je podobné farmakokinetickým vlastnostiam pozorovaným u dospelých. Na základe analýzy populačného farmakokinetického modelovania bola farmakokinetika kanakinumabu u detí vo veku 2 až < 4 roky podobná ako
u pacientov vo veku 4 roky a starších. Odhaduje sa, že rýchlosť subkutánnej absorpcie klesá s vekom a zdá sa, že je najvyššia u najmladších pacientov. V súlade s tým bol Tmax kratší (3,6 dní) u mladších pacientov so SJIA (2-3 roky) v porovnaní so staršími pacientmi so SJIA (12-19 rokov; Tmax 6 dní). Biologická dostupnosť (AUCss) nebola ovplyvnená.
Ďalšia analýza farmakokinetiky ukázala, že farmakokinetika kanakinumabu u 6 pediatrických pacientov s CAPS mladších ako 2 roky bola podobná ako farmakokinetika u pediatrických pacientov vo veku 2-4 roky. Na základe analýzy populačného farmakokinetického modelovania boli očakávané expozície po dávke 2 mg/kg porovnateľné vo všetkých pediatrických vekových skupinách pri CAPS, ale boli približne o 40 % nižšie u pediatrických pacientov s veľmi nízkou telesnou hmotnosťou (napr.
10 kg) v porovnaní s dospelými pacientmi (dávka 150 mg). Zhoduje sa to s pozorovaniami vyššej
expozície u pacientov s CAPS v skupinách vyššej telesnej hmotnosti.
Pri TRAPS, HIDS/MKD a FMF boli parametre expozície (minimálne koncentrácie) porovnateľné vo všetkých vekových skupinách od 2 do < 20 rokov po subkutánnom podaní kanakinumabu 2 mg/kg každé 4 týždne.
Farmakokinetické vlastnosti sú podobné u pediatrických populácií s CAPS, TRAPS, HIDS/MKD, FMF a SJIA.
Staršípacienti
Rozdiel farmakokinetických parametrov na základe klírensu alebo distribučného objemu sa nepozoroval medzi staršími pacientmi a dospelými pacientmi vo veku < 65 rokov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií skríženej reaktivity, toxicity po opakovanom podávaní, imunotoxicity, reprodukčnej toxicity a toxicity pre mláďatá vykonaných s kanakinumabom alebo myšacou protilátkou proti myšaciemu IL-1 beta neodhalili žiadne osobitné riziko pre ľudí.
Keďže kanakinumab sa viaže na IL-1 beta kozmáčov (C. jacchus) a ľudí s podobnou afinitou, bezpečnosť kanakinumabu sa skúmala u kozmáčov. Žiadne nežiaduce účinky sa nepozorovali po podávaní kanakinumabu dvakrát týždenne kozmáčom počas 26 týždňov alebo v štúdii toxicity pre embryofetálny vývoj počas gestácie u kozmáčov. Plazmatické koncentrácie, ktoré dobre tolerujú,zvieratá, prevyšujú najmenej 42-násobne (Cmax) a 78-násobne (Cavg) plazmatické koncentrácie u detí a dospievajúcich s CAPS (telesná hmotnosť 10 kg) liečených klinickými dávkami kanakinumabu do 8 mg/kg subkutánne každých 8 týždňov. Plazmatické koncentrácie, ktoré zvieratá dobre znášajú, sú minimálne 62-krát vyššie (Cmax) a 104-krát vyššie (Cavg) ako plazmatické koncentrácie u pediatrických pacientov so SJIA, ktorí dostávajú dávky do 4 mg/kg subkutánne každé
4 týždne. Navyše sa v týchto štúdiách nezistili žiadne protilátky proti kanakinumabu. Pri aplikácii kanakinumabu do normálnych ľudských tkanív sa nepreukázala nešpecifická tkanivová skrížená reaktivita.
Formálne štúdie karcinogenity s kanakinumabom sa nevykonali.
V štúdii toxicity pre embryofetálny vývoj kozmáčov kanakinumab nevykazoval pri podávaní počas
organogenézy toxicitu pre matky, embryotoxicitu ani teratogenitu.
V kompletnom súbore reprodukčných štúdií a štúdií na mláďatách myší sa nepozorovali nežiaduce účinky myšacej protilátky proti myšaciemu IL-1 beta. Protilátka proti myšaciemu IL-1 beta nevyvolala pri podávaní počas neskorej gestácie, pôrodu a laktácie nežiaduce udalosti súvisiace s rastom plodov alebo novorodencov (pozri časť 4.6). Vysoká dávka použitá v týchto štúdiách bola vyššia ako maximálna účinná dávka z hľadiska supresie IL-1 beta a aktivity.
Imunotoxikologická štúdia u myší s myšacou protilátkou proti myšaciemu IL-1 beta ukázala, že neutralizácia IL-1 beta nemá žiadne účinky na imunitné parametre a nespôsobila zhoršenie imunitnej funkcie u myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sacharóza
L-histidín
Monohydrát L-histidíniumchloridu
Polysorbát 80
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
Z mikrobiologického hľadiska sa liek má použiť okamžite po rekonštitúcii. Ak sa nepoužije okamžite, za čas a podmienky uchovávania pred použitím zodpovedá používateľ, pričom normálne nemajú presiahnuť 24 hodín pri 2°C – 8°C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom. Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
150 mg prášku na injekčný roztok v injekčnej liekovke (sklo typu I) so zátkou (chlórobutylová guma s krycou vrstvou) a odtrhávacím „flip/off“ uzáverom (hliník).
Balenia obsahujúce 1 injekčnú liekovku alebo multibalenia obsahujúce 4 (4 x 1) injekčné liekovky.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Ilaris 150 mg prášok na injekčný roztok sa dodáva v jednorazovej liekovke na individuálne použitie. Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
PokynynarekonštitúciuAseptickým postupom rekonštituujte každú injekčnú liekovku Ilarisu pri izbovej teplote (obvykle
15°C až 25°C) pomalým injikovaním 1 ml vody na injekciu z 1 ml injekčnej striekačky s ihlou
18 G x 2 palce (50 mm). Pomaly otáčajte liekovku v uhle asi 45° približne počas 1 minúty a nechajte stáť asi 5 minút. Potom liekovku pomaly desať ráz obráťte zhora nadol a späť. Podľa možnosti sa nedotýkajte prstami gumenej zátky. Nechajte stáť asi 15 minút pri izbovej teplote, aby sa získal číry až opalizujúci roztok. Nepotriasajte. Nepoužite roztok, ak sú v ňom prítomné pevné častice.
Poklopte zboku na liekovku, aby sa zo zátky odstránil zvyšok tekutiny. Roztok nemá obsahovať viditeľné častice a má byť číry až opalizujúci. Roztok má byť bezfarebný alebo môže mať slabé hnedožlté sfarbenie. Ak je roztok sfarbený výrazne dohneda, nemá sa použiť. Ak sa roztok nepoužije ihneď po rekonštitúcii, má sa uchovávať pri teplote 2°C až 8°C a použiť do 24 hodín.
Pokyny na podanieOpatrne odoberte požadovaný objem v závislosti od dávky, ktorá sa má podať (0,1 ml až 1 ml)
a subkutánne injikujte pomocou ihly 27 G x 0,5 palca (13 mm).
LikvidáciaPacienti alebo ich opatrovatelia majú dostať pokyny, ako v súlade s národnými požiadavkami správne postupovať pri likvidácii injekčných liekoviek, striekačiek a ihiel.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/09/564/001-002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. október 2009
Dátum posledného predĺženia registrácie: 19. jún 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUIlaris 150 mg prášok a rozpúšťadlo na injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEJedna injekčná liekovka obsahuje 150 mg kanakinumabu*.
Po rekonštitúcii obsahuje každý ml roztoku 150 mg kanakinumabu.
* ľudská monoklonálna protilátka produkovaná v myšacích myelómových bunkách Sp2/0
technológiou rekombinantnej DNA
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAPrášok a rozpúšťadlo na injekčný roztok. Prášok je biely.
Rozpúšťadlo je číre a bezfarebné.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieSyndrómyperiodickejhorúčkyIlaris je indikovaný na liečbu nasledujúcich autoinflamačných syndrómov periodickej horúčky
u dospelých, dospievajúcich a detí vo veku 2 roky a starších:
Periodické syndrómy asociované s kryopyrínomIlaris je indikovaný na liečbu periodických syndrómov asociovaných s kryopyrínom (cryopyrin- associated periodic syndromes, CAPS) vrátane:
· Muckleovho-Wellsovho syndrómu (MWS),
· multisystémovej zápalovej choroby novorodencov (Neonatal-onset multisystem inflammatory disease, NOMID) / chronického neurologického, kožného a kĺbového syndrómu u detí (chronic infantile neurological, cutaneous, articular syndrome, CINCA),
· ťažkých foriem familiárneho chladového autoinflamačného syndrómu (familial cold autoinflammatory syndrome, FCAS) / familiárnej chladovej urtikárie (familial cold urticaria, FCU), prejavujúcej sa príznakmi a prejavmi presahujúcimi kožný exantém typu urtikárie vyvolanej chladom.
Periodický syndróm asociovaný s receptorom pre faktor nekrotizujúci nádory (tumour necrosis factorreceptor associated periodic syndrome, TRAPS)Ilaris je indikovaný na liečbu periodického syndrómu asociovaného s receptorom pre faktor nekrotizujúci nádory (tumour necrosis factor, TNF) (TRAPS).
Syndróm hyperimunoglobulinémie D (hyperimmunoglobulin D syndrome, HIDS)/deficitu
mevalonátkinázy (mevalonate kinase deficiency, MKD)
Ilaris je indikovaný na liečbu syndrómu hyperimunoglobulinémie D (HIDS)/deficitu mevalonátkinázy
(MKD).
Fami l i árna s t re doze msk á horúč ka (f ami l i al medi t er rane an f ev er , FMF)
Ilaris je indikovaný na liečbu familiárnej stredozemskej horúčky (FMF). Ilaris sa má podávať
v kombinácii s kolchicínom, ak je to vhodné.
Ilaris je tiež indikovaný na liečbu:
Stillova choroba
Ilaris je indikovaný na liečbu aktívnej Stillovej choroby vrátane Stillovej choroby s nástupom ochorenia v dospelosti (adult-onset Still’s disease, AOSD) a systémovej juvenilnej idiopatickej artritídy (SJIA) u pacientov vo veku 2 rokov a starších, ktorí nedostatočne reagovali na predchádzajúcu liečbu nesteroidnými protizápalovými liekmi (NSAID) a systémovými kortikosteroidmi. Ilaris sa môže podávať ako monoterapia alebo v kombinácii s metotrexátom.
Dnavá artritída
Ilaris je indikovaný na symptomatickú liečbu dospelých pacientov s častými záchvatmi dnavej
artritídy (najmenej 3 záchvaty počas predošlých 12 mesiacov), u ktorých sú nesteroidné protizápalové
lieky (NSAID) a kolchicín kontraindikované, netolerované alebo nevyvolávajú primeranú odpoveď a u ktorých nie je vhodná opakovaná liečba kortikosteroidmi (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Pri CAPS, TRAPS, HIDS/MKD, FMF a Stillovej chorobe má liečbu začať a viesť odborný lekár so skúsenosťami v diagnostike a liečbe príslušnej indikácie.
Pri dnavej artritíde má mať lekár skúsenosti s používaním biologických liekov a Ilaris má podávať
zdravotnícky pracovník.
Po náležitom zaškolení o správnej injekčnej technike si môžu Ilaris podávať pacienti alebo im ho môžu podávať ich opatrovatelia, ak lekár rozhodne, že je to vhodné, a keď ich lekár podľa potreby bude ďalej sledovať (pozri časť 6.6).
Dávkovanie
CAPS: Dospelí, dospievajúci a deti vo veku 2 rok ov a s t arš i e Odporúčaná začiatočná dávka Ilarisu u pacientov s CAPS je: Dospelí, dospievajúci a deti vo veku ≥ 4 roky:
- 150 mg u pacientov s telesnou hmotnosťou > 40 kg
- 2 mg/kg u pacientov s telesnou hmotnosťou ≥ 15 kg a ≤ 40 kg
- 4 mg/kg u pacientov s telesnou hmotnosťou ≥ 7,5 kg a < 15 kg
Deti vo veku 2 až < 4 roky:
- 4 mg/kg u pacientov s telesnou hmotnosťou ≥ 7,5 kg
Podáva sa každých osem týždňov ako jednorazová dávka subkutánnou injekciou.
Ak sa u pacientov so začiatočnou dávkou 150 mg alebo 2 mg/kg nedosiahne uspokojivá klinická odpoveď (vymiznutie exantému a iných generalizovaných symptómov zápalu) 7 dní po začatí liečby, možno zvážiť druhú dávku Ilarisu – 150 mg alebo 2 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu, má sa dodržiavať zintenzívnený režim dávkovania 300 mg alebo 4 mg/kg každých
8 týždňov. Ak sa uspokojivá klinická odpoveď nedosiahne 7 dní po tejto zvýšenej dávke, možno zvážiť tretiu dávku Ilarisu 300 mg alebo 4 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu,
na základe individuálneho klinického posúdenia sa má zvážiť dodržiavanie zintenzívneného režimu
dávkovania 600 mg alebo 8 mg/kg každých 8 týždňov.
Ak sa u pacientov so začiatočnou dávkou 4 mg/kg nedosiahne uspokojivá klinická odpoveď 7 dní po začatí liečby, môže sa zvážiť druhá dávka Ilarisu 4 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu, na základe individuálneho klinického posúdenia sa má zvážiť dodržiavanie zintenzívneného režimu dávkovania 8 mg/kg každých 8 týždňov.
Klinické skúsenosti s dávkovaním v intervaloch kratších ako 4 týždne alebo s dávkami vyššími ako
600 mg alebo 8 mg/kg sú obmedzené.
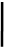 CA
PS u dospelých a detí vo veku
CA
PS u dospelých a detí vo veku
³
4 roky³
15 kgCAPS u detí vo veku 2-< 4 roky alebo detí vo veku
detí vo veku ³
4 roky ³
7,5 kg a < 15 kg
150 mg alebo 2 mg/kg
4 mg/kg
U
spokojivá klinická
odpoveď po 7 dňoch?
U
spokojivá klinická
odpoveď po 7 dňoch?
Á
no
N
i
e
Á
no
N
i
e
U
držiavacia dávka: 150 mg alebo 2 mg/kg každých
8 týždňov
Možno zvážiť ďalšiu
dávku 150 mg alebo
 2 mg/kg
U
držiavacia dávka: 4 mg/kg každých
2 mg/kg
U
držiavacia dávka: 4 mg/kg každých
 8 týždňov
Možno zvážiť ďalšiudávku
4 mg/kg
8 týždňov
Možno zvážiť ďalšiudávku
4 mg/kg
U
spokojivá klinická odpoveď po
7 dňoch?
Á
no Nie
A
k je po 7 dňoch úplná odpoveď na liečbu, udržiavacia dávka:
8 mg/kg každých
8 týždňov
U
držiavacia dávka:
 300 mg alebo 4 mg/kg
každých 8 týždňov
Možno zvážiť ďalšiu
dávku 300 mg alebo
300 mg alebo 4 mg/kg
každých 8 týždňov
Možno zvážiť ďalšiu
dávku 300 mg alebo
 4 mg/kg
4 mg/kg
 A
k je po 7 dňoch úplná odpoveď na liečbu, udržiavacia dávka:
600 mg alebo 8 mg/kg
každých 8 týždňov
A
k je po 7 dňoch úplná odpoveď na liečbu, udržiavacia dávka:
600 mg alebo 8 mg/kg
každých 8 týždňov
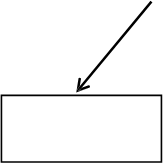
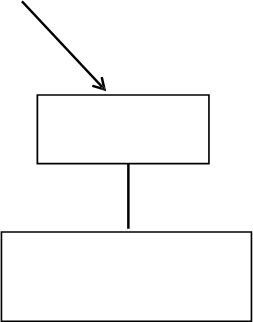
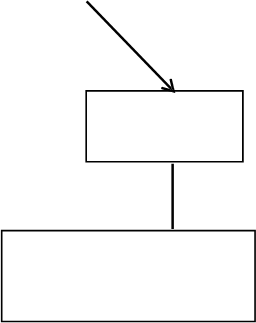
TRAPS, HIDS/MKD a FMF: Dospelí, dospievajúci a deti vo veku 2 roky a st arš i e
Odporúčaná začiatočná dávka Ilarisu u pacientov s TRAPS, HIDS/MKD a FMF je:
· 150 mg u pacientov s telesnou hmotnosťou > 40 kg
· 2 mg/kg u pacientov s telesnou hmotnosťou ≥ 7,5 kg a ≤ 40 kg
Podáva sa každé štyri týždne ako jednorazová dávka subkutánnou injekciou.
Ak sa nedosiahne uspokojivá klinická odpoveď 7 dní od začatia liečby, možno zvážiť druhú dávku Ilarisu 150 mg alebo 2 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu, má sa dodržiavať zintenzívnený režim dávkovania 300 mg (alebo 4 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) každé 4 týždne.
Ošetrujúci lekár má prehodnotiť pokračovanie liečby Ilarisom u pacientov bez klinického zlepšenia.
TRAPS, HIDS/MKD a FMF u pacientov
s telesnou hmotnosťou > 40 kg
 150 mg
TRAPS, HIDS/MKD a FMF u pacientov s telesnou hmotnosťou
150 mg
TRAPS, HIDS/MKD a FMF u pacientov s telesnou hmotnosťou
³
7,5 kg a ≤ 40 kg 2 mg/kg
2 mg/kg
U
spokojivá klinická
odpoveď po 7 dňoch?
U
spokojivá klinická
 odpoveď po 7 dňoch?
odpoveď po 7 dňoch?
Á
no
N
ie
Á
no
N
ie
 U
držiavacia dávka: 150 mg každé 4 týždne
Možno zvážiť ďalšiu dávku
U
držiavacia dávka: 150 mg každé 4 týždne
Možno zvážiť ďalšiu dávku
 150 mg
U
držiavacia dávka 2 mg/kg každé 4 týždne
Možno zvážiť ďalšiu dávku
2 mg/kg
150 mg
U
držiavacia dávka 2 mg/kg každé 4 týždne
Možno zvážiť ďalšiu dávku
2 mg/kg
A
k sa dosiahne úplná odpoveď na liečbu, udržiavacia dávka:
300 mg každé 4 týždne
A
k sa dosiahne úplná odpoveď na liečbu, udržiavacia dávka:
4 mg/kg každé 4 týždne
 Stillova choroba (AOSD a SJIA)
Stillova choroba (AOSD a SJIA)
Odporúčaná dávka Ilarisu u pacientov so Stillovou chorobou (AOSD a SJIA) s telesnou hmotnosťou
≥ 7,5 kg je 4 mg/kg (maximálne 300 mg) podávaná subkutánnou injekciou každé štyri týždne. Pokračovanie v liečbe Ilarisom u pacientov bez klinického zlepšenia má prehodnotiť ošetrujúci lekár.
Dnavá artritídaJe potrebné začať alebo optimalizovať úpravu hyperurikémie náležitou liečbou na zníženie urátu (urate lowering therapy, ULT). Ilaris sa má používať ako liečba podľa potreby na liečenie záchvatov dnavej artritídy.
Odporúčaná dávka Ilarisu u dospelých pacientov s dnavou artritídou je 150 mg podávaných subkutánne ako jednorazová dávka počas záchvatu. Na dosiahnutie maximálneho účinku sa má Ilaris podávať čo najskôr po prepuknutí záchvatu dnavej artritídy.
Pacientom, ktorí nereagujú na iniciálnu liečbu, sa Ilaris nemá opakovane podávať. U pacientov
s odpoveďou, u ktorých je potrebná opakovaná liečba, má byť interval pred podaním ďalšej dávky
Ilarisu najmenej 12 týždňov (pozri časť 5.2).
Osobitné skupiny pacientov
Pediatrická populácia
CAPS, TRAPS, HIDS/MKD a FMF
Bezpečnosť a účinnosť Ilarisu u pacientov s CAPS, TRAPS, HIDS/MKD a FMF vo veku menej ako
2 roky neboli stanovené. V súčasnosti dostupné údaje sú opísané v častiach 4.8, 5.1 a 5.2, ale
neumožňujú uviesť odporúčania na dávkovanie.
SJIA
Bezpečnosť a účinnosť Ilarisu u pacientov so SJIA vo veku menej ako 2 roky neboli stanovené. Nie je
možné uviesť odporúčania na dávkovanie.
Dnavá artritída
Použitie Ilarisu sa netýka pediatrickej populácie v indikácii dnavá artritída.
St arš í paci ent i
Nie je potrebná úprava dávky.
Porucha f unkc i e pe če ne
Ilaris sa nesledoval u pacientov s poruchou funkcie pečene.
Porucha f unkc i e obl i či ek
U pacientov s poruchou funkcie obličiek nie je potrebná úprava dávkovania. Klinické skúsenosti u takých pacientov sú však obmedzené.
Spôsob podávania
Na subkutánne použitie.
Nasledujúce miesta sú vhodné na podanie injekcie: horná časť stehna, brucho, horná časť ramena alebo sedacie svaly. Odporúča sa zvoliť pri každom podaní injekcie iné miesto vpichu, aby sa zabránilo bolesti. Je potrebné vyhnúť sa poranenej koži a miestam, v ktorých sú podliatiny alebo ktoré sú pokryté exantémom. Je potrebné vyhnúť sa podaniu injekcie do tkaniva jazvy, pretože to môže mať za následok nedostatočnú expozíciu Ilarisu.
