EVRENZO 100 MG FILMOM OBALENÉ TABLETY tbl flm 12x1x100 mg (blis.PVC/Al)

b>1,0 a +1,0 g/dl
Zvýšte dávku
o jednu úroveň
| Žiadna zmena
| Znížte dávku
o jednu úroveň
|
Tabuľka 2. Pravidlá pre úpravu dávky
Z
m
ena hladiny Hb
počas posledných
4 týždňov
1
|
A
ktuálna hladina Hb (g/dl):
|
Menej ako 10,5
|
10,5 – 11,9
|
12,0 – 12,9
|
13,0 alebo viac
|
Z
m
ena hodnoty o menej ako
−
1,0 g/dl
|
Zvýšte dávku
o jednu úroveň
|
Zvýšte dávku
o jednu úroveň
|
Žiadna zmena
|
klesne na úroveň
menej ako
12,0 g/dl
|
Dávka roxadustátu sa nemá upravovať častejšie ako raz za 4 týždne okrem prípadov, keď sa
hladina Hb zvýši o viac ako 2 g/dl kedykoľvek počas 4-týždňového obdobia, v takom prípade sa
musí dávka okamžite znížiť o jednu úroveň.
1Zmena hladiny hemoglobínu (Hb) počas posledných 4 týždňov = (aktuálna hodnota Hb) –
(predchádzajúca hodnota Hb získaná pred 4 týždňami).
Ak je potrebné ďalšie zníženie dávky u pacienta s najnižšou dávkou (20 mg trikrát týždenne),
neznižujte dávku 20 mg prelomením tablety, ale znížte frekvenciu užívania na dvakrát týždenne. Ak je potrebné ďalšie zníženie dávky, frekvenciu užívania je možné ďalej znížiť na jedenkrát týždenne.
Udržiavacia dávkaPo stabilizácii na cieľovú hladinu Hb na úrovni 10 – 12 g/dl má pokračovať pravidelné monitorovanie hladiny Hb a majú sa dodržiavať pravidlá týkajúce sa úpravy dávky (pozri Tabuľku 2).
Pacienti začínajúci dialýzu počas liečby roxadustátomPre pacientov s CKD, ktorí počas liečby roxadustátom začínajú dialýzu, nie je potrebná žiadna špecifická úprava dávky. Postupujte podľa štandardných pravidiel na úpravu dávky (pozri Tabuľku 2).
Súbežná liečba roxadustátom a induktormi alebo inhibítormiPri začiatku alebo ukončovaní súbežnej liečby silnými inhibítormi (napr. gemfibrozil) alebo
induktormi (napr. rifampicín) enzýmu CYP2C8, príp. inhibítormi (napr. probenecid) enzýmu UGT1A9: hladiny Hb sa majú pravidelne monitorovať a majú sa dodržiavať pravidlá týkajúce sa úpravy dávky (pozri Tabuľku 2, pozri aj časti 4.5 a 5.2).
Maximálna odporúčaná dávkaPacienti, ktorí nie sú na dialýze, neprekročia dávku roxadustátu 3 mg/kg telesnej hmotnosti alebo
300 mg trikrát týždenne (podľa toho, ktorá je nižšia).
Pacienti, ktorí sú na dialýze, neprekročia dávku roxadustátu 3 mg/kg telesnej hmotnosti alebo 400 mg
trikrát týždenne (podľa toho, ktorá je nižšia).
Vynechaná dávkaV prípade, že sa vynechá dávka a do nasledujúcej plánovanej dávky zostáva viac ako 1 deň,
vynechaná dávka sa musí užiť čo najskôr. Ak do nasledujúcej plánovanej dávky zostáva 1 deň alebo menej, vynechaná dávka sa musí vynechať a nasledujúca dávka sa musí užiť v nasledujúci plánovaný deň. V každom prípade sa má následne pokračovať v pravidelnom dávkovacom režime.
ŠpecificképopulácieStarší pacientiU starších pacientov nie je potrebná žiadna úprava počiatočnej dávky (pozri časť 5.2).
Pacienti s poruchou funkcie pečeneU pacientov s miernou poruchou funkcie pečene (Childova-Pughova trieda A) nie je potrebná žiadna úprava počiatočnej dávky (pozri časti 4.4 a 5.2).
Pri predpisovaní roxadustátu pacientom so stredne ťažkou poruchou funkcie pečene sa odporúča opatrnosť. Pri začiatku liečby pacientov so stredne ťažkou poruchou funkcie pečene (Childova- Pughova trieda B) sa má počiatočná dávka znížiť o polovicu alebo na úroveň dávky, ktorá sa čo najviac približuje polovičnej počiatočnej dávke. Používanie lieku Evrenzo sa neodporúča u pacientov
s ťažkou poruchou funkcie pečene (Childova-Pughova trieda C), keďže bezpečnosť a účinnosť v tejto populácii neboli vyhodnotené (pozri časti 4.4 a 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť roxadustátu u pediatrických pacientov vo veku menej ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Filmom obalené tablety Evrenzo sa užívajú perorálne s jedlom alebo bez jedla. Tablety sa majú prehltnúť celé a nemajú sa hrýzť, lámať alebo drviť z dôvodu chýbajúcich klinických údajov za týchto podmienok, a jadro tablety, ktoré je citlivé na svetlo, sa má chrániť pred fotodegradáciou..
Tablety sa majú užívať aspoň 1 hodinu po podaní látok viažucich fosfáty (okrem lantánu) alebo iných
liekov obsahujúcich multivalentné katióny, ako napr. vápnik, železo, horčík alebo hliník (pozri časti
4.5 a 5.2).
4.3 Kontraindikácie
Evrenzo je kontraindikovaný v nasledujúcich prípadoch:
• Precitlivenosť na liečivo, arašidy, sóju alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Tretí trimester gravidity (pozri časti 4.4 a 4.6).
• Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Kardiovaskulárne riziko a riziko úmrtia
Celkovo sa odhaduje, že kardiovaskulárne riziko a riziko úmrtia pri liečbe roxadustátom je
porovnateľné s kardiovaskulárnym rizikom a rizikom úmrtia pri liečbe ESA na základe údajov z priameho porovnania oboch terapií (pozri časť 5.1). Keďže riziko pre pacientov s anémiou súvisiacou
s CKD, ktorí nie sú na dialýze, nebolo možné odhadnúť s dostatočnou istotou v porovnaní s placebom,
rozhodnutie liečiť týchto pacientov roxadustátom má byť založené na podobných kritériách, ktoré by sa použili pred liečbou ESA. Taktiež bolo identifikovaných viacero prispievajúcich faktorov, ktoré môžu spôsobovať toto riziko, vrátane nedostatočnej odpovede na liečbu a prechodu stabilných pacientov liečených ESA na dialýze (pozri časti 4.2 a 5.1). V prípade nedostatočnej odpovede na liečbu nemá liečba roxadustátom pokračovať viac ako 24 týždňov od začiatku liečby (pozri časť 4.2). Prechod pacientov na dialýze, ktorí sú inak stabilní na liečbe ESA, sa má zvážiť iba v prípade oprávneného klinického dôvodu (pozri časť 4.2). U stabilných pacientov liečených ESA s anémiou spojenou s CKD, ktorí nie sú na dialýze, nie je možné odhadnúť toto riziko, pretože títo pacienti neboli sledovaní. Rozhodnutie liečiť týchto pacientov roxadustátom má byť založené na zvážení prínosu a rizika pre individuálneho pacienta.
Trombotické vaskulárne príhody
Hlásené riziko trombotických vaskulárnych príhod (TVE, z ang. Trombotic Vascular Event) sa má
pozorne zvážiť v porovnaní s prínosmi získanými liečbou roxadustátom, najmä u pacientov
s existujúcimi rizikovými faktormi TVE vrátane obezity a predchádzajúcej anamnézy TVE (napr. hlboká žilová trombóza [DVT, z ang. Deep Vein Thrombosis] a pľúcna embólia [PE, z ang. Pulmonary Embolism]). Hlboká žilová trombóza bola u pacientov v klinických štúdiách hlásená ako častá a pľúcna embólia bola hlásená ako menej častá. Väčšina udalostí DVT a PE bola závažná.
V klinických štúdiách bola hlásená trombóza v mieste cievneho prístupu (VAT, z ang. Vascular
Access Thrombosis) ako veľmi častá medzi pacienti s CKD na dialýze (pozri časť 4.8).
U pacientov s CKD na dialýze bol výskyt VAT u pacientov liečených roxadustátom najvyšší v prvých
12 týždňoch po začatí liečby, pri hladinách Hb vyšších ako 12 g/dl a pri nastavení zvyšovania hladiny
Hb o viac ako 2 g/dl počas obdobia 4 týždňov. Aby sa zabránilo hladine Hb vyššej ako 12 g/dl
a zvýšeniu hladiny Hb o viac ako 2 g/dl počas 4 týždňov, odporúča sa monitorovanie hladiny Hb a úprava dávky použitím pravidiel na úpravu dávky (pozri Tabuľku 2).
Pacienti s prejavmi a príznakmi TVE sa majú ihneď vyšetriť a liečiť v súlade so štandardmi zdravotnej starostlivosti. Rozhodnutie prerušiť alebo ukončiť liečbu má byť založené na zvážení prínosu a rizika pre individuálneho pacienta.
Záchvaty
U pacientov v klinických štúdiách, ktorí užívali roxadustát, boli hlásené záchvaty ako časté (pozri
časť 4.8). Roxadustát sa má používať opatrne u pacientov s anamnézou záchvatov (kŕčov alebo
záchvatov kŕčov), epilepsie alebo stavov spojených s predispozíciou k aktivite záchvatov, ako napr. infekcie centrálneho nervového systému (CNS). Rozhodnutie prerušiť alebo ukončiť liečbu má byť založené na zvážení prínosu a rizika pre individuálneho pacienta.
