adiny.
Hypogamaglobulinémia a opakujúce sa bakteriálne infekcie u pacientov s chronickou lymfocytovou leukémiou, u ktorých zlyhala profylaktická liečba antibiotikami; hypogamaglobulinémia a opakujúce sa bakteriálne infekcie u pacientov v plató fáze mnohopočetného myelómu, ktorí nereagovali na pneumokokovú imunizáciu; vrodený AIDS s opakujúcimi sa bakteriálnymi infekciamiOdporúčaná dávka je 0,2 – 0,4 g/kg každé tri až štyri týždne.
Hypogamaglobulinémia u pacientov po alogénnej transplantácii krvotvorných kmeňových buniek.Odporúčaná dávka je 0,2 – 0,4 g/kg každé tri až štyri týždne. Minimálne hladiny sa majú udržiavať nad 5 g/l.
Primárna imunitná trombocytopéniaExistujú dva alternatívne režimy liečby:
- 0,8 – 1 g/kg prvý deň, túto dávku je možné opakovať raz za 3 dni
- 0,4 g/kg denne po dobu dvoch až piatich dní.
V prípade relapsu je možné liečbu opakovať.
Guillainov-Barrého syndróm
0,4 g/kg/deň po dobu 5 dní.
Chronická zápalová demyelinizačná polyneuropatiaÚvodná dávka: 2 g/kg (20 ml/kg) podaná v rozdelených dávkach po dobu 2 až 4 po sebe nasledujúcich dní. Udržiavacia dávka 1 g/kg podaná v priebehu jedného dňa (10 ml/kg) alebo rozdelená do dvoch dávok po 0,5 g/kg (5 ml/kg) podaných po dobu dvoch po sebe nasledujúcich dní každé 3 týždne.
K dispozícii sú len obmedzené skúsenosti s podaním intravenóznych imunoglobulínov u detí s chronickou zápalovou demyelinizačnou polyneuropatiou.
Klinické štúdie s Gamunexom 10 %nezahŕňajú dostatočný počet pacientov vo veku 65 rokov a viac, aby bolo možné jednoznačne určiť účinok liečby.
Kawasakiho choroba
Je potrebné podať 1,6 – 2,0 g/kg v rozdelených dávkach po dobu dvoch až piatich dní alebo 2,0 g/kg ako jednorazovú dávku. Pacienti majú byť súbežne liečení kyselinou acetylsalicylovou.
Odporúčané dávkovania sú zhrnuté v nasledujúcej tabuľke:
Indikácia | Dávka | Frekvencia podania
|
Substitučná liečba primárnej imunodeficiencie
Substitučná liečba sekundárnej imunodeficiencie
Vrodený AIDS
Hypogamaglobulinémia (< 4 g/l) u pacientov po alogénnej transplantácii krvotvorných kmeňových buniek
| - úvodná dávka:
0,4 – 0,8 g/kg
- potom:
0,2 – 0,8 g/kg
0,2 – 0,4 g/kg
0,2 – 0,4 g/kg
0,2 ‑ 0,4 g/kg
|
každé 3 – 4 týždne, aby sa dosiahla minimálna hladina IgG v sére najmenej 5 – 6 g/l
každé 3 – 4 týždne, aby sa dosiahla minimálna hladina IgG v sére najmenej 5 – 6 g/l
každé 3 – 4 týždne
každé 3 –4 týždne, aby sa dosiahla minimálna hladina IgG v sére nad 5 g/l
|
Imunomodulácia:
- Primárna imunitná trombocytopénia
- Guillainov-Barrého syndróm
- Chronická zápalová demyelinizačná polyneuropatia#
- Kawasakiho choroba
|
0,8 – 1 g/kg
alebo
0,4 g/kg/deň
0,4 g/kg/deň
úvodná dávka 2 g/kg
udržiavacia dávka
1 g/kg
1,6 – 2 g/kg
alebo
2 g/kg
|
1.deň, prípadne opakovať jedenkrát v priebehu 3 dní
po dobu 2 – 5 dní
po dobu 5 dní
v rozdelených dávkach po dobu 2 až 4 po sebe nasledujúcich dní
podaná v priebehu 1 dňa alebo rozdelená do dvoch dávok po 0,5 g/kg (5 ml/kg) podaných v dvoch po sebe nasledujúcich dňoch, každé 3 týždne
v rozdelených dávkach po dobu 2 ‑ 5 dní spolu s kyselinou acetylsalicylovou
ako jedna dávka spolu s kyselinou acetylsalicylovou
|
| | | |
# Dávka je stanovená na základe dávky použitej v klinickej štúdii vykonanej s Gamunexom 10 %.
Trvanie liečby dlhšie ako 48 týždňov je na uvážení lekára na základe odpovede pacienta na začiatku a v priebehu dlhodobej terapie.
Dávkovanie a intervaly sa môžu prispôsobiť podľa individuálneho priebehu ochorenia.
Pediatrická populáciaDávkovanie u detí a dospievajúcich (0 – 18 rokov) sa nelíši od dávkovania u dospelých pacientov, keďže je pre každú indikáciu určené podľa telesnej hmotnosti a upravené podľa klinickej odpovede za vyššie uvedených podmienok.
Spôsob podávania
Na intravenózne podanie.
Normálny ľudský imunoglobulín sa má podávať intravenóznou infúziou so začiatočnou rýchlosťou 0,6 – 1,2 ml/kg/h v priebehu 30 minút. Ak pacient infúziu dobre znáša (pozri časť 4.4), je možné postupne zvýšiť rýchlosť podávania na maximálnu hodnotu 4,8 – 8,4 ml/kg/h.
4.3 Kontraindikácie Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok (pozri časť 6.1), najmä u pacientov s protilátkami proti IgA.
4.4 Osobitné upozornenia a opatrenia pri používaníAk je infúzia podávaná vysokou rýchlosťou (8,4 ml/kg/h) musia byť všetci pacienti pozorne sledovaní. U detí alebo pacientov s rizikom renálneho zlyhania by maximálna rýchlosť podávania infúzie nemala presiahnuť 4,8 ml/kg/h.
Gamunex 10 %sa nesmie miešať s inými infúznymi roztokmi (napr. s fyziologickým roztokom) a inými liekmi. Ak je pred podaním infúzie potrebné zriedenie, môže sa pre tento účel použiť roztok glukózy s koncentráciou 50 mg/ml. Avšak pacientov s latentným diabetom (kde sa môže objaviť prechodná glykozúria), pacientov s diabetom alebo pacientov, ktorí majú diétu s nízkym obsahom cukru, je potrebné v prípade podania roztoku glukózy 50 mg/ml starostlivo sledovať. Tiež pozri nižšie upozornenie týkajúce sa akútneho renálneho zlyhania.
Gamunex 10 %sa nesmie podávať súčasne s heparínom v jednej infúznej súprave.
Niektoré závažné nežiaduce reakcie môžu súvisieť s rýchlosťou podávania infúzie. Je potrebné presne dodržiavať odporúčanú rýchlosť podávania infúzie, ktorá je uvedená v časti 4.2. Počas doby podávania infúzie musia byť pacienti dôkladne monitorovaní a starostlivo sledovaní pre prípad výskytu akýchkoľvek príznakov.
Niektoré nežiaduce reakcie sa môžu vyskytnúť častejšie:
- v prípade vysokej rýchlosti podania infúzie,
- u pacientov, ktorým bol normálny ľudský imunoglobulín aplikovaný prvýkrát, alebo v zriedkavých prípadoch, keď bol zmenený prípravok normálneho ľudského imunoglobulínu, alebo ak uplynul dlhý časový interval od poslednej aplikácie infúzie.
Aby sa predišlo možným komplikáciám, ubezpečte sa, že:
- pacienti nie sú citliví na normálny ľudský imunoglobulín pomocou pomalej úvodnej infúzie lieku (0,1 ml/kg/h)
- pacienti sú starostlivo sledovaní pre prípad výskytu akýchkoľvek príznakov počas celej doby podávania infúzie. Predovšetkým pacienti, ktorým je normálny ľudský imunoglobulín aplikovaný prvýkrát, pacienti, ktorým bola zmenená liečba z alternatívneho IVIg lieku alebo pacienti, u ktorých uplynul dlhý časový interval od predchádzajúcej infúzie, musia byť sledovaní počas prvej aplikácie infúzie a hodinu po aplikácii prvej infúzie, aby sa mohli zachytiť potenciálne prejavy nežiaducich reakcií. Všetci ostatní pacienti musia byť sledovaní najmenej po dobu 20 minút po podaní.
V prípade nežiaducej reakcie je potrebné buď znížiť rýchlosť podania infúzie alebo infúziu zastaviť. Potrebná liečba závisí od povahy a závažnosti nežiaducej reakcie. V prípade šoku je nutné pristúpiť k štandardnej protišokovej terapii.
U všetkých pacientov sa pri podávaní IVIg vyžaduje:
- adekvátna hydratácia pred začatím podávania infúzie IVIg
- sledovanie objemu vylúčeného moču
- sledovanie hladín sérového kreatinínu
- zamedzenie súbežného použitia kľučkových diuretík.
Tento liek obsahuje zanedbateľné množstvo sodíka.
PrecitlivenosťPravé hypersenzitívne reakcie sú zriedkavé. Môžu sa vyskytnúť u pacientov s anti-IgA protilátkami.
IVIg nie sú indikované u pacientov so selektívnym deficitom IgA, pri ktorom je deficit IgA jedinou abnormalitou.
Zriedkavo môže normálny ľudský imunoglobulín spôsobiť pokles krvného tlaku s anafylaktickou reakciou, a to dokonca aj u pacientov, ktorí predchádzajúcu liečbu normálnym ľudským imunoglobulínom tolerovali.
TromboembolizmusJe klinicky dokázaná spojitosť medzi podaním IVIg a tromboembolickými príhodami ako infarkt myokardu, cerebrovaskulárna príhoda (vrátane mŕtvice), pľúcna embólia a hlboká žilová trombóza. Predpokladá sa, že to súvisí s relatívnym zvýšením viskozity krvi počas infúzie vysokých dávok imunoglobulínu u rizikových pacientov. Opatrnosť je potrebná pri predpisovaní a aplikácii IVIg obéznym pacientom a pacientom s už existujúcimi rizikovými faktormi trombotických príhod (ako sú pokročilý vek, hypertenzia, diabetes mellitus a anamnéza cievneho ochorenia alebo trombotických príhod, pacienti so získanými alebo zdedenými trombofilnými poruchami, pacienti s dlhými obdobiami imobilizácie, pacienti s ťažkou hypovolémiou, pacienti s ochoreniami, ktoré zvyšujú viskozitu krvi).
U pacientov s rizikom tromboembolických nežiaducich reakcií sa majú IVIg lieky podávať najnižšou možnou rýchlosťou infúzie a v najnižšej prípustnej dávke.
Akútne renálne zlyhanieU pacientov liečených IVIg boli zaznamenané prípady akútneho renálneho zlyhania. Vo väčšine prípadov boli identifikované rizikové faktory, ako napr. už existujúca renálna insuficiencia, diabetes mellitus, hypovolémia, nadváha, súčasná aplikácia nefrotoxických liekov alebo vek nad 65 rokov.
V prípade poškodenia funkcie obličiek je potrebné zvážiť prerušenie podávania IVIg. I keď tieto prípady renálnej dysfunkcie a akútneho renálneho zlyhania boli spojené s užívaním mnohých registrovaných imunoglobulínových liekov obsahujúcich rôzne pomocne látky ako sú sacharóza, glukóza a maltóza, prípravky obsahujúce sacharózu ako stabilizátor majú v celkovom počte nepomerne vysoké zastúpenie. U rizikových pacientov by sa malo uvažovať o použití IVIg liekov, ktoré neobsahujú tieto pomocné látky. Gamunex 10% neobsahuje sacharózu, maltózu ani glukózu.
U pacientov s rizikom akútneho renálneho zlyhania sa majú IVIg lieky podávať najnižšou možnou rýchlosťou infúzie a v najnižšej prípustnej dávke.
Syndróm aseptickej meningitídy (AMS)V súvislosti s liečbou IVIg bol hlásený syndróm aseptickej meningitídy. Prerušenie liečby IVIg viedlo v priebehu niekoľkých dní k remisii AMS bez následkov. Syndróm obyčajne začína v priebehu niekoľkých hodín až 2 dni po liečbe IVIg. Vyšetrenia cerebrospinálnej tekutiny sú často pozitívne s pleocytózou až niekoľko tisíc buniek/mm
3, predovšetkým granulocytárneho typu a zvýšenou hladinou proteínov až niekoľko sto mg/dl. AMS sa môže vyskytovať častejšie pri liečbe vysokými dávkami IVIg (2 g/kg).
Hemolytická anémiaIVIg lieky môžu obsahovať protilátky krvnej skupiny, ktoré môžu pôsobiť ako hemolyzíny a indukovať
in vivo povlak imunoglobulínu na červených krvinkách, čo vedie k pozitívnej priamej antiglobulínovej reakcii (Coombsov test) a zriedkavo k hemolýze. Ako následok liečby IVIg sa môže vyvinúť hemolytická anémia v dôsledku zvýšenej sekvestrácie červených krviniek (ERY). U príjemcov IVIg sa majú sledovať klinické prejavy a symptómy hemolýzy. (pozri časť 4.8).
Nasledujúce rizikové faktory sú spojené s rozvojom hemolýzy: vysoké dávky, či už podané jednorazovo alebo rozdelené v priebehu niekoľkých dní; iná krvná skupina ako skupina 0; prítomný zápalový stav. Odporúča sa zvýšená opatrnosť u pacientov s inou krvnou skupinou ako 0, ktorí dostávajú vysoké dávky z dôvodu inej indikácie ako je primárna imunodeficiencia (nonPID). Hemolýza bola zriedkavo hlásená u pacientov, ktorým bol liek podaný ako substitučná liečba primárnej imunodeficiencie.
V súvislosti s hemolýzou sa vyskytli ojedinelé prípady poruchy funkcie obličiek/renálneho zlyhania s fatálnymi následkami.
Ovplyvnenie sérologických testovPo aplikácii imunoglobulínu môže v krvi pacienta dôjsť k prechodnému vzostupu pasívne prenášaných protilátok, a tým ku vzniku falošne pozitívnych výsledkov v sérologických testoch.
Pasívny prenos protilátok k antigénom erytrocytov, ako napr. A, B, D, môže ovplyvniť niektoré sérologické testy na protilátky proti červeným krvinkám, napríklad priamy antiglobulínový test (DAT, priamy Coombsov test).
Prenosné činidláŠtandardné opatrenia, ktoré slúžia na prevenciu infekcií v súvislosti s používaním liekov vyrobených z ľudskej krvi alebo plazmy zahŕňajú výber darcov, testovanie jednotlivých odberov a plazmatických poolov na špecifické markery infekcií a zaradenie účinných výrobných krokov na inaktiváciu/odstránenie vírusov. Napriek tomu, pri podávaní liekov vyrobených z ľudskej krvi alebo plazmy nie je možné celkom vylúčiť možnosť prenosu infekčných činiteľov. To platí i pre neznáme alebo vznikajúce vírusy a iné patogény.
Tieto opatrenia sa považujú za účinné pri vírusoch s obalom ako sú HIV, HBV a HCV. Prijaté opatrenia môžu mať obmedzenú účinnosť pri vírusoch bez obalu ako sú HAV a parvovírus B19.
Klinické skúsenosti opakovane potvrdzujú absenciu prenosu hepatitídy A alebo parvovírusu B19 imunoglobulínmi. Tiež sa predpokladá, že obsah protilátok vo veľkej miere prispieva k ochrane pred vírusmi.
Pri každom podaní Gamunexu 10 % pacientovi sa dôrazne odporúča zaznamenať názov a číslo šarže lieku za účelom uchovania spojenia medzi pacientom a číslom šarže lieku.
Pediatrická populáciaNapriek tomu, že k dispozícii sú iba obmedzené údaje, predpokladá sa, že pre pediatrickú populáciu platia rovnaké upozornenia, opatrenia a rizikové faktory. V postregistračných hláseniach sa pozorovalo, že indikácie pre vysokú dávku IVIg u detí, obzvlášť Kawasakiho choroba, sú spojené so zvýšeným počtom hlásení hemolytických reakcií v porovnaní s ostatnými indikáciami IVIg u detí.
V prípade podozrenia na hemolýzu lekári musia dôkladne zvážiť monitorovanie hladín hemoglobínu 24 až 48 hodín po ukončení podania IVIg. Ak je potrebné liečbu opakovať a je podozrenie na hemolýzu, dôrazne sa odporúča monitorovať hladiny hemoglobínu jeden týždeň po následnom podaní IVIg . Rodinní príslušníci majú byť poučení, aby sa obrátili na lekára, ak sa u ich dieťaťa rozvinú symptómy hemolýzy ako sú bledosť, letargia, tmavý moč, dyspnoe alebo palpitácie.
4.5 Liekové a iné interakcieŽivé oslabené vírusové vakcíny
Podanie imunoglobulínu môže po dobu najmenej 6 týždňov až 3 mesiacov znižovať účinnosť živých oslabených vírusových vakcín, ako sú vakcíny proti osýpkam, rubeole, mumpsu alebo ovčím kiahňam. Medzi podaním tohto lieku a očkovaním živou oslabenou vírusovou vakcínou musí uplynúť interval 3 mesiacov. V prípade vakcíny proti osýpkam môže toto zníženie účinnosti trvať až 1 rok. Z tohto dôvodu sa má u pacientov očkovaných vakcínou proti osýpkam skontrolovať hladina protilátok.
Pediatrická populácia Aj keď sa neuskutočnili špecifické interakčné štúdie v pediatrickej populácii, neočakávajú sa žiadne rozdiely medzi dospelými a deťmi.
4.6 Fertilita, gravidita a laktáciaGraviditaBezpečnosť použitia tohto lieku v tehotenstve nebola stanovená v kontrolovaných klinických štúdiách, preto sa má gravidným ženám a dojčiacim matkám podávať len s veľkou opatrnosťou. U IVIg liekov bol potvrdený prestup cez placentu, vo zvýšenej miere v priebehu tretieho trimestra. Klinické skúsenosti s imunoglobulínmi nepredpokladajú škodlivé účinky na priebeh gravidity alebo na plod a novorodenca.
DojčenieImunoglobulíny sa vylučujú do ľudského mlieka a môžu prispieť k ochrane novorodenca pred patogénmi vstupujúcimi do organizmu cez sliznicu.
FertilitaNa základe klinických skúseností s imunoglobulínmi sa nepredpokladajú škodlivé účinky na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeSchopnosť viesť vozidlá a obsluhovať stroje môže byť znížená v súvislosti s niektorými nežiaducimi reakciami spojenými s Gamunexom 10%. Pacienti, ktorí zaznamenajú nežiaduce reakcie počas liečby, majú pred vedením vozidla alebo obsluhou strojov počkať, pokým neodznejú.
4.8 Nežiaduce účinkyZhrnutie bezpečnostného profiluPríležitostne sa môžu vyskytnúť nežiaduce reakcie ako sú zimnica, bolesť hlavy, závraty, horúčka, vracanie, alergické reakcie, nauzea, artralgia, nízky krvný tlak a mierna bolesť v dolnej časti chrbtice.
Zriedkavo môžu normálne ľudské imunoglobulíny spôsobiť náhly pokles krvného tlaku a v ojedinelých prípadoch anafylaktický šok, dokonca i keď sa u pacienta pri predchádzajúcich aplikáciách nevyskytli žiadne známky precitlivenosti.
Pri podaní normálneho ľudského imunoglobulínu boli pozorované prípady reverzibilnej aseptickej meningitídy a zriedkavé prípady prechodných kožných reakcií. Reverzibilné hemolytické reakcie boli pozorované hlavne u pacientov s krvnými skupinami A, B a AB. Zriedkavo sa po podaní vysokej dávky IVIg môže vyvinúť hemolytická anémia vyžadujúca transfúziu (pozri tiež časť 4.4).
Bolo pozorované zvýšenie hladiny kreatinínu v sére a/alebo akútne renálne zlyhanie.
Veľmi zriedkavo: tromboembolické reakcie ako infarkt myokardu, mŕtvica, pľúcna embólia, hlboká žilová trombóza.
Informácie o bezpečnosti v súvislosti s prenosnými činiteľmi pozri časť 4.4.
Tabuľkový prehľad nežiaducich reakciíNasledujúca tabuľka je v súlade s klasifikáciou orgánových systémov MedDRA (SOC – system organ clasification a preferovaná úroveň terminológie). Frekvencie sa posudzovali na základe nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až <1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Nežiaduce liekové reakcie (ADRs), ktoré boli v klinických štúdiách s liekom Gamunex 10%hlásené zriedkavo:
Hemolytická anémia, dyspnoe, sínusitída, exfoliácia kože, úzkosť, myalgia, pokles hemoglobínu, dyspepsia, kontúzia, dermatitída, sčervenanie, muskuloskeletálna stuhnutosť, palmárny erytém, afónia.
Frekvencia nežiaducich liekových reakcií (ADRs) v klinických štúdiách s Gamunexom 10 %MedDRA trieda orgánových systémov | Preferovaný názov podľa MedDRA | Kategória frekvencie ADR
|
Laboratórne a funkčné vyšetrenia
| Znížený počet bielych krviniek
| Menej časté
|
Poruchy nervového systému
| Bolesť hlavy
| Časté
|
Závraty
| Menej časté
|
Poruchy kože a podkožného tkaniva
| Urtikária, dermatitída, pruritus, vyrážka
| Menej časté
|
Poruchy gastrointestinálneho traktu
| Bolesť brucha, hnačka, nauzea, vracanie,
| Menej časté
|
Poruchy ciev
| Hypertenzia, hypotenzia
| Menej časté
|
Poruchy dýchacej sústavy, hrudníka a mediastína
| Faryngitída, kašeľ, nazálna kongescia, sipot
| Menej časté
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Artralgia, bolesť chrbta, bolesť krku, bolesť ramena
| Menej časté
|
Poruchy srdca a srdcovej činnosti
| Bolesť na hrudníku
| Menej časté
|
Celkové poruchy a reakcie v mieste podania
| Horúčka
| Časté
|
Ochorenie podobné chrípke, nevoľnosť, únava, zimnica, asténia, reakcie v mieste vpichu
| Menej časté
|
Pediatrická populácia Očakáva sa, že frekvencia, typ a závažnosť nežiaducich reakcií u detí budú rovnaké ako u dospelých.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.'
4.9 PredávkovaniePredávkovanie môže viesť k preťaženiu obehu tekutinami a hyperviskozite, obzvlášť u rizikových pacientov, vrátane pacientov v staršom veku a pacientov s poruchou funkcie srdca a obličiek.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunoséra a imunoglobulíny: normálne ľudské imunoglobulíny na intravenózne použitie, ATC kód: J06BA02.
Normálny ľudský imunoglobulín obsahuje nemodifikovaný imunoglobulín G (IgG) so širokým spektrom protilátok proti rôznym infekčným činiteľom.
Normálny ľudský imunoglobulín obsahuje protilátky IgG prítomné v bežnej populácii. Je vyrobený z plazmy od minimálne 1 000 darcov. Distribúcia podtried imunoglobulínu G zodpovedá ich distribúcií v prirodzenej ľudskej plazme.Primeranými dávkami tohto lieku je možné upraviť abnormálne nízke hladiny imunoglobulínu G do normálneho rozmedzia. Mechanizmus účinku v iných indikáciách ako je substitučná liečba, nie je celkom objasnený, ale zahŕňa imunomodulačné účinky.
Klinické štúdie s Gamunexom 10%u pacientov s chronickou zápalovou demyelinizačnou polyneuropatiou (CIPD):Štúdia IVIG-C CIDP efficacy trial (ICE štúdia), dvojito zaslepená, randomizovaná, placebom kontrolovaná štúdia skúmala účinnosť a bezpečnosť Gamunexu 10 %v liečbe CIDP. Celkovo 117 pacientov s CIDP bolo randomizovaných tak, že dostávali buď Gamunex 10 %alebo placebo každé tri týždne. Úvodná dávka bola 2 g/kg telesnej hmotnosti, udržiavacia dávka 1 g/kg telesnej hmotnosti.
V skupine pacientov užívajúcich Gamunex 10% bola terapeutická odpoveď (určená zlepšením skóre invalidity „Inflammatory Neuropathy Cause And Treatment“ (INCAT) a pretrvaním zlepšenia ≥ 1 počas 24- týždňového obdobia, kedy sa sledovala účinnosť) u signifikantne vyššieho percenta pacientov (54 %) v porovnaní so skupinou pacientov, ktorí užívali placebo(21 %, p=0,0002). Svalová sila hodnotená pomocou skóre MRC, sila stisku, ako aj citlivosť podľa skóre ISS sa signifikantne viac zlepšili v skupine pacientov užívajúcich Gamunex 10 %v porovnaní so skupinou s placebom.
Vzhľadom na obmedzený počet pacientov vo veku ≥ 65 rokov zaradených do štúdie nebolo možné u týchto pacientov určiť presné účinky liečby hodnotené pomocou INCAT skóre. Štatisticky významné zlepšenie však u týchto pacientov nastalo v sile stisku, pokiaľ boli liečení liekom Gamunex 10%.
Z pacientov, ktorí odpovedali na liečbu, menej ako polovica vykázala zlepšenie už po úvodnej dávke (do 3. týždňa), ale väčšina po druhej dávke (do 6. týždňa). Pacienti, ktorí na liečbu nereagovali, prešli na alternatívnu liečbu na dobu maximálne 24 týždňov.
Všetci pacienti, ktorí odpovedali na liečbu Gamunexom 10%, boli znovu randomizovaní do predĺženej fázy štúdie na obdobie ďalších 6 mesiacov udržiavacej liečby buď s Gamunexom 10 %alebo s placebom. U pacientov, ktorí pôvodne odpovedali na Gamunex 10% sa miera relapsov významne zvýšila v skupine pacientov ďalej randomizovaných do skupiny s placebom (42 %) než v skupine pacientov ďalej randomizovaných do skupiny s Gamunexom 10 %(13 %, p=0,012).
Štúdia ICE preukázala krátkodobú i dlhodobú účinnosť Gamunexu 10 %v liečbe CIDP. Výsledky sú zhrnuté v nasledujúcej tabuľke.
Primárny cieľový ukazovateľ a ostatné výsledky štúdie ICE
| Gamunex 10 %
| Placebo
| p
|
Percentuálne zastúpenie odpovedajúcich pacientov počas obdobia sledovania účinnosti (primárny cieľový ukazovateľ)
| 54 %
| 21 %
| 0,0002
|
Pravdepodobnosť relapsu v predĺženej fáze sledovania
| 13 %
| 45 %
| 0,013
|
Sila stisku (kPA)1 (zmena v porovnaní s východiskovou hodnotou)
|
|
|
|
Dominantná ruka
| 13,2
| 1,5
| 0,0008
|
Nedominantná ruka
| 13,3
| 4,3
| 0,005
|
Svalová sila (MRC3sumárne skóre)1 (zmena v porovnaní s východiskovou hodnotou)
| 3,3
| 0,2
| 0,001
|
Citlivosť (ISS4 skóre)2 (zmena v porovnaní s východiskovou hodnotou)
| -1,2
| 0,2
| 0,021
|
1 Zlepšenie indikuje pozitívne číslo
2 Zlepšenie indikuje negatívne číslo
3 MRC: Medical Research Council
4 ISS: INCAT Sensory Sum Score
Vlastný prípravok je upravený na slabo kyslé pH. Keďže Gamunex 10 %má nízku pufrovaciu kapacitu, počas infúzie sa v krvi rýchlo neutralizuje. Aj po podaní vysokých dávok Gamunexu 10 %sa nezaznamenala žiadna zmena pH krvi
. Osmolalita roztoku je 258 mOsmol/kg a tak sa približuje k normálnemu rozsahu (285 –295 mOsmol/kg).
5.2 Farmakokinetické vlastnostiPo intravenóznom podaní je normálny ľudský imunoglobulín v obehu príjemcu ihneď a v plnom rozsahu biologicky dostupný. Relatívne rýchlo sa distribuuje medzi plazmou a extravaskulárnou tekutinou; rovnováha medzi intra- a extravaskulárnym priestorom sa dosiahne približne po 3 – 5 dňoch.
Biologický polčas normálneho ľudského imunoglobulínu stanovený u pacientov s primárnym syndrómom deficiencie protilátok je približne 35 dní, a preto prevyšuje dobu 21 dní opísanú v literatúre u zdravých jedincov. Tento polčas sa môže medzi jednotlivými pacientmi líšiť, a to najmä u pacientov s primárnou imunodeficienciou.
IgG a komplexy IgG sa odbúravajú v bunkách retikuloendotelového systému.
Pediatrická populácia U pediatrickej populácie sa neočakávajú žiadne rozdiely vo farmakokinetických vlastnostiach.
5.3 Predklinické údaje o bezpečnostiImunoglobulíny sú normálnou súčasťou ľudského tela. Keďže podanie imunoglobulínov v štúdiách u zvierat môže viesť k tvorbe protilátok, predklinické údaje o bezpečnosti sú obmedzené. V uskutočnených akútnych a subakútnych štúdiách u zvierat, sa pri Gamunexe 10% nepreukázalo žiadne zvláštne rizikopre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokGlycín.
6.2 InkompatibilityTento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti3 roky
6.4 Špeciálne upozornenia na uchovávanieUchovávajte v chladničke (2 °C – 8 °C ). Neuchovávajte v mrazničke.
Tento liek sa môže jednorazovo uchovávať pri teplote miestnosti (neprevyšujúcej 25 °C) vo vonkajšom obale až po dobu 6 mesiacov. V takom prípade sa čas použiteľnosti lieku skončí na konci tohto 6 mesačného obdobia. Nový dátum exspirácie sa musí vyznačiť na vonkajšom obale a na štítku injekčnej liekovky. Nový dátum exspirácie nesmie byť neskorší ako dátum exspirácie vytlačený na obale. Potom sa musí liek použiť alebo zlikvidovať. Následné uchovávanie v chladničke nie je možné.
6.5 Druh obalu a obsah balenia Roztok na intravenóznu infúziu v sklenených injekčných liekovkách typu I alebo II s chlórbutylovými zátkami.
Veľkosti balenia: 10 ml, 50 ml, 100 ml, 200 ml, 400 ml; balenia pre nemocnice. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomPred použitím je potrebné liek zahriať na teplotu miestnosti alebo telesnú teplotu. Roztok má byť číry alebo slabo opaleskujúci a bezfarebný alebo bledožltý. Roztoky, ktoré sú zakalené alebo obsahujú častice sa nesmú použiť. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami. Po otvorení balenia sa má obsah okamžite podať infúziou. Následné uchovávanie, dokonca ani v chladničke, nie je dovolené z dôvodu možnej mikrobiálnej kontaminácie.
Ak je pred infúziou potrebné zriedenie, je možné na tento účel použiť roztok glukózy 50 mg/ml. Na riedenie nepoužívajte fyziologický roztok.
Gamunex 10 % sa nesmie podávať súčasne s heparínom v jednej infúznej súprave.
Infúzna súprava na podanieGamunexu 10 %sa môže prepláchnuť roztokom glukózy 50 mg/ml alebo roztokom chloridu sodného (9 mg/ml) a nesmie sa preplachovať heparínom.
Heparínová zátka, cez ktorú sa podáva Gamunex 10
%, sa má vypláchnuť roztokomglukózy 50 mg/ml alebo roztokom chloridu sodného (9 mg/ml) a nesmie sa preplachovať heparínom.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGrifols Deutschland GmbH
Colmarer Straße 22
60528 Frankfurt
Nemecko
Tel.:+49 69-660 593 100
8. REGISTRAČNÉ ČÍSLO59/0230/17-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTU08/2017
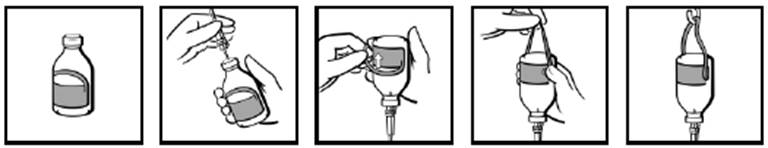
Inštrukcie na použitie injekčných liekoviek (len pre injekčné liekovky 50 ml, 100 ml, 200 ml a 400 ml)Injekčné liekovky sú dodávané so štítkom na zavesenie (obr. 1). Po zasunutí infúznej súpravy (obr. 2) obráťte injekčnú liekovku dnom hore a ohnite časť štítku so slučkou (obr. 3).
Pevným tlakom prstov vytvorte záhyb na každej strane v mieste, kde sa slučka spája so zvyškom štítku (obr. 4). Zaveste injekčnú liekovku na infúzny stojan pomocou vzniknutej slučky (obr. 5).
Obr. 1 Obr. 2 Obr. 3 Obr. 4 Obr. 5