je rizikovým pacientkám.
K dispozícii nie sú žiadne dlhodobé údaje o vplyve fulvestrantu na kosti. Vzhľadom na mechanizmus účinku fulvestrantu existuje potenciálne riziko osteoporózy.
Pediatrická populáciaFulvestrant Teva sa neodporúča používať u detí a dospievajúcich, pretože jeho bezpečnosť a účinnosť v tejto skupine pacientov neboli stanovené (pozri časť 5.1).
Fulvestrant Teva obsahuje etanol (96 %) (alkohol)Tento liek obsahuje etanol (alkohol), t.j. až do 1 000 mg na dávku, čo zodpovedá 20 ml piva alebo 8 ml vína na dávku. Je škodlivý pre osoby, ktoré trpia alkoholizmom. Má sa to vziať do úvahy u tehotných alebo dojčiacich žien a u vysokorizikových skupín, akými sú pacientky s ochorením pečene alebo s epilepsiou.
Fulvestrant Teva obsahuje benzylalkoholTento liek obsahuje benzylalkohol. Množstvo benzylalkoholu na dávku je 500 mg v 5 ml (100 mg v 1 ml) a môže vyvolať anafylaktoidné reakcie.
Fulvestrant Teva obsahuje rafinovaný ricínový olejTento liek obsahuje rafinovaný ricínový olej, ktorý môže vyvolať závažné alergické reakcie.
4.5 Liekové a iné interakcieKlinická interakčná štúdia s midazolamom (substrát pre CYP3A4) preukázala, že fulvestrant neinhibuje CYP3A4. Klinické interakčné štúdie s rifampicínom (induktor CYP3A4) a s ketokonazolom (inhibítor CYP3A4) nepreukázali žiadnu klinicky významnú zmenu klírensu fulvestrantu. U pacientok, ktorým sa fulvestrant podáva súbežne s inhibítormi alebo induktormi CYP3A4, preto nie je potrebná úprava dávky.
4.6 Fertilita, gravidita a laktáciaŽeny vo fertilnom vekuPacientkám vo fertilnom veku sa má odporučiť, aby počas liečby používali účinnú antikoncepciu.
GraviditaFulvestrant Teva je kontraindikovaný v gravidite (pozri časť 4.3). Po intramuskulárnom podaní jednorazových dávok potkanom a králikom sa preukázalo, že fulvestrant prechádza placentou. Štúdie na zvieratách preukázali reprodukčnú toxicitu zahŕňajúcu zvýšený výskyt abnormalít a úmrtí plodov (pozri časť 5.3). Ak počas liečby Fulvestrantom Teva dôjde k otehotneniu, pacientka musí byť informovaná o možnom riziku pre plod a o možnom riziku spontánneho potratu.
DojčeniePočas liečby Fulvestrantom Teva sa dojčenie musí ukončiť. Fulvestrant sa vylučoval do mlieka samíc potkanov v období laktácie. Nie je známe, či sa fulvestrant vylučuje do ľudského mlieka. Vzhľadom na možnosť závažných nežiaducich účinkov fulvestrantu u dojčených detí je použitie počas laktácie kontraindikované (pozri časť 4.3).
FertilitaÚčinky Fulvestrantu Teva na fertilitu ľudí sa nesledovali.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeFulvestrant Teva nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje. Počas liečby Fulvestrantom Teva však bola veľmi často hlásená asténia, a preto musia byť pacientky, u ktorých sa táto nežiaduca reakcia vyskytne, obozretné pri vedení vozidiel alebo obsluhe strojov.
4.8 Nežiaduce účinkyTáto časť poskytuje informácie založené na údajoch o všetkých nežiaducich reakciách zistených v klinických skúšaniach, v štúdiách uskutočnených v období po uvedení lieku na trh alebo zo spontánnych hlásení. Najčastejšie hlásené nežiaduce reakcie sú reakcie v mieste vpichu, asténia, nauzea a zvýšené hladiny pečeňových enzýmov (ALT, AST, ALP).
Nasledujúce kategórie frekvencie výskytu nežiaducich reakcií na liek (adverse drug reactions, ADR) sa vypočítali na základe údajov o liečebnej skupine s fulvestrantom v dávke 500 mg, ktoré boli zahrnuté v súhrnných analýzach bezpečnosti vykonaných v štúdiách CONFIRM (štúdia D6997C00002), FINDER 1 (štúdia D6997C00004), FINDER 2 (štúdia D6997C00006) a NEWEST (štúdia D6997C00003), ktoré porovnávali fulvestrant v dávke 500 mg s fulvestrantom v dávke 250 mg. Frekvencie výskytu uvedené v nasledujúcej tabuľke vychádzajú zo všetkých hlásených nežiaducich reakcií na liek, bez ohľadu na to, ako skúšajúci lekári hodnotili ich kauzalitu.
Nežiaduce reakcie uvedené nižšie sú klasifikované podľa frekvencie výskytu a triedy orgánových systémov (System Organ Class, SOC). Skupiny frekvencií sú definované podľa nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1 Nežiaduce reakcie na liekNežiaduce reakcie podľa triedy orgánových systémov a frekvencie
|
Infekcie a nákazy
| Časté
| Infekcie močových ciest
|
Poruchy krvi a lymfatického systému
| Menej časté
| Znížený počet krvných doštičiek
|
Poruchy imunitného systému
| Časté
| Reakcie z precitlivenosti
|
Poruchy metabolizmu a výživy
| Časté
| Anorexiaa
|
Poruchy nervového systému
| Časté
| Bolesť hlavy
|
Poruchy ciev
| Časté
| Žilový tromboembolizmusa, návaly tepla
|
Poruchy gastrointestinálneho traktu
| Veľmi časté
| Nauzea
|
Časté
| Vracanie, hnačka
|
Poruchy pečene a žlčových ciest
| Veľmi časté
| Zvýšené hladiny pečeňových enzýmov (ALT, AST, ALP)a
|
Časté
| Zvýšená hladina bilirubínua
|
Menej časté
| Zlyhanie pečenec, hepatitídac, zvýšená hladina gama‑GT
|
Poruchy kože a podkožného tkaniva
| Časté
| Vyrážka
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Časté
| Bolesť chrbtaa
|
Poruchy reprodukčného systému a prsníkov
| Menej časté
| Vaginálna moniliáza, leukorea, vaginálne krvácanie
|
Celkové poruchy a reakcie v mieste podania
| Veľmi časté
| Asténiaa, reakcie v mieste vpichub
|
Menej časté
| Krvácanie v mieste vpichu, hematóm v mieste vpichu
|
a Vrátane nežiaducich reakcií na liek, pri ktorých sa kvôli základnému ochoreniu nedá presne stanoviť, v akej miere sa na ich vzniku podieľa fulvestrant.
b Výraz reakcie v mieste vpichu nezahŕňa výraz krvácanie v mieste vpichu a výraz hematóm v mieste vpichu.
c Táto nežiaduca udalosť sa nepozorovala v hlavných klinických štúdiách (CONFIRM, FINDER 1, FINDER 2, NEWEST). Frekvencia sa vypočítala pomocou hornej hranice 95 % intervalu spoľahlivosti pre bodový odhad. Vypočítaná je ako 3/560 (kde 560 je počet pacientov v hlavných klinických štúdiách), čo zodpovedá kategórii frekvencie „menej časté“.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.
4.9 PredávkovanieNie sú skúseností s predávkovaním u ľudí. Štúdie na zvieratách svedčia o tom, že pri vyšších dávkach fulvestrantu sa nepreukázali žiadne iné účinky okrem tých, ktoré priamo či nepriamo súvisia s jeho antiestrogénnym pôsobením (pozri časť 5.3). Ak dôjde k predávkovaniu, odporúča sa symptomatická podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Endokrinná liečba, antiestrogény, ATC kód: L02BA03
Mechanizmus účinku a farmakodynamické účinkyFulvestrant je kompetitívny antagonista estrogénového receptora (ER), ktorý má afinitu porovnateľnú s afinitou estradiolu. Fulvestrant blokuje trofický účinok estrogénov bez toho, že by vykazoval akékoľvek parciálne agonistické (estrogénové) pôsobenie. Mechanizmus účinku je spájaný so znížením hladiny expresie (tzv. downreguláciou) ER proteínu . Klinické skúšania u postmenopauzálnych žien s primárnym karcinómom prsníka preukázali, že fulvestrant v porovnaní s placebom významne znižuje expresiu ER proteínu v nádoroch s pozitivitou ER. Zistilo sa aj významné zníženie expresie progesterónových receptorov, čo je v zhode s absenciou vnútorného estrogénového agonistického pôsobenia. Pri neoadjuvantnej liečbe postmenopauzálnych žien sa tiež preukázalo, že fulvestrant v dávke 500 mg znižuje expresiu ER a proliferačného markera Ki67 v nádoroch prsníka vo väčšej miere ako fulvestrant v dávke 250 mg.
Klinická účinnosť a bezpečnosť pri pokročilom karcinóme prsníkaKlinické skúšanie fázy III sa vykonalo u 736 postmenopauzálnych žien s pokročilým karcinómom prsníka, u ktorých došlo k recidíve ochorenia počas alebo po adjuvantnej endokrinnej liečbe alebo k progresii ochorenia po endokrinnej liečbe pokročilého ochorenia. Klinické skúšanie zahŕňalo 423 pacientok, u ktorých došlo k recidíve alebo progresii ochorenia počas antiestrogénovej liečby (podskupina s AE), a 313 pacientok, u ktorých došlo k recidíve alebo progresii ochorenia počas liečby inhibítorom aromatázy (podskupina s AI). Toto klinické skúšanie porovnávalo účinnosť a bezpečnosť fulvestrantu v dávke 500 mg (n = 362) s fulvestrantom v dávke 250 mg (n = 374). Primárnym cieľovým ukazovateľom bolo prežívanie bez progresie ochorenia (progression‑free survival, PFS); kľúčové sekundárne ukazovatele účinnosti zahŕňali výskyt objektívnej odpovede na liečbu (objective response rate, ORR), výskyt klinickej prospešnosti (clinical benefit rate, CBR) a celkové prežívanie (overall survival, OS). Výsledky účinnosti zo štúdie CONFIRM sú zhrnuté v tabuľke 2.
Tabuľka 2 Zhrnutie výsledkov týkajúcich sa primárneho cieľového ukazovateľa účinnosti (PFS) a kľúčových sekundárnych cieľových ukazovateľov účinnosti zo štúdie CONFIRMPremenná
| Typ odhadu;
porovnanie liečieb
| Fulvestrant
500 mg
(N = 362)
| Fulvestrant
250 mg
(N = 374)
| Porovnanie medzi skupinami
(Fulvestrant 500 mg/Fulvestrant 250 mg)
Hazard ratio 95 % IS p‑hodnota
|
PFS
| K-M medián
v mesiacoch;
hazard ratio
|
|
|
|
|
|
Všetky pacientky
- podskupina s AE (n = 423)
- podskuina s AI (n = 313)a
| 6,5
8,6
5,4
| 5,5
5,8
4,1
| 0,80
0,76
0,85
| 0,68; 0,94
0,62; 0,94
0,67; 1,08
| 0,006
0,013
0,195
|
OSb
| K-M medián
v mesiacoch;
hazard ratio
|
26,4
30,6
24,1
|
22,3
23,9
20,8
|
0,81
0,79
0,86
|
0,69; 0,96
0,63; 0,99
0,67; 1,11
|
0,016c
0,038c
0,241c
|
Všetky pacientky
- podskupina s AE (n = 423)
- podskupina s AI (n = 313)a
|
Premenná
| Typ odhadu;
porovnanie liečieb
| Fulvestrant
500 mg
(N = 362)
| Fulvestrant
250 mg
(N = 374)
| Porovnanie medzi skupinami
(Fulvestrant 500 mg/Fulvestrant 250 mg)
Absolútny 95 % IS
rozdiel v %
|
ORRd
| % pacientok s OR;
absolútny rozdiel v %
|
13,8
18,1
7,3
|
14,6
19,1
8,3
|
|
-0,8
-1,0
-1,0
|
-5,8; 6,3
-8,2; 9,3
-5,5; 9,8
|
Všetky pacientky
- podskupina s AE (n = 296)
- podskupina s AI (n = 205)a
|
CBRe
| % pacientok s CB;
absolútny rozdiel v %
|
|
|
|
6,0
7,3
3,9
|
-1,1; 13,3
-2,2; 16,6
-6,1; 15,2
|
Všetky pacientky
- podskupina s AE (n = 423)
- podskupina s AI (n = 313)a
| 45,6
52,4
36,2
| 39,6
45,1
32,3
|
a Fulvestrant je indikovaný pacientkám, u ktorých došlo k recidíve alebo progresii ochorenia počas antiestrogénovej liečby. Výsledky v podskupine s AI sú nejednoznačné.
b Uvedené OS sa preukázalo v záverečnej analýze prežívania vykonanej po dosiahnutí 75 % „zrelosti“ údajov o prežívaní (t.j. po tom, ako došlo k úmrtiu u 75 % všetkých randomizovaných pacientov).
c Nominálna p‑hodnota, ktorá nie je upravená vzhľadom na multiplicitu medzi úvodnou analýzou celkového prežívania vykonanou v čase 50 % „zrelosti“ údajov o prežívaní a aktualizovanou analýzou celkového prežívania vykonanou po dosiahnutí 75 % „zrelosti“ údajov o prežívaní.
d ORR sa hodnotil u pacientok, ktoré boli pri zaradení do štúdie klasifikované ako vhodné na hodnotenie odpovede na liečbu (t.j. pacientky s merateľným ochorením pri zaradení do štúdie: 240 pacientok v skupine s fulvestrantom v dávke 500 mg a 261 pacientok v skupine s fulvestrantom v dávke 250 mg).
e Pacientky s najlepšou objektívnou odpoveďou na liečbu, ktorou bola kompletná remisia, parciálna remisia alebo stabilizácia ochorenia trvajúca ≥ 24 týždňov.
PFS: prežívanie bez progresie (progression-free survival); ORR: výskyt objektívnej odpovede na liečbu (objective response rate); OR: objektívna odpoveď na liečbu (objective response); CBR: výskyt klinickej prospešnosti (clinical benefit rate); CB: klinická prospešnosť (clinical benefit); OS: celkové prežívanie (overall survival); K‑M: Kaplanov‑Meierov; IS: interval spoľahlivosti; AI: inhibítor aromatázy (aromatase inhibitor); AE: antiestrogén.
Dve klinické skúšania fázy III sa vykonali u celkovo 851 postmenopauzálnych žien s pokročilým karcinómom prsníka, u ktorých došlo k recidíve ochorenia počas alebo po adjuvantnej endokrinnej liečbe alebo k progresii ochorenia po endokrinnej liečbe pokročilého ochorenia. 77 % pacientok zaradených v týchto klinických štúdiách malo karcinóm prsníka s pozitivitou estrogénových receptorov. Tieto klinické skúšania porovnávali bezpečnosť a účinnosť fulvestrantu v dávke 250 mg podávanej raz za mesiac oproti anastrozolu (inhibítor aromatázy) v dávke 1 mg podávanej denne. Celkovo je možné konštatovať, že fulvestrant v dávke 250 mg podávanej raz za mesiac bol prinajmenšom rovnako účinný ako anastrozol z hľadiska prežívania bez progresie ochorenia, objektívnej odpovede na liečbu a času do úmrtia. Medzi týmito dvomi liečebnými skupinami sa nezistili štatisticky významné rozdiely v žiadnom z uvedených cieľových ukazovateľov. Primárnym cieľovým ukazovateľom bolo prežívanie bez progresie ochorenia. Kombinovaná analýza obidvoch klinických skúšaní ukázala, že k progresii ochorenia došlo u 83 % pacientok liečených fulvestrantom v porovnaní s 85 % pacientok liečených anastrozolom. Kombinovaná analýza obidvoch klinických skúšaní ukázala, že pokiaľ ide o prežívanie bez progresie ochorenia, pomer rizík pri fulvestrante v dávke 250 mg v porovnaní s anastrozolom bolo 0,95 (95 % IS: 0,82 až 1,10). Výskyt objektívnej odpovede na liečbu bol 19,2 % pri fulvestrante v dávke 250 mg v porovnaní so 16,5 % pri anastrozole. Medián času do úmrtia bol 27,4 mesiaca u pacientok liečených fulvestrantom a 27,6 mesiaca u pacientok liečených anastrozolom. Pokiaľ ide o čas do úmrtia, pomer rizík pri fulvestrante v dávke 250 mg v porovnaní s anastrozolom bolo 1,01 (95 % IS: 0,86 až 1,19).
Účinky na postmenopauzálne endometriumPredklinické údaje nesvedčia o tom, že by fulvestrant mal stimulačný účinok na postmenopauzálne endometrium (pozri časť 5.3). Dvojtýždňová štúdia u zdravých postmenopauzálnych dobrovoľníčok liečených 20 μg etinylestradiolu denne ukázala, že predliečebné podávanie fulvestrantu v dávke 250 mg v porovnaní s predliečebným podávaním placeba viedlo k významne zníženej stimulácii postmenopauzálneho endometria, čo sa posudzovalo pomocou merania hrúbky endometria ultrazvukom.
Neoadjuvantná liečba trvajúca až 16 týždňov, v rámci ktorej sa pacientkám s karcinómom prsníka podával buď fulvestrant v dávke 500 mg, alebo fulvestrant v dávke 250 mg, neviedla ku klinicky významným zmenám hrúbky endometria, čo poukazuje na absenciu agonistického účinku. U sledovaných pacientok s karcinómom prsníka sa nepreukázali nežiaduce účinky na endometrium. K dispozícii nie sú žiadne údaje týkajúce sa morfológie endometria.
V dvoch krátkodobých štúdiách (v 1‑týždňovej a v 12‑týždňovej) u premenopauzálnych pacientok s benígnym gynekologickým ochorením sa medzi skupinou s fulvestrantom a skupinou s placebom nepozorovali významné rozdiely v hrúbke endometria meranej ultrazvukom.
Účinky na kostiK dispozícii nie sú žiadne dlhodobé údaje o vplyve fulvestrantu na kosti. Neoadjuvantná liečba trvajúca až 16 týždňov, v rámci ktorej sa pacientkám s karcinómom prsníka podával buď fulvestrant v dávke 500 mg, alebo fulvestrant v dávke 250 mg, neviedla ku klinicky významným zmenám markerov kostného obratu stanovovaných v sére.
Pediatrická populáciaFulvestrant nie je indikovaný na použitie u detí. Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s fulvestrantom vo všetkých podskupinách pediatrickej populácie s karcinómom prsníka (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Otvorená štúdiá fázy II skúmala bezpečnosť, účinnosť a farmakokinetiku fulvestrantu u 30 dievčat s progresívnou predčasnou pubertou spojenou s McCuneovým‑Albrightovým syndrómom (MAS), ktoré boli vo veku 1 až 8 rokov. Pediatrickým pacientkám sa fulvestrant podával intramuskulárne v dávke 4 mg/kg raz za mesiac. Táto 12‑mesačná štúdia skúmala rôzne cieľové ukazovatele súvisiace s MAS a preukázala zníženie frekvencie vaginálneho krvácania a zníženie stupňa urýchlenia kostného veku. Minimálne (trough) koncentrácie fulvestrantu v rovnovážnom stave zistené u detí v tejto štúdii sa zhodovali s tým, ktoré sa zistili u dospelých (pozri časť 5.2). Z tejto malej štúdie nevyplynuli žiadne nové obavy týkajúce sa bezpečnosti, ale 5‑ročné údaje ešte nie sú k dispozícii.
5.2 Farmakokinetické vlastnostiAbsorpciaPo podaní intramuskulárnej injekcie dlhodobo pôsobiaceho fulvestrantu sa fulvestrant pomaly absorbuje a maximálne plazmatické koncentrácie (C
max) sa dosiahnu približne po 5 dňoch. Pri podávaní fulvestrantu v dávke 500 mg sa hladiny expozície na úrovni alebo blízko úrovne rovnovážneho stavu dosiahnu v priebehu prvého mesiaca podávania (priemer [CV]: AUC 475 [33,4 %] ng.dni/ml, C
max25,1 [35,3 %] ng/ml, C
min 16,3 [25,9 %] ng/ml). Po dosiahnutí rovnovážneho stavu sa plazmatické koncentrácie fulvestrantu udržiavajú v relatívne úzkom rozmedzí, pričom rozdiel medzi maximálnou a minimálnou koncentráciou je približne 3‑násobný. Po intramuskulárnom podaní je expozícia približne úmerná dávke, keď sa podávajú dávky v rozmedzí od 50 do 500 mg.
DistribúciaFulvestrant podlieha rozsiahlej a rýchlej distribúcii. Veľký zdanlivý distribučný objem (Vd
ss) v ustálenom stave rovný približne 3 až 5 l/kg svedčí o tom, že distribúcia je prevažne extravaskulárna. Fulvestrant sa vo vysokej miere (99 %) viaže na plazmatické bielkoviny. Viaže sa hlavne na frakcie lipoproteínov s veľmi nízkou hustotou (very low density lipoprotein, VLDL), lipoproteínov s nízkou hustotou (low density lipoprotein, LDL) a lipoproteínov s vysokou hustotou (high density lipoprotein, HDL). Neuskutočnili sa žiadne interakčné štúdie skúmajúce kompetitívnu väzbu na bielkoviny. Úloha globulínu viažuceho pohlavné hormóny (sex hormone‑binding globulin, SHBG) nebola stanovená.
BiotransformáciaMetabolizmus fulvestrantu nie je úplne zhodnotený, ale zahŕňa kombináciu niekoľkých možných biotransformačných dráh obdobných tým, ktoré sa zistili pri endogénnych steroidoch. Identifikované metabolity (zahŕňajúce ketónový metabolit na 17. pozícii steroidného jadra, sulfónové metabolity, sulfátový metabolit na 3. pozícii steroidného jadra, glukuronidové metabolity na 3. a 17. pozícii steroidného jadra) sú v antiestrogénových modeloch buď menej účinné, alebo vykazujú podobný účinok ako fulvestrant. Štúdie, v ktorých sa použili preparáty pochádzajúce z ľudskej pečene a rekombinantné ľudské enzýmy, poukazujú na to, že CYP3A4 je jediným izoenzýmom cytochrómu P450, ktorý sa podieľa na oxidácii fulvestrantu; v podmienkach
in vivo sú však zrejme rozhodujúcejšie dráhy nesprostredkované cytochrómom P450.
In vitro údaje svedčia o tom, že fulvestrant neinhibuje izoenzýmy CYP450.'
ElimináciaFulvestrant sa eliminuje hlavne v metabolizovanej forme. Hlavnou cestou exkrécie je stolica, pričom močom sa vylučuje menej ako 1 %. Fulvestrant má vysoký klírens, 11 ± 1,7 ml/min/kg, čo poukazuje na vysoký pomer hepatálnej extrakcie. Terminálny polčas (t
1/2) po intramuskulárnom podaní je determinovaný rýchlosťou absorpcie a odhadol sa na 50 dní.
Osobitné skupiny pacientokV populačnej farmakokinetickej analýze údajov zo štúdií fázy III sa nezistil žiadny rozdiel vo farmakokinetickom profile fulvestrantu v súvislosti s vekom (rozmedzie 33 až 89 rokov), telesnou hmotnosťou (40 ‑ 127 kg) alebo rasou.
Porucha funkcie obličiekMierna až stredne ťažká porucha funkcie obličiek neovplyvnila farmakokinetiku fulvestrantu v klinicky významnej miere.
Porucha funkcie pečeneFarmakokinetika fulvestrantu sa hodnotila v klinickom skúšaní s jednorazovou dávkou uskutočnenom u osôb s miernou až stredne ťažkou poruchou funkcie pečene (trieda A a B podľa Childovej‑Pughovej klasifikácie). Použila sa vysoká dávka krátkodobejšie pôsobiaceho fulvestrantu podaná intramuskulárnou injekciou. U osôb s poruchou funkcie pečene došlo v porovnaní so zdravými osobami približne k 2,5‑násobnému zvýšeniu hodnoty AUC. Očakáva sa, že u pacientok liečených fulvestrantom bude takéto zvýšenie expozície dobre tolerované. Osoby s ťažkou poruchou funkcie pečene (triedy C podľa Childovej‑Pughovej klasifikácie) sa nehodnotili.
Pediatrická populáciaFarmakokinetika fulvestrantu sa hodnotila v klinickom skúšaní uskutočnenom u 30 dievčat s progresívnou predčasnou pubertou spojenou s McCuneovým‑Albrightovým syndrómom (pozri časť 5.1). Pediatrické pacientky boli vo veku 1 až 8 rokov a fulvestrant sa im podával intramuskulárne v dávke 4 mg/kg raz za mesiac. Geometrický priemer (štandardná odchýlka) minimálnej koncentrácie v rovnovážnom stave (C
min,ss)a AUC
ss boli 4,2 (0,9) ng/ml a 3 680 (1 020) ng*h/ml v uvedenom poradí. Hoci bolo množstvo zozbieraných údajov obmedzené, minimálne koncentrácie fulvestrantu v rovnovážnom stave zistené u detí sa zrejme zhodovali s tými, ktoré sa zistili u dospelých.
5.3 Predklinické údaje o bezpečnostiAkútna toxicita fulvestrantu je nízka.
V štúdiách s opakovaným podávaním boli iné formy fulvestrantu dobre tolerované u rôznych živočíšnych druhov. Lokálne reakcie vrátane myozitídy a granulómu v mieste vpichu boli pripisované vehikulu, ale závažnosť myozitídy u králikov bola vyššia pri fulvestrante v porovnaní s kontrolným fyziologickým roztokom. V štúdiách toxicity s opakovaným intramuskulárnym podávaním fulvestrantu potkanom a psom bolo antiestrogénne pôsobenie fulvestrantu zodpovedné za väčšinu pozorovaných účinkov, najmä tých, ktoré postihovali reprodukčný systém samíc, ale aj iné orgány citlivé na hormóny u oboch pohlaví. U niektorých psov sa po dlhodobom (12‑mesačnom) podávaní pozorovala arteritída zasahujúca celú škálu rôznych tkanív.
V štúdiách na psoch sa po perorálnom a intravenóznom podávaní pozorovali účinky na kardiovaskulárny systém (mierne elevácie ST segmentu na EKG [po perorálnom podávaní] a zastavenie sínusu u jedného psa [po intravenóznom podávaní]). Tieto účinky sa vyskytli pri hladinách expozície vyšších ako sú tie, ktoré sa dosahujú u pacientov (viac ako 15‑násobok C
max), a pravdepodobne majú obmedzený význam pre bezpečnosť u ľudí pri podávaní klinickej dávky.
Fulvestrant nevykazoval genotoxický potenciál.
Pri podávaní fulvestrantu v dávkach podobných klinickej dávke sa preukázali účinky na reprodukciu a embryonálny/fetálny vývoj, ktoré zodpovedali jeho antiestrogénnemu pôsobeniu. U potkanov sa pozorovalo reverzibilné zníženie fertility samíc a zníženie miery prežívania plodov, dystokia a zvýšený výskyt abnormalít u plodov vrátane ohnutia priehlavkových kostí. Králiky, ktorým bol podávaný fulvestrant, neboli schopné donosiť plody. Pozorovalo sa zvýšenie hmotnosti placenty a zvýšenie počtu postimplantačných strát plodov. U králikov sa zistil zvýšený výskyt anatomických odchýlok u plodov (posunutie panvového pletenca smerom dozadu a výskyt 27 presakrálnych stavcov).
Dvojročná štúdia onkogenity na potkanoch (s intramuskulárnym podávaním fulvestrantu) preukázala zvýšený výskyt benígnych nádorov z granulóznych buniek ovárií u samíc potkanov, ktorým bola podávaná vysoká dávka, 10 mg na potkana/15 dní, a zvýšený výskyt testikulárnych nádorov z Leydigových buniek u samcov. V dvojročnej štúdii onkogenity na myšiach (s denným perorálnym podávaním) sa zistil zvýšený výskyt ovariálnych nádorov (benígnych aj malígnych) zo sex cord stromálnych buniek po podávaní dávok 150 a 500 mg/kg/deň. Pri dávke, pri ktorej tieto nálezy ešte neboli pozorované (no observed effect level, NOAEL), boli hladiny systémovej expozície (AUC) u potkanov približne 1,5‑násobne vyššie ako sú predpokladané hladiny expozície u žien a 0,8‑násobne vyššie ako sú predpokladané hladiny expozície u mužov a hladiny systémovej expozície (AUC) u myší boli približne 0,8‑násobne vyššie ako sú predpokladané hladiny expozície u mužov aj žien. Tvorba takýchto nádorov je dôsledkom farmakologicky podmienených zmien v sekrécii gonádotropínov regulovanej mechanizmom spätnej väzby na úrovni endokrinných žliaz, ktoré boli u cyklujúcich zvierat spôsobené podávaním antiestrogénov. Preto sa tieto nálezy nepovažujú za relevantné pre použitie fulvestrantu u postmenopauzálnych žien s pokročilým karcinómom prsníka.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokEtanol (96 %)
Benzylalkohol
Benzylbenzoát
Rafinovaný ricínový olej
6.2 InkompatibilityNevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti2 roky.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte a prepravujte v chladničke (2 °C ‑ 8 °C).
Je potrebné obmedziť výskyt teplotných odchýlok mimo rozmedzia 2 °C ‑ 8 °C. V tomto zmysle sa treba vyhnúť uchovávaniu lieku pri teplotách prevyšujúcich 25 °C a pokiaľ je priemerná teplota uchovávania lieku nižšia ako 25 °C (ale vyššia ako 2 °C ‑ 8 °C), takéto uchovávanie nemá trvať dlhšie ako 28 dní. Po výskyte teplotnej odchýlky sa treba ihneď vrátiť k odporúčaným podmienkam uchovávania lieku (uchovávanie a prepravovanie v chladničke pri 2 °C ‑ 8 °C). Teplotné odchýlky majú kumulatívny vplyv na kvalitu lieku a nesmú trvať súvisle dlhšie ako 28 dní počas celého 2-ročného času použiteľnosti (pozri časť 6.3). Vystavenie lieku teplotám nižším ako 2 °C nespôsobí jeho poškodenie, pokiaľ nie je uchovávaný pri teplote nižšej ako ‑20 °C.
Naplnenú injekčnú striekačku uchovávajte v pôvodnom balení na ochranu pred svetlom.
6.5 Druh obalu a obsah baleniaBalenie s naplnenou injekčnou striekačkou obsahuje:
Jednu naplnenú injekčnú striekačku z číreho skla typu 1 s polypropylénovým piestom, vybavenú poistným uzáverom, ktorá obsahuje 5 ml injekčného roztoku Fulvestrantu Teva.
Poskytnutá je aj bezpečnostná ihla, ktorá sa má pripojiť k valcu injekčnej striekačky.
Alebo
Dve naplnené injekčné striekačky z číreho skla typu 1 s polypropylénovým piestom, vybavené poistným uzáverom, z ktorých každá obsahuje 5 ml injekčného roztoku Fulvestrantu Teva. Poskytnuté sú aj bezpečnostné ihly, ktoré sa majú pripojiť k valcom injekčných striekačiek.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu ainé zaobchádzanie sliekomPokyny na podávanieUpozornenie ‑ bezpečnostnú ihlu pred použitím neautoklávujte.
Počas používania a pri likvidácii musia ruky stále zostať za ihlou.
Pri každej z dvoch injekčných striekačiek:
· Vyberte sklenený valec injekčnej striekačky z vaničky a skontrolujte, či nie je poškodený.
· Zlomte plombu priehľadného plastového uzávera na prípojke Luer‑Lock (t.j. na kónuse so závitom) injekčnej striekačky a snímte uzáver s pripojenou gumovou zátkou (pozri obrázok 1).
| Obrázok 1

|
· Odstráňte vonkajší obal bezpečnostnej ihly. Nasaďte bezpečnostnú ihlu na prípojku Luer‑Lock (pozri obrázok 2).
· Otáčajte ihlu, pokým pevne nedosadne.
|
Obrázok 2
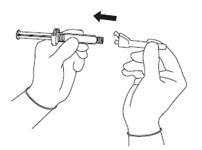
|
· Pootočením ju zafixujte v prípojke Luer-Lock.
· Stiahnite puzdro z ihly v priamom smere, aby ste zabránili poškodeniu hrotu ihly (pozri obrázok 3).
| Obrázok 3
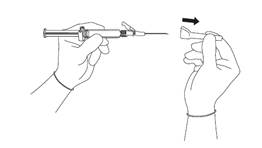
|
· Preneste naplnenú injekčnú striekačku na miesto, kde ju budete podávať.
· Parenterálne podávané roztoky sa pred podaním musia zrakom skontrolovať na prítomnosť cudzorodých častíc a zmenu farby.
· Vytlačte z injekčnej striekačky prebytočný vzduch.
· Podajte pomaly intramuskulárne (1 ‑ 2 minúty/injekcia) do sedacieho svalu. Pri aplikácii je pre používateľa výhodnejšie, ak je skosená hrana ihly orientovaná smerom k páčke ramena (pozri obrázok 4).
| Obrázok 4
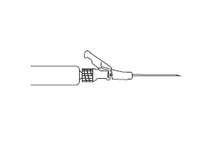
|
· Ihneď po podaní injekcie využite ťah jedného prsta na aktiváciu ramena páčky, čím aktivujete ochranný (bezpečnostný) mechanizmus ihly (pozri obrázok 5).
POZNÁMKA: Aktivujte mechanizmus smerom od seba a iných. Musíte počuť cvaknutie a zrakom sa presvedčte, že hrot ihly je úplne zakrytý.
|
Obrázok 5
|
LikvidáciaNaplnené injekčné striekačky sú určené
len na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIITEVA Pharmaceuticals Slovakia s.r.o.
Teslova 26, 821 02 Bratislava
Slovenská republika
8. REGISTRAČNÉ ČÍSLO34/0179/16-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUApríl 2016