GAZYVARO 1 000 MG INFÚZNY KONCENTRÁT con inf 1x40 ml (liek inj.skl.)

/>
Všetci pacienti Perorálne analgetikum/antipyretikum
Pacienti s IRR (1. alebo
infúziou Gazyvara
1100 mg prednizónu/prednizolónu alebo 20 mg dexametazónu alebo 80 mg metylprednizolónu
Hydrokortizón sa nemá použiť, keďže nie je účinný v znižovaní výskytu IRR.
2 napr. 1 000 mg acetaminofenu/paracetamolu
3 napr. 50 mg difenhydramínu
Dávka
Odporúčaná dávka Gazyvara je uvedená v tabuľke 2.
1. cyklus
Odporúčaná dávka Gazyvara je 1 000 mg podaná počas 1. a 2. dňa, v 8. deň a 15. deň počas prvého
28-dňového liečebného cyklu. Na infúziu v 1. a 2. deň majú byť pripravené 2 infúzne vaky (100 mg v deň 1 a 900 mg v deň 2). Ak je podávanie prvého vaku ukončené bez úpravy rýchlosti infúzie alebo bez jej prerušenia, druhý infúzny vak môže byť podaný v ten istý deň (nie je potrebné oddialenie dávky, nie je potrebné zopakovať premedikáciu) za predpokladu, že je k dispozícii dostatočný čas, podmienky a lekársky dohľad počas podávania infúzie. Ak sú počas podávania prvých 100 mg potrebné úpravy rýchlosti infúzie alebo prerušenia infúzie, druhý vak sa musí podať na nasledujúci deň.
2. - 6. cyklus
Odporúčaná dávka Gazyvara je 1 000 mg podaná v 1. deň.
Tabuľka 2 Dávka Gazyvara, ktorá sa má podávať počas 6 liečebných cyklov každých 28 dní
Cyklus
|
Deň liečby
|
Dávka Gazyvara
|
1. cyklus
|
1. deň
|
100 mg
|
2. deň
(alebo pokračujúci 1. deň)
|
900 mg
|
8. deň
|
1 000 mg
|
15. deň
|
1 000 mg
|
2. - 6. cyklus
|
1. deň
|
1 000 mg
|
Dĺžka liečby
Šesť liečebných cyklov, z ktorých každý trvá 28 dní.
Odložené alebo vynechané dávkyAk sa vynechá plánovaná dávka Gazyvara, má sa podať čo najskôr; nemá sa čakať do ďalšej plánovanej dávky. Medzi dávkami Gazyvara sa má dodržiavať plánovaný liečebný interval.
Úpravy dávky počas liečbyNeodporúčajú sa žiadne zníženia dávky Gazyvara.
Osobitné skupiny pacientovStaršie osobyU starších pacientov nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiekU pacientov s miernou až stredne ťažkou poruchou funkcie obličiek (klírens kreatinínu [CrCl]
30 - 89 ml/min) nie je potrebná žiadna úprava dávky (pozri časť 5.2). Bezpečnosť a účinnosť Gazyvara u pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu [CrCl] < 30 ml/min) neboli stanovené.
Porucha funkcie pečeneBezpečnosť a účinnosť Gazyvara u pacientov s poruchou funkcie pečene neboli stanovené. Nie je možné uviesť odporúčania na dávkovanie.
Pediatrická populáciaBezpečnosť a účinnosť Gazyvara u detí a dospievajúcich mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávaniaGazyvaro je na intravenózne použitie. Má sa po nariedení (pozri časť 6.6) podávať intravenóznou
infúziou cez osobitnú infúznu hadičku. Infúzny roztok Gazyvara sa nemá podávať formou pretlakovej infúzie (tzv. i.v.
push) ani intravenóznej bolusovej injekcie.
Pokyny na riedenie Gazyvara pred podaním, pozri časť 6.6. Pokyny týkajúce sa rýchlosti infúzie sú uvedené v tabuľke 3.
Tabuľka 3 Štandardná rýchlosť infúzie pri neprítomnosti reakcií na infúziu/precitlivenosti
Cyklus
|
Deň liečby
|
Rýchlosť infúzie
|
1. cyklus
|
1. deň
(100 mg)
|
Podávajte rýchlosťou 25 mg/h počas 4 hodín. Rýchlosť infúzie sa nemá zvyšovať.
|
2. deň
(alebo pokračujúci
1. deň) (900 mg)
|
Podávajte rýchlosťou 50 mg/h.
Rýchlosť infúzie sa môže zvyšovať
v prírastkoch o 50 mg/h každých 30 minút do maximálnej rýchlosti 400 mg/h.
|
8. deň
|
Infúzia sa môže začať podávať rýchlosťou
100 mg/h a zvyšovať v prírastkoch o 100 mg/h každých 30 minút do maximálnej rýchlosti
400 mg/h.
|
15. deň
|
2. - 6. cyklus
|
1. deň
|
Zvládnutie IRR si môže vyžiadať dočasné prerušenie podávania infúzie, zníženie rýchlosti jej
podávania alebo ukončenie liečby Gazyvarom, ako je uvedené nižšie (pozri aj časť 4.4).
• 4. stupeň (život ohrozujúca): Podávanie infúzie sa musí zastaviť a liečba sa musí natrvalo ukončiť.
• 3. stupeň (závažná): Podávanie infúzie sa musí dočasne zastaviť a príznaky sa musia liečiť.
Po odznení príznakov sa infúzia môže znovu začať podávať najviac polovičnou rýchlosťou (s použitím rýchlosti v čase, keď sa vyskytla IRR) a ak sa u pacienta nevyskytnú žiadne príznaky IRR, môže sa znovu začať so zvyšovaním rýchlosti podávania infúzie v prírastkoch a intervaloch vhodných pre liečebnú dávku (pozri tabuľku 3). Rýchlosť infúzie v 1. deň
(1. cyklu) sa môže až na 25 mg/h po 1 hodine, ale ďalej sa už nesmie zvyšovať. Podávanie infúzie sa musí zastaviť a liečba natrvalo ukončiť, ak sa u pacienta druhýkrát vyskytne IRR
3. stupňa.
• 1. - 2. stupeň (mierna až stredne závažná): Rýchlosť podávania infúzie sa musí znížiť
a príznaky sa musia liečiť. V podávaní infúzie sa môže pokračovať po odznení príznakov a ak sa u pacienta nevyskytnú žiadne príznaky IRR, môže sa znovu začať so zvyšovaním rýchlosti podávania infúzie v prírastkoch a intervaloch vhodných pre liečebnú dávku (pozri tabuľku 3). V
1. deň (1. cyklu) sa rýchlosť infúzie môže znovu zvýšiť až na 25 mg/h po 1 hodine, ale ďalej sa už nesmie zvyšovať.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníNa zlepšenie sledovateľnosti biologických liekov sa má v zdravotnej dokumentácii pacienta jasne zaznamenať (alebo uviesť) obchodný názov podaného lieku.
Reakcie súvisiace s infúziou (IRR)Najčastejšie pozorované nežiaduce reakcie na liek (
adverse drug reactions, ADR) u pacientov
liečených Gazyvarom boli IRR, ktoré sa vyskytovali hlavne počas infúzie prvých 1 000 mg. U pacientov, u ktorých sa vykonali kombinované opatrenia na prevenciu IRR (adekvátny glukokortikoid, perorálne analgetikum/antihistaminikum, vynechanie antihypertenzívneho lieku ráno v deň prvej infúzie a podávanie dávky určenej na 1. deň 1. cyklu počas 2 dní) tak, ako sú opísané
v časti 4.2, sa pozoroval znížený výskyt IRR všetkých stupňov. Výskyt IRR 3. - 4. stupňa (ktorý sa
zakladal na relatívne malom počte pacientov) bol pred zavedením zmierňujúcich opatrení a po ich zavedení podobný. Zmierňujúce opatrenia na zníženie výskytu IRR sa majú dodržiavať (pozri
časť 4.2). Výskyt a závažnosť príznakov súvisiacich s infúziou sa podstatne znížili po infúznom podaní prvých 1 000 mg, pričom väčšina pacientov nemala žiadne IRR počas nasledujúcich podaní Gazyvara (pozri časť 4.8).