Závažnéinfekcie
Najčastejšie hlásenými závažnými infekciami boli pneumónia a infekcie močových ciest. Pacienti s prejavmi a príznakmi infekcie sa majú ihneď vyšetriť a liečiť v súlade so štandardmi zdravotnej starostlivosti.
Sepsa
Sepsa bola jednou z najčastejšie hlásených závažných infekcií a zahŕňala smrteľné udalosti. Pacienti
s prejavmi a príznakmi sepsy (napr. infekcia, ktorá sa šíri po tele s nízkym krvným tlakom
a možnosťou zlyhania orgánov) sa majú ihneď vyšetriť a liečiť v súlade so štandardmi zdravotnej starostlivosti.
Nedostatočnáodpoveďnaliečbu
Nedostatočná odpoveď na liečbu roxadustátom má viesť k hľadaniu príčinných faktorov. Má sa upraviť nedostatok živín. Pridružené infekcie, skryté krvácanie, hemolýza, závažná toxicita spôsobená
hliníkom, základné hematologické ochorenia alebo fibróza kostnej drene môžu mať tiež vplyv na
odpoveď na liečbu erytropoetínom. Má sa zvážiť vyšetrenie počtu retikulocytov ako súčasť hodnotenia. Ak sa vylúčia štandardné príčiny neprítomnosti odpovede a pacient má retikulocytopéniu, má sa zvážiť vyšetrenie kostnej drene. Ak nie je možné zistiť príčinu nedostatočnej odpovede na liečbu, v liečbe liekom Evrenzo sa nemá pokračovať dlhšie ako 24 týždňov.
Poruchafunkciepečene
Pri podávaní roxadustátu pacientom so stredne ťažkou poruchou funkcie pečene (Childova-Pughova trieda B) je potrebná opatrnosť. Používanie lieku Evrenzo sa neodporúča u pacientov s ťažkou
poruchou funkcie pečene (Childova-Pughova trieda C) (pozri časť 5.2).
Gravidita a antikoncepcia
Liečba roxadustátom sa nemá začínať u žien, ktoré plánujú otehotnieť, počas gravidity alebo v prípade diagnostikovania anémie súvisiacej s CKD počas gravidity. V takýchto prípadoch sa má začať
alternatívna liečba, ak je to vhodné. V prípade otehotnenia počas podávania roxadustátu sa má liečba ukončiť a má sa začať alternatívna liečba, ak je to vhodné. Ženy vo fertilnom veku musia počas liečby
a minimálne jeden týždeň po poslednej dávke lieku Evrenzo používať vysokoúčinnú antikoncepciu
(pozri časti 4.3 a 4.6).
Nesprávnepoužitie
Nesprávne použitie môže spôsobiť nadmerné zvýšenie objemu červených krviniek. To môže byť spojené so život ohrozujúcimi komplikáciami kardiovaskulárneho systému.
Pomocné látky
Evrenzo obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek. Evrenzo obsahuje hliníkový lak červene Allura AC (pozri časť 6.1), ktorá môže vyvolať alergické reakcie.
Evrenzo obsahuje stopové množstvo sójového lecitínu. Pacienti, ktorí sú alergickí na arašidy alebo sóju, nesmú používať tento liek.
4.5 Liekové a iné interakcie
Vplyv iných liekov na roxadustát
Látky viažuce fosfáty a iné produkty obsahujúce multivalentné katióny
Súbežné podávanie roxadustátu s látkami viažucimi fosfáty, sevelamérium-karbonátom alebo octanom
vápenatým zdravým účastníkom znížilo AUC roxadustátu o 67 %, resp. 46 % a koncentráciu Cmax
o 66 %, resp. 52 %. Roxadustát môže vytvárať chelát s multivalentnými katiónmi, ako napríklad
v látkach viažucich fosfáty alebo iných produktoch obsahujúcich vápnik, železo, horčík alebo hliník. Nesúbežné podávanie látok viažucich fosfáty (s odstupom aspoň 1 hodiny) nemalo žiadny klinicky významný účinok na expozíciu roxadustátu u pacientov s CKD. Roxadustát sa má užívať aspoň
1 hodinu po podaní látok viažucich fosfáty alebo iných liekov alebo výživových doplnkov obsahujúcich multivalentné katióny (pozri časť 4.2). Toto obmedzenie neplatí pre uhličitan lantnatý, pretože súbežné podávanie roxadustátu a uhličitanu lantnatého nespôsobilo klinicky významnú zmenu expozície roxadustátu v plazme.
Modifikátory aktivity enzýmov CYP2C8 alebo UGT1A9
Roxadustát je substrátom enzýmov CYP2C8 a UGT1A9. Súbežné podávanie roxadustátu
s gemfibrozilom (inhibítor enzýmov CYP2C8 a OATP1B1) alebo probenecidom (inhibítor UGT a OAT1/OAT3) zdravým osobám zvýšilo AUC roxadustátu 2,3-násobne a Cmax 1,4-násobne. Pri začiatku alebo ukončovaní súbežnej liečby gemfibrozilom, probenecidom, inými silnými inhibítormi alebo induktormi enzýmu CYP2C8 alebo inými silnými inhibítormi enzýmu UGT1A9 monitorujte hladiny Hb. Na základe monitorovania hladiny Hb upravte dávku roxadustátu podľa pravidiel pre úpravu dávky (pozri Tabuľku 2).
Vplyv roxadustátu na iné lieky
Substráty OATP1B1 alebo BCRP
Roxadustát je inhibítorom BCRP a OATP1B1. Tieto transportéry hrajú dôležitú úlohu pri absorpcii
a efluxe statínov v črevách a pečeni. Súbežné podávanie dávky 200 mg roxadustátu so simvastatínom zdravým osobám zvýšilo AUC a Cmax simvastatínu 1,8-násobne, resp. 1,9-násobne, a AUC a Cmax kyseliny simvastatínovej (aktívny metabolit simvastatínu) 1,9-násobne, resp. 2,8-násobne. Koncentrácie simvastatínu a kyseliny simvastatínovej sa tiež zvýšili, keď sa simvastatín podal
2 hodiny pred alebo 4 alebo 10 hodín po podaní roxadustátu. Súbežné podávanie 200 mg roxadustátu
s rosuvastatínom zvýšilo AUC a Cmax rosuvastatínu 2,9-násobne, resp. 4,5-násobne. Súbežné podávanie 200 mg roxadustátu s atorvastatínom zvýšilo AUC a Cmax atorvastatínu 2,0-násobne, resp. 1,3-násobne.
Očakávajú sa tiež interakcie s inými statínmi. Pri súbežnom podávaní s roxadustátom zvážte túto interakciu, monitorujte výskyt nežiaducich reakcií súvisiacich so statínmi a potrebu zníženia dávky statínu. Pri rozhodovaní o vhodnej dávke statínu pre konkrétnych pacientov si prečítajte informácie o predpisovaní statínu.
Roxadustát môže zvýšiť plazmatickú expozíciu iných liekov, ktoré sú substrátmi BCRP alebo OATP1B1. Monitorujte možné nežiaduce reakcie súbežne podávaných liekov a primerane upravte dávkovanie.
Roxadustát a ESA
Neodporúča sa kombinovať podávanie roxadustátu a ESA, keďže táto kombinácia nebola študovaná.
4.6 Fertilita, gravidita a laktácia
G
r
avidita,
ženy
v
o
fertilnom
veku
a antikoncepcia
Nie sú k dispozícii žiadne údaje o použití roxadustátu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
Roxadustát je kontraindikovaný počas tretieho trimestra gravidity (pozri časti 4.3 a 4.4). Roxadustát sa neodporúča užívať počas prvého a druhého trimestra gravidity (pozri časť 4.4).
V prípade otehotnenia počas podávania lieku Evrenzo sa má liečba ukončiť a má začať alternatívna
liečba, ak je to vhodné (pozri časť 4.3).
Dojčenie
Nie je známe, či sa roxadustát/metabolity vylučuje/vylučujú do ľudského mlieka. Dostupné údaje u zvierat preukázali vylučovanie roxadustátu do mlieka (pre podrobné informácie pozri časť 5.3). Evrenzo je kontraindikovaný počas dojčenia (pozri časti 4.3 a 5.3).
Fertilita
V štúdiách na zvieratách sa nevyskytli žiadne účinky roxadustátu na mužskú a ženskú fertilitu. Boli však pozorované zmeny reprodukčných orgánov samcov potkanov. Možné účinky roxadustátu na mužskú fertilitu u ľudí v súčasnosti nie sú známe. Pri toxickej dávke pre matku bola pozorovaná zvýšená úmrtnosť embryí (pozri časť 5.3). Ženy vo fertilnom veku musia používať vysokoúčinnú antikoncepciu počas trvania liečby a aspoň jeden týždeň po poslednej dávke lieku Evrenzo.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Roxadustát má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Počas liečby liekom Evrenzo boli hlásené záchvaty (pozri časť 4.4). Pri vedení vozidiel alebo obsluhovaní strojov je preto potrebná opatrnosť.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Bezpečnosť lieku Evrenzo bola hodnotená u 3 542 pacientov, ktorí neboli závislí od dialýzy (non dialysis dependent, NDD), a u 3353 pacientov závislých od dialýzy (dialysis dependent, DD)
s anémiou a CKD, ktorí užili minimálne jednu dávku roxadustátu.
Najčastejšími (≥ 10 %) nežiaducimi reakciami súvisiacimi s roxadustátom sú hypertenzia (13,9 %), trombóza v mieste cievneho prístupu (12,8 %), hnačka (11,8 %), periférny edém (11,7 %), hyperkaliémia (10,9 %) a nevoľnosť (10,2 %).