Každá injekčná liekovka Ilarisu je určená na jednorazové použitie u jedného pacienta, a to na jednu dávku.
Pokyny o použití a manipulácii s rekonštituovaným roztokom, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívne, závažné infekcie (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Infekcie
Ilaris sa spája so zvýšenou incidenciou závažných infekcií. Preto je u pacientov potrebné dôsledne sledovať príznaky a prejavy infekcií počas liečby Ilarisom a po nej. Lekári majú byť opatrní pri podávaní Ilarisu pacientom s infekciami, opakovanými infekciami v anamnéze alebo základnými ochoreniami, ktoré ich môžu predisponovať na infekcie.
Liečba CAPS, TRAPS, HIDS/MKD, FMF a Stillovej choroby (AOSD a SJIA)
Liečba Ilarisom sa nemá začať alebo pokračovať u pacientov počas aktívnej infekcie, ktorá vyžaduje
zásah lekára.
Liečba dnavej artritídy
Ilaris sa nemá podávať počas aktívnej infekcie.
Súbežné použitie Ilarisu s inhibítormi faktora nekrotizujúceho nádory (TNF) sa neodporúča, pretože to môže zvyšovať riziko závažných infekcií (pozri časť 4.5).
Počas liečby Ilarisom boli hlásené ojedinelé prípady neobvyklých alebo oportúnnych infekcií (vrátane
aspergilózy, atypických mykobakteriálnych infekcií, herpes zoster). Príčinný vzťah medzi Ilarisom
a týmito udalosťami nemožno vylúčiť.
Asi u 12 % pacientov s CAPS testovaných v klinických skúšaniach kožným testom PPD (purifikovaným proteínovým derivátom) bol výsledok ďalšieho testovania počas liečby Ilarisom pozitívny bez klinických dôkazov latentnej alebo aktívnej infekcie tuberkulózou.
Nie je známe, či používanie inhibítorov interleukínu-1 (IL-1), ako je Ilaris, zvyšuje riziko reaktivácie tuberkulózy. Pred začatím liečby sa u všetkých pacientov musí vyšetriť prítomnosť aktívnej a latentnej infekcie tuberkulózou. Najmä u dospelých pacientov má toto vyšetrenie zahŕňať podrobnú anamnézu. Príslušné skríningové testy (napr. tuberkulínový kožný test, stanovenie uvoľňovania interferónu gama alebo röntgen hrudníka) je potrebné vykonať u všetkých pacientov (môžu platiť národné odporúčania). U pacientov sa musia dôsledne sledovať príznaky a prejavy tuberkulózy počas liečby Ilarisom a po nej. Všetkých pacientov je potrebné poučiť, aby sa poradili s lekárom, ak sa
počas liečby Ilarisom vyskytnú príznaky a prejavy svedčiace o tuberkulóze (napr. pretrvávajúci kašeľ, úbytok telesnej hmotnosti, subfebrilná teplota). V prípade zmeny z negatívneho na pozitívny test PPD, najmä u pacientov s vysokým rizikom, je potrebné zvážiť alternatívne spôsoby skríningu na infekciu tuberkulózou.
Neutropénia a leukopénia
Neutropénia (absolútny počet neutrofilov [absolute neutrophil count, ANC] < 1,5 x 109/l)
a leukopénia sa pozorovali pri liekoch, ktoré inhibujú IL-1, vrátane Ilarisu. Liečba Ilarisom sa nemá začať u pacientov s neutropéniou alebo leukopéniou. Odporúča sa stanoviť počet bielych krviniek (white blood cell, WBC) vrátane počtu neutrofilov pred začatím liečby a znovu po 1 až 2 mesiacoch. Pri chronickej alebo opakovanej liečbe sa odporúča stanoviť počet WBC tiež pravidelne počas liečby. Ak sa u pacienta objaví neutropénia alebo leukopénia, počet WBC sa má dôsledne sledovať a má sa zvážiť ukončenie liečby.
Malignity
U pacientov liečených Ilarisom boli hlásené prípady malignít. Riziko vzniku malignít pri liečbe anti- interleukínom (IL)-1 nie je známe.
Reakcie z precitlivenosti
Pri liečbe Ilarisom boli hlásené reakcie z precitlivenosti. Závažnosť väčšiny týchto udalostí bola mierna. Počas klinického vývoja Ilarisu u viac ako 2 600 pacientov neboli hlásené žiadne anafylaktoidné alebo anafylaktické reakcie. Avšak riziko závažných reakcií z precitlivenosti, ktoré nie je neobvyklé pri injekčne podávaných proteínoch, nemožno vylúčiť (pozri časť 4.3).
Funkciapečene
V klinických skúšaniach sa zaznamenali prechodné a asymptomatické prípady zvýšenia hladín
aminotransferáz alebo bilirubínu v sére (pozri časť 4.8).
V
akcinácie
Nie sú dostupné údaje o riziku sekundárneho prenosu infekcie živými (oslabenými) vakcínami
u pacientov, ktorí dostávajú Ilaris. Preto sa živé vakcíny nemajú podávať súčasne s Ilarisom, pokiaľ prínos nie je jednoznačne väčší ako riziká (pozri časť 4.5).
Odporúča sa, aby dospelí a pediatrickí pacienti dostali pred začatím liečby Ilarisom podľa potreby všetky vakcinácie, vrátane pneumokokovej vakcíny a inaktivovanej chrípkovej vakcíny (pozri
časť 4.5).
Mutácia génu NLRP3 u pacientov s CAPS
Klinické skúsenosti u pacientov s CAPS bez potvrdenej mutácie génu NLRP3 sú obmedzené.
Syndróm aktivácie makrofágov u pacientov so Stillovou chorobou
Syndróm aktivácie makrofágov (MAS) je známa, život ohrozujúca porucha, ktorá sa môže vyvinúť
u pacientov s reumatickými chorobami, najmä so Stillovou chorobou. Pri výskyte alebo podozrení na MAS sa má začať čo najskôr s hodnotením stavu a liečbou. Lekári si majú pozorne všímať prejavy infekcie alebo zhoršenie Stillovej choroby, pretože sú to známe spúšťače MAS. Na základe skúseností z klinických skúšaní sa nezdá, že by Ilaris zvyšoval incidenciu MAS u pacientov so SJIA, ale definitívny záver nie je možné urobiť.
4.5 Liekové a iné interakcie
Interakcie medzi Ilarisom a inými liekmi sa nesledovali vo formálnych štúdiách.
Zvýšená incidencia závažných infekcií bola spojená s podávaním iného blokátora IL-1 v kombinácii s inhibítormi TNF. Použitie Ilarisu s inhibítormi TNF sa neodporúča, pretože to môže zvýšiť riziko závažných infekcií.
Expresiu pečeňových enzýmov CYP450 môžu potlačiť cytokíny, ktoré stimulujú chronický zápal, napr. interleukín-1 beta (IL-1 beta). Expresia CYP450 sa preto môže zvrátiť, keď sa začne účinná liečba inhibítormi cytokínov, napr. kanakinumabom. Je to klinicky významné pri substrátoch CYP450 s úzkym terapeutickým indexom, ktorých dávka sa individuálne upravuje. Pri začatí liečby kanakinumabom u pacientov, ktorí dostávajú liek tohto typu, sa má sledovať účinok liečby alebo koncentrácia liečiva a individuálna dávka lieku sa má podľa potreby upraviť.
Nie sú dostupné údaje ani o účinkoch vakcinácie živou vakcínou, ani o sekundárnom prenose infekcie
živými vakcínami u pacientov, ktorí dostávajú Ilaris. Preto sa živé vakcíny nemajú podávať súčasne s Ilarisom, pokiaľ prínos nie je jednoznačne väčší ako riziká. Ak je po začatí liečby Ilarisom vakcinácia živými vakcínami potrebná, odporúča sa počkať aspoň 3 mesiace po poslednej injekcii Ilarisu a pred ďalšou injekciou (pozri časť 4.4).
Výsledky štúdie so zdravými dospelými ukázali, že jednorazová dávka 300 mg Ilarisu neovplyvnila vyvolanie a trvanie protilátkovej odpovede po podaní vakcíny proti chrípke alebo vakcíny proti meningokokom založenej na glykozylovaných proteínoch.
Výsledky otvorenej štúdie trvajúcej 56 týždňov u pacientov s CAPS vo veku 4 rokov a mladších ukázali, že u všetkých pacientov, ktorí dostali neživú vakcínu štandardne používanú u detí, sa vyvinuli ochranné hladiny protilátok.
4.6 Fertilia, gravidita a laktácia
Ž
eny
v
o
fertilnom veku / Antikoncepcia u mužov
a
žien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby Ilarisom a počas
3 mesiacov od poslednej dávky.
Gravidita
O použití kanakinumabu u gravidných žien je obmedzené množstvo údajov. Štúdie na zvieratách nenaznačujú priame alebo nepriame škodlivé účinky vzhľadom na reprodukčnú toxicitu (pozri časť 5.3). Riziko pre plod/matku nie je známe. Ženy, ktoré sú tehotné alebo chcú otehotnieť, preto majú byť liečené len po dôkladnom zhodnotení prínosu a rizika liečby.
Štúdie na zvieratách naznačujú, že kanakinumab prechádza placentou a je preukázateľný v plode. Nie sú k dispozícii žiadne údaje u ľudí, ale keďže je kanakinumab imunoglobulín triedy G (IgG1), predpokladá sa transplacentárny prenos u žien. Klinický dopad tohto zistenia nie je známy. Podávanie živých vakcín novorodencom, vystaveným kanakinumabu v maternici sa však neodporúča 16 týždňov od podania poslednej dávky Ilarisu matke pred pôrodom. Ženy, ktoré dostali kanakinumab počas gravidity, je potrebné poučiť, aby o tom informovali detského lekára pred akoukoľvek vakcináciou ich novorodenca.
Dojčenie
Nie je známe, či sa kanakinumab vylučuje do ľudského mlieka. Rozhodnutie, či dojčiť počas liečby
Ilarisom, sa preto má urobiť len po dôkladnom zhodnotení prínosu a rizika liečby.
Štúdie na zvieratách ukázali, že myšacia protilátka proti myšaciemu IL-1 beta nemala nežiaduce účinky na vývoj dojčených myšacích mláďat a že táto protilátka sa na ne preniesla (pozri časť 5.3).
Fertilita
Formálne štúdie možného účinku Ilarisu na ľudskú plodnosť sa nevykonali.
Kanakinumab nemal účinok na parametre plodnosti u samcov opíc kozmáčov (C. jacchus). Myšacia protilátka proti myšaciemu IL-1 beta nemala nežiaduce účinky na plodnosť samcov ani samíc myši (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ilaris má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Liečba Ilarisom môže vyvolať závraty/vertigo alebo asténiu (pozri časť 4.8). Pacienti, u ktorých sa počas liečby Ilarisom vyskytnú takéto symptómy, majú počkať do ich úplného vymiznutia, kým budú viesť vozidlo alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Viac ako 2 600 osôb, vrátane približne 480 detí (vo veku 2 až 17 rokov), bolo liečených Ilarisom v intervenčných klinických skúšaniach s pacientmi s CAPS, TRAPS, HIDS/MKD, FMF, SJIA, dnavou artritídou alebo s inými ochoreniami sprostredkovanými IL-1 beta, ako aj zdravých dobrovoľníkov. Pozorovali sa závažné infekcie. Najčastejšími nežiaducimi reakciami na liek boli infekcie predovšetkým horného dýchacieho traktu. Pri dlhodobejšej liečbe sa nezaznamenal žiadny vplyv na typ ani frekvenciu nežiaducich reakcií na liek.
U pacientov liečených Ilarisom boli hlásené reakcie z precitlivenosti (pozri časti 4.3 a 4.4). U pacientov liečených Ilarisom boli hlásené oportúnne infekcie (pozri časť 4.4).
C
APS
V klinických skúšaniach dostávalo Ilaris celkovo 211 dospelých a pediatrických pacientov s CAPS (vrátane FCAS/FCU, MWS a NOMID/CINCA). Bezpečnosť Ilarisu v porovnaní s placebom sa skúmala v pivotnom skúšaní fázy III, ktoré pozostávalo z 8-týždňového obdobia otvoreného podávania (časť I), 24-týždňového randomizovaného, dvojito zaslepeného a placebom kontrolovaného obdobia vysadenia lieku (časť II) a 16-týždňového obdobia otvoreného podávania Ilarisu (časť III). Všetci pacienti dostávali subkutánne Ilaris v dávke 150 mg alebo 2 mg/kg, ak ich telesná hmotnosť bola
≥ 15 kg a ≤ 40 kg.
TRAPS, HIDS/MKD, FMF
V jednom pivotnom klinickom skúšaní fázy III dostávalo Ilaris celkovo 169 dospelých a pediatrických pacientov s TRAPS, HIDS/MKD a FMF vo veku 2 roky a starších. Bezpečnosť Ilarisu v porovnaní
s placebom sa skúmala v tomto skúšaní, ktoré pozostávalo z 12-týždňového obdobia skríningu (časť I) a 16-týždňového randomizovaného, dvojito zaslepeného, placebom kontrolovaného obdobia liečby (časť II). Pacientom, ktorí dostávali Ilaris, sa podávalo subkutánne 150 mg, alebo 2 mg/kg, ak ich telesná hmotnosť bola ≤ 40 kg (pozri časť 5.1).
Stillova choroba
Celkovo 324 pacientov so SJIA vo veku 2 až < 20 rokov dostávalo Ilaris v klinických skúšaniach, vrátane 293 pacientov vo veku 2 až < 16 rokov, 21 pacientov vo veku 16 až < 18 rokov a 10 pacientov vo veku 18 až < 20 rokov. Bezpečnosť Ilarisu sa skúmala v porovnaní s placebom v dvoch pivotných skúšaniach fázy III (pozri časť 5.1).
Dnavá artritída
Viac ako 700 pacientov s dnavou artritídou bolo liečených Ilarisom v dávkach od 10 mg do 300 mg v randomizovaných, dvojito zaslepených a účinným liekom kontrolovaných klinických skúšaniach trvajúcich do 24 týždňov. Viac ako 250 pacientov dostávalo odporúčanú dávku 150 mg v klinických skúšaniach fázy II a III (pozri časť 5.1).
T
abuľkový
z
oznam
nežiaducich
reakcií
Nežiaduce reakcie sú usporiadané podľa tried orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa kategórie frekvencie, najčastejšie ako prvé. Kategórie frekvencie sú definované pomocou nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až< 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov).V rámci každej skupiny frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 1 Tabuľkový zoznam nežiaducich reakcií pri CAPS, TRAPS, HIDS/MKD, FMF, SJIA a dnavej artritíde
T
rieda
orgánových systémov MedDRA
Infekcie a nákazy
V
šetky indikácie:
CA
P
S, TRAPS, HIDS/MKD, FMF, SJIA, dnavá artritída
Veľmi časté Infekcie dýchacích ciest (vrátane pneumónie, bronchitídy, chrípky, vírusovej infekcie, sínusitídy, rinitídy, faryngitídy, tonzilitídy, nazofaryngitídy, infekcie horných dýchacích ciest)
Infekcia ucha Celulitída Gastroenteritída
Infekcia močových ciest Časté Vulvovaginálna kandidóza
Poruchy nervového systémuČasté Závraty/vertigo
Poruchy gastrointestinálneho traktuVeľmi časté Bolesť hornej časti brucha 1
Menej časté Gastroezofágová refluxná choroba 2
Poruchy kože a podkožného tkanivaVeľmi časté Reakcia v mieste vpichu
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté Artralgia 1
Časté Bolesť svalov a kostí 1
Bolesť chrbta 2
Celkové poruchy a reakcie v mieste podaniaČasté Únava/asténia 2
Laboratórne a funkčné vyšetreniaVeľmi časté Zníženie obličkového klírensu kreatinínu 1,3
Proteinúria 1,4
Leukopénia 1,5
Časté Neutropénia 5
Menej časté Pokles počtu trombocytov 5
1 Pri SJIA
2 Pri dnavej artritíde
3 Založené na odhadovanom klírense kreatinínu, väčšinou prechodné
4 Väčšinu predstavovali prechodné stopy až 1+ pozitivita bielkovín v moči, stanovené indikátorovým papierikom
5 Pozri ďalšie informácie nižšie
V podskupine mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov (N=31) bol profil
bezpečnosti Ilarisu v súlade s tým, ktorý sa pozoroval u pacientov so SJIA mladších ako 16 rokov. Na základe literárnych údajov sa očakáva podobný profil bezpečnosti u AOSD pacientov ako u pacientov so SJIA.
Popis
vy
braných
nežiaducich
reakcií
D
l
hodobé údaje a laboratórne abnormality u pacientov s CAPS
Počas klinických skúšaní Ilarisu u pacientov s CAPS sa priemerné hodnoty hemoglobínu zvýšili
a hodnoty leukocytov, neutrofilov a trombocytov znížili.
Zvýšenie hladín aminotransferáz sa u pacientov s CAPS pozorovalo zriedkavo.
U pacientov s CAPS liečených Ilarisom sa pozorovali asymptomatické a mierne zvýšenia bilirubínu
v sére, bez súčasného zvýšenia aminotransferáz.
V dlhodobých, otvorených klinických skúšaniach so zvyšovaním dávky boli ako udalosti častejšie hlásené infekcie (gastroenteritída, infekcia dýchacieho traktu, infekcia horného dýchacieho traktu), vracanie a závraty v skupine s dávkou 600 mg alebo 8 mg/kg ako v skupinách s inými dávkami.
Laboratórne abnormality u pacientov s TRAPS, HIDS/MKD a FMF Neutrofily
Hoci sa zníženie počtu neutrofilov ≥ stupňa 2 vyskytlo u 6,5 % pacientov (časté) a zníženie stupňa 1 sa vyskytlo u 9,5 % pacientov, zníženie je všeobecne prechodné a infekcia spojená s neutropéniou nebola identifikovaná ako nežiaduca reakcia.
Trombocyty
Hoci sa zníženie počtu trombocytov (≥ stupňa 2) vyskytlo u 0,6 % pacientov, krvácanie nebolo identifikované ako nežiaduca reakcia. Mierne a prechodné zníženie trombocytov stupňa 1 sa vyskytlo u 15,9 % pacientov bez akýchkoľvek súvisiacich krvácavých nežiaducich príhod.
Laboratórne abnormality u pacientov so SJIA Hematológia
V celkovom programe SJIA bol prechodný pokles počtu bielych krviniek (WBC) ≤ 0,8 x LLN hlásený u 33 pacientov (16,5 %).
V celkovom programe SJIA bol prechodný pokles absolútneho počtu neutrofilov (ANC) na menej ako
1 x 109/l hlásený u 12 pacientov (6,0 %).
V celkovom programe SJIA sa prechodný pokles trombocytov (< LLN) pozoroval u 19 pacientov
(9,5 %).
ALT/AST
V celkovom programe SJIA boli vysoké hodnoty ALT a/alebo AST > 3 x hornej hranice normálneho rozmedzia (ULN) hlásené u 19 pacientov (9,5 %).
Laboratórne abnormality u pacientov s dnavou artritídou
Hematológia
Znížený počet leukocytov (white blood cell counts, WBC) ≤ 0,8 x dolná hranica normálu (lower limit of normal, LLN) bol hlásený u 6,7 % pacientov liečených Ilarisom v porovnaní s 1,4 % pacientov liečených triamcinolónacetonidom. V porovnávacích klinických skúšaniach bol u 2 % pacientov hlásený pokles absolútneho počtu neutrofilov (ANC) na menej ako 1 x 109/l. Pozorovali sa aj ojedinelé prípady ANC < 0,5 x 109/l (pozri časť 4.4).
V klinických skúšaniach kontrolovaných účinným liekom sa u pacientov s dnavou artritídou pozorovalo mierne (< LLN a > 75 x 109/l) a prechodné zníženie počtu trombocytov s vyššou incidenciou (12,7 %) pri Ilarise oproti komparátoru (7,7 %).
K
yselina močová
V porovnávacích klinických skúšaniach pri dnavej artritíde sa po liečbe Ilarisom pozorovalo zvýšenie koncentrácie kyseliny močovej (0,7 mg/dl po 12 týždňoch a 0,5 mg/dl po 24 týždňoch). V inom klinickom skúšaní sa nepozorovalo zvýšenie kyseliny močovej u pacientov, ktorí začínali ULT. Zvýšenie kyseliny močovej sa nepozorovalo v klinických skúšaniach u populácií bez dnavej artritídy (pozri časť 5.1).
ALT/ASTZvýšenie priemeru o 3,0 U/l a mediánu o 2,0 U/l pri alanínaminotransferáze (ALT) a o 2,7 U/l
a o 2,0 U/l pri aspartátaminotransferáze (AST) sa pozorovalo oproti východiskovým hodnotám do konca klinického skúšania v skupinách liečených Ilarisom oproti skupinám liečeným triamcinolónacetonidom, avšak incidencia klinicky významných zmien (≥ 3 x horná hranica normálu) bola vyššia u pacientov liečených triamcinolónacetonidom (2,5 % pri AST aj ALT) v porovnaní s pacientmi liečenými Ilarisom (1,6 % pri ALT a 0,8 % pri AST).
TriacylglycerolyV klinických skúšaniach kontrolovaných účinným liekom pri dnavej artritíde sa triacylglyceroly zvýšili v priemere o 33,5 mg/dl u pacientov liečených Ilarisom v porovnaní s miernym poklesom o -3,1 mg/dl pri triamcinolónacetonide. Incidencia pacientov so zvýšením triacylglycerolov > 5 x horná hranica normálu (ULN) bola 2,4 % pri Ilarise a 0,7 % pri triamcinolónacetonide. Klinická významnosť tohto pozorovania nie je známa.
