mediánu sledovania 6,0 rokov bola incidencia myopatie pri ezetimib/simvastatín 0,2 % a pri simvastatíne 0,1 %, pričom myopatia bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN alebo dve konsekutívne pozorovania hladiny CK ≥ 5 a < 10-násobok HHN. Incidencia rabdomyolýzy bola pri ezetimibe/simvastatíne 0,1 % a pri simvastatíne 0,2 %, pričom rabdomyolýza bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN s dôkazom poškodenia obličiek, ≥ 5-násobok HHN a < 10-násobok HHN počas dvoch konsekutívnych udalostí s dôkazom poškodenia obličiek alebo hladinou CK ≥ 10 000 IU/l bez dôkazu poškodenia obličiek. (Pozri časť 4.8).
V klinickom skúšaní, v ktorom bolo viac ako 9 000 pacientov s chronickým ochorením obličiek randomizovaných na užívanie kombinácie ezetimib/simvastatín v dávke 10 mg/20 mg denne (n = 4 650) alebo placeba (n = 4 620) (medián sledovania 4,9 rokov), bola incidencia myopatie 0,2 % pri ezetimibe/simvastatíne a 0,1 % pri placebe. (Pozri časť 4.8).
V klinickom skúšaní, v ktorom boli pacienti s vysokým rizikom kardiovaskulárneho ochorenia liečení simvastatínom 40 mg/deň (medián sledovania 3,9 rokov), bola incidencia myopatie približne 0,05 % u pacientov iného ako čínskeho pôvodu (n = 7 367) v porovnaní s 0,24 % u pacientov čínskeho pôvodu (n = 5 468). Hoci jediná ázijská populácia hodnotená v tomto klinickom skúšaní bola čínska, pri predpisovaní Ezetimibu/simvastatínu Teva B.V. pacientom ázijského pôvodu je potrebná opatrnosť a má sa použiť najnižšia potrebná dávka.
Znížená funkcia transportných proteínovZnížená funkcia pečeňových transportných proteínov OATP môže zvýšiť systémovú expozíciu simvastatínu a zvýšiť riziko myopatie a rabdomyolýzy. Znížená funkcia sa môže vyskytnúť v dôsledku inhibície interagujúcimi liekmi (napr. cyklosporín) alebo u pacientov, ktorí sú nositeľmi genotypu SLCO1B1 c.521T > C.
Pacienti, ktorí sú nositeľmi alely génu SLCO1B1 (c.521T > C) kódujúcej menej aktívny proteín OATP1B1, majú zvýšenú systémovú expozíciu simvastatínu a zvýšené riziko myopatie. Riziko myopatie súvisiacej s vysokou dávkou simvastatínu (80 mg) je vo všeobecnosti, bez genetického testovania, asi 1 %. Na základe výsledkov skúšania SEARCH, homozygotní nositelia alely C (nazývaní tiež CC), liečení 80 mg majú 15 % riziko myopatie v priebehu jedného roka, zatiaľ čo riziko u heterozygotných nositeľov alely C (CT) je 1,5 %. U pacientov, ktorí majú najčastejší genotyp (TT) je zodpovedajúce riziko 0,3 % (pozri časť 5.2). Tam, kde je to možné, sa má ako súčasť hodnotenia pomeru prínosu a rizika pred predpísaním 80 mg simvastatínu u jednotlivých pacientov zvážiť genotypizácia na prítomnosť alely C a vyhnúť sa vysokým dávkam u tých pacientov, u ktorých sa zistilo, že sú nositeľmi genotypu CC. Absencia tohto génu pri genotypizácii však nevylučuje možnosť výskytu myopatie.
Meranie hladiny kreatínkinázyHladina kreatínkinázy (CK) sa nemá merať po namáhavom cvičení alebo ak je prítomná akákoľvek možná alternatívna príčina zvýšenia hladiny CK, pretože to sťažuje interpretáciu hodnoty. Ak sú východiskové hladiny CK signifikantne zvýšené (> 5‑násobok HHN), hladiny sa majú znovu zmerať po 5 až 7 dňoch, aby sa výsledky potvrdili.
Pred liečbouVšetci pacienti, u ktorých sa začína s liečbou Ezetimibom/simvastatínom Teva B.V. alebo pacienti, ktorým sa dávka Ezetimibu/simvastatínu Teva B.V. zvyšuje, majú byť oboznámení s rizikom myopatie a upozornení, aby okamžite hlásili akúkoľvek nevysvetliteľnú svalovú bolesť, citlivosť alebo slabosť.
Opatrnosť je potrebná u pacientov s predispozičnými faktormi pre vznik rabdomyolýzy. Kvôli stanoveniu základnej referenčnej hodnoty sa má hladina CK merať pred začiatkom liečby v nasledujúcich prípadoch:
- starší pacienti (vek ≥ 65 rokov),
- ženské pohlavie,
- porucha funkcie obličiek,
- nekontrolovaná hypotyreóza,
- dedičné muskulárne poruchy v osobnej alebo rodinnej anamnéze,
- muskulárna toxicita pri statíne alebo fibráte v anamnéze,
- závislosť od alkoholu.
V takýchto situáciách treba zvážiť riziko liečby vo vzťahu k možnému prínosu a odporúča sa klinické sledovanie. Ak mal pacient v minulosti poruchu svalstva pri liečbe fibrátom alebo statínom, liečba akýmkoľvek liekom obsahujúcim statín (ako Ezetimib/simvastatín Teva B.V.) sa má začať jedine s opatrnosťou. Ak sú východiskové hladiny CK významne zvýšené (> 5‑násobok HHN), liečba sa nesmie začať.
Počas liečbyAk sa počas liečby Ezetimibom/simvastatínom Teva B.V. vyskytne u pacienta svalová bolesť, slabosť alebo kŕče, je potrebné zmerať jeho hladiny CK. Ak sú namerané hladiny aj bez namáhavého cvičenia významne zvýšené (> 5‑násobok HHN), liečba sa má ukončiť. Ak sú svalové príznaky závažné a spôsobujú každodenné nepríjemnosti, a to aj v prípade, ak sú hladiny CK < 5‑násobok HHN, môže sa zvážiť prerušenie liečby. Ak je podozrenie na myopatiu z akéhokoľvek iného dôvodu, liečba sa má prerušiť.
Ak príznaky pominú a hladiny CK sa vrátia do normálu, je možné uvažovať o opätovnom začatí liečby Ezetimibom/simvastatínom Teva B.V. alebo o začatí liečby iným liekom obsahujúcim statín v najnižšej dávke a pri dôkladnom sledovaní.
U pacientov nastavených na dávku 80 mg simvastatínu sa pozorovala vyššia miera myopatie (pozri časť 5.1). Odporúčajú sa pravidelné merania hladiny CK, keďže môžu pomôcť identifikovať subklinické prípady myopatie. Neexistuje však záruka, že takýmto sledovaním sa predíde myopatii.
Nekrotizujúca myopatia sprostredkovaná imunitným systémomPočas liečby alebo po liečbe niektorými statínmi boli hlásené veľmi zriedkavé prípady nekrotizujúcej myopatie sprostredkovanej imunitným systémom (immune-mediated necrotizing myopathy, IMNM). IMNM je klinicky charakterizovaná pretrvávajúcou slabosťou proximálnych svalov a zvýšenou sérovou hladinou kreatínkinázy, ktoré pretrvávajú napriek ukončeniu liečby statínmi (pozri časť 4.8).
Liečba Ezetimibom/simvastatínom Teva B.V. sa má dočasne prerušiť na niekoľko dní pred plánovanou väčšou operáciou a v prípade vážnejšieho stavu vyžadujúceho lekársku alebo chirurgickú starostlivosť.
Opatrenia na zníženie rizika myopatie zapríčinenej liekovými interakciami (pozri tiež časť 4.5)Riziko myopatie a rabdomyolýzy je signifikantne zvýšené pri súbežnom užívaní ezetimibu/simvastatínu so silnými inhibítormi CYP3A4 (ako napr. itrakonazol, ketokonazol, posakonazol, vorikonazol, erytromycín, klaritromycín, telitromycín, inhibítory HIV proteázy (napr. nelfinavir), boceprevir, telaprevir, nefazodón), ako aj cyklosporínom, danazolom a gemfibrozilom. Použitie týchto liekov je kontraindikované (pozri časť 4.3).
Vzhľadom na to, že zložkou Ezetimibu/simvastatínu Teva B.V. je simvastatín, riziko myopatie a rabdomyolýzy sa zvyšuje tiež pri súbežnom užívaní iných fibrátov, kyseliny nikotínovej v dávkach znižujúcich lipidy (³ 1 g/deň) alebo pri súbežnom užívaní amiodarónu, amlodipínu, verapamilu alebo diltiazemu s určitými dávkami Ezetimibu/simvastatínu Teva B.V. (pozri časti 4.2 a 4.5). Riziko myopatie vrátane rabdomyolýzy sa môže zvýšiť pri súbežnom podávaní kyseliny fusidovej s Ezetimibom/simvastatínom Teva B.V. U pacientov s HoFH sa môže toto riziko zvýšiť súbežným užitím Ezetimibu/simvastatínu Teva B.V. s lomitapidom (pozri časť 4.5).
Preto, čo sa týka inhibítorov CYP3A4, je užívanie Ezetimibu/simvastatínu Teva B.V. súbežne s itrakonazolom, ketokonazolom, posakonazolom, vorikonazolom, inhibítormi HIV proteázy (napr. nelfinavir), boceprevirom, telaprevirom, erytromycínom, klaritromycínom, telitromycínom, nefazodónom a liekmi obsahujúcimi kobicistát kontraindikované (pozri časti 4.3 a 4.5). Ak sa liečbe so silnými inhibítormi CYP3A4 (látky zvyšujúce AUC približne 5-násobne alebo viac) nie je možné vyhnúť, počas trvania tejto liečby musí byť liečba Ezetimibom/simvastatínom Teva B.V. pozastavená (a má sa zvážiť použitie alternatívneho statínu). Okrem toho je potrebná zvýšená opatrnosť pri kombinovaní Ezetimibu/simvastatínu Teva B.V. s niektorými inými menej silnými inhibítormi CYP3A4: flukonazolom, verapamilom, diltiazemom (pozri časti4.2 a 4.5). Súbežnej konzumácii grapefruitového džúsu s Ezetimibom/simvastatínom Teva B.V. sa treba vyhnúť.
Simvastatín sa nesmie súbežne podávať so systémovými formami kyseliny fusidovej alebo počas 7 dní od ukončenia liečby kyselinou fusidovou. U pacientov u ktorých sa systémové použitie kyseliny fusidovej považuje za nevyhnutné, sa má liečba statínom prerušiť počas trvania liečby kyselinou fusidovou. U pacientov užívajúcich kombináciu kyseliny fusidovej a statínov sa vyskytli prípady rabdomyolýzy (vrátane niektorých fatálnych prípadov) (pozri časť 4.5). Pacient má byť poučený, aby vyhľadal lekársku pomoc ihneď, ak sa u neho objavia akékoľvek príznaky svalovej slabosti, bolesti alebo citlivosti.
Liečba statínom môže byť obnovená sedem dní po poslednej dávke kyseliny fusidovej.
Vo výnimočných prípadoch, kedy je potrebná dlhodobá systémová liečba kyselinou fusidovou, napr. pri liečbe ťažkých infekcií, má byť súbežné podávanie Ezetimibu/simvastatínu Teva B.V. a kyseliny fusidovej posúdené individuálne pod starostlivým lekárskym dohľadom.
Kombinovanému užívaniu ezetimibu/simvastatínu v dávkach vyšších ako 10 mg/20 mg denne s kyselinou nikotínovou v dávkach znižujúcich lipidy (³ 1 g/deň) sa treba vyhnúť, pokiaľ možný klinický prínos nepreváži zvýšené riziko myopatie (pozri časti 4.2 a 4.5).
Zriedkavé prípady myopatie/rabdomyolýzy boli spojené so súbežným podávaním inhibítorov HMG-CoA reduktázy a kyseliny nikotínovej v dávkach upravujúcich lipidy (≥ 1 g/deň), pričom jedno aj druhé liečivo môže spôsobovať myopatiu, ak sa podáva samostatne.
V klinickom skúšaní (medián sledovania 3,9 roka) zahŕňajúcom pacientov s vysokým rizikom kardiovaskulárneho ochorenia, ktorí mali dostatočne kontrolované hladiny LDL-C simvastatínom 40 mg/deň s ezetimibom 10 mg alebo bez neho, nemalo pridanie kyseliny nikotínovej v dávkach upravujúcich lipidy (³ 1 g/deň) žiadny dodatočný prínos pre kardiovaskulárne riziko. Preto, lekári zvažujúci kombinovanú liečbu simvastatínom a kyselinou nikotínovou v dávkach upravujúcich lipidy (≥ 1 g/deň) alebo liekmi obsahujúcimi kyselinu nikotínovú majú starostlivo zvážiť potenciálne prínosy a riziká a majú u pacientov starostlivo sledovať akékoľvek prejavy a príznaky svalovej bolesti, citlivosti alebo slabosti, obzvlášť počas prvých mesiacov liečby a pri zvýšení dávky niektorého z liekov.
V tomto skúšaní bola navyše incidencia myopatie približne 0,24 % u pacientov čínskeho pôvodu užívajúcich simvastatín 40 mg alebo ezetimib/simvastatín 10 mg/40 mg v porovnaní s 1,24 % pacientov čínskeho pôvodu užívajúcich simvastatín 40 mg alebo ezetimib/simvastatín 10 mg/40 mg súbežne s kyselinou nikotínovou/laropiprantom 2 000 mg/40 mg s riadeným uvoľňovaním. Hoci jediná ázijská populácia hodnotená v tomto klinickom skúšaní bola čínska, z dôvodu, že incidencia myopatie je vyššia u pacientov čínskeho pôvodu v porovnaní s pacientmi iného ako čínskeho pôvodu, sa súbežné podávanie Ezetimibu/simvastatínu Teva B.V. s kyselinou nikotínovou v dávkach upravujúcich lipidy (³ 1 g/deň) neodporúča u pacientov ázijského pôvodu.
Acipimox je štrukturálne podobný kyseline nikotínovej. Hoci acipimox nebol skúmaný, riziko toxických účinkov na svalstvo môže byť podobné ako u kyseliny nikotínovej.
Kombinovanému užívaniu ezetimibu/simvastatínu v dávkach vyšších ako 10 mg/20 mg denne s amiodarónom, amlodipínom, verapamilom alebo diltiazemom sa treba vyhnúť. U pacientov s HoFH sa treba vyhnúť kombinovanému užívaniu Ezetimibu/simvastatínu Teva B.V. v dávkach vyšších ako 10 mg/40 mg denne s lomitapidom (pozri časti 4.2, 4.3 a 4.5).
U pacientov užívajúcich iné lieky, ktoré sa označujú ako lieky so stredne silným inhibičným účinkom na CYP3A4 v terapeutických dávkach, súbežne s Ezetimibom/simvastatínom Teva B.V. , najmä vyššími dávkami Ezetimibu/simvastatínu Teva B.V., sa môže vyskytovať zvýšené riziko myopatie. Pri súbežnom podávaní Ezetimibu/simvastatínu Teva B.V. so stredne silným inhibítorom CYP3A4 (látky zvyšujúce AUC približne 2,5-násobne) môže byť potrebná úprava dávky. Pre určité stredne silné inhibítory CYP3A4, napr. diltiazem, sa odporúča maximálna dávka 10 mg/20 mg Ezetimibu/simvastatínu Teva B.V. (pozri časť 4.2).
Simvastatín je substrátom efluxného transportéra proteínu rezistencie voči rakovine prsníka (Breast Cancer Resistant Protein – BCRP). Súbežné podávanie liekov, ktoré sú inhibítormi BCRP (napr. elbasvir a grazoprevir) môže viesť k zvýšeným plazmatickým koncentráciám simvastatínu a k zvýšenému riziku myopatie; preto sa má zvážiť úprava dávky simvastatínu v závislosti od predpísanej dávky. Súbežné podávanie elbasviru a grazopreviru so simvastatínom sa neskúmalo, dávka simvastatínu však nemá u pacientov súbežne užívajúcich lieky obsahujúce elbasvir alebo grazoprevir presiahnuť 20 mg denne (pozri časť 4.5).
Bezpečnosť a účinnosť ezetimibu/simvastatínu podávaného s fibrátmi sa neštudovala. Existuje zvýšené riziko myopatie, keď sa simvastatín užíva súbežne s fibrátmi (zvlášť s gemfibrozilom). Preto je súbežné užívanie Ezetimibu/simvastatínu Teva B.V. s gemfibrozilom kontraindikované (pozri časť 4.3) a súbežné užívanie s inými fibrátmi sa neodporúča (pozri časť 4.5).
Pečeňové enzýmyV kontrolovaných skúšaniach súbežného podávania ezetimibu a simvastatínu sa u pacientov pozorovali konsekutívne zvýšenia transamináz (³ 3‑násobok HHN) (pozri časť 4.8).
V skúšaní IMPROVE-IT bolo 18 144 pacientov s koronárnou chorobou srdca a príhodou AKS v anamnéze randomizovaných na užívanie ezetimibu/simvastatínu v dávke 10 mg/40 mg denne (n = 9 067) alebo simvastatínu 40 mg denne (n = 9 077). Počas mediánu sledovania 6,0 rokov bola incidencia konsekutívnych zvýšení transamináz (³ 3‑násobok HHN) pri ezetimibe/simvastatíne 2,5 % a pri simvastatíne 2,3 % (pozri časť 4.8).
V kontrolovanej klinickej štúdii, v ktorej bolo viac ako 9 000 pacientov s chronickým ochorením obličiek randomizovaných na užívanie ezetimibu/simvastatínu v dávke 10 mg/20 mg denne (n = 4 650) alebo placeba (n = 4 620) (medián sledovaného obdobia 4,9 roka), bola incidencia konsekutívnych zvýšení transamináz (> 3‑násobok HHN) 0,7 % pri ezetimibe/simvastatíne a 0,6 % pri placebe (pozri časť 4.8).
Funkčné vyšetrenia pečene sa odporúča urobiť pred začiatkom liečby Ezetimibom/simvastatínom Teva B.V. a potom, keď je to klinicky indikované. Pacientom, u ktorých bola vytitrovaná dávka 10 mg/80 mg, sa má urobiť dodatočné vyšetrenie pred titráciou, 3 mesiace po vytitrovaní dávky na 10 mg/80 mg a potom periodicky (napr. polročne) počas prvého roku liečby. Osobitnú pozornosť treba venovať pacientom, u ktorých došlo k zvýšeniu hladín sérových transamináz, a u týchto pacientov sa majú vyšetrenia čo najskôr zopakovať, a potom robiť častejšie. Ak hladiny transamináz prejavujú známky progresie, najmä ak vystúpia na 3‑násobok HHN a na tejto úrovni pretrvávajú, liečbu je potrebné ukončiť. Majte na pamäti, že ALT sa môže vyplavovať zo svalu, preto zvyšovanie hladiny ALT s CK môže poukazovať na myopatiu (pozri vyššie „Myopatia/rabdomyolýza“).
Po uvedení lieku na trh boli u pacientov užívajúcich statíny vrátane simvastatínu zaznamenané zriedkavé hlásenia fatálneho a nefatálneho hepatálneho zlyhania. Ak sa počas liečby Ezetimibom/simvastatínom Teva B.V. vyskytne závažné poškodenie pečene s klinickými symptómami a/alebo hyperbilirubinémia alebo žltačka, liečbu ihneď prerušte. Ak sa nezistí iná etiológia, liečbu Ezetimibom/simvastatínom Teva B.V. znova nezačínajte.
Ezetimib/simvastatín Teva B.V. sa má používať s opatrnosťou u pacientov, ktorí konzumujú nadmerné množstvá alkoholu.
Porucha funkcie pečeneV dôsledku neznámych účinkov zvýšenej expozície ezetimibu u pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene sa u nich Ezetimib/simvastatín Teva B.V. neodporúča (pozri časť 5.2).
Diabetes mellitusNiektoré dôkazy naznačujú, že statíny ako skupina zvyšujú hladinu glukózy v krvi a u niektorých pacientov s vysokým rizikom vzniku diabetu môžu vyvolať úroveň hyperglykémie, pri ktorej je potrebná štandardná diabetická liečba. Nad týmto rizikom však prevažuje zníženie vaskulárneho rizika statínmi, a preto nemá byť dôvodom na ukončenie liečby statínmi. U rizikových pacientov (glykémia nalačno 5,6 – 6,9 mmol/l, BMI > 30 kg/m
2, zvýšená hladina triglyceridov, hypertenzia) je potrebné sledovať klinický stav aj biochemické parametre v súlade s národnými odporúčaniami.
Pediatrická populáciaÚčinnosť a bezpečnosť ezetimibu podávaného súbežne so simvastatínom u pacientov vo veku 10 až 17 rokov s heterozygotnou familiárnou hypercholesterolémiou sa hodnotili v kontrolovanom klinickom skúšaní u dospievajúcich chlapcov (II. Tannerovo štádium alebo vyššie) a u dievčat minimálne jeden rok po menarché.
V tejto limitovanej kontrolovanej štúdii sa vo všeobecnosti nezaznamenal žiadny detegovateľný účinok na rast alebo sexuálne dozrievanie u dospievajúcich chlapcov alebo dievčat alebo akýkoľvek účinok na dĺžku menštruačného cyklu u dievčat. Účinky ezetimibu na rast a sexuálne dozrievanie počas liečebného obdobia > 33 týždňov sa však neštudovali (pozri časti 4.2 a 4.8).
Bezpečnosť a účinnosť ezetimibu podávaného súbežne so simvastatínom v dávkach nad 40 mg denne sa u pediatrických pacientov vo veku 10 až 17 rokov neskúmali.
Ezetimib sa neskúmal u pacientov mladších ako 10 rokov alebo u dievčat pred menarché. (Pozri časti 4.2 a 4.8).
Dlhodobá účinnosť liečby ezetimibom na zníženie morbidity a mortality v dospelosti sa u pacientov mladších ako 17 rokov neštudovala.
FibrátyBezpečnosť a účinnosť ezetimibu podávaného s fibrátmi neboli stanovené (pozri vyššie a časti 4.3 a 4.5).
AntikoagulanciáAk sa Ezetimib/simvastatín Teva B.V. pridáva k warfarínu, k inému kumarínovému antikoagulantu alebo k fluindiónu, je potrebné náležite sledovať medzinárodný normalizovaný pomer (International Normalised Ratio, INR) (pozri časť 4.5).
Intersticiálne ochorenie pľúcPri užívaní niektorých statínov vrátane simvastatínu, hlavne pri dlhodobej liečbe, boli hlásené prípady intersticiálneho ochorenia pľúc (pozri časť 4.8). Prejavy môžu zahŕňať dyspnoe, neproduktívny kašeľ a zhoršenie celkového zdravotného stavu (únava, chudnutie a horúčka). Ak je podozrenie, že sa u pacienta objavilo intersticiálne ochorenie pľúc, liečba Ezetimibom/simvastatínom Teva B.V. sa má prerušiť.
Pomocné látkyLaktózaPacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo‑galaktózovou malabsorpciou nesmú užívať tento liek.
SodíkTento liek obsahuje menej ako 1 mmol sodíka (23 mg) v tablete, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcieFarmakodynamické interakcieInterakcie s liekmi znižujúcimi lipidy, ktoré môžu spôsobiť myopatiu, keď sa podávajú samostatnePočas súbežného podávania simvastatínu s fibrátmi je zvýšené riziko myopatie vrátane rabdomyolýzy. Okrem toho, existuje farmakokinetická interakcia simvastatínu s gemfibrozilom, ktorá vedie k zvýšeným plazmatickým hladinám simvastatínu (pozri nižšie „Farmakokinetické interakcie“ a časti 4.3 a 4.4). So simvastatínom podávaným súbežne s kyselinou nikotínovou v dávkach upravujúcich lipidy (≥ 1 g/deň) sa spájali zriedkavé prípady myopatie/rabdomyolýzy (pozri časť 4.4).
Fibráty môžu zvýšiť vylučovanie cholesterolu do žlče, čo vedie k cholelitiáze. V predklinickej štúdii na psoch ezetimib zvýšil hladinu cholesterolu v žlči uskladnenej v žlčníku (pozri časť 5.3). Hoci význam tohto predklinického zistenia pre ľudí nie je známy, súbežné podávanie Ezetimibu/simvastatínu Teva B.V. s fibrátmi sa neodporúča (pozri časť 4.4).
Farmakokinetické interakciePreskripčné odporúčania pre interagujúce liečivá sú zosumarizované v tabuľke uvedenej nižšie (ďalšie detaily sú uvedené v texte, pozri tiež časti 4.2, 4.3 a 4.4).
Liekové interakcie spojené so zvýšeným rizikom myopatie/rabdomyolýzyInteragujúce liečivá
| Preskripčné odporúčania
|
Silné inhibítory CYP3A4, napr.
Itrakonazol
Ketokonazol
Posakonazol
Vorikonazol
Erytromycín
Klaritromycín
Telitromycín
Inhibítory HIV proteázy (napr. nelfinavir)
Boceprevir
Telaprevir
Nefazodón
Kobicistát
Cyklosporín
Danazol
Gemfibrozil
| Kontraindikované s Ezetimibom/simvastatínom Teva B.V.
|
Ostatné fibráty
Kyselina fusidová
| Neodporúčajú sa s Ezetimibom/simvastatínom Teva B.V.
|
Niacín (kyselina nikotínová) (≥ 1 g/deň)
| Neodporúča sa s Ezetimibom/simvastatínom Teva B.V. u pacientov ázijského pôvodu
|
Amiodarón
Amlodipín
Verapamil
Diltiazem
Elbasvir
Grazoprevir
Niacín (kyselina nikotínová) (≥ 1 g/deň)
| Neprekročiť 10 mg/20 mg Ezetimibu/simvastatínu Teva B.V. denne
|
Lomitapid
| U pacientov s HoFH neprekročiť 10 mg/40 mg Ezetimibu/simvastatínu Teva B.V. denne
|
Grapefruitový džús
| Vyhnúť sa grapefruitovému džúsu pri užívaní Ezetimibu/simvastatínu Teva B.V.
|
Ezetimib/simvastatín Teva B.V.
Niacín (Kyselina nikotínová): V štúdii 15 zdravých dospelých spôsobilo súbežné podávanie ezetimibu/simvastatínu (10 mg/20 mg denne počas 7 dní) malé zvýšenie v priemerných AUC kyseliny nikotínovej (22 %) a kyseliny nikotinúrovej (19 %) podávaných ako NIASPAN tablety s predĺženým uvoľňovaním (1 000 mg počas 2 dní a 2 000 mg počas 5 dní po raňajkách s nízkym obsahom tuku). V tej istej štúdii súbežne podávaný NIASPAN mierne zvýšil priemerné AUC ezetimibu (9 %), celkového ezetimibu (26 %), simvastatínu (20 %) a kyseliny simvastatínovej (35 %). (Pozri časti 4.2 a 4.4)
Štúdie liekových interakcií s vyššími dávkami simvastatínu sa neskúmali.
EzetimibAntacidá: Súbežné podávanie antacíd znížilo mieru absorpcie ezetimibu, ale nemalo žiadny vplyv na biologickú dostupnosť ezetimibu. Toto zníženie miery absorpcie sa nepovažuje za klinicky významné.
Cholestyramín: Súbežné podávanie cholestyramínu znížilo priemernú plochu pod krivkou (AUC) celkového ezetimibu (ezetimib + glukuronid ezetimibu) približne o 55 %. Prírastok zníženia LDL‑C v dôsledku pridania ezetimibu/simvastatínu k cholestyramínu môže byť touto interakciou znížený (pozri časť 4.2).
Cyklosporín: V štúdii, ktorej sa zúčastnilo osem pacientov po transplantácii obličky s klírensom kreatinínu > 50 ml/min na stabilnej dávke cyklosporínu, mala jednorazová 10 mg dávka ezetimibu za následok 3,4‑násobné (rozsah 2,3‑ až 7,9‑násobné) zvýšenie priemernej hodnoty AUC celkového ezetimibu v porovnaní so zdravou kontrolnou populáciou z inej štúdie, ktorá dostávala ezetimib samotný (n = 17). V inej štúdii pacient po transplantácii obličky s ťažkou poruchou funkcie obličiek, ktorý dostával cyklosporín a viacero iných liekov, preukázal 12‑násobne vyššiu expozíciu celkovému ezetimibu v porovnaní so súbežnou kontrolnou skupinou pacientov, ktorí dostávali ezetimib samotný. V dvojitej skríženej štúdii u dvanástich zdravých osôb viedlo denné podávanie ezetimibu 20 mg počas 8 dní spolu s jednorazovou 100 mg dávkou cyklosporínu na 7. deň k priemernému nárastu AUC cyklosporínu o 15 % (rozsah 10 % pokles až 51 % nárast) v porovnaní s jednorazovou 100 mg dávkou samotného cyklosporínu. Kontrolovaná štúdia účinku súbežného podávania ezetimibu na expozíciu cyklosporínu u pacientov po transplantácii obličky sa nevykonala. Súbežné podávanie Ezetimibu/simvastatínu Teva B.V. s cyklosporínom je kontraindikované (pozri časť 4.3).
Fibráty: Súbežné podávanie fenofibrátu alebo gemfibrozilu zvýšilo koncentrácie celkového ezetimibu približne 1,5‑ a 1,7‑násobne v uvedenom poradí. Hoci sa tieto zvýšenia nepovažujú za klinicky významné, súbežné podávanie Ezetimibu/simvastatínu Teva B.V. s gemfibrozilom je kontraindikované a s ostatnými fibrátmi sa neodporúča (pozri časti 4.3 a 4.4).
SimvastatínSimvastatín je substrát cytochrómu P450 3A4. Silné inhibítory cytochrómu P450 3A4 zvyšujú riziko myopatie a rabdomyolýzy zvýšením koncentrácie inhibičnej aktivity na HMG‑CoA reduktázu v plazme počas liečby simvastatínom. K týmto inhibítorom patrí itrakonazol, ketokonazol, posakonazol, vorikonazol, erytromycín, klaritromycín, telitromycín, inhibítory HIV proteázy (napr. nelfinavir), boceprevir, telaprevir, nefazodón a lieky obsahujúce kobicistát. Súbežné podávanie itrakonazolu viedlo k viac ako 10‑násobnému zvýšeniu v expozícii kyseline simvastatínovej (aktívny beta-hydroxykyselinový metabolit). Telitromycín spôsobil 11‑násobné zvýšenie v expozícii kyseline simvastatínovej.
Kombinácia s itrakonazolom, ketokonazolom, posakonazolom, vorikonazolom, inhibítormi HIV proteázy (napr. nelfinavir), boceprevirom, telaprevirom, erytromycínom, klaritromycínom, telitromycínom, nefazodónom a liekmi obsahujúcimi kobicistát, ako aj s gemfibrozilom, cyklosporínom a danazolom je kontraindikovaná (pozri časť 4.3). Ak sa liečbe so silnými inhibítormi CYP3A4 (látky zvyšujúce AUC približne 5-násobne alebo viac) nie je možné vyhnúť, počas trvania tejto liečby musí byť liečba Ezetimibom/simvastatínom Teva B.V. pozastavená (a má sa zvážiť použitie alternatívneho statínu). Opatrnosť je potrebná pri kombinovaní Ezetimibu/simvastatínu Teva B.V. s niektorými inými menej silnými inhibítormi CYP3A4: flukonazolom, verapamilom alebo diltiazemom (pozri časti 4.2 a 4.4).
Flukonazol: V súvislosti so súbežným podaním simvastatínu a flukonazolu boli hlásené zriedkavé prípady rabdomyolýzy (pozri časť 4.4).
Cyklosporín: Riziko myopatie/rabdomyolýzy je zvýšené pri súbežnom podávaní cyklosporínu s Ezetimibom/simvastatínom Teva B.V.; preto je užívanie s cyklosporínom kontraindikované (pozri časti 4.3 a 4.4). Hoci mechanizmus nie je celkom objasnený, ukázalo sa, že cyklosporín zvyšuje AUC inhibítorov HMG‑CoA reduktázy. Zvýšenie AUC kyseliny simvastatínovej je pravdepodobne z časti následkom inhibície CYP3A4 a/alebo OATP1B1.
Danazol: Riziko myopatie a rabdomyolýzy je zvýšené pri súbežnom podávaní danazolu s Ezetimibom/simvastatínom Teva B.V.; preto je užívanie s danazolom kontraindikované (pozri časti 4.3 a 4.4).
Gemfibrozil: Gemfibrozil zvyšuje AUC kyseliny simvastatínovej 1,9‑násobne, pravdepodobne v dôsledku inhibície glukuronidačnej dráhy (pozri časti 4.3 a 4.4). Súbežné podávanie s gemfibrozilom je kontraindikované.
Kyselina fusidová: Pri súbežnom systémovom podávaní kyseliny fusidovej so statínmi môže byť zvýšené riziko myopatie vrátane rabdomyolýzy. Súbežné podávanie tejto kombinácie môže spôsobiť zvýšenie koncentrácií oboch látok v plazme. Mechanizmus tejto interakcie (či je farmakodynamický alebo farmakokinetický, alebo oboje) nie je ešte známy. U pacientov užívajúcich túto kombináciu boli hlásené prípady rabdomyolýzy (vrátane fatálnych prípadov).
Ak je liečba kyselinou fusidovou nevyhnutná, liečba Ezetimibom/simvastatínom Teva B.V. sa má prerušiť počas liečby kyselinou fusidovou.
Pozri tiež časť 4.4.Amiodarón: Pri súbežnom podávaní amiodarónu so simvastatínom je zvýšené riziko myopatie a rabdomyolýzy (pozri časť 4.4). V klinickom skúšaní bola myopatia hlásená u 6 % pacientov, ktorí dostávali simvastatín 80 mg a amiodarón. U pacientov liečených súbežne amiodarónom nemá preto dávka Ezetimibu/simvastatínu Teva B.V. prekročiť 10 mg/20 mg denne.
Blokátory kalciového kanála:-
Verapamil: Súbežným užívaním verapamilu so simvastatínom 40 mg alebo 80 mg sa zvyšuje riziko myopatie a rabdomyolýzy (pozri časť 4.4). Vo farmakokinetickej štúdii viedlo súbežné podávanie simvastatínu s verapamilom k 2,3‑násobnému zvýšeniu v expozícii kyseline simvastatínovej, pravdepodobne čiastočne v dôsledku inhibície CYP3A4. Preto u pacientov liečených súbežne verapamilom nemá dávka Ezetimibu/simvastatínu Teva B.V.prekročiť 10 mg/20 mg denne.
-
Diltiazem: Súbežným užívaním diltiazemu so simvastatínom 80 mg sa zvyšuje riziko myopatie a rabdomyolýzy (pozri časť 4.4). Vo farmakokinetickej štúdii spôsobilo súbežné podávanie diltiazemu so simvastatínom 2,7‑násobné zvýšenie v expozícii kyseline simvastatínovej, pravdepodobne v dôsledku inhibície CYP3A4. Preto u pacientov liečených súbežne diltiazemom nemá dávka Ezetimibu/simvastatínu Teva B.V. prekročiť 10 mg/20 mg denne.
AmlodipínPacienti na amlodipíne liečení súbežne simvastatínom majú zvýšené riziko myopatie. Vo farmakokinetickej štúdii spôsobilo súbežné podávanie amlodipínu 1,6‑násobné zvýšenie v expozícii kyseline simvastatínovej. Preto u pacientov súbežne liečených amlodipínom, nemá dávka Ezetimibu/simvastatínu Teva B.V. prekročiť 10 mg/20 mg denne.
Lomitapid: Pri súbežnom podávaní lomitapidu so simvastatínom môže byť zvýšené riziko myopatie a rabdomyolýzy (pozri časti 4.3 a 4.4)
. Preto u pacientov s HoFH pri súbežnej liečbe s lomitapidom nesmie dávka Ezetimibu/simvastatínu Teva B.V. prekročiť 10 mg/40 mg denne.
Stredne silné inhibítory CYP3A4: Pacienti užívajúci iné lieky, ktoré sa označujú ako lieky so stredne silným inhibičným účinkom na CYP3A4, súbežne s Ezetimibom/simvastatínom Teva B.V., najmä vyššími dávkami Ezetimibu/simvastatínu Teva B.V., môžu mať zvýšené riziko myopatie (pozri časť 4.4).
Inhibítory transportného proteínu OATP1B1: Kyselina simvastatínová je substrátom transportného proteínu OATP1B1. Súbežné podávanie liekov, ktoré sú inhibítormi transportného proteínu OATP1B1 môže viesť k zvýšeným plazmatickým koncentráciám kyseliny simvastatínovej a k zvýšenému riziku myopatie (pozri časti 4.3 a 4.4).
Inhibítory proteínu rezistencie voči rakovine prsníka (BCRP): Súbežné podávanie liekov, ktoré sú inhibítormi BCRP, vrátane liekov obsahujúcich elbasvir alebo grazoprevir, môže viesť k zvýšeným plazmatickým koncentráciám simvastatínu a k zvýšenému riziku myopatie (pozri časti 4.2 a 4.4).
Grapefruitový džús: Grapefruitový džús inhibuje cytochróm P450 3A4. Súbežné požitie veľkého množstva (viac ako 1 liter denne) grapefruitového džúsu a simvastatínu viedlo k 7‑násobnému zvýšeniu v expozícii kyseline simvastatínovej. Požitie 240 ml grapefruitového džúsu ráno a podanie simvastatínu večer viedlo tiež k 1,9‑násobnému zvýšeniu. Preto sa treba konzumácii grapefruitového džúsu počas liečby Ezetimibom/simvastatínom Teva B.V. vyhnúť.
Kolchicín: Pri súbežnom podaní kolchicínu a simvastatínu sa vyskytli hlásenia myopatie a rabdomyolýzy u pacientov s poruchou funkcie obličiek. Odporúča sa, aby boli pacienti užívajúci takúto kombináciu pozorne klinicky sledovaní.
Rifampicín: Pretože rifampicín je silný induktor CYP3A4, u pacientov podstupujúcich dlhodobú liečbu rifampicínom (napr. liečba tuberkulózy) môže dôjsť k strate účinnosti simvastatínu. Vo farmakokinetickej štúdii u zdravých dobrovoľníkov bola plocha pod krivkou plazmatickej koncentrácie (AUC) pre kyselinu simvastatínovú pri súbežnom podávaní s rifampicínom znížená o 93 %.
Niacín (kyselina nikotínová): Pri súbežnom podávaní simvastatínu a kyseliny nikotínovej v dávkach upravujúcich lipidy (³ 1 g/deň) boli pozorované prípady myopatie a rabdomyolýzy (pozri časť 4.4).
Účinky Ezetimibu/simvastatínu Teva B.V. na farmakokinetiku iných liekovEzetimibV predklinických štúdiách sa zistilo, že ezetimib neindukuje enzýmy cytochrómu P450, ktoré metabolizujú liečivá. Nepozorovali sa žiadne klinicky významné farmakokinetické interakcie medzi ezetimibom a liečivami, o ktorých je známe, že sú metabolizované cytochrómami P450 1A2, 2D6, 2C8, 2C9 a 3A4 alebo N-acetyltransferázou.
Antikoagulanciá:V skúšaní, ktorého sa zúčastnilo dvanásť zdravých dospelých mužov, nemalo súbežné podávanie ezetimibu (10 mg jedenkrát denne) významný účinok na biologickú dostupnosť warfarínu a protrombínový čas. Po uvedení lieku na trh sa však vyskytli hlásenia o zvýšení medzinárodného normalizovaného pomeru (INR) u pacientov, ktorým bol ezetimib pridaný k warfarínu alebo k fluindiónu. Ak sa Ezetimib/simvastatín Teva B.V. pridáva k warfarínu, k inému kumarínovému antikoagulantu alebo k fluindiónu, má sa primerane monitorovať INR (pozri časť 4.4).
SimvastatínSimvastatín nemá inhibičný účinok na cytochróm P450 3A4. Preto sa neočakáva, že simvastatín ovplyvní plazmatické koncentrácie látok metabolizovaných cytochrómom P450 3A4.
Perorálne antikoagulanciá: V dvoch klinických štúdiách, jednej u zdravých dobrovoľníkov a druhej u pacientov s hypercholesterolémiou, simvastatín 20 – 40 mg/deň mierne potencoval účinok kumarínových antikoagulancií: protrombínový čas vyjadrený medzinárodným normalizovaným pomerom (INR) sa zvýšil z východiskovej hodnoty 1,7 na 1,8 v štúdii u dobrovoľníkov a z 2,6 na 3,4 v štúdii u pacientov. Boli hlásené veľmi zriedkavé prípady zvýšenia INR. U pacientov užívajúcich kumarínové antikoagulanciá sa má protrombínový čas stanoviť pred začiatkom liečby Ezetimibom/simvastatínom Teva B.V., a potom dostatočne často na začiatku liečby, aby bolo isté, že nedochádza k signifikantnej zmene protrombínového času. Po preukázaní stabilného protrombínového času môže byť protrombínový čas monitorovaný v intervaloch obvykle odporúčaných pre pacientov liečených kumarínovými antikoagulanciami. Ak sa dávka Ezetimibu/simvastatínu Teva B.V. zmení alebo liečba preruší, je potrebné zopakovať rovnaký postup. U pacientov, ktorí neužívali antikoagulanciá, nebola liečba simvastatínom spojená s krvácaním alebo so zmenami v protrombínovom čase.
Pediatrická populáciaInterakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktáciaGraviditaAteroskleróza je chronický proces a bežné prerušenie užívania liekov znižujúcich lipidy počas gravidity by malo mať len malý vplyv na dlhodobé riziko spojené s primárnou hypercholesterolémiou.
Ezetimib/simvastatín Teva B.V.Ezetimib/simvastatín Teva B.V. je kontraindikovaný počas gravidity. K dispozícii nie sú žiadne klinické údaje o použití Ezetimibu/simvastatínu Teva B.V. počas gravidity. Štúdie kombinovanej liečby na zvieratách preukázali reprodukčnú toxicitu. (Pozri časť 5.3.)
SimvastatínBezpečnosť simvastatínu u gravidných žien nebola stanovená. U gravidných žien sa neuskutočnili žiadne kontrolované klinické skúšania so simvastatínom. Zriedkavo boli hlásené kongenitálne anomálie po intrauterinnej expozícii inhibítorom HMG‑CoA reduktázy. V analýze približne 200 prospektívne sledovaných gravidít s expozíciou simvastatínu alebo inému blízko príbuznému inhibítoru HMG‑CoA reduktázy počas prvého trimestra bola však incidencia kongenitálnych anomálií porovnateľná s incidenciou v bežnej populácii. Tento počet gravidít bol štatisticky dostatočný na vylúčenie 2,5‑násobného alebo vyššieho nárastu kongenitálnych anomálií v porovnaní s incidenciou v celkovej populácii.
Hoci nie je dokázané, že sa incidencia kongenitálnych anomálií u potomkov pacientok užívajúcich simvastatín alebo iný blízko príbuzný inhibítor HMG‑CoA reduktázy líši od incidencie pozorovanej v bežnej populácii, liečba nastávajúcich matiek simvastatínom môže u plodu znížiť hladiny mevalonátu, ktorý je prekurzorom biosyntézy cholesterolu. Z tohto dôvodu sa Ezetimib/simvastatín Teva B.V. nesmie používať u žien, ktoré sú gravidné, pokúšajú sa otehotnieť alebo predpokladajú, že môžu byť tehotné. Liečba Ezetimibom/simvastatínom Teva B.V. sa musí prerušiť počas trvania gravidity alebo kým sa nepotvrdí, že žena nie je tehotná. (Pozri časť 4.3.)
EzetimibK dispozícii nie sú žiadne klinické údaje o použití ezetimibu počas gravidity.
DojčenieEzetimib/simvastatín Teva B.V. je počas dojčenia kontraindikovaný. Štúdie na potkanoch preukázali, že ezetimib sa vylučuje do materského mlieka. Nie je známe, či sa liečivá Ezetimibu/simvastatínu Teva B.V. vylučujú do ľudského mlieka. (Pozri časť 4.3.)
FertilitaEzetimibK dispozícii nie sú žiadne údaje z klinického skúšania účinkov ezetimibu na fertilitu u ľudí. Ezetimib nemal žiadny účinok na fertilitu samcov alebo samíc potkana (pozri časť 5.3).
SimvastatínK dispozícii nie sú žiadne údaje z klinického skúšania účinkov simvastatínu na fertilitu u ľudí. Simvastatín nemal žiadny účinok na fertilitu samcov a samíc potkana (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeNeuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Pri vedení vozidiel alebo obsluhe strojov treba však brať do úvahy, že bol hlásený závrat.
4.8 Nežiaduce účinkyBezpečnosť ezetimibu/simvastatínu bola hodnotená v klinických skúšaniach u približne 12 000 pacientov.
Frekvencie nežiaducich udalostí sú zoradené nasledovne: veľmi časté (³ 1/10), časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100), zriedkavé (³ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) zahŕňajúce jednotlivé hlásenia, neznáme (nemožno odhadnúť z dostupných údajov).
U pacientov liečených ezetimibom/simvastatínom (n = 2 404) boli s vyššou incidenciou ako pri placebe (n = 1 340) pozorované nasledujúce nežiaduce reakcie.
Nežiaduce reakcie pri ezetimibe/simvastatíne pozorované s vyššou incidenciou ako pri placebeTrieda orgánových systémov
| Nežiaduce reakcie
| Frekvencia
|
Psychické poruchy
| porucha spánku
| menej časté
|
Poruchy nervového systému
| závrat, bolesť hlavy
| menej časté
|
Poruchy gastrointestinálneho traktu
| bolesť brucha, abdominálny diskomfort, bolesť v hornej časti brucha, dyspepsia, nadúvanie, nevoľnosť, vracanie
| menej časté
|
Poruchy kože a podkožného tkaniva
| pruritus, vyrážka
| menej časté
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| artralgia, svalové spazmy, svalová slabosť, muskuloskeletárny diskomfort, bolesť krku, bolesť v končatine
| menej časté
|
Celkové poruchy a reakcie v mieste podania
| asténia, únava, celkový pocit nepohodlia, periférny edém
| menej časté
|
Laboratórne a funkčné vyšetrenia
| zvýšenie ALT a/alebo AST; zvýšenie CK v krvi
| časté
|
zvýšenie bilirubínu v krvi, zvýšenie kyseliny močovej v krvi, zvýšenie gamaglutamyltransferázy, zvýšenie medzinárodného normalizovaného pomeru (INR, international normalized ratio), prítomnosť bielkoviny v moči, zníženie hmotnosti
| menej časté
|
U pacientov liečených ezetimibom/simvastatínom (n = 9 595) boli s vyššou incidenciou ako pri samostatne podávaných statínoch (n = 8 883) pozorované nasledujúce nežiaduce reakcie.
Nežiaduce reakcie pri ezetimibe/simvastatíne s vyššou incidenciou ako pri statínochTrieda orgánových systémov
| Nežiaduce reakcie
| Frekvencia
|
Psychické poruchy
| insomnia
| menej časté
|
Poruchy nervového systému
| bolesť hlavy, parestézia
| menej časté
|
Poruchy gastrointestinálneho traktu
| abdominálna distenzia, hnačka, sucho v ústach, dyspepsia, nadúvanie, gastroezofagálna refluxná choroba, vracanie
| menej časté
|
Poruchy kože a podkožného tkaniva
| pruritus, vyrážka, urtikária
| menej časté
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| myalgia
| časté
|
artralgia, bolesť chrbta, svalové spazmy, svalová slabosť, muskuloskeletárna bolesť, bolesť v končatine
| menej časté
|
Celkové poruchy a reakcie v mieste podania
| asténia, bolesť na hrudi, únava, periférny edém
| menej časté
|
Laboratórne a funkčné vyšetrenia
| zvýšenie ALT a/alebo AST
| časté
|
zvýšenie bilirubínu v krvi, zvýšenie CK v krvi, zvýšenie gamaglutamyltransferázy
| menej časté
|
Pediatrická populáciaV štúdii zahŕňajúcej dospievajúcich pacientov (vo veku 10 až 17 rokov) s heterozygotnou familiárnou hypercholesterolémiou (n = 248) sa zvýšenia ALT a/alebo AST (³ 3-násobok HHN, konsekutívne) pozorovali u 3 % pacientov (4 pacienti) v skupine s ezetimibom/simvastatínom v porovnaní s 2 % (2 pacienti) v skupine so simvastatínom v monoterapii; pre zvýšenie CK (≥ 10-násobok HHN) boli hodnoty v skupine s ezetimibom/simvastatínom 2 % (2 pacienti) a v skupine so simvastatínom v monoterapii 0 %. Neboli hlásené žiadne prípady myopatie.
Toto skúšanie nebolo vhodné na porovnanie zriedkavých nežiaducich liekových reakcií.
Pacienti s koronárnou chorobou srdca a príhodou AKS v anamnézeV štúdii IMPROVE-IT (pozri časť 5.1) zahŕňajúcej 18 144 pacientov liečených ezetimibom/simvastatínom v dávke 10 mg/40 mg (n = 9 067, z ktorých 6 % bolo vytitrovaných na ezetimib/simvastatín 10 mg/80 mg) alebo simvastatínom 40 mg (n = 9 077, z ktorých 27 % bolo vytitrovaných na simvastatín 80 mg), boli bezpečnostné profily počas mediánu sledovaného obdobia 6,0 rokov podobné. Miery prerušenia liečby z dôvodu nežiaducich účinkov boli 10,6 % u pacientov liečených ezetimibom/simvastatínom a 10,1 % u pacientov liečených simvastatínom. Incidencia myopatie bola u pacientov liečených ezetimibom/simvastatínom 0,2 % a u pacientov liečených simvastatínom 0,1 %, pričom myopatia bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN alebo dve konsekutívne pozorovania hladiny CK ≥ 5 a < 10-násobok HHN. Incidencia rabdomyolýzy bola pri ezetimibe/simvastatíne 0,1 % a pri simvastatíne 0,2 %, pričom rabdomyolýza bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN s dôkazom poškodenia obličiek, ≥ 5‑násobok HHN a < 10-násobok HHN počas dvoch konsekutívnych udalostí s dôkazom poškodenia obličiek alebo hladinou CK ≥ 10 000 IU/l bez dôkazu poškodenia obličiek. Incidencia konsekutívnych zvýšení transamináz (≥ 3‑násobok HHN) bola 2,5 % pri ezetimibe/simvastatíne a 2,3 % pri simvastatíne. (Pozri časť 4.4). Nežiaduce účinky súvisiace so žlčníkom boli hlásené u 3,1 % pacientov s ezetimibom/simvastatínom oproti 3,5 % pacientov so simvastatínom. Incidencia hospitalizácií z dôvodu cholecystektómie bola v obidvoch liečebných skupinách 1,5 %. Rakovina (definovaná ako akákoľvek nová malignita) bola počas skúšania diagnostikovaná u 9,4 % pacientov s ezetimibom/simvastatínom oproti 9,5 % pacientov so simvastatínom.
Pacienti s chronickým ochorením obličiekV štúdii Study of Heart and Renal Protection (SHARP) (pozri časť 5.1) zahŕňajúcej viac ako 9 000 pacientov liečených ezetimibom/simvastatínom v dávke 10 mg/20 mg denne (n = 4 650) alebo placebom (n = 4 620) boli bezpečnostné profily počas mediánu sledovaného obdobia 4,9 roka porovnateľné. V tomto skúšaní sa zaznamenávali len závažné nežiaduce udalosti a prerušenia liečby z dôvodu akýchkoľvek nežiaducich udalostí. Miery prerušenia liečby z dôvodu nežiaducich udalostí boli porovnateľné (10,4 % u pacientov liečených ezetimibom/simvastatínom, 9,8 % u pacientov, ktorí dostávali placebo). Incidencia myopatie/rabdomyolýzy bola u pacientov s ezetimibom/simvastatínom 0,2 % a u pacientov, ktorí dostávali placebo 0,1 %. Konsekutívne zvýšenia transamináz (> 3‑násobok HHN) sa objavili u 0,7 % pacientov liečených ezetimibom/simvastatínom v porovnaní s 0,6 % pacientov, ktorí dostávali placebo. V tomto skúšaní sa neobjavili žiadne štatisticky významné zvýšenia incidencie vopred stanovených nežiaducich udalostí vrátane rakoviny (9,4 % pri ezetimibe/simvastatíne, 9,5 % pri placebe), hepatitídy, cholecystektómie alebo komplikácií cholelitiázy alebo pankreatitídy.
Výsledky laboratórnych vyšetreníV skúšaniach súbežného podávania bola incidencia klinicky významných zvýšení hladín sérových transamináz (ALT a/alebo AST ≥ 3‑násobok HHN, konsekutívne) 1,7 % u pacientov liečených ezetimibom/simvastatínom. Tieto zvýšenia boli vo všeobecnosti asymptomatické, neboli spojené s cholestázou a vrátili sa na východiskovú hodnotu po prerušení liečby alebo pri jej pokračovaní. (Pozri časť 4.4).
Klinicky významné zvýšenia CK (≥ 10‑násobok HHN) sa pozorovali u 0,2 % pacientov liečených ezetimibom/simvastatínom.
Skúsenosti po uvedení lieku na trhPri používaní ezetimibu/simvastatínu po uvedení na trh alebo počas klinických štúdií alebo pri používaní jednej z jeho zložiek po uvedení lieku na trh boli hlásené nasledujúce ďalšie nežiaduce reakcie.
Poruchy krvi a lymfatického systému:trombocytopénia, anémia.
Poruchy nervového systému:periférna neuropatia, porucha pamäti.
Poruchy dýchacej sústavy, hrudníka a mediastína:kašeľ, dyspnoe, intersticiálne ochorenie pľúc (pozri časť 4.4).
Poruchy gastrointestinálneho traktu: zápcha, pankreatitída, gastritída.
Poruchy kože a podkožného tkaniva:alopécia, multiformný erytém, vyrážka, urtikária, angioedém.
Poruchy imunitného systému: hypersenzitivita, vrátane anafylaktických reakcií; anafylaxia (veľmi zriedkavé)
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva: svalové kŕče, myopatia* (vrátane myozitídy), rabdomyolýza s akútnym renálnym zlyhaním alebo bez neho (pozri časť 4.4), tendinopatia, niekedy komplikovaná ruptúrou, nekrotizujúca myopatia sprostredkovaná imunitným systémom (IMNM) (frekvencia neznáma)**.
* V klinickom skúšaní sa u pacientov liečených simvastatínom 80 mg/deň v porovnaní s pacientmi liečenými 20 mg/deň vyskytovala myopatia často (1,0 % oproti 0,02 %) (pozri časti 4.4 a 4.5).
**Veľmi zriedkavo sa počas liečby alebo po liečbe niektorými statínmi hlásila nekrotizujúca myopatia sprostredkovaná imunitným systémom (IMNM), autoimunitná myopatia. IMNM je klinicky charakterizovaná: pretrvávajúcou slabosťou proximálneho svalstva a zvýšenou hladinou kreatínkinázy v sére, ktoré pretrvávajú napriek prerušeniu liečby statínmi; biopsiou svalu preukazujúcu nekrotizujúcou myopatiou bez výrazného zápalu; zlepšením vplyvom imunosupresívnych liekov (pozri časť 4.4).
Poruchy metabolizmu a výživy:zníženie chuti do jedla.
Poruchy ciev:nával tepla, hypertenzia.
Celkové poruchy a reakcie v mieste podania: bolesť.
Poruchy pečene a žlčových ciest: hepatitída/žltačka, fatálne a nefatálne hepatálne zlyhanie, cholelitiáza, cholecystitída.
Poruchy reprodukčného systému a prsníkov: erektilná dysfunkcia.
Psychické poruchy: depresia, insomnia.
Zriedkavo bol hlásený zjavný syndróm z precitlivenosti, ktorý zahŕňal niektoré z nasledujúcich znakov: angioedém, syndróm podobný lupusu, reumatickú polymyalgiu, dermatomyozitídu, vaskulitídu, trombocytopéniu, eozinofíliu, zvýšenú sedimentáciu erytrocytov, artritídu a artralgiu, urtikáriu, fotosenzitívnu reakciu, pyrexiu, začervenanie, dyspnoe a celkový pocit nepohodlia.
Výsledky laboratórnych vyšetrení:zvýšenie hladiny alkalickej fosfatázy, abnormálne výsledky funkčných vyšetrení pečene.
Pri statínoch vrátane simvastatínu bolo hlásené zvýšenie HbA1c a sérových hladín glukózy nalačno.
Po uvedení lieku na trh boli v súvislosti s použitím statínov vrátane simvastatínu zaznamenané zriedkavé hlásenia poškodenia kognitívnych funkcií (napr. strata pamäti, zábudlivosť, amnézia, poškodenie pamäti, zmätenosť). Hlásenia nie sú vo všeobecnosti závažné a po prerušení liečby statínmi sú reverzibilné s variabilným časom pre vznik (1 deň až roky) a vymiznutie symptómov (medián 3 týždne).
Nasledujúce ďalšie nežiaduce udalosti boli hlásené v súvislosti s niektorými statínmi:
- poruchy spánku, vrátane nočných môr,
- sexuálna dysfunkcia,
- diabetes mellitus: frekvencia bude závisieť od prítomnosti alebo neprítomnosti rizikových faktorov (glykémia nalačno ≥ 5,6 mmol/l, BMI > 30 kg/m
2, zvýšená hladina triglyceridov, hypertenzia v anamnéze).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 PredávkovanieKombinácia ezetimibu/simvastatínuV prípade predávkovania treba vykonať symptomatické a podporné opatrenia. Súbežné podávanie ezetimibu (1 000 mg/kg) a simvastatínu (1 000 mg/kg) bolo dobre tolerované v štúdiách akútnej perorálnej toxicity na myšiach a potkanoch. U týchto zvierat sa nepozorovali žiadne klinické prejavy toxicity. Odhadovaná perorálna LD
50 pre oba druhy bola ezetimib ≥ 1 000 mg/kg/simvastatín ≥ 1 000 mg/kg.
EzetimibV klinických štúdiách bolo podávanie ezetimibu 50 mg/deň 15 zdravým osobám až po dobu 14 dní alebo 40 mg/deň 18 pacientom s primárnou hypercholesterolémiou až po dobu 56 dní vo všeobecnosti dobre tolerované. Bolo hlásených niekoľko prípadov predávkovania; väčšina nebola spojená s nežiaducimi účinkami. Hlásené nežiaduce účinky neboli závažné. U zvierat sa nepozorovala žiadna toxicita po jednorazových perorálnych dávkach 5 000 mg/kg ezetimibu u potkanov a myší a 3 000 mg/kg u psov.
SimvastatínBolo hlásených niekoľko prípadov predávkovania; maximálna užitá dávka bola 3,6 g. Všetci pacienti sa zotavili bez následkov.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Hypolipidemiká, inhibítory HMG‑CoA reduktázy v kombinácii s inými hypolipidemikami, ATC kód: C10BA02.
Ezetimib/simvastatín Teva B.V. je liek znižujúci lipidy, ktorý selektívne inhibuje črevnú absorpciu cholesterolu a príbuzných rastlinných sterolov a inhibuje endogénnu syntézu cholesterolu.
Mechanizmus účinkuEzetimib/simvastatín Plazmatický cholesterol pochádza z črevnej absorpcie a endogénnej syntézy. Ezetimib/simvastatín Teva B.V. obsahuje ezetimib a simvastatín, dve látky znižujúce lipidy s komplementárnymi mechanizmami účinku. Ezetimib/simvastatín Teva B.V. znižuje zvýšený celkový cholesterol (celkový‑C), LDL‑C, apolipoproteín B (Apo B), triglyceridy (TG) a cholesterol nízkodenzitných lipoproteínov (non-HDL‑C) a zvyšuje cholesterol vysokodenzitných lipoproteínov (HDL‑C) duálnou inhibíciou absorpcie a syntézy cholesterolu.
EzetimibEzetimib inhibuje črevnú absorpciu cholesterolu. Ezetimib je perorálne aktívny a má mechanizmus účinku, ktorý sa líši od iných skupín látok znižujúcich cholesterol (napr. statíny, sekvestranty žlčových kyselín [živice], deriváty kyseliny fibrovej a rastlinné stanoly). Molekulovým cieľom ezetimibu je sterolový transportér Niemann-Pick C1-Like 1 (NPC1L1), ktorý je zodpovedný za intestinálne vychytávanie cholesterolu a fytosterolov.
Ezetimib sa lokalizuje na kefkovitý lem tenkého čreva a inhibuje absorpciu cholesterolu, čo vedie k zníženiu dodávky črevného cholesterolu do pečene; statíny znižujú syntézu cholesterolu v pečeni a spoločne tieto odlišné mechanizmy zabezpečujú komplementárnu redukciu cholesterolu. V 2‑týždňovej klinickej štúdii u 18 pacientov s hypercholesterolémiou ezetimib inhiboval črevnú absorpciu cholesterolu o 54 % v porovnaní s placebom.
Na určenie selektivity ezetimibu na inhibíciu absorpcie cholesterolu sa vykonala séria predklinických štúdií. Ezetimib inhiboval absorpciu [
14C]‑cholesterolu bez účinku na absorpciu triglyceridov, mastných kyselín, žlčových kyselín, progesterónu, etinylestradiolu alebo v tukoch rozpustných vitamínov A a D.
SimvastatínPo perorálnom užití je simvastatín, ktorý je inaktívny laktón, hydrolyzovaný v pečeni na príslušnú aktívnu formu β‑hydroxykyseliny, ktorá má silnú inhibičnú aktivitu na HMG‑CoA reduktázu (3 hydroxy – 3 metylglutaryl CoA reduktázu). Tento enzým katalyzuje premenu HMG‑CoA na mevalonát, počiatočný a rýchlosť limitujúci krok v biosyntéze cholesterolu.
Zistilo sa, že simvastatín znižuje normálne, aj zvýšené koncentrácie LDL‑C. LDL je tvorený z veľmi nízkodenzitného proteínu (VLDL) a je katabolizovaný prevažne vysoko afinitným LDL receptorom. Mechanizmus LDL-znižujúceho účinku simvastatínu môže zahŕňať zníženie koncentrácie VLDL‑cholesterolu (VLDL‑C) aj indukciu LDL receptoru, čo vedie k zníženej tvorbe a zvýšenému katabolizmu LDL‑C. Počas liečby simvastatínom značne klesá aj apolipoproteín B. Okrem toho simvastatín mierne zvyšuje HDL‑C a znižuje plazmatické TG. Výsledkom týchto zmien je zníženie pomerov celkového cholesterolu k HDL‑C a LDL‑C k HDL‑C.
Klinická účinnosť a bezpečnosťV kontrolovaných klinických štúdiách tablety ezetimibu/simvastatínu signifikantne znížili celkový cholesterol, LDL‑C, Apo B, TG a non-HDL‑C a zvýšil HDL‑C u pacientov s hypercholesterolémiou.
Prevencia kardiovaskulárnych príhodPreukázalo sa, že ezetimib/simvastatín znižuje veľké vaskulárne príhody u pacientov s koronárnou chorobou srdca a príhodou AKS v anamnéze.
Skúšanie IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) bolo multicentrickou, randomizovanou, dvojito zaslepenou štúdiou s aktívnym komparátorom, zahŕňajúcou 18 144 pacientov zaradených v priebehu 10 dní od hospitalizácie z dôvodu akútneho koronárneho syndrómu (AKS, buď akútny infarkt myokardu [IM] alebo nestabilná angína pektoris [NAP]). Pacienti mali v čase AKS LDL‑C ≤ 125 mg/dl (≤ 3,2 mmol/l), ak neužívali liečbu na zníženie tukov alebo ≤ 100 mg/dl (≤ 2,6 mmol/l), ak užívali liečbu na zníženie tukov. Všetci pacienti boli randomizovaní v pomere 1:1 na užívanie ezetimibu/simvastatínu v dávke 10 mg/40 mg (n = 9 067) alebo simvastatínu 40 mg (n = 9 077) a boli sledovaní počas mediánu 6,0 rokov.
Priemerný vek pacientov bol 63,6 rokov; 76 % bolo mužov, 84 % belochov a 27 % diabetikov. Priemerná hodnota LDL‑C v čase príhody spĺňajúcej podmienky na zaradenie do štúdie bola 80 mg/dl (2,1 mmol/l) u pacientov užívajúcich liečbu na zníženie tukov (n = 6 390) a 101 mg/dl (2,6 mmol/l) u pacientov, ktorí v minulosti neužívali liečbu na zníženie tukov (n = 11 594). Pred hospitalizáciou z dôvodu príhody AKS spĺňajúcej podmienky na zaradenie do štúdie bolo 34 % pacientov na liečbe statínom. U pacientov pokračujúcich v liečbe bola po jednom roku priemerná LDL-C 53,2 mg/dl (1,4 mmol/l) v skupine užívajúcej ezetimib/simvastatín a 69,9 mg/dl (1,8 mmol/l) v skupine užívajúcej simvastatín v monoterapii. Hodnoty lipidov sa vo všeobecnosti určovali pre pacientov, ktorí pokračovali v skúšanej liečbe.
Primárny cieľový ukazovateľ bol zložený z kardiovaskulárneho úmrtia, veľkých koronárnych príhod (MCE [major coronary events], definované ako nefatálny infarkt myokardu, dokumentovaná nestabilná angína pektoris vyžadujúca hospitalizáciu alebo akýkoľvek koronárny revaskularizačný výkon, ktorý sa uskutočnil aspoň 30 dní po randomizovanom priradení liečby) a nefatálnej cievnej mozgovej príhody. Štúdia preukázala, že liečba ezetimibom/simvastatínom poskytla dodatočný prínos pri znižovaní primárneho cieľového ukazovateľa zloženého z kardiovaskulárneho úmrtia, MCE a nefatálnej cievnej mozgovej príhody v porovnaní so samotným simvastatínom (zníženie relatívneho rizika 6,4 %, p = 0,016). Primárny cieľový ukazovateľ sa vyskytol u 2 572 z 9 067 pacientov (7-ročná Kaplanova-Meierova [KM] hodnota 32,72 %) v skupine užívajúcej ezetimib/simvastatín a u 2 742 z 9 077 pacientov (7-ročná KM hodnota 34,67 %) v skupine užívajúcej samotný simvastatín. (Pozri obrázok 1 a tabuľku 1). Celková úmrtnosť sa v tejto vysoko rizikovej skupine nezmenila (pozri tabuľku 1).
Pre všetky typy cievnej mozgovej príhody sa pozoroval celkový prínos, avšak v skupine užívajúcej ezetimib so simvastatínom sa pozoroval nesignifikantný nárast hemoragickej cievnej mozgovej príhody v porovnaní so skupinou so samotným simvastatínom (pozri tabuľku 1). Riziko hemoragickej cievnej mozgovej príhody pri podávaní ezetimibu súbežne s účinnejšími statínmi sa v dlhodobých štúdiách nehodnotilo.
Účinok liečby ezetimibom/simvastatínom bol vo všeobecnosti zhodný s celkovými výsledkami medzi mnohými podskupinami zahŕňajúcimi pohlavie, vek, rasu, diabetes mellitus v anamnéze, východiskové hladiny lipidov, predchádzajúcu liečbu statínom, predchádzajúcu cievnu mozgovú príhodu a hypertenziu.
Obrázok 1: Účinok ezetimibu/simvastatínu na primárny zložený cieľový ukazovateľ kardiovaskulárneho úmrtia, veľkej koronárnej príhody alebo nefatálnej cievnej mozgovej príhody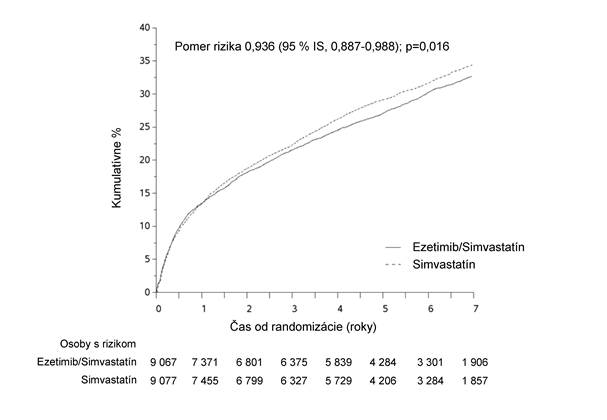 Tabuľka 1Veľké kardiovaskulárne príhody podľa liečebnej skupiny u všetkých randomizovaných pacientov v štúdii IMPROVE-IT
Tabuľka 1Veľké kardiovaskulárne príhody podľa liečebnej skupiny u všetkých randomizovaných pacientov v štúdii IMPROVE-ITVýsledok
| Ezetimib/ simvastatín 10/40 mga
(N = 9 067)
| Simvastatín 40 mgb
(N = 9 077)
| Pomer rizika
(95 % IS)
| Hodnota p
|
| n
| K-M %c
| n
| K-M % c
|
|
|
Primárny zložený cieľový ukazovateľ účinnosti
|
(KV úmrtie, veľké koronárne príhody a nefatálna cievna mozgová príhoda)
| 2 572
| 32,72 %
| 2 742
| 34,67 %
| 0,936 (0,887; 0,988)
| 0,016
|
Sekundárny zložený cieľový ukazovateľ účinnosti
|
Úmrtie z dôvodu KCHS, nefatálny IM, naliehavá koronárna revaskularizácia po 30 dňoch
| 1 322
| 17,52 %
| 1 448
| 18,88 %
| 0,912 (0,847; 0,983)
| 0,016
|
MCE, nefatálna cievna mozgová príhoda, úmrtie (všetky príčiny)
| 3 089
| 38,65 %
| 3 246
| 40,25 %
| 0,948 (0,903; 0,996)
| 0,035
|
KV úmrtie, nefatálny IM, nestabilná angína pektoris vyžadujúca hospitalizáciu, akákoľvek revaskularizácia, nefatálna cievna mozgová príhoda/
| 2 716
| 34,49 %
| 2 869
| 36,20 %
| 0,945 (0,897; 0,996)
| 0,035
|
Zložky primárneho zloženého cieľového ukazovateľa a vybrané cieľové ukazovatele účinnosti (prvý výskyt špecifikovanej príhody v akomkoľvek čase)
|
Kardiovaskulárne úmrtie
| 537
| 6,89 %
| 538
| 6,84 %
| 1,000 (0,887; 1,127)
| 0,997
|
Veľká koronárna príhoda:
|
|
|
|
|
|
|
Nefatálny IM
| 945
| 12,77 %
| 1 083
| 14,41 %
| 0,871 (0,798; 0,950)
| 0,002
|
Nestabilná angína pektoris vyžadujúca hospitalizáciu
| 156
| 2,06 %
| 148
| 1,92 %
| 1,059 (0,846; 1,326)
| 0,618
|
Koronárna revaskularizácia po 30 dňoch
| 1 690
| 21,84 %
| 1 793
| 23,36 %
| 0,947 (0,886; 1,012)
| 0,107
|
Nefatálna cievna mozgová príhoda
| 245
| 3,49 %
| 305
| 4,24 %
| 0,802 (0,678; 0,949)
| 0,010
|
IM (fatálny a nefatálny)
| 977
| 13,13 %
| 1 118
| 14,82 %
| 0,872 (0,800; 0,950)
| 0,002
|
Cievna mozgová príhoda (fatálna a nefatálna)
| 296
| 4,16 %
| 345
| 4,77 %
| 0,857 (0,734; 1,001)
| 0,052
|
Nehemoragická cievna mozgová príhodad
| 242
| 3,48 %
| 305
| 4,23 %
| 0,793 (0,670; 0,939)
| 0,007
|
Hemoragická cievna mozgová príhoda
| 59
| 0,77 %
| 43
| 0,59 %
| 1,377 (0,930; 2,040)
| 0,110
|
Úmrtie z akejkoľvek príčiny
| 1 215
| 15,36 %
| 1 231
| 15,28 %
| 0,989 (0,914; 1,070)
| 0,782
|
6 % bolo vytitrovaných na ezetimib/simvastatín 10/80 mg.
b 27 % bolo vytitrovaných na simvastatín 80 mg.
c Odhad v 7. roku podľa Kaplana-Meiera.
d Zahŕňa ischemickú cievnu mozgovú príhodu alebo cievnu mozgovú príhodu neurčeného typu.
Primárna hypercholesterolémiaV dvojito zaslepenej, placebom kontrolovanej, 8-týždňovej štúdii bolo 240 pacientov s hypercholesterolémiou, ktorí už dostávali monoterapiu simvastatínom a podľa National Cholesterol Education Program (NCEP) nedosiahli cieľový LDL‑C (2,6 až 4,1 mmol/l [100 až 160 mg/dl] v závislosti od vstupných hodnôt), randomizovaných tak, aby k prebiehajúcej liečbe simvastatínom dostávali buď ezetimib 10 mg alebo placebo. Medzi pacientmi liečenými simvastatínom, ktorí pri vstupe do štúdie nemali cieľovú hladinu LDL‑C (~80%), signifikantne viac pacientov randomizovaných na ezetimib podávaný súbežne so simvastatínom dosiahlo cieľovú hladinu LDL‑C na konci štúdie v porovnaní s pacientmi randomizovanými na placebo podávané súbežne so simvastatínom, 76 % a 21,5 % v uvedenom poradí.
Príslušné zníženia LDL‑C pri ezetimibe alebo placebe podávaných súbežne so simvastatínom boli tiež signifikantne rozdielne (27 % alebo 3 %, v uvedenom poradí). Okrem toho ezetimib podávaný súbežne so simvastatínom signifikantne znížil celkový‑C, Apo B a TG v porovnaní s placebom podávaným súbežne so simvastatínom.
V multicentrickom, dvojito zaslepenom, 24‑týždňovom skúšaní bolo 214 pacientov s diabetes mellitus typu 2 liečených tiazolidíndiónmi (rosiglitazón alebo pioglitazón) minimálne 3 mesiace a simvastatínom 20 mg minimálne 6 týždňov s priemerným LDL‑C 2,4 mmol/l (93 mg/dl) randomizovaných tak, aby dostávali buď simvastatín 40 mg alebo súbežne podávané liečivá ekvivalentné kombinácii ezetimib/simvastatín 10 mg/20 mg. Ezetimib/simvastatín 10 mg/20 mg bol signifikantne účinnejší ako zdvojnásobenie dávky simvastatínu na 40 mg v ďalšom znižovaní LDL‑C (-21 % oproti 0 %), celkového‑C (-14 % oproti -1 %), Apo B (-14 % oproti -2 %) a non-HDL‑C (-20 % oproti -2 %) nad zníženia pozorované pri simvastatíne 20 mg. Výsledné hodnoty HDL‑C a TG medzi týmito dvomi terapeutickými skupinami sa signifikantne nelíšili. Výsledky neboli ovplyvnené typom tiazolidíndiónovej liečby.'
Účinnosť jednotlivých dávkových síl kombinácie ezetimib/simvastatín (10/10 až 10/80 mg/deň) bola preukázaná v multicentrickom, dvojito zaslepenom, placebom kontrolovanom 12-týždňovom skúšaní, ktoré zahŕňalo všetky dostupné dávky kombinácie ezetimib/simvastatín a všetky relevantné dávky simvastatínu. Pri porovnaní pacientov dostávajúcich všetky dávky kombinácie ezetimib/simvastatín s pacientmi dostávajúcimi všetky dávky simvastatínu sa zistilo, že ezetimib/simvastatín signifikantne znížil celkový‑C, LDL‑C a TG (pozri tabuľku 2) ako aj Apo B (-42 % oproti -29 %), non-HDL‑C (-49 % oproti -34 %) a C-reaktívny proteín (-33 % oproti -9 %). Účinky ezetimibu/simvastatínu na HDL‑C boli podobné ako účinky pozorované pri simvastatíne. Ďalšia analýza ukázala, že ezetimib/simvastatín signifikantne zvýšil HDL‑C v porovnaní s placebom.
Tabuľka 2Odpoveď na ezetimib/simvastatín u pacientov s primárnou hypercholesterolémiou(Priemernáa % zmena oproti východiskovej hodnote bez liečbyb)Liečba
|
|
|
|
|
|
(denná dávka)
| N
| Celkový‑C
| LDL‑C
| HDL‑C
| TGa
|
Súhrnná analýza (ezetimib/simvastatín všetky dávky)c
| 353
| -38
| -53
| +8
| -28
|
Súhrnná analýza (simvastatín všetky dávky)c
| 349
| -26
| -38
| +8
| -15
|
Ezetimib 10 mg
| 92
| -14
| -20
| +7
| -13
|
Placebo
| 93
| +2
| +3
| +2
| -2
|
ezetimib/simvastatín v dávke
|
|
|
|
|
|
10/10
| 87
| -32
| -46
| +9
| -21
|
10/20
| 86
| -37
| -51
| +8
| -31
|
10/40
| 89
| -39
| -55
| +9
| -32
|
10/80
| 91
| -43
| -61
| +6
| -28
|
Simvastatín v dávke
|
|
|
|
|
|
10 mg
| 81
| -21
| -31
| +5
| -4
|
20 mg
| 90
| -24
| -35
| +6
| -14
|
40 mg
| 91
| -29
| -42
| +8
| -19
|
80 mg
| 87
| -32
| -46
| +11
| -26
|
Pri triglyceridoch medián % zmeny oproti východiskovej hodnote
b Východisková hodnota – bez liečby liekom znižujúcim lipidy
c ezetimib/simvastatín združené dávky (10/10 – 10/80) signifikantne znížili celkový‑C, LDL‑C a TG v porovnaní so simvastatínom a signifikantne zvýšili HDL‑C v porovnaní s placebom.
V podobne navrhnutej štúdii sa výsledky pre všetky lipidové parametre vo všeobecnosti zhodovali. V združenej analýze týchto dvoch štúdií bola odpoveď lipidov na ezetimib/simvastatín tablety podobná u pacientov s hladinami TG väčšími alebo menšími než 200 mg/dl.
V multicentrickej, dvojito zaslepenej, kontrolovanej klinickej štúdii (ENHANCE) bolo 720 pacientov s heterozygotnou familiárnou hypercholesterolémiou randomizovaných na ezetimib 10 mg v kombinácii so simvastatínom 80 mg (n = 357) alebo na simvastatín 80 mg (n = 363) počas 2 rokov. Primárnym cieľom štúdie bolo preskúmať účinok kombinovanej liečby ezetimibom/simvastatínom na hrúbku vrstvy intima-média (intima-media thickness, IMT) krčnej tepny v porovnaní s monoterapiou simvastatínom. Vplyv tohto zástupného markera na kardiovaskulárnu morbiditu a mortalitu nie je stále preukázaný.
Primárny cieľový ukazovateľ, zmena strednej hodnoty IMT všetkých šiestich segmentov krčnej tepny meraná ultrazvukom v B móde, sa medzi dvoma liečebnými skupinami signifikantne nelíšil (p = 0,29). Počas 2-ročného trvania štúdie sa hrúbka vrstvy intima-média zväčšila o 0,0111 mm pri ezetimibe 10 mg v kombinácii so simvastatínom 80 mg (východisková stredná hodnota karotickej IMT 0,68 mm) a o 0,0058 mm pri samotnom simvastatíne 80 mg (východisková stredná hodnota karotickej IMT 0,69 mm).
Ezetimib 10 mg v kombinácii so simvastatínom 80 mg znížil LDL-C, celkový-C, Apo B a TG signifikantne viac ako simvastatín 80 mg. Percentuálne zvýšenie HDL-C bolo podobné pre obe liečebné skupiny. Nežiaduce reakcie hlásené pre ezetimib 10 mg v kombinácii so simvastatínom 80 mg boli zhodné s jeho známym bezpečnostným profilom.
Ezetimib/simvastatín Teva B.V. obsahuje simvastatín. V dvoch veľkých placebom kontrolovaných klinických skúšaniach, Scandinavian Simvastatin Survival Study (20 - 40 mg, N = 4 444 pacientov) a Heart Protection Study (40 mg, N = 20 536 pacientov), sa hodnotili účinky liečby simvastatínom u pacientov s vysokým rizikom koronárnych príhod v dôsledku existujúcej koronárnej choroby srdca, diabetu, ochorenia periférnych ciev, cievnej mozgovej príhody alebo iného cerebrovaskulárneho ochorenia v anamnéze. Dokázalo sa, že simvastatín znižuje: riziko celkovej mortality znížením úmrtí v dôsledku KCHS, riziko nefatálneho infarktu myokardu a cievnej mozgovej príhody a potrebu koronárnych a nekoronárnych revaskularizačných výkonov.
Štúdia účinnosti ďalších znížení cholesterolu a homocysteínu (The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH)) hodnotila účinok liečby simvastatínom 80 mg oproti 20 mg (medián sledovania 6,7 rokov) na veľké vaskulárne príhody (MVE, definované ako fatálna KCHS, nefatálny IM, výkon koronárnej revaskularizácie, nefatálna alebo fatálna cievna mozgová príhoda, alebo výkon periférnej revaskularizácie) u 12 064 pacientov s infarktom myokardu v anamnéze. V incidencii veľkých vaskulárnych príhod nebol medzi 2 skupinami signifikantný rozdiel; simvastatín 20 mg (n = 1 553; 25,7 %) oproti simvastatínu 80 mg (n = 1 477; 24,5 %), RR 0,94, 95 % IS: 0,88 až 1,01. Absolútny rozdiel v LDL-C medzi dvoma skupinami počas trvania štúdie bol 0,35 ± 0,01 mmol/l. Bezpečnostné profily boli medzi dvoma liečebnými skupinami podobné, okrem toho, že incidencia myopatie bola u pacientov liečených simvastatínom 80 mg približne 1,0 % v porovnaní s 0,02 % u pacientov s 20 mg. Približne polovica z týchto prípadov myopatie sa vyskytla počas prvého roku liečby. Incidencia myopatie počas každého nasledujúceho roku liečby bola približne 0,1 %.
Pediatrická populáciaV multicentrickej dvojito zaslepenej kontrolovanej štúdii bolo 142 chlapcov (II. Tannerovo štádium a vyššie) a 106 dievčat jeden rok po menarché vo veku 10 až 17 rokov (priemerný vek 14,2 roka) s heterozygotnou familiárnou hypercholesterolémiou (HeFH) s východiskovými hladinami LDL-C v rozmedzí 4,1 a 10,4 mmol/l randomizovaných buď do skupiny s ezetimibom 10 mg súbežne podávaným so simvastatínom (10, 20 alebo 40 mg) alebo do skupiny so samotným simvastatínom (10, 20 alebo 40 mg) počas 6 týždňov, do skupiny s ezetimibom súbežne podávaným so simvastatínom 40 mg alebo do skupiny so samotným simvastatínom 40 mg počas nasledujúcich 27 týždňov a následne počas 20 týždňov otvoreného usporiadania sa súbežne podával ezetimib a simvastatín (10 mg, 20 mg alebo 40 mg).
Ezetimib súbežne podávaný so simvastatínom (všetky sily) v 6. týždni signifikantne znížil hladinu celkového cholesterolu (38 % oproti 26 %), LDL-C (49 % oproti 34 %), Apo B (39 % oproti 27 %) a non-HDL-C (47 % oproti 33 %) v porovnaní so samotným simvastatínom (všetky sily). Výsledky pre TG a HDL-C boli pre dve liečebné skupiny podobné (‑17 % oproti ‑12 % pre TG a +7 % oproti +6 % pre HDL-C). V 33. týždni boli výsledky zhodné s výsledkami v 6. týždni a signifikantne viac pacientov užívajúcich ezetimib a 40 mg simvastatínu (62 %) dosiahlo ideálne cieľové hodnoty podľa NCEP AAP (< 2,8 mmol/l[110 mg/dl] pre LDL-C v porovnaní s pacientmi, ktorí užívali 40 mg simvastatínu (25 %). V 53. týždni, na konci odslepeného predĺženia, boli účinky na parametre lipidov zachované.
Bezpečnosť a účinnosť ezetimibu súbežne podávaného so simvastatínom v dávkach nad 40 mg denne neboli študované u pediatrických pacientov vo veku 10 až 17 rokov. Dlhodobá účinnosť liečby ezetimibom na morbiditu a mortalitu v dospelosti nebola u pacientov mladších ako 17 rokov študovaná.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Ezetimib/simvastatín Teva B.V. vo všetkých podskupinách pediatrickej populácie v liečbe hypercholesterolémie (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Homozygotná familiárna hypercholesterolémia (HoFH)Dvojito zaslepená, randomizovaná, 12-týždňová štúdia sa uskutočnila u pacientov s klinickou a/alebo genotypovou diagnózou HoFH. Analyzovali sa údaje podskupiny pacientov (n = 14), ktorí dostávali pri vstupe do štúdie simvastatín 40 mg. Zvýšenie dávky simvastatínu zo 40 na 80 mg (n = 5) viedlo k zníženiu LDL‑C o 13 % oproti východiskovej hodnote pri užívaní simvastatínu 40 mg. Súbežné podanie ezetimibu a simvastatínu ekvivalentné ku kombinácii ezetimib/simvastatín (10 mg/40 mg a 10 mg/80 mg združené, n = 9) viedlo k zníženiu LDL‑C o 23 % oproti východiskovej hodnote pri užívaní simvastatínu 40 mg. U pacientov, ktorým bol podávaný súbežne ezetimib a simvastatín ekvivalentný ku kombinácii ezetimib/simvastatín (10 mg/80 mg, n = 5), došlo k redukcii LDL‑C o 29 % oproti východiskovej hodnote pri užívaní simvastatínu 40 mg.
Prevencia veľkých vaskulárnych príhod pri chronickom ochorení obličiek (CKD)Štúdia Study of Heart and Renal Protection (SHARP) bola mnohonárodná, randomizovaná, placebom kontrolovaná, dvojito zaslepená štúdia vykonaná u 9 438 pacientov s chronickým ochorením obličiek, z ktorých jedna tretina podstupovala pri zaradení do štúdie dialýzu. Celkovo bolo 4 650 pacientov randomizovaných na užívanie ezetimibu/simvastatínu 10 mg/20 mg a 4 620 na placebo a pacienti sa sledovali počas mediánu 4,9 roka. Priemerný vek pacientov bol 62 rokov a 63 % boli muži, 72 % boli belosi, 23 % boli diabetici a u tých pacientov, ktorí nepodstupovali dialýzu, bola priemerná odhadovaná rýchlosť glomerulárnej filtrácie (eGFR) 26,5 ml/min/1,73 m
2. Neboli udané žiadne vstupné kritéria pre hladiny lipidov. Priemerná východisková hladina LDL‑C bola 108 mg/dl. V porovnaní s placebom sa po jednom roku znížila hladina LDL‑C samotným simvastatínom 20 mg o 26 % a ezetimibom/simvastatínom 10 mg/20 mg o 38 % aj u pacientov, ktorí už viac neužívali skúšaný liek.
Primárnym porovnaním, ktoré bolo špecifikované protokolom štúdie SHARP, bola analýza podľa liečebného zámeru
(intention to treat analysis) „veľkých vaskulárnych príhod“ (MVE, definované ako nefatálny infarkt myokardu alebo kardiálne úmrtie, cievna mozgová príhoda alebo akýkoľvek revaskularizačný výkon) len u tých pacientov, ktorí boli na začiatku randomizovaní do skupiny s ezetimibom/simvastatínom (n = 4 193) alebo do skupiny s placebom (n = 4 191). Sekundárne analýzy zahŕňali rovnaký súbor analyzovaný v celej kohorte randomizovanej (na začiatku štúdie alebo v 1. roku) na ezetimib/simvastatín (n = 4 650) alebo na placebo (n = 4 620), ako aj zložky tohto súboru.
Analýza primárneho cieľového ukazovateľa preukázala, že ezetimib/simvastatín signifikantne znížil riziko veľkých vaskulárnych príhod (749 pacientov s príhodami v skupine s placebom oproti 693 v skupine s ezetimibom/simvastatínom) s relatívnym znížením rizika o 16 % (p = 0,001).
Tento dizajn štúdie však neumožnil sledovať separátny podiel samotného ezetimibu na účinnosti vedúcej k signifikantnému zníženiu rizika veľkých vaskulárnych príhod u pacientov s CKD.
Jednotlivé zložky MVE u všetkých randomizovaných pacientov sú uvedené v tabuľke 2. Kombinácia ezetimib/simvastatín signifikantne znížila riziko cievnej mozgovej príhody a akejkoľvek revaskularizácie, s nesignifikantnými číselnými rozdielmi v prospech ezetimibu/simvastatínu pri nefatálnom infarkte myokardu a kardiálnom úmrtí.
Tabuľka 3Veľké vaskulárne príhody podľa liečebnej skupiny u všetkých randomizovaných pacientov v štúdii SHARPa
Výsledok
| Ezetimib/
simvastatín 10/20
(N = 4 650)
| Placebo
(N = 4 620)
| Pomer rizika
(95 % IS)
| Hodnota p
|
Veľké vaskulárne príhody
| 701 (15,1 %)
| 814 (17,6 %)
| 0,85 (0,77-0,94)
| 0,001
|
Nefatálny infarkt myokardu
| 134 (2,9 %)
| 159 (3,4 %)
| 0,84 (0,66-1,05)
| 0,12
|
Kardiálne úmrtie
| 253 (5,4 %)
| 272 (5,9 %)
| 0,93 (0,78-1,10)
| 0,38
|
Akákoľvek cievna mozgová príhoda
| 171 (3,7 %)
| 210 (4,5 %)
| 0,81 (0,66-0,99)
| 0,038
|
Nehemoragická cievna mozgová príhoda
| 131 (2,8 %)
| 174 (3,8 %)
| 0,75 (0,60-0,94)
| 0,011
|
Hemoragická cievna mozgová príhoda
| 45 (1,0 %)
| 37 (0,8 %)
| 1,21 (0,78-1,86)
| 0,40
|
Akákoľvek revaskularizácia
| 284 (6,1 %)
| 352 (7,6 %)
| 0,79 (0,68-0,93)
| 0,004
|
Veľké aterosklerotické príhody (MAE)b
| 526 (11,3 %)
| 619 (13,4 %)
| 0,83 (0,74-0,94)
| 0,002
|
a Analýza podľa liečebného zámeru (intention to treat analysis) u všetkých pacientov v štúdii SHARP randomizovaných na ezetimib/simvastatín alebo placebo buď na začiatku alebo po 1. roku.
b MAE; definované ako súbor nefatálneho infarktu myokardu, koronárneho úmrtia, nehemoragickej cievnej mozgovej príhody alebo akejkoľvek revaskularizácie.
|
Absolútne zníženie LDL cholesterolu dosiahnuté pri ezetimibe/simvastatíne medzi pacientmi s nižšou východiskovou hladinou LDL‑C (< 2,5 mmol/l) a pacientmi podstupujúcimi pri vstupe do štúdie dialýzu bolo nižšie ako u ostatných pacientov a zodpovedajúce zníženia rizika v týchto dvoch skupinách boli zmenšené.
Aortálna stenózaMulticentrická, dvojito zaslepená, placebom kontrolovaná štúdia „Simvastatín a ezetimib na liečbu aortálnej stenózy“ (Simvastatin and Ezetimibe for the Treatment of Aortic Stenosis, SEAS) s mediánom trvania 4,4 roka sa uskutočnila u 1 873 pacientov s asymptomatickou aortálnou stenózou (AS) zdokumentovanou pomocou Dopplerovho merania maximálnej rýchlosti prúdenia aortou v rozmedzí 2,5 až 4,0 m/s. Do štúdie boli zaradení len tí pacienti, u ktorých sa zvážilo, že nie je potrebná liečba statínmi za účelom zníženia rizika aterosklerotického kardiovaskulárneho ochorenia. Pacienti boli randomizovaní v pomere 1:1 a dostávali placebo alebo im bol denne súbežne podávaný ezetimib 10 mg a simvastatín 40 mg.
Primárnym cieľovým ukazovateľom bol súbor veľkých kardiovaskulárnych udalostí (major cardiovascular events, MCE) pozostávajúci z kardiovaskulárneho úmrtia, chirurgickej náhrady aortálnej chlopne (aortic valve replacement, AVR), kongestívneho srdcového zlyhania (congestive heart failure, CHF) v dôsledku progresie AS, nefatálneho infarktu myokardu, bypassu koronárnej artérie (coronary artery bypass grafting, CABG), perkutánnej koronárnej intervencie (percutaneous coronary intervention, PCI), hospitalizácie pre nestabilnú angínu pektoris, a nehemoragickej cievnej mozgovej príhody. Sekundárnymi cieľovými ukazovateľmi boli súbory podskupín kategórií udalostí primárneho cieľového ukazovateľa.
Ezetimib/simvastatín 10 mg/40 mg v porovnaní s placebom signifikantne neznížili riziko MCE.
Primárny výsledok sa vyskytol u 333 pacientov (35,3 %) v skupine ezetimib/simvastatín a u 355 pacientov (38,2 %) v skupine s placebom (pomer rizika v skupine ezetimib/simvastatín 0,96; 95 % interval spoľahlivosti 0,83 až 1,12; p = 0,59). Náhrada aortálnej chlopne sa uskutočnila u 267 pacientov (28,3 %) v skupine ezetimib/simvastatín a u 278 pacientov (29,9 %) v skupine s placebom (pomer rizika 1,00; 95 % IS 0,84 až 1,18; p = 0,97). V skupine ezetimib/simvastatín (n = 148) malo menej pacientov ischemické kardiovaskulárne príhody ako v skupine s placebom (n = 187) (pomer rizika 0,78; 95 % IS 0,63 až 0,97; p = 0,02) predovšetkým kvôli menšiemu počtu pacientov, ktorí podstúpili bypass koronárnej artérie.
V skupine užívajúcej ezetimib/simvastatín sa častejšie vyskytovala rakovina (105 oproti 70, p = 0,01). Klinický význam tohto zistenia nie je jasný, pretože vo väčšom skúšaní SHARP sa celkový počet pacientov s akýmkoľvek výskytom rakoviny (438 pri ezetimibe/simvastatíne oproti 439 v skupine s placebom) nelíšil. Okrem toho, v skúšaní IMPROVE-IT sa celkový počet pacientov s akoukoľvek novou malignitou (853 v skupine užívajúcej ezetimib/simvastatín oproti 863 v skupine so simvastatínom) signifikantne nelíšil a preto zistenia zo skúšania SEAS nie je možné potvrdiť skúšaním SHARP alebo IMPROVE-IT.
5.2 Farmakokinetické vlastnostiPri súbežnom podávaní ezetimibu so simvastatínom neboli pozorované žiadne klinicky významné farmakokinetické interakcie.
AbsorpciaEzetimib/simvastatín Teva B.V.Ezetimib/simvastatín Teva B.V. je bioekvivalentný so súbežne podávaným ezetimibom a simvastatínom.
EzetimibPo perorálnom podaní je ezetimib rýchlo absorbovaný a extenzívne konjugovaný na farmakologicky aktívny fenolový glukuronid (ezetimib‑glukuronid). Priemerné maximálne plazmatické koncentrácie (C
max) ezetimib‑glukuronidu sa dosiahnu za 1 až 2 hodiny a ezetimibu za 4 až 12 hodín. Absolútnu biologickú dostupnosť ezetimibu nemožno stanoviť, pretože zlúčenina je prakticky nerozpustná vo vodných médiách vhodných na injekciu.
Súbežné podanie jedla (jedál s vysokým obsahom tukov alebo bez tuku) nemalo žiadny vplyv na perorálnu biologickú dostupnosť ezetimibu podávaného vo forme 10‑mg tabliet.
SimvastatínZistilo sa, že dostupnosť aktívnej β‑hydroxykyseliny v systémovej cirkulácii po perorálnej dávke simvastatínu bola menej ako 5 % dávky, čo zodpovedá rozsiahlej extrakcii pri prvom prechode pečeňou. Hlavné metabolity simvastatínu prítomné v ľudskej plazme sú β‑hydroxykyselina a štyri ďalšie aktívne metabolity.
Čo sa týka stavu nalačno, plazmatické profily ani aktívnych, ani celkových inhibítorov neboli ovplyvnené, keď bol simvastatín podaný bezprostredne pred testovacím jedlom.
DistribúciaEzetimib99,7 % ezetimibu a 88 až 92 % ezetimib‑glukuronidu sa viaže na proteíny ľudskej plazmy.
SimvastatínSimvastatín aj β‑hydroxykyselina sa viažu na proteíny ľudskej plazmy (95 %).
Farmakokinetika jednorazových a opakovaných dávok simvastatínu ukázala, že po opakovanom podaní nedošlo ku kumulácii lieku. Vo všetkých vyššie uvedených fakmakokinetických štúdiách došlo k maximálnej plazmatickej koncentrácii inhibítorov za 1,3 až 2,4 hodín po podaní dávky.
BiotransformáciaEzetimibEzetimib je primárne metabolizovaný v tenkom čreve a v pečeni konjugáciou na glukuronid (reakcia fázy II) s následným vylučovaním do žlče. U všetkých hodnotených druhov sa pozoroval minimálny oxidatívny metabolizmus (reakcia fázy I). Ezetimib a ezetimib-glukuronid sú hlavné od liečiva odvodené látky detegované v plazme, pričom ezetimib tvoril približne 10 až 20 % a ezetimib‑glukuronid približne 80 až 90 % celkového liečiva v plazme. Ezetimib aj ezetimib-glukuronid sú pomaly eliminované z plazmy s dôkazom signifikantnej enterohepatálnej cirkulácie. Biologický polčas ezetimibu a ezetimib‑glukuronidu je približne 22 hodín.
SimvastatínSimvastatín je inaktívny laktón, ktorý sa
in vivo rýchlo hydrolyzuje na príslušnú β‑hydroxykyselinu, silný inhibítor HMG‑CoA reduktázy. Hydrolýza prebieha najmä v pečeni; rýchlosť hydrolýzy v ľudskej plazme je veľmi pomalá.
U človeka sa simvastatín dobre absorbuje a podlieha rozsiahlej extrakcii pri prvom prechode pečeňou. Extrakcia v pečeni závisí od prietoku krvi pečeňou. Pečeň je jeho primárnym miestom účinku s následným vylučovaním ekvivalentov liečiva do žlče. V dôsledku toho je dostupnosť aktívneho liečiva pre systémovú cirkuláciu nízka.
Po intravenóznej injekcii metabolitu β‑hydroxykyseliny bol jeho priemerný biologický polčas 1,9 hodiny.
ElimináciaEzetimibPo perorálnom podaní
14C‑ezetimibu (20 mg) osobám tvoril celkový ezetimib približne 93 % celkovej rádioaktivity v plazme. Počas 10-dňového obdobia zberu sa izolovalo približne 78 % podanej rádioaktivity v stolici a 11 % v moči. Po 48 hodinách neboli v plazme detegovateľné hladiny rádioaktivity.
SimvastatínKyselina simvastatínová je aktívne vychytávaná do hepatocytov transportérom OATP1B1.
Simvastatín je substrátom efluxného transportéra BCRP.
Po perorálnej dávke rádioaktívneho simvastatínu človeku bolo 13 % rádioaktivity vylúčenej v moči a 60 % v stolici v priebehu 96 hodín. Množstvo izolované v stolici predstavuje ekvivalenty absorbovaného liečiva vylúčeného žlčou ako aj neabsorbované liečivo. Po intravenóznom podaní metabolitu β‑hydroxykyseliny bolo v priemere iba 0,3 % i. v. dávky vylúčenej v moči vo forme inhibítorov.
Osobitné skupiny pacientovPediatrická populáciaAbsorpcia a metabolizmus ezetimibu u detí a dospievajúcich (10 až 18 rokov) sú podobné ako u dospelých. Vychádzajúc z celkového ezetimibu, medzi dospievajúcimi a dospelými nie sú žiadne farmakokinetické rozdiely. Farmakokinetické údaje pre pediatrickú populáciu vo veku < 10 rokov nie sú k dispozícii. Klinické skúsenosti u detských a dospievajúcich pacientov zahŕňajú pacientov s HoFH, HeFH alebo sitosterolémiou. (Pozri časť 4.2).
Staršie osobyPlazmatické koncentrácie celkového ezetimibu sú u starších pacientov (≥ 65 rokov) približne 2‑násobne vyššie ako u mladých pacientov (18 až 45 rokov). Zníženie LDL‑C a bezpečnostný profil sú u starších a mladších osôb liečených ezetimibom porovnateľné. (Pozri časť 4.2).
Porucha funkcie pečenePo jednorazovej 10 mg dávke ezetimibu sa priemerná AUC celkového ezetimibu zvýšila približne 1,7‑násobne u pacientov s miernou poruchou funkcie pečene (Childovo-Pughovo skóre 5 alebo 6) v porovnaní so zdravými osobami. V 14‑dňovej štúdii opakovaných dávok (10 mg denne) u pacientov so stredne ťažkou poruchou funkcie pečene (Childovo-Pughovo skóre 7 až 9) bola priemerná AUC celkového ezetimibu zvýšená približne 4‑násobne 1. a 14. deň v porovnaní so zdravými osobami. U pacientov s miernou poruchou funkcie pečene nie je potrebná úprava dávkovania. Vzhľadom na neznáme účinky zvýšenej expozície ezetimibu u pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene (Childovo-Pughovo skóre > 9) sa u týchto pacientov ezetimib neodporúča (pozri časti 4.2 a 4.4).
Porucha funkcie obličiekEzetimibPo jednorazovej 10 mg dávke ezetimibu sa priemerná AUC celkového ezetimibu zvýšila približne 1,5‑násobne u pacientov s ťažkým renálnym ochorením (n = 8; priemerný klírens kreatinínu CrCl ≤ 30 ml/min) v porovnaní so zdravými osobami (n = 9). (Pozri časť 4.2).
Ďalší pacient v tejto štúdii (po transplantácii obličky, ktorý dostával mnohopočetnú liečbu vrátane cyklosporínu) mal 12‑násobne väčšiu expozíciu celkovému ezetimibu.
SimvastatínV štúdii pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) boli plazmatické koncentrácie celkových inhibítorov po jednorazovej dávke príbuzného inhibítora HMG‑CoA reduktázy približne dvojnásobne vyššie ako u zdravých dobrovoľníkov.
PohlaviePlazmatické koncentrácie celkového ezetimibu sú mierme vyššie (približne 20 %) u žien ako u mužov. Zníženie LDL‑C a bezpečnostný profil sú u mužov a žien liečených ezetimibom porovnateľné.
Polymorfizmus SLCO1B1Nositelia alely c.521T > C génu SLCO1B1 majú nižšiu aktivitu OATP1B1. Priemerná expozícia (AUC) hlavného aktívneho metabolitu, kyseliny simvastatínovej, je 120 % u heterozygotných nositeľov alely C (CT) a 221 % u homozygotných nositeľov (CC) v porovnaní s pacientmi, ktorí majú najčastejší genotyp (TT). Frekvencia výskytu alely C v európskej populácii je 18 %. U pacientov s polymorfizmom SLCO1B1 je riziko zvýšenej expozície kyseliny simvastatínovej, čo môže viesť k zvýšenému riziku rabdomyolýzy (pozri časť 4.4).
5.3 Predklinické údaje o bezpečnostiEzetimib/simvastatín V štúdiách súbežného podávania ezetimibu a simvastatínu boli pozorované toxické účinky v podstate tie isté, aké sú typicky spojené so statínmi. Niektoré z toxických účinkov boli výraznejšie ako účinky pozorované počas liečby samotnými statínmi. Toto sa pripisuje farmakokinetickým a/alebo farmakodynamickým interakciám po súbežnom podaní. V klinických štúdiách sa takéto interakcie nevyskytli. Myopatie sa objavili u potkanov až po expozícii dávkam, ktoré boli niekoľkokrát vyššie, ako je terapeutická dávka pre ľudí (približne 20‑násobok hodnoty AUC pre simvastatín a 1 800‑násobok hodnoty AUC pre aktívny metabolit). Nezistilo sa, že by súbežné podávanie ezetimibu ovplyvnilo myotoxický potenciál simvastatínu.
U psov, ktorým sa súbežne podával ezetimib a statíny, sa pri nízkych expozíciách (< 1‑násobok AUC u ľudí) pozorovali niektoré účinky na pečeň. Zistil sa výrazný vzostup pečeňových enzýmov (ALT, AST) bez prítomnosti nekrózy tkaniva. U psov, ktorým sa súbežne podával ezetimib a simvastatín, sa pozorovali histopatologické pečeňové nálezy (hyperplázia žlčovodu, kumulácia pigmentu, infiltrácia mononukleárnymi bunkami a malé hepatocyty). Pri dlhšom podávaní v trvaní do 14 mesiacov tieto zmeny neprogredovali. Po prerušení podávania sa pozorovala celková úprava pečeňových nálezov. Tieto zistenia zodpovedali zisteniam opísaným pri inhibítoroch HMG‑CoA reduktázy alebo sa prisúdili veľmi nízkym hladinám cholesterolu dosiahnutým u ovplyvnených psov.
Súbežné podávanie ezetimibu a simvastatínu nebolo u potkanov teratogénne. U gravidných králikov sa pozoroval malý počet deformít skeletu (spojené kaudálne stavce, znížený počet kaudálnych stavcov).
V sérii
in vivo a
in vitro hodnotení ezetimib podávaný samostatne alebo súbežne so simvastatínom nemal žiadny genotoxický potenciál.
EzetimibŠtúdie chronickej toxicity ezetimibu na zvieratách neidentifikovali žiadne cieľové orgány pre toxické účinky. U psov liečených štyri týždne ezetimibom (≥ 0,03 mg/kg/deň) sa koncentrácia cholesterolu v žlčníkovej žlči zvýšila 2,5 až 3,5-násobne. V jednoročnej štúdii na psoch, ktorým sa podávali dávky do 300 mg/kg/deň, sa však nepozorovalo zvýšenie incidencie cholelitiázy alebo iné hepatobiliárne účinky. Významnosť týchto údajov pre ľudí nie je známa. Riziko litogenity spojené s terapeutickým užívaním ezetimibu nemožno vylúčiť.
Dlhodobé testy karcinogenicity ezetimibu boli negatívne.
Ezetimib nemal žiadny účinok na fertilitu samcov a samíc potkanov, ani sa nezistilo, že by bol teratogénny u potkanov alebo králikov a nemal vplyv ani na prenatálny a postnatálny vývoj. Ezetimib prechádzal cez placentárnu bariéru u gravidných potkanov a králikov, ktorým boli podané mnohonásobné dávky 1 000 mg/kg/deň.
SimvastatínNa základe obvyklých štúdií na zvieratách týkajúcich sa farmakodynamiky, toxicity po opakovanom podaní, genotoxicity a karcinogenicity nie sú žiadne iné riziká pre pacienta, než aké sa dajú očakávať v súvislosti s farmakologickým mechanizmom. Pri maximálne tolerovaných dávkach u potkanov aj králikov simvastatín nespôsobil žiadne fetálne malformácie a nemal žiadne účinky na fertilitu, reprodukčnú funkciu alebo neonatálny vývoj.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokmonohydrát laktózy
hypromelóza
sodná soľ kroskarmalózy
mikrokryštalická celulóza
kyselina askorbová
kyselina citrónová
butylhydroxyanizol
propylgalát
stearan horečnatý
pigmentová zmes, PB-220001 žltá obsahujúca monohydrát laktózy, žltý oxid železitý (E172), červený oxid železitý (E172), čierny oxid železitý (E172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnostiOPA/hliníkové/PVC blistre s hliníkovou fóliou a HDPE fľaše:
2 roky
PVC/Aclar blistre s hliníkovou fóliou:
18 mesiacov
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote neprevyšujúcej 25 °C.
6.5 Druh obalu a obsah baleniaOPA/hliníkové/PVC blistre s hliníkovou fóliou:
Veľkosti balenia: 30, 98 a 100 tabliet.
PVC/Aclar blistre s hliníkovou fóliou:
Veľkosti balenia: 30 a 100 tabliet.
HDPE obal na tablety s polypropylénovým (PP) viečkom:
Veľkosti balenia: 100 a 180 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIITeva B.V.
Swensweg 5
2031 GA Haarlem
Holandsko
8. REGISTRAČNÉ ČÍSLAEzetimib/simvastatín Teva B.V. 10 mg/20 mg: 31/0475/16-S
Ezetimib/simvastatín Teva B.V. 10 mg/40 mg: 31/0476/16-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 14. novembra 2016
Dátum posledného predĺženia registrácie: 01. augusta 2019
10. DÁTUM REVÍZIE TEXTU09/2019