U väčšiny pacientov boli IRR mierne až stredne závažné a bolo ich možné zvládnuť spomalením alebo dočasným zastavením podávania prvej infúzie, ale hlásené boli aj závažné a život ohrozujúce IRR vyžadujúce symptomatickú liečbu. IRR môžu byť klinicky nerozoznateľné od alergických reakcií sprostredkovaných imunoglobulínom E (IgE) (napr. anafylaxie). Pacienti s vysokou nádorovou
záťažou (t.j. vysoký počet lymfocytov v periférnej krvi pri CLL (> 25 x 109/l) môžu mať zvýšené riziko závažných IRR. Pacienti s poruchou funkcie obličiek (CrCl < 50 ml/min) a pacienti
so súčasným skóre komorbidity Cumulative Illness Rating Scale (CIRS) > 6 a CrCl < 70 ml/min majú zvýšené riziko IRR, vrátane závažných IRR (pozri časť 4.8)
Pri použití Gazyvara boli hlásené aj prípady syndrómu z uvoľnenia cytokínov. Informácie o profylaxii, pozri časť 4.2.
Ak sa u pacienta vyskytne IRR, podávanie infúzie sa má riadiť stupňom reakcie. Pri IRR 4. stupňa sa musí podávanie infúzie zastaviť a liečba natrvalo ukončiť. Pri IRR 3. stupňa sa musí podávanie infúzie dočasne prerušiť a podať vhodný liek na liečbu príznakov. Pri IRR 1. - 2. stupňa sa musí podávanie infúzie spomaliť a príznaky sa musia náležite liečiť. Po odznení príznakov sa infúzia môže znovu
začať podávať, okrem IRR 4. stupňa, a to najviac polovičnou rýchlosťou oproti predchádzajúcej rýchlosti a ak sa u pacienta nevyskytne rovnaká nežiaduca udalosť s rovnakou závažnosťou, môže sa znovu začať so zvyšovaním rýchlosti podávania infúzie v prírastkoch a intervaloch vhodných pre liečebnú dávku. Ak predchádzajúca rýchlosť infúzie nebola dobre znášaná, majú sa použiť pokyny pre rýchlosť infúzie v 1. a 2. deň 1. cyklu (pozri tabuľku 3 v časti 4.2).
Pacienti nesmú dostať ďalšie infúzie Gazyvara, ak sa u nich objavia:
• akútne život ohrozujúce respiračné príznaky,
• IRR 4. stupňa (t.j. život ohrozujúca) alebo
• druhýkrát sa vyskytujúca (dlhotrvajúca/opakovaná) IRR 3. stupňa (po znovu začatí podávania prvej infúzie alebo počas nasledujúcej infúzie).
Pacienti, ktorí majú už existujúce ochorenie srdca alebo pľúcne ochorenie, majú byť starostlivo sledovaní počas celej doby podávania infúzie a v období po podaní infúzie. Počas intravenóznych infúzií Gazyvara sa môže vyskytnúť hypotenzia. Preto sa má uvažovať o tom, že sa antihypertenzívne lieky dočasne vysadia 12 hodín pred každou infúziou Gazyvara, počas celej doby jej podávania a prvú hodinu po jej podaní. U pacientov s akútnym rizikom hypertenznej krízy sa majú zhodnotiť prínosy
a riziká dočasného vysadenia ich antihypertenzívneho lieku.
Reakcie z precitlivenosti vrátane anafylaxie
U pacientov liečených Gazyvarom bola hlásená anafylaxia. Môže byť ťažké rozoznať precitlivenosť
od IRR. Ak počas podávania infúzie vznikne podozrenie na reakciu z precitlivenosti (napr. príznaky typicky sa vyskytujúce po predchádzajúcej expozícii a veľmi zriedkavo pri prvej infúzii), musí sa podávanie infúzie zastaviť a liečba natrvalo ukončiť. Pacienti so známou IgE sprostredkovanou precitlivenosťou na obinutuzumab sa nesmú liečiť (pozri časť 4.3).
Syndróm rozpadu nádoru (TLS)
Počas liečby Gazyvarom bol hlásený syndróm rozpadu nádoru (TLS). Pacienti, ktorí sa považujú
za rizikových z hľadiska vzniku TLS (napr. pacienti s veľkou nádorovou záťažou alebo s vysokým počtom cirkulujúcich lymfocytov [> 25 x 109/l]), majú 12 - 24 hodín pred infúziou Gazyvara začať dostávať adekvátnu profylaxiu proti rozpadu nádoru urikostatikami (napr. alopurinolu) a hydratácii (pozri časť 4.2). Pri liečbe TLS je potrebné korigovať abnormality elektrolytov, sledovať funkciu obličiek a rovnováhu tekutín a podať podpornú liečbu vrátane dialýzy, ak je to indikované.
Neutropénia
Počas liečby Gazyvarom bola hlásená závažná a život ohrozujúca neutropénia vrátane febrilnej
neutropénie. Pacienti, u ktorých sa vyskytne neutropénia, sa majú pozorne sledovať pravidelnými laboratórnymi vyšetreniami až do odznenia. Ak je potrebná liečba, má sa podávať v súlade
s miestnymi odporúčaniami a má sa zvážiť podávanie faktorov stimulujúcich kolónie granulocytov. Akékoľvek prejavy sprievodnej infekcie sa majú náležite liečiť. V prípade závažnej alebo život ohrozujúcej neutropénie sa má zvážiť odloženie podania dávky. Dôrazne sa odporúča, aby pacienti so závažnou a dlhotrvajúcou (> 1 týždeň) neutropéniou počas obdobia liečby dostávali antimikrobiálnu
profylaxiu, pokým sa nezmierni na 1. alebo 2. stupeň. Má sa zvážiť antivírusová a antimykotická
profylaxia. Hlásené boli aj prípady neutropénie s oneskoreným nástupom (vyskytujúcej sa 28 dní po ukončení liečby) alebo dlhotrvajúcej neutropénie (trvajúcej dlhšie ako 28 dní po ukončení/zastavení liečby). Pacienti s poruchou funkcie obličiek (CrCl < 50 ml/min) majú zvýšené riziko vzniku neutropénie (pozri časť 4.8).
Trombocytopénia
Počas liečby Gazyvarom bola pozorovaná závažná a život ohrozujúca trombocytopénia vrátane
akútnej trombocytopénie (vyskytujúcej sa do 24 hodín po infúzii). Pacienti s poruchou funkcie obličiek (CrCl < 50 ml/min) majú zvýšené riziko vzniku trombocytopénie (pozri časť 4.8).
U pacientov liečených Gazyvarom boli hlásené aj fatálne hemoragické príhody v 1. cykle. Jasná súvislosť medzi trombocytopéniou a hemoragickými príhodami sa nestanovila.
Pacienti majú byť pozorne sledovaní na trombocytopéniu, najmä počas prvého cyklu; majú sa vykonávať pravidelné laboratórne vyšetrenia až do odznenia udalosti a v prípade závažnej alebo život ohrozujúcej trombocytopénie sa má zvážiť odloženie podania dávky. Podanie transfúzie krvných preparátov (t.j. transfúzia krvných doštičiek) v súlade s postupmi pracoviska je na zvážení ošetrujúceho lekára. Do úvahy treba tiež vziať všetky súbežne užívané lieky, ktoré by mohli zhoršiť udalosti súvisiace s trombocytopéniou, ako napríklad inhibítory agregácie krvných doštičiek alebo antikoagulanciá, najmä počas prvého cyklu.
Zhoršenie už existujúcich ochorení srdca
U pacientov so základným ochorením srdca sa počas liečby Gazyvarom vyskytli arytmie (ako je
atriálna fibrilácia a tachyarytmia), angína pektoris, akútny koronárny syndróm, infarkt myokardu
a srdcové zlyhávanie (pozri časť 4.8). Tieto kardiálne príhody sa môžu vyskytnúť ako súčasť IRR
a môžu byť fatálne. Preto pacienti s ochorením srdca v anamnéze majú byť pozorne sledovaní. Okrem toho majú byť títo pacienti hydratovaní s obozretnosťou, aby sa predišlo možnému preťaženiu tekutinami.
Infekcie
Gazyvaro sa nemá podávať v prítomnosti aktívnej infekcie a je potrebná obozretnosť, keď sa uvažuje
o použití Gazyvara u pacientov s opakujúcimi sa alebo chronickými infekciami v anamnéze. Počas liečby Gazyvarom a po jej ukončení sa môžu vyskytnúť závažné bakteriálne, mykotické a novo vzniknuté alebo reaktivované vírusové infekcie. Hlásené boli fatálne infekcie. Pacienti so súčasným skóre komorbidity Cumulative Illness Rating Scale (CIRS) > 6 a CrCl < 70 ml/min majú zvýšené riziko infekcií, vrátane závažných infekcií (pozri časť 4.8).
Reaktivácia vírusu hepatitídy B
U pacientov liečených protilátkami proti CD20 vrátane Gazyvara môže dôjsť k reaktivácii vírusu
hepatitídy B (HBV), ktorá v niektorých prípadoch vedie k fulminantnej hepatitíde, k zlyhaniu pečene a k smrti (pozri časť 4.8). Pred začiatkom liečby Gazyvarom sa má u všetkých pacientov vykonať skríning na vírus hepatitídy B. Má pozostávať minimálne z vyšetrenia povrchového antigénu hepatitídy B (HBsAg) a jadrového antigénu hepatitídy B (HBcAb). Tieto môžu byť doplnené inými vhodnými markermi podľa miestnych odporúčaní. Pacienti s aktívnou hepatitídou B sa nemajú liečiť
Gazyvarom. Liečba pacientov so sérologickou pozitivitou hepatitídy B má byť pred začiatkom konzultovaná s hepatológom a pacienti majú byť prísne sledovaní a liečení v súlade s lokálnymi postupmi, aby sa predišlo reaktivácii hepatitídy.
Progresívna multifokálna leukoencefalopatia (PML)
U pacientov liečených Gazyvarom bola hlásená progresívna multifokálna leukoencefalopatia (PML)
(pozri časť 4.8). Diagnóza PML sa má zvážiť u každého pacienta, u ktorého sa objavia nové neurologické prejavy alebo zmeny v už existujúcich neurologických prejavoch. Príznaky PML sú nešpecifické a môžu varírovať v závislosti od postihnutej oblasti mozgu. Časté sú motorické príznaky s nálezmi súvisiacimi s kortikospinálnym traktom (napr. svalová slabosť, paralýza a senzorické poruchy), senzorické abnormality, cerebelárne príznaky a poruchy zrakového poľa. Môžu sa vyskytnúť niektoré prejavy/príznaky považované za „kortikálne“ (napr. afázia alebo
zrakovo-priestorová dezorientácia). Vyšetrovanie možnej PML zahŕňa, ale neobmedzuje sa len na, konzultáciu s neurológom, zobrazenie mozgu magnetickou rezonanciou (MRI) a lumbálnu punkciu (vyšetrenie mozgovomiechového moku na prítomnosť DNA vírusu JC (pomenovaného podľa Johna Cunninghama). Liečba Gazyvarom sa má počas vyšetrovania potenciálnej PML prerušiť a v prípade potvrdenej PML natrvalo ukončiť. Má sa zvážiť aj ukončenie podávania alebo zníženie dávok akejkoľvek súbežnej chemoterapie alebo imunosupresívnej liečby. Pacient sa má odoslať
k neurológovi na vyšetrenie a liečbu PML.
Imunizácia
Bezpečnosť imunizácie živými alebo oslabenými vírusovými očkovacími látkami po liečbe
Gazyvarom sa nesledovala a očkovanie živými vírusovými očkovacími látkami sa neodporúča počas liečby a až do obnovy počtu B-buniek.
Expozícia obinutuzumabu in utero a očkovanie novorodencov živou vírusovou očkovacou látkou
Pre možnú depléciu B-buniek u novorodencov po vystavení obinutuzumabu počas tehotenstva má byť u novorodencov monitorovaná deplécia B-buniek a očkovanie živými vírusovými očkovacími látkami má byť odložené až do obnovy počtu B-buniek (pozri časť 4.6).
4.5 Liekové a iné interakcie Neuskutočnili sa žiadne interakčné štúdie. Farmakokinetické interakcie
Obinutuzumab nie je substrátom, inhibítorom ani induktorom cytochrómu P450 (CYP450), enzýmov
uridín difosfát glukuronyltransferázy (UGT) a transportérov ako P-glykoproteín. Preto sa nepredpokladá žiadna farmakokinetická interakcia s liekmi, o ktorých je známe, že sú metabolizované týmito enzymatickými systémami.
Farmakodynamické interakcie
Očkovanie živými vírusovými očkovacími látkami sa kvôli imunosupresívnemu účinku
obinutuzumabu počas liečby a až do obnovy B-buniek neodporúča (pozri časť 4.4).
Kombinácia obinutuzumabu s chlorambucilom môže zvýšiť neutropéniu (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby Gazyvarom a počas
18 mesiacov po liečbe Gazyvarom.
Gravidita
Reprodukčná štúdia na opiciach rodu Cynomolgus nepreukázala žiadne známky embryofetálnej
toxicity alebo teratogénnych účinkov, viedla však k úplnej deplécii B-lymfocytov u potomkov. Počet
B-buniek sa u potomkov vrátil do referenčných hodnôt a imunologická funkcia sa obnovila
do 6 mesiacov od narodenia. Okrem toho, sérové koncentrácie obinutuzumabu u potomkov boli zhodné so sérovými koncentráciami u matiek v 28. deň po pôrode, svedčiac o prechode obinutuzumabu placentou (pozri časť 5.3). K dispozícii nie sú žiadne údaje o použití obinutuzumabu u gravidných žien. Gazyvaro sa nemá podávať gravidným ženám, pokiaľ možný prínos neprevažuje možné riziko.
V prípade expozície počas tehotenstva môže byť v dôsledku farmakokinetických vlastností lieku očakávaná u novorodencov deplécia B-buniek. Preto má byť u novorodencov monitorovaná deplécia B-buniek a očkovanie živými vírusovými očkovacími látkami má byť odložené až do obnovy počtu B-buniek (pozri časť 4.4).
Laktácia
Štúdie na zvieratách preukázali vylučovanie obinutuzumabu do materského mlieka (pozri časť 5.3).
Keďže ľudský imunoglobulín G (IgG) sa vylučuje do ľudského mlieka a možnosť absorpcie
a poškodenia dôjčaťa je neznáma, ženám sa má odporučiť, aby ukončili dojčenie počas liečby
Gazyvarom a počas 18 mesiacov po poslednej dávke Gazyvara (pozri časť 5.3).
Fertilita
Neuskutočnili sa žiadne špecifické štúdie na zvieratách hodnotiace vplyv obinutuzumabu na fertilitu.
V štúdiách toxicity po opakovanom podávaní na opiciach rodu Cynomolgus sa nepozorovali žiadne nežiaduce účinky na reprodukčné orgány samcov a samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Gazyvaro nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. IRR sú počas prvej infúzie Gazyvarom veľmi časté a pacientom, u ktorých sa vyskytnú reakcie súvisiace s infúziou, sa má odporučiť, aby neviedli vozidlá alebo neobsluhovali stroje, pokým príznaky neustúpia.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Nežiaduce reakcie na liek (ADR) opísané v tejto časti sa zistili počas liečby a v období sledovania
v pilotnej klinickej štúdii, BO21004/CLL11, v ktorej sa Gazyvaro podával v kombinácii
s chlorambucilom v porovnaní so samotným chlorambucilom (štádium 1) alebo s rituximabom plus chlorambucilom (štádium 2). V skupine pacientov liečených Gazyvarom v kombinácii
s chlorambucilom podstúpilo všetkých 6 liečebných cyklov 81 % pacientov v porovnaní
s 89 % pacientov v skupine s rituximabom plus chlorambucilom a 67 % pacientov v skupine so samotným chlorambucilom.
Najčastejšie pozorované ADR u pacientov liečených Gazyvarom boli IRR, ktoré sa vyskytli u väčšiny pacientov počas prvého cyklu (pozri časť 4.4). Výskyt príznakov súvisiacich s infúziou sa podstatne znížil zo 65 % pri infúzii prvých 1 000 mg Gazyvara na menej ako 3 % pri nasledujúcich infúziách.
V pilotnej štúdii sa neutropénia vyskytla u 41 % pacientov a trombocytopénia u 15 % pacientov
a výskyt infekcie 3. - 5. stupňa v skupine s Gazyvarom plus chlorambucilom bol 16 % (pozri časť 4.4).
Ďalšie závažné ADR hlásené počas klinického vývoja zahŕňajú syndróm rozpadu nádoru, kardiálne príhody a veľmi zriedkavo PML (pozri časť 4.4).
V tabuľke 4 sú zhrnuté ADR, ktoré sa vyskytli s vyšším výskytom (s ≥ 2 % rozdielom) u pacientov liečených Gazyvarom plus chlorambucilom v porovnaní so samotným chlorambucilom alebo
s rituximabom plus chlorambucilom.
Frekvencie sú definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000
až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) a veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky uvedené v poradí podľa klesajúcej závažnosti.
Tabuľkový súhrn nežiaducich reakcií
Tabuľka 4 Súhrn ADR hlásených s vyšším výskytom (s ≥ 2 % rozdielom) u pacientov liečených
Gazyvarom plus chlorambucilom v porovnaní so samotným chlorambucilom alebo s rituximabom plus chlorambucilom (štúdia BO21004/CLL11)*
Frekvencia
| Všetky stupne závažnosti % Gazyvaro + chlorambucil
| 3. - 5. stupeň závažnosti† % Gazyvaro + chlorambucil
|
Infekcie a nákazy
|
Časté
| Infekcia močových ciest,
nazofaryngitída, orálny herpes, rinitída‡, faryngitída
| Infekcia močových ciest
|
Menej časté
|
| Nazofaryngitída
|
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
|
Časté
| Spinocelulárny karcinóm kože
| Spinocelulárny karcinóm kože
|
Poruchy krvi a lymfatického systému
|
Veľmi časté
| Neutropénia, trombocytopénia,
anémia
| Neutropénia, trombocytopénia
|
Časté
| Leukopénia
| Anémia, leukopénia
|
Poruchy metabolizmu a výživy
|
Časté
| Syndróm rozpadu nádoru,
hyperurikémia
| Syndróm rozpadu nádoru
|
Menej časté
|
| Hyperurikémia
|
Poruchy srdca a srdcovej činnosti
|
Časté
| Atriálna fibrilácia
|
|
Menej časté
|
| Atriálna fibrilácia
|
Poruchy ciev
|
Časté
| Hypertenzia
| Hypertenzia
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Časté
| Kašeľ
|
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
| Hnačka
|
|
Časté
| Zápcha
| Hnačka
|
Poruchy kože a podkožného tkaniva
|
Alopécia
| Alopécia
|
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Časté
| Artralgia, bolesť chrbta, bolesť
svalov a kostí hrudníka
|
|
Menej časté
|
| Artralgia, bolesť chrbta, bolesť
svalov a kostí hrudníka
|
Celkové poruchy a reakcie v mieste podania
|
Veľmi časté
| Pyrexia
|
|
Menej časté
|
| Pyrexia
|
Laboratórne a funkčné vyšetrenia
|
Časté
| Znížený počet bielych krviniek‡,
znížený počet neutrofilov, zvýšená telesná hmotnosť
| Znížený počet bielych krviniek‡,
znížený počet neutrofilov
|
Úrazy, otravy a komplikácie liečebného postupu
|
Veľmi časté
| Reakcie súvisiace s infúziou
| Reakcie súvisiace s infúziou
|
* Pri všetkých stupňoch alebo pri 3. - 5. stupni.
† Nepozorovali sa žiadne nežiaduce reakcie 5. stupňa s ≥ 2 % rozdielom vo výskyte medzi liečebnými skupinami
‡ Hoci bola táto nežiaduca udalosť hlásená s ≥ 2 % rozdielom vo výskyte medzi liečebnými skupinami v štádiu 1 štúdie, pri aktualizácii údajov zo štádia 1 alebo v údajoch zo štádia 2 už viac nebola hlásená s ≥ 2 % rozdielom vo výskyte medzi
liečebnými skupinami.
Popis vybraných nežiaducich reakciíReakcie súvisiace s infúziou (IRR)Výskyt IRR bol vyšší v skupine s Gazyvarom plus chlorambucilom v porovnaní so skupinou
s rituximabom plus chlorambucilom. Výskyt IRR bol 65 % pri infúzii prvých 1 000 mg Gazyvara
(u 20 % pacientov sa vyskytli IRR 3. - 5. stupňa, bez hlásených fatálnych udalostí). U celkovo
7 % pacientov sa vyskytla IRR vedúca k ukončeniu liečby Gazyvarom. Výskyt IRR pri nasledujúcich infúziách bol 3 % pri druhej 1 000 mg dávke a 1 % pri ďalších infúziách. Neboli hlásené žiadne IRR
3. - 5. stupňa okrem tých, ktoré sa vyskytli pri infúzii prvých 1 000 mg v 1. cykle.
Najčastejšie hlásené príznaky súvisiace s IRR boli nauzea, triaška, hypotenzia, pyrexia, vracanie, dyspnoe, návaly tepla, hypertenzia, bolesť hlavy, tachykardia a hnačka. Hlásené boli aj respiračné a kardiálne príznaky ako je bronchospazmus, podráždenie hrtana a hrdla, pískavé dýchanie, edém hrtana a atriálna fibrilácia (pozri časť 4.4).
Neutropénia a infekcie
Výskyt neutropénie bol vyšší v skupine s Gazyvarom plus chlorambucilom v porovnaní so skupinou
s rituximabom plus chlorambucilom, pričom neutropénia odznela spontánne alebo s použitím faktorov stimulujúcich kolónie granulocytov. Výskyt infekcie bol 38 % v skupine s Gazyvarom plus chlorambucilom a 37 % v skupine s rituximabom plus chlorambucilom (udalosti 3. - 5. stupňa boli hlásené u 12 % a 14 % v uvedenom poradí a fatálne udalosti boli hlásené u < 1 % v oboch liečebných skupinách). Hlásené boli aj prípady dlhotrvajúcej neutropénie (2 % v skupine s Gazyvarom plus chlorambucilom a 4 % v skupine s rituximabom plus chlorambucilom) a neutropénia s oneskoreným nástupom (16 % v skupine s Gazyvarom plus chlorambucilom a 12 % v skupine s rituximabom plus chlorambucilom) (pozri časť 4.4).
Trombocytopénia
Výskyt trombocytopénie bol vyšší v skupine s Gazyvarom plus chlorambucilom v porovnaní
so skupinou s rituximabom plus chlorambucilom, hlavne počas prvého cyklu. U 4 % pacientov liečených Gazyvarom plus chlorambucilom sa vyskytla akútna trombocytopénia (vyskytujúca sa
do 24 hodín po infúzii Gazyvara) (pozri časť 4.4). Celkový výskyt hemoragických príhod bol medzi skupinou liečenou Gazyvarom a skupinou liečenou rituximabom podobný. Počet fatálnych hemoragických príhod bol medzi liečebnými skupinami vyvážený; avšak všetky príhody u pacientov liečených Gazyvarom boli hlásené v 1. cykle. Jasná súvislosť medzi trombocytopéniou
a hemoragickými príhodami sa nestanovila.
Osobitné skupiny pacientov
Staršie osoby
V pilotnej štúdii bolo 46 % (156 z 336) pacientov s CLL liečených Gazyvarom plus chlorambucilom
75-ročných alebo starších (priemerný vek bol 74 rokov). U týchto pacientov sa vyskytlo viac závažných nežiaducich udalostí a nežiaducich udalostí vedúcich k smrti ako u pacientov vo veku < 75 rokov.
Porucha funkcie obličiek
V pilotnej štúdii malo 27 % (90 z 336) pacientov s CLL liečených Gazyvarom plus chlorambucilom stredne ťažkú poruchu funkcie obličiek (klírens kreatinínu CrCl < 50 ml/min). U týchto pacientov sa
vyskytlo viac závažných nežiaducich udalostí a nežiaducich udalostí vedúcich k smrti ako u pacientov
s klírenskom kreatinínu CrCl ≥ 50 ml/min.
Ďalšie údaje o bezpečnosti zo skúseností z klinických štúdií
Progresívna multifokálna leukoencefalopatia (PML)
U pacientov liečených Gazyvarom boli hlásené prípady PML (pozri časť 4.4).
Reaktivácia vírusu hepatitídy B
U pacientov liečených Gazyvarom boli hlásené prípady reaktivácie vírusu hepatitídy B (pozri
časť 4.4).
Zhoršenie už existujúcich ochorení srdca
U pacientov liečených Gazyvarom sa vyskytli prípady arytmie (ako napríklad atriálna fibrilácia
a tachyarytmia), angína pektoris, akútny koronárny syndróm, infarkt myokardu a srdcové zlyhávanie
(pozri časť 4.4). Tieto udalosti sa môžu vyskytnúť ako súčasť IRR a môžu byť fatálne.
Laboratórne abnormality
Krátko po prvej infúzii Gazyvara bol pozorovaný prechodný vzostup hladín pečeňových enzýmov
(aspartátaminotransferáza [AST], alanínaminotransferáza [ALT], alkalická fosfatáza).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieZ klinických štúdií s ľuďmi nie sú k dispozícii žiadne skúsenosti s predávkovaním. V klinických štúdiách s Gazyvarom boli podávané dávky v rozmedzí od 50 mg až do a vrátane 2 000 mg v jednej infúzii. Výskyt a intenzita nežiaducich reakcií hlásených v týchto štúdiách sa nezdali byť závislé
od dávky.
U pacientov, u ktorých dôjde k predávkovaniu, sa má ihneď prerušiť podávanie infúzie alebo zredukovať jej podávanie a pacienti sa majú pozorne sledovať. Má sa zvážiť potreba pravidelného kontrolovania krvného obrazu a sledovania kvôli zvýšenému riziku infekcií v období, keď majú pacienti depléciu B-buniek.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, monoklonálne protilátky, ATC kód: L01XC15
Mechanizmus účinkuObinutuzumab je rekombinantná monoklonálna humanizovaná anti-CD20 monoklonová protilátka
izotypu IgG1 II. typu pripravená pomocou génového inžinierstva. Špecificky sa zameriava na extracelulárnu slučku transmembránového antigénu CD20 na povrchu nemalígnych a malígnych pre-B a zrelých B-lymfocytov, ale nie na hematopoetických kmeňových bunkách, pro-B bunkách, normálnych plazmatických bunkách alebo na bunkách iných normálnych tkanív. Upravenie Fc fragmentu obinutuzumabu technikou génového inžinierstva vedie k vyššej afinite k receptoru FcɣRIII
na efektorových bunkách imunitného systému akými sú NK bunky, magrofágy a monocyty v porovnaní s génovým inžinierstvom neupravenými protilátkami.
V predklinických štúdiách obinutuzumab indukuje priamu smrť buniek a sprostredkováva od protilátok závislú cytotoxicitu (ADCC) a od protilátok závislú fagocytózu (ADCP) prostredníctvom
zapojenia FcɣRIII pozitívnych efektorových buniek imunitného systému. Okrem toho,
in vivo, obinutuzumab sprostredkováva nízky stupeň od komplementu závislej cytotoxicity (CDC).
V porovnaní s protilátkami I. typu sa obinutuzumab, protilátka II. typu, vyznačuje zvýšenou indukciou priamej smrti buniek so súbežným znížením CDC, keď sa podávajú v rovnakej dávke. Obinutuzumab, génovým inžinierstvom upravená protilátka, sa vyznačuje zvýšenou od protilátky závislou cytotoxicitou (ADCC) v porovnaní s génovými inžinierstvom neupravenými protilátkami, keď sa podávajú v rovnakej dávke. U zvieracích modelov obinutuzumab sprostredkováva účinnú depléciu
B-buniek a protinádorový efekt.
V pilotnej klinickej štúdii BO21004/CLL11 dosiahlo 91 % (40 zo 44) hodnotiteľných pacientov liečených Gazyvarom depléciu B-buniek (definovanú ako počet CD19+ B-buniek < 0,07 x 109/l)
na konci obdobia liečby a deplécia sa udržala počas prvých 6 mesiacov sledovania. Obnova B-buniek
sa pozorovala v priebehu 12 - 18 mesiacov sledovania až u 35 % (14 zo 40) pacientov bez progresie ochorenia a 13 % (5 zo 40) s progresiou ochorenia.
Klinická účinnosť a bezpečnosť
Medzinárodná, multicentrická, otvorená, randomizovaná klinická štúdia fázy III s dvoma štádiami
a s tromi skupinami (BO21004/CLL11) skúmajúca účinnosť a bezpečnosť Gazyvara plus chlorambucilu (GClb) v porovnaní s rituximabom plus chlorambucilom (RClb) alebo so samotným chlorambucilom (Clb) sa uskutočnila u pacientov s dovtedy neliečenou chronickou lymfocytovou leukémiou s komorbiditami.
Pred zaradením do štúdie musela byť u pacientov zdokumentovaná CD20+ CLL a jedno alebo obidve z nasledujúcich hodnotení koexistujúcich ochorení: skóre komorbidity (CIRS), vyššie ako 6 alebo znížená funkcia obličiek pod hodnotou CrCl < 70 ml/min. Pacienti s nedostatočnou funkciou pečene (National Cancer Institute - Common Terminology Criteria for Adverse Events 3. stupňa hodnôt pečeňových funkcií (AST, ALT > 5-násobok hornej hranice referenčného rozpätia (ULN)
počas > 2 týždňov; bilirubín > 3-násobok ULN)) a s nedostatočnou funkciou obličiek
(CrCl < 30 ml/min) boli vylúčení. Pacienti s poškodením jedného alebo viacerých jednotlivých orgánov/orgánových systémov ohodnoteným bodovým skóre 4 podľa definície CIRS s výnimkou očí, uší, nosa, hrdla a hrtanu boli z účasti na štúdii vylúčení.
Celkovo 781 pacientov bolo randomizovaných v pomere 2:2:1 na podávanie Gazyvara plus chlorambucilu, rituximabu plus chlorambucilu alebo samotného chlorambucilu. V štádiu 1a bol porovnávaný Gazyvaro plus chlorambucil so samotným chlorambucilom u 356 pacientov a v štádiu 2 bol porovnávaný Gazyvaro plus chlorambucil s rituximabom plus chlorambucilom u 663 pacientov. Výsledky účinnosti sú zhrnuté v tabuľke 5 a v grafoch 1 - 3.
U väčšiny pacientov sa Gazyvaro podal intravenózne vo forme 1 000 mg úvodnej dávky podanej v 1. deň, 8. deň a 15. deň prvého liečebného cyklu. Aby sa u pacientov znížil výskyt reakcií súvisiacich s rýchlosťou infúzie, schéma podávania bola pozmenená a 140 pacientom sa prvá dávka Gazyvara podávala počas 2 dní (1. deň [100 mg] a 2. deň [900 mg]) (pozri časť 4.2 a 4.4). V každom
ďalšom liečebnom cykle (2. až 6. cyklus) sa Gazyvaro podal pacientom v dávke 1 000 mg len v 1. deň. Chlorambucil sa podával perorálne v dávke 0,5 mg/kg telesnej hmotnosti v 1. deň a 15. deň všetkých liečebných cyklov (1. až 6).
Demografické údaje a východiskové charakteristiky boli medzi liečebnými skupinami dobre
vyvážené. Väčšinu pacientov tvorili belosi (95 %) a muži (61 %). Priemerný vek bol 73 rokov, pričom
44 % bolo 75-ročných alebo starších. Pred začiatkom liečby (baseline) malo 22 % pacientov ochorenie v štádiu A podľa Bineta, 42 % malo ochorenie v štádiu B podľa Bineta a 36 % malo ochorenie
v štádiu C podľa Bineta.
Priemerné skóre komorbidity bol 8 a 76 % zaradených pacientov malo skóre komorbidy nad 6. Medián odhadovanej hodnoty CrCl bol 62 ml/min a 66 % zo všetkých pacientov malo hodnotu CrCl < 70 ml/min. Štyridsaťdva percent zaradených pacientov malo hodnotu CrCl < 70 ml/min
a zároveň aj skóre komorbidity > 6. Tridsaťštyri percent pacientov bolo zaradených len na základe samotného skóre komorbidity a 23 % pacientov bolo zaradených len na základe samotnej poruchy funkcie obličiek.
Najčastejšie hlásené koexistujúce ochorenia (s použitím 30 % alebo vyššej hraničnej hodnoty)
v orgánových systémoch podľa MedDRA sú: poruchy ciev (73 %), poruchy srdca a srdcovej činnosti (46 %), poruchy gastrointestinálneho traktu (38 %), poruchy metabolizmu a výživy (40 %), poruchy obličiek a močových ciest (38 %), poruchy kostrovej a svalovej sústavy a spojivového tkaniva (33 %).
Tabuľka 5 Súhrn účinnosti zo štúdie BO21004/CLL11
|
Štádium 1a
|
Štádium 2
|
|
Chlorambucil
N = 118
|
Gazyvaro +
chlorambucil
N = 238
|
Rituximab +
chlorambucil
N = 330
|
Gazyvaro +
chlorambucil
N = 333
|
|
22,8-mesačný medián času pozorovania
|
18,7-mesačný medián času pozorovania
|
Primárny cieľový ukazovateľ
|
PFS hodnotené skúšajúcimi lekármi
(PFS-INV)
a
Počet (%) pacientov s udalosťou
Medián trvania PFS (mesiace) Hazard ratio (95 % IS)
p-hodnota (stratifikovaný log-rank testb)
|
96 (81,4 %) 93 (39,1 %)
11,1 26,7
0,18 [0,13; 0,24]
< 0,0001
|
199 (60,3 %) 104 (31,2 %)
15,2 26,7
0,39 [0,31; 0,49]
< 0,0001
|
Kľúčové sekundárne cieľové ukazovatele
|
PFS hodnotené nezávislou posudkovou komisiou (PFS-IRC)
a
Počet (%) pacientov s udalosťou Medián trvania PFS (mesiace) Hazard ratio (95 % IS)
p-hodnota (stratifikovaný log-rank testb)
|
'
90 (76,3 %) 89 (37,4 %)
11,2 27,2
0,19 [0,14; 0,27]
< 0,0001
|
183 (55,5 %) 103 (30,9 %)
14,9 26,7
0,42 [0,33; 0,54]
< 0,0001
|
V
ýskyt odpovede pri ukončení liečby
Počet pacientov zahrnutých v analýze Pacienti, ktorí odpovedali na liečbu (%)
Pacienti, ktorí neodpovedali na liečbu
(%)
Rozdiel vo výskyte odpovede na liečbu, (95 % IS)
p-hodnota (Chí-kvadrátový test) Počet pacientov, ktorí dosiahli úplnú
odpoveď na liečbuc (%)
|
118 238
37 (31,4 %) 184 (77,3 %)
81 (68,6 %) 54 (22,7 %)
45,95 [35,6; 56,3]
< 0,0001
0 (0,0 %) 53 (22,3 %)
|
329 333
214 (65,0 %) 261 (78,4 %)
115 (35,0 %) 72 (21,6 %)
13,33 [6,4; 20,3]
0,0001
23 (7,0 %) 69 (20,7 %)
|
Molekulová remisia na konci liečby
d
Počet pacientov zahrnutých v analýze
MRD negatívni pacientie (%) MRD pozitívni pacientif (%)
Rozdiel vo výskyte MRD, (95 % IS)
|
90 168
0 (0 %) 45 (26,8 %)
90 (100 %) 123 (73,2 %)
26,79 [19,5; 34,1]
|
244 239
6 (2,5 %) 61 (25,5 %)
238 (97,5 %) 178 (74,5 %)
23,06 [17,0; 29,1]
|
Prežívanie bez výskytu udalosti
Počet (%) pacientov s udalosťou
Medián času do vzniku udalosti
(mesiace)
Hazard ratio (95 % IS)
p-hodnota (stratifikovaný log-rank testb)
|
103 (87,3 %) 104 (43,7 %)
10,8 26,1
0,19 [0,14; 0,25]
< 0,0001
|
208 (63,0 %) 118 (35,4 %)
14,3 26,1
0,43 [0,34; 0,54]
< 0,0001
|
|
|
|
|
|
|
|
Štádium 1a
|
Štádium 2
|
|
Chlorambucil
N = 118
|
Gazyvaro +
chlorambucil
N = 238
|
Rituximab +
chlorambucil
N = 330
|
Gazyvaro +
chlorambucil
N = 333
|
|
22,8-mesačný medián času pozorovania
|
18,7-mesačný medián času pozorovania
|
Čas do novej protileukemickej liečby
Počet (%) pacientov s udalosťou Medián trvania udalosti (mesiace) Hazard ratio (95 % IS)
p-hodnota (stratifikovaný log-rank testb)
|
65 (55,1 %) 51 (21,4 %)
14,8 -
0,24 [0,16; 0,35]
< 0,0001
|
86 (26,1 %) 55 (16,5 %)
30,8 -
0,59 [0,42; 0,82]
< 0,0018
|
Celkové prežívanie
Počet (%) pacientov s udalosťou
Medián času do vzniku udalosti
(mesiace)
Hazard ratio (95 % IS)
p-hodnota (stratifikovaný log-rank testb)
|
24 (20,3 %) 22 (9,2 %) NR NR
0,41 [0,23; 0,74]
0,0022
|
41 (12,4 %) 28 (8,4 %) NR** NR**
0,66 [0,41; 1,06] **
0,0849**
|
|
|
|
|
|
|
PFS: prežívanie bez progresie ochorenia (
progression-free survival); HR: Hazard ratio; IS: interval
spoľahlivosti, MRD: minimálne reziduálne ochorenie (
Minimal Residual Disease)
a Definované ako čas od randomizácie do prvého výskytu progresie ochorenia, relapsu alebo smrti z akejkoľvek príčiny hodnotené skúšajúcim lekárom
b stratifikovaný podľa štádia ochorenia podľa Bineta pred začiatkom liečby
c Zahŕňa 11 pacientov v skupine s GClb s úplnou odpoveďou na liečbu a s neúplnou obnovou kostnej drene
d Hodnotená kombinovane v krvi a v kostnej dreni
e Negativita MRD je definovaná ako výsledok pod 0,0001
f Zahŕňa MRD pozitívnych pacientov a pacientov, u ktorých došlo k progresii ochorenia alebo ktorí zomreli pred koncom liečby
NR = Nedosiahnuté
** Údaje ešte nie sú konečné
Celkové prežívanie v štádiu 1a je zobrazené v grafe 2. Celkové prežívanie v štádiu 2 bude naďalej sledované a údaje o ňom ešte nie sú konečné. Výsledky analýzy PFS v podskupinách (t.j. pohlavie, vek, štádium ochorenia podľa Bineta, CrCl, CIRS skóre, beta2-mikroglobulín, IGVH status, chromozómové abnormality, počet lymfocytov pred začiatkom liečby) sa zhodovali s výsledkami pozorovanými v celkovej populácii (
Intent-to-Treat population). Riziko progresie ochorenia alebo smrti bolo znížené v skupine GClb v porovnaní so skupinou RClb a so skupinou Clb vo všetkých podskupinách s výnimkou podskupiny pacientov s deléciou 17p. V malej podskupine pacientov
s deléciou 17p bol pozorovaný len pozitívny trend v porovnaní s Clb (HR = 0,42, p = 0,0892); nebol pozorovaný žiaden benefit v porovnaní s RClb. Zníženie rizika progresie ochorenia alebo smrti sa
v podskupinách pohybovala od 92 % do 58 % pre GClb verzus Clb a od 72 % do 29 % pre GClb verzus RClb.
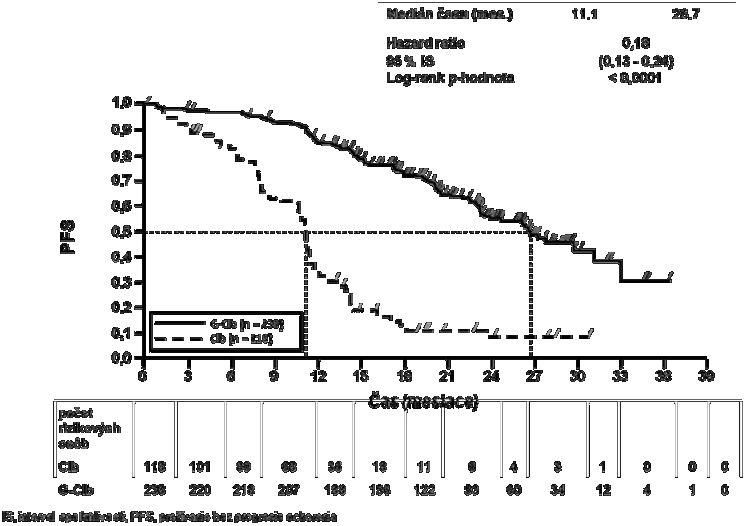 Graf 1 Kaplanova-Meierova krivka prežívania bez progresie ochorenia hodnoteného skúšajúcimi lekármi zo štádia 1a
Graf 1 Kaplanova-Meierova krivka prežívania bez progresie ochorenia hodnoteného skúšajúcimi lekármi zo štádia 1a

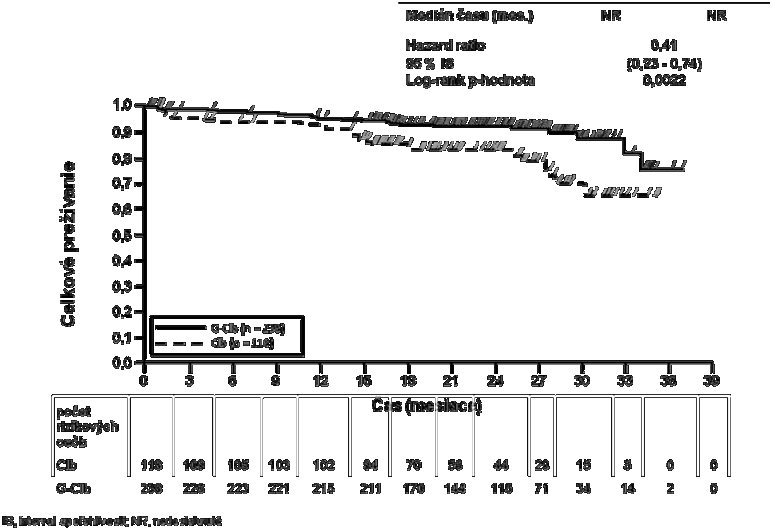 Graf 2 Kaplanova-Meierova krivka celkového prežívania zo štádia 1a
Graf 2 Kaplanova-Meierova krivka celkového prežívania zo štádia 1a

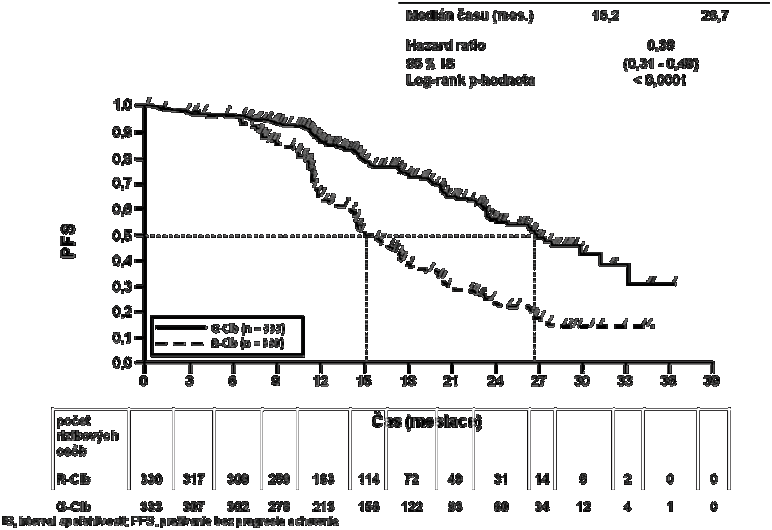 Graf 3 Kaplanova-Meierova krivka prežívania bez progresie ochorenia hodnoteného skúšajúcimi lekármi zo štádia 2
Graf 3 Kaplanova-Meierova krivka prežívania bez progresie ochorenia hodnoteného skúšajúcimi lekármi zo štádia 2
 Kvalita života
Kvalita života
Nepozoroval sa žiaden podstatný rozdiel v niektorej z podškál dotazníkov QLQC30 a QLQ-CLL-16
vyplnených počas obdobia liečby. Údaje získané v období sledovania sú obmedzené, najmä pokiaľ ide o skupinu so samotným chlorambucilom. Doteraz sa však v období sledovania nezistili žiadne význačné rozdiely v kvalite života.
Hodnotenia kvality života súvisiacej so zdravím, ktoré boli zamerané na únavu počas celého obdobia liečby, neukázali štatisticky významný rozdiel poukazujúci na to, že by pridanie Gazyvara k režimu na báze chlorambucilu zvyšovalo výskyt únavy u pacientov.
ImunogenicitaPacienti v pilotnej štúdii BO21004/CLL11 boli vo viacerých časových bodoch testovaní na prítomnosť protilátok proti lieku Gazyvaru (
anti-therapeutic antibodies, ATA). U pacientov liečených Gazyvarom sa u 8 zo 140 pacientov v randomizovanej fáze a u 2 zo 6 pacientov v úvodnej fáze zistila pozitivita
ATA v 12 mesačnom sledovaní. Z týchto pacientov sa u žiadneho sa nevyskytli anafylaktické reakcie
alebo reakcie z precitlivenosti, ktoré by sa považovali za súvisiace s ATA, ani u nich nebola ovplyvnená klinická odpoveď na liečbu.
Výsledky testu imunogenicity vo vysokej miere závisia od niekoľkých faktorov vrátane senzitivity a špecificity testu, metodológie testu, citlivosti testu na kvantitu Gazyvara v cirkulácii, zaobchádzania so vzorkami, času odberu vzorky, súbežne podávaných liekov a základného ochorenia. Z týchto dôvodov môže byť porovnanie výskytu protilátok proti Gazyvaru s výskytom protilátok proti iným liekom zavádzajúce.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Gazyvarom
vo všetkých podskupinách pediatrickej populácie s chronickou lymfocytovou leukémiou (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Vyvinutý bol populačný farmakokinetický model na analyzovanie FK údajov získaných
od 678 pacientov s non-Hodgkinovým lymfómom (NHL) a s CLL zo štúdií fázy I, fázy II a fázy III, ktorým bol podávaný obinutuzumab. Tento populačný FK model sa použil na opísanie FK vlastností obinutuzumabu u pacientov s CLL.
Absorpcia
Obinutuzumab sa podáva intravenózne, a preto je hodnotenie absorpcie neaplikovateľné.
Neuskutočnili sa žiadne štúdie s inými spôsobmi podávania. Na základe populačného FK modelu sa stanovilo, že u pacientov s CLL bol po infúzii podanej v 1. deň 6. cyklu odhadovaný medián hodnoty Cmax 473,2 μg/ml a hodnoty AUC(τ) 9 516 μg•d/ml.
Distribúcia
Po intravenóznom podaní sa hodnota distribučného objemu centrálneho kompartmentu (2,76 l)
približuje hodnote objemu séra, čo naznačuje, že distribúcia sa z veľkej miery obmedzuje na plazmu a intersticiálnu tekutinu.
Biotransformácia
Metabolizmus obinutuzumabu sa priamo neskúmal. Protilátky podliehajú výlučne katabolizmu.
Eliminácia
U pacientov s CLL je klírens obinutuzumabu v 6. cykle približne 0,083 l/deň s priemerom eliminácie
t½ 30,3 dňa. Eliminácia obinutuzumabu pozostáva z modelu časovo premenlivého klírensu s dvoma paralelnými cestami popisujúcimi klírens, a to cestou lineárneho klírensu a cestou nelineárneho klírensu, ktorý sa mení v závislosti od času. Na začiatku liečby prevláda cesta nelineárneho časovo premenlivého klírensu a predstavuje hlavnú cestu klírensu. Ako liečba napreduje, vplyv tejto cesty sa zmenšuje a prevláda cesta lineárneho klírensu. To naznačuje vylučovanie liečiva sprostredkované farmakologickým cieľom (target mediated drug disposition, TMDD), keď počiatočné veľké množstvo CD20 pozitívnych buniek spôsobuje rýchlu depléciu obinutuzumabu. Avšak len čo sa väčšina CD20 pozitívnych buniek naviaže na obinutuzumab, dochádza k zníženiu vplyvu TMDD na FK.
Farmakokinetický(é)/farmakodynamický(é) vzťah(y)
V populačnej farmakokinetickej analýze sa zistilo, že pohlavie je kovariantom, ktorý v určitej miere
vysvetľuje interindividuálnu variabilitu s u mužov o 22 % vyšším klírensom v rovnovážnom stave (CLss) a distribučným objemom (V) vyšším o 18 % . Výsledky populačnej farmakokinetickej analýzy však ukázali, že rozdiely v expozícii nie sú významné (s odhadovaným mediánom AUC 11 282
µg•d/ml a Cmax 578,9 µg/ml u žien a 8 451 µg•d/ml a 432,5 µg/ml u mužov v uvedenom poradí, v 6. cykle), čo svedčí o tom, že nie je potrebné upravovať dávku na základe pohlavia.
Staršie osoby
Populačná farmakokinetická analýza obinutuzumabu ukázala, že vek neovplyvnil farmakokinetiku
obinutuzumabu. Nepozorovali sa žiadne významné rozdiely vo farmakokinetike obinutuzumabu medzi pacientmi vo veku < 65 rokov (n = 265), pacientmi vo veku medzi 65 až 75 rokov (n = 197)
a pacientmi vo veku > 75 rokov (n = 128).
Pediatrická populácia
Neuskutočnili sa žiadne štúdie skúmajúce farmakokinetiku obinutuzumabu u pediatrických pacientov.
Porucha funkcie obličiek
Populačná farmakokinetická analýza obinutuzumabu ukázala, že klírens kreatinínu neovplyvňuje
farmakokinetiku obinutuzumabu. Farmakokinetika obinutuzumabu u pacientov s miernou
(CrCl 50 - 89 ml/min, n = 306) alebo stredne ťažkou (CrCl 30 až 49 ml/min, n = 72) poruchou funkcie obličiek je podobná ako u pacientov s normálnou funkciou obličiek (CrCl ≥ 90 ml/min, n = 207). Farmakokinetické údaje týkajúce sa pacientov s ťažkou poruchou funkcie obličiek
(CrCl 15 - 29 ml/min) sú obmedzené (n = 5), preto nie je možné uviesť odporúčanie pre dávkovanie.
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene sa neuskutočnila žiadna oficiálna farmakokinetická štúdia.
5.3 Predklinické údaje o bezpečnosti
Neuskutočnili sa žiadne štúdie stanovujúce karcinogénny potenciál obinutuzumabu.
Neuskutočnili sa žiadne špecifické štúdie na zvieratách hodnotiace vplyv obinutuzumabu na fertilitu. V štúdiách toxicity po opakovanom podávaní na opiciach rodu Cynomolgus nemal obinutuzumab žiadne nežiaduce účinky na reprodukčné orgány samcov a samíc.
V rozšírenej štúdii prenatálnej a postnatálnej vývojovej (enhanced pre and postnatal development, ePPND) toxicity na gravidných opiciach rodu Cynomolgus nebol dokázaný teratogénny efekt. Aj keď obinutuzumab podávaný intravenózne raz týždenne v dávke 25 a 50 mg/kg (2- až 5-násobok klinickej expozície založenej na Cmax a AUC) od 20. dňa post-coitum až do pôrodu viedol ku kompletnej deplécii B-buniek u dojčených mláďat. Expozícia u potomkov zistená na 28. deň po pôrode svedčí
o tom, že obinutuzumab prechádza placentárnou bariérou. Koncentrácie v sére dojčených mláďat zistené na 28. deň po pôrode boli v rozmedzí koncentrácií v sére samičích matiek, zatiaľ čo koncentrácie v mlieku v ten istý deň boli veľmi nízke (menej ako 0,5 % zodpovedajúcich hladín v sére samičích matiek), čo poukazuje na to, že k expozícii dojčeného mláďaťa muselo dôjsť in utero. Počet B-buniek sa vrátil do referenčných hodnôt a imunologická funkcia sa obnovila do 6 mesiacov po pôrode.
V 26-týždňovej štúdii na opiciach rodu Cynomolgus sa pozorovali reakcie z precitlivenosti
a prisudzovali sa rozpoznaniu humanizovanej protilátky ako cudzorodej látky u opíc rodu Cynomolgus
(0,7- až 6-násobok klinickej expozície založenej na hodnotách Cmax a AUC v rovnovážnom stave
po podávaní 5, 25 a 50 mg/kg raz týždenne. Nálezy zahŕňali akútne anafylaktické alebo anafylaktoidné
reakcie a zvýšenú prevalenciu systémového zápalu a infiltrátov, ktoré zodpovedali reakciám z precitlivenosti sprostredkovaným imunokomplexami, akými sú arteritída/periarteritída, glomerulonefritída a zápal seróznych blán/spojiva. Tieto reakcie viedli k neplánovanému utrateniu
6/36 zvierat liečených obinutuzumabom počas fázy podávania lieku a fázy zotavovania sa; tieto zmeny boli čiastočne reverzibilné. U ľudí sa nepozorovala žiadna renálna toxicita s príčinným vzťahom k obinutuzumabu.
6
. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
Monohydrát L-histidíniumchloridu
Dihydrát trehalózy
Poloxamér 188
Voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
3 roky
Po nariedení
Po nariedení je chemická a fyzikálna stabilita lieku riedeného v 0,9 % (9 mg/ml) injekčnom roztoku chloridu sodného na koncentrácie od 0,4 mg/ml do 20 mg/ml preukázaná na 24 hodín pri teplote
od 2 °C do 8 °C a následne na 48 hodín (vrátane času podania infúzie) pri teplote ≤ 30 °C.
Z mikrobiologického hľadiska sa má pripravený infúzny roztok použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky uchovávania pred použitím je zodpovedný používateľ a za normálnych okolností to nemá byť dlhšie ako 24 hodín pri teplote 2 °C - 8 °C, pokiaľ sa riedenie nevykonalo
za kontrolovaných a validovaných aseptických podmienok.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšej škatuľke na ochranu pred svetlom.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
40 ml koncentrátu v 50 ml injekčnej liekovke (číre sklo typu I) so zátkou (butylkaučuk). Veľkosť
balenia s 1 injekčnou liekovkou.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Návod na riedenie
Gazyvaro má pripraviť zdravotnícky pracovník za použitia aseptickej techniky. Injekčnou liekovkou netraste.
Odoberte 40 ml koncentrátu z injekčnej liekovky a zrieďte ich v polyvinylchloridových (PVC) infúznych vakoch alebo polyolefínových infúznych vakoch bez obsahu PVC obsahujúcich 0,9 % (9 mg/ml) injekčný roztok chloridu sodného.
Aby sa zaistilo odlíšenie dvoch infúznych vakov obsahujúcich úvodnú 1 000 mg dávku, odporúča sa použiť vaky rôznej veľkosti, aby sa rozoznala 100 mg dávka určená na 1. deň 1. cyklu od 900 mg dávky určenej na (pokračujúci) 1. deň 1. cyklu alebo na 2. deň 1. cyklu. Pripravte 2 infúzne vaky tak, že odoberiete 40 ml koncentrátu z injekčnej liekovky a zriedite 4 ml v 100 ml PVC infúznom vaku alebo polyolefínovom infúznom vaku bez obsahu PVC a zvyšných 36 ml v 250 ml PVC infúznom vaku alebo polyolefínovom infúznom vaku bez obsahu PVC obsahujúcom 0,9 % (9 mg/ml) injekčný roztok chloridu sodného. Každý infúzny vak jasne označte. Podmienky na uchovávanie infúznych vakov, pozri časť 6.3.
Dávka Gazyvara, ktorá sa má podať
| Potrebné množstvo koncentrátu Gazyvara
| Veľkosť PVC infúzneho vaku alebo polyolefínového
infúzneho vaku bez obsahu
PVC
|
100 mg
| 4 ml
| 100 ml
|
900 mg
| 36 ml
| 250 ml
|
1 000 mg
| 40 ml
| 250 ml
|
Nepoužívajte iné roztoky na riedenie ako je (5 %) roztok glukózy (pozri časť 6.2).
Vak sa má jemne prevrátiť, aby sa roztok premiešal a aby sa predišlo nadmernému speneniu. Zriedený roztok sa nemá pretrepávať ani mraziť.
Parenterálne lieky sa majú pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc alebo zmenu sfarbenia.
Neboli pozorované žiadne inkompatibility medzi Gazyvarom v rozsahu koncentrácií od 0,4 mg/ml do 20,0 mg/ml po zriedení Gazyvara s 0,9 % (9 mg/ml) injekčným roztokom chloridu sodného a:
- PVC, polyetylénovými (PE), polypropylénovými alebo polyolefínovými vakmi
- PVC, polyuretánovými (PUR) alebo PE infúznymi súpravami
- voliteľnými
in-line filtrami s kontaktným povrchom z polyétersulfónu (PES), 3-cestným uzatváracím kohútikom pre infúzne súpravy vyrobeným z polykarbonátu (PC) a katétrami vyrobenými z polyuretánu (PEU).
LikvidáciaNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/14/937/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.