Pediatrická populáciaV štúdiách bolo 80 pediatrických pacientov s CAPS (vo veku 2–17 rokov), ktorí dostávali kanakinumab. Celkovo sa nevyskytli žiadne klinicky významné rozdiely v profiloch bezpečnosti a znášanlivosti Ilarisu u pediatrických pacientov v porovnaní s celkovou populáciou s CAPS
(tvorenou dospelými aj pediatrickými pacientmi, N=211), vrátane celkovej frekvencie a závažnosti
epizód infekcií. Infekcie horného dýchacieho traktu boli najčastejšie hlásenými udalosťami infekcií.
Okrem toho sa 6 pediatrických pacientov mladších ako 2 roky vyhodnotilo v malej, otvorenej klinickej štúdii. Ukázalo sa, že profil bezpečnosti Ilarisu je podobný ako u pacientov vo veku 2 rokov a starších.
V 16-týždňovej štúdii bolo 102 pacientov s TRAPS, HIDS/MKD a FMF (vo veku 2–17 rokov), ktorí dostávali kanakinumab. Celkovo sa nevyskytli žiadne klinicky významné rozdiely v profiloch bezpečnosti a znášanlivosti kanakinumabu u pediatrických pacientov v porovnaní s celkovou populáciou.
StaršiapopuláciaV profile bezpečnosti pozorovanom u pacientov vo veku ≥ 65 rokov nie je významný rozdiel.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieZaznamenané skúsenosti s predávkovaním sú obmedzené. V počiatočných klinických skúšaniach dostali pacienti a zdraví dobrovoľníci intravenózne alebo subkutánne podané dávky až 10 mg/kg bez preukázanej akútnej toxicity.
V prípade predávkovania sa odporúča u pacienta sledovať akékoľvek príznaky a prejavy nežiaducich
reakcií a okamžite začať náležitú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu, ATC kód: L04AC08
Mechanizmus účinku
Kanakinumab je ľudská monoklonálna protilátka proti ľudskému interleukínu-1 beta (IL-1 beta) izotypu IgG1/κ. Kanakinumab sa s vysokou afinitou viaže špecificky na ľudský IL-1 beta a neutralizuje biologickú aktivitu ľudského IL-1 beta blokovaním jeho interakcie s receptormi IL-1, čím bráni aktivácii génu vyvolanej IL-1 beta a tvorbe zápalových mediátorov.
Farmakodynamickéúčinky
CAPS, TRAPS, HIDS/MKD a FMF
V klinických štúdiách pacienti s CAPS, TRAPS, HIDS/MKD a FMF, ktorí majú nekontrolovanú nadmernú tvorbu IL-1 beta, vykazujú rýchlu a pretrvávajúcu odpoveď na liečbu kanakinumabom, t.j. laboratórne parametre, ako vysoké hladiny C-reaktívneho proteínu (CRP) a sérového amyloidu A (SAA), vysoký počet neutrofilov a trombocytov a leukocytóza, sa rýchlo vrátili na normálne hodnoty.
Stillova choroba (AOSD a SJIA)
Stillova choroba s nástupom ochorenia v dospelosti (AOSD) a systémová juvenilná idiopatická artritída sú závažné autoinflamačné choroby spôsobené vrodenou imunitou a sprostredkovaná prozápalovými cytokínmi, z ktorých kľúčový je IL-1-beta.
Časté príznaky AOSD a SJIA sú horúčka, exantém, hepatosplenomegália, lymfadenopatia, polyserozitída a artritída. Liečba kanakinumabom spôsobila rýchle a pretrvávajúce zmiernenie kĺbových aj systémových príznakov SJIA s významným poklesom počtu zapálených kĺbov, rýchlym odznením horúčky a znížením reaktantov akútnej fázy u väčšiny pacientov (pozri časť Klinická účinnosť a bezpečnosť).
Dnavá artritída
Záchvat dnavej artritídy je spôsobený kryštálmi urátu (monohydrát mononátriumurátu) v kĺbe a v okolitých tkanivách, čo spúšťa tvorbu IL-1 beta rezidentnými makrofágmi prostredníctvom
„inflamazómového komplexu NALP3“. Aktivácia makrofágov a súčasná nadmerná tvorba IL-1 beta vyvolávajú akútnu bolestivú zápalovú reakciu. Iné aktivátory vrodeného imunitného systému, ako sú endogénne agonisty „toll-like“ receptorov, môžu prispieť k transkripčnej aktivácii génu IL-1 beta a vyvolať záchvat dnavej artritídy. Po liečbe kanakinumabom rýchlo poklesli zápalové markery CRP alebo SAA a ustúpili prejavy akútneho zápalu (napr. bolesť, opuch, sčervenanie) v postihnutom kĺbe.
Klinickáúčinnosťabezpečnosť
CAPS
Účinnosť a bezpečnosť Ilarisu sa preukázali u pacientov s rôznymi stupňami závažnosti ochorenia a rôznymi fenotypmi CAPS (vrátane FCAS/FCU, MWS a NOMID/CINCA). Do hlavnej štúdie boli zaradení len pacienti s potvrdenou mutáciou NLRP3.
V štúdii fázy I/II mala liečba Ilarisom rýchly nástup účinku, s vymiznutím alebo klinicky významným zlepšením symptómov do jedného dňa po podaní. Laboratórne parametre, ako vysoké hladiny CRP a SAA, vysoký počet neutrofilov a trombocytov, sa normalizovali rýchlo, počas niekoľkých dní po injekcii Ilarisu.
Hlavná štúdia pozostávala zo 48-týždňovej multicentrickej štúdie zloženej z troch častí, t.j. 8- týždňového otvoreného obdobia (časť I), 24-týždňového randomizovaného, dvojito zaslepeného a placebom kontrolovaného obdobia vysadenia lieku (časť II), po ktorom nasledovalo 16-týždňové otvorené obdobie (časť III). Cieľom tejto štúdie bolo hodnotenie účinnosti, bezpečnosti a znášanlivosti Ilarisu (150 mg alebo 2 mg/kg každých 8 týždňov) u pacientov s CAPS.
- Časť I: Úplná klinická odpoveď a odpoveď biomarkerov na Ilaris (definovaná ako súhrn celkového hodnotenia autoinflamačného a kožného ochorenia lekárom ≤ minimálne a hodnôt CRP alebo SAA < 10 mg/l) sa pozorovala u 97 % pacientov a objavila sa do 7 dní od začatia liečby. V klinickom hodnotení aktivity autoinflamačného ochorenia lekárom sa pozorovali významné zlepšenia: pri celkovom hodnotení aktivity autoinflamačného ochorenia, hodnotení kožného ochorenia (kožný exantém typu urtikárie), artralgie, myalgie, bolesti hlavy/migrény, konjunktivitídy, únavy/pocitu nepohody, hodnotení iných súvisiacich symptómov a hodnotení symptómov pacientom.
- Časť II: V období vysadenia lieku počas hlavnej štúdie bol primárny koncový parameter definovaný ako podiel pacientov s relapsom/vzplanutím ochorenia: žiadny (0 %) z pacientov randomizovaných do skupiny Ilarisu nemal vzplanutie ochorenia, v porovnaní s 81 % pacientov randomizovaných do skupiny placeba.
- Časť III: Pacienti, ktorí v časti II dostávali placebo a u ktorých došlo k vzplanutiu ochorenia, po zaradení do otvorenej extenzie štúdie Ilarisu opäť dosiahli a udržali si klinickú a sérologickú odpoveď.
Tabuľka 2 Tabuľkové zhrnutie účinnosti v klinickom skúšaní fázy III, hlavné obdobie
vysadenia lieku kontrolované placebom (časť II)
Klinické skúšanie fázy III, hlavné obdobie vysadenia lieku kontrolované placebom (časť II)
Ilaris N=15 n(%)
Placebo N=16 n(%)
Hodnota p
P
rimárny parameter (vzplanutie ochorenia)
Podiel pacientov so vzplanutím ochorenia v
časti II
0 (0 %) 13 (81 %) < 0,001
Z
ápalové markery*
C-reaktívny proteín, mg/l 1,10 (0,40) 19,93 (10,50) < 0,001
Sérový amyloid A, mg/l 2,27 (-0,20) 71,09 (14,35) 0,002
* priemerná (medián) zmena od začiatku časti II
Vykonali sa dve otvorené, nekontrolované dlhodobé klinické skúšania fázy III. Jedno z nich bolo
skúšanie bezpečnosti, znášanlivosti a účinnosti kanakinumabu u pacientov s CAPS. Celkové trvanie liečby bolo v rozmedzí od 6 mesiacov do 2 rokov. Druhé bolo otvorené skúšanie kanakinumabu, ktoré hodnotilo účinnosť a bezpečnosť u japonských pacientov s CAPS počas 24 týždňov, s fázou extenzie až do 48 týždňov. Primárnym cieľom bolo stanovenie podielu pacientov, ktorí boli bez relapsov po
24. týždni, vrátane tých pacientov, ktorým sa dávka zvýšila.
Podľa združenej analýzy účinnosti týchto dvoch skúšaní dosiahlo 65,6 % pacientov, ktorí sa predtým neliečili kanakinumabom, úplnú odpoveď pri dávke 150 mg alebo 2 mg/kg, zatiaľ čo 85,2 % pacientov dosiahlo úplnú odpoveď pri akejkoľvek dávke. Z pacientov, ktorí dostávali 600 mg alebo
8 mg/kg (alebo ešte vyššiu dávku), 43,8 % dosiahlo úplnú odpoveď. Menej pacientov vo veku 2 až
< 4 roky dosiahlo úplnú odpoveď (57,1 %) v porovnaní so staršími pediatrickými a dospelými pacientmi. Z pacientov, ktorí dosiahli úplnú odpoveď, si 89,3 % odpoveď udržalo bez relapsov.
Skúsenosti u jednotlivých pacientov, ktorí dosiahli úplnú odpoveď po zvýšení dávky na 600 mg
(8 mg/kg) každých 8 týždňov, naznačujú, že vyššia dávka môže byť prínosom pre pacientov, ktorí nedosiahnu úplnú odpoveď alebo si neudržia úplnú odpoveď pri odporúčaných dávkach (150 mg alebo 2 mg/kg u pacientov ≥ 15 kg a ≤ 40 kg). Zvýšená dávka sa častejšie podávala pacientom vo veku 2 až < 4 roky a pacientom so symptómami NOMID/CINCA v porovnaní s pacientmi s FCAS alebo MWS.
Pediatrická populácia
Do klinických skúšaní Ilarisu pri CAPS bolo zaradených celkovo 80 pediatrických pacientov vo vekovom rozmedzí od 2 do 17 rokov (približne polovica z nich bola liečená na základe mg/kg). Celkovo sa nezistili žiadne klinicky významné rozdiely v účinnosti, bezpečnosti a znášanlivosti Ilarisu u detí a dospievajúcich v porovnaní s celkovou populáciou s CAPS. Väčšina pediatrických pacientov dosiahla zlepšenie v klinických príznakoch a v objektívnych zápalových markeroch (napr. SAA
a CRP).
Na vyhodnotenie účinnosti, bezpečnosti a znášanlivosti Ilarisu u pediatrických pacientov s CAPS vo
veku ≤ 4 roky sa vykonala otvorená štúdia trvajúca 56 týždňov. Vyhodnotilo sa 17 pacientov (vrátane
6 pacientov vo veku menej ako 2 roky), ktorí v závislosti od telesnej hmotnosti dostali začiatočné dávky 2-8 mg/kg. Štúdia tiež vyhodnotila účinok kanakinumabu na vznik protilátok proti vakcínam štandardne podávaným deťom. Nepozorovali sa rozdiely v bezpečnosti alebo účinnosti u pacientov mladších ako 2 roky v porovnaní s pacientmi vo veku 2 roky a staršími. U všetkých pacientov, ktorí dostali neživú vakcínu štandardne používanú u detí (N=7), sa vyvinuli ochranné hladiny protilátok.
TRAPS, HIDS/MKD a FMF
Účinnosť a bezpečnosť Ilarisu v liečbe TRAPS, HIDS/MKD a FMF sa preukázali v jednej pivotnej
štúdii fázy III so štyrmi časťami (N2301), ktorá pozostávala z troch samostatných skupín chorôb.
- Časť I: Pacienti v každej skupine chorôb vo veku 2 roky a starší sa zúčastnili 12-týždňového obdobia skríningu, počas ktorého sa u nich hodnotil nástup vzplanutia ochorenia.
- Časť II: Pacienti boli pri nástupe vzplanutia randomizovaní do 16-týždňového, dvojito zaslepeného, placebom kontrolovaného obdobia liečby, počas ktorého dostávali buď 150 mg Ilarisu (2 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) subkutánne (s.c.), alebo placebo každé 4 týždne. Pacientom vo veku > 28 dní, ale < 2 roky bolo povolené vstúpiť do štúdie priamo do skupiny otvorenej liečby v časti II ako nerandomizovaní pacienti (a boli vylúčení
z primárnej analýzy účinnosti).
- Časť III: Pacienti, ktorí ukončili 16 týždňov liečby a boli klasifikovaní ako pacienti odpovedajúci na liečbu, boli opäť randomizovaní do 24-týždňového, dvojito zaslepeného obdobia vysadenia lieku, počas ktorého dostávali 150 mg Ilarisu (2 mg/kg u pacientov
s telesnou hmotnosťou ≤ 40 kg) s.c., alebo placebo každých 8 týždňov.
- Časť IV: Všetci pacienti z časti III liečení Ilarisom boli vhodní na účasť v 72-týždňovom období predĺženia otvorenej liečby.
Celkovo 185 pacientov vo veku 28 dní a starších bolo zaradených a celkovo 181 pacientov vo veku
2 roky a starších bolo randomizovaných do časti II tejto štúdie.
Primárnym parametrom účinnosti randomizovaného obdobia liečby (časť II) bol podiel pacientov
s odpoveďou na liečbu v rámci každej skupiny, u ktorých došlo k poklesu indexu vzplanutia ochorenia na 15. deň a u ktorých sa nevyskytlo nové vzplanutie počas zostávajúceho 16-týždňového obdobia liečby (definované ako úplná odpoveď). Pokles indexu vzplanutia ochorenia bolo definovaný ako
skóre aktivity ochorenia < 2 („minimálne alebo žiadne ochorenie“) podľa globálneho hodnotenia
lekárom (Physician´s Global Assessment, PGA) a CRP v rozmedzí normy (≤ 10 mg/l) alebo zníženie
≥ 70 % oproti východiskovej hodnote. Nové vzplanutie bolo definované ako skóre ≥ 2 („mierne, stredne ťažké alebo ťažké ochorenie“) podľa PGA a CRP ≥ 30 mg/ml. Sekundárne parametre, všetky založené na výsledkoch v 16. týždni (koniec časti II), zahŕňali podiel pacientov, ktorí dosiahli skóre
< 2 podľa PGA, podiel pacientov so sérologickou remisiou (definovaná ako CRP ≤ 10 mg/ml)
a podiel pacientov s normalizovanou hladinou SAA (definovaná ako SAA ≤ 10 mg/ml).
Z hľadiska primárneho parametra účinnosti bol Ilaris účinnejší ako placebo vo všetkých troch skupinách chorôb. Ilaris tiež preukázal vyššiu účinnosť v porovnaní s placebom pre sekundárne parametre PGA < 2 a CRP ≤ 10 mg/ml vo všetkých troch skupinách. Vyššie podiely pacientov mali normalizovanú SAA (≤ 10 mg/ml) v 16. týždni liečby Ilarisom v porovnaní s placebom vo všetkých troch skupinách, so štatisticky významným rozdielom pozorovaným u pacientov s TRAPS (pozri nižšie Tabuľku 3 s výsledkami štúdie).
Tabuľka 3 Tabuľkové zhrnutie účinnosti v klinickom skúšaní fázy III, pivotné,
randomizované, placebom kontrolované obdobie liečby (časť II)
Klinické skúšanie fázy III, pivotné, randomizované placebom kontrolované obdobie liečby
(časť II)
Ilaris n/N (%)
Placebo
n/N (%) Hodnota p
P
rimárny parameter (vzplanutie ochorenia) – podiel pacientov, u ktorých došlo k poklesu indexu vzplanutia ochorenia na 15. deň a u ktorých sa nevyskytlo nové vzplanutie počas zostávajúceho 16- týždňového obdobia liečby
FMF 19/31 (61,29) 2/32 (6,25) < 0,0001* HIDS/MKD 13/37 (35,14) 2/35 (5,71) 0,0020* TRAPS 10/22 (45,45) 2/24 (8,33) 0,0050*
Sekundárne parametre (ochorenie a zápalové markery)Globálne hodnotenie lekárom < 2
FMF 20/31 (64,52) 3/32 (9,38) < 0,0001** HIDS/MKD 17/37 (45,95) 2/35 (5,71) 0,0006** TRAPS 10/22 (45,45) 1/24 (4,17) 0,0028**
C-reaktívny proteín ≤ 10 mg/l
FMF 21/31 (67,74) 2/32 (6,25) < 0,0001** HIDS/MKD 15/37 (40,54) 2/35 (5,71) 0,0010** TRAPS 8/22 (36,36) 2/24 (8,33) 0,0149**
Sérový amyloid A ≤ 10 mg/l
FMF 8/31 (25,81) 0/32 (0,00) 0,0286
HIDS/MKD 5/37 (13,51) 1/35 (2,86) 0,0778
TRAPS 6/22 (27,27) 0/24 (0,00) 0,0235**
n=počet pacientov s odpoveďou na liečbu; N=počet vyhodnotiteľných pacientov
* indikuje štatistickú významnosť (jednostranná) s hladinou 0,025 na základe Fisherovho presného testu
** indikuje štatistickú významnosť (jednostranná) s hladinou 0,025 na základe modelu logistickej regresie so skupinou liečby a východiskovými hodnotami PGA, CRP alebo SAA ako vysvetľujúce premenné pre každú skupinu
Titrácia nahorV časti II štúdie dostali pacienti liečení Ilarisom, ktorí mali pretrvávajúcu aktivitu ochorenia, ďalšiu dávku 150 mg (alebo 2 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) počas prvého mesiaca. Táto ďalšia dávka sa mohla podať už 7 dní po prvej liečebnej dávke. Všetci pacienti s dávkou
titrovanou nahor zostali pri zvýšenej dávke 300 mg (alebo 4 mg/kg u pacientov s telesnou hmotnosťou
≤ 40 kg) každé 4 týždne.
V exploračnej analýze primárneho parametra sa pozorovalo, že u pacientov s nedostatočnou odpoveďou na liečbu po prvej dávke titrácia nahor počas prvého mesiaca na dávku 300 mg (alebo
4 mg/kg) každé 4 týždne ďalej zlepšila kontrolu vzplanutia, znížila aktivitu ochorenia a normalizovala hladiny CRP a SAA.
Pediatrickí pacienti:
Dvaja nerandomizovaní pacienti s HIDS/MKD vo veku > 28 dní, ale < 2 roky, boli zaradení do štúdie a dostávali kanakinumab. Jeden pacient mal pokles indexu vzplanutia na 15. deň po podaní jednej jednorazovej dávky kanakinumabu 2 mg/kg, ale liečbu prerušil po tejto prvej dávke pre závažné nežiaduce udalosti (pancytopénia a zlyhanie pečene). Tento pacient mal pri vstupe do štúdie imunitnú trombocytopenickú purpuru v anamnéze a aktívne ochorenie s abnormálnou funkciou pečene. Druhý pacient dostal začiatočnú dávku kanakinumabu 2 mg/kg a doplnkovú dávku 2 mg/kg v 3. týždni,
s titráciou dávky nahor v 5. týždni na podávanie 4 mg/kg každé 4 týždne až do konca časti II štúdie. Vymiznutie vzplanutia ochorenia sa dosiahlo v 5. týždni a u pacienta sa nevyskytlo žiadne nové vzplanutie na konci časti II štúdie (16. týždeň).
Stillova choroba
SJIA
Účinnosť Ilarisu v liečbe aktívnej SJIA sa vyhodnotila v dvoch pivotných klinických skúšaniach (G2305 a G2301). Pacienti zaradení do skúšaní boli vo veku 2 až < 20 rokov (priemerný vek 8,5 roka a priemerné trvanie choroby v čase zaradenia do skúšania 3,5 roka) a mali aktívnu chorobu definovanú ako ≥ 2 kĺby s aktívnou artritídou, horúčkou a zvýšeným CRP.
Klinické skúšanie G2305
G2305 bolo randomizované, dvojito zaslepené, placebom kontrolované, 4 týždne trvajúce skúšanie na stanovenie krátkodobej účinnosti Ilarisu u 84 pacientov randomizovaných na jednorazovú dávku
4 mg/kg (do 300 mg) Ilarisu alebo na placebo. Primárnym ukazovateľom bol podiel pacientov, u ktorých sa na 15. deň dosiahlo zlepšenie minimálne o 30 % v zmysle kritérií odpovede podľa pediatrickej American College of Rheumatology (ACR) upravených tak, aby zahŕňali neprítomnosť horúčky. Liečba Ilarisom zlepšila všetky skóre odpovede podľa pediatrickej ACR v porovnaní
s placebom na 15. aj 29. deň (Tabuľka 4).
Tabuľka 4 Odpoveď podľa pediatrickej ACR a stav choroby na 15. a 29. deň
15. deň 29. deň
Ilaris
N=43
Placebo
N=41
Ilaris
N=43
Placebo
N=41
ACR30 84 % 10 % 81 % 10 % ACR50 67 % 5 % 79 % 5 % ACR70 61 % 2 % 67 % 2 % ACR90 42 % 0 % 47 % 2 % ACR100 33 % 0 % 33 % 2 % Neaktívna choroba 33 % 0 % 30 % 0 % Rozdiel v liečbe pri všetkých skóre ACR bol významný (p ≤ 0,0001)
Výsledky zložiek upravenej odpovede podľa pediatrickej ACR, ktoré zahŕňali systémové a artritické
zložky, boli v súlade s celkovými výsledkami odpovede podľa ACR. Na 15. deň bol medián zmien oproti východiskovej hodnote v počte kĺbov s aktívnou artritítou -67 % a s obmedzeným rozsahom pohybu -73 % pri Ilarise (N=43) v porovnaní s mediánom zmien pri placebe 0 % a 0 % (N=41). Priemerná zmena v skóre bolesti podľa pacienta (vizuálna analogická škála 0-100 mm) na 15. deň bola pri Ilarise -50,0 mm (N=43) v porovnaní s placebom +4,5 mm (N=25). Priemerná zmena v skóre bolesti u pacientov liečených Ilarisom pretrvávala na 29. deň.
Klinické skúšanie G2301G2301 bolo randomizované, dvojito zaslepené, placebom kontrolované skúšanie Ilarisu na prevenciu vzplanutí pri vysadení liečby. Skúšanie pozostávalo z dvoch častí s dvoma nezávislými primárnymi ukazovateľmi (úspešné zníženie dávky steroidov a čas do vzplanutia). Do časti I (otvorená liečba) bolo zaradených 177 pacientov, ktorí dostávali 4 mg/kg (do 300 mg) Ilarisu každé 4 týždne počas najviac 32 týždňov. Pacienti v časti II (dvojito zaslepená liečba) dostávali buď 4 mg/kg Ilarisu, alebo placebo každé 4 týždne, až do výskytu 37 udalostí vzplanutia.
Znižovanie dávky kortikosteroidov:
Z celkového počtu 128 pacientov, ktorí pri vstupe do časti I užívali kortikosteroidy, sa 92 pokúsilo o zníženie dávky kortikosteroidov. Päťdesiatsedem (62 %) z 92 pacientov, ktorí sa pokúsili znížiť svoju dávku, bolo pri znížení dávky kortikosteroidov úspešných a 42 (46 %) kortikosteroidy vysadilo.
Čas do vzplanutia:
Pacienti, ktorí používali Ilaris v časti II, mali riziko udalosti vzplanutia znížené o 64 % v porovnaní so skupinou placeba (pomer rizika 0,36; 95 % IS: 0,17 až 0,75; p=0,0032). U 63 zo 100 pacientov, ktorí vstúpili do časti II, bez ohľadu na to, či boli pridelení na placebo alebo kanakinumab, sa nevyskytlo vzplanutie počas sledovaného obdobia (najviac 80 týždňov).
Výsledky klinických skúšaní G2305 a G2301 týkajúce sa zdravia a kvality života
Liečba Ilarisom vyvolala klinicky významné zlepšenie telesných funkcií a kvality života pacientov. V skúšaní G2305 bolo zlepšenie podľa dotazníka na hodnotenie zdravia v detskom veku (Childhood Health Assessment Questionnaire) metódou najmenších štvorcov pri Ilarise 0,69 oproti placebu, čo predstavuje 3,6-násobok minimálneho klinicky významného rozdielu 0,19 (p=0,0002). Medián zlepšenia oproti východiskovej hodnote do konca časti I skúšania G2301 bol 0,88 (79 %). Štatisticky významné zlepšenie skóre v dotazníku zdravia dieťaťa (Child Health Questionnaire-PF50) pri Ilarise v porovnaní s placebom bolo hlásené v skúšaní G2305 (telesné p=0,0012; psychosociálne zdravie p=0,0017).
Združená analýza účinnosti
Údaje z prvých 12 týždňov liečby Ilarisom v skúšaniach G2305, G2301 a v extenzii klinického skúšania sa združili na vyhodnotenie zachovania účinnosti. Tieto údaje preukázali do 12. týždňa oproti východiskovým hodnotám podobné zlepšenie podľa upravených odpovedí pediatrickej ACR a ich zložiek, aké sa pozorovalo v skúšaní kontrolovanom placebom (G2305). Po 12. týždni boli upravené odpovede podľa pediatrickej ACR30, 50, 70, 90 a 100: 70 %, 69 %, 61 %, 49 % a 30 %, choroba bola neaktívna u 28 % pacientov (N=178).
Účinnosť pozorovaná v skúšaniach G2305 a G2301 sa udržala počas pokračujúcej otvorenej, dlhodobej extenzie (údaje sú dostupné pre medián 49 týždňov následného sledovania). V tomto skúšaní sa u 25 pacientov, ktorí mali silnú odpoveď na liečbu podľa ACR počas najmenej 5 mesiacov, znížila dávka Ilarisu na 2 mg/kg každé 4 týždne, pričom odpoveď si títo pacienti udržali podľa pediatrickej ACR100 počas celého obdobia, keď sa im podávala znížená dávka (medián 32 týždňov,
8-124 týždňov).
Hoci sú údaje obmedzené, dôkazy z klinických skúšaní naznačujú, že pacienti, ktorí nereagujú na
tocilizumab alebo anakinru, môžu reagovať na kanakinumab.
SJIA u mladých dospelých a AOSD
Účinnosť Ilarisu v podskupine mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov bola v súlade s účinnosťou, pozorovanou u pacientov mladších ako 16 rokov. Na základe literárnych údajov sa očakáva podobný profil účinnosti u AOSD pacientov ako u pacientov so SJIA.
Dnavá artritída
Účinnosť Ilarisu v liečbe akútnych záchvatov dnavej artritídy sa preukázala v dvoch multicentrických, randomizovaných, dvojito zaslepených klinických skúšaniach kontrolovaných účinným liekom u pacientov s častou dnavou artritídou (≥ 3 záchvaty počas predošlých 12 mesiacov), ktorí nemohli používať NSAID alebo kolchicín (pre kontraindikáciu, neznášanlivosť alebo nedostatočnú účinnosť). Klinické skúšania trvali 12 týždňov, po ktorých nasledovala 12-týždňová dvojito zaslepená extenzia. Celkovo 225 pacientov bolo liečených subkutánne podaným Ilarisom 150 mg a 229 pacientov bolo liečených intramuskulárne podaným triamcinolónacetonidom (TA) 40 mg pri zaradení do klinického skúšania a neskôr pri výskyte nového záchvatu. Priemerný počet záchvatov dnavej artritídy počas predošlých 12 mesiacov bol 6,5. Viac ako 85 % pacientov malo sprievodné choroby vrátane hypertenzie (60 %), diabetu (15 %), ischemickej choroby srdca (12 %) a chronickej choroby obličiek
v štádiu ≥ 3 (25 %). Približne u jednej tretiny zaradených pacientov (76 [33,8 %] v skupine Ilarisu a
84 [36,7 %] v skupine triamcinolónacetonidu) bolo zdokumentované, že nemôžu (pre intoleranciu, kontraindikáciu alebo nedostatočnú odpoveď) používať NSAID ani kolchicín. Súbežnú liečbu ULT hlásilo 42 % pacientov pri zaradení do klinického skúšania.
Súbežné primárne parametre boli: (i) intenzita bolesti pri dnavej artritíde (vizuálna analógová škála,
VAS) 72 hodín po podaní, a (ii) čas do prvého nového záchvatu dnavej artritídy.
V celkovej populácii v klinickom skúšaní bola intenzita bolesti po 72 hodinách štatisticky významne nižšia pri Ilarise 150 mg v porovnaní s triamcinolónacetonidom. Ilaris tiež znížil riziko ďalších záchvatov (pozri Tabuľku 5).
Výsledky účinnosti u pacientov, ktorí nemohli užívať NSAID ani kolchicín a ktorí dostávali ULT,
u ktorých ULT nebola účinná alebo u ktorých bola ULT kontraindikovaná (N=101), sa zhodovali s výsledkami celkovej populácie v klinickom skúšaní, so štatisticky významným rozdielom oproti triamcinolónacetonidu pri intenzite bolesti po 72 hodinách (-10,2 mm, p=0,0208) a pri znížení rizika ďalších záchvatov (pomer rizika 0,39, p=0,0047 po 24 týždňoch).
Výsledky účinnosti v užšej podskupine obmedzenej na súčasných používateľov ULT (N=62) sú uvedené v Tabuľke 5. Liečba Ilarisom vyvolala zmiernenie bolesti a znížila riziko ďalších záchvatov u pacientov, ktorí používali ULT a nemohli používať NSAID ani kolchicín, hoci rozdiel pozorovaný pri liečbe v porovnaní s triamcinolónacetonidom bol menej výrazný ako v celkovej populácii v klinickom skúšaní.
Tabuľka 5 Účinnosť u celkovej populácie v klinickom skúšaní a u podskupiny pacientov v súčasnosti používajúcich ULT, ktorí nemôžu používať NSAID ani kolchicín
P
arameter účinnosti Celková populácia
v klinickom skúšaní; N=454
N
emožnosť používať NSAID
ani kolchicín; používanie
ULT
N=62
Liečbazáchvatovdnavej
artritídy
m
eraná ako intenzita bolesti (VAS) po 72 hodinách
Priemerný rozdiel oproti triamcinolónacetonidu odhadovaný metódou najmenších štvorcov
IS
Hodnota p, 1-stranná
-10,7
(-15,4, -6,0)
p < 0,0001*
-3,8
(-16,7, 9,1)
p=0,2798
Z
níženie rizika ďalších záchvatov dnavej artritídy merané ako čas do prvého nového vzplanutia
(
24 týždňov)
Pomer rizika oproti triamcinolón- acetonidu
IS
Hodnota p, 1-stranná
* Označuje významnú hodnotu p < 0,025
0,44
(0,32, 0,60)
p < 0,0001*
0,71
(0,29, 1,77)
p=0,2337
Výsledky bezpečnosti ukázali zvýšenú incidenciu nežiaducich udalostí pri kanakinumabe v porovnaní
s triamcinolónacetonidom, keď počas 24 týždňov akúkoľvek nežiaducu udalosť hlásilo 66 % oproti
53 % pacientov a infekciu ako nežiaducu udalosť hlásilo 20 % oproti 10 % pacientov.
Starší pacienti
Celkovo bol profil účinnosti, bezpečnosti a znášanlivosti Ilarisu u starších pacientov vo veku
≥ 65 rokov a vo veku < 65 rokov porovnateľný.
Pacienti, ktorí dostávajú liečbu znižujúcu urát (ULT)
V klinických skúšaniach sa Ilaris bezpečne podával s ULT. V celkovej populácii v klinickom skúšaní bol u pacientov, ktorí dostávali ULT, pri liečbe menej výrazný rozdiel v zmiernení bolesti aj znížení rizika ďalších záchvatov dnavej artritídy v porovnaní s pacientmi, ktorí nedostávali ULT.
Im
unogenita
Protilátky proti Ilarisu sa pozorovali u približne 1,5 % pacientov liečených Ilarisom pre CAPS, u 3% pacientov liečených pre SJIA a u 2 % pacientov liečených pre dnavú artritídu. Nezistili sa žiadne neutralizujúce protilátky. Nepozorovala sa žiadna zjavná korelácia medzi tvorbou protilátok
a klinickou odpoveďou alebo nežiaducimi udalosťami.
Nepozorovali sa žiadne protilátky proti Ilarisu u pacientov s TRAPS, HIDS/MKD a FMF, ktorí dostávali dávky 150 mg a 300 mg počas 16 týždňov liečby.
Tento liek bol registrovaný pre CAPS za tzv. mimoriadnych okolností. To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku. Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
Pediatrická populácia
Držiteľ rozhodnutia o registrácii uskutočnil štyri výskumné pediatrické plány pre Ilaris (pre CAPS, SJIA, FMF - HIDS/MKD a TRAPS). Táto informácia o lieku bola aktualizovaná, aby zahrnula výsledky štúdií s Ilarisom v pediatrickej populácii.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Ilarisom vo všetkých podskupinách pediatrickej populácie pre dnavú artritídu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
CAPS
Absorpcia
Maximálna koncentrácia kanakinumabu v sére (Cmax) sa dosiahla asi 7 dní po jednorazovom subkutánnom podaní 150 mg dospelým pacientom s CAPS. Priemerný terminálny polčas bol 26 dní. Priemerné hodnoty Cmax a AUCinf po jednorazovej subkutánnej dávke 150 mg u typického dospelého pacienta s CAPS (70 kg) boli 15,9 µg/ml a 708 µg*deň/ml. Odhad absolútnej biologickej dostupnosti subkutánne podaného kanakinumabu bol 66 %. Parametre expozície (napr. AUC a Cmax) sa zvyšovali úmerne dávke v rozmedzí dávok 0,30 až 10,0 mg/kg podávaných intravenóznou infúziou alebo od 150 do 600 mg subkutánnou injekciou. Predpokladané hodnoty expozície v rovnovážnom stave (Cmin,ss, Cmax,ss, AUC,ss,8w) po subkutánnom podávaní 150 mg (alebo 2 mg/kg) každých 8 týždňov boli o niečo vyššie v hmotnostnej kategórii 40-70 kg (6,6 µg/ml, 24,3 µg/ml, 767 µg*d/ml) v porovnaní
s hmotnostnými kategóriami < 40 kg (4,0 µg/ml, 19,9 µg/ml, 566 µg*d/ml) a > 70 kg (4,6 µg/ml,
17,8 µg/ml, 545 µg*d/ml). Očakávaný pomer akumulácie bol 1,3-násobný po 6 mesiacoch subkutánneho podávania 150 mg kanakinumabu každých 8 týždňov.
Distribúcia
Kanakinumab sa viaže na sérový IL-1 beta. Distribučný objem (Vss) kanakinumabu sa líšil podľa
telesnej hmotnosti. U pacienta s CAPS a telesnou hmotnosťou 70 kg sa odhadol na 6,2 l.
Eliminácia
Zdanlivý klírens (CL/F) kanakinumabu sa zvyšuje s telesnou hmotnosťou. U pacienta s CAPS
s telesnou hmotnosťou 70 kg sa odhadol na 0,17 l/d a u pacienta so SJIA s telesnou hmotnosťou 33 kg na 0,11 l/d. Po zohľadnení rozdielov v telesnej hmotnosti sa nepozorovali žiadne klinicky významné rozdiely vo farmakokinetických vlastnostiach kanakinumabu medzi pacientmi s CAPS a SJIA.
Po opakovanom podávaní kanakinumabu sa nezistil žiadny náznak zrýchleného klírensu alebo zmeny farmakokinetických vlastností v závislosti od času. Po korekcii na telesnú hmotnosť sa nepozorovali žiadne farmakokinetické rozdiely v súvislosti s pohlavím alebo vekom.
T
RA
PS, HIDS/MKD a FMF
Biologická dostupnosť sa nestanovila nezávisle u pacientov s TRAPS, HIDS/MKD a FMF. Zdanlivý klírens (CL/F) bol porovnateľný medzi populáciami s TRAPS, HIDS/MKD a FMF s telesnou hmotnosťou 55 kg (0,14 l/d) a s CAPS s telesnou hmotnosťou 70 kg (0,17 l/d). Zdanlivý distribučný objem (V/F) bol 4,96 l pri telesnej hmotnosti 55 kg.
Po opakovanom subkutánnom podávaní 150 mg každé 4 týždne bola minimálna koncentrácia kanakinumabu v 16. týždni (Cmin) odhadovaná na 15,4 ± 6,6 µg/ml. Odhadovaný rovnovážny stav AUCtau bol 636,7 ± 260,2 µg*d/ml.
Stillova choroba (AOSD a SJIA)
Biologická dostupnosť sa nestanovila nezávisle u pacientov so SJIA. Zdanlivý klírens na kg telesnej hmotnosti (CL/F na kg) bol porovnateľný medzi populáciami so SJIA a s CAPS (0,004 l/d na kg). Zdanlivý distribučný objem na kg (V/F na kg) bol 0,14 l/kg.
Po opakovanom podávaní 4 mg/kg každé 4 týždne pacientom so SJIA bol pomer akumulácie kanakinumabu 1,6-násobný. Rovnovážny stav sa dosiahol po 110 dňoch. Celkové predpokladané priemerné (±SD) hodnoty boli pre Cmin,ss 14,7±8,8 μg/ml, Cmax,ss 36,5 ± 14,9 μg/ml a AUC,ss4w 696,1 ±
326,5 μg*d/ml.
AUCss4w bola vo vekovej skupine 2-3 roky 692 µg*d/ml, 4-5 rokov 615 µg*d/ml, 6-11 rokov
707 µg*d/ml a 12-19 rokov 742 µg*d/ml. Po stratifikácii podľa telesnej hmotnosti sa pozoroval nižší
(30-40%) medián expozície pre Cmin,ss (11,4 oproti 19 µg/ml) a AUCss (594 oproti 880 µg*d/ml) pre
kategóriu nižšej telesnej hmotnosti (≤ 40 kg) oproti kategórii vyššej telesnej hmotnosti (> 40 kg).
Na základe analýzy populačného farmakokinetického modelovania bola farmakokinetika kanakinumabu u mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov podobná ako
u pacientov mladších ako 16 rokov. Predpokladané expozície kanakinumabu v rovnovážnom stave pri
veľkosti dávky 4 mg/kg (maximálne 300 mg) boli porovnateľné u pacientov nad 20 rokov a u pacientov so SJIA mladších ako 20 rokov.
Populácia s dnavou artritídou
Biologická dostupnosť u pacientov s dnavou artritídou sa nezávisle nestanovila. Zdanlivý klírens na
kg telesnej hmotnosti (CL/F na kg) bol porovnateľný medzi populáciami s dnavou artritídou a s CAPS
(0,004 l/deň/kg). Priemerná expozícia u typického pacienta s dnavou artritídou (93 kg) po
jednorazovej subkutánnej dávke 150 mg (Cmax: 10,8 µg/ml a AUCinf: 495 µg*deň/ml) bola nižšia ako u typického pacienta s CAPS s telesnou hmotnosťou 70 kg (15,9 µg/ml a 708 µg*deň/ml). Zhoduje sa to s pozorovaným zvyšovaním CL/F s telesnou hmotnosťou.
Očakávaný pomer akumulácie bol 1,1-násobný po subkutánnom podávaní 150 mg kanakinumabu
každých 12 týždňov.
Pediatrická populácia
Maximálne koncentrácie kanakinumabu u pediatrických pacientov vo veku 4 rokov a starších sa dosiahli medzi 2. a 7. dňom (Tmax) po jednorazovom subkutánnom podaní 150 mg alebo 2 mg/kg kanakinumabu. Terminálny polčas bol v rozmedzí 22,9 až 25,7 dní, čo je podobné farmakokinetickým vlastnostiam pozorovaným u dospelých. Na základe analýzy populačného farmakokinetického modelovania bola farmakokinetika kanakinumabu u detí vo veku 2 až < 4 roky podobná ako
u pacientov vo veku 4 roky a starších. Odhaduje sa, že rýchlosť subkutánnej absorpcie klesá s vekom a zdá sa, že je najvyššia u najmladších pacientov. V súlade s tým bol Tmax kratší (3,6 dní) u mladších pacientov so SJIA (2-3 roky) v porovnaní so staršími pacientmi so SJIA (12-19 rokov; Tmax 6 dní). Biologická dostupnosť (AUCss) nebola ovplyvnená.
Ďalšia analýza farmakokinetiky ukázala, že farmakokinetika kanakinumabu u 6 pediatrických pacientov s CAPS mladších ako 2 roky bola podobná ako farmakokinetika u pediatrických pacientov vo veku 2-4 roky. Na základe analýzy populačného farmakokinetického modelovania boli očakávané expozície po dávke 2 mg/kg porovnateľné vo všetkých pediatrických vekových skupinách pri CAPS, ale boli približne o 40 % nižšie u pediatrických pacientov s veľmi nízkou telesnou hmotnosťou (napr.
10 kg) v porovnaní s dospelými pacientmi (dávka 150 mg). Zhoduje sa to s pozorovaniami vyššej
expozície u pacientov s CAPS v skupinách vyššej telesnej hmotnosti.
Pri TRAPS, HIDS/MKD a FMF boli parametre expozície (minimálne koncentrácie) porovnateľné vo všetkých vekových skupinách od 2 do < 20 rokov po subkutánnom podaní kanakinumabu 2 mg/kg každé 4 týždne.
Farmakokinetické vlastnosti sú podobné u pediatrických populácií s CAPS, TRAPS, HIDS/MKD, FMF a SJIA.
Staršípacienti
Rozdiel farmakokinetických parametrov na základe klírensu alebo distribučného objemu sa nepozoroval medzi staršími pacientmi a dospelými pacientmi vo veku < 65 rokov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií skríženej reaktivity, toxicity po opakovanom podávaní, imunotoxicity, reprodukčnej toxicity a toxicity pre mláďatá vykonaných s kanakinumabom alebo myšacou protilátkou proti myšaciemu IL-1 beta neodhalili žiadne osobitné riziko pre ľudí.
Keďže kanakinumab sa viaže na IL-1 beta kozmáčov (C. jacchus) a ľudí s podobnou afinitou, bezpečnosť kanakinumabu sa skúmala u kozmáčov. Žiadne nežiaduce účinky sa nepozorovali po podávaní kanakinumabu dvakrát týždenne kozmáčom počas 26 týždňov alebo v štúdii toxicity pre embryofetálny vývoj počas gestácie u kozmáčov. Plazmatické koncentrácie, ktoré dobre tolerujú,zvieratá, prevyšujú najmenej 42-násobne (Cmax) a 78-násobne (Cavg) plazmatické koncentrácie u detí a dospievajúcich s CAPS (telesná hmotnosť 10 kg) liečených klinickými dávkami kanakinumabu do 8 mg/kg subkutánne každých 8 týždňov. Plazmatické koncentrácie, ktoré zvieratá dobre znášajú, sú minimálne 62-krát vyššie (Cmax) a 104-krát vyššie (Cavg) ako plazmatické koncentrácie u pediatrických pacientov so SJIA, ktorí dostávajú dávky do 4 mg/kg subkutánne každé
4 týždne. Navyše sa v týchto štúdiách nezistili žiadne protilátky proti kanakinumabu. Pri aplikácii kanakinumabu do normálnych ľudských tkanív sa nepreukázala nešpecifická tkanivová skrížená reaktivita.
Formálne štúdie karcinogenity s kanakinumabom sa nevykonali.
V štúdii toxicity pre embryofetálny vývoj kozmáčov kanakinumab nevykazoval pri podávaní počas
organogenézy toxicitu pre matky, embryotoxicitu ani teratogenitu.
V kompletnom súbore reprodukčných štúdií a štúdií na mláďatách myší sa nepozorovali nežiaduce účinky myšacej protilátky proti myšaciemu IL-1 beta. Protilátka proti myšaciemu IL-1 beta nevyvolala pri podávaní počas neskorej gestácie, pôrodu a laktácie nežiaduce udalosti súvisiace s rastom plodov alebo novorodencov (pozri časť 4.6). Vysoká dávka použitá v týchto štúdiách bola vyššia ako maximálna účinná dávka z hľadiska supresie IL-1 beta a aktivity.
Imunotoxikologická štúdia u myší s myšacou protilátkou proti myšaciemu IL-1 beta ukázala, že neutralizácia IL-1 beta nemá žiadne účinky na imunitné parametre a nespôsobila zhoršenie imunitnej funkcie u myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok na injekčný roztok:
Sacharóza
L-histidín
Monohydrát L-histidíniumchloridu
Polysorbát 80
Rozpúšťadlo:
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
Z mikrobiologického hľadiska sa liek má použiť okamžite po rekonštitúcii. Ak sa nepoužije okamžite, za čas a podmienky uchovávania pred použitím zodpovedá používateľ, pričom normálne nemajú presiahnuť 24 hodín pri 2°C – 8°C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom. Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Prášok: 150 mg prášku na injekčný roztok v injekčnej liekovke (sklo typu I) s objemom 6 ml so zátkou (chlórobutylová guma s krycou vrstvou) a odtrhávacím „flip/off“ uzáverom (hliník/plastový disk).
Rozpúšťadlo: 5 ml vody na injekciu v injekčnej liekovke (sklo typu I) s objemom 6 ml so zátkou (chlórobutylová guma pokrytá fluórovaným polymérom) a odtrhávacím „flip/off“ uzáverom (hliník/plastový disk).
Jedna injekčná súprava Ilarisu obsahuje 1 injekčnú liekovku s práškom na injekčný roztok, 1 injekčnú liekovku s rozpúšťadlom, 1 injekčnú striekačku s objemom 1 ml, 1 bezpečnostnú ihlu, 2 adaptéry na injekčnú liekovku a 4 čistiace tampóny.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Ilaris 150 mg prášok a rozpúšťadlo na injekčný roztok sa dodáva v jednorazovej liekovke na individuálne použitie. Použite len obsah injekčnej súpravy Ilarisu. Nepoužitý liek alebo odpad vzniknutý z lieku alebo injekčnú striekačku treba vrátiť do lekárne.
PokynynarekonštitúciuAseptickým postupom pripevnite adaptéry na injekčné liekovky Ilarisu a rozpúšťadla. Vtlačte 1 ml
vzduchu do injekčnej liekovky s rozpúšťadlom, potom odoberte 1 ml vody na injekciu z liekovky s rozpúšťadlom pomocou injekčnej striekačky, ktorá je súčasťou súpravy. Rekonštituujte obsah injekčnej liekovky Ilarisu pri izbovej teplote (obvykle 15°C až 25°C) pomalým injikovaním 1 ml vody na injekciu odobranej z liekovky s rozpúšťadlom. Pomaly otáčajte liekovku v uhle asi 45° približne počas 1 minúty a nechajte stáť asi 5 minút. Potom liekovku pomaly desať ráz obráťte zhora nadol
a späť. Nechajte stáť asi 15 minút pri izbovej teplote. Nepotriasajte. Nepoužite roztok, ak sú v ňom prítomné pevné častice.
Poklopte zboku na liekovku, aby sa zo zátky odstránil zvyšok tekutiny. Roztok nemá obsahovať viditeľné častice a nemá byť zakalený. Roztok má byť bezfarebný alebo môže mať slabé hnedožlté sfarbenie. Ak je roztok sfarbený výrazne dohneda, nemá sa použiť. Ak sa roztok nepoužije ihneď po rekonštitúcii, má sa uchovávať pri teplote 2°C až 8°C a použiť do 24 hodín.
Pokyny na podanieOpatrne odoberte požadovaný objem v závislosti od dávky, ktorá sa má podať (0,1 ml až 1 ml)
a subkutánne injikujte pomocou bezpečnostnej ihly, ktorá je súčasťou súpravy.
LikvidáciaZvyškový objem sa má zlikvidovať ihneď po podaní injekcie. Pacienti alebo ich opatrovatelia majú dostať pokyny, ako v súlade s národnými požiadavkami správne postupovať pri likvidácii injekčných liekoviek, striekačiek a ihiel.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/09/564/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. október 2009
Dátum posledného predĺženia registrácie: 19. jún 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUIlaris 150 mg/ml injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Jedna injekčná liekovka obsahuje 150 mg kanakinumabu*. Každý ml roztoku obsahuje 150 mg kanakinumabu.
* ľudská monoklonálna protilátka produkovaná v myšacích myelómových bunkách Sp2/0
technológiou rekombinantnej DNA
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný roztok.
Roztok je číry až opalizujúci a bezfarebný až slabo hnedožltý.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieSyndrómyperiodickejhorúčkyIlaris je indikovaný na liečbu nasledujúcich autoinflamačných syndrómov periodickej horúčky
u dospelých, dospievajúcich a detí vo veku 2 roky a starších:
Periodické syndrómy asociované s kryopyrínomIlaris je indikovaný na liečbu periodických syndrómov asociovaných s kryopyrínom (cryopyrin- associated periodic syndromes, CAPS) vrátane:
· Muckleovho-Wellsovho syndrómu (MWS),
· multisystémovej zápalovej choroby novorodencov (Neonatal-onset multisystem inflammatory disease, NOMID) / chronického neurologického, kožného a kĺbového syndrómu u detí (chronic infantile neurological, cutaneous, articular syndrome, CINCA),
· ťažkých foriem familiárneho chladového autoinflamačného syndrómu (familial cold autoinflammatory syndrome, FCAS) / familiárnej chladovej urtikárie (familial cold urticaria, FCU), prejavujúcej sa príznakmi a prejavmi presahujúcimi kožný exantém typu urtikárie vyvolanej chladom.
Periodický syndróm asociovaný s receptorom pre faktor nekrotizujúci nádory (tumour necrosis factorreceptor associated periodic syndrome, TRAPS)Ilaris je indikovaný na liečbu periodického syndrómu asociovaného s receptorom pre faktor nekrotizujúci nádory (tumour necrosis factor, TNF) (TRAPS).
Syndróm hyperimunoglobulinémie D (hyperimmunoglobulin D syndrome, HIDS)/deficitu
mevalonátkinázy (mevalonate kinase deficiency, MKD)
Ilaris je indikovaný na liečbu syndrómu hyperimunoglobulinémie D (HIDS)/deficitu mevalonátkinázy
(MKD).
Fami l i árna s t re doze msk á horúč ka (f ami l i al medi t er rane an fever, FMF)
Ilaris je indikovaný na liečbu familiárnej stredozemskej horúčky (FMF). Ilaris sa má podávať
v kombinácii s kolchicínom, ak je to vhodné.
Ilaris je tiež indikovaný na liečbu:
Stillova choroba
Ilaris je indikovaný na liečbu aktívnej Stillovej choroby vrátane Stillovej choroby s nástupom ochorenia v dospelosti (adult-onset Still’s disease, AOSD) a systémovej juvenilnej idiopatickej artritídy (SJIA) u pacientov vo veku 2 rokov a starších, ktorí nedostatočne reagovali na predchádzajúcu liečbu nesteroidnými protizápalovými liekmi (NSAID) a systémovými kortikosteroidmi. Ilaris sa môže podávať ako monoterapia alebo v kombinácii s metotrexátom.
Dnavá artritída
Ilaris je indikovaný na symptomatickú liečbu dospelých pacientov s častými záchvatmi dnavej artritídy (najmenej 3 záchvaty počas predošlých 12 mesiacov), u ktorých sú nesteroidné protizápalové lieky (NSAID) a kolchicín kontraindikované, netolerované alebo nevyvolávajú primeranú odpoveď a
u ktorých nie je vhodná opakovaná liečba kortikosteroidmi (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Pri CAPS, TRAPS, HIDS/MKD, FMF a Stillovej chorobe má liečbu začať a viesť odborný lekár so skúsenosťami v diagnostike a liečbe príslušnej indikácie.
Pri dnavej artritíde má mať lekár skúsenosti s používaním biologických liekov a Ilaris má podávať
zdravotnícky pracovník.
Po náležitom zaškolení o správnej injekčnej technike si môžu Ilaris podávať pacienti alebo im ho môžu podávať ich opatrovatelia, ak lekár rozhodne, že je to vhodné, a keď ich lekár podľa potreby bude ďalej sledovať (pozri časť 6.6).
Dávkovanie
CAPS: Dospelí, dospievajúci a deti vo veku 2 rok ov a s t arš i e Odporúčaná začiatočná dávka Ilarisu u pacientov s CAPS je: Dospelí, dospievajúci a deti vo veku ≥ 4 roky:
- 150 mg u pacientov s telesnou hmotnosťou > 40 kg
- 2 mg/kg u pacientov s telesnou hmotnosťou ≥ 15 kg a ≤ 40 kg
- 4 mg/kg u pacientov s telesnou hmotnosťou ≥ 7,5 kg a < 15 kg
Deti vo veku 2 až < 4 roky:
- 4 mg/kg u pacientov s telesnou hmotnosťou ≥ 7,5 kg
Podáva sa každých osem týždňov ako jednorazová dávka subkutánnou injekciou.
Ak sa u pacientov so začiatočnou dávkou 150 mg alebo 2 mg/kg nedosiahne uspokojivá klinická odpoveď (vymiznutie exantému a iných generalizovaných symptómov zápalu) 7 dní po začatí liečby, možno zvážiť druhú dávku Ilarisu – 150 mg alebo 2 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu, má sa dodržiavať zintenzívnený režim dávkovania 300 mg alebo 4 mg/kg každých
8 týždňov. Ak sa uspokojivá klinická odpoveď nedosiahne 7 dní po tejto zvýšenej dávke, možno
zvážiť tretiu dávku Ilarisu 300 mg alebo 4 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu,
na základe individuálneho klinického posúdenia sa má zvážiť dodržiavanie zintenzívneného režimu
dávkovania 600 mg alebo 8 mg/kg každých 8 týždňov.
Ak sa u pacientov so začiatočnou dávkou 4 mg/kg nedosiahne uspokojivá klinická odpoveď 7 dní po začatí liečby, môže sa zvážiť druhá dávka Ilarisu 4 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu, na základe individuálneho klinického posúdenia sa má zvážiť dodržiavanie zintenzívneného režimu dávkovania 8 mg/kg každých 8 týždňov.
Klinické skúsenosti s dávkovaním v intervaloch kratších ako 4 týždne alebo s dávkami vyššími ako
600 mg alebo 8 mg/kg sú obmedzené.
CA
PS u dospelých a detí vo veku
³
4 roky³
15 kgCAPS u detí vo veku 2-< 4 roky alebodetí vo veku ³
4 roky ³
7,5 kg a < 15 kg
150 mg alebo 2 mg/kg
4 mg/kg
U
spokojivá klinická
odpoveď po 7 dňoch?
'
U
spokojivá klinická
odpoveď po 7 dňoch?
Á
no
N
i
e
Á
no
N
i
e
U
držiavacia dávka: 150 mg alebo 2 mg/kg každých
8 týždňov
Možno zvážiť ďalšiu
dávku 150 mg alebo
2 mg/kg
U
držiavacia dávka: 4 mg/kg každých
8 týždňov
Možno zvážiť ďalšiudávku
4 mg/kg
U
spokojivá klinická odpoveď po
7 dňoch?
Á
no Nie
A
k je po 7 dňoch úplná odpoveď na liečbu, udržiavacia dávka:
8 mg/kg každých
8 týždňov
U
držiavacia dávka:
300 mg alebo 4 mg/kg
každých 8 týždňov
Možno zvážiť ďalšiu
dávku 300 mg alebo
4 mg/kg
A
k je po 7 dňoch úplná odpoveď na liečbu, udržiavacia dávka:
600 mg alebo 8 mg/kg
každých 8 týždňov
TRAPS, HIDS/MKD a FMF: Dospelí, dospievajúci a deti vo veku 2 roky a st arš i e
Odporúčaná začiatočná dávka Ilarisu u pacientov s TRAPS, HIDS/MKD a FMF je:
· 150 mg u pacientov s telesnou hmotnosťou > 40 kg
· 2 mg/kg u pacientov s telesnou hmotnosťou ≥ 7,5 kg a ≤ 40 kg
Podáva sa každé štyri týždne ako jednorazová dávka subkutánnou injekciou.
Ak sa nedosiahne uspokojivá klinická odpoveď 7 dní od začatia liečby, možno zvážiť druhú dávku Ilarisu 150 mg alebo 2 mg/kg. Ak sa následne dosiahne úplná odpoveď na liečbu, má sa dodržiavať zintenzívnený režim dávkovania 300 mg (alebo 4 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) každé 4 týždne.
Ošetrujúci lekár má prehodnotiť pokračovanie liečby Ilarisom u pacientov bez klinického zlepšenia.
TRAPS, HIDS/MKD a FMF u pacientov
s telesnou hmotnosťou > 40 kg
150 mg
TRAPS, HIDS/MKD a FMF u pacientov s telesnou hmotnosťou
³
7,5 kg a ≤ 40 kg2 mg/kg
U
spokojivá klinická
odpoveď po 7 dňoch?
U
spokojivá klinická
odpoveď po 7 dňoch?
Á
no
N
ie
Á
no
N
ie
U
držiavacia dávka: 150 mg každé 4 týždne
Možno zvážiť ďalšiu dávku
150 mg
U
držiavacia dávka 2 mg/kg každé 4 týždne
Možno zvážiť ďalšiu dávku
2 mg/kg
A
k sa dosiahne úplná odpoveď na liečbu, udržiavacia dávka:
300 mg každé 4 týždne
A
k sa dosiahne úplná odpoveď na liečbu, udržiavacia dávka:
4 mg/kg každé 4 týždne
Stillova choroba (AOSD a SJIA)
Odporúčaná dávka Ilarisu u pacientov so Stillovou chorobou (AOSD a SJIA) s telesnou hmotnosťou
≥ 7,5 kg je 4 mg/kg (maximálne 300 mg) podávaná subkutánnou injekciou každé štyri týždne. Pokračovanie v liečbe Ilarisom u pacientov bez klinického zlepšenia má prehodnotiť ošetrujúci lekár.
Dnavá artritídaJe potrebné začať alebo optimalizovať úpravu hyperurikémie náležitou liečbou na zníženie urátu (urate lowering therapy, ULT). Ilaris sa má používať ako liečba podľa potreby na liečenie záchvatov dnavej artritídy.
Odporúčaná dávka Ilarisu u dospelých pacientov s dnavou artritídou je 150 mg podávaných subkutánne ako jednorazová dávka počas záchvatu. Na dosiahnutie maximálneho účinku sa má Ilaris podávať čo najskôr po prepuknutí záchvatu dnavej artritídy.
Pacientom, ktorí nereagujú na iniciálnu liečbu, sa Ilaris nemá opakovane podávať. U pacientov
s odpoveďou, u ktorých je potrebná opakovaná liečba, má byť interval pred podaním ďalšej dávky
Ilarisu najmenej 12 týždňov (pozri časť 5.2).
Osobitné skupiny pacientov
Pediatrická populácia
CAPS, TRAPS, HIDS/MKD a FMF
Bezpečnosť a účinnosť Ilarisu u pacientov s CAPS, TRAPS, HIDS/MKD a FMF vo veku menej ako
2 roky neboli stanovené. V súčasnosti dostupné údaje sú opísané v častiach 4.8, 5.1 a 5.2, ale
neumožňujú uviesť odporúčania na dávkovanie.
SJIA
Bezpečnosť a účinnosť Ilarisu u pacientov so SJIA vo veku menej ako 2 roky neboli stanovené. Nie je
možné uviesť odporúčania na dávkovanie.
Dnavá artritída
Použitie Ilarisu sa netýka pediatrickej populácie v indikácii dnavá artritída.
St arš í paci ent i
Nie je potrebná úprava dávky.
Porucha f unkc i e pe če ne
Ilaris sa nesledoval u pacientov s poruchou funkcie pečene.
Porucha funkcie obl i či ek
U pacientov s poruchou funkcie obličiek nie je potrebná úprava dávkovania. Klinické skúsenosti
u takých pacientov sú však obmedzené.
Spôsob podávania
Na subkutánne použitie.
Nasledujúce miesta sú vhodné na podanie injekcie: horná časť stehna, brucho, horná časť ramena alebo sedacie svaly. Odporúča sa zvoliť pri každom podaní injekcie iné miesto vpichu, aby sa zabránilo bolesti. Je potrebné vyhnúť sa poranenej koži a miestam, v ktorých sú podliatiny alebo ktoré sú pokryté exantémom. Je potrebné vyhnúť sa podaniu injekcie do tkaniva jazvy, pretože to môže mať za následok nedostatočnú expozíciu Ilarisu.
Každá injekčná liekovka Ilarisu je určená na jednorazové použitie u jedného pacienta, a to na jednu dávku.
Pokyny o použití a manipulácii s rekonštituovaným roztokom, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívne, závažné infekcie (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
I
nfekcie
Ilaris sa spája so zvýšenou incidenciou závažných infekcií. Preto je u pacientov potrebné dôsledne sledovať príznaky a prejavy infekcií počas liečby Ilarisom a po nej. Lekári majú byť opatrní pri podávaní Ilarisu pacientom s infekciami, opakovanými infekciami v anamnéze alebo základnými ochoreniami, ktoré ich môžu predisponovať na infekcie.
Liečba CAPS, TRAPS, HIDS/MKD, FMF a Stillovej choroby (AOSD a SJIA)
Liečba Ilarisom sa nemá začať alebo pokračovať u pacientov počas aktívnej infekcie, ktorá vyžaduje
zásah lekára.
Liečba dnavej artritídy
Ilaris sa nemá podávať počas aktívnej infekcie.
Súbežné použitie Ilarisu s inhibítormi faktora nekrotizujúceho nádory (TNF) sa neodporúča, pretože to môže zvyšovať riziko závažných infekcií (pozri časť 4.5).
Počas liečby Ilarisom boli hlásené ojedinelé prípady neobvyklých alebo oportúnnych infekcií (vrátane aspergilózy, atypických mykobakteriálnych infekcií, herpes zoster). Príčinný vzťah medzi Ilarisom
a týmito udalosťami nemožno vylúčiť.
Asi u 12 % pacientov s CAPS testovaných v klinických skúšaniach kožným testom PPD (purifikovaným proteínovým derivátom) bol výsledok ďalšieho testovania počas liečby Ilarisom pozitívny bez klinických dôkazov latentnej alebo aktívnej infekcie tuberkulózou.
Nie je známe, či používanie inhibítorov interleukínu-1 (IL-1), ako je Ilaris, zvyšuje riziko reaktivácie tuberkulózy. Pred začatím liečby sa u všetkých pacientov musí vyšetriť prítomnosť aktívnej a latentnej infekcie tuberkulózou. Najmä u dospelých pacientov má toto vyšetrenie zahŕňať podrobnú anamnézu. Príslušné skríningové testy (napr. tuberkulínový kožný test, stanovenie uvoľňovania interferónu gama alebo röntgen hrudníka) je potrebné vykonať u všetkých pacientov (môžu platiť národné odporúčania). U pacientov sa musia dôsledne sledovať príznaky a prejavy tuberkulózy počas liečby Ilarisom a po nej. Všetkých pacientov je potrebné poučiť, aby sa poradili s lekárom, ak sa
počas liečby Ilarisom vyskytnú príznaky a prejavy svedčiace o tuberkulóze (napr. pretrvávajúci kašeľ, úbytok telesnej hmotnosti, subfebrilná teplota). V prípade zmeny z negatívneho na pozitívny test PPD, najmä u pacientov s vysokým rizikom, je potrebné zvážiť alternatívne spôsoby skríningu na infekciu tuberkulózou.
Neutropénia a leukopénia
Neutropénia (absolútny počet neutrofilov [absolute neutrophil count, ANC] < 1,5 x 109/l)
a leukopénia sa pozorovali pri liekoch, ktoré inhibujú IL-1, vrátane Ilarisu. Liečba Ilarisom sa nemá začať u pacientov s neutropéniou alebo leukopéniou. Odporúča sa stanoviť počet bielych krviniek (white blood cell, WBC) vrátane počtu neutrofilov pred začatím liečby a znovu po 1 až 2 mesiacoch. Pri chronickej alebo opakovanej liečbe sa odporúča stanoviť počet WBC tiež pravidelne počas liečby. Ak sa u pacienta objaví neutropénia alebo leukopénia, počet WBC sa má dôsledne sledovať a má sa zvážiť ukončenie liečby.
Malignity
U pacientov liečených Ilarisom boli hlásené prípady malignít. Riziko vzniku malignít pri liečbe anti- interleukínom (IL)-1 nie je známe.
Reakcie z precitlivenosti
Pri liečbe Ilarisom boli hlásené reakcie z precitlivenosti. Závažnosť väčšiny týchto udalostí bola mierna. Počas klinického vývoja Ilarisu u viac ako 2 600 pacientov neboli hlásené žiadne anafylaktoidné alebo anafylaktické reakcie. Avšak riziko závažných reakcií z precitlivenosti, ktoré nie je neobvyklé pri injekčne podávaných proteínoch, nemožno vylúčiť (pozri časť 4.3).
Funkcia
pečene
V klinických skúšaniach sa zaznamenali prechodné a asymptomatické prípady zvýšenia hladín
aminotransferáz alebo bilirubínu v sére (pozri časť 4.8).
Vakcinácie
Nie sú dostupné údaje o riziku sekundárneho prenosu infekcie živými (oslabenými) vakcínami
u pacientov, ktorí dostávajú Ilaris. Preto sa živé vakcíny nemajú podávať súčasne s Ilarisom, pokiaľ prínos nie je jednoznačne väčší ako riziká (pozri časť 4.5).
Odporúča sa, aby dospelí a pediatrickí pacienti dostali pred začatím liečby Ilarisom podľa potreby všetky vakcinácie, vrátane pneumokokovej vakcíny a inaktivovanej chrípkovej vakcíny (pozri
časť 4.5).
Mutácia génu NLRP3 u pacientov s CAPS
Klinické skúsenosti u pacientov s CAPS bez potvrdenej mutácie génu NLRP3 sú obmedzené.
Syndróm aktivácie makrofágov u pacientov so Stillovou chorobou
Syndróm aktivácie makrofágov (MAS) je známa, život ohrozujúca porucha, ktorá sa môže vyvinúť
u pacientov s reumatickými chorobami, najmä so Stillovou chorobou. Pri výskyte alebo podozrení na MAS sa má začať čo najskôr s hodnotením stavu a liečbou. Lekári si majú pozorne všímať prejavy infekcie alebo zhoršenie Stillovej choroby, pretože sú to známe spúšťače MAS. Na základe skúseností z klinických skúšaní sa nezdá, že by Ilaris zvyšoval incidenciu MAS u pacientov so SJIA, ale definitívny záver nie je možné urobiť.
4.5 Liekové a iné interakcie
Interakcie medzi Ilarisom a inými liekmi sa nesledovali vo formálnych štúdiách.
Zvýšená incidencia závažných infekcií bola spojená s podávaním iného blokátora IL-1 v kombinácii s inhibítormi TNF. Použitie Ilarisu s inhibítormi TNF sa neodporúča, pretože to môže zvýšiť riziko závažných infekcií.
Expresiu pečeňových enzýmov CYP450 môžu potlačiť cytokíny, ktoré stimulujú chronický zápal, napr. interleukín-1 beta (IL-1 beta). Expresia CYP450 sa preto môže zvrátiť, keď sa začne účinná liečba inhibítormi cytokínov, napr. kanakinumabom. Je to klinicky významné pri substrátoch CYP450 s úzkym terapeutickým indexom, ktorých dávka sa individuálne upravuje. Pri začatí liečby kanakinumabom u pacientov, ktorí dostávajú liek tohto typu, sa má sledovať účinok liečby alebo koncentrácia liečiva a individuálna dávka lieku sa má podľa potreby upraviť.
Nie sú dostupné údaje ani o účinkoch vakcinácie živou vakcínou, ani o sekundárnom prenose infekcie
živými vakcínami u pacientov, ktorí dostávajú Ilaris. Preto sa živé vakcíny nemajú podávať súčasne s Ilarisom, pokiaľ prínos nie je jednoznačne väčší ako riziká. Ak je po začatí liečby Ilarisom vakcinácia živými vakcínami potrebná, odporúča sa počkať aspoň 3 mesiace po poslednej injekcii Ilarisu a pred ďalšou injekciou (pozri časť 4.4).
Výsledky štúdie so zdravými dospelými ukázali, že jednorazová dávka 300 mg Ilarisu neovplyvnila vyvolanie a trvanie protilátkovej odpovede po podaní vakcíny proti chrípke alebo vakcíny proti meningokokom založenej na glykozylovaných proteínoch.
Výsledky otvorenej štúdie trvajúcej 56 týždňov u pacientov s CAPS vo veku 4 rokov a mladších ukázali, že u všetkých pacientov, ktorí dostali neživú vakcínu štandardne používanú u detí, sa vyvinuli ochranné hladiny protilátok.
4.6 Fertilia, gravidita a laktácia
Ž
eny
v
o
fertilnom veku / Antikoncepcia u mužov
a
žien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby Ilarisom a počas
3 mesiacov od poslednej dávky.
Gravidita
O použití kanakinumabu u gravidných žien je obmedzené množstvo údajov. Štúdie na zvieratách
nenaznačujú priame alebo nepriame škodlivé účinky vzhľadom na reprodukčnú toxicitu (pozri
časť 5.3). Riziko pre plod/matku nie je známe. Ženy, ktoré sú tehotné alebo chcú otehotnieť, preto majú byť liečené len po dôkladnom zhodnotení prínosu a rizika liečby.
Štúdie na zvieratách naznačujú, že kanakinumab prechádza placentou a je preukázateľný v plode. Nie sú k dispozícii žiadne údaje u ľudí, ale keďže je kanakinumab imunoglobulín triedy G (IgG1), predpokladá sa transplacentárny prenos u žien. Klinický dopad tohto zistenia nie je známy. Podávanie živých vakcín novorodencom, vystaveným kanakinumabu v maternici sa však neodporúča 16 týždňov od podania poslednej dávky Ilarisu matke pred pôrodom. Ženy, ktoré dostali kanakinumab počas gravidity, je potrebné poučiť, aby o tom informovali detského lekára pred akoukoľvek vakcináciou ich novorodenca.
Dojčenie
Nie je známe, či sa kanakinumab vylučuje do ľudského mlieka. Rozhodnutie, či dojčiť počas liečby
Ilarisom, sa preto má urobiť len po dôkladnom zhodnotení prínosu a rizika liečby.
Štúdie na zvieratách ukázali, že myšacia protilátka proti myšaciemu IL-1 beta nemala nežiaduce účinky na vývoj dojčených myšacích mláďat a že táto protilátka sa na ne preniesla (pozri časť 5.3).
Fertilita
Formálne štúdie možného účinku Ilarisu na ľudskú plodnosť sa nevykonali.
Kanakinumab nemal účinok na parametre plodnosti u samcov opíc kozmáčov (C. jacchus). Myšacia protilátka proti myšaciemu IL-1 beta nemala nežiaduce účinky na plodnosť samcov ani samíc myši (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ilaris má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Liečba Ilarisom môže vyvolať závraty/vertigo alebo asténiu (pozri časť 4.8). Pacienti, u ktorých sa počas liečby Ilarisom vyskytnú takéto symptómy, majú počkať do ich úplného vymiznutia, kým budú viesť vozidlo alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn
bezpečnostného
profilu
Viac ako 2 600 osôb, vrátane približne 480 detí (vo veku 2 až 17 rokov), bolo liečených Ilarisom v intervenčných klinických skúšaniach s pacientmi s CAPS, TRAPS, HIDS/MKD, FMF, SJIA, dnavou artritídou alebo s inými ochoreniami sprostredkovanými IL-1 beta, ako aj zdravých dobrovoľníkov. Pozorovali sa závažné infekcie. Najčastejšími nežiaducimi reakciami na liek boli infekcie predovšetkým horného dýchacieho traktu. Pri dlhodobejšej liečbe sa nezaznamenal žiadny vplyv na typ ani frekvenciu nežiaducich reakcií na liek.
U pacientov liečených Ilarisom boli hlásené reakcie z precitlivenosti (pozri časti 4.3 a 4.4). U pacientov liečených Ilarisom boli hlásené oportúnne infekcie (pozri časť 4.4).
CAPS
V klinických skúšaniach dostávalo Ilaris celkovo 211 dospelých a pediatrických pacientov s CAPS (vrátane FCAS/FCU, MWS a NOMID/CINCA). Bezpečnosť Ilarisu v porovnaní s placebom sa skúmala v pivotnom skúšaní fázy III, ktoré pozostávalo z 8-týždňového obdobia otvoreného podávania (časť I), 24-týždňového randomizovaného, dvojito zaslepeného a placebom kontrolovaného obdobia vysadenia lieku (časť II) a 16-týždňového obdobia otvoreného podávania Ilarisu (časť III). Všetci pacienti dostávali subkutánne Ilaris v dávke 150 mg alebo 2 mg/kg, ak ich telesná hmotnosť bola
≥ 15 kg a ≤ 40 kg.
TRAPS, HIDS/MKD, FMF
V jednom pivotnom klinickom skúšaní fázy III dostávalo Ilaris celkovo 169 dospelých a pediatrických pacientov s TRAPS, HIDS/MKD a FMF vo veku 2 roky a starších. Bezpečnosť Ilarisu v porovnaní
s placebom sa skúmala v tomto skúšaní, ktoré pozostávalo z 12-týždňového obdobia skríningu (časť I) a 16-týždňového randomizovaného, dvojito zaslepeného, placebom kontrolovaného obdobia liečby (časť II). Pacientom, ktorí dostávali Ilaris, sa podávalo subkutánne 150 mg, alebo 2 mg/kg, ak ich telesná hmotnosť bola ≤ 40 kg (pozri časť 5.1).
Stillova choroba
Celkovo 324 pacientov so SJIA vo veku 2 až < 20 rokov dostávalo Ilaris v klinických skúšaniach, vrátane 293 pacientov vo veku 2 až < 16 rokov, 21 pacientov vo veku 16 až < 18 rokov a 10 pacientov vo veku 18 až < 20 rokov. Bezpečnosť Ilarisu sa skúmala v porovnaní s placebom v dvoch pivotných skúšaniach fázy III (pozri časť 5.1).
Dnavá artritída
Viac ako 700 pacientov s dnavou artritídou bolo liečených Ilarisom v dávkach od 10 mg do 300 mg v randomizovaných, dvojito zaslepených a účinným liekom kontrolovaných klinických skúšaniach trvajúcich do 24 týždňov. Viac ako 250 pacientov dostávalo odporúčanú dávku 150 mg v klinických skúšaniach fázy II a III (pozri časť 5.1).
T
abuľkový
z
oznam
nežiaducich
reakcií
Nežiaduce reakcie sú usporiadané podľa tried orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa kategórie frekvencie, najčastejšie ako prvé. Kategórie frekvencie sú definované pomocou nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až< 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov).V rámci každej skupiny frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 1 Tabuľkový zoznam nežiaducich reakcií pri CAPS, TRAPS, HIDS/MKD, FMF, SJIA a dnavej artritíde
T
rieda
orgánových systémov MedDRA
Infekcie a nákazy
V
šetky indikácie:
CA
P
S, TRAPS, HIDS/MKD, FMF, SJIA, dnavá artritída
Veľmi časté Infekcie dýchacích ciest (vrátane pneumónie, bronchitídy, chrípky, vírusovej infekcie, sínusitídy, rinitídy, faryngitídy, tonzilitídy, nazofaryngitídy, infekcie horných dýchacích ciest)
Infekcia ucha Celulitída Gastroenteritída
Infekcia močových ciest Časté Vulvovaginálna kandidóza
Poruchy nervového systémuČasté Závraty/vertigo
Poruchy gastrointestinálneho traktuVeľmi časté Bolesť hornej časti brucha 1
Menej časté Gastroezofágová refluxná choroba 2
Poruchy kože a podkožného tkanivaVeľmi časté Reakcia v mieste vpichu
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté Artralgia 1
Časté Bolesť svalov a kostí 1
Bolesť chrbta 2
Celkové poruchy a reakcie v mieste podaniaČasté Únava/asténia 2
Laboratórne a funkčné vyšetreniaVeľmi časté Zníženie obličkového klírensu kreatinínu 1,3
Proteinúria 1,4
Leukopénia 1,5
Časté Neutropénia 5
Menej časté Pokles počtu trombocytov 5
1 Pri SJIA
2 Pri dnavej artritíde
3 Založené na odhadovanom klírense kreatinínu, väčšinou prechodné
4 Väčšinu predstavovali prechodné stopy až 1+ pozitivita bielkovín v moči, stanovené indikátorovým papierikom
5 Pozri ďalšie informácie nižšie
V podskupine mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov (N=31) bol profil
bezpečnosti Ilarisu v súlade s tým, ktorý sa pozoroval u pacientov so SJIA mladších ako 16 rokov. Na základe literárnych údajov sa očakáva podobný profil bezpečnosti u AOSD pacientov ako u pacientov so SJIA.
Popis
vy
braných
nežiaducich
reakcií
D
l
hodobé údaje a laboratórne abnormality u pacientov s CAPS
Počas klinických skúšaní Ilarisu u pacientov s CAPS sa priemerné hodnoty hemoglobínu zvýšili
a hodnoty leukocytov, neutrofilov a trombocytov znížili.
Zvýšenie hladín aminotransferáz sa u pacientov s CAPS pozorovalo zriedkavo.
U pacientov s CAPS liečených Ilarisom sa pozorovali asymptomatické a mierne zvýšenia bilirubínu
v sére, bez súčasného zvýšenia aminotransferáz.
V dlhodobých, otvorených klinických skúšaniach so zvyšovaním dávky boli ako udalosti častejšie hlásené infekcie (gastroenteritída, infekcia dýchacieho traktu, infekcia horného dýchacieho traktu), vracanie a závraty v skupine s dávkou 600 mg alebo 8 mg/kg ako v skupinách s inými dávkami.
Laboratórne abnormality u pacientov s TRAPS, HIDS/MKD a FMF Neutrofily
Hoci sa zníženie počtu neutrofilov ≥ stupňa 2 vyskytlo u 6,5 % pacientov (časté) a zníženie stupňa 1 sa vyskytlo u 9,5 % pacientov, zníženie je všeobecne prechodné a infekcia spojená s neutropéniou nebola identifikovaná ako nežiaduca reakcia.
Trombocyty
Hoci sa zníženie počtu trombocytov (≥ stupňa 2) vyskytlo u 0,6 % pacientov, krvácanie nebolo identifikované ako nežiaduca reakcia. Mierne a prechodné zníženie trombocytov stupňa 1 sa vyskytlo u 15,9 % pacientov bez akýchkoľvek súvisiacich krvácavých nežiaducich príhod.
Laboratórne abnormality u pacientov so SJIA Hematológia
V celkovom programe SJIA bol prechodný pokles počtu bielych krviniek (WBC) ≤ 0,8 x LLN hlásený u 33 pacientov (16,5 %).
V celkovom programe SJIA bol prechodný pokles absolútneho počtu neutrofilov (ANC) na menej ako
1 x 109/l hlásený u 12 pacientov (6,0 %).
V celkovom programe SJIA sa prechodný pokles trombocytov (< LLN) pozoroval u 19 pacientov
(9,5 %).
ALT/AST
V celkovom programe SJIA boli vysoké hodnoty ALT a/alebo AST > 3 x hornej hranice normálneho rozmedzia (ULN) hlásené u 19 pacientov (9,5 %).
Laboratórne abnormality u pacientov s dnavou artritídou
Hematológia
Znížený počet leukocytov (white blood cell counts, WBC) ≤ 0,8 x dolná hranica normálu (lower limit of normal, LLN) bol hlásený u 6,7 % pacientov liečených Ilarisom v porovnaní s 1,4 % pacientov liečených triamcinolónacetonidom. V porovnávacích klinických skúšaniach bol u 2 % pacientov hlásený pokles absolútneho počtu neutrofilov (ANC) na menej ako 1 x 109/l. Pozorovali sa aj ojedinelé prípady ANC < 0,5 x 109/l (pozri časť 4.4).
V klinických skúšaniach kontrolovaných účinným liekom sa u pacientov s dnavou artritídou pozorovalo mierne (< LLN a > 75 x 109/l) a prechodné zníženie počtu trombocytov s vyššou incidenciou (12,7 %) pri Ilarise oproti komparátoru (7,7 %).
K
yselina močová
V porovnávacích klinických skúšaniach pri dnavej artritíde sa po liečbe Ilarisom pozorovalo zvýšenie koncentrácie kyseliny močovej (0,7 mg/dl po 12 týždňoch a 0,5 mg/dl po 24 týždňoch). V inom klinickom skúšaní sa nepozorovalo zvýšenie kyseliny močovej u pacientov, ktorí začínali ULT. Zvýšenie kyseliny močovej sa nepozorovalo v klinických skúšaniach u populácií bez dnavej artritídy (pozri časť 5.1).
ALT/ASTZvýšenie priemeru o 3,0 U/l a mediánu o 2,0 U/l pri alanínaminotransferáze (ALT) a o 2,7 U/l
a o 2,0 U/l pri aspartátaminotransferáze (AST) sa pozorovalo oproti východiskovým hodnotám do konca klinického skúšania v skupinách liečených Ilarisom oproti skupinám liečeným triamcinolónacetonidom, avšak incidencia klinicky významných zmien (≥ 3 x horná hranica normálu) bola vyššia u pacientov liečených triamcinolónacetonidom (2,5 % pri AST aj ALT) v porovnaní s pacientmi liečenými Ilarisom (1,6 % pri ALT a 0,8 % pri AST).
TriacylglycerolyV klinických skúšaniach kontrolovaných účinným liekom pri dnavej artritíde sa triacylglyceroly zvýšili v priemere o 33,5 mg/dl u pacientov liečených Ilarisom v porovnaní s miernym poklesom o -3,1 mg/dl pri triamcinolónacetonide. Incidencia pacientov so zvýšením triacylglycerolov > 5 x horná hranica normálu (ULN) bola 2,4 % pri Ilarise a 0,7 % pri triamcinolónacetonide. Klinická významnosť tohto pozorovania nie je známa.
Pediatrická populáciaV štúdiách bolo 80 pediatrických pacientov s CAPS (vo veku 2–17 rokov), ktorí dostávali kanakinumab. Celkovo sa nevyskytli žiadne klinicky významné rozdiely v profiloch bezpečnosti a znášanlivosti Ilarisu u pediatrických pacientov v porovnaní s celkovou populáciou s CAPS
(tvorenou dospelými aj pediatrickými pacientmi, N=211), vrátane celkovej frekvencie a závažnosti
epizód infekcií. Infekcie horného dýchacieho traktu boli najčastejšie hlásenými udalosťami infekcií.
Okrem toho sa 6 pediatrických pacientov mladších ako 2 roky vyhodnotilo v malej, otvorenej klinickej štúdii. Ukázalo sa, že profil bezpečnosti Ilarisu je podobný ako u pacientov vo veku 2 rokov a starších.
V 16-týždňovej štúdii bolo 102 pacientov s TRAPS, HIDS/MKD a FMF (vo veku 2–17 rokov), ktorí dostávali kanakinumab. Celkovo sa nevyskytli žiadne klinicky významné rozdiely v profiloch bezpečnosti a znášanlivosti kanakinumabu u pediatrických pacientov v porovnaní s celkovou populáciou.
StaršiapopuláciaV profile bezpečnosti pozorovanom u pacientov vo veku ≥ 65 rokov nie je významný rozdiel.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieZaznamenané skúsenosti s predávkovaním sú obmedzené. V počiatočných klinických skúšaniach dostali pacienti a zdraví dobrovoľníci intravenózne alebo subkutánne podané dávky až 10 mg/kg bez preukázanej akútnej toxicity.
V prípade predávkovania sa odporúča u pacienta sledovať akékoľvek príznaky a prejavy nežiaducich
reakcií a okamžite začať náležitú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu, ATC kód: L04AC08
Mechanizmus účinku
Kanakinumab je ľudská monoklonálna protilátka proti ľudskému interleukínu-1 beta (IL-1 beta) izotypu IgG1/κ. Kanakinumab sa s vysokou afinitou viaže špecificky na ľudský IL-1 beta a neutralizuje biologickú aktivitu ľudského IL-1 beta blokovaním jeho interakcie s receptormi IL-1, čím bráni aktivácii génu vyvolanej IL-1 beta a tvorbe zápalových mediátorov.
Farmakodynamickéúčinky
CAPS, TRAPS, HIDS/MKD a FMF
V klinických štúdiách pacienti s CAPS, TRAPS, HIDS/MKD a FMF, ktorí majú nekontrolovanú nadmernú tvorbu IL-1 beta, vykazujú rýchlu a pretrvávajúcu odpoveď na liečbu kanakinumabom, t.j. laboratórne parametre, ako vysoké hladiny C-reaktívneho proteínu (CRP) a sérového amyloidu A (SAA), vysoký počet neutrofilov a trombocytov a leukocytóza, sa rýchlo vrátili na normálne hodnoty.
Stillova choroba (AOSD a SJIA)
Stillova choroba s nástupom ochorenia v dospelosti (AOSD) a systémová juvenilná idiopatická artritída sú závažné autoinflamačné choroby spôsobené vrodenou imunitou a sprostredkovaná prozápalovými cytokínmi, z ktorých kľúčový je IL-1-beta.
Časté príznaky AOSD a SJIA sú horúčka, exantém, hepatosplenomegália, lymfadenopatia, polyserozitída a artritída. Liečba kanakinumabom spôsobila rýchle a pretrvávajúce zmiernenie kĺbových aj systémových príznakov SJIA s významným poklesom počtu zapálených kĺbov, rýchlym odznením horúčky a znížením reaktantov akútnej fázy u väčšiny pacientov (pozri časť Klinická účinnosť a bezpečnosť).
Dnavá artritída
Záchvat dnavej artritídy je spôsobený kryštálmi urátu (monohydrát mononátriumurátu) v kĺbe a v okolitých tkanivách, čo spúšťa tvorbu IL-1 beta rezidentnými makrofágmi prostredníctvom
„inflamazómového komplexu NALP3“. Aktivácia makrofágov a súčasná nadmerná tvorba IL-1 beta vyvolávajú akútnu bolestivú zápalovú reakciu. Iné aktivátory vrodeného imunitného systému, ako sú endogénne agonisty „toll-like“ receptorov, môžu prispieť k transkripčnej aktivácii génu IL-1 beta a vyvolať záchvat dnavej artritídy. Po liečbe kanakinumabom rýchlo poklesli zápalové markery CRP alebo SAA a ustúpili prejavy akútneho zápalu (napr. bolesť, opuch, sčervenanie) v postihnutom kĺbe.
Klinickáúčinnosťabezpečnosť
CAPS
Účinnosť a bezpečnosť Ilarisu sa preukázali u pacientov s rôznymi stupňami závažnosti ochorenia a rôznymi fenotypmi CAPS (vrátane FCAS/FCU, MWS a NOMID/CINCA). Do hlavnej štúdie boli zaradení len pacienti s potvrdenou mutáciou NLRP3.
V štúdii fázy I/II mala liečba Ilarisom rýchly nástup účinku, s vymiznutím alebo klinicky významným zlepšením symptómov do jedného dňa po podaní. Laboratórne parametre, ako vysoké hladiny CRP a SAA, vysoký počet neutrofilov a trombocytov, sa normalizovali rýchlo, počas niekoľkých dní po injekcii Ilarisu.
Hlavná štúdia pozostávala zo 48-týždňovej multicentrickej štúdie zloženej z troch častí, t.j. 8- týždňového otvoreného obdobia (časť I), 24-týždňového randomizovaného, dvojito zaslepeného a placebom kontrolovaného obdobia vysadenia lieku (časť II), po ktorom nasledovalo 16-týždňové otvorené obdobie (časť III). Cieľom tejto štúdie bolo hodnotenie účinnosti, bezpečnosti a znášanlivosti Ilarisu (150 mg alebo 2 mg/kg každých 8 týždňov) u pacientov s CAPS.
- Časť I: Úplná klinická odpoveď a odpoveď biomarkerov na Ilaris (definovaná ako súhrn celkového hodnotenia autoinflamačného a kožného ochorenia lekárom ≤ minimálne a hodnôt CRP alebo SAA < 10 mg/l) sa pozorovala u 97 % pacientov a objavila sa do 7 dní od začatia liečby. V klinickom hodnotení aktivity autoinflamačného ochorenia lekárom sa pozorovali významné zlepšenia: pri celkovom hodnotení aktivity autoinflamačného ochorenia, hodnotení kožného ochorenia (kožný exantém typu urtikárie), artralgie, myalgie, bolesti hlavy/migrény, konjunktivitídy, únavy/pocitu nepohody, hodnotení iných súvisiacich symptómov a hodnotení symptómov pacientom.
- Časť II: V období vysadenia lieku počas hlavnej štúdie bol primárny koncový parameter definovaný ako podiel pacientov s relapsom/vzplanutím ochorenia: žiadny (0 %) z pacientov randomizovaných do skupiny Ilarisu nemal vzplanutie ochorenia, v porovnaní s 81 % pacientov randomizovaných do skupiny placeba.
- Časť III: Pacienti, ktorí v časti II dostávali placebo a u ktorých došlo k vzplanutiu ochorenia, po zaradení do otvorenej extenzie štúdie Ilarisu opäť dosiahli a udržali si klinickú a sérologickú odpoveď.
Tabuľka 2 Tabuľkové zhrnutie účinnosti v klinickom skúšaní fázy III, hlavné obdobie
vysadenia lieku kontrolované placebom (časť II)
Klinické skúšanie fázy III, hlavné obdobie vysadenia lieku kontrolované placebom (časť II)
Ilaris N=15 n(%)
Placebo N=16 n(%)
Hodnota p
P
rimárny parameter (vzplanutie ochorenia)
Podiel pacientov so vzplanutím ochorenia v
časti II
0 (0 %) 13 (81 %) < 0,001
Z
ápalové markery*
C-reaktívny proteín, mg/l 1,10 (0,40) 19,93 (10,50) < 0,001
Sérový amyloid A, mg/l 2,27 (-0,20) 71,09 (14,35) 0,002
* priemerná (medián) zmena od začiatku časti II
Vykonali sa dve otvorené, nekontrolované dlhodobé klinické skúšania fázy III. Jedno z nich bolo
skúšanie bezpečnosti, znášanlivosti a účinnosti kanakinumabu u pacientov s CAPS. Celkové trvanie liečby bolo v rozmedzí od 6 mesiacov do 2 rokov. Druhé bolo otvorené skúšanie kanakinumabu, ktoré hodnotilo účinnosť a bezpečnosť u japonských pacientov s CAPS počas 24 týždňov, s fázou extenzie až do 48 týždňov. Primárnym cieľom bolo stanovenie podielu pacientov, ktorí boli bez relapsov po
24. týždni, vrátane tých pacientov, ktorým sa dávka zvýšila.
Podľa združenej analýzy účinnosti týchto dvoch skúšaní dosiahlo 65,6 % pacientov, ktorí sa predtým neliečili kanakinumabom, úplnú odpoveď pri dávke 150 mg alebo 2 mg/kg, zatiaľ čo 85,2 % pacientov dosiahlo úplnú odpoveď pri akejkoľvek dávke. Z pacientov, ktorí dostávali 600 mg alebo
8 mg/kg (alebo ešte vyššiu dávku), 43,8 % dosiahlo úplnú odpoveď. Menej pacientov vo veku 2 až
< 4 roky dosiahlo úplnú odpoveď (57,1 %) v porovnaní so staršími pediatrickými a dospelými pacientmi. Z pacientov, ktorí dosiahli úplnú odpoveď, si 89,3 % odpoveď udržalo bez relapsov.
Skúsenosti u jednotlivých pacientov, ktorí dosiahli úplnú odpoveď po zvýšení dávky na 600 mg
(8 mg/kg) každých 8 týždňov, naznačujú, že vyššia dávka môže byť prínosom pre pacientov, ktorí nedosiahnu úplnú odpoveď alebo si neudržia úplnú odpoveď pri odporúčaných dávkach (150 mg alebo 2 mg/kg u pacientov ≥ 15 kg a ≤ 40 kg). Zvýšená dávka sa častejšie podávala pacientom vo veku 2 až < 4 roky a pacientom so symptómami NOMID/CINCA v porovnaní s pacientmi s FCAS alebo MWS.
Pediatrická populácia
Do klinických skúšaní Ilarisu pri CAPS bolo zaradených celkovo 80 pediatrických pacientov vo vekovom rozmedzí od 2 do 17 rokov (približne polovica z nich bola liečená na základe mg/kg). Celkovo sa nezistili žiadne klinicky významné rozdiely v účinnosti, bezpečnosti a znášanlivosti Ilarisu u detí a dospievajúcich v porovnaní s celkovou populáciou s CAPS. Väčšina pediatrických pacientov dosiahla zlepšenie v klinických príznakoch a v objektívnych zápalových markeroch (napr. SAA
a CRP).
Na vyhodnotenie účinnosti, bezpečnosti a znášanlivosti Ilarisu u pediatrických pacientov s CAPS vo
veku ≤ 4 roky sa vykonala otvorená štúdia trvajúca 56 týždňov. Vyhodnotilo sa 17 pacientov (vrátane
6 pacientov vo veku menej ako 2 roky), ktorí v závislosti od telesnej hmotnosti dostali začiatočné dávky 2-8 mg/kg. Štúdia tiež vyhodnotila účinok kanakinumabu na vznik protilátok proti vakcínam štandardne podávaným deťom. Nepozorovali sa rozdiely v bezpečnosti alebo účinnosti u pacientov mladších ako 2 roky v porovnaní s pacientmi vo veku 2 roky a staršími. U všetkých pacientov, ktorí dostali neživú vakcínu štandardne používanú u detí (N=7), sa vyvinuli ochranné hladiny protilátok.
TRAPS, HIDS/MKD a FMF
Účinnosť a bezpečnosť Ilarisu v liečbe TRAPS, HIDS/MKD a FMF sa preukázali v jednej pivotnej
štúdii fázy III so štyrmi časťami (N2301), ktorá pozostávala z troch samostatných skupín chorôb.
- Časť I: Pacienti v každej skupine chorôb vo veku 2 roky a starší sa zúčastnili 12-týždňového obdobia skríningu, počas ktorého sa u nich hodnotil nástup vzplanutia ochorenia.
- Časť II: Pacienti boli pri nástupe vzplanutia randomizovaní do 16-týždňového, dvojito zaslepeného, placebom kontrolovaného obdobia liečby, počas ktorého dostávali buď 150 mg Ilarisu (2 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) subkutánne (s.c.), alebo placebo každé 4 týždne. Pacientom vo veku > 28 dní, ale < 2 roky bolo povolené vstúpiť do štúdie priamo do skupiny otvorenej liečby v časti II ako nerandomizovaní pacienti (a boli vylúčení
z primárnej analýzy účinnosti).
- Časť III: Pacienti, ktorí ukončili 16 týždňov liečby a boli klasifikovaní ako pacienti odpovedajúci na liečbu, boli opäť randomizovaní do 24-týždňového, dvojito zaslepeného obdobia vysadenia lieku, počas ktorého dostávali 150 mg Ilarisu (2 mg/kg u pacientov
s telesnou hmotnosťou ≤ 40 kg) s.c., alebo placebo každých 8 týždňov.
- Časť IV: Všetci pacienti z časti III liečení Ilarisom boli vhodní na účasť v 72-týždňovom období predĺženia otvorenej liečby.
Celkovo 185 pacientov vo veku 28 dní a starších bolo zaradených a celkovo 181 pacientov vo veku
2 roky a starších bolo randomizovaných do časti II tejto štúdie.
Primárnym parametrom účinnosti randomizovaného obdobia liečby (časť II) bol podiel pacientov
s odpoveďou na liečbu v rámci každej skupiny, u ktorých došlo k poklesu indexu vzplanutia ochorenia na 15. deň a u ktorých sa nevyskytlo nové vzplanutie počas zostávajúceho 16-týždňového obdobia liečby (definované ako úplná odpoveď). Pokles indexu vzplanutia ochorenia bolo definovaný ako
skóre aktivity ochorenia < 2 („minimálne alebo žiadne ochorenie“) podľa globálneho hodnotenia lekárom (Physician´s Global Assessment, PGA) a CRP v rozmedzí normy (≤ 10 mg/l) alebo zníženie
≥ 70 % oproti východiskovej hodnote. Nové vzplanutie bolo definované ako skóre ≥ 2 („mierne, stredne ťažké alebo ťažké ochorenie“) podľa PGA a CRP ≥ 30 mg/ml. Sekundárne parametre, všetky založené na výsledkoch v 16. týždni (koniec časti II), zahŕňali podiel pacientov, ktorí dosiahli skóre
< 2 podľa PGA, podiel pacientov so sérologickou remisiou (definovaná ako CRP ≤ 10 mg/ml)
a podiel pacientov s normalizovanou hladinou SAA (definovaná ako SAA ≤ 10 mg/ml).
Z hľadiska primárneho parametra účinnosti bol Ilaris účinnejší ako placebo vo všetkých troch skupinách chorôb. Ilaris tiež preukázal vyššiu účinnosť v porovnaní s placebom pre sekundárne parametre PGA < 2 a CRP ≤ 10 mg/ml vo všetkých troch skupinách. Vyššie podiely pacientov mali normalizovanú SAA (≤ 10 mg/ml) v 16. týždni liečby Ilarisom v porovnaní s placebom vo všetkých troch skupinách, so štatisticky významným rozdielom pozorovaným u pacientov s TRAPS (pozri nižšie Tabuľku 3 s výsledkami štúdie).
Tabuľka 3 Tabuľkové zhrnutie účinnosti v klinickom skúšaní fázy III, pivotné,
randomizované, placebom kontrolované obdobie liečby (časť II)
Klinické skúšanie fázy III, pivotné, randomizované placebom kontrolované obdobie liečby
(časť II)
Ilaris n/N (%)
Placebo
n/N (%) Hodnota p
P
rimárny parameter (vzplanutie ochorenia) – podiel pacientov, u ktorých došlo k poklesu indexu vzplanutia ochorenia na 15. deň a u ktorých sa nevyskytlo nové vzplanutie počas zostávajúceho 16- týždňového obdobia liečby
FMF 19/31 (61,29) 2/32 (6,25) < 0,0001* HIDS/MKD 13/37 (35,14) 2/35 (5,71) 0,0020* TRAPS 10/22 (45,45) 2/24 (8,33) 0,0050*
Sekundárne parametre (ochorenie a zápalové markery)Globálne hodnotenie lekárom < 2
FMF 20/31 (64,52) 3/32 (9,38) < 0,0001** HIDS/MKD 17/37 (45,95) 2/35 (5,71) 0,0006** TRAPS 10/22 (45,45) 1/24 (4,17) 0,0028**
C-reaktívny proteín ≤ 10 mg/l
FMF 21/31 (67,74) 2/32 (6,25) < 0,0001** HIDS/MKD 15/37 (40,54) 2/35 (5,71) 0,0010** TRAPS 8/22 (36,36) 2/24 (8,33) 0,0149**
Sérový amyloid A ≤ 10 mg/l
FMF 8/31 (25,81) 0/32 (0,00) 0,0286
HIDS/MKD 5/37 (13,51) 1/35 (2,86) 0,0778
TRAPS 6/22 (27,27) 0/24 (0,00) 0,0235**
n=počet pacientov s odpoveďou na liečbu; N=počet vyhodnotiteľných pacientov
* indikuje štatistickú významnosť (jednostranná) s hladinou 0,025 na základe Fisherovho presného testu
** indikuje štatistickú významnosť (jednostranná) s hladinou 0,025 na základe modelu logistickej regresie so skupinou liečby a východiskovými hodnotami PGA, CRP alebo SAA ako vysvetľujúce premenné pre každú skupinu
Titrácia nahorV časti II štúdie dostali pacienti liečení Ilarisom, ktorí mali pretrvávajúcu aktivitu ochorenia, ďalšiu dávku 150 mg (alebo 2 mg/kg u pacientov s telesnou hmotnosťou ≤ 40 kg) počas prvého mesiaca. Táto ďalšia dávka sa mohla podať už 7 dní po prvej liečebnej dávke. Všetci pacienti s dávkou
titrovanou nahor zostali pri zvýšenej dávke 300 mg (alebo 4 mg/kg u pacientov s telesnou hmotnosťou
≤ 40 kg) každé 4 týždne.
V exploračnej analýze primárneho parametra sa pozorovalo, že u pacientov s nedostatočnou odpoveďou na liečbu po prvej dávke titrácia nahor počas prvého mesiaca na dávku 300 mg (alebo
4 mg/kg) každé 4 týždne ďalej zlepšila kontrolu vzplanutia, znížila aktivitu ochorenia a normalizovala hladiny CRP a SAA.
Pediatrickí pacienti:
Dvaja nerandomizovaní pacienti s HIDS/MKD vo veku > 28 dní, ale < 2 roky, boli zaradení do štúdie a dostávali kanakinumab. Jeden pacient mal pokles indexu vzplanutia na 15. deň po podaní jednej jednorazovej dávky kanakinumabu 2 mg/kg, ale liečbu prerušil po tejto prvej dávke pre závažné nežiaduce udalosti (pancytopénia a zlyhanie pečene). Tento pacient mal pri vstupe do štúdie imunitnú trombocytopenickú purpuru v anamnéze a aktívne ochorenie s abnormálnou funkciou pečene. Druhý pacient dostal začiatočnú dávku kanakinumabu 2 mg/kg a doplnkovú dávku 2 mg/kg v 3. týždni,
s titráciou dávky nahor v 5. týždni na podávanie 4 mg/kg každé 4 týždne až do konca časti II štúdie. Vymiznutie vzplanutia ochorenia sa dosiahlo v 5. týždni a u pacienta sa nevyskytlo žiadne nové vzplanutie na konci časti II štúdie (16. týždeň).
Stillova choroba
SJIA
Účinnosť Ilarisu v liečbe aktívnej SJIA sa vyhodnotila v dvoch pivotných klinických skúšaniach (G2305 a G2301). Pacienti zaradení do skúšaní boli vo veku 2 až < 20 rokov (priemerný vek 8,5 roka a priemerné trvanie choroby v čase zaradenia do skúšania 3,5 roka) a mali aktívnu chorobu definovanú ako ≥ 2 kĺby s aktívnou artritídou, horúčkou a zvýšeným CRP.
Klinické skúšanie G2305
G2305 bolo randomizované, dvojito zaslepené, placebom kontrolované, 4 týždne trvajúce skúšanie na stanovenie krátkodobej účinnosti Ilarisu u 84 pacientov randomizovaných na jednorazovú dávku
4 mg/kg (do 300 mg) Ilarisu alebo na placebo. Primárnym ukazovateľom bol podiel pacientov, u ktorých sa na 15. deň dosiahlo zlepšenie minimálne o 30 % v zmysle kritérií odpovede podľa pediatrickej American College of Rheumatology (ACR) upravených tak, aby zahŕňali neprítomnosť horúčky. Liečba Ilarisom zlepšila všetky skóre odpovede podľa pediatrickej ACR v porovnaní
s placebom na 15. aj 29. deň (Tabuľka 4).
Tabuľka 4 Odpoveď podľa pediatrickej ACR a stav choroby na 15. a 29. deň
15. deň 29. deň
Ilaris
N=43
Placebo
N=41
Ilaris
N=43
Placebo
N=41
ACR30 84 % 10 % 81 % 10 % ACR50 67 % 5 % 79 % 5 % ACR70 61 % 2 % 67 % 2 % ACR90 42 % 0 % 47 % 2 % ACR100 33 % 0 % 33 % 2 % Neaktívna choroba 33 % 0 % 30 % 0 % Rozdiel v liečbe pri všetkých skóre ACR bol významný (p ≤ 0,0001)
Výsledky zložiek upravenej odpovede podľa pediatrickej ACR, ktoré zahŕňali systémové a artritické
zložky, boli v súlade s celkovými výsledkami odpovede podľa ACR. Na 15. deň bol medián zmien oproti východiskovej hodnote v počte kĺbov s aktívnou artritítou -67 % a s obmedzeným rozsahom pohybu -73 % pri Ilarise (N=43) v porovnaní s mediánom zmien pri placebe 0 % a 0 % (N=41). Priemerná zmena v skóre bolesti podľa pacienta (vizuálna analogická škála 0-100 mm) na 15. deň bola pri Ilarise -50,0 mm (N=43) v porovnaní s placebom +4,5 mm (N=25). Priemerná zmena v skóre bolesti u pacientov liečených Ilarisom pretrvávala na 29. deň.
Klinické skúšanie G2301G2301 bolo randomizované, dvojito zaslepené, placebom kontrolované skúšanie Ilarisu na prevenciu vzplanutí pri vysadení liečby. Skúšanie pozostávalo z dvoch častí s dvoma nezávislými primárnymi ukazovateľmi (úspešné zníženie dávky steroidov a čas do vzplanutia). Do časti I (otvorená liečba) bolo zaradených 177 pacientov, ktorí dostávali 4 mg/kg (do 300 mg) Ilarisu každé 4 týždne počas najviac 32 týždňov. Pacienti v časti II (dvojito zaslepená liečba) dostávali buď 4 mg/kg Ilarisu, alebo placebo každé 4 týždne, až do výskytu 37 udalostí vzplanutia.
Znižovanie dávky kortikosteroidov:
Z celkového počtu 128 pacientov, ktorí pri vstupe do časti I užívali kortikosteroidy, sa 92 pokúsilo o zníženie dávky kortikosteroidov. Päťdesiatsedem (62 %) z 92 pacientov, ktorí sa pokúsili znížiť svoju dávku, bolo pri znížení dávky kortikosteroidov úspešných a 42 (46 %) kortikosteroidy vysadilo.
Čas do vzplanutia:
Pacienti, ktorí používali Ilaris v časti II, mali riziko udalosti vzplanutia znížené o 64 % v porovnaní so skupinou placeba (pomer rizika 0,36; 95 % IS: 0,17 až 0,75; p=0,0032). U 63 zo 100 pacientov, ktorí vstúpili do časti II, bez ohľadu na to, či boli pridelení na placebo alebo kanakinumab, sa nevyskytlo vzplanutie počas sledovaného obdobia (najviac 80 týždňov).
Výsledky klinických skúšaní G2305 a G2301 týkajúce sa zdravia a kvality života
Liečba Ilarisom vyvolala klinicky významné zlepšenie telesných funkcií a kvality života pacientov. V skúšaní G2305 bolo zlepšenie podľa dotazníka na hodnotenie zdravia v detskom veku (Childhood Health Assessment Questionnaire) metódou najmenších štvorcov pri Ilarise 0,69 oproti placebu, čo predstavuje 3,6-násobok minimálneho klinicky významného rozdielu 0,19 (p=0,0002). Medián zlepšenia oproti východiskovej hodnote do konca časti I skúšania G2301 bol 0,88 (79 %). Štatisticky významné zlepšenie skóre v dotazníku zdravia dieťaťa (Child Health Questionnaire-PF50) pri Ilarise v porovnaní s placebom bolo hlásené v skúšaní G2305 (telesné p=0,0012; psychosociálne zdravie p=0,0017).
Združená analýza účinnosti
Údaje z prvých 12 týždňov liečby Ilarisom v skúšaniach G2305, G2301 a v extenzii klinického skúšania sa združili na vyhodnotenie zachovania účinnosti. Tieto údaje preukázali do 12. týždňa oproti východiskovým hodnotám podobné zlepšenie podľa upravených odpovedí pediatrickej ACR a ich zložiek, aké sa pozorovalo v skúšaní kontrolovanom placebom (G2305). Po 12. týždni boli upravené odpovede podľa pediatrickej ACR30, 50, 70, 90 a 100: 70 %, 69 %, 61 %, 49 % a 30 %, choroba bola neaktívna u 28 % pacientov (N=178).
Účinnosť pozorovaná v skúšaniach G2305 a G2301 sa udržala počas pokračujúcej otvorenej, dlhodobej extenzie (údaje sú dostupné pre medián 49 týždňov následného sledovania). V tomto skúšaní sa u 25 pacientov, ktorí mali silnú odpoveď na liečbu podľa ACR počas najmenej 5 mesiacov, znížila dávka Ilarisu na 2 mg/kg každé 4 týždne, pričom odpoveď si títo pacienti udržali podľa pediatrickej ACR100 počas celého obdobia, keď sa im podávala znížená dávka (medián 32 týždňov,
8-124 týždňov).
Hoci sú údaje obmedzené, dôkazy z klinických skúšaní naznačujú, že pacienti, ktorí nereagujú na tocilizumab alebo anakinru, môžu reagovať na kanakinumab.
SJIA u mladých dospelých a AOSD
Účinnosť Ilarisu v podskupine mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov bola v súlade s účinnosťou, pozorovanou u pacientov mladších ako 16 rokov. Na základe literárnych údajov sa očakáva podobný profil účinnosti u AOSD pacientov ako u pacientov so SJIA.
Dnavá artritída
Účinnosť Ilarisu v liečbe akútnych záchvatov dnavej artritídy sa preukázala v dvoch multicentrických, randomizovaných, dvojito zaslepených klinických skúšaniach kontrolovaných účinným liekom u pacientov s častou dnavou artritídou (≥ 3 záchvaty počas predošlých 12 mesiacov), ktorí nemohli používať NSAID alebo kolchicín (pre kontraindikáciu, neznášanlivosť alebo nedostatočnú účinnosť). Klinické skúšania trvali 12 týždňov, po ktorých nasledovala 12-týždňová dvojito zaslepená extenzia. Celkovo 225 pacientov bolo liečených subkutánne podaným Ilarisom 150 mg a 229 pacientov bolo liečených intramuskulárne podaným triamcinolónacetonidom (TA) 40 mg pri zaradení do klinického skúšania a neskôr pri výskyte nového záchvatu. Priemerný počet záchvatov dnavej artritídy počas predošlých 12 mesiacov bol 6,5. Viac ako 85 % pacientov malo sprievodné choroby vrátane hypertenzie (60 %), diabetu (15 %), ischemickej choroby srdca (12 %) a chronickej choroby obličiek
v štádiu ≥ 3 (25 %). Približne u jednej tretiny zaradených pacientov (76 [33,8 %] v skupine Ilarisu a
84 [36,7 %] v skupine triamcinolónacetonidu) bolo zdokumentované, že nemôžu (pre intoleranciu, kontraindikáciu alebo nedostatočnú odpoveď) používať NSAID ani kolchicín. Súbežnú liečbu ULT hlásilo 42 % pacientov pri zaradení do klinického skúšania.
Súbežné primárne parametre boli: (i) intenzita bolesti pri dnavej artritíde (vizuálna analógová škála,
VAS) 72 hodín po podaní, a (ii) čas do prvého nového záchvatu dnavej artritídy.
V celkovej populácii v klinickom skúšaní bola intenzita bolesti po 72 hodinách štatisticky významne nižšia pri Ilarise 150 mg v porovnaní s triamcinolónacetonidom. Ilaris tiež znížil riziko ďalších záchvatov (pozri Tabuľku 5).
Výsledky účinnosti u pacientov, ktorí nemohli užívať NSAID ani kolchicín a ktorí dostávali ULT, u ktorých ULT nebola účinná alebo u ktorých bola ULT kontraindikovaná (N=101), sa zhodovali s výsledkami celkovej populácie v klinickom skúšaní, so štatisticky významným rozdielom oproti triamcinolónacetonidu pri intenzite bolesti po 72 hodinách (-10,2 mm, p=0,0208) a pri znížení rizika ďalších záchvatov (pomer rizika 0,39, p=0,0047 po 24 týždňoch).
Výsledky účinnosti v užšej podskupine obmedzenej na súčasných používateľov ULT (N=62) sú uvedené v Tabuľke 5. Liečba Ilarisom vyvolala zmiernenie bolesti a znížila riziko ďalších záchvatov u pacientov, ktorí používali ULT a nemohli používať NSAID ani kolchicín, hoci rozdiel pozorovaný pri liečbe v porovnaní s triamcinolónacetonidom bol menej výrazný ako v celkovej populácii v klinickom skúšaní.
Tabuľka 5 Účinnosť u celkovej populácie v klinickom skúšaní a u podskupiny pacientov v súčasnosti používajúcich ULT, ktorí nemôžu používať NSAID ani kolchicín
P
arameter účinnosti Celková populácia
v klinickom skúšaní; N=454
N
emožnosť používať NSAID
ani kolchicín; používanie
ULT
N=62
Liečbazáchvatovdnavej
artritídy
m
eraná ako intenzita bolesti (VAS) po 72 hodinách
Priemerný rozdiel oproti triamcinolónacetonidu odhadovaný metódou najmenších štvorcov
IS
Hodnota p, 1-stranná
-10,7
(-15,4, -6,0)
p < 0,0001*
-3,8
(-16,7, 9,1)
p=0,2798
Z
níženie rizika ďalších záchvatov dnavej artritídy merané ako čas do prvého nového vzplanutia
(
24 týždňov)
Pomer rizika oproti triamcinolón- acetonidu
IS
Hodnota p, 1-stranná
* Označuje významnú hodnotu p < 0,025
0,44
(0,32, 0,60)
p < 0,0001*
0,71
(0,29, 1,77)
p=0,2337
Výsledky bezpečnosti ukázali zvýšenú incidenciu nežiaducich udalostí pri kanakinumabe v porovnaní
s triamcinolónacetonidom, keď počas 24 týždňov akúkoľvek nežiaducu udalosť hlásilo 66 % oproti
53 % pacientov a infekciu ako nežiaducu udalosť hlásilo 20 % oproti 10 % pacientov.
Starší pacienti
Celkovo bol profil účinnosti, bezpečnosti a znášanlivosti Ilarisu u starších pacientov vo veku
≥ 65 rokov a vo veku < 65 rokov porovnateľný.
Pacienti, ktorí dostávajú liečbu znižujúcu urát (ULT)
V klinických skúšaniach sa Ilaris bezpečne podával s ULT. V celkovej populácii v klinickom skúšaní bol u pacientov, ktorí dostávali ULT, pri liečbe menej výrazný rozdiel v zmiernení bolesti aj znížení rizika ďalších záchvatov dnavej artritídy v porovnaní s pacientmi, ktorí nedostávali ULT.
Im
unogenita
Protilátky proti Ilarisu sa pozorovali u približne 1,5 % pacientov liečených Ilarisom pre CAPS, u 3% pacientov liečených pre SJIA a u 2 % pacientov liečených pre dnavú artritídu. Nezistili sa žiadne neutralizujúce protilátky. Nepozorovala sa žiadna zjavná korelácia medzi tvorbou protilátok
a klinickou odpoveďou alebo nežiaducimi udalosťami.
Nepozorovali sa žiadne protilátky proti Ilarisu u pacientov s TRAPS, HIDS/MKD a FMF, ktorí dostávali dávky 150 mg a 300 mg počas 16 týždňov liečby.
Tento liek bol registrovaný pre CAPS za tzv. mimoriadnych okolností. To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku. Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
Pediatrická populácia
Držiteľ rozhodnutia o registrácii uskutočnil štyri výskumné pediatrické plány pre Ilaris (pre CAPS, SJIA, FMF - HIDS/MKD a TRAPS). Táto informácia o lieku bola aktualizovaná, aby zahrnula výsledky štúdií s Ilarisom v pediatrickej populácii.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Ilarisom vo všetkých podskupinách pediatrickej populácie pre dnavú artritídu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
CAPS
Absorpcia
Maximálna koncentrácia kanakinumabu v sére (Cmax) sa dosiahla asi 7 dní po jednorazovom subkutánnom podaní 150 mg dospelým pacientom s CAPS. Priemerný terminálny polčas bol 26 dní. Priemerné hodnoty Cmax a AUCinf po jednorazovej subkutánnej dávke 150 mg u typického dospelého pacienta s CAPS (70 kg) boli 15,9 µg/ml a 708 µg*deň/ml. Odhad absolútnej biologickej dostupnosti subkutánne podaného kanakinumabu bol 66 %. Parametre expozície (napr. AUC a Cmax) sa zvyšovali úmerne dávke v rozmedzí dávok 0,30 až 10,0 mg/kg podávaných intravenóznou infúziou alebo od 150 do 600 mg subkutánnou injekciou. Predpokladané hodnoty expozície v rovnovážnom stave (Cmin,ss, Cmax,ss, AUC,ss,8w) po subkutánnom podávaní 150 mg (alebo 2 mg/kg) každých 8 týždňov boli o niečo vyššie v hmotnostnej kategórii 40-70 kg (6,6 µg/ml, 24,3 µg/ml, 767 µg*d/ml) v porovnaní
s hmotnostnými kategóriami < 40 kg (4,0 µg/ml, 19,9 µg/ml, 566 µg*d/ml) a > 70 kg (4,6 µg/ml,
17,8 µg/ml, 545 µg*d/ml). Očakávaný pomer akumulácie bol 1,3-násobný po 6 mesiacoch subkutánneho podávania 150 mg kanakinumabu každých 8 týždňov.
Distribúcia
Kanakinumab sa viaže na sérový IL-1 beta. Distribučný objem (Vss) kanakinumabu sa líšil podľa
telesnej hmotnosti. U pacienta s CAPS a telesnou hmotnosťou 70 kg sa odhadol na 6,2 l.
Eliminácia
Zdanlivý klírens (CL/F) kanakinumabu sa zvyšuje s telesnou hmotnosťou. U pacienta s CAPS
s telesnou hmotnosťou 70 kg sa odhadol na 0,17 l/d a u pacienta so SJIA s telesnou hmotnosťou 33 kg na 0,11 l/d. Po zohľadnení rozdielov v telesnej hmotnosti sa nepozorovali žiadne klinicky významné rozdiely vo farmakokinetických vlastnostiach kanakinumabu medzi pacientmi s CAPS a SJIA.
Po opakovanom podávaní kanakinumabu sa nezistil žiadny náznak zrýchleného klírensu alebo zmeny farmakokinetických vlastností v závislosti od času. Po korekcii na telesnú hmotnosť sa nepozorovali žiadne farmakokinetické rozdiely v súvislosti s pohlavím alebo vekom.
T
RA
PS, HIDS/MKD a FMF
Biologická dostupnosť sa nestanovila nezávisle u pacientov s TRAPS, HIDS/MKD a FMF. Zdanlivý klírens (CL/F) bol porovnateľný medzi populáciami s TRAPS, HIDS/MKD a FMF s telesnou hmotnosťou 55 kg (0,14 l/d) a s CAPS s telesnou hmotnosťou 70 kg (0,17 l/d). Zdanlivý distribučný objem (V/F) bol 4,96 l pri telesnej hmotnosti 55 kg.
Po opakovanom subkutánnom podávaní 150 mg každé 4 týždne bola minimálna koncentrácia kanakinumabu v 16. týždni (Cmin) odhadovaná na 15,4 ± 6,6 µg/ml. Odhadovaný rovnovážny stav AUCtau bol 636,7 ± 260,2 µg*d/ml.
Stillova choroba (AOSD a SJIA)
Biologická dostupnosť sa nestanovila nezávisle u pacientov so SJIA. Zdanlivý klírens na kg telesnej hmotnosti (CL/F na kg) bol porovnateľný medzi populáciami so SJIA a s CAPS (0,004 l/d na kg). Zdanlivý distribučný objem na kg (V/F na kg) bol 0,14 l/kg.
Po opakovanom podávaní 4 mg/kg každé 4 týždne pacientom so SJIA bol pomer akumulácie kanakinumabu 1,6-násobný. Rovnovážny stav sa dosiahol po 110 dňoch. Celkové predpokladané priemerné (±SD) hodnoty boli pre Cmin,ss 14,7±8,8 μg/ml, Cmax,ss 36,5 ± 14,9 μg/ml a AUC,ss4w 696,1 ±
326,5 μg*d/ml.
AUCss4w bola vo vekovej skupine 2-3 roky 692 µg*d/ml, 4-5 rokov 615 µg*d/ml, 6-11 rokov
707 µg*d/ml a 12-19 rokov 742 µg*d/ml. Po stratifikácii podľa telesnej hmotnosti sa pozoroval nižší
(30-40%) medián expozície pre Cmin,ss (11,4 oproti 19 µg/ml) a AUCss (594 oproti 880 µg*d/ml) pre
kategóriu nižšej telesnej hmotnosti (≤ 40 kg) oproti kategórii vyššej telesnej hmotnosti (> 40 kg).
Na základe analýzy populačného farmakokinetického modelovania bola farmakokinetika kanakinumabu u mladých dospelých pacientov so SJIA vo veku 16 až 20 rokov podobná ako
u pacientov mladších ako 16 rokov. Predpokladané expozície kanakinumabu v rovnovážnom stave pri veľkosti dávky 4 mg/kg (maximálne 300 mg) boli porovnateľné u pacientov nad 20 rokov a
u pacientov so SJIA mladších ako 20 rokov.
Populácia s dnavou artritídou
Biologická dostupnosť u pacientov s dnavou artritídou sa nezávisle nestanovila. Zdanlivý klírens na
kg telesnej hmotnosti (CL/F na kg) bol porovnateľný medzi populáciami s dnavou artritídou a s CAPS
(0,004 l/deň/kg). Priemerná expozícia u typického pacienta s dnavou artritídou (93 kg) po
jednorazovej subkutánnej dávke 150 mg (Cmax: 10,8 µg/ml a AUCinf: 495 µg*deň/ml) bola nižšia ako u typického pacienta s CAPS s telesnou hmotnosťou 70 kg (15,9 µg/ml a 708 µg*deň/ml). Zhoduje sa to s pozorovaným zvyšovaním CL/F s telesnou hmotnosťou.
Očakávaný pomer akumulácie bol 1,1-násobný po subkutánnom podávaní 150 mg kanakinumabu
každých 12 týždňov.
Pediatrická populácia
Maximálne koncentrácie kanakinumabu u pediatrických pacientov vo veku 4 rokov a starších sa dosiahli medzi 2. a 7. dňom (Tmax) po jednorazovom subkutánnom podaní 150 mg alebo 2 mg/kg kanakinumabu. Terminálny polčas bol v rozmedzí 22,9 až 25,7 dní, čo je podobné farmakokinetickým vlastnostiam pozorovaným u dospelých. Na základe analýzy populačného farmakokinetického modelovania bola farmakokinetika kanakinumabu u detí vo veku 2 až < 4 roky podobná ako
u pacientov vo veku 4 roky a starších. Odhaduje sa, že rýchlosť subkutánnej absorpcie klesá s vekom a zdá sa, že je najvyššia u najmladších pacientov. V súlade s tým bol Tmax kratší (3,6 dní) u mladších pacientov so SJIA (2-3 roky) v porovnaní so staršími pacientmi so SJIA (12-19 rokov; Tmax 6 dní). Biologická dostupnosť (AUCss) nebola ovplyvnená.
Ďalšia analýza farmakokinetiky ukázala, že farmakokinetika kanakinumabu u 6 pediatrických pacientov s CAPS mladších ako 2 roky bola podobná ako farmakokinetika u pediatrických pacientov vo veku 2-4 roky. Na základe analýzy populačného farmakokinetického modelovania boli očakávané expozície po dávke 2 mg/kg porovnateľné vo všetkých pediatrických vekových skupinách pri CAPS, ale boli približne o 40 % nižšie u pediatrických pacientov s veľmi nízkou telesnou hmotnosťou (napr.
10 kg) v porovnaní s dospelými pacientmi (dávka 150 mg). Zhoduje sa to s pozorovaniami vyššej
expozície u pacientov s CAPS v skupinách vyššej telesnej hmotnosti.
Pri TRAPS, HIDS/MKD a FMF boli parametre expozície (minimálne koncentrácie) porovnateľné vo všetkých vekových skupinách od 2 do < 20 rokov po subkutánnom podaní kanakinumabu 2 mg/kg každé 4 týždne.
Farmakokinetické vlastnosti sú podobné u pediatrických populácií s CAPS, TRAPS, HIDS/MKD, FMF a SJIA.
Staršípacienti
Rozdiel farmakokinetických parametrov na základe klírensu alebo distribučného objemu sa nepozoroval medzi staršími pacientmi a dospelými pacientmi vo veku < 65 rokov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií skríženej reaktivity, toxicity po opakovanom podávaní, imunotoxicity, reprodukčnej toxicity a toxicity pre mláďatá vykonaných s kanakinumabom alebo myšacou protilátkou proti myšaciemu IL-1 beta neodhalili žiadne osobitné riziko pre ľudí.
Keďže kanakinumab sa viaže na IL-1 beta kozmáčov (C. jacchus) a ľudí s podobnou afinitou, bezpečnosť kanakinumabu sa skúmala u kozmáčov. Žiadne nežiaduce účinky sa nepozorovali po podávaní kanakinumabu dvakrát týždenne kozmáčom počas 26 týždňov alebo v štúdii toxicity pre embryofetálny vývoj počas gestácie u kozmáčov. Plazmatické koncentrácie, ktoré dobre tolerujú,zvieratá, prevyšujú najmenej 42-násobne (Cmax) a 78-násobne (Cavg) plazmatické koncentrácie u detí a dospievajúcich s CAPS (telesná hmotnosť 10 kg) liečených klinickými dávkami kanakinumabu do 8 mg/kg subkutánne každých 8 týždňov. Plazmatické koncentrácie, ktoré zvieratá dobre znášajú, sú minimálne 62-krát vyššie (Cmax) a 104-krát vyššie (Cavg) ako plazmatické koncentrácie u pediatrických pacientov so SJIA, ktorí dostávajú dávky do 4 mg/kg subkutánne každé
4 týždne. Navyše sa v týchto štúdiách nezistili žiadne protilátky proti kanakinumabu. Pri aplikácii kanakinumabu do normálnych ľudských tkanív sa nepreukázala nešpecifická tkanivová skrížená reaktivita.
Formálne štúdie karcinogenity s kanakinumabom sa nevykonali.
V štúdii toxicity pre embryofetálny vývoj kozmáčov kanakinumab nevykazoval pri podávaní počas
organogenézy toxicitu pre matky, embryotoxicitu ani teratogenitu.
V kompletnom súbore reprodukčných štúdií a štúdií na mláďatách myší sa nepozorovali nežiaduce účinky myšacej protilátky proti myšaciemu IL-1 beta. Protilátka proti myšaciemu IL-1 beta nevyvolala pri podávaní počas neskorej gestácie, pôrodu a laktácie nežiaduce udalosti súvisiace s rastom plodov alebo novorodencov (pozri časť 4.6). Vysoká dávka použitá v týchto štúdiách bola vyššia ako maximálna účinná dávka z hľadiska supresie IL-1 beta a aktivity.
Imunotoxikologická štúdia u myší s myšacou protilátkou proti myšaciemu IL-1 beta ukázala, že neutralizácia IL-1 beta nemá žiadne účinky na imunitné parametre a nespôsobila zhoršenie imunitnej funkcie u myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol
L-histidín
Monohydrát L-histidíniumchloridu
Polysorbát 80
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
Z mikrobiologického hľadiska sa liek má použiť okamžite po prvom otvorení.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
1 ml injekčného roztok v injekčnej liekovke (sklo typu I) so zátkou (chlórobutylová guma s laminátovou vrstvou) a odtrhávacím „flip/off“ uzáverom (hliník).
Balenia obsahujúce 1 injekčnú liekovku.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Ilaris 150 mg/ml injekčný roztok sa dodáva v jednorazovej liekovke na individuálne použitie. Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
Pokyny na podanie
Nechajte injekčnú liekovku zohriať na izbovú teplotu pred podaním injekcie. Roztok má byť prakticky
bez viditeľných častíc a číry až opalizujúci. Roztok má byť bezfarebný alebo môže mať slabé hnedožlté sfarbenie. Pomocou ihly s veľkosťou 18 G alebo 21 G x 2 palce (alebo podobnou ihlou, ako je dostupná na trhu) a 1 ml injekčnej striekačky opatrne odoberte požadovaný objem v závislosti od dávky, ktorá sa má podať. Po odobratí požadovaného objemu znova nasaďte kryt na ihlu a odstráňte ihlu na odobratie roztoku z injekčnej striekačky a nasaďte ihlu 27 G x 0,5 palca (alebo podobnú ihlu, ako je dostupná na trhu), aby ste roztok ihneď subkutánne injikovali.
Likvidácia
Injekčná liekovka je určená na jednorazové použitie. Zvyškový objem sa má zlikvidovať ihneď po podaní injekcie. Pacienti alebo ich opatrovatelia majú dostať pokyny, ako v súlade s národnými požiadavkami správne postupovať pri likvidácii injekčných liekoviek, striekačiek a ihiel.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLAEU/1/09/564/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. október 2009
Dátum posledného predĺženia registrácie: 19. jún 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu