
antu u dojčených detí je používanie počas laktácie kontraindikované (pozri časť 4.3).
Fertilita
Účinky fulvestrantu na plodnosť ľudí sa nehodnotili.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Fulvestrant nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje. Vzhľadom na to, že sa počas užívania fulvestrantu veľmi často vyskytli prípady asténie, musí sa venovať zvýšená opatrnosť pacientkam, u ktorých sa vyskytol tento nežiaduci účinok počas vedenia motorových vozidiel alebo obsluhy strojov.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Monoterapia
V tejto časti sa nachádzajú informácie, vychádzajúce zo všetkých nežiaducich reakcií z klinických štúdií, postmarketingových štúdií alebo zo spontánnych hlásení. V súhrnnom súbore údajov
z monoterapie fulvestrantom boli najčastejšie hlásenými nežiaducimi reakciami reakcie v mieste podania injekcie, asténia, nauzea a zvýšenie hepatálnych enzýmov (ALT, AST, ALP).
V tabuľke 1 boli zistené nasledujúce kategórie frekvencie nežiaducich reakcií (adverse drug reactions, ADRs), ktoré sú založené na združených analýzach bezpečnosti v liečebnej skupine užívajúcej fulvestrant 500 mg v štúdiách porovnávajúcich fulvestrant 500 mg s fulvestrantom 250 mg [CONFIRM (Štúdia D6997C00002), FINDER 1 (Štúdia D6997C00004), FINDER 2 (Štúdia D6997C00006)
a NEWEST (Štúdia D6997C00003)], alebo zo samotnej štúdie FALCON (Štúdia D699BC00001), ktorá porovnávala fulvestrant 500 mg s anastrozolom 1 mg.
Pri rozdieloch vo frekvenciách pri združených analýzach bezpečnosti a štúdii FALCON sa uvádza
najvyššia frekvencia. V tabuľke 1 sú frekvencie založené na všetkých hlásených nežiaducich reakciách bez ohľadu na hodnotenie kauzality skúšajúcim. Medián trvania liečby fulvestrantom 500 mg v združenom súbore údajov (vrátane štúdií uvedených vyššie plus FALCON) bol 6,5 mesiacov.
Tabuľkovýzoznamnežiaducichreakcií
Nižšie uvedené nežiaduce reakcie sú klasifikované podľa frekvencie a triedy orgánových systémov
(System Organ Class – SOC). Skupiny frekvencií sú definované nasledovne: veľmi časté (≥ 1/10),
sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1 Nežiaduce reakcie hlásené pacientkami liečenými fulvestrantom v monoterapiiNežiaduce reakcie podľa orgánových systémov a frekvencie
|
Infekcie a nákazy
| Časté
| Infekcie močového systému
|
Poruchy krvi a lymfatického systému
| Časté
| Zníženie počtu krvných doštičiek e
|
Poruchy imunitného systému
| Veľmi časté
| Hypersenzitívne reakcie e
Anafylaktické reakcie
|
Menej časté
|
Poruchy metabolizmu a výživy
| Časté
| Anorexiaa
|
Poruchy nervového systému
| Časté
| Bolesť hlavy
|
Poruchy ciev
| Veľmi časté
| Návaly tepla e
Venózna tromboembólia a
|
Časté
|
Poruchy gastrointestinálneho traktu
| Veľmi časté
| Nauzea
|
Časté
| Vracanie, hnačka
|
Poruchy pečene a žlčových ciest
| Veľmi časté
| Zvýšenie hladín pečeňových enzýmov (ALT, AST, ALP)a
|
Časté
| Zvýšený bilirubína
|
Menej časté
| Zlyhanie pečenec,f, hepatitídaf, zvýšené
gama-GTf
|
Poruchy kože a podkožného tkaniva
| Veľmi časté
| Vyrážkae
|
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
| Veľmi časté
| Bolesť kĺbov a muskuloskeletálna bolesť d
Bolesť chrbtaa
|
Časté
|
Poruchy reprodukčného systému a prsníkov
| Časté
| Vaginalne krvácanie e
Vaginálna moniliáza f, leukorea f
|
Menej časté
|
Celkové poruchy a reakcie v mieste
podania
| Veľmi časté
| Asténiaa, reakcie v mieste podania injekcieb
Periférna neuropatia e, ischias e
|
Časté
|
Menej časté
| Krvácanie v mieste podania injekcie f, hematóm v mieste podania injekcie f, neuralgia cf
|
a Vrátane nežiaducich reakcií, pri ktorých nie je možné stanoviť podiel fulvestrantu na ich vzniku
vzhľadom na základné ochorenie.
b Termín reakcie v mieste podania injekcie nezahŕňa termíny krvácanie v mieste podania injekcie, hematóm v mieste podania injekcie, ischias, neuralgia a periférna neuropatia.
c Udalosť sa nepozorovala vo veľkých klinických štúdiách (CONFIRM, FINDER 1, FINDER 2.
NEWEST). Frekvencia sa vypočítala pomocou hornej hranice 95% intervalu spoľahlivosti pre odhad bodu. Počíta sa ako 3/560 (kde 560 je počet pacientok vo veľkých klinických štúdiách), čo zodpovedá
kategórii frekvencie "menej časté“.
d Zahŕňa: artralgiu a menej častú muskuloskeletálnu bolesť, myalgiu a bolesť v končatinách.
e Kategória frekvencie je rozdielna medzi údajmi zo združených analýz bezpečnosti a štúdiou FALCON.
f Nežiaduca reakcia sa v štúdii FALCON nepozorovala.
OpisvybranýchnežiaducichreakciíOpis uvádzaný nižšie je založený na bezpečnostnej analýze súboru 228 pacientok, ktoré dostali aspoň jednu (1) dávku fulvestrantu a 232 pacientok, ktoré dostali aspoň jednu (1) dávku anastrozolu vo fáze
3 štúdie FALCON.
Bolesť kĺbov a muskuloskeletálna bolesťV štúdii FALCON bol počet pacientok, ktoré hlásili nežiaducu reakciu bolesť kĺbov
a muskuloskeletálnu bolesť 65 (31,2%) v skupine s fulvestrantom a 48 (24,1%) v skupine
s anastrozolom. Zo 65 pacientok v skupine s fulvestrantom 40% (26/65) pacientok hlásilo bolesť kĺbov a muskuloskeletálnu bolesť počas prvého mesiaca liečby a 66,2% (43/65) pacientok počas prvých 3
mesiacov liečby. Žiadna pacientka nehlásila udalosť stupňa ≥ 3 podľa CTCAE, alebo udalosť
vyžadujúcu zníženie dávky, prerušenie dávkovania alebo ukončenie liečby z dôvodu týchto nežiaducich reakcií.
Kombinovaná liečba s palbociklibomCelkový bezpečnostný profil fulvestrantu pri použití v kombinácii s palbociklibom je založený na údajoch od 517 pacientok s HR-pozitívnym, HER2-negatívnym pokročilým alebo metastatickým
karcinómom prsníka v randomizovanej štúdii PALOMA3 (pozri časť 5.1). Najčastejšími (≥ 20%)
nežiaducimi reakciami akéhokoľvek stupňa, hlásenými u pacientok dostávajúcich fulvestrant v kombinácii s palbociklibom, boli neutropénia, leukopénia, infekcie, únava, nauzea, anémia,
stomatitída, hnačka, trombocytopénia a vracanie. Najčastejšími (≥ 2%) nežiaducimi reakciami stupňa
≥ 3 boli neutropénia, leukopénia, anémia, infekcie, zvýšená hladina AST, trombocytopénia a únava.
V tabuľke 2 sú uvedené nežiaduce reakcie zo štúdie PALOMA3.
Medián trvania expozície fulvestrantu bol v skupine s fulvestrantom + palbociklibom 11,2 mesiacov
a 4,8 mesiacov v skupine s fulvestrantom + placebo. Medián trvania expozície palbociklibu v skupine s fulvestrantom + palbociklib bol 10,8 mesiacov.
Tabuľka 2 Nežiaduce reakcie zo štúdie PALOMA3 (N=517)
Trieda orgánových systémov Frekvencia
Preferovaný termín a
| Faslodex + Palbociklib
(N=345)
| Faslodex + placebo
(N=172)
|
Všetky stupne n (%)
|
Stupeň ≥ 3
n (%)
| Všetky stupne n (%)
|
Stupeň ≥ 3
n (%)
|
Infekcie a nákazy
|
Veľmi časté
|
|
|
|
|
Infekcieb
| 188 (54,5)
| 19 (5,5)
| 60 (34,9)
| 6 (3,5)
|
Poruchy krvi a lymfatického systému
|
Veľmi časté
|
|
|
|
|
Neutropéniac
| 290 (84,1)
| 240 (69,6)
| 6 (3,5)
| 0
|
Leukopéniad
| 207 (60,0)
| 132 (38,3)
| 9 (5,2)
| 1 (0,6)
|
Anémiae
|
109 (31,6)
|
15 (4,3)
|
24 (14,0)
|
4 (2,3)
|
Trombocytopéniaf
|
88 (25,5)
|
10 (2,9)
|
0
|
0
|
M
enej časté
|
|
|
|
|
Febrilná neutropénia
|
3 (0,9)
|
3 (0,9)
|
0
|
0
|
Poruchy metabolizmu a výživy
|
Veľmi časté
|
|
|
|
|
Znížená chuť do jedla
|
60 (17,4)
|
4 (1,2)
|
18 (10,5)
|
1 (0,6)
|
Poruchy nervového systému
|
Č
asté
|
|
|
|
|
Dysgeúzia
|
27 (7,8)
|
0
|
6 (3,5)
|
0
|
Poruchy oka
|
Č
asté
|
|
|
|
|
Zvýšené slzenie
|
25 (7,2)
|
0
|
2 (1,2)
|
0
|
Rozmazané videnie
|
24 (7,0)
|
0
|
3 (1,7)
|
0
|
Suché oko
|
15 (4,3)
|
0
|
3 (1,7)
|
0
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Č
asté
|
|
|
|
|
Epistaxa
|
25 (7,2)
|
0
|
4 (2,3)
|
0
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
|
|
|
|
|
Nauzea
|
124 (35,9)
|
2 (0,6)
|
53 (30,8)
|
1 (0,6)
|
Stomatitídag
|
104 (30,1)
|
3 (0,9)
|
24 (14,0)
|
0
|
Hnačka
|
94 (27,2)
|
0
|
35 (20,3)
|
2 (1,2)
|
Vracanie
|
75 (21,7)
|
2 (0,6)
|
28 (16,3)
|
1 (0,6)
|
Poruchy kože a podkožného tkaniva
|
Veľmi časté
|
|
|
|
|
Alopécia
|
67 (19,4)
|
NA
|
11 (6,4)
|
NA
|
Vyrážkah
|
63 (18,3)
|
3 (0,9)
|
10 (5,8)
|
0
|
Č
asté
|
|
|
|
|
Suchá koža
|
28 (8,1)
|
0
|
3 (1,7)
|
0
|
C
elkové poruchy a reakcie v mieste podania
|
Veľmi časté
|
|
|
|
|
Únava
|
152 (44,1)
|
9 (2,6)
|
54 (31,4)
|
2 (1,2)
|
Pyrexia
|
47 (13,6)
|
1 (0,3)
|
10 (5,8)
|
0
|
Č
asté
|
|
|
|
|
Asténia
|
27 (7,8)
|
1 (0,3)
|
13 (7,6)
|
2 (1,2)
|
Laboratórne vyšetrenia
|
Veľmi časté
|
|
|
|
|
Zvýšená hladina AST
|
40 (11,6)
|
11 (3,2)
|
13 (7,6)
|
4 (2,3)
|
Č
asté
|
|
|
|
|
Zvýšená hladina ALT
|
30 (8,7)
|
7 (2,0)
|
10 (5,8)
|
1 (0,6)
|
ALT=alanínaminotransferáza; AST=aspartátaminotransferáza; N/n=počet pacientov; NA=neaplikovateľné
a Preferované termíny (Preferred Terms, PTs) sú uvedené podľa MedDRA 17.1.
b Infekcie zahŕňajú všetky PTs, ktoré sú súčasťou triedy orgánových systémov „Infekcie a nákazy”.
c Neutropénia zahŕňa nasledujúce PTs: neutropénia, znížený počet neutrofilov.
d Leukopénia zahŕňa nasledujúce PTs: leukopénia, znížený počet bielych krviniek.
e Anémia zahŕňa nasledujúce PTs: anémia, znížený hemoglobín, znížený hematokrit.
f Trombocytopénia zahŕňa nasledujúce PTs: trombocytopénia, znížený počet krvných doštičiek.
g Stomatitída zahŕňa nasledujúce PTs: aftózna stomatitída, cheilitída, glositída, glosodýnia, ulcerácia v ústach, zápal sliznice, bolesť v ústach, orofaryngeálny diskomfort, orofaryngeálna bolesť, stomatitída.
h Vyrážka zahŕňa nasledujúce PTs: vyrážka, makulopapulárna vyrážka, svrbiaca vyrážka, erytematózna
vyrážka, papulárna vyrážka, dermatitída, akneiformná dermatitída, toxická kožná erupcia.
OpisvybranýchnežiaducichreakciíNeutropéniaU pacientok, ktoré v štúdii PALOMA3 dostávali fulvestrant v kombinácii s palbociklibom sa neutropénia akéhokoľvek stupňa hlásila u 290 (84,1%) pacientok, neutropénia stupňa 3 sa hlásila u 200 (58,0%) pacientok a neutropénia stupňa 4 sa hlásila u 40 (11,6%) pacientok. V skupine s fulvestrantom + placebo (n=172) sa neutropénia akéhokoľvek stupňa hlásila u 6 (3,5%) pacientok. V skupine s fulvestrantom + placebo sa nehlásila žiadna neutropénia stupňa 3 a 4.
U pacientok dostávajúcich fulvestrant v kombinácii s palbociklibom bol medián času do prvej epizódy neutropénie akéhokoľvek stupňa 15 dní (rozsah: 13-512 dní) a medián trvania neutropénie stupňa ≥ 3 bol
16 dní. Febrilná neutropénia sa hlásila u 3 (0,9%) pacientok dostávajúcich fulvestrant v kombinácii s
palbociklibom.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSú známe jednotlivé hlásenia predávkovania fulvestrantom u ľudí. V prípade predávkovania sa odporúča symptomatická podporná liečba. Štúdie na zvieratách udávajú, že okrem účinkov priamo alebo nepriamo spojených s antiestrogénnou aktivitou, sa pri vyšších dávkach fulvestrantu nepreukázali žiadne iné účinky (pozri časť 5.3).
5 FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Endokrinná liečba, antiestrogény, ATC kód: L02BA03
Mechanizmus účinkuafarmakodynamickéúčinkyFulvestrant je kompetitívny antagonista estrogénového receptora (ER) s afinitou porovnateľnou
s estradiolom. Fulvestrant blokuje trofický účinok estrogénov bez akejkoľvek čiastočnej agonistickej
(estrogénu podobnej) aktivity. Mechanizmus účinku je spojený s potlačením hladín proteínu estrogénového receptora.
Klinické skúšky u postmenopauzálnych žien s primárnym karcinómom prsníka preukázali, že fulvestrant
významne znižuje hladinu proteínu ER v ER pozitívnych nádoroch v porovnaní s placebom. Taktiež sa pozorovalo významné zníženie expresie progesterónového receptora v súlade s chýbajúcim vnútorným
agonistickým estrogénovým účinkom Tiež sa preukázalo, že fulvestrant 500 mg znižuje počet ER a proliferáciu markeru Ki67, vo väčšej miere ako fulvestrant 250 mg v nádoroch prsníka v postmenopauzálnej neoadjuvantnej liečbe.
Klinická bezpečnosťaúčinnosťpripokročilomkarcinómeprsníka
Monoterapia
Klinickú štúdiu fázy 3 ukončilo 736 postmenopauzálnych žien s pokročilým karcinómom prsníka, ktoré mali rekurenciu ochorenia počas alebo po adjuvantnej endokrinnej liečbe alebo progresii následnej endokrinnej liečby pokročilého ochorenia. Štúdia zahŕňala 423 pacientov, ktorí recidivovali alebo mali progresiu počas antiestrogénnej liečby (AE podskupina) a 313 pacientov, ktorí recidivovali alebo mali progresiu počas liečby inhibítorom aromatázy (AI podskupina). Táto štúdia porovnávala účinnosť a bezpečnosť fulvestrantu 500 mg (n=362) s fulvestrantom 250 mg (n=374). Prežitie bez
progresie (Progression-free survival,PFS) bolo primárnym cieľom, kľúčové sekundárne ciele bezpečnosti zahŕňali objektívnu mieru odpovede (objective response rate, ORR), mieru klinického úžitku (clinical
benefit rate, CBR) a celkové prežívanie (overall survival, OS). Výsledky účinnosti pre CONFIRM štúdiu sú zhrnuté pre všetkých pacientov a podskupinou pacientov, ktorí mali nedostatočnú liečbu antiestrogén
verzus inhibítor aromatázy v tabuľke 3.
Tabuľka 3 Prehľad výsledkov primárneho cieľa účinnosti (PFS – progression free survival)
a kľúčovýchsekundárnychcieľovúčinnostivštúdiiCONFIRM
R
ôzne Typ odhadu; porovnanie liečby
f
u
l
vestrant
500 mg
(
N=
362)
f
u
l
vestrant
250 mg
(
N=
374)
P
orovnanie medzi skupinami
 (
f
ulvestrant 500 mg/fulvestrant 250 mg)
H
azard ratio 95% CI p-hodnota
(
f
ulvestrant 500 mg/fulvestrant 250 mg)
H
azard ratio 95% CI p-hodnota
PFS
|
K
M medián v
|
|
|
|
|
|
|
m
esiacoch;
|
|
|
|
|
|
|
hazard ratio
|
|
|
|
|
|
V
šetci pacienti
|
|
6,5
|
5,5
|
0,80
|
0,68; 0,94
|
0,006
|
-
A
E podskupina
|
(
n=423)
|
8,6
|
5,8
|
0,76
|
0,62; 0,94
|
0,013
|
-
A
I podskupina
|
(
n=313)
a
|
5,4
|
4,1
|
0,85
|
0,67; 1,08
|
0,195
|
OSb
|
K-M medián v
|
|
|
|
|
|
|
mesiacoch;
|
|
|
|
|
|
|
hazard ratio
|
|
|
|
|
|
Všetci pacienti
|
26,4
|
22,3
|
0,81
|
0,69; 0,96
|
0,016c
|
-AE podskupina (n=423)
|
30,6
|
23,9
|
0,79
|
0,63; 0,99
|
0,038c
|
-AI podskupina (n=313)a
|
24,1
|
20,8
|
0,86
|
0,67; 1,11
|
0,241c
|
|
|
|
|
|
|
|
|
|
|
|
|
Rôzne Typ odhadu;
porovnanie
fulvestrant
500 mg
fulvestrant
250 mg
Porovnanie medzi skupinami
(fulvestrant 500 mg / fulvestrant 250 mg)
liečby

ORRd % pacientov s OR; absolútny rozdiel v %
(N=362)
(N=374)
Absolútny
rozdiel v %
95% CI
Všetci pacienti
|
13,8
|
14,6
|
-0,8
|
-5,8; 6,3
|
-AE podskupina (n=296)
|
18,1
|
19,1
|
-1,0
|
-8,2; 9,3
|
-AI podskupina (n=205)a
|
7,3
|
8,3
|
-1,0
|
-5,5; 9,8
|

CBRe % pacientov s CB; absolútny rozdiel v %
Všetci pacienti
| 45,6
| 39,6
| 6,0
| -1,1; 13,3
|
-AE podskupina (n=423)
| 52,4
| 45,1
| 7,3
| -2,2; 16,6
|
-AI podskupina (n=313)a
| 36,2
| 32,3
| 3,9
| -6,1; 15,2
|
a Fulvestrant je indikovaný u pacientok, ktoré recidivovali alebo mali progresiu počas antiestrogénnej
liečby. Výsledky v AI podskupine sú nepresvedčivé.
b OS je uvádzané pre finálnu analýzu celkového prežitia pri 75% úplnosti dát.
c Nominálna hodnota p bez úpravy na opakované hodnoty medzi pôvodnou analýzou celkového prežitia
pri 50% úplnosti dát a aktualizovanou analýzou celkového prežitia pri 75% úplnosti dát.
d ORR bola stanovená u pacientok, u ktorých bola dostupná základná odpoveď (t.j., u ktorých bolo merateľné základné ochorenie: skupina 240 pacientov na fulvestrante 500 mg a skupina 261 pacientov
na fulvestrante 250 mg).
e Pacientky s najlepšou objektívnou odpoveďou kompletnej odpovede, čiastočnou odpoveďou alebo stabilizovaným ochorením ≥24 týždňov.
PFS: Prežitie bez progresie (progresion- free survival); ORR: Objektívna miera odpovede (objective response rate); OR: Objektívna odpoveď (objective response); CBR: Miera klinickej prospešnosti (clinical benefit rate); CB: Klinická prospešnosť (clinical benefit); OS: Celkové prežívanie (overall survival); K-
M:Kaplan-Meier; CI: Interval spoľahlivosti (confidence interval); AI: inhibítor aromatázy (aromatese inhibitor); AE: antiestrogén.
Randomizovaná, dvojito zaslepená „double-dummy“ multicentrická štúdia fázy 3 fulvestrant 500 mg oproti anastrozolu 1 mg sa uskutočnila u postmenopauzálnych žien s lokálne pokročilým alebo metastatickým karcinómom prsníka s pozitivitou estrogénových a/alebo progesterónových receptorov, ktoré neboli v minulosti liečené žiadnou hormonálnou liečbou. Celkovo 462 pacientok bolo postupne randomizovaných 1:1 na podávanie fulvestrantu 500 mg alebo na užívanie anastrozolu 1 mg.
Randomizácia bola stratifikovaná podľa typu ochorenia (lokálne pokročilé alebo metastatické), predchádzajúcej chemoterapie pokročilého ochorenia a merateľného ochorenia.
Primárnym cieľom účinnosti v štúdii bolo prežívanie bez progresie (PFS) hodnotené skúšajúcim pomocou kritérií na hodnotenie odpovede pri solídnych nádoroch (Response Evaluation Criteria in Solid Tumours, RECIST verzia 1.1). Kľúčové sekundárne ciele účinnosti zahŕňali celkové prežívanie (OS) a objektívnu mieru odpovede (ORR).
Medián veku pacientok zaradených do tejto štúdie bol 63 rokov (rozsah 36-90). Väčšina z týchto pacientok (87,0%) mala metastatické ochorenie na začiatku. Päťdesiatpäť percent (55,0%) pacientok malo viscerálne metastázy na začiatku. Celkovo 17,1% z pacientok dostalo predchádzajúci režim chemoterapie na liečbu pokročilého ochorenia; 84,2% z pacientok malo merateľné ochorenie.
Konzistentné výsledky sa pozorovali vo väčšine vopred špecifikovaných podskupín pacientok.
V podskupine pacientok s len neviscerálnymi metastázami (n=208) bol HR 0,592 (95% CI: 0,419;
0,837) v skupine s fulvestrantom oproti skupine s anastrozolom. V podskupine pacientok s viscerálnymi metastázami (n=254) bol HR 0,993 (95% CI: 0,740; 1,331) v skupine s fulvestrantom oproti skupine
s anastrozolom. Výsledky účinnosti zo štúdie FALCON sú uvedené v tabuľke 4 a na obrázku 1.
Tabuľka 4 Súhrn výsledkov primárneho cieľa účinnosti (PFS) a kľúčových sekundárnych cieľov účinnosti (hodnotené skúšajúcim v populácii podľa liečebného zámeru) ─ štúdia FALCON
|
F
u
l
vestrant
500 mg
(
N=
230)
|
A
nastrozol
1 mg
(
N=
232)
|
P
r
ežívanie bez progresie
|
P
očet PFS udalostí (%)
|
143 (62.2%)
|
166 (71.6%)
|
PFS pomer rizika (95% CI)
a hodnota p
|
HR 0.797 (0.637 - 0.999)
p = 0.0486
|
Medián PFS [mesiace (95%
C
I)]
|
16.6 (13.8, 21.0)
|
13.8 (12.0, 16.6)
|
P
očet OS udalostí*
|
67 (29.1%)
|
75 (32.3%)
|
O
S pomer rizika (95% CI)
a hodnota p
|
HR 0.875 (0.629 – 1.217)
p = 0.4277
|
O
RR
**
|
89 (46.1%)
|
88 (44.9%)
|
O
R
R Odds Ratio (95% CI) a hodnota p
|
OR 1.074 (0.716 – 1.614)
p = 0.7290
|
Medián DoR (mesiace)
|
20.0
|
13.2
|
C
B
R
|
180 (78.3%)
|
172 (74.1%)
|
C
B
R Odds Ratio (95% CI) a hodnota p
|
OR 1.253 (0.815 – 1.932)
p = 0.3045
|
*(31% úplnosť dát) – nie konečná analýza OS
**u pacientok s merateľným ochorením
Obrázok 1 Kaplanova-Meierova krivka prežívania bez progresie (hodnotené skúšajúcim v populácii podľa liečebného zámeru) ─ štúdia FALCON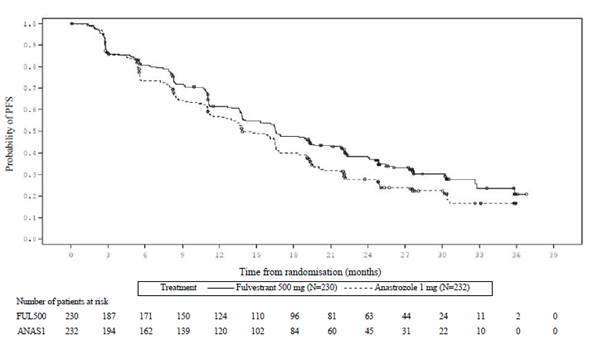
Probability of PFS = Pravdepodobnosť PFS
Čas od randomizácie (mesiace)

Liečba ——— Fulvestrant 500 mg (N=230) - - - - - - Anastrozol 1 mg (N=232)
Počet pacientov v riziku:
FUL500
| 230
| 187
| 171
| 150
| 124
| 110
| 96
| 81
| 63
| 44
| 24
| 11
| 2
| 0
|
ANAS1
| 232
| 194
| 162
| 139
| 120
| 102
| 84
| 60
| 45
| 31
| 22
| 10
| 0
| 0
|
Vykonali sa dve klinické štúdie fázy 3 u celkovo 851 postmenopauzálnych žien s pokročilým
karcinómom prsníka, u ktorých došlo k návratu ochorenia počas alebo po adjuvantnej endokrinnej liečbe, alebo k progresii ochorenia po endokrinnej liečbe pokročilého ochorenia. Sedemdesiatsedem
percent (77%) populácie v štúdii malo karcinóm prsníka s pozitivitou estrogénových receptorov.
V týchto štúdiách sa porovnávala bezpečnosť a účinnosť podávania fulvestrantu 250 mg raz do mesiaca s každodenným podávaním 1 mg anastrozolu (inhibítora aromatázy). Celkovo bol fulvestrant
v mesačnej dávke 250 mg minimálne rovnako účinný ako anastrozol z hľadiska prežívania bez
progresie, objektívnej reakcie a doby do úmrtia. Medzi oboma liečebnými skupinami neboli
v konečných ukazovateľoch zaznamenané žiadne štatisticky významné rozdiely. Primárnym cieľom štúdie bolo prežívanie bez progresie. Kombinovaná analýza oboch štúdií preukázala, že k progresii došlo u 83% pacientok, ktorým sa podával fulvestrant oproti 85% pacientok, ktoré užívali anastrozol. Kombinovaná analýza oboch štúdií poukázala na to, že pomer rizika fulvestrantu 250 mg a anastrozolu z hľadiska prežívania bez progresie predstavoval 0,95 (95% CI 0,82 až 1,10). Objektívna miera odpovede bola pre fulvestrant 250 mg 19,2% v porovnaní so 16,5% pre anastrozol. Medián času do úmrtia bol 27,4 mesiacov u pacientok liečených fulvestrantom a 27,6 mesiacov u pacientok liečených anastrozolom. Pomer rizika pre fulvestrant 250 mg k anastrozolu v čase do úmrtia bol 1,01 (95% CI
0,86 až 1,19).
Kombinovaná liečba s palbociklibomMedzinárodná, randomizovaná, dvojito-zaslepená, multicentrická štúdia fázy 3 s paralelnými skupinami porovnávajúca fulvestrant 500 mg plus palbociklib 125 mg oproti fulvestrantu 500 mg plus
placebo sa vykonala u žien s HR-pozitívnym, HER2-negatívnym lokálne pokročilým karcinómom
prsníka nevhodným na resekciu alebo rádioterapiu s kuratívnym zámerom alebo s metastatickým karcinómom prsníka, nezávisle od ich menopauzálneho stavu, s progresiou ochorenia po predchádzajúcej endokrinnej liečbe v (neo)adjuvantnom použití alebo použití pri metastatickom ochorení.
Celkovo 521 pre/perimenopauzálnych žien, ktorých ochorenie progredovalo v priebehu 12 mesiacov od ukončenia adjuvantnej endokrinnej liečby alebo počas nej, či v rámci 1 mesiaca po endokrinnej liečbe pokročilého ochorenia, alebo počas nej, bolo randomizovaných v pomere 2:1 do skupín fulvestrant plus palbociklib alebo fulvestrant plus placebo a stratifikovaných podľa zdokumentovanej citlivosti na predchádzajúcu hormonálnu liečbu, menopauzálneho stavu pri vstupe do štúdie (pre/perimenopauzálne oproti postmenopauzálnym) a prítomnosti viscerálnych metastáz. Pre/perimenopauzálne ženy dostali agonistu LHRH, goserelín. Pacientky s pokročilým, symptomatickým, viscerálnym rozsevom metastáz, ktoré mali riziko život ohrozujúcich komplikácií
v krátkej dobe (vrátane pacientok s masívnymi nekontrolovanými výpotkami [pleurálny, perikardiálny, peritoneálny], pľúcnou lymfangitídou a viac ako 50% postihnutím pečene), neboli vhodné pre zaradenie do štúdie.
Pacientky pokračovali v užívaní priradenej liečby, kým nedošlo k objektívnej progresii ochorenia, symptomatickému zhoršeniu, neprijateľnej toxicite, úmrtiu alebo zrušeniu súhlasu, podľa toho, čo sa vyskytlo ako prvé. Prestup z jednej liečebnej skupiny do druhej nebol povolený.
Rozdelenie pacientok podľa vstupných demografických a prognostických charakteristík do skupiny fulvestrant plus palbociklib a skupiny fulvestrant plus placebo bolo vyvážené. Medián veku pacientok zaradených do tejto štúdie bol 57 rokov (rozsah 29 až 88). V každej liečebnej skupine bola väčšina
pacientok bielej rasy s dokumentovanou citlivosťou na predchádzajúcu hormonálnu liečbu a po menopauze. Približne 20% pacientok bolo pre/perimenopauzálnych. Všetky pacientky dostali predchádzajúcu systémovú liečbu a väčšina pacientok v každej z liečebných skupín dostala predchádzajúcu chemoterapiu kvôli primárnej diagnóze. ECOG skóre PS = 0 mala viac ako polovica (62%) pacientok, 60% malo viscerálne metastázy a 60% dostalo viac ako 1 predchádzajúcu hormonálnu liečbu kvôli ich primárnej diagnóze.
Primárnym cieľom štúdie bolo skúšajúcim lekárom hodnotené PFS podľa kritérií RECIST 1.1. Podporné PFS analýzy boli založené na nezávislej centrálnej rádiologickej kontrole. Sekundárne ciele zahŕňali OR, CBR, celkové prežívanie (overall survival, OS), bezpečnosť a čas do zhoršenia bolesti (time- to-deterioration, TTD).
Štúdia splnila svoj primárny cieľ, predĺženie PFS hodnotené skúšajúcim lekárom pri predbežnej analýze vykonanej pri 82% plánovaných PFS udalostí; výsledky prekročili vopred špecifikovanú Haybittle-Petovu hranicu účinnosti (α = 0,00135) dokazujúc tak štatisticky významné predĺženie PFS a klinicky významný účinok liečby. Novšia aktualizácia údajov o účinnosti je uvedená v tabuľke 5.'
Finálna analýza OS sa uskutočnila po uplynutí mediánu doby sledovania 45 mesiacov vychádzajúc z 310
udalostí (60% randomizovaných pacientov). V skupine s palbociklibom plus fulvestrant sa
pozoroval 6,9-mesačný rozdiel v mediáne OS v porovnaní s placebom plus fulvestrant; tento výsledok nebol štatisticky významný vo vopred špecifikovanej hladine významnosti 0,235 (1-stranný).
V skupine s placebom plus fulvestrant 15,5% randomizovaných pacientov dostávalo palbociklib a iné inhibítory CDK ako následnú liečbu po progresii.
Výsledky PFS a finálneho OS hodnotené skúšajúcim zo štúdie PALOMA3 sú uvedené v tabuľke 5. Príslušné Kaplanove-Meierove krivky sú znázornené na obrázku 2 a 3.
| Aktualizovaná analýza
(ukončenie zberu údajov 23. októbra 2015)
| Fulvestrant plus palbociklib (N=347)
| Fulvestrant plus placebo (N=174)
| Prežívanie bez progresie
|
| Medián [mesiace (95% IS)]
| 11,2 (9,5; 12,9)
| 4,6 (3,5; 5,6)
| Pomer rizika (95% IS) a
hodnota p
| 0,497 (0,398; 0,620), p <0,000001
| Sekundárne ciele
| OR [% (95% IS)]
| 26,2 (21,7; 31,2)
| 13,8 (9,0; 19,8)
| OR (merateľné ochorenie) [%
(95% IS)]
| 33,7 (28,1; 39,7)
| 17,4 (11,5; 24,8)
| CBR [% (95% IS)]
| 68,0 (62,8; 72,9)
| 39,7 (32,3; 47,3)
| Finálne celkové prežívanie (OS) (ukončenie zberu údajov 13. apríla 2018)
| Počet udalostí (%)
| 201 (57,9)
| 109 (62,6)
| Medián [mesiace (95% IS)]
| 34,9 (28,8; 40,0)
| 28,0 (23,6; 34,6)
| Pomer rizika (95% IS)
a p-hodnota†
| 0,814 (0,644; 1,029)
p=0,0429†*
|
|
|
Tabuľka 5 Výsledky účinnosti – štúdia PALOMA3 (hodnotené skúšajúcim v populácii podľa liečebného zámeru)CBR = miera klinického prínosu; IS = interval spoľahlivosti; N = počet pacientok; OR = objektívna
odpoveď Výsledky sekundárnych cieľov založené na potvrdených a nepotvrdených odpovediach podľa
RECIST 1.1.
* Nie je štatistický významný.
† 1-stranná p-hodnota z log-rank testu stratifikovaného na základe prítomnosti viscerálnych metastáz a
senzitivity na predchádzajúcu endokrinnú liečbu pri randomizácii.
Obrázok 2. Kaplanova-Meierova krivka prežívania bez progresie ochorenia (hodnotenie skúšajúcim lekárom v populácii podľa liečebného zámeru) – štúdia PALOMA3 (ukončenie zberu údajov 23. októbra 2015)FUL=fulvestrant; PAL=palbociklib; PCB=placebo.
Zníženie rizika progresie ochorenia alebo úmrtia v skupine fulvestrant plus palbociklib sa pozorovalo vo všetkých podskupinách pacientok definovaných stratifikačnými faktormi a vstupnými charakteristikami. Bolo to evidentné u pre/perimenopauzálnych žien (HR 0,46 [95% CI: 0,28; 0,75]) a postmenopauzálnych žien (HR 0,52 [95% CI: 0,40; 0,66]) a pacientok s viscerálnymi metastatickými ložiskami (HR 0,50 [95% CI: 0,38; 0,65]) a pacientok s neviscerálnymi metastatickými ložiskami (HR 0,48 [95% CI: 0,33; 0,71]). Prínos bol tiež pozorovaný nezávisle od počtu línií predchádzajúcej liečby pre metastatické ochorenie, či bol počet 0 (HR 0,59 [95% CI: 0,37; 0,93]), 1 (HR 0,46 [95% CI: 0,32; 0,64]), 2 (HR 0,48 [95% CI: 0,30;
0,76]) alebo ≥ 3 línie (HR 0,59 [95% CI: 0,28; 1,22]).
Obrázok 3. Kaplanova-Meierova krivka celkového prežívania (populácia podľa liečebného zámeru)– štúdia PALOMA3 (ukončenie zberu údajov 13. apríla 2018)

Overall Survival Probability (%) = Pravdepodobnosť celkového prežívania (%)
P
očet pacientov v riziku
Č
as (mesiace)
P
AL+
F
U
L 347 321 286 247 209 165 148 126 17
PCB+FUL 174 155 135 115 86 68 57 43 7
FUL=fulvestrant; PAL=palbociclib; PCB=placebo.
Ďalšie ukazovatele účinnosti (OR a TTR) hodnotené v podskupinách pacientok s viscerálnym ochorením alebo bez neho sú zobrazené v tabuľke 6.
Tabuľka 6 Výsledky účinnosti na viscerálne a neviscerálne ochorenie zo štúdie PALOMA3 (populácia podľa liečebného zámeru)
| Viscerálne ochorenie
| Neviscerálne ochorenie
|
| Fulvestrant plus palbociclib
(N=206)
| Fulvestrant plus placebo
(N=105)
| Fulvestrant plus palbociclib
(N=141)
| Fulvestrant plus placebo
(N=69)
|
OR [% (95% CI)]
| 35.0
(28.5, 41.9)
| 13.3
(7.5, 21.4)
| 13.5
(8.3, 20.2)
| 14.5
(7.2, 25.0)
|
TTR*, Medián
[mesiace (rozsah)]
| 3.8
(3.5, 16.7)
| 5.4
(3.5, 16.7)
| 3.7
(1.9, 13.7)
| 3.6
(3.4, 3.7)
|
*Výsledky odpovedí založené na potvrdených a nepotvrdených odpovediach.
N = počet pacientok; CI = interval spoľahlivosti; OR = objektívna odpoveď; TTR = čas do prvej odpovede
nádoru.
Pacientkami hlásené príznaky boli hodnotené pomocou dotazníka kvality života (quality of life questionnaire, QLQ)-C30 Európskej organizácie pre výskum a liečbu rakoviny a jeho modulu rakoviny prsníka (EORTC QLQ BR23). Celkovo 335 pacientok v skupine fulvestrant plus palbociklib
a 166 pacientok v skupine fulvestrant plus placebo vyplnilo dotazník pri vstupe do štúdie a aspoň raz na ďalšej návšteve.
Čas do zhoršenia bol vopred špecifikovaný ako čas medzi vstupom do štúdie a prvým výskytom
≥ 10-bodového vzostupu oproti počiatočnej hodnote skóre príznakov bolesti. Pridanie palbociklibu
k fulvestrantu viedlo k prínosu, pokiaľ ide o príznaky, pretože významne predĺžilo čas do zhoršenia príznakov bolesti v porovnaní so skupinou fulvestrant plus placebo (medián 8,0 mesiacov oproti 2,8
mesiacov; HR = 0,64 [95% CI: 0,49; 0,85]; p < 0,001).
Účinky na endometrium po menopauze
Predklinické údaje nenaznačujú stimulačný účinok fulvestrantu na endometrium po menopauze (pozri časť 5.3). Dvojtýždňová štúdia u zdravých postmenopauzálnych dobrovoľníčok liečených 20 μg etinylestradiolu denne preukázala, že predliečenie fulvestrantom 250 mg malo za následok signifikantne redukovanú stimuláciu postmenopauzálneho endometria v porovnaní s predliečením placebom, hodnotenú na základe ultrasonografického merania hrúbky endometria.
Neoadjuvantná liečba trvajúca až 16 týždňov u pacientiek s karcinómom prsníka liečených buď fulvestrantom 500 mg alebo 250 mg fulvestrantom neviedla ku klinicky významným zmenám v hrúbke endometria, čo ukazuje na nedostatočný agonistický účinok. Neexistujú žiadne dôkazy o nepriaznivých účinkoch na endometrium u sledovaných pacientok s karcinómom prsníka. Nie sú k dispozícii údaje týkajúce sa endometriálnej morfológie.
V dvoch krátkodobých štúdiách (1 a 12 týždňov) u premenopauzálnych pacientiek s benígnym gynekologickým ochorením sa nepozorovali žiadne signifikantné zmeny v hrúbke endometria ultrazvukovým meraním pri porovnaní skupín, ktorým sa podával fulvestrant a placebo.
Účinky na kosti
Nie sú k dispozícii dlhodobé údaje o účinku fulvestrantu na kosti. Neoadjuvantná liečba trvajúca až 16
týždňov u pacientok s karcinómom prsníka buď fulvestrantom 500 mg alebo 250 mg fulvestrantom neviedla ku klinicky významným zmenám markerov kostného obratu v sére.
Pediatrickápopulácia
Fulvestrant STADA nie je indikovaný na použitie u detí. Európska lieková agentúra udelila výnimku z
povinnosti predložiť výsledky štúdii s fulvestrantom vo všetkých podskupinách pediatrickej populácie pri
karcinóme prsníka (pozri časť 4.2 pre informáciu o pediatrickom použití).
Otvorená štúdia fázy 2 skúmala bezpečnosť, účinnosť a farmakokinetiku fulvestrantu u 30 dievčat vo veku od 1 do 8 rokov s progresívnou predčasnou pubertou spojenou s McCune Albrightovým syndrómom (MAS). Detské pacientky dostali intramuskulárnu dávku fulvestrantu 4 mg/kg mesačne. Táto 12 mesačná štúdia skúmala škálu cieľov MAS a ukázala zníženie frekvencie vaginálneho krvácania a zníženie rýchlosti vzostupu kostného veku. V tejto štúdii bol u detí rovnovážny stav koncentrácie fulvestrantu v súlade s dospelými (pozri časť 5.2). Z tejto malej štúdie sa nezistili žiadne nové vyplývajúce obavy týkajúce sa bezpečnosti, ale 5-ročné údaje zatiaľ nie sú k dispozícii.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po podaní fulvestrantu vo forme dlhodobo pôsobiacej intramuskulárnej injekcie sa fulvestrant pomaly
vstrebáva a maximálne plazmatické koncentrácie (Cmax) sa dosiahnu približne po 5 dňoch. Podaním fulvestrantu v režime 500 mg sa dosiahnu hladiny expozície zodpovedajúce alebo blízke rovnovážnemu stavu počas prvého mesiaca dávkovania (priemer [CV]: AUC 475 [33,4%] ng.dní/ml, Cmax 25,1
[35,3%] ng/ml, Cmin 16,3 [25,9%] ng/ml). V rovnovážnom stave sa plazmatické koncentrácie fulvestrantu udržiavajú v relatívne úzkom rozmedzí s až približne 3-násobným rozdielom medzi
maximálnou a minimálnou koncentráciou. Po intramuskulárnom podaní je expozícia v rozsahu dávok 50
až 500 mg približne úmerná dávke.
Distribúcia
Fulvestrant sa extenzívne a rýchlo distribuuje. Veľký zdanlivý distribučný objem v ustálenom stave
(volume of distribution at steady state Vdss) je približne 3 až 5 l/kg, čo naznačuje, že distribúcia je väčšinou extravaskulárna. Fulvestrant sa vo vysokej miere (99%) viaže na plazmatické bielkoviny. Hlavnými zložkami väzby sú lipoproteínové frakcie veľmi nízkej denzity (very low density lipoprotein VLDL), nízkej denzity (low density lipoprotein LDL) a vysokej denzity (high density lipoprotein HDL). Neboli uskutočnené žiadne štúdie interakcie ohľadom kompetitívnej väzby na bielkoviny. Úloha globulínu, ktorý viaže pohlavné hormóny (sex hormone-binding globulin SHBG), nebola stanovená.
Biotransformácia
Metabolizmus fulvestrantu nebol plne hodnotený, ale zahŕňa kombinácie celého radu možných biotransformačných ciest, ktoré sú analogické cestám endogénnych steroidov. Identifikované
metabolity (vrátane metabolitov typu 17-ketón, sulfón, 3-síran, 3- a 17-glukuronid) sú v
antiestrogénových modeloch buď menej účinné, alebo vykazujú podobný účinok ako fulvestrant. Skúšky na preparátoch ľudskej pečene a rekombinantných ľudských enzýmoch udávajú, že na oxidácii
fulvestrantu sa podieľa z P-450 izoenzýmov iba CYP 3A4, in vivo sa však zdá, že prevládajú cesty
nevyužívajúce P-450. Údaje in vitro naznačujú, že fulvestrant neinhibuje izoenzýmy CYP450.
Eliminácia
Fulvestrant sa eliminuje predovšetkým v metabolizovanej forme. Hlavnou cestou vylučovania je stolica, močom sa vylučuje menej ako 1%. Fulvestrant má vysoký klírens, 11±1,7ml/min/kg, čo naznačuje vysoký podiel extrakcie pečeňou. Terminálny polčas (t1/2) po intramuskulárnom podaní sa riadi rýchlosťou absorpcie a odhaduje sa na 50 dní.
Osobitné skupiny
Farmakokinetická analýza populácie, podľa údajov zo štúdií fázy 3, nezaznamenala pri fulvestrante
žiadne rozdiely vo farmakokinetickom profile pokiaľ ide o vek (rozsah 33 až 89 rokov), hmotnosť (40-
127 kg) alebo rasu.
Porucha funkcie obličiek
Ľahká až stredne ťažká porucha funkcie obličiek neovplyvnila v žiadnom klinicky významnom
rozsahu farmakokinetiku fulvestrantu.
Porucha funkcie pečene
Farmakokinetika fulvestrantu bola hodnotená v klinickom skúšaní s podaním jednorazovej dávky
pacientom s ľahkou až stredne ťažkou poruchou funkcie pečene (v štádiu A a B podľa Child-
Pughovej klasifikácie). Bola použitá vysoká dávka lieku v krátkodobo pôsobiacej intramuskulárnej injekcii. U žien s poruchou funkcie pečene bolo až 2,5-násobné zvýšenie AUC v porovnaní so zdravými subjektami. Predpokladá sa, že u pacientok, ktorým sa podáva Fulvestrant, bude takéto zvýšenie expozície dobre tolerované. Ženy s ťažkou poruchou funkcie pečene (štádium C podľa Child-Pughovej klasifikácie) neboli hodnotené.
Pediatrická populácia
Farmakokinetika fulvestrantu sa hodnotila v klinickom skúšaní vykonanom u 30 dievčat s
progresívnou predčasnou pubertou spojenou s McCune Albrightovým syndrómom (pozri časť 5.1). Detské pacientky boli vo veku 1 až 8 rokov a dostali intramuskulárnu dávku fulvestrantu 4 mg/kg mesačne. Geometrický priemer (smerodajná odchýlka) rovnovážneho stavu koncentrácie (Cmin, ss)
a AUCss bol 4,2 (0,9) ng/ml a 3680 (1020) ng*hod/ml, v uvedenom poradí. Hoci sú zozbierané údaje obmedzené, rovnovážny stav koncentrácie fulvestrantu u detí sa zdá byť v súlade s dospelými.
5.3 Predklinické údaje o bezpečnosti
Akútna toxicita fulvestrantu je nízka.
Fulvestrant bol dobre tolerovaný u všetkých zvieracích druhov v skúškach s podávaním opakovaných dávok. Miestne reakcie vrátane myozitídy a tvorby granulómov v mieste podania injekcie sa pripisovali vehikulu, avšak závažnosť myozitídy u králikov sa pri fulvestrante zvyšovala v porovnaní so skupinou, ktorej bol podávaný fyziologický roztok. V skúškach toxicity s opakovanými intramuskulárnymi dávkami fulvestrantu potkanom a psom bola antiestrogénová aktivita fulvestrantu zodpovedná za väčšinu prejavených účinkov, a to predovšetkým na ženský reprodukčný systém, ale aj na iné orgány, citlivé na hormóny u obidvoch pohlaví.U niektorých psov sa pozorovala arteritída v rade rôznych tkanív po chronickom (12 mesačnom) dávkovaní.
V skúškach na psoch pri perorálnom a intravenóznom podaní boli pozorované účinky na kardiovaskulárny systém (mierne zvýšenie S-T segmentu EKG pri perorálnom podaní
a zastavenie sínusového uzla u jedného psa pri intravenóznom podaní). Tieto účinky sa vyskytli pri
koncentrácii fulvestrantu vyššej ako u pacientov (Cmax > 15 krát), a pre bezpečnosť u človeka majú pri podaní klinickej dávky pravdepodobne len obmedzený význam.
Fulvestrant nepreukázal žiadny genotoxický potenciál.
Účinky fulvestrantu na reprodukciu a vývoj embrya/plodu preukázali pri dávkach podobných dávkam klinickým jeho antiestrogénny účinok. U potkanov sa pozoroval reverzibilný pokles plodnosti samíc a prežití embryí, dystokia a zvýšený výskyt abnormalít plodu vrátane tarzálnej flexúry. Fulvestrant podávaný králikom mal za následok potrat. Bolo pozorované zvýšenie hmotnosti placenty a postimplantačná strata plodov. U králikov došlo k zvýšenému výskytu zmien plodu (dorzálny posun panvového pletenca a 27. presakrálneho stavca).
Dvojročná štúdia onkogenicity na potkanoch (intramuskulárne podanie fulvestrantu) ukázala zvýšený výskyt benígnych bunkových nádorov granulózy vaječníkov u potkaních samíc pri vysokých dávkach
10 mg raz za 15 dní a zvýšený výskyt testikulárnych Leydigových bunkových nádorov u samcov. V
dvojročnej štúdii onkogenicity na myšiach (perorálne podanie denne) bola zvýšená incidencia zväzkových stromálnych nádorov vaječníkov u samíc (benígnych aj malígnych) v dávkach 150
a 500 mg/kg/deň. S ohľadom na tieto nálezy pre úroveň nulového účinku ('the no-effect level') bola
systémová expozícia (AUC) u potkanov približne 1,5-krát vyššia než očakávaná expozícia u žien
a 0,8-krát vyššia než expozícia u mužov a u myší približne 0,8-krát vyššia než očakávaná expozícia u mužov či žien. Vyvolanie týchto nádorov zodpovedá farmakologicky vyvolaným endokrinným
spätne väzbovým zmenám v hladinách gonadotropínov spôsobených antiestrogénmi u cyklujúcich zvierat. Preto tieto poznatky nie sú považované za dôležité pre použitie fulvestrantu
u postmenopauzálnych žien s pokročilým karcinómom prsníka.
Hodnotenieenvironmentálnehorizika(ERA)
Štúdie hodnotenia environmentálneho rizika preukázali, že fulvestrant má potenciál nepriaznivo ovplyvňovať vodné prostredie (pozri časť 6.6).
6 FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
etanol 96 % benzylalkohol benzylbenzoát
ricínový olej, rafinovaný
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
4 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte a prepravujte v chladničke (2 °C – 8 °C).
Naplnenú injekčnú striekačku uchovávajte v pôvodnom obale na ochranu pred svetlom.
Má sa zamedziť teplotným odchýlkam mimo rozsahu 2 °C – 8 °C. Vyhnite sa uchovávaniu pri teplote prevyšujúcej 30 °C a v prípade, uchovávania tohto lieku pri teplote nižšej ako 25 °C v priemere (ale nad rozsah 2 °C - 8 °C) uchovávanie nemá presiahnuť 28-dňové obdobie. Po teplotných odchýlkach sa má liek ihneď vrátiť do odporúčaných podmienok uchovávania (uchovávanie a preprava v chladničke
2 °C – 8 °C). Teplotné odchýlky majú kumulatívny účinok na kvalitu lieku a 28-dňová lehota sa nesmie prekročiť počas štvorročného času použiteľnosti Fulvestrantu STADA (pozri časť 6.3). Vystavením
teplotám nižším ako 2 °C nedôjde k poškodeniu lieku za predpokladu, že nie je uchovávaný pod -20 °C.
6.5 Druh obalu a obsah balenia
Balenie s naplnenou injekčnou striekačkou obsahuje:
Jednu naplnenú injekčnú striekačku z číreho skla typu 1 s plastovým uzáverom injekčnej striekačky,
s brómbutylovou gumenou zátkou, polypropylénovou piestovou tyčinkou a spätnou zátkou s poistným uzáverom obsahujúcu 5 ml injekčného roztoku.
Fulvestrant STADA má tri typy balenia:
- Papierová škatuľa s blistrom s jednou naplnenou injekčnou striekačkou, jednou hypodermickou
sterilnou ihlou (BD SafetyGlide) a jednou písomnou informáciou.
alebo
- Papierová škatuľa s dvoma blistrami v každom s naplnenou injekčnou striekačkou, dvoma hypodermickými sterilnými ihlami (BD SafetyGlide) a jednou písomnou informáciou.
alebo
- Papierová škatuľa so šiestimi blistrami v každom s naplnenou injekčnou striekačkou, šiestimi hypodermickými sterilnými ihlami (BD SafetyGlide) a jednou písomnou informáciou.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Spôsob zaobchádzania a likvidácie musí korešpondovať s inými chemoterapeutikami a byť v súlade s národnými požiadavkami. Tehotné zdravotnícke pracovníčky nesmú manipulovať a/alebo podávať Fulvestrant STADA 250 mg injekčný roztok v naplnenej injekčnej striekačke.
PokynynapodávanieInjekciu podávajte podľa lokálnych postupov na podávanie intramuskulárnych injekcií s veľkým
objemom.
POZNÁMKA: Vzhľadom na blízkosť sedacieho nervu je potrebná opatrnosť pri injekčnom podávaní
Fulvestrantu STADA do dorzogluteálnej oblasti (pozri časť 4.4).
Upozornenie: Bezpečnostnú ihlu (BD SafetyGlide chránená hypodermická ihla) pred použitím neautoklávujte. Počas použitia aj pri likvidácii musia byť ruky stále za ihlou.
Pre každú z oboch injekčnýchstriekačiek:· Vyberte sklenenú injekčnú striekačku z blistra a skontrolujte, či nie je poškodená.
· Opatrne vyberte bezpečnostnú ihlu (SafetyGlide) z vonkajšieho obalu.
· Parenterálne roztoky sa musia pred podávaním vizuálne skontrolovať, či neobsahujú častice a či nedošlo k zmene ich zafarbenia.
· Injekčnú striekačku držte vo zvislej polohe
· Druhou rukou držte kryt a opatrným otáčaním ho odstráňte. Na zachovanie sterility sa
nedotýkajte hrotu injekčnej striekačky (pozri obrázok 1).
Obrázok 1
· Pripojte bezpečnostnú ihlu k luerovej koncovke injekčnej striekačky a otáčajte ňou, kým pevne nezapadne (pozri obrázok 2).
Obrázok 2
· Skontrolujte, či je ihla zafixovaná k luerovému konektoru, predtým ako ju otočíte z vertikálnej
roviny.
· Priamym pohybom stiahnite z ihly kryt tak, aby sa nepoškodil hrot ihly.
· Preneste naplnenú injekčnú striekačku na miesto podania.
· Odstráňte kryt z ihly.
· Vytlačte z injekčnej striekačky prebytočný plyn.
· Podávajte pomaly intramuskulárne (1-2 minúty/injekcia) do sedacieho svalu (gluteálna oblasť). Na uľahčenie podávania je skosená strana ihly orientovaná k ramenu páčky (pozri obrázok 3).
Obrázok 3
· Po podaní injekcie ihneď využite ťah jedného prsta na aktiváciu ramena páčky, ktorá aktivuje ochranný mechanizmus (pozri obrázok 4).
POZNÁMKA: Aktivujte mechanizmus smerom od seba a iných ľudí. Dávajte pozor na kliknutie a
vizuálne sa presvedčte, že hrot ihly je úplne zakrytý.
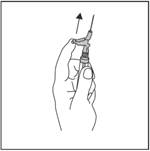
Obrázok 4
LikvidáciaNaplnené injekčné striekačky sú určené
len na jednorazové použitie.
Tento liek môže predstavovať riziko pre vodné prostredie. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami (pozri časť 5.3).
7 DRŽITEĽ ROZHODNUTIA O REGISTRÁCIISTADA Arzneimittel AG Stadastrasse 2-18
61118 Bad Vilbel
Nemecko
8 REGISTRAČNÉ ČÍSLO34/0224/19-S
9 DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 12. júla 2019
10. DÁTUM REVÍZIE TEXTU08/2022