Najčastejšími (≥ 1 %) závažnými nežiaducimi reakciami súvisiacimi s roxadustátom boli sepsa
(3,4 %), hyperkaliémia (2,5 %), hypertenzia (1,4 %) a hlboká žilová trombóza (1,2 %).
Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie pozorované počas klinických štúdií sú uvedené v tejto časti podľa kategórie
frekvencie.
Kategórie frekvencie sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
T
abuľka 3. Nežiaduce reakcie
T
rieda orgánových
systémov (SOC) podľa
MedDRA
|
K
ategória frekvencie
|
N
ežiaduca reakcia
|
Infekcie a nákazy
|
Časté
|
Sepsa
|
Poruchy metabolizmu a výživy
|
Veľmi časté
|
Hyperkaliémia
|
Psychické poruchy
|
Časté
|
Insomnia
|
Poruchy nervového systému
|
Časté
|
Záchvaty, bolesť hlavy
|
Poruchy ciev
|
Veľmi časté
|
Hypertenzia, trombóza v mieste cievneho prístupu (VAT)1
|
Časté
|
Hlboká žilová trombóza (DVT)
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
|
Nevoľnosť, hnačka
|
Časté
|
Zápcha, vracanie
|
Poruchy pečene a žlčových
ciest
|
Menej časté
|
Hyperbilirubinémia
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Menej časté
|
Pľúcna embólia
|
Celkové poruchy a reakcie v mieste podania
|
Veľmi časté
|
Periférny edém
|
1Táto nežiaduca reakcia je spájaná s pacientmi s CKD, ktorí boli počas užívania roxadustátu na dialýze.
PopisvybranýchnežiaducichreakciíTrombotické vaskulárne príhodyU pacientov s CKD, ktorí neboli na dialýze, bol výskyt DVT menej častý s frekvenciou výskytu 1,0 % (výskyt u 0,6 pacienta na 100 pacientorokov expozície) v skupine s roxadustátom a 0,2 % (výskyt
0,2 pacienta na 100 pacientorokov expozície) v skupine s placebom. U pacientov s CKD na dialýze sa
DVT vyskytla u 1,3 % (výskyt u 0,8 pacienta na 100 pacientorokov expozície) v skupine
s roxadustátom a 0,3 % (výskyt 0,1 pacienta na 100 pacientorokov expozície) v skupine s ESA (pozri
časť 4.4).
U pacientov s CKD, ktorí neboli na dialýze, bola pľúcna embólia pozorovaná u 0,4 % (výskyt
u 0,2 pacienta na 100 pacientorokov expozície) v skupine s roxadustátom v porovnaní s 0,2 % (výskyt u 0,1 pacienta na 100 pacientorokov expozície) v skupine s placebom. U pacientov s CKD na dialýze
bola pľúcna embólia pozorovaná u 0,6 % (výskyt u 0,3 pacienta na 100 pacientorokov expozície)
v skupine s roxadustátom v porovnaní s 0,5 % (výskyt u 0,3 pacienta na 100 pacientorokov expozície)
v skupine s ESA (pozri časť 4.4).
U pacientov s CKD na dialýze, bola trombóza v mieste cievneho prístupu pozorovaná u 12,8 % (výskyt u 7,6 pacienta na 100 pacientorokov expozície) v skupine s roxadustátom v porovnaní
s 10,2 % (výskyt u 5,4 pacienta na 100 pacientorokov expozície) v skupine s ESA (pozri časť 4.4).
ZáchvatyU pacientov s CKD, ktorí neboli na dialýze, sa záchvaty vyskytli u 1,1 % (výskyt u 0,6 pacienta na
100 pacientorokov expozície) v skupine s roxadustátom a 0,2 % (výskyt u 0,2 pacienta na
100 pacientorokov expozície) v skupine s placebom (pozri časť 4.4).
U pacientov s CKD na dialýze sa záchvaty vyskytli u 2,0 % (výskyt u 1,2 pacienta na
100 pacientorokov expozície) v skupine s roxadustátom a 1,6 % (výskyt 0,8 pacienta na
100 pacientorokov expozície) v skupine s ESA (pozri časť 4.4).
Sepsa
U pacientov s CKD, ktorí neboli na dialýze, bola sepsa pozorovaná u 2,1 % (výskyt u 1,3 pacienta na
100 pacientorokov expozície) v skupine s roxadustátom v porovnaní s 0,4 % (výskyt u 0,3 pacienta na
100 pacientorokov expozície) v skupine s placebom. U pacientov na dialýze bola sepsa pozorovaná
u 3,4 % (výskyt u 2,0 pacienta na 100 pacientorokov expozície) v skupine s roxadustátom v porovnaní s 3,4 % (výskyt u 1,8 pacienta na 100 pacientorokov expozície) v skupine s ESA (pozri časť 4.4).
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieJednotlivé supraterapeutické dávky roxadustátu 5 mg/kg (až do 510 mg) u zdravých osôb boli spájané s prechodným zvýšením srdcovej frekvencie, zvýšenou frekvenciou miernej až stredne závažnej muskuloskeletálnej bolesti, bolesťami hlavy, sínusovou tachykardiou a menej často s nízkym krvným tlakom, žiadne z uvedených zistení neboli závažné. Predávkovanie roxadustátom môže zvýšiť hladiny Hb nad požadovanú úroveň (10 – 12 g/dl), čo sa má vyriešiť ukončením liečby alebo znížením dávky roxadustátu (pozri časť 4.2) a pozorným monitorovaním a liečbou na základe klinickej indikácie. Hemodialýza nemá podstatný vplyv na odstraňovanie roxadustátu a jeho metabolitov (pozri časť 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antianemiká, iné antianemiká, ATC kód: B03XA05.
MechanizmusúčinkuRoxadustát je inhibítorom prolylhydroxylázy (HIF-PHI), hypoxiou indukovateľného faktora. Aktivita enzýmov HIF-PH riadi vnútrobunkové hladiny HIF, čo je transkripčný faktor, ktorý reguluje expresiu
génov podieľajúcich sa na erytropoéze. Aktivácia dráhy HIF je dôležitá pri adaptívnej odpovedi na
hypoxiu na zvýšenie produkcie červených krviniek. Reverzibilnou inhibíciou HIF-PH roxadustát stimuluje koordinovanú erytropoetickú odpoveď, čo zahŕňa zvýšenie hladiny endogénneho
erytropoetínu (EPO) v plazme, reguláciu transportných proteínov železa a zníženie hepcidínu (proteín
regulujúci železo, ktorý sa zvyšuje počas zápalu pri CKD). Výsledkom je zlepšená biologická dostupnosť železa, zvýšená produkcia Hb a zvýšená hmota červených krviniek.
FarmakodynamickéúčinkyÚčinky na QTc a srdcovú frekvenciuDôkladná štúdia QT (TQT) u zdravých účastníkov s roxadustátom podávaným ako jednorazová terapeutická dávka 2,75 mg/kg a jednorazová supraterapeutická dávka 5 mg/kg (až do 510 mg) nepreukázala predĺženie intervalu QTc. Tá istá dôkladná štúdia QT preukázala placebom upravené zvýšenie srdcovej frekvencie až 9 – 10 úderov za minútu v čase 8 – 12 hodín po podaní dávky
2,75 mg/kg a 15 – 18 úderov za minútu v čase 6 – 12 hodín po podaní dávky 5 mg/kg.
KlinickáúčinnosťabezpečnosťVývojový program pri anémii s CKDÚčinnosť a bezpečnosť roxadustátu boli hodnotené aspoň 52 týždňov v globálne uskutočňovanom programe fázy 3, ktorý pozostával z 8 multicentrických a randomizovaných štúdií s pacientmi trpiacimi anémiou s CKD, ktorí neboli závislí od dialýzy (NDD) a pacientmi závislými od dialýzy (DD) (pozri Tabuľku 4).
Tri štúdie s pacientmi NDD s CKD v štádiu 3 – 5 boli dvojito zaslepené a placebom kontrolované
štúdie (ALPS, 1517-CL-0608; ANDES, FGCL-4592-060; OLYMPUS, D5740C00001) a jedna štúdia
bola otvorená, ESA kontrolovaná (DOLOMITES, 1517-CL-0610) s použitím darbepoetínu alfa ako komparátora. Všetky štúdie s pacientmi NDD vyhodnocovali účinnosť a bezpečnosť u pacientov, ktorí neboli liečení ESA, úpravou a následným udržiavaním hladiny Hb v cieľovom rozsahu
10 – 12 g/dl (korekčné nastavenie Hb).
Štyri otvorené štúdie pacientov DD kontrolované ESA (kontrola: epoetín alfa a/alebo darbepoetín alfa)
u pacientov na hemodialýze alebo na peritoneálnej dialýze vyhodnocovali účinnosť a bezpečnosť
v rôznych podmienkach:
• v prostredí úpravy hladiny Hb (HIMALAYAS, FGCL-4592-063),
• v prostredí prechodu z ESA, kde pacienti prechádzali z liečby ESA na udržiavanie hladiny Hb v cieľovom rozsahu (PYRENEES, 1517-CL-0613; SIERRAS, FGCL-4592-064),
• alebo kombinácie prístupov úpravy hladiny Hb a prechodu z ESA (ROCKIES, D5740C00002).
Pacienti v NDD štúdiách mali CKD v štádiu 3 – 5 a nedostávali dialýzu. U všetkých pacientov bola priemerná hladina Hb ≤ 10,0 g/dl okrem pacientov v štúdii DOLOMITES (1517-CL-0610), u ktorých bola povolená priemerná hladina Hb ≤ 10,5 g/dl. Požadované hladiny feritínu boli ≥ 30 ng/ml
(ALPS, 1517-CL-0608; ANDES, FGCL-4592-060), ≥ 50 ng/ml (OLYMPUS, D5740C00001) alebo
≥ 100 ng/ml (DOLOMITES, 1517-CL-0610). Okrem pacientov v štúdii (OLYMPUS, D5740C00001), v ktorej bola povolená liečba ESA až do doby 6 týždňov pred randomizáciou, pacienti nemohli dostávať žiadnu liečbu ESA v období 12 týždňov pred randomizáciou.
Pacienti v DD štúdiách museli byť na dialýze: stabilnej pre pacientov DD v štúdii PYRENEES (1517-CL-0613), ktorá bola definovaná ako dialýza trvajúca viac ako 4 mesiace, alebo incidentnej (ID) pre pacientov DD v štúdii HIMALAYAS (FGCL-4592-063), ktorá bola definovaná ako dialýza
≥ 2 týždne ale ≤ 4 mesiace. Pacientmi v štúdiách SIERRAS (FGCL-4592-064) a ROCKIES (D5740C00002) boli pacienti DD na stabilnej (približne 80 – 90 %) aj incidentnej dialýze (približne
10 – 20 %). Požadovaná hladina feritínu pre všetkých pacientov bola ≥ 100 ng/ml. Požiadavkou pre
všetkých pacientov bolo intravenózne alebo subkutánne podávanie ESA minimálne 8 týždňov pred randomizáciou, okrem pacientov v štúdii HIMALAYAS (FGCL-4592-063), v ktorej boli vylúčení pacienti, ktorí dostali akúkoľvek liečbu ESA v období 12 týždňov pred randomizáciou.
Liečba roxadustátom prebiehala podľa pokynov týkajúcich sa dávkovania, ako je uvedené v časti 4.2. Demografické a všetky východiskové charakteristiky v rámci jednotlivých štúdií boli porovnateľné medzi skupinami s roxadustátom a kontrolnými skupinami. Medián veku pri randomizácii bol
55 – 69 rokov, z toho 16,6 – 31,1 % vo vekovej skupine 65 – 74 rokov a 6,8 – 35 % vo veku
≥ 75 rokov. Percentuálny podiel pacientov ženského pohlavia bol v rozsahu 40,5 – 60,7 %.
Najčastejšie zastúpenou rasou v rámci všetkých štúdií bola belošská, černošská alebo afroamerická
a ázijská rasa. Najčastejšou príčinou CKD boli diabetická a hypertenzná nefropatia. Medián hladiny Hb sa pohyboval v rozsahu 8,60 – 10,78 g/dl. Približne 50 – 60 % pacientov NDD a 80 – 90 % pacientov DD mali na začiatku dostatok železa.
Údaje zo siedmich štúdií fázy 3 boli spojené do dvoch samostatných populácií (tri NDD a štyri DD)
(pozri Tabuľku 4).
Do súboru NDD boli zahrnuté tri placebom kontrolované NDD štúdie (2 386 pacientov užívajúcich roxadustát, 1 884 pacientov užívajúcich placebo). Údaje zo štúdie DOLOMITES fázy 3 s pacientmi NDD kontrolovanej ESA (1517-CL-0610; 323 pacientov užívajúcich roxadustát a 293 pacientov užívajúcich darbepoetín alfa) nie sú zahrnuté do analýz spojených NDD údajov, keďže ide o jedinú otvorenú, aktívne kontrolovanú štúdiu v populácii pacientov NDD.
Štyri štúdie pacientov DD kontrolované ESA (2 354 pacientov užívajúcich roxadustát, 2 360 pacientov
užívajúcich ESA [epoetín alfa a/alebo darbepoetín alfa]) boli zahrnuté do súboru pacientov DD. V súbore pacientov DD boli vytvorené dve podskupiny s cieľom zohľadniť dve rôzne nastavenia
liečby:
• Pacienti v populácii DD, ktorí boli na dialýze viac ako 2 týždne a menej ako 4 mesiace, boli
označení ako incidentní (ID) pacienti DD (súbor ID DD), čo odráža nastavenie korekcie Hb.
• Pacienti DD, ktorí boli na dialýze po tejto hraničnej hodnote 4 mesiacov, boli označení ako
stabilní pacienti DD (stabilný súbor DD), čo odráža nastavenie prechodu z ESA.
Tabuľka 4. Prehľad vývojového programu roxadustátu fázy 3 u pacientov s anémiou s CKDŠtúdie u pacientov NDD
|
| Placebom kontrolované štúdie (súbor NDD)
| Kontrola ESA (darbepoetín
alfa)
|
Nastavenie
| Úprava hladiny Hb
|
Štúdia
| ALPS (1517-CL-0608)
| ANDES (FGCL-4592-060)
| OLYMPUS (D5740C00001)
| DOLOMITES (1517-CL-0610)
|
Randomizované
(roxadustát/
komparátor)
| 594 (391/203)
| 916 (611/305)
| 2 760
(1 384/1 376)
| 616 (323/293)
|
Štúdie u pacientov DD
|
| Štúdie kontrolované ESA (súbor DD) (epoetín alfa alebo darbepoetín alfa)
|
Nastavenie
| Prechod z ESA
| Úprava hladiny
Hb
| Prechod z ESA
a úprava hladiny Hb
|
Štúdia
| PYRENEES
(1517-CL-0613)
| SIERRAS
(FGCL-4592-064)
| HIMALAYAS
(FGCL-4592-063)
| ROCKIES
(D5740C00002)
|
Randomizované
(roxadustát/
komparátor)
| 834
(414/420)
| 740
(370/370)
| 1 039
(522/517)
| 2 101
(1 048/1 053)
|
DD: závislý od dialýzy; ESA: látka stimulujúca erytropoézu; Hb: hemoglobín; NDD: nezávislý od dialýzy
.Pacienti NDD s CKDVýsledky účinnostiVývoj hladiny Hb počas liečbyV klinických štúdiách bol roxadustát účinný pri dosahovaní a udržiavaní cieľovej hladiny Hb
(10 – 12 g/dl) u pacientov s anémiou s CKD, ktorí neboli na dialýze (pozri Obrázok 1).
O
brázok 1. Priemerná (SE) hladina Hb (g/dl) v priebehu času do 52. týždňa (FAS); súbor NDD (úprava hladiny Hb)
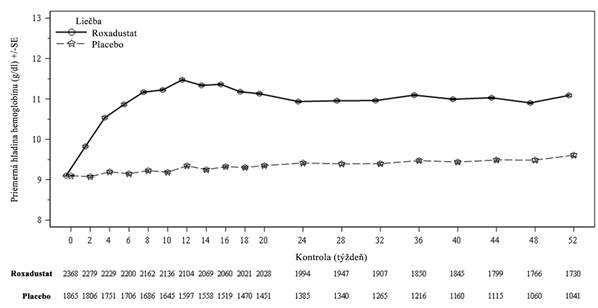
FAS: analýza celého súboru; Hb: hemoglobín; NDD: nezávislý od dialýzy; SE: štandardná chyba.
Kľúčové cieľové ukazovatele účinnosti Hb u pacientov NDD s CKDU pacientov NDD, ktorí potrebovali liečbu anémie na úpravu hladiny Hb, bola časť pacientov, ktorá dosiahla odpoveď Hb počas prvých 24 týždňov, vyššia v skupine s roxadustátom (80,2 %) v porovnaní s placebom (8,7 %). Bolo zaznamenané štatisticky významné zvýšenie hladiny Hb v porovnaní
s východiskovou hodnotou do 28. – 36. týždňa v skupine s roxadustátom (1,91 g/dl) v porovnaní
s placebom (0,14 g/dl) a dolný limit 95 % intervalu spoľahlivosti je vyšší ako 1. V štúdiách NDD bolo dosiahnuté zvýšenie hladiny Hb o minimálne 1 g/dl s mediánom času na úrovni 4,1 týždňa (pozri Tabuľku 5).
V otvorenej štúdii NDD pacientov kontrolovanej ESA s názvom DOLOMITES (1517-CL-0610) bol podiel pacientov, ktorí dosiahli odpoveď Hb počas prvých 24 týždňov, vyšší v skupine s roxadustátom (89,5 %) v porovnaní s darbepoetínom alfa (78 %) (pozri Tabuľku 5).
Populácia
| Pacienti NDD s CKD
| Nastavenie
| Úprava hladiny Hb
| Úprava hladiny Hb
|
Cieľový ukazovateľ/
Parameter
| Súbor NDD (FAS)
| DOLOMITES (PPS)
1517-CL-0610
| Roxadustát n = 2 368
| Placebo n = 1 865
| Roxadustát n = 286
| Darbepoetín alfa
n = 273
| Časť pacientov, ktorí dosiahli odpoveď Hb1
|
Pacienti reagujúci na liečbu, n (%)
[95 % CI]
|
1 899 (80,2)
[78,5; 81,8]
|
163 (8,7)
[7,5; 10,1]
|
256 (89,5)
[85,4; 92,8]
|
213 (78,0)
[72,6; 82,8]
| Rozdiel v podieloch [95 % CI]
| 71,5 [69,40; 73,51]
| 11,51 [5,66; 17,36]
| Pomer pravdepodobnosti
| 40,49 [33,01; 49,67]
| 2,48 [1,53; 4,04]
| Hodnota p
| < 0,0001
| ND
| Zmena hladiny Hb od východiskovej hodnoty (g/dl)2
| Priemer (SD), východisková hodnota
|
9,10 (0,74)
|
9,10 (0,73)
|
9,55 (0,76)
|
9,54 (0,69)
| Priemer (SD), CFB
| 1,85 (1,07)
| 0,17 (1,08)
| 1,85 (1,08)
| 1,84 (0,97)
|
|
|
Tabuľka 5. Kľúčové cieľové ukazovatele účinnosti Hb (NDD)
P
opulácia
|
P
acienti NDD s CKD
|
N
astavenie
|
Ú
prava hladiny Hb
|
Ú
prava hladiny Hb
|
C
i
eľový ukazovateľ/
Parameter
|
Súbor NDD (FAS)
|
D
O
LO
MITES (PPS)
1517-CL-0610
|
R
oxadustát n = 2 368
|
Placebo n = 1 865
|
R
oxadustát n = 286
|
D
arbepoetín alfa
n = 273
|
Priemer LS
|
1,91
|
0,14
|
1,85
|
1,84
|
Rozdiel priemeru LS [95 % CI]
|
1,77 [1,69; 1,84]
|
0,02 [−0,13; 0,16]
|
Hodnota p
|
< 0,0001
|
0,844
|
CFB: zmena oproti východiskovej hodnote; CI: interval spoľahlivosti; CKD: chronické ochorenie obličiek; FAS: analýza celého súboru; Hb: hemoglobín; LS: metóda najmenších štvorcov; ND: neuskutočnené; NDD: nezávislý od dialýzy; PPS: súbor podľa protokolu; SD: štandardná odchýlka.
1Odpoveď Hb v období prvých 24 týždňov
2Zmena hladiny Hb od východiskovej hodnoty do 28. – 36. týždňa
Pacienti DD s CKDVývoj hladiny Hb počas liečbyV klinických štúdiách bol roxadustát účinný pri dosahovaní a udržiavaní cieľovej hladiny Hb (10 – 12 g/dl) u pacientov s CKD na dialýze, bez ohľadu na predchádzajúcu liečbu ESA (pozri Obrázok 2 a 3).
Obrázok 2. Priemerná (SE) hladina Hb do 52. týždňa (FAS); spojená skupina ID DD (úprava hladiny Hb)
DD: závislý od dialýzy; FAS: analýza celého súboru; Hb: hemoglobín; ID: incidentná; SE: štandardná
chyba.
O
brázok 3. Priemerná (SE) hladina Hb (g/dl) v priebehu času do 52. týždňa (FAS); stabilná podskupina DD (prechod z ESA)

DD: závislý od dialýzy; ESA: látka stimulujúca erytropoézu; FAS: analýza celého súboru; Hb:
hemoglobín; SE: štandardná chyba.
Kľúčové cieľové ukazovatele účinnosti Hb u pacientov DD s CKDU pacientov DD, ktorí potrebovali liečbu anémie na úpravu hladiny Hb a pacientov, ktorí prešli
z liečby ESA, bolo zaznamenané zvýšenie hladiny Hb v porovnaní s východiskovou hodnotou do
28. – 36. týždňa v skupine s roxadustátom; toto zvýšenie bolo porovnateľné so zvýšením
pozorovaným v skupine s ESA a bolo vyššie ako vopred špecifikovaná hranica nepodradenosti
−0,75 g/dl. Podiel pacientov, ktorí dosiahli odpoveď Hb počas prvých 24 týždňov, bol podobný
v skupinách s roxadustátom a ESA (pozri Tabuľku 6).
Tabuľka 6. Kľúčové cieľové ukazovatele účinnosti Hb (DD)Populácia
| Pacienti DD
|
Nastavenie
| Úprava hladiny Hb
| Prechod z ESA
|
Cieľový ukazovateľ/
Parameter
| Súbor ID DD (FAS/PPS)
| Stabilný súbor DD (PPS)
|
Roxadustát n = 756
| ESA
n = 759
| Roxadustát n = 1 379
| ESA
n = 1 417
|
Zmena hladiny Hb od východiskovej hodnoty (g/dl)
|
Priemerná (SD) východisková hodnota
|
8,77 (1,20)
|
8,82 (1,20)
|
10,32 (0,99)
|
10,37 (0,99)
|
Priemer (SD) CFB
| 2,37 (1,57)
| 2,12 (1,46)
| 0,65 (1,15)
| 0,36 (1,23)
|
Priemer LS
| 2,17
| 1,89
| 0,58
| 0,28
|
Rozdiel priemeru LS [95 % CI]
| 0,28 [0,110; 0,451]
| 0,30 [0,228; 0,373]
|
Hodnota p
| 0,0013
| < 0,0001
|
Podiel pacientov, ktorí dosiahli odpoveď Hb1,2
|
Pacienti reagujúci na liečbu, n (%)
[95 % CI]
| 453 (59,9)
[56,3; 63,4]
| 452 (59,6)
[56,0; 63,1]
| 978 (70,9)
[68,4; 73,3]
| 959 (67,7)
[65,2; 70,1]
|
Rozdiel v podieloch [95 % CI]
| 0,3 [−4,5; 5,1]
| 2,7 [−0,7; 6,0]
|
Pomer pravdepodobnosti [95 % CI]
| ND
| ND
|
Hodnota p
| ND
| ND
|
CFB: zmena oproti východiskovej hodnote; CI: interval spoľahlivosti; CKD: chronické ochorenie obličiek; DD: závislý od dialýzy; ESA: látka stimulujúca erytropoézu; FAS: analýza celého súboru;
Hb: hemoglobín; ID: incidentná; LS: metóda najmenších štvorcov; ND: neuskutočnené; PPS: súbor podľa protokolu; SD: štandardná odchýlka.
1Hladina Hb v cieľovom rozpätí 10,0 – 12,0 g/dl počas 28. – 36. týždňa bez použitia záchrannej
terapie v období 6 týždňov pred a počas tohto 8-týždňového obdobia hodnotenia.
2Údaje v súbore ID DD boli analyzované iba pre 28. – 52. týždeň.
Záchranná terapia, transfúzia červených krviniek a intravenózne podanie železaÚčinky liečby roxadustátom na použitie záchrannej terapie, transfúziu červených krviniek
a intravenózne podanie železa sú uvedené v Tabuľke 7 (NDD) a Tabuľke 8 (DD). V klinických štúdiách roxadustát znížil hladinu hepcidínu (regulátor metabolizmu železa), znížil hladinu feritínu, zvýšil hladinu železa v sére, zatiaľ čo saturácia transferínu bola stabilná, všetky tieto ukazovatele boli posudzované v priebehu času ako indikátory stavu železa.
LDL cholesterol (low-density lipoprotein, lipoproteín s nízkou hustotou)Účinky liečby roxadustátom na LDL cholesterol sú uvedené v Tabuľke 7 a Tabuľke 8. Bolo
zaznamenané zníženie priemernej hodnoty hladín LDL a HDL cholesterolu (high density lipoprotein, lipoproteín s vysokou hustotou) u pacientov liečených roxadustátom v porovnaní s pacientmi užívajúcich placebo alebo pacientmi liečenými ESA. Účinok na LDL cholesterol bol výraznejší, viedol k zníženiu pomeru LDL/HDL a bol pozorovaný bez ohľadu na použitie statínov.
Populácia
| Pacienti NDD s CKD
| Zásah
| Úprava
| Úprava
|
Cieľový ukazovateľ/Parameter
| Súbor NDD (FAS)
| DOLOMITES (1517-CL-0610)
| Roxadustát n = 2 368
| Placebo n = 1 865
| Roxadustát n = 322
| Darbepoetín
alfa
|
Počet pacientov so záchrannou terapiou, n (%)*
|
211 (8,9)
|
580 (31,1)
|
ND
| RBC
| 118 (5,0)
| 240 (12,9)
| IV železo
| 50 (2,1)
| 90 (4,8)
| ESA
| 48 (2,0)
| 257 (13,8)
|
IR
|
10,4
|
41,0
| Pomer rizík
| 0,19
|
ND
| 95 % CI
| 0,16; 0,23
| Hodnota p
| < 0,0001
| Počet pacientov s IV
železom, n (%)2
|
ND
|
20 (6,2)
|
37 (12,7)
|
IR
|
9,9
|
21,2
| Pomer rizík
| 0,45
| 95 % CI
| 0,26; 0,78
| Hodnota p
| 0,004
| Zmena LDL cholesterolu od východiskovej hodnoty (mmol/l) do 12. – 28. týždňa3
|
|
|
Tabuľka 7. Ostatné cieľové ukazovatele účinnosti: použitie záchrannej terapie, mesačné intravenózne podanie železa a zmena LDL cholesterolu od východiskovej hodnoty (NDD)
P
opulácia
|
P
acienti NDD s CKD
|
Z
ásah
|
Ú
prava
|
Ú
prava
|
C
i
eľový ukazovateľ/Parameter
|
Súbor NDD (FAS)
|
D
O
LO
MITES (1517-CL-0610)
|
R
oxadustát n = 2 368
|
Placebo n = 1 865
|
R
oxadustát n = 322
|
D
arbepoetín
alfa
|
A
nalýza použitím ANCOVA
|
Priemer LS
|
−0,446
|
0,066
|
−0,356
|
0,047
|
95 % CI
|
−0,484; −0,409
|
0,017; 0,116
|
−0,432; −0,280
|
−0,033; 0,127
|
Rozdiel priemeru
LS (komparátor R)
|
−0,513
|
−0,403
|
95 % CI
|
−0,573; −0,453
|
−0,510; −0,296
|
Hodnota p
|
< 0,0001
|
< 0,001
|
|
|
|
|
|
|
|
Hodnoty p uvedené pre súbor NDD sú nominálne hodnoty p.
ANCOVA: analýza kovariancie; CI: interval spoľahlivosti; ESA: látka stimulujúca erytropoézu; FAS: analýza celého súboru; IR: miera výskytu (na 100 pacientorokov v riziku); IV: intravenózne;
LDL: lipoproteín s nízkou hustotou; LS: metóda najmenších štvorcov; ND: neuskutočnené; NDD:
nezávislý od dialýzy; R: roxadustát; RBC: červené krvinky;
1Pre použitie záchrannej terapie bola spojená skupina NDD analyzovaná do 52. týždňa.
2Počas 1. – 36. týždňa.
3Zmena LDL cholesterolu od východiskovej hodnoty sa posudzovala iba do 24. týždňa pre štúdiu
OLYMPUS (D5740C00001).
Tabuľka 8. Ostatné cieľové ukazovatele účinnosti: použitie záchrannej terapie, mesačnéintravenózne podanie železa a zmena LDL cholesterolu od východiskovej hodnoty (DD)Populácia
| Pacienti DD s CKD
|
Zásah
| Úprava
| Prechod
|
Cieľový ukazovateľ/
Parameter
|
Súbor ID DD (FAS)
|
Stabilný súbor DD (FAS)
|
Roxadustát n = 756
| ESA
n = 759
| Roxadustát n = 1 586
| ESA
n = 1 589
|
Priemerné množstvo IV železa počas 28. – 52. týždňa (mg)1
|
n
| 606
| 621
| 1 414
| 1 486
|
Priemer (SD)
| 53,57 (143,097)
| 70,22 (173,33)
| 42,45 (229,80)
| 61,99 (148,02)
|
Zmena LDL cholesterolu od východiskovej hodnoty (mmol/l) do 12. – 28. týždňa
|
Analýza použitím ANCOVA
|
Priemer LS
| −0,610
| −0,157
| −0,408
| −0,035
|
95 % CI
| −0,700; −0,520
| −0,245; −0,069
| −0,449; −0,368
| −0,074; 0,003
|
Rozdiel priemeru LS (Komparátor R)
|
−0,453
|
−0,373
|
95 % CI
| −0,575; −0,331'
| −0,418; −0,328
|
Hodnota p
| < 0,0001
| < 0,0001
|
Hodnoty p uvedené pre spojené skupiny ID DD a stabilnú DD sú nominálne hodnoty p. ANCOVA: analýza kovariancie; CI: interval spoľahlivosti; CKD: chronické ochorenie obličiek; DD: závislý od dialýzy; ESA: látka stimulujúca erytropoézu; FAS: analýza celého súboru; ID: incidentná dialýza; IV: intravenózne; LDL: lipoproteín s nízkou hustotou; LS: metóda najmenších štvorcov; R: roxadustát.
1Časové obdobie pre štúdiu PYRENEES (1517-CL-0613) bolo do 36. týždňa a časové obdobie pre štúdiu ROCKIES (D5740C0002) bolo od 36. týždňa do ukončenia štúdie.
V štúdii pacientov na dialýze SIERRAS (FGCL-4592-064) dostalo podstatne nižšie množstvo pacientov transfúziu červených krviniek počas liečby v skupine s roxadustátom v porovnaní so skupinou na EPO-alfa (12,5 % oproti 21,1 %); v štúdii ROCKIES (D5740C00002) nebolo číselné zníženie štatisticky významné (9,8 % oproti 13,2 %).
Výsledky hlásené pacientmi, ktorí neboli na dialýze
V štúdii DOLOMITES (1517-CL-0610) bola stanovená nepodradenosť roxadustátu voči darbepoetínu,
čo sa týka SF-36 PF a SF-36 VT.
Výsledky hlásené pacientmi, ktorí boli na dialýze
V štúdii PYRENEES (1517-CL-0613) bola stanovená nepodradenosť roxadustátu voči ESA, čo sa
týka zmien SF-36 PF a SF-36 VT od východiskovej hodnoty do 12. až 28. týždňa.
Klinická bezpečnosť
Metaanalýza spojených, posudzovaných kardiovaskulárnych udalostí
Metaanalýza posudzovaných závažných nežiaducich kardiovaskulárnych udalostí (major adverse cardiovascular events, MACE; kombinácia všetkých príčin úmrtnosti [all-cause mortality, ACM],
infarktu myokardu, mozgovej príhody) a MACE+ (kombinácia ACM, infarktu myokardu, mozgovej príhody a hospitalizácie z dôvodu nestabilnej anginy pectoris alebo kongestívneho zlyhávania srdca) z
programu štúdie fázy 3 sa uskutočnila u 8 984 pacientov.
Výsledky MACE, MACE+ a ACM sú uvedené pre tri súbory údajov použitím spojeného pomeru rizík
(HR) a jeho 95 % intervalu spoľahlivosti (CI). Týmito tromi súbormi údajov sú:
• Spojený súbor údajov placebom kontrolovanej úpravy hladiny Hb u pacientov NDD [vrátane pacientov zo štúdií OLYMPUS (D5740C00001), ANDES (FGCL-4592-060) a ALPS (1517- CL-0608); pozri Tabuľku 4]
• Spojený súbor údajov ESA kontrolovanej úpravy hladiny Hb u pacientov NDD a ID-DD
[vrátane pacientov zo štúdií DOLOMITES (1517-CL-0610), HIMALAYAS (FGCL-4592-
063) a pacientov ID-DD zo štúdií SIERRAS (FGCL-4592-064) a ROCKIES (D5740C00002);
pozri Tabuľku 4]
• Spojený súbor údajov ESA kontrolovaného prechodu z ESA u stabilných pacientov DD
[vrátane pacientov zo štúdie PYRENEES (1517-CL-0613) a stabilných pacientov DD zo
štúdií ROCKIES (D5740C00002) a SIERRAS (FGCL-4592-064); pozri Tabuľku 4]
MACE, MACE+ a ACM v placebom kontrolovanom súbore s úpravou hladiny Hb u pacientov s CKD, ktorí nie sú závislí od dialýzy
U pacientov NDD zahŕňali analýzy MACE, MACE+ a ACM analýz počas liečby všetky údaje od
začiatku skúšanej liečby až do 28 dní od ukončenia sledovania liečby. Pri analýzach počas liečby sa použil Coxov model nepriamo vážený pre pravdepodobnosť cenzúry (metóda IPCW), ktorého cieľom je opraviť rozdiely doby sledovania medzi roxadustátom a placebom vrátane identifikovaných prispievajúcich faktorov k zvýšenému riziku a predčasnému ukončeniu liečby, najmä činiteľov pre odhadovanú rýchlosť glomerulárnej filtrácie (estimated glomerular filtration rate, eGFR) a Hb na začiatku liečby a v priebehu času. Možnosť prítomnosti zvyškového skreslenia pri tomto modeli zostáva neistá. Hodnoty HR pre analýzy počas liečby boli 1,26; 1,17 a 1,16 (pozri Tabuľku 9). Súčasťou analýz ITT boli všetky údaje od začiatku liečby v štúdii až do konca sledovania bezpečnosti po ukončení liečby. Analýza ITT bola zahrnutá s cieľom poukázať na nevyváženosť rozdelenia rizika v prospech placeba v analýze vykonanej počas liečby, analýzy ITT však vo všeobecnosti ukazujú zoslabenie účinku liečby skúmaného lieku a v týchto analýzach ITT nie je možné úplne vylúčiť skreslenie, najmä keď po ukončení skúšanej liečby bola nasadená záchranná liečba ESA. Hodnoty HR boli 1,10; 1,07 a 1,08 s hornými limitmi 95 % CI 1,27; 1,21 a 1,26; v tomto poradí.
T
abuľka 9. Bezpečnosť a úmrtnosť CV v placebom kontrolovanom súbore pacientov NDD
s úpravou hladiny Hb
|
MACE
|
MACE+
|
ACM
|
|
R
oxadustát
n = 2 386
|
Placebo
n = 1 884
|
R
oxadustát
n = 2 386
|
Placebo
n = 1 884
|
R
oxadustát
n = 2 386
|
Placebo
n = 1 88
|
P
o
čas liečby
|
Počet pacientov s udalosťami (%)
|
344 (14,4)
|
166 (8,8)
|
448 (18,8)
|
242 (12,8)
|
260 (10,9)
|
122 (6,5)
|
FAIR
|
8,7
|
6,8
|
11,6
|
10,1
|
6,4
|
5,0
|
HR (95 % CI)
|
1,26 (1,02; 1,55)
|
1,17 (0,99; 1,40)
|
1,16 (0,90; 1,50)
|
ITT
|
Počet pacientov
s udalosťami (%)
|
480 (20,1)
|
350 (18,6)
|
578 (24,2)
|
432 (22,9)
|
400 (16,8)
|
301 (16)
|
FAIR
|
10,6
|
10,3
|
13,2
|
13,2
|
8,3
|
8,1
|
HR (95 % CI)
|
1,10 (0,96; 1,27)
|
1,07 (0,94; 1,21)
|
1,08 (0,93; 1,26)
|
ACM: všetky príčiny úmrtnosti (z ang. all-cause mortality); ACM je kombináciou MACE/MACE+; CI: interval spoľahlivosti; FAIR: miera výskytu upravená podľa následného sledovania (počet pacientov s udalosťou/100 pacientorokov); HR: pomer rizík; ITT: všetci randomizovaní pacienti; MACE: závažné nežiaduce kardiovaskulárne udalosti (smrť, nefatálny infarkt myokardu a/mozgová príhoda); MACE+: závažná nežiaduca kardiovaskulárna udalosť vrátane hospitalizácií z dôvodu nestabilnej anginy pectoris a/alebo kongestívneho zlyhávania srdca.
MACE, MACE+ a ACM v ESA kontrolovanom súbore s úpravou hladiny Hb u pacientov, ktorí nie súzávislí od dialýzy, a pacientov s CKD závislých od incidentnej dialýzyV skupine pacientov NDD a ID-DD s úpravou hladiny Hb boli východiskové charakteristiky a miery
ukončenia liečby porovnateľné medzi pacientmi v spojenej skupine užívajúcej roxadustát a spojenej skupine užívajúcej ESA. Analýza MACE, MACE+ a ACM pozorovaných pri liečbe ukázala hodnoty HR 0,79; 0,78, resp. 0,78 s hornými limitmi 95 % CI 1,02; 0,98; resp. 1,05 (pozri Tabuľku 10). Analýzy počas liečby podporujú tvrdenie, že neexistujú dôkazy zvýšeného rizika kardiovaskulárnej bezpečnosti alebo úmrtnosti pri užívaní roxadustátu v porovnaní s ESA u pacientov s CKD vyžadujúcich úpravu hladiny Hb.
Tabuľka 10. Bezpečnosť a úmrtnosť CV v ESA kontrolovanej skupine s úpravou hladiny Hb
| MACE
| MACE+
| ACM
|
Roxadustát
n = 1 083
| ESA
n = 1 059
| Roxadustát
n = 1 083
| ESA
n = 1 059
| Roxadustát
n = 1 083
| ESA
n = 1 059
|
Počas liečby
|
Počet
pacientov s udalosťami
(%)
|
105 (9,7)
|
136 (12,8)
|
134 (12,4)
|
171 (16,1)
|
74 (6,8)
|
99 (9,3)
|
IR
| 6,5
| 8,2
| 8,3
| 10,3
| 4,6
| 6,0
|
HR (95 % CI)
| 0,79 (0,61; 1,02)
| 0,78 (0,62; 0,98)
| 0,78 (0,57; 1,05)
|
ACM: všetky príčiny úmrtnosti; ACM je kombináciou MACE/MACE+; CI: interval spoľahlivosti; ESA:
látka stimulujúca erytropoézu; HR: pomer rizík; IR: miera výskytu (počet pacientov
s udalosťou/100 pacientorokov); MACE: závažné nežiaduce kardiovaskulárne udalosti (smrť, nefatálny
infarkt myokardu a/alebo mozgová príhoda); MACE+: závažná nežiaduca kardiovaskulárna udalosť
vrátane hospitalizácií z dôvodu nestabilnej anginy pectoris a/alebo kongestívneho zlyhávania srdca.
MACE, MACE+ a ACM v ESA kontrolovanej skupine s prechodom z ESA u stabilných pacientov sCKD, ktorí sú závislí od dialýzyU stabilných DD pacientov prechádzajúcich z liečby ESA výsledky analýzy MACE, MACE+ a ACM
pozorované pri liečbe ukázali hodnoty HR 1,18; 1,03; resp. 1,23 s hornými limitmi spoľahlivosti 95 %
pre HR na úrovni 1,38; 1,19; resp. 1,49 (pozri Tabuľku 11). Výsledky uvedené v Tabuľke 11 sa majú
vysvetľovať opatrne, keďže pacienti užívajúcich rodaxustat prešli z liečby ESA na začiatku štúdie
a vplyv inherentného rizika pri prechode na akúkoľvek inú liečbu v porovnaní so zotrvaním na liečbe so stabilizovanou hladinou Hb môže ovplyvniť pozorované výsledky, a preto nie je možné spoľahlivo stanoviť porovnanie odhadov účinku liečby.
Tabuľka 11. Bezpečnosť a úmrtnosť CV v ESA kontrolovanej skupine s prechodom z ESAu stabilných pacientov DD
| MACE
| MACE+
| ACM
|
Roxadustát
n = 1 594
| ESA
n = 1 594
| Roxadustát
n = 1 594
| ESA
n = 1 59
4
| Roxadustát
n = 1 594
| ESA
n = 1 594
|
Počas liečby
|
Počet
pacientov
s udalosťami
(%)
|
297 (18,6)
|
301 (18,9)
|
357 (22,4)
|
403 (25,
3)
|
212 (13,3)
|
207 (13,0)
|
IR
| 10,4
| 9,2
| 12,5
| 12,3
| 7,4
| 6,3
|
HR (95 % CI)
| 1,18 (1,00; 1,38)
| 1,03 (0,90; 1,19)
| 1,23 (1,02; 1,49)
|
ACM: všetky príčiny úmrtnosti; ACM je kombináciou MACE/MACE+; CI: interval spoľahlivosti;
ESA: látka stimulujúca erytropoézu; HR: pomer rizík; IR: miera výskytu (počet pacientov
s udalosťou/100 pacientorokov); MACE: závažné nežiaduce kardiovaskulárne udalosti (smrť, nefatálny infarkt myokardu a/mozgová príhoda); MACE+: závažná nežiaduca kardiovaskulárna udalosť vrátane
hospitalizácií z dôvodu nestabilnej anginy pectoris a/alebo kongestívneho zlyhávania srdca.
5.2 Farmakokinetické vlastnostiExpozícia roxadustátu v plazme (plocha pod krivkou koncentrácie lieku v plazme v priebehu času
[AUC] a maximálne koncentrácie v plazme [Cmax]) je úmerná dávke v rámci odporúčaného rozsahu terapeutickej dávky. Pri dávkovacom režime trikrát týždenne sa dosiahnu ustálené koncentrácie
roxadustátu v plazme do jedného týždňa (3 dávky) s minimálnou akumuláciou. Farmakokinetika roxadustátu sa v priebehu času nemení.
AbsorpciaMaximálne koncentrácie v plazme (Cmax) sa zvyčajne dosiahnu v priebehu 2 hodín po podaní dávky
nalačno.
Podanie roxadustátu s jedlom znížilo koncentráciu Cmax o 25 %, nemalo však vplyv na AUC
v porovnaní so stavom nalačno. Roxadustát sa preto môže užívať s jedlom alebo bez jedla (pozri
časť 4.2).
DistribúciaRoxadustát sa vo veľkej miere viaže na ľudské proteíny v plazme (približne 99 %), prevažne na albumín. Pomer roxadustátu v krvi a v plazme je 0,6. Zdanlivý objem distribúcie v rovnovážnom stave je 24 l.
BiotransformáciaNa základe údajov
in vitro je roxadustát substrátom pre enzýmy CYP2C8 a UGT1A9 aj pre BCRP, OATP1B1, OAT1 a OAT3. Roxadustát nie je substrátom pre OATP1B3 alebo proteín P-gp. Roxadustát sa primárne metabolizuje na hydroxyroxadustát a roxadustát-
O-glukuronid. Nezmenený roxadustát bol hlavnou cirkulujúcou zložkou v ľudskej plazme, žiadny z detekovateľných metabolitov v ľudskej plazme nepredstavoval viac ako 10 % celkovej expozície materiálu súvisiacemu s liekom
a neboli pozorované žiadne metabolity špecifické pre ľudí.
ElimináciaPriemerný účinný polčas (t1/2) roxadustátu je približne 15 hodín u pacientov s CKD.
Zdanlivý celkový telesný klírens (CL/F) roxadustátu je 1,1 l/h u pacientov s CKD, ktorí nie sú na
dialýze, a 1,4 l/h u pacientov s CKD na dialýze. Roxadustát a jeho metabolity nie sú významne odstránené hemodialýzou.
Keď bol rádioaktívne označený roxadustát podaný perorálne zdravým osobám, priemerná eliminácia rádioaktivity bola 96 % (50 % v stolici, 46 % v moči). V stolici sa 28 % dávky vylúčilo ako nezmenený roxadustát. Menej ako 2 % dávky sa vylúčili v moči ako nezmenený roxadustát.
Špecificképopulácie
Účinky veku, pohlavia, telesnej hmotnosti a rasy
Neboli pozorované žiadne klinicky relevantné rozdiely vo farmakokinetike roxadustátu na základe
veku (≥ 18 rokov), pohlavia, rasy, telesnej hmotnosti, funkcie obličiek (eGFR) alebo stavu dialýzy
u dospelých pacientov s anémiou spôsobenou CKD.
Hemodialýza
U pacientov s CKD závislých od dialýzy neboli pozorované žiadne výrazné rozdiely hodnôt farmakokinetických parametrov pri podaní roxadustátu 2 hodiny pred alebo 1 hodinu po hemodialýze.
Dialýza je zanedbateľná cesta celkového klírensu roxadustátu.
Porucha funkcie pečene
Po podaní jednorazovej dávky 100 mg roxadustátu bola priemerná AUC o 23 % vyššia a priemerná koncentrácia Cmax bola o 16 % nižšia u osôb s miernou poruchou funkcie pečene (Childova-Pughova trieda B) a normálnou funkciou obličiek v porovnaní s osobami s normálnou funkciou pečene
a obličiek. U osôb so strednou poruchou funkcie pečene (Childova-Pughova trieda B) a normálnou
funkciou obličiek sa prejavilo zvýšenie AUCinf neviazaného roxadustátu (+70 %) v porovnaní so zdravými osobami.
Farmakokinetika roxadustátu u osôb s ťažkou poruchou funkcie pečene (Childova-Pughova trieda C)
nebola študovaná.
Liekové interakcie
Na základe údajov in vitro je roxadustát inhibítorom CYP2C8, BCRP, OATP1B1 a OAT3 (pozri
časť 4.5). Farmakokinetika rosiglitazónu (mierne citlivý substrát CYP2C8) nebola ovplyvnená súbežným podávaním roxadustátu. Roxadustát môže byť inhibítorom UGT1A1 v črevách, ale nie v pečeni. Roxadustát nevykázal žiadnu inhibíciu iných metabolizujúcich CYP enzýmov alebo
prenášačov ani indukciu enzýmov CYP v klinicky významných koncentráciách. Neexistuje žiadny klinicky významný účinok perorálne podávaného aktívneho uhlia alebo omeprazolu na
farmakokinetiku roxadustátu. Klopidogrel nemá žiadny účinok na expozíciu roxadustátu u pacientov s CKD.
5.3 Predklinické údaje o bezpečnosti
Štúdie toxicity po opakovanej dávke
V 26-týždňovej štúdii s prerušovanou opakovanou dávkou roxadustátu potkanom druhu
Sprague-Dawley alebo Fisher vo výške približne 4- až 6-násobku celkovej AUC pri maximálnej
odporúčanej dávke pre ľudí (Maximum Recommended Human Dose, MRHD) boli výsledkom histopatologické nálezy vrátane valvulopatií aortálnych a atrioventrikulárnych chlopní (A-V). Tieto zistenia boli prítomné u zvierat, ktoré v čase ukončenia štúdie žili, ako aj u zvierat, ktorých účasť sa ukončila predčasne z dôvodu uhynutia. Tieto zistenia navyše neboli úplné zvratné, keďže boli prítomné aj u zvierat na konci 30-dňového obdobia zotavenia.
V štúdiách toxicity s opakovanými dávkami so zdravými zvieratami bol pozorovaný nadmerný farmakologický účinok, ktorý mal za následok nadmernú erytropoézu.
U potkanov boli zaznamenané hematologické zmeny, ako napr. zníženia počtu cirkulujúcich trombocytov a zvýšenia aktivovaného parciálneho tromboplastínového času a protrombínového času, od približne 2-násobku hodnôt celkovej AUC pri MRHD. Tromby boli zaznamenané v kostnej dreni (systémové expozície vo výške približne 7-násobku celkovej AUC pri MRHD u potkanov), obličkách (systémové expozície vo výške približne 5- až 6-násobku celkovej AUC pri MRHD u potkanov), pľúcach (systémové expozície približne vo výške 8-násobku celkovej AUC pri MRHD u potkanov
a 2-násobku celkovej AUC pri MRHD u makakov dlhochvostých) a v srdci (systémové expozície vo
výške približne 4- až 6-násobku celkovej AUC pri MRHD u potkanov).
Bezpečnosť mozgu
V 26-týždňovej štúdii s prerušovanou opakovanou dávkou u potkanov druhu Sprague-Dawley pri
približne 6-násobku celkovej AUC pri MRHD sa u jedného zvieraťa vyskytli histologické nálezy mozgovej nekrózy a gliózy. U potkanov druhu Fisher, liečených rovnakú dobu, bola zaznamenaná nekróza mozgu/hipokampu celkovo u štyroch zvierat pri približne 3- až 5-násobku celkovej AUC pri MRHD.
U makakov dlhochvostých, ktorým bol prerušovane podávaný roxadustát v období 22 alebo
52 týždňov, sa nevyskytli podobné zistenia pri systémových expozíciách až do približne 2-násobku celkovej AUC pri MRHD.
Karcinogenita a mutagenita
Roxadustát bol negatívny v in vitro Amesovom teste mutagenity, in vitro teste chromozómových aberácií v lymfocytoch ľudskej periférnej krvi a in vivo mikronukleovom teste u myší s dávkou vo výške 40-násobku MRHD na základe ekvivalentnej dávky pre ľudí.
V štúdiách karcinogenity na myšiach a potkanoch bol zvieratám podaný roxadustát s klinickým režimom dávkovania trikrát týždenne. Z dôvodu rýchleho klírensu roxadustátu u hlodavcov neboli systémové expozície počas obdobia podávania kontinuálne. Z toho dôvodu môžu byť možné mimocieľové karcinogénne účinky podcenené.
V 2-ročnej štúdii karcinogenity na myšiach boli zaznamenané významné zvýšenia výskytu pľúcneho bronchoalveolárneho karcinómu v skupinách, kde bola podávaná nízka a vysoká dávka (systémové expozície vo výške približne 1-násobku a približne 3-násobku celkovej AUC pri MRHD). U samíc
v skupine s vysokou dávkou bol pozorovaný významný nárast výskytu podkožného fibrosarkómu
(systémové expozície vo výške približne 3-násobku celkovej AUC pri MRHD).
V 2-ročnej štúdii karcinogenity na potkanoch bolo zaznamenané významné zvýšenie výskytu adenómu mliečnych žliaz v skupine, kde bola podávaná stredne vysoká dávka (systémová expozícia vo výške menej ako 1-násobku celkovej AUC pri MRHD). Toto zistenie však nemalo súvislosť
s dávkou a výskyt tohto typu tumoru bol nižší v testovanej skupine, kde bola podávaná najvyššia dávka (systémová expozícia vo výške približne 2-násobku celkovej AUC pri MRDH), preto sa
nepovažovalo za súvisiace s predmetom testovania.
Podobné zistenia zo štúdií karcinogenity u myší a potkanov neboli pozorované v klinických štúdiách.
Reprodukčná a vývojová toxicita
Roxadustát nemal žiadne účinky na párenie ani fertilitu liečených samcov alebo samíc potkanov, ktorým bola podávaná dávka vo výške približne 4-násobku ľudskej expozície pri MRHD. Na úrovni
hladiny, pri ktorej nebol pozorovaný žiadny nežiaduci účinok (NOAEL), sa však u samcov potkanov vyskytli poklesy hmotnosti nadsemenníkov a semenných mechúrikov (s tekutinou) bez účinkov na
fertilitu samca. Hladina, pri ktorej nebol pozorovaný žiadny účinok (NOAEL), pre zistenia týkajúce sa
akéhokoľvek samčieho reprodukčného orgánu, bola 1,6-násobok MRHD. U samíc potkanov sa zistili
zvýšenia počtu neživých embryí a postimplantačné straty pri tejto úrovni dávky v porovnaní s kontrolnými zvieratami.
Výsledky zo štúdií reprodukčnej a vývojovej toxicity na potkanoch a králikoch preukázali zníženie priemernej telesnej hmotnosti plodu alebo mláďaťa, priemerné zvýšenie placentárnej hmotnosti, potrat a úmrtnosť mláďat.
U gravidných potkanov druhu Sprague-Dawley, ktorým bol podávaný roxadustát denne od implantácie až do uzatvorenia tvrdého podnebia (7. – 17. gestačný deň), sa vyskytol pokles telesnej hmotnosti plodu a zvýšené zmeny skeletu pri výške približne 6-násobku celkovej AUC pri MRHD. Roxadustát nemal žiadny účinok na prežitie plodu po implantácii.
Gravidným novozélandským králikom bol podávaný roxadustát denne od 7. do 19. gestačného dňa
a v 29. gestačný deň bol vykonaný cisársky rez. Podávanie roxadustátu pri systémových expozíciách až do výšky približne 3-násobku celkovej AUC pri MRHD neukázalo žiadne zistenia na embryu alebo plode. Jedna samica však potratila pri dávke vo výške približne 1-násobku celkovej AUC pri MRHD
a 2 samice potratili pri dávke vo výške približne 3-násobku celkovej AUC pri MRHD, samice, ktoré potratili, boli vychudnuté.
V štúdii prenatálneho/postnatálneho vývoja u potkanov druhu Sprague-Dawley bol tehotným
samiciam podávaný roxadustát denne od 7. gestačného dňa do 20. dňa laktácie. Počas obdobia laktácie sa u mláďat od samíc, ktorým bol podávaný roxadustát vo výške približne 2-násobku celkovej koncentrácie Cmax pri MRHD, vyskytla vysoká mortalita počas obdobia pred odstavením a pri odstavení boli utratené. U mláďat od samíc, ktorým bol podávaný roxadustát v dávkach vedúcich
k systémovej expozícii vo výške približne 3-násobku ľudskej expozície pri MRHD, sa vyskytlo významné zníženie 21-dňového prežitia po pôrode (laktačný index) v porovnaní s mláďatami
z kontrolných vrhov.
V tzv. cross-fostering štúdii (v ktorej sa odobrali mláďatá vlastným matkám) boli najvýraznejšie účinky na životaschopnosť mláďat potkanov zaznamenané u mláďat vystavených roxadustátu iba postnatálne. Životaschopnosť mláďat vystavených roxadustátu do pôrodu bola nižšie ako u mláďat, ktoré neboli roxadustátu vystavené.
V štúdii cross-fostering, v ktorej mláďatá potkanov nevystavených roxadustátu boli adoptované samicami, ktorým bol podaný roxadustát (ekvivalent ľudskej dávky vo výške približne 2-násobku MRHD), bol roxadustát prítomný v plazme mláďat, čo naznačuje prenos lieku mliekom. V mlieku týchto samíc bol prítomný roxadustát. U mláďat, ktoré boli vystavené mlieku, v ktorom bol prítomný roxadustát, bola nižšia miera prežitia (85,1 %) v porovnaní s mláďatami samíc, ktorým nebol roxadustát podaný, ktoré boli adoptované samicami, ktorým nebol roxadustát podaný (miera prežitia
98,5 %). Priemerná telesná hmotnosť prežívajúcich mláďat vystavených roxadustátu počas obdobia laktácie bola tiež nižšia ako u kontrolných mláďat (žiadna expozícia in utero – žiadna expozícia
v mlieku).
Kardiovaskulárna bezpečnosť
Farmakologická štúdia kardiovaskulárnej bezpečnosti ukázala zvýšenie srdcovej frekvencie po jednorazovom podaní 100 mg/kg roxadustátu opiciam. Nebol pozorovaný žiadny účinok na hERG alebo EKG. Doplnkové farmakologické štúdie bezpečnosti u potkanov ukázali, že roxadustát znížil celkovú periférnu rezistenciu po reflexnom zvýšení srdcovej frekvencie od výšky približne 6-násobku expozície pri MRHD.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
monohydrát laktózy mikrokryštalická celulóza (E460 (i)) sodná soľ kroskarmelózy (E468) povidón (E1201)
stearát horečnatý (E470b)
Filmový obal polyvinylalkohol (E1203) mastenec (E553b) makrogol (E1521)
červeň Allura AC, hliníkový lak (E129)
oxid titaničitý (E171)
lecitín (sójový) (E322)
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti4 roky
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaPVC/hliníkový perforovaný blister s jednotlivými dávkami v škatuľke obsahujúcej 12 x 1 filmom obalených tabliet.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky na likvidáciu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAstellas Pharma Europe B.V. Sylviusweg 62
2333 BE Leiden
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/21/1574/001
EU/1/21/1574/002
EU/1/21/1574/003
EU/1/21/1574/004
EU/1/21/1574/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu